

Abstract
Hypertrophic scars are proliferative diseases of dermal fibroblasts that produce abundant amounts of collagen and extracellular matrix in the skin after severe burns, inflammation and trauma. Hypertrophic scars affect the daily life of patients and cause a series of problems. The biological mechanism of hypertrophic scar formation is still unclear and has received much attention in plastic surgery. Therefore, we hypothesized that LPS can activate TLR4 signaling, leading to the overexpression of collagen I and TGF-β and the induction of hypertrophic scar formation. In the present study, we used LPS to validate the role of the TLR4 signaling pathway in 3T3-L1 cells in vitro and hypertrophic scar mouse models to determine the role of the TLR4 signaling pathway in proliferative scar formation in vivo. The results suggested that LPS leads to the activation of the TLR4 pathway in fibroblasts, and inhibitor experiments confirmed that TLR4 is involved in the expression of collagen I by regulating the NF-κB pathway. The mouse skin wound model experiments demonstrated that TLR4 is involved in wound healing and scar formation. Our experiments demonstrated that the TLR4-IRAK4-NF-κB pathway is involved in the production of hypertrophic scars and wound healing.
Keywords: TLR4, LPS, fibroblast, scar formation
Introduction
Hypertrophic scars are dermal fibroblast proliferative disorders that are characterized by highly abundant collagen and extracellular matrix (ECM) after severe burns, inflammation and trauma [1]. Hypertrophic scars can increase morbidity that affects the daily lives of patients and can also lead to psychological issues, such as anxiety or depression. The biological mechanism of hypertrophic scar formation remains unclear and has gained much attention in plastic surgery [2]. Therefore, the specific mechanism needs to be fully understood, and effective management strategies to inhibit hypertrophic scar formation should be used. Excess collagen synthesis by fibroblasts is considered a key feature of the hypertrophic scar formation process [3]. Therefore, regulating the proliferation of fibroblasts, reducing the formation of collagen and accelerating the decomposition of collagen in scar tissue have been the main research strategies. Recent studies have shown that fibroblasts regulate the fibrotic process through two signaling pathways, CD 40/CD 40L signaling [4] and toll-like receptor (TLR) signaling [5].
TLRs are transmembrane proteins that are located on the cell membrane and have been shown to recognize endogenous ligands or exogenous pathogens. TLR4 can activate nuclear factor-κB (NF-κB) through myeloid differentiation factor 88 (MYD88)-dependent or non-MYD88-dependent pathways leading to cytokine transcription and costimulatory molecule expression. Recent observations have indicated the upregulation of toll-like receptor 4 (TLR4) in a hypertrophic scar model [6]. TGF-β-mediated signaling plays an important role in the fibrotic process, and lipopolysaccharide (LPS) can activate TGF-β, leading to hepatic fibrosis. However, the effect of TLR4 signaling activation on fibroblast fibrosis warrants further research.
Therefore, we hypothesized that LPS can activate TLR4 signaling, leading to collagen I and TGF-β overexpression, and can induce hypertrophic scar formation. In this study, we used LPS to verify the role of the TLR4 signaling pathway in 3T3-L1 cells in vitro and in a hypertrophic scar mouse model to determine the role of the TLR4 signaling pathway in the process of hypertrophic scar formation in vivo.
Materials and methods
Materials
LPS was purchased from Sigma-Aldrich (St. Louis, MO, USA). Dulbecco’s-modified Eagle’s medium (DMEM) and fetal bovine serum were purchased from Gibco (Carlsbad, CA, USA). Anti-TLR4, anti-Myd88, anti-IRAK4, anti-NF-κB and anti-phospho-NF-κB p65 antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). An anti-beta-actin antibody was purchased from Beijing Ray Antibody Biotech (Beijing, China). Goat anti-mouse and anti-rabbit IgG (H+L) HRP-conjugated antibodies were purchased from Dingguo (Beijing, China). Anti-rabbit and anti-mouse IgG (H+L) Alexa Fluor 555-conjugated antibodies were purchased from Cell Signaling Technology (Danvers, MA, USA). An anti-collagen I antibody was purchased from Santa Cruz Biotechnology, Inc. An anti-TGF-beta antibody was purchased from Bioss Technology (Beijing, China). TAK-242 (C15H17ClFNO4S) was purchased from Sigma-Aldrich (St. Louis, MO, USA). A Super ECL Assay was purchased from Thermo Fisher Scientific (Waltham, MA, USA). The other reagents were purchased from Sigma-Aldrich unless specifically mentioned (St. Louis, MO, USA).
Cell culture
3T3-L1 cells, a mouse cell line, were purchased from the cell bank of Shanghai Institutes for Biological Sciences, Chinese Academy of Science (Shanghai, China). 3T3-L1 cells have a fibroblast-like morphology. The cells were cultured in DMEM medium with 10% fetal bovine serum and 50 units/ml penicillin G at 37°C in a humidified atmosphere with 5% CO2. The culture medium was changed every 1 or 2 days. The cells were passaged when they reached approximately 80 to 90% confluency.
