

Abstract
Background: Multiple previous studies have indicated miR-516a-3p was associated with carcinogenesis in lung cancer. However, its biologic functions in lung adenocarcinoma remain unknown. The aim of this study was to investigate the expression of miR-516a-3p in lung adenocarcinoma, its molecular mechanisms of miR-516a-3p, and its effects on cell proliferation, migration, invasion, and apoptosis. Methods: The expression of miR-516a-3p and PTPRD was tested by reverse transcription-quantitative polymerase chain reaction. Cell migration and invasion assays were used to evaluate the migration and invasion ability of cells. Flow cytometry was performed to observe the effects of miR-516a-3p on the cell apoptosis. Western blot analysis was used to assess the protein levels of PTPRD. Luciferase reporter assay was utilized to identify whether PTPRD was a direct target of miR-516a-3p. Results: There was upregulated expression of miR-516a-3p in lung adenocarcinoma tissues as well as cell lines. In addition, miR-516a-3p expression knock-down could inhibit cell proliferation, invasion, and migration, but promote apoptosis in lung adenocarcinoma. By contrast, overexpression of miR-516a-3p resulted in the opposite effect. Dual luciferase assay, RT-PCR and western blot analysis results confirmed that PTPRD was a direct target for miR-516a-3p. Further studies also found PTPRD was down-regulated in lung adenocarcinoma and there was a negative correlation between miR-516a-3p and PTPRD expression in lung adenocarcinoma. Moreover, miR-516a-3p and PTPRD were significantly correlated with the clinical stage of lung adenocarcinoma. Conclusions: Our current findings showed that miR-516a-3p was up-regulated in lung adenocarcinoma, functioning as a tumor-promoting gene by targeting PTPRD.
Keywords: microRNA, miR-516a-3p, lung adenocarcinoma, tumor-promoting, protein tyrosine phosphatase, Receptor Type D
Introduction
The pathologic classification of lung cancer includes non-small cell lung cancer (NSCLC) as well as small cell lung cancer (SCLC). The prevalence of lung adenocarcinoma, the main histological type of NSCLC, has been recently rising worldwide, with low overall survival (OS) rate [1].
MicroRNAs (miRNAs), endogenous small-RNA molecules of 23 nt in length, can negatively modulate target gene expression by translationally suppressing or degrading its target mRNA [2]. The critical roles of miRNAs in all biological processes have been widely reported previously, which are correlated with multiple human disorders, including lung cancer [3,4]. miR-30b and miR-30c are able to suppress the proliferation of NSCLC cells by targeting Rab18 [5]. The supplementation with let-7b and miR-34a could increase therapeutic sensitivity of erlotinib in NSCLC cells [6]. miRNA-199b could suppress cell proliferation, invasion, as well as migration in NSCLC by targeting ZEB1 [7]. miR-32 has been reported to inhibit proliferation, epithelial-mesenchymal transition (EMT) and subsequent metastasis by targeting TWIST1 in NSCLC [8]. miRNA-223 may induce apoptosis of NSCLC through the PI3K/AKT pathway by targeting EGFR [9].
In this research, we revealed the up-regulated expression of miR-516a-3p in lung adenocarcinoma tissuesand cells in comparison to matched normal lung tissues and human bronchial epithelial cell line (HBEpC). In addition, miR-516a-3p expression knock-down could inhibit cell proliferation, invasion, and migration, but promote apoptosis in lung adenocarcinoma. By contrast, overexpression of miR-516a-3p resulted in the opposite effect. Moreover, PTPRD was shown as a direct target for miR-516a-3p in lung adenocarcinoma, and the expression of PTPRD was down-regulated in lung adenocarcinoma tissues and cell lines. We also revealed that there was a negative correlation between the expression of miR-516a-3p and PTPRD in lung adenocarcinoma and the expression of miR-516a-3p, and PTPRD was significantly correlated with the clinical stage of lung adenocarcinoma. In conclusion, miR-516a-3p was shown to might act as a tumor-promoter gene by targeting PTPRD in lung adenocarcinoma.
Materials and methods
Patients and specimen collection and cell culture
Tissues of lung adenocarcinoma and normal lung were collected from 57 patients with lung adenocarcinoma (age 37-71 years; mean age, 57 years; 28 males and 29 females) in the Department of Thoracic Surgery Ward II at the Third Affiliated Hospital of Kunming Medical University from August 2018 to November 2018. Tissues were immediately stored in RNAlater (Sigma, USA) at -80°C after surgical resection. Adjuvant radiochemotherapy was not performed on any patients. Clinicopathological features were extracted from all patients, including smoking status, age, tumor size, lymph node (LN) involvement, gender, tumor-node-metastasis (TNM) classification.
