

Abstract
MicroRNAs (miRNAs) contribute to multiple cellular processes in embryonic development and disorders. Several miRNAs are strongly associated with the progression of peripheral vascular disease. Recently, it was reported that miRNA (miR)-92 is one of the most upregulated miRNAs in vascular injury after intervention in the lower extremities; however, the function of miR-92 during proliferative vascular diseases remains unclear and the potential targets of miR-92 are poorly characterized. In this study, we investigated the expression of miR-92 in vitro and in vivo, and explored the associated underlying mechanism. qRT-PCR analysis showed that miR-92 expression was highly upregulated in patients with artery restenosis compared to those without restenosis. Platelet-derived growth factor (PDGF)-BB is regarded as a critical regulator of the phenotypic switch of VSMCs. miR-92 expression was significantly upregulated in PDGF-BB-stimulated VSMCs. Introducing an miR-92 mimic into VSMCs enhanced cell proliferation and migration, and induced S-phase arrest. The luciferase reporter assay revealed that KLF4 is a downstream target gene of miR-92. In vivo, miR-92 overexpression promoted neointimal formation, and thus resulted in a dramatic increase in intimal-medial area and thickness. These results reveal that miR-92 regulates VSMC function by directly targeting KLF4, which may be an important finding useful for the diagnosis and treatment of vascular restenosis and injury, arteriosclerosis, and other proliferative vascular diseases.
Keywords: microRNA-92, vascular smooth muscle cell, Kruppel like factor 4, platelet-derived growth factor-BB, vascular restenosis, vascular injury
Introduction
Arteriosclerosis obliterans (ASO) of the lower extremities is a common vascular disease and remains the main cause of adult limb loss worldwide [1,2]. Several risk factors have been reported to be involved in the development of ASO following the progression of atherosclerosis in a lower extremity causing the arterial canal to narrow, and even a complete block of blood flow. Although surgery is the leading therapeutic strategy for ASO, many patients develop vascular restenosis, which is regarded as the major limiting factor for the long-term success of revascularization approaches [3]. Recently, a number of reports have indicated that vascular smooth muscle cells (VSMCs) have an important role in the pathogenesis of ASO [4,5]. Generally, during ASO, quiescent VSMCs migrate from the medial arterial wall into the intimal space and gradually form a neointima. This formation combined with the presence of multiple extracellular matrix proteins results in vascular stenosis [6]. However, the precise molecular underlying mechanisms implicated in the modulation of proliferation and migration of arterial smooth muscle cells (ASMCs) in ASO and vascular restenosis is still unclear.
MicroRNAs (miRNAs) are a class of short noncoding RNAs that can serve as important regulators of human gene expression [7]. More than 2,000 human miRNAs have been cloned and sequenced, and typically they are considered to act as negative regulators of gene expression at the post-transcriptional level [8]. miRNAs affect diverse cellular processes in embryonic development and disease conditions [9-11]. Recently, several miRNAs have been reported to regulate the functions of ASMCs and participate in the development of atherosclerosis, including miR-21, miR-221, and miR-14 [12-14].
miR-92, a member of the miR-17-92 cluster, is highly expressed in young healthy human endothelial cells compared with senescent endothelial cells that have higher levels of oxidative stress and apoptosis [15]. Lan et al [16] reported that overexpression of miR-92 enhances the viability of endothelial cells under oxidative stress by targeting the protein kinase B (Akt) signaling pathway. Conversely, miR-92 was revealed as a negative regulator of angiogenesis by binding to a5 integrin subunit mRNA [17]. However, the potential role of miR-92 in ASMC function and peripheral vascular diseases remains largely unknown. The aim of this study was to determine the effect of miR-92 in vascular restenosis of patients with ASO after surgery, and to explore the relevant underlying mechanisms involved.
