

Abstract
The acquired resistance to epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI) is the major reason for the failure of target therapy in advanced non small cell lung cancer (NSCLC) patients, the mechanism of which has not been fully elucidated yet. The present study aimed to investigate the different DNA methylation profile before and after acquired EGFR-TKI resistance, and explore the influence of the DNA demethylater, decitabine, on EGFR-TKI resistance. The DNA methylation chip was used to screen the genes whose DNA methylation status were changed in the EGFR-TKI sensitive human NSCLC cell line PC9, and the induced EGFR-TKI resistant NSCLC cell line PC9/GR (harboring T790M mutation). According to the results and literature reports, the tumor suppressor genes, RASSF1A and GADD45β were selected for further research. Methylation specific PCR (MSP) and western blot further confirmed that the promoters of these two genes were methylated, and the protein expressions were significantly inhibited in PC9/GR cells. Additionally, decitabine, the DNA methyl transferase inhibitor, could reverse the methylation status of RASSF1A and GADD45β promoters, elevate protein expression, and partially restore the sensitivity of PC9/GR cells to EGFR-TKI. To conclude, our results suggested that the DNA methylation of RASSF1 and GADD45β may play a role in EGFR-TKI resistance, and epigenetic intervention might be an effective strategy to reverse EGFR-TKI resistance, suggesting further study.
Keywords: Lung adenocarcinoma, gefitinib resistance, methylation, decitabine
Introduction
Gefitinib, a small molecule epidermal growth factor receptor-tyrosine kinase inhibitor (EGFR-TKI), has been used successfully in the treatment of EGFR-mutated advanced non-small cell lung cancer (NSCLC) [1]. However, nearly all the patients who initially responded to the drug will acquire resistance after the treatment, with a median progression free survival (mPFS) of about 10 months [2]. The mechanism of acquired EGFR-TKI resistance has not been fully elucidated yet. However, the secondary T790M mutation at the 20 exon of EGFR gene is considered to be the major mechanism [3], which has been reported in 60% of NSCLC patients with acquired resistance to EGFR-TKI [4].
DNA methylation is an important epigenetic modification process, which leads to inactivation of gene. DNA methylation has been found to be abnormal in various types of cancer, and it is widely involved in the process of tumorigenesis, metastasis, and drug resistance [5]. It has been demonstrated that the methylation of tumor suppressor genes and DNA damage repair genes is involved in the resistance to chemotherapy drugs [6]. The hypermethylation of certain genes is also found to contribute to EGFR-TKI resistance [7,8]. 5-Aza-CdR, a DNA methylation transferase inhibitor, is found to be effective in reversing EGFR-TKI resistance [8]. However, the overall perspective of genome methylation abnormality and critical gene involved in the process has not been fully elucidated yet. The present research detected the status of gene promoter methylation in human lung adenocarcinoma cell line PC9 (harboring 19-Del of EGFR), and gefitinib resistant human lung adenocarcinoma cell line PC9/GR (harboring 19-Del and T790M mutation of EGFR) by DNA methylation chip technique. Screening enabled confirming of the possible genes whose methylation may relate to gefitinib acquired resistance. Further, we intervened PC9 and PC9/GR cell lines by decitabine, a DNA demethylation drug, to evaluate the effect of DNA demethylation on gefitinib acquired resistance.
Material and method
Cell culture and treatment
The gefitinib-sensitive human lung adenocarcinoma cell line PC9 (harboring EGFR 19 exon deletion) and gefitinib-resistant human lung adenocarcinoma cell line PC9/GR (harboring EGFR 19 exon deletion and 20 exon T790M mutation) were donated by Guangzhou Insititute of Respiratory Disease. All cells were cultured at 37°C in a humidified atmosphere of 5% CO2 and maintained in a defined medium (1:1 mixture of DMEM and F12 supplemented with 10% FBS). In the first panel of experiments, gefitinib (AstraZenac) was dissolved in double-distilled water. PC9 and PC9/GR cells were treated with different concentrations of gefitinib. In the second panel of experiments, PC9 and PC9/GR cells were treated by different concentrations of decitabine. Cells were then collected for the DNA methylation array, cell viability assay, real-time RT-PCR, methylation specific PCR (MSP), and western blot analysis.
Cell viability assay
Cell viability was assessed by trypan blue exclusion and methylthiazol tetrazolium (MTT) assay. In the first panel of experiments, PC9 and PC9/GR cells were seeded in 96-well culture plates and then synchronously cultured in serum-free medium for 16 h. Cells were then incubated in medium containing different concentrations of gefitinib (0-10.0 μM for PC9 and 0-40.0 μM for PC9/GR) for 48 h. In the second panel of experiments, PC9/GR cells were seeded in 96-well culture plates and then, cells were incubated in medium containing different concentrations of decitabine (0-80.0 μM) for 48 h. In the third panel of experiments, PC9 and PC9/GR cells were seeded in 96-well culture plates and then incubated in medium with or without decitabine for 48 h. Then, both PC9 and PC9/GR cells were treated by different concentrations of geftinib. After all interventions, a volume of 20 μl of methylthiazol tetrazolium was then added to each well. The supernatant was then discarded and DMSO (150 μl) was added to each well to dissolve the blue formazan crystals. The optical density (OD) was measured using a microplate reader (SpectraMax 340, Molecular Devices, Japan) at a wavelength of 490 nm. The arithmetic mean of the OD of each group of wells was calculated, and the OD of the cells in the normal group was assigned a relative value of 100.