LPS and TLR4 inhibitor treatment
When the 3T3-L1 cells reached approximately 80% confluency in 6-well plates, the culture medium was changed to serum-free medium, and the cells were stimulated with 0, 0.5, 1.0 or 2.0 mg/L LPS for 24 h. This concentration range was selected based on the LC25 results, and this concentration range is similar to those reported in other studies [7-9]. In the experiments with TAK-242, an inhibitor of TLR4, the cells were precultured for 3 h with 100 nM TAK-242 and then incubated with 2.0 mg/L LPS for 24 h. The concentration of TAK-242 was selected based on previous studies [10-13], and this concentration had similar inhibition effects in our study.
Western blotting
Cells and skin samples from mice were lysed in an ice-cold RIPA buffer with 1% PMSF protease inhibitor. Protein concentrations were determined using a BCA protein quantitative analysis kit. The samples were separated using a sodium dodecyl sulfate polyacrylamide gel (SDS-PAGE). Then, the gel was transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore, Billerica, MA, USA). Then, the PVDF membrane was incubated in a blocking buffer, which consisted of 5% nonfat milk in TBS-T buffer (Tris-buffered saline supplemented with 0.1% Tween 20), for 2 h at room temperature. After blocking, the membrane was incubated with primary antibodies at 4°C overnight. The membrane was washed three times in a TBS-T buffer and incubated with secondary antibodies at room temperature for 1 h. After washing three times with TBS-T, the membrane was incubated with an ECL assay, and chemiluminescent immunoreactivity complexes were collected using a Tanon imaging system (Shanghai, China). Beta-actin immunoreactivity was used as a control.
Immunofluorescence
The cells were grown on a confocal dish. When the cells reached confluency, the medium and debris were washed away using a PBS buffer, and then the cells were fixed with 4% paraformaldehyde for 10 min at room temperature. After washing three times with a PBS buffer, the cells were incubated with 5% BSA containing 0.5% Triton-100 for 30 min and then incubated with primary antibodies at 4°C overnight. After washing three times with a PBS buffer, the cells were incubated with secondary antibodies at room temperature for 30 min. DAPI staining was conducted for 5 min after washing with PBS. Then, the cells were observed and imaged using confocal microscopy.
Animal protocol
Six- to eight-week-old C57BL/6 male mice were obtained from the Laboratory Animal Center of Southern Medical University (Guangzhou, China) and were singly housed in tub cages in a temperature-controlled room with a 12 h light/dark cycle. All animal protocols in this study were approved by the Institutional Animal Care and Use Committee of Southern Medical University and followed the most recent NIH Guidelines for the Care and Use of Laboratory Animals (NIH, 2011). The mice were randomly divided into 3 groups (n = 3 mice/group) as follows: the saline control group, the wound group, and the wound + TAK-242 exposure group. The animals were habituated for 1 week in the animal facility before use. For the mice in the wound group, the skin was cut 1 cm on the side of the back of the spine with a scalpel, and a linear incision was made. To simulate the formation of hypertrophic scars, mechanical stretching devices were applied using sutures to cover the incisions. The devices were used for 10 days. In the inhibitor group, the mice were injected intraperitoneally (i.p.) with TAK-242 (five injections, 3 mg/kg/injection, at 24 h intervals) before surgery. Five days later, the wound group underwent their operations simultaneously. The control mice were not treated. After the wounds healed, all the mice were sacrificed by cervical dislocation, and the skin of the back scar was collected for analysis.
Hematoxylin and eosin (HE) staining and Masson’s trichrome staining
The skin tissue was fixed with 4% formaldehyde, dehydrated, embedded, and cut into 3 μm sections. The sections were placed in a 65°C oven to melt the paraffin, and the paraffin was removed by xylene. The sections were then rehydrated with absolute ethanol and 70% alcohol. The sections were placed in a hematoxylin staining solution for 5 min at room temperature, washed with water, digested with hydrochloric acid for 1 s, and rinsed with running water for 30 min to make the blue color more vivid. Then, the sections were placed in an eosin staining solution for 2 min, dehydrated with increasing concentrations of ethanol, made transparent with xylene, dried and sealed.
The initial steps of Masson’s trichrome staining were consistent with HE staining. After the sections were stained with hematoxylin, the cells were stained with Masson’s red acid solution for 5 min, temporarily immersed in 2% aqueous glacial acetic acid, and then differentiated with 1% aqueous phosphomolybdate for 3 min. Without washing, the sections were directly stained with aniline blue for 5 min and temporarily immersed in a 0.2% glacial acetic acid aqueous solution. Then, the sections were dehydrated with alcohol and anhydrous alcohol, made transparent with xylene, dried, and sealed.