Materials and methods
Peripheral blood sample collection
Peripheral blood samples were collected from 100 patients with ASO who underwent lower limb artery occlusion intervention (55 females and 45 males; mean age, 52.5 ± 5.6 years). All patients underwent peripheral blood collection before and after arterial intervention and 6 months after discharge. 3 ml peripheral venous blood was collected in the early morning following fasting which was placed in an anticoagulant tube containing EDTA and rapidly mixed upside down. The present study was approved by the Ethics Committee of Pudong New Area Peoples’ Hospital Affiliated to Shanghai University of Medicine & Health Sciences and informed consent was obtained from all patients.
Reverse transcription-quantitative polymerase chain reaction (qRT-PCR) to detect miR-92 in peripheral blood
Total RNA was extracted from the separation of white cells from the blood, using TRIzol reagent (Thermo Fisher Scientific, Inc., Waltham, MA, USA) following the manufacturer’s instructions, and reverse transcription was performed to synthesize cDNA. miR-92 expression in white cells was evaluated by qRT-PCR. The assay was carried out as previously described [18].
Culture and transfection of VSMCs
Human VSMCs were purchased from the American Type Culture Collection (Manassas, VA, USA) and cultured in Dulbecco’s modified Eagle’s medium with 10% fetal bovine serum and 1% penicillin/streptomycin, at 37°C in a humidified incubator containing 5% CO2. When cultured cells reached >75% confluence, they were sub-cultured at a 1:2 ratio. VSMCs were plated in a 6-well plate and cultured for 24 h, then transfected with control mimic (miR-NC) or miR-92 mimic, and induced with platelet-derived growth factor (PDGF)-BB (4 ng/ml, Oncogene, USA) using a reagent kit (cat. no. PMG0041; Thermo Fisher Scientific, Inc.) according to the manufacturer’s protocol. miR-92, internal primers and miR-92 analogs were designed and synthesized by Shanghai Bioengineering Co., Ltd. (Shanghai, China).
VSMC proliferation
After VSMCs were transfected for 24 h, proliferation was determined using a CCK-8 kit according to the manufacturer’s protocol (Invitrogen; Thermo Fisher Scientific, Inc.). Cell proliferation was analyzed by measuring absorbance at 450 nm using a microplate reader. CCK-8 and cell counting were performed to detect cell proliferation as previously described [19,20].
Flow cytometry
VSMCs were suspended and collected at 48 h after PDGF and/or miR-92 treatment, washed with saline twice, fixed with 70% ethyl alcohol, and treated with 1% Triton X-100. Propidium iodide (50 mg/l) was added for staining at 37°C away from light. The cell cycle of VSMCs was detected by FACSCalibur (BD Biosciences, San Jose, CA, USA) as previously described [21]. Data were analyzed by FlowJo software (FlowJo LLC, Ashland, OR, USA).
Cell migration
After transfection for 48 h, VSMCs were treated with pancreatin, seeded onto 12-well plates, and cultured under standard conditions for 24 h. A wound was made by scraping the cell monolayer with a 200 μl pipette tip. Cell migration was determined by measuring the movement of cells into the scraped area. The process of wound closure was monitored and cells were imaged at 24 h after wounding under a microscope. The number of migratory cells was quantified under a microscope, and the data for statistical analyses were obtained from three independent cell migration experiments.
Dual luciferase assays
The targets and binding sites of miR-92 were predicted by TargetScan (http://www.targetscan.org/). To determine the interaction between miR-92 and Kruppel like factor 4 (KLF4) VSMCs were plated in a 24-well plate and co-transfected with 200 ng pMIR-KLF4-wt or pMIR-KLF4-mut, together with 100 nM miR-101 mimics or negative control mimics (NControl), and 10 ng pRL-TK vector containing the Renilla luciferase gene (Promega Corporation, Madison, WI, USA) using Lipofectamine 2000 (Gibco; Thermo Fisher Scientific, Inc.). After 48 h, reporter activity was determined using the Dual-Glo luciferase assay system according to the manufacturer’s instruction (Promega Corporation).
