

Abstract
Tamoxifen is recommended as a first line treatment for estrogen receptor positive breast cancer. However, the acquisition of endocrine resistance remains the biggest hurdle to achieving treatment success. We, therefore, designed the present study to disclose the relationship between autophagy and endocrine resistance and to provide some insight into overcoming tamoxifen resistance. Experiments were performed using TAM-sensitive (MCF-7) cell lines and TAM-resistant (TAM-R) cell lines. Western blot, real-time PCR, and immunofluorescence analyses were conducted to detect autophagy and apoptosis related proteins and to evaluate pathways that stimulated autophagy in the two targeted cell lines. Higher LC3 and Beclin-1 levels were found in the TAM-R cell lines compared with the MCF-7 cell lines, suggesting that the degree of autophagy was higher in the TAM-R cells. Other proteins kinases such as pAMPK, BAX, and p-p70S6K also proved the involvement of autophagy in the process of developing tamoxifen resistance. Lower levels of microRNA-101 were detected in the TAM-R cells, indicating a negative correlation between microRNA-101 and autophagy. Based on the findings presented in this study, autophagy is a major cause of tamoxifen resistance in breast cancer patients. Inhibiting autophagy could improve the therapeutic efficacy of TAM by overcoming endocrine resistance in estrogen receptor positive breast cancers.
Keywords: Autophagy, acquired resistance, tamoxifen, breast cancers
Introduction
Breast cancer is reported to be the second highest killer among all cancers in women, with up to a 30% death rate in diagnosed patients [1,2]. It is estimated that approximately 1.2 million newly diagnosed patients appear each year, with an increased occurrence in younger-aged women [3,4]. The expression of hormone receptors is a threshold for categorizing breast cancer into estrogen receptor (ER), progesterone receptor (PR), and human epidermal growth receptor 2 (HER2). The treatment options for breast cancer patients are varied according to the breast cancer phenotypes. Since almost 70% of patients express ER, endocrine therapies targeting estrogen and its receptor are the most common strategy used in treating breast cancer [5].
Tamoxifen (TAM), as an antiestrogen agent, has been considered the “gold standard” and been used as a first-line endocrine therapy since it was approved by the Food and Drug Administration [6]. The function of TAM is to competitively bind to ERs, thereby inhibiting the proliferation of estrogen-stimulated breast cancer cells [7]. The application of TAM therapy has been proved to be effective by reducing the risk of recurrence and death [8]. However, obstacles and limitations are still in the blocking the hope of achieving the best possible performance of TAM. Data show that around one third of patients do not respond to TAM treatment at the very beginning, and the majority of patients with an initial response develop resistance over time [9]. Several mechanisms responsible for TAM resistance have been explored, such as pharmacologic mechanisms, loss or modification in ER expression, regulation of the various signaling pathways that engage in different cellular processes, and the inhibition of apoptosis [10]. Recently, accumulating evidence suggests that the induction of autophagy in breast cancer cells is related to the development of therapeutic resistance [11,12].
Autophagy is a conserved process initiated as a response to stress or nutrient deprivation conditions in an attempt to maintain metabolic homeostasis by degrading damaged or superfluous proteins and subcellular organelles through delivery to lysosomes [13,14]. Autophagy, with its dual role as either pro-survival or pro-death, on the one hand, acts as a tumor suppressor; on the other hand, it delays and limits breast cancer cells death in response to stressors, thereby protecting the tumor cells [15-17]. There are five sequential steps that comprise the procedure of autophagy (induction, nucleation, elongation, maturation, and degradation) [13,14], and each stage is tightly controlled by specific complexes and involved with pathways [18]. Previous research has considered autophagy as an important mechanism of TAM resistance and demonstrated that the re-sensitization of breast cancer cells with acquired resistance can be achieved by inhibiting autophagy [11,19]. However, the exact mechanisms of protective autophagy and TAM resistant autophagy in breast cancer cells are still confusing and largely unknown. To better understand the correlation between autophagy and acquired resistance in breast cancer cell to TAM, we have compared the proteins related to the process of autophagy as well as the signaling pathways controlling autophagy between ER+ and estrogen/TAM-sensitive breast cancer cells (MCF-7) and TAM-resistant breast cancer cells (TAM-R).