Immunohistochemistry
The sections were rehydrated as mentioned in the HE staining procedure. The sections were placed in a citrate solution and the antigen was fixed in the microwave for 15 min. Notably, the solution should not be dried. After the sections and the solution reached room temperature, the sections were washed three times with PBS, and the sections were incubated with 10% H2O2 for 15 min. After washing with PBS, the sections were blocked with normal goat serum for 30 min at room temperature. Then, the cells were incubated with primary antibodies at 4°C overnight. After washing three times with PBS, the sections were incubated with secondary antibodies at room temperature for 30 min. After washing with PBS, the color was developed with diaminobenzidine (DAB) under a microscope. Then, the sections were dehydrated with alcohol and anhydrous alcohol, made transparent with xylene, dried and sealed.
Statistics
A statistical analysis of data was performed using the Bonferroni post hoc test based on a one-way analysis of variance (one-way ANOVA) using SPSS 19.0 software (SPSS Inc, Chicago, IL). The data were expressed as the mean ± SD, and statistical significance was set at P<0.05.
Results
LPS increases TLR4 and associated signaling pathway molecules in 3T3-L1 cells
To determine whether LPS affects TLR4 and its associated signaling molecules, 3T3-L1 cells were treated with a dose range (0.5-2 mg/L) of LPS for 24 h. The western blot results showed that LPS increased the expression levels of TLR4, IRAK4, MYD88 and P-NF-κB in a dose-dependent manner (Figure 1A and 1B). For example, the expression levels of TLR4, IRAK4, MYD88 and P-NF-κB proteins after 24 h of LPS exposure significantly increased by 2.9-fold, 3.6-fold, 4.5-fold and 2.2-fold, respectively (n=3, *P<0.05). However, IRAK4 and phosphor-NF-κB protein expression did not significantly change after 24 h of exposure to 0.5 mg/L LPS, nor did NF-κB protein expression by any concentration of LPS after 24 h of exposure. The immunofluorescence staining results showed that TLR4 protein expression levels increased in the LPS-treated cells (1 mg/L, 24 h; Figure 1C), but no change in NF-κB protein expression was observed in the treated cells (1 mg/L, 24 h; Figure 1D).
Figure 1.
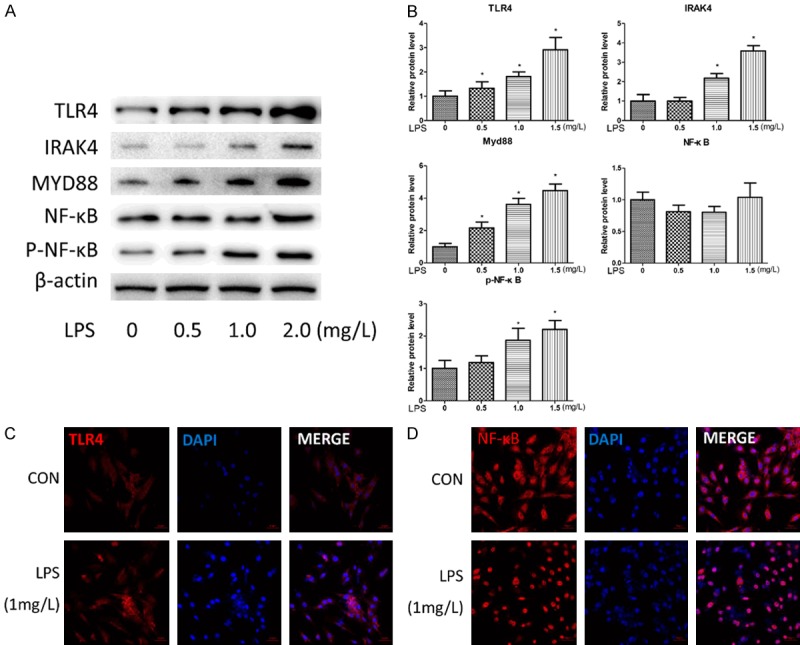
LPS causes activation of the TLR4-NF-κB pathway in a dose-dependent manner. (A and B) 3T3-L1 cells were stimulated with 0, 0.5, 1.0 or 2.0 mg/L LPS for 24 h. The protein expressions of TLR4, IRAK4, Myd88, NF-κB, phospho-NF-κB and B-actin were analyzed by western blotting and quantitative analyses. The fold induction relative to the control group is shown. *P<0.05 compared with the saline treatment group. The data were analyzed with one-way ANOVA followed by least significant difference (LSD) post hoc analysis. (C and D) The morphological changes of the 3T3-L1 cells following treatment with LPS (1 mg/L); TLR4 (C) and NF-κB (D) were stained and exhibited bright red fluorescence, and the nuclei were stained blue with DAPI. Scale bar, 50 μm.