Western blot analysis
VSMCs were transfected with miR-NC or miR-92 mimic for 36 h, and then were lysed with IP solution and collected. Total protein samples were separated by 12% SDS-PAGE and then transferred to a PVDF membrane. The membrane was probed with primary antibody for 2 h at room temperature. Rabbit polyclonal antibody against KLF4 (1:1,000; Wuhan Boster Biological Technology, Ltd., Wuhan, China) and a mouse monoclonal antibody for GAPDH (1:4,000; Cell Signaling Technology, Inc., Danvers, MA, USA) were used as primary antibodies. The membranes were then probed with a goat anti-rabbit or a goat anti-mouse horseradish peroxidase-conjugated secondary antibody (1:20,000; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Signals were detected by incubation with a SuperSignal West Pico Chemiluminescent Substrate (Pierce; Thermo Fisher Scientific, Inc.), imaged using a ChemiDoc XRS (BioRad Laboratories, Inc., Hercules, CA, USA) and quantified using Quantity One software (Bio-Rad Laboratories, Inc.).
Mouse carotid artery injury model
The mouse carotid artery injury was performed as previously described [18]. C57BL/6 mice at 8-12 weeks of age were purchased from Charles River Laboratories (Wilmington, MA, USA). After housing for 7 days, mice were randomly divided into three groups: sham surgery group with intraperitoneal injection of 0.2 ml normal saline as control, and carotid artery injury models with intraperitoneal injection of miR-92 negative control or miR-92 agomir at doses of 10 nmol/(kg·ml), respectively (n=4). Mice were sacrificed after treatment for 21 days; the carotid artery was separated and injuries were analyzed using hematoxylin and eosin (H&E) staining. All the procedures were performed in accordance with national (D.L.n.26, March 4, 2014) and international (directive 2010/63/EU) laws and policies, and were approved by the Animal Experimental Ethics Committee of the Pudong New Area Peoples’ Hospital Affiliated to Shanghai University of Medicine & Health Sciences.
Statistical analyses
All quantitative data for statistical analyses were obtained from at least three independent experiments. The data are presented as the mean ± standard deviation. Comparison between two groups was performed using Student’s t-test and comparison between three or more groups was performed using ANOVA test with Bonferroni as a post hoc test. P<0.05 was considered significant.
Results
miRNA-92 expression in ASO patients with lower limb artery intervention
Previous reports have revealed that miR-92 was significantly upregulated in ASO arteries compared with normal arteries [22]. In this study, we performed a qRT-PCR to determine miR-92 expression in ASO patients before and after intervention in a lower limb artery in white cells of blood. As shown in Figure 1A, miR-92 expression was significantly downregulated after surgery compared with pre-surgery. To further investigate the distribution of miR-92 expression post-surgery, follow-up was conducted with 100 patients for 6 months, and miR-92 expression was highly upregulated in the 37 cases with artery restenosis compared with the 63 cases without restenosis after intervention (Figure 1B). Taken together, these results suggest that miR-92 is associated with vascular injury and restenosis caused by intervention.
Figure 1.
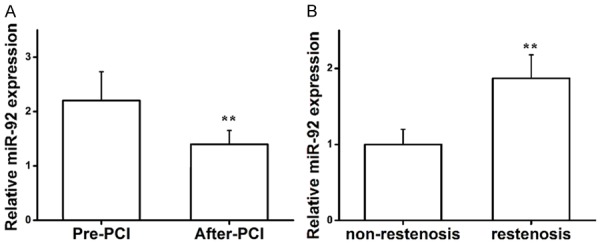
qRT-PCR analysis of miR-92 expression in peripheral blood. (A) miR-92 expression of ASO patients pre- and post-intervention within the lower limb artery. **P<0.01 vs. pre-PCI. (B) miR-92 expression in patients with and without restenosis. **P<0.01 vs. non-restenosis. Student’s T-test was used in (A and B). qRT-PCR, reverse transcription-quantitative polymerase chain reaction; miR, microRNA; ASO, arteriosclerosis obliterans.