RT-PCR
Total RNA extraction was performed with TRIZOL (Invitrogen). RNA purity was determined by the A 260 to A 280 ratio. cDNA was synthesized according to the instructions in the RevertAidTM First Strand cDNA Synthesis Kit (Fermentas). mRNA expression was assessed by real-time PCR using the SYBR green PCR reagent kit (Applied Biosystems, Foster City, CA). PCR thermal cycling parameters consisted of one cycle of 2 min at 50°C and 10 min at 95°C, followed by 40 cycles of 10 min at 72°C. The mRNA levels of various genes were calculated after normalizing with glyceraldehyde 3-phosphate dehydrogenase or β-actin, and the 2ΔΔCt method was used for analyzing target gene expression level. All the oligonucleotide primers were designed by Primer Premier 5.0.
Methylation specific PCR
Genomic DNA was extracted from PC9 and PC9/GR cells using the GenElute Mammalian Genomic DNA Miniprep Kit (G1N10; Sigma) as described above. The DNA was then bisulfite converted using EZ DNAMethylation-Gold Ki (D5005; Zymo Research). The DNA was incubated with sodium bisulphite that converts unmethylated cytosines to uracil but leaves 5-MeC intact. The RASSF1A and GADD45β promoter has previously been established to be located around the ATG start site53; we examined this region for CpG islands, and primers were designed using the MethPrimer software.54 The converted DNA was then used in PCR with primers specific for methylated DNA and unmethylated DNA of the RASSF1A and GADD45β promoter region. The Epitect Control DNA Set (59695; Qiagen) was used for positive and negative control DNA in the PCR. Water-only was used as a negative control. Methylated primer sequences are: RASSF1A-m-F: 5’-GTGTTAACGCGTTGCGTATC-3’, RASSF1A-m-R: 5’-AACCCCGCGAACTAAAAACGA-3’, GADD45β-m-F: 5’-CGGAATTGTGTTTTGGTCG-3’, GADD45β-m-R: 5’-ACCAACCTATATAAAAACGCG-3’. Unmethylated primer sequences are: RASSF1A-u-F: 5’-TTTGGTTGGAGTGTGTTAATGTG-3’, RASSF1A-u-R: 5’-CAAACCCCACAAACTAAAAACAAA-3’, GADD45β-u-F: 5’-ATGTGGTTTTTTGGTATGAGTT-3’, GADD45β-u-R: 5’-CACCAACCTATAAAAAACACA-3’. The PCR conditions were: a cycle of denaturation at 95°C for 7 minutes, then denaturation at 95°C for 10 seconds, annealing at 55°C for 10 seconds, and elongation at 72°C for 8 seconds. This was repeated from the second denaturation step for 45 cycles followed by a final elongation step at 72°C for 1 minute. Three normal and 3 glaucoma donors were used for all experiments. Independent triplicate experiments were carried out for each donor.
DNA methylation analysis
The DNA methylation data for PC9 and PC9/GR cell were generated in Shanghai Kangcheng Bio-technology cooperation, using the NimbleGen Human DNA Methylation 3x720K Promoter Plus CpG Island Array Methylation (NimbleGem) according to the manufacturer’s instructions. The hybridization signal was detected by chip scanner and analysis by software. According to the literature reports, the DNA methylation status of various genes including tumor suppresor gene family (p16, P15, P18, P19, LKB1, PTEN, 4.1B/DAL-1, GPRC5A, GATA, PTPRO, RASSF1, GADD45β, CIP-KIP family, nm23, KAI1, APH, MCC, APC), oncogene family (KRAS, NRAS, HRAS, MYC, SRC), tyrosine receptor kinase (EGFR, ERBB2, PDGFRβ, FLT1, KDR), DNA damage repaire genes (MGMT, ERCC, RRM1, hMSH, hMLH, hPMS, RAD), and DNMT, were selected for further analysis.
Western blot
Cells were collected and homogenized in 1 ml of lysis buffer (50 mmol/l Hepes, pH 7.5, 150 mmol/l NaCl, 1.5 mmol/l MgCl2, 1 mmol/l ethylene glycol tetraacetic acid, 10% glycerol, 1% Triton X-100, 1 g/ml aprotinin, 1 g/ml leupeptin, 1 mmol/l phenylmethyl sulfonyl fluoride, 0.1 mmol/l sodium orthovanadate) at 4°C. After centrifugation, soluble lysates (60-80 μg) were loaded in each lane and separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. Membranes were blocked with 5% nonfat dry milk in Tris-buffered saline/0.5% Tween-20 for 1 h, washed with Tris-buffered saline/Tween-20, and incubated with rabbit polyclonal RASSF1A antibody (1:2,000; PLlabs, US) or rabbit polyclonal GADD45β antibody (1:2,000; Abcam, UK). Blots were rinsed with Tris-buffered saline/Tween-20 and subsequently incubated with HRP goat anti-rabbit IgG (1:10,000; Kang wei shi ji, China). After washing with Tris-buffered saline/Tween-20, the blots were developed with the enhanced chemiluminescence method (Amersham, UK). The intensity of the identified bands was quantified by densitometry. Results were expressed as arbitrary densitometric units.