Materials and methods
Materials
The MCF-7 and TAM-R cell lines for breast cancer we used were kind gifts from Professor Wei Yue (University of Virginia, VA, USA). Tamoxifen was purchased from Sigma (St. Louis, MO). The cell culture medium, Improved Minimum Essential Medium (IMEM), was obtained from Cellgro through Fisher Scientific (Pittsburgh, PA). The fetal bovine serum (FBS), glutamine, and trypsin were obtained from Invitrogen (Carlsbad, CA). The antibodies against LC3, Beclin-1, p62, Survivin, pAMPK, total AMPK, Bax, and p-p70S7K were obtained from Cell Signaling Technology (Beverly, MA). Near infrared dye conjugated secondary antibodies for the western blot analysis were purchased from LI-COR INC (Lincoln, NE). MitoTracker Red CMXRos, Hoechst 33324, and Alex Fluor 488 goat anti-rabbit IgG were obtained from Molecular Probes (Eugene, Oregon USA). All other chemicals were obtained from Sigma, unless indicated otherwise.
Cell culture
The culture medium IMEM containing 5% FBS was used to maintain the MCF-7 cells. TAM-R cells were cultured in the same medium as MCF-7 cells and further supplemented with 10-7 M tamoxifen.
Determination of cell number
MCF-7 cells and TAM-R cells were seeded in six-well plates at a density of 3 × 104 per well in 5% FBS IMEM. Tamoxifen was then added to both cell lines, and the cells were treated for 5 days. The medium was changed on day 3. After culturing, MCF-7 cells and TAM-R cells were rinsed twice with saline. Then, 2 mL HEPES MgCl2 solution (0.01 mol/L HEPES and 1.5 mmol/L MgCl2) and 0.2 mL ZAP solution [0.13 mol/L ethylhexadecyldimethylammonium bromide in 3% glacial acetic acid (v/v)] were sequentially added for the preparation of the nuclei. The number of cells in both cell lines was counted with a model Z1 Coulter counter.
Immunoblotting
MCF-7 cells and TAM-R cells were grown in 60-mm dishes with 5% FBS IMEM. Tamoxifen (10-7 and 10-6 M), and rapamycin (10 nM) were imported as vehicles in the culture medium when the cells were about 80% of confluence, and the cells were treated for 24 hours. Cells plated in 60-mm dishes were washed with phosphate buffered saline (PBS), incubated on ice for 5 min with 0.5 mL lysis buffer [20 mmol/L Tris (pH 7.5), 150 mmol/L NaCl, 1 mmol/L EDTA, 1 mmol/L EGTA, 1 mmol/L sodium orthovanadate, 2.5 mmol/L sodium pyrophosphate, 1% Triton X-100, 1 mmol/L β-glycerophosphate, 1 μg/mL leupeptin and aprotinin, and 1 mmol/L phenylmethylsulfonyl fluoride (PMSF)] pulse sonicated, and centrifuged at 14,000 rpm for 10 min at 4°C. Before the analysis, the cell lysates were stored at -80°C. The total content of the protein in the lysate was measured by a standard Bradford assay utilizing a reagent from Bio-Rad Laboratories (Hercules, CA). Fifty micrograms of total protein was extracted on a 10% Sodium dodecyl sulfate (SDS) polyacrylamide gel. Then, the protein was transferred to a nitrocellulose membrane. Primary antibodies (Cell Signaling) dissolved in tris-buffered saline (TBS) with 5% bovine serum albumin was added to the membrane followed by IRDye conjugated secondary antibody (Li-Cor Inc., Lincoln, NE) for incubation. An Odyssey imaging scanner (Li-Cor Inc., Lincoln, NE) was applied to visualize and quantify the protein bands.
Real time PCR analysis
The extraction and purification of the total RNA was conducted using Qiagen RNeasy Mini Kit. The expression of mature miR-101 was determined by the TaqMan miRNA-assay (Applied Biosystems, Foster City, CA, USA) following the manufacturer’s instructions. Relative miR-101 copies were compared using the DCt method. (There is no endogenous housekeeping control for this assay).
Immunofluorescent staining
MCF-7 and TAM-R cells were cultivated on a sterile glass cover slip in 6-well plates in IMEM-5% FBS. They were treated with rapamycin 10 nM and chloroquine 10 mM for 24 h. At the appropriate time, the cells were rinsed briefly with PBS after the culture medium was removed. Then they were fixed in PBS containing 4% formaldehyde for 15 min at room temperature. Later, the cells were rinsed 3 times with PBS and permeabilized with cold methanol for 10 min in a -20°C freezer. Afterwards, the cells were rinsed again with PBS for 5 min. The background was blocked in 5% normal goat serum in PBS/Triton for 1 h. The cells were incubated with a primary antibody against LC3 (1:200 in PBS/Triton) at 4°C overnight, then rinsed 3 times with PBS and incubated with a fluorochrome-conjugated secondary antibody (10 μg/ml in PBS/Triton) at room temperature for 1 h in the dark. Then Hoechst 33342 was added, and the cells were incubated at room temperature for 15 min in darkness. Finally, the cells were rinsed 3 times with PBS and covered with Fluoromount-G. The immunofluorescence staining was visualized using an Olympus (IX81) inverted fluorescence microscope.