Expression of miR-92 stimulated by FBS and PDGF-BB
To determine the distribution of miR-92 in different phase of vascular injuries, the miR-92 expression was analyzed. miR-92 was increased in patients with vascular injuries than in those without injuries, with a positive time-dependent relationship (Figure 2A). Furthermore, VSMCs were cultured with FBS, and the expression of miR-92 was significantly upregulated compared with serum-starved VSMCs (Figure 2B). It has been previously reported that PDGF-BB is involved in the growth of VSMCs and is regarded as a proliferative agent [23,24]. To further explore the association between miR-92 expression and PDGF-BB, PDGF-BB was used to stimulate VSMCs, which significantly upregulated miR-92 expression compared with the control group (Figure 2C). These results reveal that the expression of miR-92 is positively associated with FBS and PDGF-BB stimulation of VSMCs.
Figure 2.
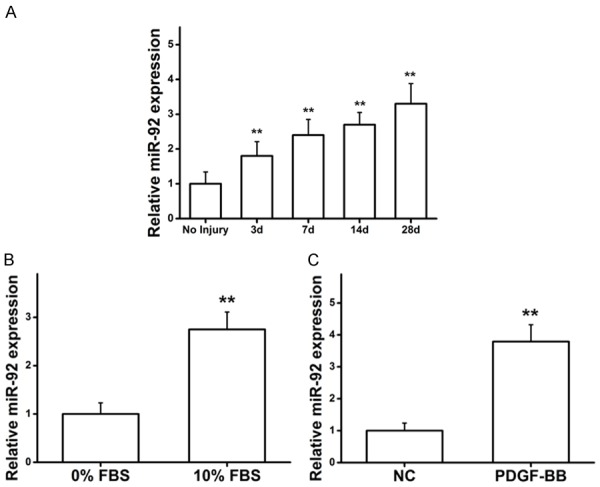
FBS and PDGF-BB stimulates the expression of miR-92. (A) The expression of miR-92 in patients with vascular injuries in a time-dependent manner. **P<0.01 vs. no injury. (B) and (C) miR-92 was upregulated under the induction of FBS and PDGF. **P<0.01 vs. control. Two-way ANOVA test with Bonferroni as a post hoc test was used in (A), and Student’s T-test was used in (B and C). FBS, fetal bovine serum; PDGF-BB, platelet-derived growth factor-BB; miR, microRNA.
Effects of miR-92 on VSMC proliferation and migration
Excessive VSMC proliferation and migration are critical events in the development of arteriosclerosis [25]. To determine the effects of miR-92 on the phenotype of VSMCs, a miR-92 mimic was introduced into PDGF-BB-stimulated VSMCs. The CCK8 assay showed that VSMCs transfected with miR-92 exhibited significantly increased proliferation compared with the miR-NC group (Figure 3A). Flow cytometry analysis demonstrated that miR-92 significantly enhanced the PDGF-stimulated acceleration of cell cycle progression from the G0/G1 phase to the S phase, suggesting that miR-92 induced S-phase arrest in VSMCs (Figure 3B). There was no significant difference in the G1-phase or G2M-phase (data not shown). Furthermore, a wound closure assay indicated that miR-92 significantly increased the PDGF-stimulated migration of VSMCs based on the quantification of migrated cells for both groups (Figure 3C). The above results suggest that miR-92 contributes to growth of VSMCs, including proliferation, migration, and S-phase arrest.
Figure 3.