Statistical analysis
SPSS 13.0 was used for statistical analysis. Data are presented as the mean ± SD. Student t test was used for mean comparison, while Chi-square test was use for rate comparison. P<0.05 was considered significant.
Results
Different DNA methylation status between PC9 and PC9/GR
NimbleGen Human DNA Methylation 3x720K Promoter Plus CpG Island Array Methylation CHIP was used to detect the different gene promotor methylation status between PC9 and PC9/GR. The results showed that there are 2818 genes whose promotor was methylated in PC9/GR cells, compared with that in PC9 cells (Supplementary 1). As shown in Table 1, the tumor suppressor gene, RASSF1, GADD45β and PTPRO, the oncogene HRAS, and those genes which coded for tyrosine kinase receptors, including FLT1, KDR and ERBB2, were unmethylated in PC9, while methylated after secondary T790M mutation. Meanwhile, GADD45γ was methylated in PC9 cell and demethylated in PC9/GR cell. The methylation status of GADD45α has not been changed in PC9 and PC9/GR. The tumor suppressor genes, RASSF1 and GADD45β, were chosen for further experiments.
Table 1.
Different DNA methylation status of important genes in PC9 and PC9/GR
| Gene | Methylation status in PC9 | Methylation status in PC9/GR |
|---|---|---|
| RASSF1 | - | + |
| GADD45β | - | + |
| GADD45α | - | - |
| GADD45γ | + | - |
| PTPRO | - | + |
| HRAS | - | + |
| FLT1 | - | + |
| KDR | - | + |
| ERBB2 | - | + |
-: unmethylated, +: methylated.
RASSF1A and GADD45β promoter was methylated and their expression were inhibited in PC9/GR
Methylation specific PCR (MSP) was used to further confirm the gene promoter methylation status of RASSF1A and GADD45β. The results showed that the promoters of RASSF1A and GADD45β were under unme-thylated status in PC9 cells while totally methylated in PC9/GR cells (Figures 1 and 2). Further, western blot showed that the expression of RASSF1A and GADD45β were down-regulated due to the transcription inhibition caused by promoter methylation (Figure 3).
Figure 1.
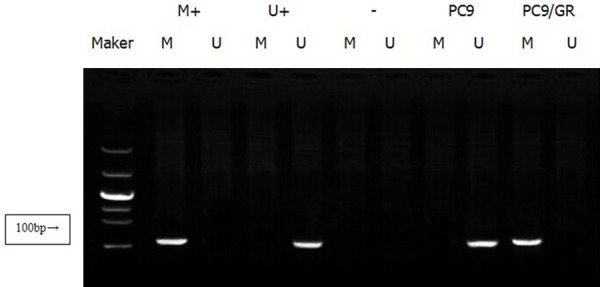
The methylation status of RASSF1A promoter in PC9 and PC9/GR. MSP results showed that the RASSF1A promoter was unmethylated in PC9 cells, and methylated in PC9/GR cells. (M+methylated positive control, U+: unmethylated positive control, -: negative control).
Figure 2.
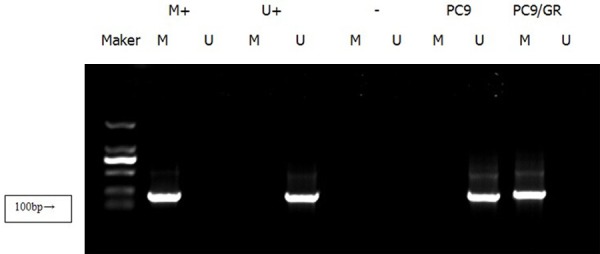
The methylation status of GADD45β promoter in PC9 and PC9/GR. MSP results showed that GADD45β promoter was unmethylated in PC9 cells while methylated in PC9/GR cells (M+: methylated positive control, U+: unmethylated positive control, -: negative control).
Figure 3.
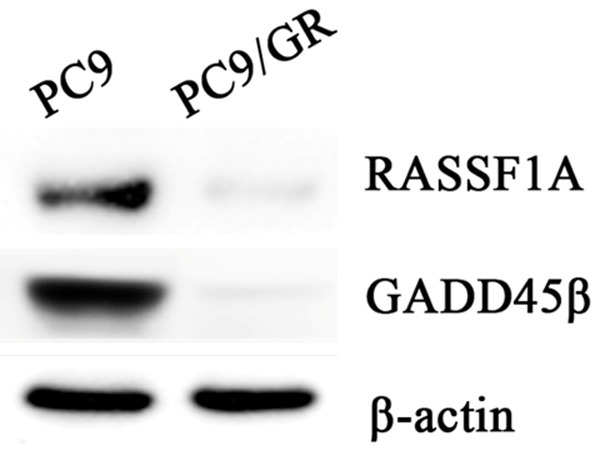
RASSF1A and GADD45β expression in PC9 and PC9/GR. Western blot results show that the promoter methylation of RASSF1A and GADD45β leads to downregulation of their protein expression.