Statistical analysis
All reported data are presented as the means ± SD. Statistical comparisons are conducted with two-tailed Student’s t tests. A P value < 0.05 is considered as statistically significant.
Results
Effect of tamoxifen on the growth of TAM-R and MCF-7 cells
To investigate whether autophagy participated in the drug resistance of TAM-R cells, we established a model where TAM-R cells manifested a stable resistance. A stepwise drug selection was applied, and comparisons of the cytotoxicity of TAM between the TAM-R and MCF-7 cells were performed to verify the efficacy of the established models. According to the cell count, the cell death caused by TAM in both TAM-R and MCF-7 cells was dose-dependent. At the concentration of 10-10 to 10-8, TAM only exhibited a weak inhibitory effect on TAM-R and MCF-7 cells. Tam at a concentration of 10-7 to 10-6 induced significant cell death in the MCF-7 cells; however, cell death in the TAM-R cells did not reach statistical significance until TAM reached 10-6. These results further proved that TAM-R is resistant to TAM (Figure 1).
Figure 1.

The effect of tamoxifen (5 days) on the growth of TAM-R and MCF-7 cells. The TAM-R and MCF-7 cells were treated with tamoxifen for 5 days in IMEM-5% FBS 5% DCC; seeding cells number: 3*10^4/well in 6-well plate.
The effects of tamoxifen on autophagy markers in MCF-7 and TAM-R cells
Autophagy was deemed to induce antiestrogen resistance in breast cancer cells [11]. Thus, a comparison was performed regarding Tam induced autophagy between the drug-resistance cell lines and the wild-type cell lines. Endogenous LC3 can be used to measure the induction of autophagy. It is suggested that LC3 is the first mammalian protein that is recruited to the autophagosome membranes and involved in the formation of autophagosome [20]. A Western blot analysis revealed higher levels of LC3 in the TAM-R cells compared with the MCF-7 cells in the TAM free environment (control), suggesting a process of acquired resistance is involved with autophagy (Figure 2A). The interference high-dose TAM (10-6) brought a lower expression of LC3, which inhibits autophagy. Immunofluorescent staining localizing the LC3 protein in TAM-R and MCF-7 cells further confirmed the presence of autophagy. The localization of LC3 appeared to be diffuse in the control MCF-7 cells under immunofluorescence microscopy. However, the expression of LC3 in the TAM-R cells was markedly higher than it was in the MCF-7 cells (Figure 3). The level of p62 is also an indicator for autophagy turnover, as the p62 protein directly binds to LC3 and is degraded by autophagy [21]. Evidence has shown that the inhibition of autophagy leads to the accumulation of p62. However, from our analysis, the basal level of p62 is higher in TAM-R than it is in MCF-7 (Figure 2B). This contradictory result may be a reflection of the reduction of autophagy when TAM-R cells were shifted from a routine tamoxifen containing medium to a vehicle containing medium. Another explanation could be that there is increased oxidative stress in the TAM-R cells because p62 is also involved in this process.
Figure 2.
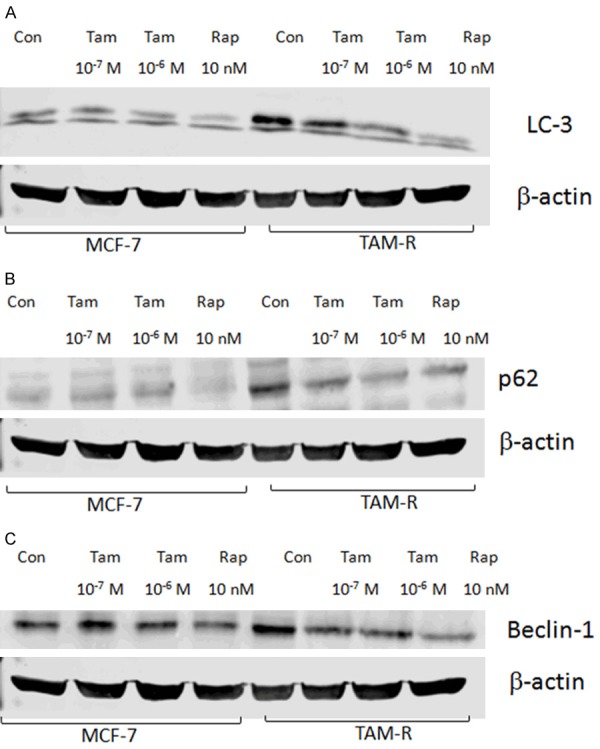
Protective autophagy in MCF-7 cells and TAM-R cells. MCF-7 cells and TAM-R cells were plated in 60-mm dishes with 5% FBS IMEM, and treated with tamoxifen (10-7 and 10-6 M), and rapamycin (10 nM) in a culture medium for 24 hours when the cells were about 80% of confluence before immunoblotting. The protein bands were visualized and quantified using an Odyssey imaging scanner. A: The effects of tamoxifen on autophagy marker LC-3 in MCF-7 and TAM-R cells; B: The effects of tamoxifen on autophagy marker Beclin-1 in the MCF-7 and TAM-R cells; C: The effects of tamoxifen on autophagy marker p62 in the MCF-7 and TAM-R cells.