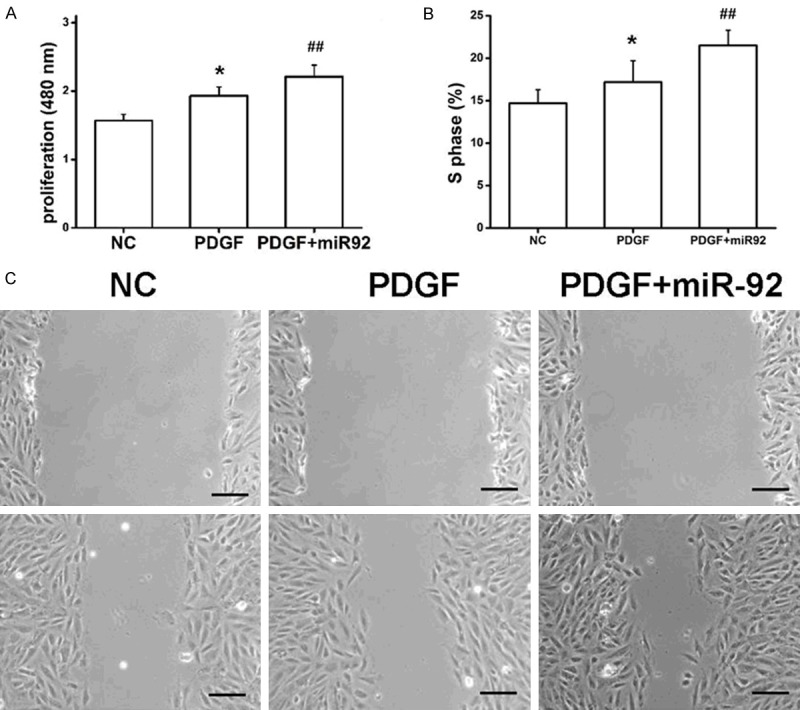
miR-92 affects VSMC proliferation, migration, and the cell cycle. (A) miR-92 was into cultured VSMCs and proliferation of PDGF-BB-stimulated cells was determined. (B) miR-92 enhanced the PDGF-stimulated acceleration of cell cycle progression to S phase. The results of G1, G2 and M phases were not shown. (C) miR-92 increased the PDGF-stimulated migration of VSMCs. One-way ANOVA test with Bonferroni as a post hoc test was used in (A and B). *P<0.05 vs. NC and PDGF + miR92; ##P<0.01 vs. NC and PDGF. miR, microRNA; VMSC, vascular smooth muscle cell; PDGF-BB, platelet-derived growth factor-BB.
miR-92 regulates KLF4 expression via targeting of the KLF4 3’-UTR
The KLF4 3’-UTR was identified by screening for potential miRNA binding sites using TargetScan. A potential binding site for miR-92 was identified at the 3’-UTR repressor element (Figure 4A). To determine whether miR-92 regulates expression of KLF4, the miR-92 binding site was mutated in a luciferase reporter (pMIR-KLF4-wt; wild type reporter), resulting in a mutant reporter (pMIR-KLF4-mut; Figure 4B). The pMIR-KLF4-wt or pMIR-KLF4-mut reporter was co-transfected into VSMCs together with miR-92 mimics or a negative control miRNA mimic (Ncontrol), and luciferase assays were performed. Compared with empty pMIR-REPORTER vector, the luciferase activity of pMIR-KLF4-wt was significantly attenuated due to endogenous miR-92; however, miR-92 did not affect the luciferase activity of pMIR-KLF4-mut reporter, wherein the miR-92 binding site was mutated (Figure 4C). Together, these data suggest that miR-92 downregulates the expression of KLF4 by directly targeting the 3’-UTR of KLF4.
Figure 4.