Decitabine inhibited PC9/GR cell proliferation
MTT assay was used to evaluate the decitabine’s effect on PC9/GR cells. The results showed that the proliferation of PC9/GR cells was inhibited after 48 h decitabine intervention. And the effect was dose-dependent. The IC50, IC20, IC10 values were: 56.8 μmol/l, 13.2 μmol/l, 8.7 μmol/l (P<0.01) (Figure 4). 13.2 μM decitabine was selected for further experiments.
Figure 4.
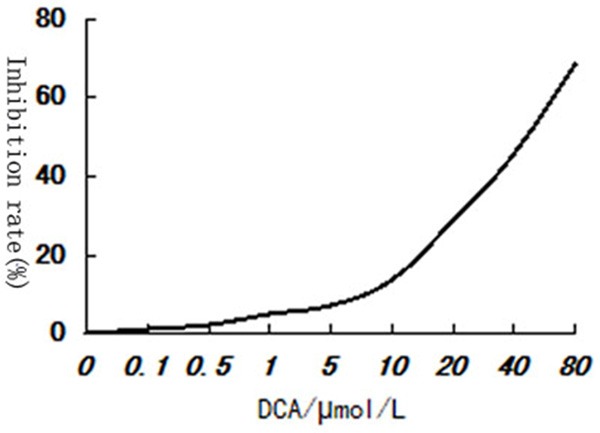
Decitabine dose-dependently inhibits the proliferation of PC9/GR cells.
Decitabine increased the sensitivity of PC9/GR to gefitinib
After incubation with or without 13.2 μM decitabine (DCA) for 48 h, PC9 and PC9/GR cells were treated by different concentrations of gefitinib. MTT assay was used to evaluate the proliferation inhibition effect of gefitinib on PC9 and PC9/GR cells. The results showed that gefitinib could dose-dependently inhibit cell proliferation in both the control group and the DCA group. The IC50 value of gefitinib on PC9 cells with and without DCA were 0.08 μM and 0.07 μM. While the IC50 value of gefitinib on PC9/GR cells with and without DCA were 4.62 μM and 7.80 μM. The resistance index (RI) were 66.00 and 97.50 separately (Figure 5; Table 2) (P<0.01) (RI = IC50 of gefitinib on PC9/GR/IC50 of gefitinib on PC9). These results indicated that PC9/GR resistance to gefitinib could be partially reversed by decitabine.
Figure 5.
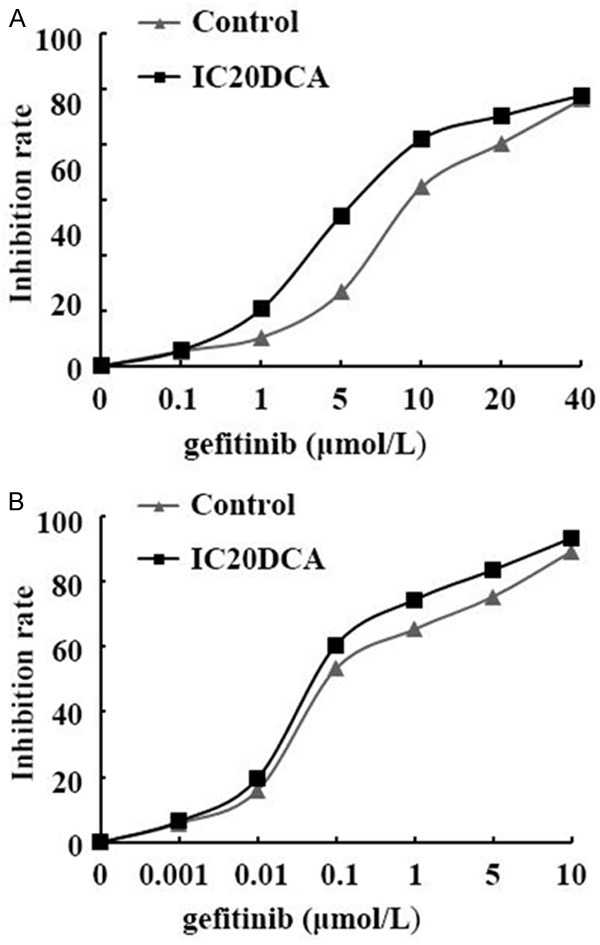
The inhibition rate of gefitinib in PC9 (A) and PC9/GR (B) cells before and after DCA treatment. MTT assay showed that IC20DCA intervention could sensitize PC9/GR cells to gefitinib, but the sensitivity to gefitinib in PC9 cells was not changed before and after IC20DCA treatment.
Table 2.