Human lung adenocarcinoma cell lines (H1299, SPC-A1, A549) as well as human bronchial epithelial cell line including (16HBE, BEAS-2B) were commercially obtained from Shanghai Institute of Cell Biology (Academia Sinica, China), and then maintained in RPMI-1640 medium (HyClone, USA) containing 10% FBS (HyClone), penicillin (100 U/ml) and streptomycin (100 mg/ml) (Gibco) at 37°C with 5% CO2.
Total RNA extraction and reverse transcription-quantitative polymerase chain reaction (RT-qPCR)
Trizol (Sigma) reagent was employed for total RNA extraction from tissues and cells. cDNA was prepared from total RNA for mRNA and miRNA quantification using miRcute Plus miRNA First-strand cDNA Synthesis kit (Tiangen Biotech Co, Ltd.) as well as RevertAid First Strand cDNA Synthesis Kit (Thermo ScientificTM). miRcute Plus miRNA qPCR Detection Kit (TIANGEN) and FastStart Universal SYBR Green Master (ROX) (Roche Diagnostics) were purchased to assess miR-516a-3p and PTPRD expression, respectively, followed by analysis on 7500 Real-Time PCR system (Applied Biosystems). Primers were designed as: miR-516a-3p, forward, 5’-ACCCTCTGAAAGGAAGCA-3’; U6, forward, 5’-CTCGCTTCGGCAGCACATATACT-3’; PTPRD, forward, 5’-CTCCAAGGTTTACACGAACACC-3’ and reverse, 5’-AGTCCGTAAGGGTTGTATTCTGA-3’; GAPDH, forward, 5’-CAAGGTCATCCATGACAACTTTG-3’ and reverse, 5’-GTCCACCACCCTGTTGCTGTAG-3’. The 2-ΔΔCT method was employed for calculation of the relative expression of miR-516a-3p and PTPRD, followed by normalization to U6-snRNA and β-actin expression, respectively. Melt curve was used to verify the specificity of amplification.
Transfection
miR-516a-3p mimics, inhibitor, and NC were all commercially obtained from GenePharma Co, Ltd. (Shanghai, China). The sequences of miR-516a-3p mimics, miR-516a-3p inhibitor and NC sequence were 5’-UGCUUCCUUUCAGAGGGU-3’, 5’-ACCCUCUGAAAGGAAGCA-3’ and 5’-UUCUCCGAACGUGUCACGUTT-3’, respectively. Briefly, H1299 and SPC-A1 cells were inoculated and incubated in 6-well plates for 24 h, followed by transfection of these small RNA molecules (50 nM) using Lipofectamine 3000 (Invitrogen) in line with standard protocol. Western blot and qRT-PCR were employed to assess transfection efficiency after 48 h.
Wound-healing assay
After transfection with miR-516a-3p inhibitor, mimics, or NC for 48 h, H1299 and SPC-A1 cells were inoculated in 6-well plate, followed by incubation for 12 h. After transfected and untransfected cells reached to 90% confluence, a blue P1000 pipette tip (Corning) was employed to scratch the cell monolayer, followed by incubation for 24 h. Cells were photographed under a phase contrast microscope (magnification, × 100) for the evaluation of healing rate at 0, 24 and 48 h. ImageJ (version 1.51; NIH, USA) was used to plot the wound areas. Alterations in wound areas during different time points were shown as the percentage of original ones.
Cell proliferation assay
After transfection with miR-516a-3p inhibitor, mimics, or NC for 24 h, H1299 and SPC-A1 were inoculated into 96-well plates (2 × 103 cells/well, 200 µl/well), incubated for 24, 48, 72 and 96 h. Afterwards, CCK-8 assay (Beyotime, China) was conducted in line with the manufacturer’s instructions using a microplate reader (Thermo Fisher Scientific) at 450 nm.
Cell migration and invasion assays
Transwell chambers without or with Matrigel (Corning) were employed for the assessment of the migration or invasion ability of transfected cells. In brief, H1299 and SPC-A1 cells transfected with miR-516a-3p inhibitor, mimics or NC (5 × 104 cells/Transwell) in 200 µl FBS-free 1640 medium were inoculated in the upper chamber (8-μm pore size; BD Falcon). A total of 600 µl 1640 medium supplemented with 10% FBS was added to the lower chamber. After incubation for 24 h, a cotton swab was used to remove cells in the outside surface of upper chamber, while those in the inside surface was fixed in methanol, stained with 0.1% crystal violet. Afterwards, we counted the number of cells penetrating the membrane under microscope (magnification, × 100) in five random areas.