Collagen I protein expression in LPS-treated 3T3-L1 cells is downregulated by inhibiting the TLR4-dependent signaling pathway
Collagen I has been shown to be involved in hypertrophic scarring formation [14]. To determine whether the TLR4 signaling pathway influences collagen I expression, we used a TLR4 inhibitor, TAK242, to inhibit the expression of TLR4 and then examined the downstream molecules and collagen I expression. The western blot results showed that TAK242 pretreatment suppressed TLR4, IRAK4, MYD88 and P-NF-κB upregulation induced by LPS (Figure 2A, 2B; n=3, *P<0.05). The immunofluorescence staining results showed that TAK242 pretreatment prevented collagen I upregulation induced by LPS (Figure 2C).
Figure 2.
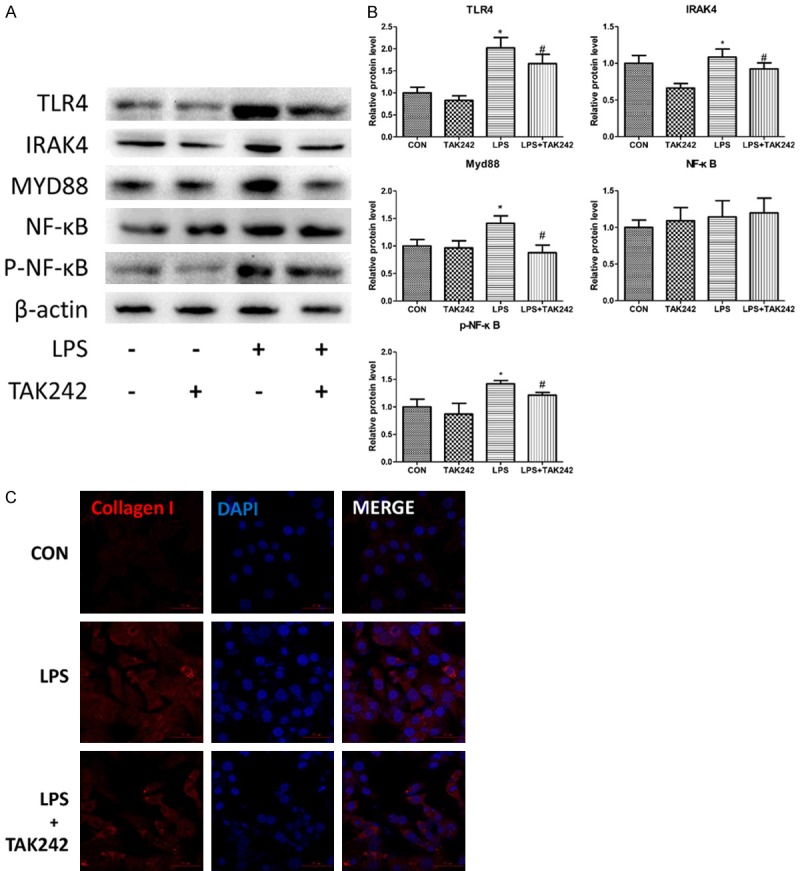
The inhibitor of TLR4, TAK242, inhibits the NF-κB pathway and causes downregulation of collagen I expression. 3T3-L1 cells were exposed to TAK242 for 2 h prior to 1.0 mg/L LPS treatment as indicated. A and B. The protein expressions of TLR4, IRAK4, Myd88, NF-κB, phospho-NF-κB and B-actin were analyzed by western blotting and quantitative analyses. The fold induction relative to the control group is shown. *P<0.05 compared with the saline treatment group. #P<0.05 compared with the LPS treatment group. The data were analyzed using a one-way ANOVA followed by a least significant difference (LSD) post hoc analysis. C. The morphological changes of the 3T3-L1 cells following treatment with LPS and TAK242; collagen I was stained and exhibited bright red fluorescence, and the nuclei were stained blue with DAPI. Scale bar, 50 μm.
Inhibiting TLR-4 ameliorates hypertrophic scarring
To evaluate the effects of TAK-242 on hypertrophic scarring, tissue sections were stained with HE and Masson’s trichrome to analyze the tissue morphology. The HE staining results showed a thicker epidermis and increased collagen degradation in the wound group compared with normal skin. However, the fibrotic tissue in the dermal layer was flatter in the wound + TAK242 group than in the wound group (Figure 3A). The Masson’s trichrome staining results showed increased collagen deposition in the wound group than in the normal skin. TAK242 alleviated the collagen deposition induced by the wound (Figure 3B). Since TGF-β affects the fibrotic process by TGF-β/SMAD signaling [15], we analyzed TGF-β protein expression using immunohistochemical staining to determine the effect of TAK242 on TGF-β protein expression. In comparison with the high expression level of the TGF-β protein in the wound group, the expression level of the TGF-β protein decreased in the wound + TAK242 group (Figure 4A). Similarly, TAK242 suppressed collagen I upregulation induced by the wound (Figure 4B).
Figure 3.