IC50 of gefitinib on PC9 and PC9/GR with and without DCA intervention
| Gefitinib | PC9 | PC9/GR | RI |
|---|---|---|---|
|
| |||
| IC50 (μmol/L) | IC50 (μmol/L) | ||
| Control | 0.08 | 7.80 | 97.50 |
| IC20DCA | 0.07 | 4.62 | 66.00 |
Decitabine demethylated RASSF1A and GADD45β promotor and elevated their expression in PC9/GR cells
Methylation specific PCR (MSP) was used to detect RASSF1A and GADD45β promoter methylation status in PC9/GR cells after 48 h ICD20 decitabine intervention. As shown in Figure 6, the promoter of RASSF1A was unmethylated in PC9, while totally methylated in PC9/GR cells. After 48 h intervention of ICD20 decitabine, there were both methylated and unmethylated bands, indicating that decitabine could partially demethylate the RASSF1A promoter. On the other hand, for GADD45β, its promoter was unmethylated in PC9 cells, while totally methylated in PC9/GR cells. However, ICD20 decitabine could totally demethylate GADD45β promoter. The western blot results further showed that the protein expressions of these genes were elevated after decitabine intervention (Figure 7).
Figure 6.
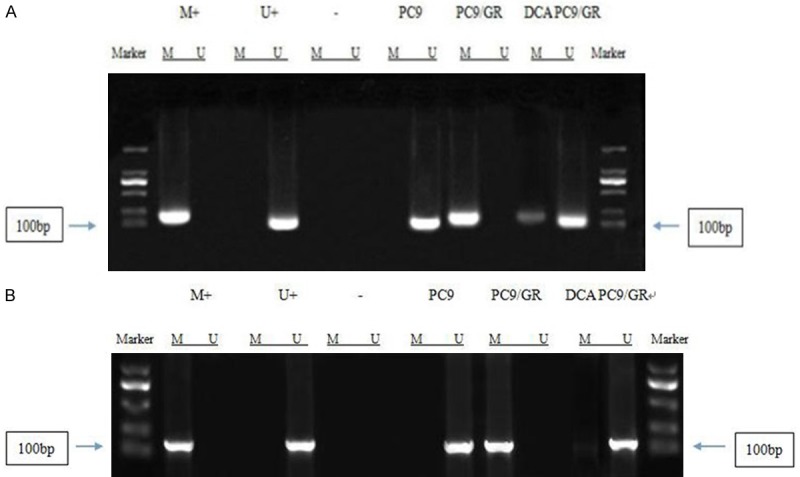
The status of RASSF1A and GADD45β promoter methylation in PC9, PC9/GR, and PC9/GR after decitabine intervention. A. RASSF1A promoter was demethylated by DCA in PC9/GR cells; B. GADD45β promoter was demethylated by DCA in PC9/GR cells.
Figure 7.

The expression of RASSF1A and GADD45β protein in PC9, PC9/GR and PC9/GR with decitabine intervention. Western blot results further showed that the protein expression of RASSF1A and GADD45β were partially restored after DCA treatment.
Discussion
The epidermal growth factor receptor tyrosine kinase inhibitor (EGFR-TKI) significantly improves the prognosis of EGFR mutant advanced non-small cell lung cancer (NSCLC). However, nearly all patients will acquire resistance to the drug, which causes failure of treatment. The major mechanism of acquired gefitinib resistance is the missense mutation on the 2369 nucleotide of EGFR exon 20, the T790M mutation. The secondary T790M mutation induces a conformational change of EGFR protein and increases its affinity for ATP, which subsequently inhibits the combination between gefitinib and EGFR kinase domain, and facilitates the auto-activation signal transduction and cell proliferation [9,10]. Besides T790M mutation, there are many other mechanisms involved in the acquired resistance to EGFR-TKI, including c-Met over-expression, EMT, and transforming to SCLC. It has been reported that the gene promoter methylation status of certain pathways may affect the efficiency of EGFR-TKI in NSCLC patients [11]. Also, gene methylation maybe an incentive of T790M formation [12]. So far, the research on the role of epigenetic mechanism in the process of EGFR-TKI resistance is rare. There has been no report on the change of gene methylation profile in T790M-mediated EGFR-TKI resistance in NSCLC cells.
RASSF1A is a tumor suppressor gene, and in vivo and in vitro research has demonstrated that it can inhibit cell proliferation and induce cell apoptosis [13,14]. Abnormal methylation and allelic deletion are the major mechanisms of RASSF1A deactivation, while the point mutation is rare [15]. RASSF1A promoter methylation can be detected in 63% of NSCLC cell lines and 30% of NSCLC tumor tissues, but it cannot be detected in normal lung tissue [16]. It has been reported that the incidence of RASSF1A promoter methylation in NSCLC tissue was about 47% [17,18], but its correlation with pathological type is still controversial [19,20]. Another multivariate analysis reported that the incidence of RASSF1A promoter methylation is not correlated with the incidence of K-ras, P53 and EGFR gene mutation, after age, sex, smoking status, pathological type of NSCLC, and staging adjustment, which indicated that RASSF1A promoter methylation might be an independent issue in the pathogenesis of NSCLC. The role of gene promoter methylation in the multidrug resistance to chemotherapy drugs in NSCLC has been reported. Guo et al. [21] reported that RASSF1, MT1G and GPR56 promoter methylation is involved in the cisplatin resistance in human adenocarcinoma cell line A549. However, the role of RASSF1A promoter methylation in acquired EGFR-TKI resistance in NSCLC has not been reported yet.