Flow cytometry analyses
FITC Annexin V Apoptosis Detection Kit (BD Pharmingen) was purchased for apoptosis assay. After washing with cold PBS twice, cells were resuspended in 1 × Binding Buffer (1 × 106 cells/ml), followed by transferring 100 µl solution to a 5 ml culture tube. Then, 5 µl of FITC Annexin V as well as 5 µl PI were added, followed by gentle vortexing as well as incubation in dark at room temperature (RT) for 15 min. Finally, apoptosis was analyzed by flow cytometry (BD FACSAria II, USA) after adding 400 µl of Binding Buffer.
Western blot analysis
Cells were detached with trypsin, centrifuged, and washed with cold PBS twice, then lysed in 1% RIPA (Solarbio) with PMSF (Solarbio). After quantification by BCA assay kit (Tiangen), equal amount of protein was subjected 10% SDS-PAGE, transferred to PVDF membrane (Millipore), blocked with 5% skimmed-milk at RT for 1 h. Afterwards, the membranes were reacted with proper primary antibodies (PTPRD (1:1000, #ab233806, Abcam), β-actin (1:2,000, #E-AB-20031, Elabscience)) at 4°C overnight. After washing with TBST three times (10 min each), membranes were reacted with HRP-conjugated secondary antibodies for 2 h at RT, followed by visualization using ECL detection kit (Solarbio). Finally, Image lab software was utilized for analysis.
Luciferase reporter assay
In order to determine the mechanism underlying the promoting role of miR-516a-3p on cell invasion, migration and proliferation, TargetScan (http://www.targetscan.org) was used to explore the miRNA target. As a result, a highly conserved putative miR-516a-3p binding site was detected in the 3’UTR of PTPRD. Luciferase reporter assay was further conducted. 293 cells were co-transfected with pRL-PTPRD-3’UTR wild type (WT) or pRL-PTPRD-3’UTR mutant (MUT) (synthesized and validated by Shanghai Genomeditech Co., Ltd.), and miR-516a-3p mimics or NC using Lipofectamine 2000 (Invitrogen). Co-transfection of pRL-TK 3’-UTR target plasmid (Promega, 10 ng) and pRL-TK 3’-UTR negative control plasmid (10 ng) into 293 cells was taken as control. Reportable gene detection kit (Genomeditech Co., Ltd.) was purchased to analyze final results, Dual Luciferase Reporter assay (Promega) was used to analyze luciferase activities.
Statistical analysis
SPSS 16.0 software (SPSS, Chicago, USA) was employed for statistical analysis. Assays were conducted in duplicate, with data shown as mean ± SD. Student’s t-test and one-way ANOVA followed by Tukey’s test were employed for two-group comparison and multi-group comparison, respectively. χ2 test was utilized to analyze clinicopathologic data and the expression of miR-516a-3p and PTPRD. A P<0.05 was considered significant.
Results
The increased miR-516a-3p expression but decreased PTPRD expression in lung adenocarcinoma
RT-qPCR was utilized to measure miR-516a-3p expression in tissues as well as cells, revealing the up-regulation of miR-516a-3p expression in lung adenocarcinoma tissues and cell lines (H1299, SPC-A1, A549) compared to paired normal lung tissues and HBEpC lines (16HBE and BEAS-2B) (Figure 1A and 1B). However, the expression of PTPRD was down-regulated in lung adenocarcinoma tissues as well as cell lines in comparison to paired normal lung tissues as well as HBEpC lines (Figure 1B and 1D). Thus, the expression of miR-516a-3p and of PTPRD were negatively correlated in lung adenocarcinoma (Figure 1E). Furthermore, miR-516a-3p and PTPRD expression was significantly related to lung adenocarcinoma stage, but not other clinicopathologic features (including tumor size, gender, LN involvement, age, or smoking status) (Tables 1 and 2).
Figure 1.