Figure 3.
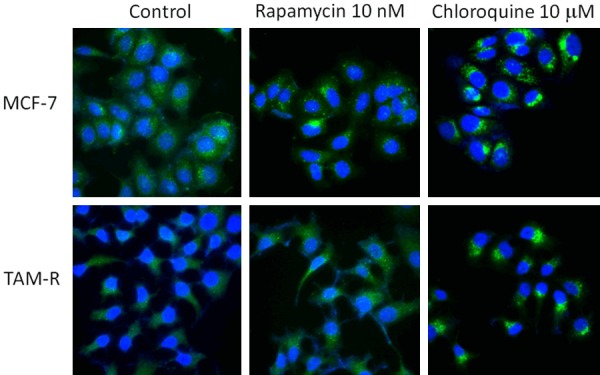
Autophagy fluorescent microscopy of LC-3 in MCF-7 and TAM-R cells in the control, rapamycin, and chloroquine. MCF-7 and TAM-R cells grown on sterile glass cover slip in 6-well plates in IMEM-5%FBS were treated with rapamycin 10 nM and chloroquine 10 mM for 24 h and then stained with CytoID green dye for autophagosome and Hoechst 33342 for nuclei. The immunofluorescence staining was visualized with an Olympus (IX81) inverted fluorescent microscope. Imaging: FITC and DAPI (60 ×).
Beclin-1 is one of the critical markers responsible for both autophagosome formation and autolysosome fusion [22]. A higher Beclin-1 level was detected in the TAM-R cells compared with the MCF-7 cells among the control, low-dose TAM, and high dose TAM groups, indicating a stronger autophagy in the TAM-R cells (Figure 2C).
Evidence of mTOR inhibition induces autophagy
As a highly conserved Ser/Thr protein kinase, AMPK can balance energy homeostasis and metabolic stress [23]. It is also involved in many basic biological processes, including cell growth, proliferation, apoptosis, autophagy [24]. Previous studies demonstrated AMPK could positively mediate autophagy through the regulation of the mammalian target of the rapamycin (mTOR) complex [25]. Numerous signaling pathways can trigger autophagy, and the PI3K/Akt-mTOR signaling pathway is regarded as a crucial negative regulator for the formation of autophagosomes [26]. The PI3K/Akt signaling pathway controls the activity of mTOR and activated mTOR can phosphorylate the ribosomal protein S6 kinase (P70S6K). Evidence has shown that AMPK activation may stimulate autophagy through the negative regulation of mTOR [14,27]. Western blot imaging from our analysis revealed that the TAM-R cells had higher basal levels of pAMPK (Figure 4A) and lower levels of p-p70S6K (Figure 4B) than the MCF-7 cells, indicating the exhibition of autophagy in TAM resistance. In the intervention of low-dose and high-dose TAM treatment, the expression of pAMPK was negatively correlated with p-p70S6K.
Figure 4.