Growth arrest and DNA damage inducible protein 45β (GADD45β) is a downstream gene of p53 and BRCA1, which encodes a conservative nucleoprotein regulating P53 and JNK signal pathways [22-24]. It has been found that GADD45β, interacted with other genes, is involved in DNA damage repair [22]. The incidence of GADD45β promoter methylation in NSCLC is very low. One research found that GADD45β was expressed in normal lung tissue and no gene promoter methylation was detected in 5 NSCLC cell lines [25]. Na et al [26] detected GADD45β promoter methylation in 139 NSCLC patients’ tissues, and reported that the incidence of GADD45β promoter methylation was 7.2%. Therefore, the role of GADD45β promoter methylation was not very clear.
In this research, a DNA methylation chip was used to detect the genomic DNA of PC9 cell and PC9/GR cell. The results showed that, compared with PC9 cells, there are large amount of gene promoter methylations in PC9/GR cells. After referring to the reference, we chose two tumor suppressor genes, RASSF1A and GADD45β, for further research. Consistent with the results of DNA methylation chips, methylation specific PCR (MSP) results also showed that RASSF1A and GADD45β promoter were unmethylated in PC9 cells, while methylated in PC9/GR cells, indicating that the T790M mutation may induce the methylation of these genes’ promoters. Western blot results further confirmed that gene promoter methylation of these genes inhibited their expression. However, the relationship between the methylation silencing of these two genes and the acquired gefitinib resistance of PC9 cells still needs further research.
The mechanism of acquired EGFR-TKI resistance is so complex that, besides some major mechanisms, there may be other mechanisms involved in this process. Epigenetic mechanisms may be an important aspect. So far, it has been reported that epigenetic mechanisms are involved in the drug resistance of cancer cells. In the realm of chemotherapy drug resistance, it is reported that, the hypermethylation induced silence of tumor suppressor genes RASSF1 and BRCA1 is related to the resistance to carboplatin in ovarian carcinoma cells [27]. Also, the combination of DNA methyltransferase inhibitor, decitabine, and carboplatin could demethylate RASSF1 and BRCA1, and reverse the resistance to carboplatin [27].
In the realm of EGFR-TKI resistance, there is research demonstrating that the deletion of PTEN phosphatase function and the sustained activation of Akt pathway play an important role. The expression of PTEN is closely related to DNA methylation [28]. In the acquired gefitinib resistant lung adenocarcinoma cell lines PC9/f9 and PC9/f14, the expression of PTEN mRNA and protein were obviously inhibited, compared with the maternal cell line, gefitinib sensitive PC9. And the epigenetic modulators, including DNA demethylation drugs, 5-aza-2’-deoxycytidine, and histone deacetylase inhibitor, TSA, could elevate the expression of PTEN mRNA and protein, and restore the sensitivity of the cells to gefitinib.
In this research, we demonstrated that decitabine could demethylate the DNA promoters of RASSF1A and GADD45β in acquired gefitinib resistant lung adenocarcinoma cell line PC9/GR, induce a partial restoration of RASSF1A and GADD45β protein expression, and partially reverse the gefitinib resistance of the cell, which indicated that, besides T790M mutation, the methylation silencing of RASSF1A and GADD45β may be a concurrent non-mainstream mechanism in the process of gefitinib resistance in PC9/GR cells. It is rational to hypothesize that the epigenetic drug intervention at the early stage of EGFR-TKI treatment may be helpful to delay the onset of EGFR-TKI resistance, which brings us a new insight into the prevention and reversion of EGFR-TKI acquired resistance.
Acknowledgements
This work was supported by The National Natural Science Foundation of China (No. 81771896).
Disclosure of conflict of interest
None.