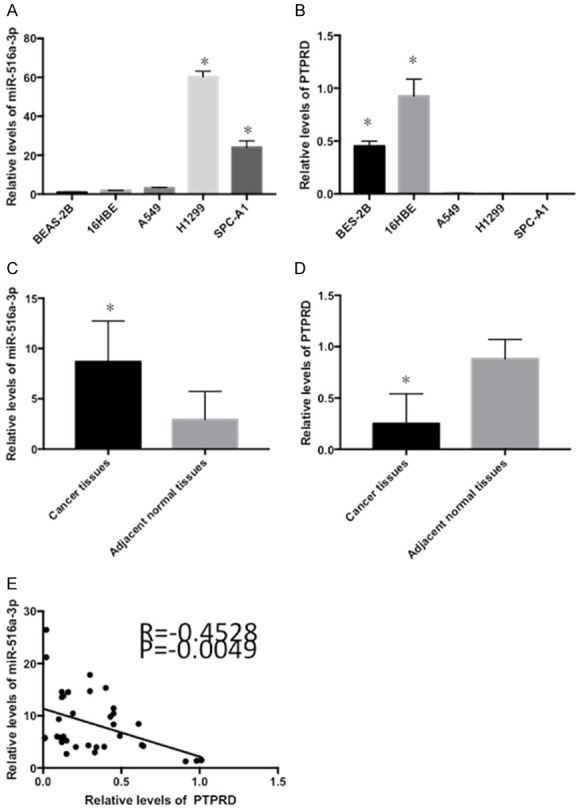
Expression levels of miR-516a-3p and PTPRD in lung adenocarcinoma. A and B: The expression of miR-516a-3p and PTPRD was assessed in different lung adenocarcinoma cell lines (A549, H1299 and SPC-A1) and human bronchial epithelial cell lines (BEAS-2B and 16HBE). C and D: miR-516a-3p was upregulated and PTPRD was downregulated in lung adenocarcinoma tissues compared with matched adjacent normal tissues. E: Negative correlation between miR-516a-3p and PTPRD expression in lung adenocarcinoma. *P<0.05 vs. BES-2B, 16HBE, the negative control and adjacent normal tissues. miR, microRNA; neg, negative.
Table 1.
Comparison analysis of clinicopathological characteristics for miR-516a-3p in tissue
| Characteristics | N | miR-516a-3p | χ2 | P-value | |
|---|---|---|---|---|---|
|
| |||||
| High | Low | ||||
| Gender | |||||
| Male | 16 | 10 | 6 | 0.304 | 0.581 |
| Female | 24 | 17 | 7 | ||
| Age | |||||
| <60 | 22 | 14 | 8 | 0.333 | 0.564 |
| ≥60 | 18 | 13 | 5 | ||
| Smoking status | |||||
| Yes | 13 | 8 | 5 | 0.312 | 0.576 |
| No | 27 | 19 | 8 | ||
| pT | |||||
| T1 | 24 | 16 | 8 | 0.019 | 0.890 |
| T2-4 | 16 | 11 | 5 | ||
| pN | |||||
| N0 | 32 | 21 | 11 | 0.256 | 0.613 |
| N1-3 | 8 | 6 | 2 | ||
| TNM stage | |||||
| I | 28 | 22 | 6 | 3.543 | 0.022 |
| II-III | 12 | 5 | 7 | ||
N, number; p, pathological staging; pT, pathologic tumor staging; pN, pathologic lymph node staging; TNM, tumor-nodes-metastasis.
Table 2.
Comparison analysis of clinicopathological characteristics for PTPRD in tissue
| Characteristics | N | PTPRD | χ2 | P-value | |
|---|---|---|---|---|---|
|
| |||||
| High | Low | ||||
| Gender | |||||
| Male | 22 | 3 | 19 | 0.305 | 0.580 |
| Female | 20 | 4 | 16 | ||
| Age | |||||
| <60 | 29 | 5 | 24 | 0.022 | 0.881 |
| ≥60 | 13 | 2 | 11 | ||
| Smoking status | |||||
| Yes | 25 | 5 | 20 | 0.724 | 0.395 |
| No | 17 | 2 | 15 | ||
| pT | |||||
| T1 | 23 | 6 | 17 | 3.249 | 0.071 |
| T2-4 | 19 | 1 | 18 | ||
| pN | |||||
| N0 | 33 | 7 | 26 | 2.291 | 0.130 |
| N1-3 | 9 | 0 | 9 | ||
| TNM stage | |||||
| I | 28 | 7 | 21 | 4.200 | 0.040 |
| II-III | 14 | 0 | 14 | ||
N, number; p, pathological staging; TNM, tumor-nodes-metastasis.