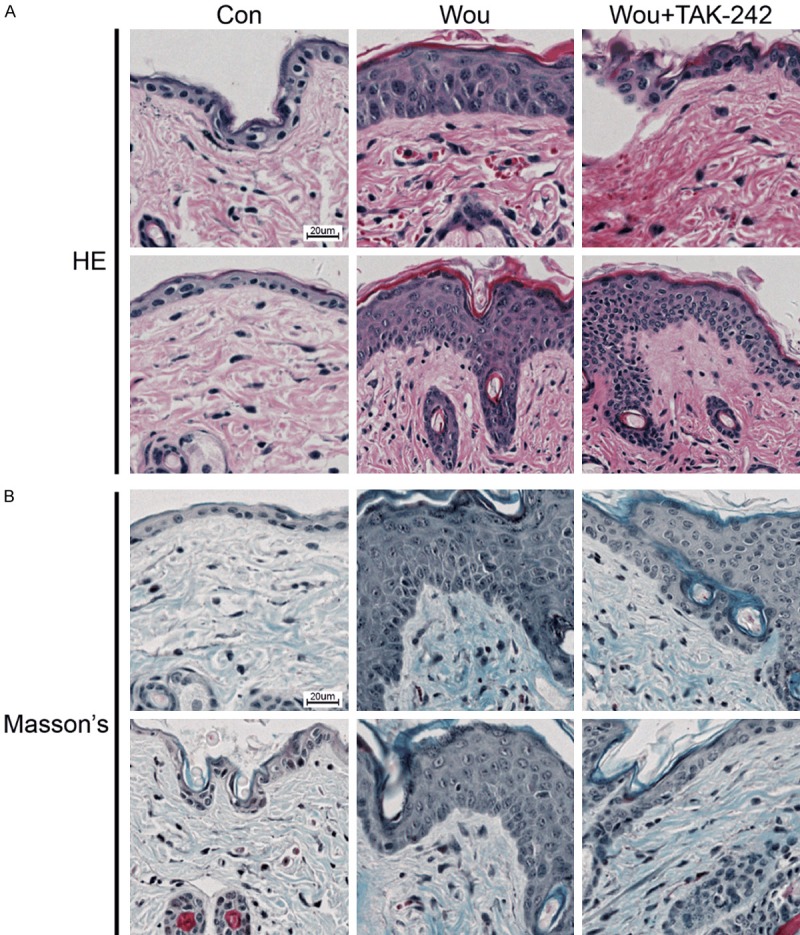
TAK242 ameliorates hypertrophic scar formation and decreases collagen deposition in mice. C57BL/6 mice were randomly divided into three groups as follows: the control group, the wound group, and the wound and TAK242-treated group (n = 3 mice/group). The mice were injected intraperitoneally (i.p.) with TAK242 (five injections, 3 mg/kg/injection, at 24 h intervals) or saline. A. HE staining of the skin. Scale bar, 20 μm. B. Masson’s trichrome staining. Scale bar, 20 μm.
Figure 4.
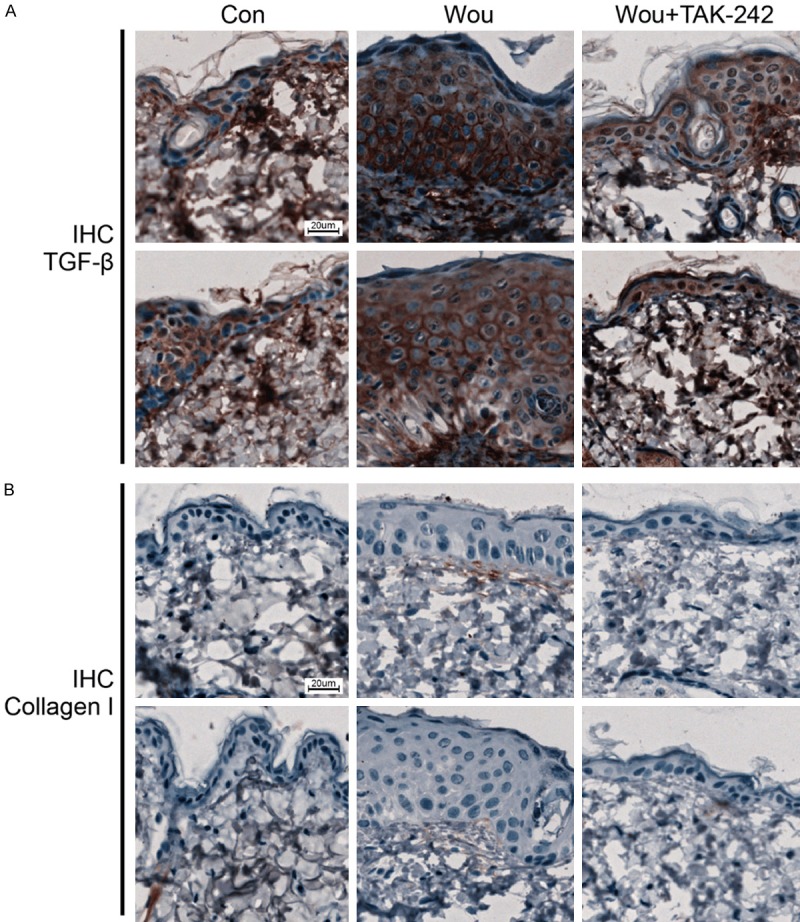
TAK242 decreases the expression of Collagen I and TGFβ. C57BL/6 mice were randomly divided into three groups as follows: the control group, the wound group, and the wound and TAK242-treated group (n = 3 mice/group). The mice were injected intraperitoneally (i.p.) with TAK242 (five injections, 3 mg/kg/injection, at 24 h intervals) or saline. Immunohistochemical staining of collagen I (A) and TGFβ (B) in the mouse skin. Scale bar, 20 μm.