Supporting Information
References
- 1.Mok TS, Wu YL, Thongprasert S, Yang CH, Chu DT, Saijo N, Sunpaweravong P, Han B, Margono B, Ichinose Y, Nishiwaki Y, Ohe Y, Yang JJ, Chewaskulyong B, Jiang H, Duffield EL, Watkins CL, Armour AA, Fukuoka M. Gefitinib or carboplatin-paclitaxel in pulmonary adenocarcinoma. N Engl J Med. 2009;361:947–957. doi: 10.1056/NEJMoa0810699. [DOI] [PubMed] [Google Scholar]
- 2.Maemondo M, Inoue A, Kobayashi K, Sugawara S, Oizumi S, Isobe H, Gemma A, Harada M, Yoshizawa H, Kinoshita I, Fujita Y, Okinaga S, Hirano H, Yoshimori K, Harada T, Ogura T, Ando M, Miyazawa H, Tanaka T, Saijo Y, Hagiwara K, Morita S, Nukiwa T North-East Japan Study Group. Gefitinib or chemotherapy for non-small-cell lung cancer with mutated EGFR. N Engl J Med. 2010;362:2380–2388. doi: 10.1056/NEJMoa0909530. [DOI] [PubMed] [Google Scholar]
- 3.Kobayashi S, Boggon TJ, Dayaram T, Jänne PA, Kocher O, Meyerson M, Johnson BE, Eck MJ, Tenen DG, Halmos B. EGFR mutation and resistance of non-small cell lung cancer to gefitinib. N Engl J Med. 2005;352:786–792. doi: 10.1056/NEJMoa044238. [DOI] [PubMed] [Google Scholar]
- 4.Westover D, Zugazagoitia J, Cho BC, Lovly CM, Paz-Ares L. Mechanisms of acquired resistance to first- and second-generation EGFR tyrosine kinase inhibitors. Ann Oncol. 2018;29:i10–i19. doi: 10.1093/annonc/mdx703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Klutstein M, Nejman D, Greenfield R, Cedar H. DNA methylation in cancer and aging. Cancer Res. 2016;76:3446–3450. doi: 10.1158/0008-5472.CAN-15-3278. [DOI] [PubMed] [Google Scholar]
- 6.Dejeux E, Rønneberg JA, Solvang H, Bukholm I, Geisler S, Aas T, Gut IG, Børresen-Dale AL, Lønning PE, Kristensen VN, Tost J. DNA methylation profiling in doxorubicin treated primary locally advanced breast tumours identifies novel genes associated with survival and treatment response. Mol Cancer. 2010;9:68. doi: 10.1186/1476-4598-9-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Maeda M, Murakami Y, Watari K, Kuwano M, Izumi H, Ono M. CpG hypermethylation contributes to decreased expression of PTEN during acquired resistance to gefitinib in human lung cancer cell lines. Lung Cancer. 2015;87:265–271. doi: 10.1016/j.lungcan.2015.01.009. [DOI] [PubMed] [Google Scholar]
- 8.Yang B, Yang ZG, Gao B, Shao GG, Li GH. 5-Aza-CdR can reverse geftinib resistance caused by DAPK gene promoter methylation in lung adenocarcinoma cells. Int J Clin Exp Pathol. 2015;8:12961–12966. [PMC free article] [PubMed] [Google Scholar]
- 9.Cross DA, Ashton SE, Ghiorghiu S, Eberlein C, Nebhan CA, Spitzler PJ, Orme JP, Finlay MR, Ward RA, Mellor MJ, Hughes G, Rahi A, Jacobs VN, Red Brewer M, Ichihara E, Sun J, Jin H, Ballard P, Al-Kadhimi K, Rowlinson R, Klinowska T, Richmond GH, Cantarini M, Kim DW, Ranson MR, Pao W. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discov. 2014;4:1046–1061. doi: 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Yun CH, Mengwasser KE, Toms AV, Woo MS, Greulich H, Wong KK, Meyerson M, Eck MJ. The T790M mutation in EGFR kinase causes drug resistance by increasing the affinity for ATP. Proc Natl Acad Sci U S A. 2008;105:2070–2075. doi: 10.1073/pnas.0709662105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhu J, Wang Y, Duan J, Bai H, Wang Z, Wei L, Zhao J, Zhuo M, Wang S, Yang L, An T, Wu M, Wang J. DNA methylation status of wnt antagonist SFRP5 can predict the response to the EGFR-tyrosine kinase inhibitor therapy in non-small cell lung cancer. J Exp Clin Cancer Res. 2012;31:80. doi: 10.1186/1756-9966-31-80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fujii A, Harada T, Iwama E, Ota K, Furuyama K, Ijichi K, Okamoto T, Okamoto I, Takayama K, Nakanishi Y. Hypermethylation of the CpG dinucleotide in epidermal growth factor receptor codon 790: implications for a mutational hotspot leading to the T790M mutation in non-small-cell lung cancer. Cancer Genet. 2015;208:271–8. doi: 10.1016/j.cancergen.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 13.Lai Q, Xu YH, Chen Q, Tang L, Li AG, Zhang LF, Zhang CF, Song JF, Du ZZ. The loss-of-function of DNA methyltransferase 1 by siRNA impairs the growth of non-small cell lung cancer with alleviated side effects by reactivation of RASSF1A and APC in vitro and vivo. Oncotarget. 