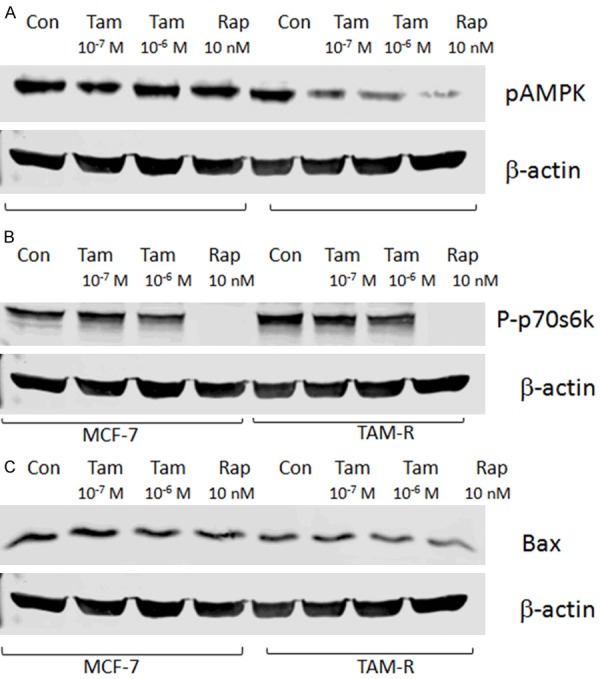
Tamoxifen therapy altered the autophagy and apoptosis markers in the MCF-7 and TAM-R cells. The MCF-7 and TAM-R cells were plated in 60-mm dishes with 5% FBS IMEM and treated with tamoxifen (10-7 and 10-6 M), and rapamycin (10 nM) in a culture medium for 24 hours when the cells were about 80% of confluence before western blotting. Bar graphs indicate the relative levels of pAMPK, Bax, and P-p70s6k normalized to β-actin. A: The effects of tamoxifen on the expression of pAMPK in the MCF-7 and TAM-R cells; B: The effects of tamoxifen on the expression of P-p70s6k in MCF-7 and TAM-R cells; C: The effects of tamoxifen on the expression of BAX in the MCF-7 and TAM-R cells.
Effects of tamoxifen on the expression of total Bax in MCF-7 and TAM-R cells
BAX is a pro-apoptotic agent, and its reduction may predispose it to enhance drug resistance in breast cancer [28]. A lower level of Bax was observed in the TAM-R cells, indicating Bax was involved in the acquired resistance process. The attendant reduction of Bax in the TAM-R cells with a different dose of TAM intervention further supported the hypothesis that the inhibition of Bax could indirectly affect autophagy (Figure 4C).
miR-101 in MCF-7 and TAM-R cells
miR-101 is considered a powerful inhibitor of autophagy. A previous study showed that the overexpression of miR-101 repressed autophagy [29]. In our study, real-time PCR revealed that miR-101 expression was lower in the Tam-R cell lines compared with the MCF-7 cell lines, which indicates that enhanced autophagy had appeared in the breast cancer cells which developed TAM resistance (Figure 5).
Figure 5.
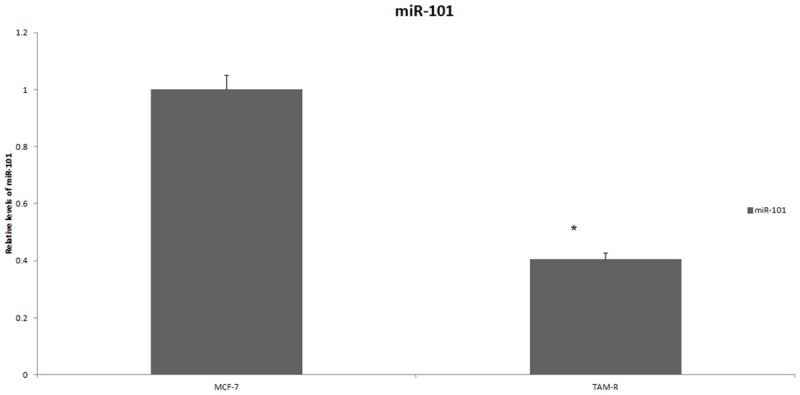
The expression of miR-101 in MCF-7 cells and TAM-R cells. Total RNA was extracted and purified using a Qiagen RNeasy Mini Kit. The expression of mature miR-101 was determined by the TaqMan miRNA-assay following the manufacturer’s instructions. Relative miR-101 copies were compared using the DCt method. *P < 0.05.
Discussion
While TAM is still the key to treating breast cancer patients, the acquisition of TAM resistance still is the biggest challenge hampering treatment success. Autophagy is considered to be one of the mechanisms causing endocrine therapy resistance. The protective and pro-survival mechanisms of autophagy were observed in previous investigations, and this mechanism is thought to play an antagonistic role against a few chemotherapeutics in human cancer cell lines [30]. A number of experiments also showed that the acquisition of TAM resistance in breast cancer cells is strongly correlated with autophagy [31]. However, a lot is still unknown regarding the relationship between autophagy and acquired resistance. Thus, we have designed the current experiment in an attempt to disclose the relevance of autophagy to TAM resistance by analyzing the related parameters.