miR-516a-3p enhanced proliferation, migration as well as invasion in lung adenocarcinoma cells
To reveal the roles of miR-516a-3p on lung adenocarcinoma cells, miR-516a-3p inhibitor, mimic, and NC were transfected into lung adenocarcinoma cells, followed by fluorescence microscopy (Figure 2A) and RT-qPCR to determine the transfection efficiency. Consequently, miR-516a-3p expression was markedly enhanced after transfection with miR-516a-3p mimic, but was markedly decreased following miR-516a-3p inhibitor transfection, in comparison with NC transfection (Figure 2B), suggesting the successful construction of transfected cells. CCK-8 assay showed significantly enhanced cell growth following transfection with miR-516a-3p mimic, which was reduced after transfection with miR-516a-3p inhibitor, compared with the NC group at various time points (Figure 2C). In addition, a wound-healing assay along with a transwell migration/invasion assay were used to assess the migration/invasion capacity of lung adenocarcinoma cells after transfection. Transfection of miR-516a-3p inhibitor led to reduced migration/invasion, while transfection of miR-516a-3p mimic resulted in enhanced migration/invasion capacity, in comparison with NC transfection (Figure 3).
Figure 2.
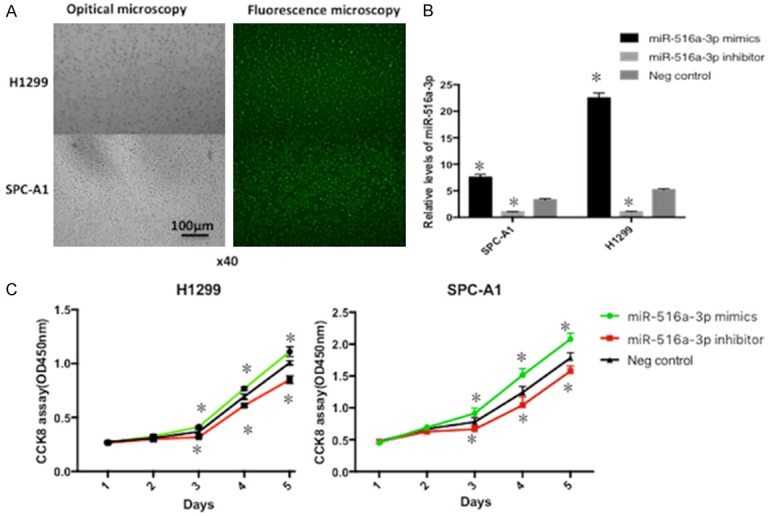
Evaluation of transfection efficiency and miR-516a-3p effect on cell proliferation. A: Observation by fluorescence microscopy following 12 h after transfection. B: The expression of miR-516a-3p in cells transfected with miR-516a-3p mimics, miR-516a-3p inhibitor, or negative control was determined by reverse transcription quantitative polymerase chain reaction assay. C: Knock-down of miR-516a-3p could inhibit cell proliferation of lung adenocarcinoma cells, while cell growth could be promoted by over-expression of miR-516a-3p.
Figure 3.

Cell migration and invasion assays of lung adenocarcinoma cells transfected with miR-516a-3p mimics, inhibitor, or negative control. A and B: Representative images of miR-516a-3p-transfected cell monolayer wound healing following 24 h (magnification, × 100). C and D: Transwell assay demonstrated that miR-516a-3p could regulate lung adenocarcinoma cell migration and invasion. The original magnification was × 200, and data represent the average cell number from three viewing fields. *P<0.05 vs. Negative control. miR, microRNA; neg, negative.
miR-516a-3p inhibited apoptosis of lung adenocarcinoma cells
Flow cytometry showed that knock-down of miR-516a-3p could promote apoptosis, while over-expression of miR-516a-3p suppressed apoptosis in lung adenocarcinoma cells, compared to the NC transfection group (Figure 4).
Figure 4.

Flow cytometry of lung adenocarcinoma cells transfected with miR-516a-3p mimics, inhibitor or negative control. A and B: Knockdown of miR-516a-3p could promote lung adenocarcinoma cells apoptosis, which was inhibited by overexpression of miR-516a-3p. *P<0.05 vs. Negative control. miR, microRNA; neg, negative.
PTPRD was a novel direct target of miR-516a-3p
To examine the mechanism underlying the promoting role of miR-516a-3p in invasion, migration and proliferation, TargetScan was utilized for searching miRNA target. Bioinformatics analysis showed that PTPRD was a candidate target of miR-516a-3p (Figure 5A). Luciferase reporter assay, RT-qPCR as well as WB were performed for further validation whether PTPRD was a direct target of miR-516a-3p. pGL3-PTPRD-3’UTR WT or pGL3-PTPRD-3’UTR MUT was co-transfected with miR-516a-3p mimics or NC in 293 cells. As a result, upregulation of miR-516a-3p repressed the luciferase activities of pGL3-PTPRD-3’UTR WT, but not pGL3-PTPRD-3’UTR MUT (Figure 5D). Knockdown of miR-516a-3p could increase mRNA levels of PTPRD in H1299 and SPC-A1 cells, while the mRNA levels of PTPRD was decreased following miR-516a-3p overexpression (Figure 5B). Knockdown of miR-516a-3p could enhance the PTPRD protein level (Figure 5C). The present findings demonstrated that miR-516a-3p could negatively modulate PTPRD expression in lung adenocarcinoma.