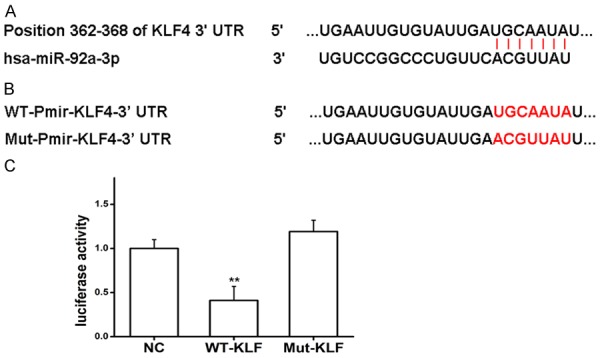
miR-92 regulates KLF4 expression via binding to KLF43’-UTR in vitro. (A) Schematic representation of KLF43’-UTR as a target for miR-92. (B) WT or Mut miR-92 target sequences of the KLF43’-UTR. (C) Luciferase activity of cells co-transfected with the WT or Mut KLF4 3’-UTR reporter genes or negative control miR mimics (pMIR-reporter). **P<0.01 vs. NC and Mut-KLF. One-way ANOVA test with Bonferroni as a post hoc test was used in (C). miR, microRNA; KLF4, Kruppel like factor 4; 3’-UTR, 3-untranslated region; WT, wild type; Mut, mutant.
miR-92 suppresses KLF4 expression at the mRNA and protein levels
To further validate the underlying mechanism that miR-92 regulates the expression of KLF4, VSMCs were transfected with a miR-92 mimic or miR-NC, and the protein and mRNA expression of KLF4 were determined using western blot and qRT-PCR, respectively. As expected, compared with miR-NC, miR-92 mimics significantly attenuated the protein expression level of KLF4 (Figure 5A). Similarly, miR-92 significantly decreased the mRNA expression of KLF4 in VSMCs compared with the control (Figure 5B). These results revealed that miR-92 can directly inhibit the expression of KLF4 in VSMCs at the mRNA and protein levels.
Figure 5.
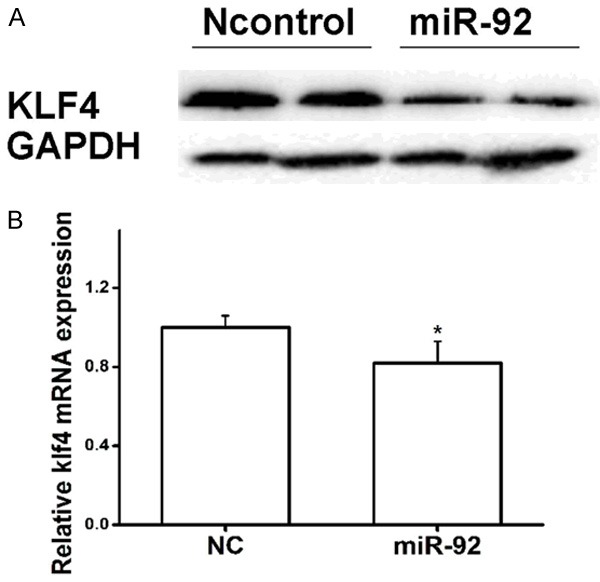
Effects of miR-92 on KLF4 expression. (A) miR-92 inhibited KLF4 expression at the protein level. (B) miR-92 inhibited KLF4 expression at the mRNA level. *P<0.05 vs. NC. Student’s T-test was used in (B). miR, microRNA; KLF4, Kruppel like factor 4.
miR-92 accelerates vascular injuries
Sullivan et al [26] reported that carotid injury led to significant increase in vascular intimal area and thickness; whereas, estrogen could suppress the proliferation of VSMCs in this model. The mouse model was used in this study to elucidate the role of miR-92 in vascular injury. Carotid artery injured mice were treated with miR-92 mimic or negative control (intraperitoneal injection). H&E staining was performed, which showed that carotid artery intimal-media area and thickness were significantly increased in the mice receiving the injection of miR-92 compared with negative control (Figure 6A, 6B), suggesting that miR-92 exerts a marked effect on vascular injuries by enhancing the proliferation and migration of VSMCs in vivo.
Figure 6.