LC3 is a key protein responsible for the major steps of autophagy. It is mainly localized in the nucleus, but it functions primarily in the cytoplasm where the autophagosomes and autolysosomes arise [32] and determines autophagosome size and membrane curvature [33]. A Western blot analysis showed higher expressions of LC-3 in TAM-R cells compared with the MCF-7 cell lines in all three groups (control, 10-7 M TAM, and 10-6 M TAM), indicating that the levels of autophagy were higher in the TAM-R cell lines. In high-dose TAM interference (10-6 M), when the cell growth of TAM-R cells was significantly inhibited, the LC3 levels subsequently dropped. This phenomenon further confirmed that autophagy was engaged in the acquired resistance process against TAM in breast cancer cells. Beclin-1, as the first identified autophagic gene in mammalian cells, can positively regulate autophagy [34]. Reports have shown that autophagy may be stimulated as the synthesis of beclin-1 is increased under treatment with TAM [35]. According to our result, the TAM-R cell lines contained higher beclin-1 levels than the MCF-7 cell lines in all three groups, suggesting that the induction of autophagy involves the regulation of beclin-1. The ability to interact with proteins in certain intracellular signaling pathways makes p62 important at the crossroads of autophagy, apoptosis, and cancer [36]. p62 is an adaptor protein that contributes to the formation of autophagosome, and it is also associated with LC3 and ubiquitinated proteins. Also, as a substrate of autophagy, p62 appears to be accumulative in autophagy-deficient cells. The highest p62 concentration clearly appeared in the control group of Tam-R cell lines which was contradictory with previous findings [37]. Many factors might have led to this discrepancy. One possible explanation is that the shift from a routine tamoxifen containing medium to a vehicle containing medium may cause a reduction of autophagy. Another explanation could be that there is increased oxidative stress in TAM-R cells because p62 is also involved in this process. Protein kinase AMPK and P70S6K were also evaluated, and the results demonstrated that levels of pAMPK and p-p70S6K were connected to the regulation of autophagy. The results also showed that the pro-apoptotic agent BAX and miR-101 may function as negative regulators of autophagy in the acquisition of TAM resistance.
It is important to reveal the correlation between autophagy and the acquired resistance of TAM therapy. By understanding which protein can positively or negatively regulate autophagy, clinicians can design specific treatments to inhibit endocrine resistance or resensitized resistance cells to therapy by modulating certain proteins. For example, by disclosing the relationship between miR-101 and autophagy, researchers can block autophagy by enhancing miR-101 in patients who acquire resistance to endocrine therapy to increase the therapeutic success.
In conclusion, the use of TAM chemotherapy could trigger protected autophagy in cancer cells. The development of drug resistance to TAM therapy is chiefly caused by autophagy. Inhibiting autophagy could improve the therapeutic effect of TAM by overcoming endocrine resistance in ER+ breast cancers.
Acknowledgements
The study was supported by the Young Scientists Fund of the National Natural Science Foundation of China (no. 81503302), and Beijing University of Traditional Chinese Medicine, 1166 Development Program for Junior Scientists.
Disclosure of conflict of interest
None.
References
- 1.Jemal A, Siegel R, Xu J, Ward E. Cancer statistics, 2010. CA Cancer J Clin. 2015;60:277–300. doi: 10.3322/caac.20073. [DOI] [PubMed] [Google Scholar]
- 2.Montero AJ, Eapen S, Gorin B, Adler P. The economic burden of metastatic breast cancer: a U.S. managed care perspective. Breast Cancer Res Treat. 2012;134:815–822. doi: 10.1007/s10549-012-2097-2. [DOI] [PubMed] [Google Scholar]
- 3.Vergne Y, Matta J, Morales L, Vargas W, Alvarez-Garriga C, Bayona M. Breast cancer and DNA repair capacity: association with use of multivitamin and calcium supplements. Integr Med. 2013;12:38–46. [PMC free article] [PubMed] [Google Scholar]