Figure 5.
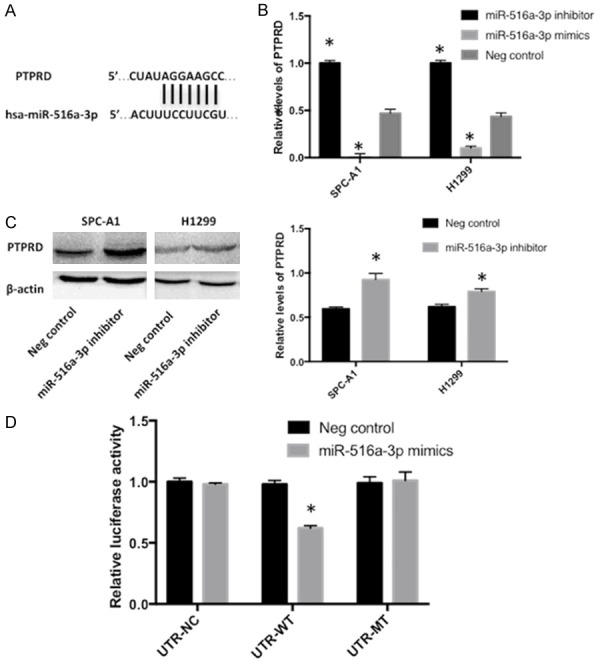
PTPRD may be a target gene of miR-516a-3p. A: The TargetScan alignment of PTPRD’s 3’UTR with hsa-miR-516a-3p indicated the highly conserved regions. B: mRNA expression of PTPRD in lung adenocarcinoma cells (H1299 and SPC-A1) transfected with miR-516a-3p mimics, miR-516a-3p inhibitor or negative control was determined using reverse transcription-quantitative polymerase chain reaction. C: Western blot was performed to measure PTPRD protein expression in H1299 and SPC-A1 cells transfected with miR-516a-3p inhibitor or negative control. D: The luciferase activity of PTPRD 3’UTR-luc-WT was decreased following transfection with miR-516a-3p mimics when compared with the control groups. *P<0.05 vs. Negative control. PTPRD, Protein Tyrosine Phosphatase, Receptor Type D; miR, microRNA; Neg, negative.
Discussion
Here, we showed that miR-516a-3p was up-regulated in lung adenocarcinoma tissues as well as cell lines compared to paired normal lung tissues and HBEpC lines, respectively. Our study also showed that PTPRD expression was down-regulated in lung adenocarcinoma tissues as well as cell lines compared to paired normal lung tissues as well as HBEpC lines. Therefore, the expression of miR-516a-3p and PTPRD were negatively correlated in lung adenocarcinoma. Furthermore, miR-516a-3p and PTPRD expression was significantly correlated with the clinical stage of lung adenocarcinoma. In addition, cell experiments were conducted to evaluate the roles of miR-516a-3p on physiologic processes of lung adenocarcinoma. In brief, CCK8, wound-healing as well as Transwell migration/invasion assays indicated that over-expression of miR-516a-3p could promote cell proliferation, migration, and invasion, which were suppressed by knock-down of miR-516a-3p in lung cancer cell lines. Bioinformatic analysis, luciferase reporter assay, RT-qPCR and WB confirmed that PTPRD was a novel direct target of miR-516a-3p. Collectively, our current outcomes demonstrated that miR-516a-3p acted as a tumor promotor through directly targeting PTPRD in lung adenocarcinoma.