Discussion
In the present study, we demonstrated that LPS activated TLR4 signaling molecules through the MYD88-dependent pathway in 3T3-L1 cells. Inhibiting TLR4 decreased collagen I upregulation induced by LPS in 3T3-L1 cells. Similarly, we demonstrated that inhibiting TLR4 alleviated hypertrophic scar formation in a mouse model.
The wound injury site is easily infected by gram-negative bacteria, which produce a large amount of LPS [16]. LPS is the ligand for TLR4, which can cause inflammation in a burned animal model [17]. Activating TLR4 can lead to the synthesis of inflammatory factors and cytokines via phosphor-NF-κB activation [18]. In the cell model used in this study, we used 0~2.0 mg/L LPS to treat fibroblast cells to simulate the action of bacteria on fibroblasts and the occurrence of inflammation during wound healing. TLR4 deficiency reduces LPS-mediated inflammation in mice, indicating that TLR4 may be a therapeutic target for hematosepsis induced by Gram-negative bacteria [19,20].
In the animal model used in this study, we used the inhibitor of TLR4, TAK242, to treat the mice. The western blot results show that TAK242 could significantly down-regulate the expression of TLR4 and its downstream pathway proteins. After the inhibition of TLR4 expression, hypertrophic scars were significantly reduced. This may be due to a decrease in proinflammatory factors downstream of TLR4-NF-κB. Other researchers [21] found that the mRNA and protein levels of IL-6, IL-8 and monocyte chemotactic protein 1 (MCP-1) were significantly down-regulated after interference with Myd88. In the study of burn scars [22], the expression levels of IL-1β and decorin in the skin were also found to increase. Detection of the serum levels of these factors can predict hypertrophic scar formation.
Our results showed that the downstream proteins of TLR4, including IRAK4, MYD88 and phosphor-NF-κB, were overexpressed after LPS exposure, indicating that LPS activated TLR4 signaling through the MYD88-dependent pathway in 3T3-L1 cells, but the TAK242 treatment inhibited TLR4 signaling activation. Collagen I protein deposition is a characteristic of hypertrophic scar formation. Our results showed that collagen I was overexpressed after TLR4 activation, and this effect was attenuated when TLR4 was inhibited. These results suggest that collagen I may be regulated by TLR4/MYD88/IRAK4 signaling in 3T3-L1 cells.
The TGF-β protein is one of the most relevant cytokines associated with hypertrophic scar formation [23]. Fibroblasts from the wound site can release TGF-β, which affects inflammation and the deposition and reconstruction of ECM [24]. TGF-β protein upregulation and excess collagen I synthesis has been observed in many types of fibrotic diseases [14,25,26]. Furthermore, recent research has shown that collagen synthesis decreases via the downregulation of TGF-β [27]. Therefore, it is unclear whether TLR4 signaling plays a role in the regulation of TGF-β and collagen I expression in the mouse hypertrophic scar model. In this study, we demonstrated that inhibiting TLR4 downregulates TGF-β expression and protects wound sites from excess collagen deposition in a mouse hypertrophic scar model. Inhibiting TLR4 also alleviates epidermal incrassation. Overall, these results indicate that TLR4 signaling activation mediates collagen I expression through TGF-β in the process of hypertrophic scar formation.
In conclusion, our results demonstrated that TAK242 inhibits TLR4 signaling in vitro and alleviates hypertrophic scar formation by means of the TLR4 signaling pathway in vivo. The data suggest that TLR4 can be considered a target to prevent hypertrophic scar formation.
Disclosure of conflict of interest
None.