2017;8:59301–59311. doi: 10.18632/oncotarget.19573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Schmidt ML, Hobbing KR, Donninger H, Clark GJ. RASSF1A deficiency enhances RAS-driven lung tumorigenesis. Cancer Res. 2018;78:2614–2623. doi: 10.1158/0008-5472.CAN-17-2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Dammann RH, Richter AM, Jiménez AP, Woods M, Küster M, Witharana C. Impact of natural compounds on DNA methylation levels of the tumor suppressor gene RASSF1A in cancer. Int J Mol Sci. 2017:18. doi: 10.3390/ijms18102160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Burbee DG, Forgacs E, Zöchbauer-Müller S, Shivakumar L, Fong K, Gao B, Randle D, Kondo M, Virmani A, Bader S, Sekido Y, Latif F, Milchgrub S, Toyooka S, Gazdar AF, Lerman MI, Zabarovsky E, White M, Minna JD. Epigenetic inactivation of RASSF1A in lung and breast cancers and malignant phenotype suppression. J Natl Cancer Inst. 2001;93:691–699. doi: 10.1093/jnci/93.9.691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu Y, Gao W, Siegfried JM, Weissfeld JL, Luketich JD, Keohavong P. Promoter methylation of RASSF1A and DAPK and mutations of K-ras, p53, and EGFR in lung tumors from smokers and never-smokers. BMC Cancer. 2007;7:74. doi: 10.1186/1471-2407-7-74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marsit CJ, Kim DH, Liu M, Hinds PW, Wiencke JK, Nelson HH, Kelsey KT. Hypermethylation of RASSF1A and BLU tumor suppressor genes in non-small cell lung cancer: implications for tobacco smoking during adolescence. Int J Cancer. 2005;114:219–23. doi: 10.1002/ijc.20714. [DOI] [PubMed] [Google Scholar]
- 19.Kim DH, Kim JS, Ji YI, Shim YM, Kim H, Han J, Park J. Hypermethylation of RASSF1A promoter is associated with the age at starting smoking and a poor prognosis in primary non-small cell lung cancer. Cancer Res. 2003;63:3743–3746. [PubMed] [Google Scholar]
- 20.Kim DH, Kim JS, Park JH, Lee SK, Ji YI, Kwon YM, Shim YM, Han J, Park J. Relationship of ras association domain family 1 methylation and K-ras mutation in primary non-small cell lung cancer. Cancer Res. 2003;63:6206–6211. [PubMed] [Google Scholar]
- 21.Guo R, Wu G, Li H, Qian P, Han J, Pan F, Li W, Li J, Ji F. Promoter methylation profiles between human lung adenocarcinoma multidrug resistant A549/cisplatin (A549/DDP) cells and its progenitor A549 cells. Biol Pharm Bull. 2013;36:1310–1316. doi: 10.1248/bpb.b13-00153. [DOI] [PubMed] [Google Scholar]
- 22.Cretu A, Sha X, Tront J, Hoffman B, Liebermann DA. Stress sensor Gadd45 genes as therapeutic targets in cancer. Cancer Ther. 2009;7:268–276. [PMC free article] [PubMed] [Google Scholar]
- 23.Papa S, Zazzeroni F, Bubici C, Jayawardena S, Alvarez K, Matsuda S, Nguyen DU, Pham CG, Nelsbach AH, Melis T, De Smaele E, Tang WJ, D’Adamio L, Franzoso G. Gadd45 beta mediates the NF-kappa B suppression JNK signaling by targeting MKK7/JNKK2. Nat Cell Biol. 2004;6:146–53. doi: 10.1038/ncb1093. [DOI] [PubMed] [Google Scholar]
- 24.De Smaele E, Zazzeroni F, Papa S, Nguyen DU, Jin R, Jones J, Cong R, Franzoso G. Induction of gadd45 beta by NF-kappa B downregulates pro-apoptotic JNK signaling. Nature. 2001;414:308–313. doi: 10.1038/35104560. [DOI] [PubMed] [Google Scholar]
- 25.Ying J, Srivastava G, Hsieh WS, Gao Z, Murray P, Liao SK, Ambinder R, Tao Q. The stress-responsive gene GADD45G is a functional tumor suppressor, with its response to environmental stresses frequently disrupted epigenetically in multiple tumors. Clin Cancer Res. 2005;11:6442–6449. doi: 10.1158/1078-0432.CCR-05-0267. [DOI] [PubMed] [Google Scholar]
- 26.Na YK, Lee SM, Hong HS, Kim JB, Park JY, Kim DS. Hypermethylation of growth arrest DNA-damage inducible gene 45 in non-small cell lung cancer and its relationship with clinicopathologic features. Mol Cells. 2010;30:89–92. doi: 10.1007/s10059-010-0092-1. [DOI] [PubMed] [Google Scholar]
- 27.Glasspool RM, Brown R, Gore ME, Rustin GJ, McNeish IA, Wilson RH, Pledge S, Paul J, Mackean M, Hall GD, Gabra H, Halford SE, Walker J, Appleton K, Ullah R, Kaye S Scottish Gynaecological Trials Group. A randomised, phase II trial of the DNA-hypomethylating agent 5-aza-2’-deoxycytidine (decitabine) in combination with carboplatin vs carboplatin alone in patients with recurrent, partially platinum-sensitive ovarian cancer. Br J Cancer. 2014;110:1923–1929. doi: 10.1038/bjc.2014.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Noro R, Gemma A, Miyanaga A, Kosaihira S, Minegishi Y, Nara M, Kokubo Y, Seike M, Kataoka K, Matsuda K, Okano T, Yoshimura A, Kudoh S. PTEN inactivation in lung cancer cells and the effect of its recovery on treatment with epidermal growth factor receptor tyrosine kinase inhibitors. Int J Oncol. 2007;31:1157–1163. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.