According to several previous studies, miR-516a-3p is critically involved in cancer. In gastric cancers, miR-516a-3p has been revealed to be associated with metastasis and function as an anti-metastamir in blocking metastatic dissemination [10]. In prostate cancer, loss of miR-516a-3p could drive its progression by up-regulation of ABCC5 [11]. miR-516a-3p has also found to have a correlation with the aggressiveness of LN-negative, ER-positive human breast malignancy [12]. In lung cancer, a novel binding site for miR-516a-3p was disrupted by SNP, causing moderately increased expression of both mRNA and protein of CXCR2, and enhanced MAPK signaling [13]. However, there were no reports concerning the expression or function of miR-516a-3p in lung adenocarcinoma.
miRNAs play diverse roles in both physiologic and pathologic processes by binding with the 3’UTR of target gene mRNA for negative regulation. Therefore, identification of miR-516a-3p target genes is important to understand the roles of miR-516a-3p in tumorigenesis, and for the investigation of new therapeutic targets. Herein, we identified PTPRD as a direct target of miR-516a-3p. Luciferase reporter assay showed that miR-516a-3p might directly target PTPRD, which was further revealed by RT-qPCR as well as WB. miR-516a-3p overexpression caused significantly reduced mRNA and protein expression of PTPRD in lung adenocarcinoma cells.
PTPRD, located on chromosome 9p, is one of the protein tyrosine phosphatase (PTP) family members [14], which is dem a tumor suppressor gene, involved in human malignancy progression [15]. High-resolution analysis of chromosomal breakpoints as well as genomic instability has confirmed PTPRD as a potential tumor suppressor gene in neuroblastoma [16]. PTPRD is also identified as a potential tumor suppressor gene in cutaneous squamous cell carcinoma, which is probably correlated with metastasis, detected by single nucleotide polymorphism microarray analysis [17]. In addition, high resolution ArrayCGH and expression profiling show PTPRD as a potential tumor suppressor gene in laryngeal squamous cell carcinoma [18]. Moreover, deleterious alterations in PTPRD could be a potential biomarker for bevacizumab resistance in metastatic colorectal cancer patients [19]. Furthermore, a study has found that the expression of PTPRD is reduced in most cell lines and surgical specimens of lung cancer [20]. Kim [14] et al. have found that the mechanism of PTPRD in cancer may be mediated by regulating STAT3 signaling. Generally, the mechanism of PTPRD in cancer needs to be further investigated. Our present outcomes have demonstrated PTPRD was down-regulated in lung adenocarcinoma tissues as well as cell lines in comparison to paired normal lung tissues as well as HBEpC lines. In addition, the expression of miR-516a-3p and PTPRD expression were inversely correlated in lung adenocarcinoma. Furthermore, miR-516a-3p and PTPRD expression was significantly related to lung adenocarcinoma stage, but not other clinicopathologic features (including tumor size, gender, LN involvement, age or smoking status). Moreover, miR-516a-3p can suppress both mRNA and protein expression of PTPRD in lung adenocarcinoma cells.
Thus, miR-516a-3p expression is increased in lung adenocarcinoma. Aberrant expression of miR-516a-3p could affect various biologic functions in lung adenocarcinoma cells, including cell migration, invasion, proliferation and apoptosis, partially through directly targeting PTPRD.
Acknowledgements
This research was funded by National Natural Science Foundation of China (No. 81460356 and 81760554).
Disclosure of conflict of interest
None.
References
- 1.Siegel RL, Miller KD, Jemal A. Cancer statistics, 2017. CA Cancer J Clin. 2017;67:7–30. doi: 10.3322/caac.21387. [DOI] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: target recognition and regulatory functions. Cell. 2009;136:215–33. doi: 10.1016/j.cell.2009.01.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Acunzo M, Croce CM. MicroRNA in cancer and cachexia--a mini-review. J Infect Dis. 2015;212(Suppl 1):S74–7. doi: 10.1093/infdis/jiv197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hammond SM. An overview of microRNAs. Adv Drug Deliv Rev. 2015;87:3–14. doi: 10.1016/j.addr.2015.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zhong K, Chen K, Han L, Li B. miR-30b/c could inhibits non-small cell lung cancer cell proliferation by targeting Rab18. BMC Cancer. 2014;12:703. doi: 10.1186/1471-2407-14-703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stahlhut C, Slack FJ. Combinatorial action of MicroRNAs let-7 and miR-34 effectively synergizes with erlotinib to suppress non-small cell lung cancer cell proliferation. Cell Cycle. 2015;14:2171–80. doi: 10.1080/15384101.2014.1003008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wang J, Zhou F, Yin L, Zhao L, Zhang Y, Wang J. MicroRNA-199b targets the regulation of ZEB1 expression to inhibit cell proliferation, migration and invasion in non-small cell lung cancer. Mol Med Rep. 2017;16:5007–5014. doi: 10.3892/mmr.2017.7195. [DOI] [PubMed] [Google Scholar]