Abbreviations
- TLR
Toll like receptor
- ECM
extracellular matrix
- NF-κB
nuclear factor-κB
- MYD88
myeloid differentiation factor 88
- LPS
lipopolysaccharide
- SDS-PAGE
sodium dodecyl sulfate polyacrylamide gel
- PVDF
polyvinylidene difluoride
References
- 1.Butzelaar L, Ulrich MM, Mink van der Molen AB, Niessen FB, Beelen RH. Currently known risk factors for hypertrophic skin scarring: a review. J Plast Reconstr Aesthet Surg. 2016;69:163–9. doi: 10.1016/j.bjps.2015.11.015. [DOI] [PubMed] [Google Scholar]
- 2.Ault P, Plaza A, Paratz J. Scar massage for hypertrophic burns scarring-a systematic review. Burns. 2018;44:24–38. doi: 10.1016/j.burns.2017.05.006. [DOI] [PubMed] [Google Scholar]
- 3.Coentro JQ, Pugliese E, Hanley G, Raghunath M, Zeugolis DI. Current and upcoming therapies to modulate skin scarring and fibrosis. Adv Drug Deliv Rev. 2018 doi: 10.1016/j.addr.2018.08.009. [Epub ahead of print] [DOI] [PubMed] [Google Scholar]
- 4.Donner AJ, Yeh ST, Hung G, Graham MJ, Crooke RM, Mullick AE. CD40 generation 2.5 antisense oligonucleotide treatment attenuates doxorubicin-induced nephropathy and kidney inflammation. Mol Ther Nucleic Acids. 2015;4:e265. doi: 10.1038/mtna.2015.40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Muñoz-Rodríguez C, Fernández S, Osorio JM, Olivares F, Anfossi R, Bolivar S, Humeres C, Boza P, Vivar R, Pardo-Jimenez V, Hemmings KE, Turner NA, Díaz-Araya G. Expression and function of TLR4-induced B1R bradykinin receptor on cardiac fibroblasts. Toxicol Appl Pharmacol. 2018;351:46–56. doi: 10.1016/j.taap.2018.05.011. [DOI] [PubMed] [Google Scholar]
- 6.Zhao J, Yu J, Xu Y, Chen L, Zhou F, Zhai Q, Wu J, Shu B, Qi S. Epidermal HMGB1 activates dermal fibroblasts and causes hypertrophic scar formation in reduced hydration. J Invest Dermatol. 2018;138:2322–2332. doi: 10.1016/j.jid.2018.04.036. [DOI] [PubMed] [Google Scholar]
- 7.Wang M, Xiu L, Diao J, Wei L, Sun J. Sparstolonin B inhibits lipopolysaccharide-induced inflammation in 3T3-L1 adipocytes. Eur J Pharmacol. 2015;769:79–85. doi: 10.1016/j.ejphar.2015.10.050. [DOI] [PubMed] [Google Scholar]
- 8.Zhang XY, Liu Y, He T, Yang TT, Wu J, Cianflone K, Lu HL. Anaphylatoxin C5a induces inflammation and reduces insulin sensitivity by activating TLR4/NF-kB/PI3K signaling pathway in 3T3-L1 adipocytes. Biomed Pharmacother. 2018;103:955–964. doi: 10.1016/j.biopha.2018.04.057. [DOI] [PubMed] [Google Scholar]
- 9.Shinjo T, Iwashita M, Yamashita A, Sano T, Tsuruta M, Matsunaga H, Sanui T, Asano T, Nishimura F. Nishimura, IL-17A synergistically enhances TNFalpha-induced IL-6 and CCL20 production in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2016;477:241–246. doi: 10.1016/j.bbrc.2016.06.049. [DOI] [PubMed] [Google Scholar]
- 10.Sun J, Luo J, Ruan Y, Xiu L, Fang B, Zhang H, Wang M, Chen H. Free fatty acids activate renin-angiotensin system in 3t3-l1 adipocytes through nuclear factor-kappa B pathway. J Diabetes Res. 2016;2016:1587594. doi: 10.1155/2016/1587594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bhattacharyya S, Wang W, Tamaki Z, Shi B, Yeldandi A, Tsukimi Y, Yamasaki M, Varga J. Pharmacological inhibition of toll-like receptor-4 signaling by TAK242 prevents and induces regression of experimental organ fibrosis. Front Immunol. 2018;9:2434. doi: 10.3389/fimmu.2018.02434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Pfalzgraff A, Heinbockel L, Su Q, Brandenburg K, Weindl G. Synthetic anti-endotoxin peptides inhibit cytoplasmic LPS-mediated responses. Biochem Pharmacol. 2017;140:64–72. doi: 10.1016/j.bcp.2017.05.015. [DOI] [PubMed] [Google Scholar]