- 4.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2016. CA Cancer J Clin. 2015;60:277–300. [Google Scholar]
- 5.Cardoso F, Bischoff J, Brain E, Zotano ÁG, Lück HJ, Tjan-Heijnen VC, Tanner M, Aapro M. A review of the treatment of endocrine responsive metastatic breast cancer in postmenopausal women. Cancer Treat Rev. 2013;39:457–465. doi: 10.1016/j.ctrv.2012.06.011. [DOI] [PubMed] [Google Scholar]
- 6.Clarke M, Collins R, Davies C. Tamoxifen for early breast cancer: an overview of the randomised trials. Lancet. 1998;351:1451–1467. [PubMed] [Google Scholar]
- 7.Mandlekar S, Kong AN. Mechanisms of tamoxifen-induced apoptosis. Apoptosis. 2001;6:469–477. doi: 10.1023/a:1012437607881. [DOI] [PubMed] [Google Scholar]
- 8.Vogel VG, Costantino JP, Wickerham DL, Cronin WM. Tamoxifen for prevention of breast cancer: report of the national surgical adjuvant breast and bowel project P-1 study. J Natl Cancer Inst. 2002;94:1504. doi: 10.1093/jnci/94.19.1504. [DOI] [PubMed] [Google Scholar]
- 9.Riggins RB, Schrecengost RS, Guerrero MS, Bouton AH. Pathways to tamoxifen resistance. Cancer Lett. 2007;256:1–24. doi: 10.1016/j.canlet.2007.03.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Viedma-Rodríguez R, Baiza-Gutman L, Salamanca-Gómez F, Diaz-Zaragoza M, Martínez-Hernández G, Ruiz Esparza-Garrido R, Velázquez-Flores MA, Arenas-Aranda D. Mechanisms associated with resistance to tamoxifen in estrogen receptor-positive breast cancer (review) Oncol Rep. 2014;32:3–15. doi: 10.3892/or.2014.3190. [DOI] [PubMed] [Google Scholar]
- 11.Samaddar JS, Gaddy VT, Duplantier J, Thandavan SP, Shah M, Smith MJ, Browning D, Rawson J, Smith SB, Barrett JT, Schoenlein PV. A role for macroautophagy in protection against 4-hydroxytamoxifen-induced cell death and the development of antiestrogen resistance. Mol Cancer Ther. 2008;7:2977–87. doi: 10.1158/1535-7163.MCT-08-0447. [DOI] [PubMed] [Google Scholar]
- 12.Yang ZJ, Cheng EC, Huang S, Sinicrope FA. The role of autophagy in cancer: therapeutic implications. Mol Cancer Ther. 2011;10:1533–1541. doi: 10.1158/1535-7163.MCT-11-0047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Antonioli M, Di RM, Piacentini M, Fimia GM. Emerging mechanisms in initiating and terminating autophagy. Trends Biochem Sci. 2016;42:28–41. doi: 10.1016/j.tibs.2016.09.008. [DOI] [PubMed] [Google Scholar]
- 14.He C, Klionsky DJ. Regulation mechanisms and signaling pathways of autophagy. Annu Rev Genet. 2009;43:67–93. doi: 10.1146/annurev-genet-102808-114910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Abedin MJ, Wang D, Mcdonnell MA, Lehmann U, Kelekar A. Autophagy delays apoptotic death in breast cancer cells following DNA damage. Cell Death Differ. 2007;14:500–510. doi: 10.1038/sj.cdd.4402039. [DOI] [PubMed] [Google Scholar]
- 16.Degenhardt K, Mathew R, Beaudoin B, Bray K, Anderson D, Chen G, Mukherjee C, Shi Y, Gélinas C, Fan Y. Autophagy promotes tumor cell survival and restricts necrosis, inflammation, and tumorigenesis. Cancer Cell. 2006;10:51–64. doi: 10.1016/j.ccr.2006.06.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bursch W, Hochegger K, Torok L, Marian B, Ellinger A, Hermann RS. Autophagic and apoptotic types of programmed cell death exhibit different fates of cytoskeletal filaments. J Cell Sci. 2000;113:1189–1198. doi: 10.1242/jcs.113.7.1189. [DOI] [PubMed] [Google Scholar]
- 18.Meijer AJ, Codogno P. Autophagy: regulation and role in disease. Crit Rev Clin Lab Sci. 2009;46:210–40. doi: 10.1080/10408360903044068. [DOI] [PubMed] [Google Scholar]
- 19.Qadir MA, Kwok B, Dragowska WH, To KH, Le D, Bally MB, Gorski SM. Macroautophagy inhibition sensitizes tamoxifen-resistant breast cancer cells and enhances mitochondrial depolarization. Breast Cancer Res Treat. 2008;112:389–403. doi: 10.1007/s10549-007-9873-4. [DOI] [PubMed] [Google Scholar]