- 8.Li L, Wu D. miR-32 inhibits proliferation, epithelial-mesenchymal transition and metastasis by targeting TWIST1 in non-small-cell lung cancer cells. Onco Targets Ther. 2016;9:1489–1498. doi: 10.2147/OTT.S99931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ma HP, Kong WX, Li XY, Li W, Zhang Y, Wu Y. miRNA-223 is an anticancer gene in human non-small cell lung cancer through the PI3K/AKT pathway by targeting EGFR. Oncol Rep. 2019;41:1549–1559. doi: 10.3892/or.2019.6983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yoshifumi T, Misato T, Keichiro M, Yuzo T, Kazuyoshi Y. The metastasis-associated microRNA miR-516a-3p is a novel therapeutic target for inhibiting peritoneal dissemination of human scirrhous gastric cancer. Cancer Res. 2011;71:1442–1453. doi: 10.1158/0008-5472.CAN-10-2530. [DOI] [PubMed] [Google Scholar]
- 11.Zhang H, Lian Z, Sun G, Liu R, Xu Y. Loss of miR-516a-3p mediates upregulation of ABCC5 in prostate cancer and drives its progression. Onco Targets Ther. 2018;11:3853–3867. doi: 10.2147/OTT.S167463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.John A, Foekens AM, Sieuwerts MS, Maxime P, Look VW, Antonius WM, Boersma JG, Erik AC, John WM. Four miRNAs associated with aggressiveness of lymph node-negative, estrogen receptor-positive human breast cancer. Proc Natl Acad Sci U S A. 2008;105:13021–13026. doi: 10.1073/pnas.0803304105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ryan BM, Robles AI, McClary AC, Haznadar M, Bowman ED, Pine SR, Brown D, Khan M, Shiraishi K, Kohno T. Identification of a functional SNP in the 3’UTR of CXCR2 that is associated with reduced risk of lung cancer. Cancer Res. 2015;7:566–75. doi: 10.1158/0008-5472.CAN-14-2101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim M, Morales LD, Jang IS, Cho YY, Kim DJ. Protein tyrosine phosphatases as potential regulators of STAT3 signaling. Int J Mol Sci. 2018;19 doi: 10.3390/ijms19092708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Veeriah S, Brennan C, Meng S, Singh B, Fagin JA, Solit DB. The tyrosine phosphatase PTPRD is a tumor suppressor that is frequently inactivated and mutated in glioblastoma and other human cancers. Proc Natl Acad Sci U S A. 2009;106:9435–40. doi: 10.1073/pnas.0900571106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Stallings RL, Nair P, Maris JM, Catchpoole D, McDermott M, O’Meara A, Breatnach F. High-resolution analysis of chromosomal breakpoints and genomic instability identifies PTPRD as a candidate tumor suppressor gene in neuroblastoma. Cancer Res. 2006;66:3673–80. doi: 10.1158/0008-5472.CAN-05-4154. [DOI] [PubMed] [Google Scholar]
- 17.Purdie KJ, Lambert SR, Teh MT, Chaplin T, Molloy G, Raghavan M, Kelsell DP, Leigh IM, Harwood CA, Proby CM. Allelic imbalances and microdeletions affecting the PTPRD gene in cutaneous squamous cell carcinomas detected using single nucleotide polymorphism microarray analysis. Genes Chromosomes Cancer. 2007;46:661–9. doi: 10.1002/gcc.20447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Giefing M, Zemke N, Brauze D, Kostrzewska-Poczekaj M, Luczak M, Szaumkessel M, Pelinska K, Kiwerska K, Tönnies H, Grenman R. High resolution ArrayCGH and expression profiling identifies PTPRD and PCDH17/PCH68 as tumor suppressor gene candidates in laryngeal squamous cell carcinoma. Genes Chromosomes Cancer. 2011;50:154–166. doi: 10.1002/gcc.20840. [DOI] [PubMed] [Google Scholar]
- 19.Hsu HC, Lapke N, Chen SJ, Lu YJ, Jhou RS, Yeh CY, Tsai WS, Hung HY, Hsieh JC, Yang TS. PTPRT and PTPRD deleterious mutations and deletion predict bevacizumab resistance in metastatic colorectal cancer patients. Cancers (Basel) 2018;10 doi: 10.3390/cancers10090314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kohno T, Otsuka A, Girard L, Sato M, Iwakawa R, Ogiwara H, Sanchez-Cespedes M, Minna JD, Yokota J. A catalog of genes homozygously deleted in human lung cancer and the candidacy of PTPRD as a tumor suppressor gene. Genes Chromosomes Cancer. 2010;49:342–52. doi: 10.1002/gcc.20746. [DOI] [PMC free article] [PubMed] [Google Scholar]