- 13.Janda J, Burkett NB, Blohm-Mangone K, Huang V, Curiel-Lewandrowski C, Alberts DS, Petricoin EF 3rd, Calvert VS, Einspahr J, Dong Z, Bode AM, Wondrak GT, Dickinson SE. Resatorvid-based pharmacological antagonism of cutaneous TLR4 blocks UV-induced NF-kappaB and AP-1 signaling in keratinocytes and mouse skin. Photochem Photobiol. 2016;92:816–825. doi: 10.1111/php.12659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Ricard-Blum S, Baffet G, Théret N. Molecular and tissue alterations of collagens in fibrosis. Matrix Biol. 2018;68-69:122–149. doi: 10.1016/j.matbio.2018.02.004. [DOI] [PubMed] [Google Scholar]
- 15.Lu J, Liu Q, Wang L, Tu W, Chu H, Ding W, Jiang S, Ma Y, Shi X, Pu W, Zhou X, Jin L, Wang J, Wu W. Increased expression of latent TGF-beta-binding protein 4 affects the fibrotic process in scleroderma by TGF-beta/SMAD signaling. Lab Invest. 2017;97:591–601. doi: 10.1038/labinvest.2017.20. [DOI] [PubMed] [Google Scholar]
- 16.Cheng ZD, Liu MY, Chen G, Zhang HM, Qin GJ, Liang G, Liu DX. Anti-vascular permeability of the cleaved reactive center loop within the carboxyl-terminal domain of C1 inhibitor. Mol Immunol. 2008;45:1743–1751. doi: 10.1016/j.molimm.2007.09.035. [DOI] [PubMed] [Google Scholar]
- 17.Chen H, Li Y, Gu J, Yin L, Bian F, Su L, Hong Y, Deng Y, Chi W. TLR4-MyD88 pathway promotes the imbalanced activation of NLRP3/NLRP6 via caspase-8 stimulation after alkali burn injury. Exp Eye Res. 2018;176:59–68. doi: 10.1016/j.exer.2018.07.001. [DOI] [PubMed] [Google Scholar]
- 18.Sun Y, Huang J, Song K. BET protein inhibition mitigates acute myocardial infarction damage in rats via the TLR4/TRAF6/NF-kappaB pathway. Exp Ther Med. 2015;10:2319–2324. doi: 10.3892/etm.2015.2789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuzmich NN, Sivak KV, Chubarev VN, Porozov YB, Savateeva-Lyubimova TN, Peri F. TLR4 signaling pathway modulators as potential therapeutics in inflammation and sepsis. Vaccines (Basel) 2017;5 doi: 10.3390/vaccines5040034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cao C, Chai Y, Shou S, Wang J, Huang Y, Ma T. Toll-like receptor 4 deficiency increases resistance in sepsis-induced immune dysfunction. Int Immunopharmacol. 2018;54:169–176. doi: 10.1016/j.intimp.2017.11.006. [DOI] [PubMed] [Google Scholar]
- 21.Wang J, Hori K, Ding J, Huang Y, Kwan P, Ladak A, Tredget EE. Toll-like receptors expressed by dermal fibroblasts contribute to hypertrophic scarring. J Cell Physiol. 2011;226:1265–73. doi: 10.1002/jcp.22454. [DOI] [PubMed] [Google Scholar]
- 22.Kwan PO, Ding J, Tredget EE. Serum decorin, interleukin-1β, and transforming growth factor-β predict hypertrophic scarring postburn. J Burn Care Res. 2016;37:356–366. doi: 10.1097/BCR.0000000000000271. [DOI] [PubMed] [Google Scholar]
- 23.van der Veer WM, Bloemen MC, Ulrich MM, Molema G, van Zuijlen PP, Middelkoop E, Niessen FB. Potential cellular and molecular causes of hypertrophic scar formation. Burns. 2009;35:15–29. doi: 10.1016/j.burns.2008.06.020. [DOI] [PubMed] [Google Scholar]
- 24.Mokoena D, Dhilip Kumar SS, Houreld NN, Abrahamse H. Role of photobiomodulation on the activation of the smad pathway via TGF-beta in wound healing. J Photochem Photobiol B. 2018;189:138–144. doi: 10.1016/j.jphotobiol.2018.10.011. [DOI] [PubMed] [Google Scholar]
- 25.Hu B, Mao Z, Jiang X, He D, Wang Z, Wang X, Zhu Y, Wang H. Role of TGF-beta1/Smad3-mediated fibrosis in drug resistance mechanism of prolactinoma. Brain Res. 2018;1698:204–212. doi: 10.1016/j.brainres.2018.07.024. [DOI] [PubMed] [Google Scholar]
- 26.Li H, Cai H, Deng J, Tu X, Sun Y, Huang Z, Ding Z, Dong L, Chen J, Zang Y, Zhang J. TGF-beta-mediated upregulation of Sox9 in fibroblast promotes renal fibrosis. Biochim Biophys Acta Mol Basis Dis. 2018;1864:520–532. doi: 10.1016/j.bbadis.2017.11.011. [DOI] [PubMed] [Google Scholar]
- 27.Seo SA, Park B, Hwang E, Park SY, Yi TH. Borago officinalis L. attenuates UVB-induced skin photodamage via regulation of AP-1 and Nrf2/ARE pathway in normal human dermal fibroblasts and promotion of collagen synthesis in hairless mice. Exp Gerontol. 2018;107:178–186. doi: 10.1016/j.exger.2018.02.017. [DOI] [PubMed] [Google Scholar]