- 20.Kabeya Y, Mizushima N, Ueno T, Yamamoto A, Kirisako T, Noda T, Kominami E, Ohsumi Y, Yoshimori T. LC3, a mammalian homologue of yeast Apg8p, is localized in autophagosome membranes after processing. EMBO J. 2000;19:5720–8. doi: 10.1093/emboj/19.21.5720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geir B, Trond L, Andreas B, Heidi O, Maria P, Aud Ø, Harald S, Terje J. p62/SQSTM1 forms protein aggregates degraded by autophagy and has a protective effect on huntingtin-induced cell death. J Cell Biol. 2005;171:603–14. doi: 10.1083/jcb.200507002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kihara A, Noda T, Ishihara N, Ohsumi Y. Two distinct Vps34 phosphatidylinositol 3-kinase complexes function in autophagy and carboxypeptidase Y sorting in saccharomyces cerevisiae. J Cell Biol. 2001;152:519–30. doi: 10.1083/jcb.152.3.519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hardie DG, Ashford ML. AMPK: regulating energy balance at the cellular and whole body levels. Physiology. 2014;29:99–107. doi: 10.1152/physiol.00050.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Song X, Kim SY, Zhang L, Tang D, Bartlett DL, Kwon YT, Lee YJ. Role of AMP-activated protein kinase in cross-talk between apoptosis and autophagy in human colon cancer. Cell Death Dis. 2014;5:e1504. doi: 10.1038/cddis.2014.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dunlop EA, Tee AR. The kinase triad, AMPK, mTORC1 and ULK1, maintains energy and nutrient homoeostasis. Biochem Soc Trans. 2013;41:939–943. doi: 10.1042/BST20130030. [DOI] [PubMed] [Google Scholar]
- 26.Yang Z, Klionsky DJ. Mammalian autophagy: core molecular machinery and signaling regulation. Curr Opin Cell Biol. 2010;22:124–31. doi: 10.1016/j.ceb.2009.11.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Papandreou I, Lim AL, Laderoute K, Denko NC. Hypoxia signals autophagy in tumor cells via AMPK activity, independent of HIF-1, BNIP3, and BNIP3L. Cell Death Differ. 2008;15:1572–1581. doi: 10.1038/cdd.2008.84. [DOI] [PubMed] [Google Scholar]
- 28.Tewari M, Krishnamurthy A, Shukla HS. Predictive markers of response to neoadjuvant chemotherapy in breast cancer. Surg Oncol. 2008;17:301–311. doi: 10.1016/j.suronc.2008.03.003. [DOI] [PubMed] [Google Scholar]
- 29.Frankel LB, Wen J, Lees M, Høyer-Hansen M, Farkas T, Krogh A, Jäättelä M, Lund AH. microRNA-101 is a potent inhibitor of autophagy. EMBO J. 2014;30:4628–4641. doi: 10.1038/emboj.2011.331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dalby KN, Tekedereli I, Lopez-Berestein G, Ozpolat B. Targeting the prodeath and prosurvival functions of autophagy as novel therapeutic strategies in cancer. Autophagy. 2010;6:322–329. doi: 10.4161/auto.6.3.11625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schoenlein PV, Periyasamy-Thandavan S, Samaddar JS, Jackson WH, Barrett JT. Autophagy facilitates the progression of ERalpha-positive breast cancer cells to antiestrogen resistance. Autophagy. 2009;5:400–403. doi: 10.4161/auto.5.3.7784. [DOI] [PubMed] [Google Scholar]
- 32.Huang R, Liu W. Identifying an essential role of nuclear LC3 for autophagy. Autophagy. 2015;11:852–853. doi: 10.1080/15548627.2015.1038016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Cook KL, Shajahan AN, Clarke R. Autophagy and endocrine resistance in breast cancer. Expert Rev Anticancer Ther. 2011;11:1283–94. doi: 10.1586/era.11.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Fu LL, Cheng Y, Liu B. Beclin-1: autophagic regulator and therapeutic target in cancer. Int J Biochem Cell Biol. 2013;45:921–924. doi: 10.1016/j.biocel.2013.02.007. [DOI] [PubMed] [Google Scholar]
- 35.Wienecke R, Fackler I, Linsenmaier U, Mayer K, Licht T, Kretzler M. Antitumoral activity of rapamycin in renal angiomyolipoma associated with tuberous sclerosis complex. Am J Kidney Dis. 2006;48:e27–9. doi: 10.1053/j.ajkd.2006.05.018. [DOI] [PubMed] [Google Scholar]
- 36.Wei H, Wang C, Croce CM, Guan JL. p62/SQSTM1 synergizes with autophagy for tumor growth in vivo. Genes Dev. 2014;28:1204–1216. doi: 10.1101/gad.237354.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lamark T, Svenning S, Johansen T. Regulation of selective autophagy: the p62/SQSTM1 paradigm. Essays Biochem. 2017;61:609–624. doi: 10.1042/EBC20170035. [DOI] [PubMed] [Google Scholar]