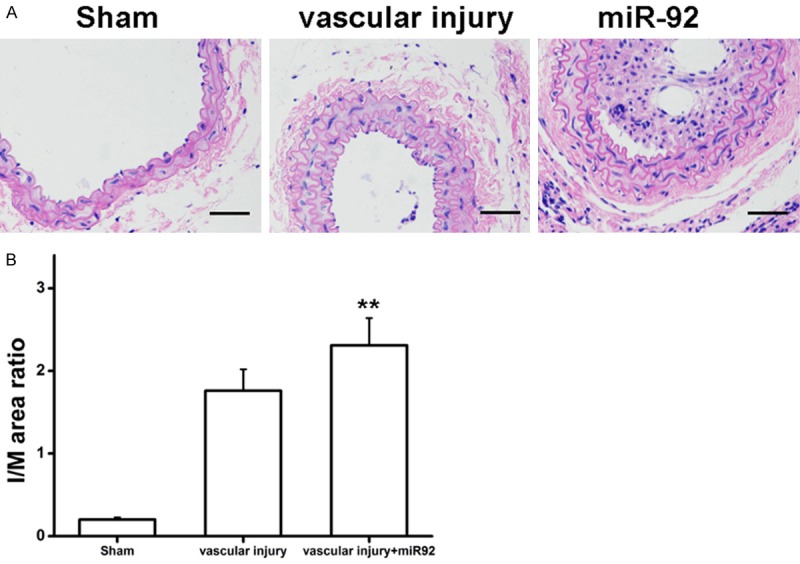
miR-92 accelerated vascular injuries. (A) H&E staining. (B) Degree of neointima formation (I/M) was significantly increased by overexpression of miR-92. One-way ANOVA test with Bonferroni as a post hoc test was used in (B). **P<0.01 vs. Sham and vascular injury. ‘vascular injury’ group vs. ‘vascular injury + miR92’ group. miR, microRNA; H&E, hematoxylin and eosin; I/M, intima-media ratio.
Discussion
As endovascular surgery is widely applied to treat ASO, vascular restenosis is the primary factor that restricts treatment success due to intimal hyperplasia after surgery. It is reported that ASO progression is associated with the activation of various genes that induce the proliferation and migration of VSMCs [27,28]. In this study, miR-92 was significantly downregulated in ASO patients post-surgery; whereas, miR-92 is one of the most upregulated miRNAs during vascular restenosis. Functional analysis revealed that miR-92 promoted the proliferation of VSMCs in vitro and in vivo. Furthermore, KLF4 was validated as a direct target of miR-92, indicating that miR-92 may exert its effects on VMSC proliferation, migration, and cell cycle by interaction with KLF4. Accordingly, miR-92 may have a pivotal role in vascular injury and arterial restenosis post-surgery.
PDGF has been identified to be involved in the pathogenesis of multiple vascular disorders. PDGF-BB was used as a proliferative agent in the study, as a previous report revealed that PDGF is an important regulator in the proliferation of VSMCs [29]. PDGF-BB can stimulate and enhance the growth of VSMCs by mediating the relevant underlying signaling pathways [30,31]. PDGF-BB binds to PDGF receptor-B and, subsequently, initiates intracellular signal transduction in VSMCs, including phosphatidylinositol 3-kinase/Akt, extracellular signal-regulated kinase and p38 mitogen-activated protein kinase pathways [32-34]. In our study, PDFD-BB increased proliferation and migration, and induced S-phase arrest in VSMCs, suggesting that VSMCs dedifferentiated into proliferative behavior in response to PDGF-BB stimulation.
Notably, the proliferation and migration of ASMCs have critical roles in the development of arteriosclerosis [35]. The proliferation and migration ability of VSMCs is closely associated with cellular phenotype. ASMCs can have contractile or synthetic phenotypes, and synthetic VSMCs proliferate faster than contractile cells. Synthetic VSMCs are present in neointima, and are associated with the process of arteriosclerosis [36]. In the present study, ASMCs were induced into a proliferative state using PDGF-BB. However, limited knowledge is available regarding the association between miR-92 expression and VSMCs. In our study, qRT-PCR analysis revealed that miR-92 was increased in ASO patients with artery injuries in a time-dependent manner. Additionally, in an established model of phenotypic regulation in vitro, VSMCs were treated with PDGF-BB and FBS to induce expression of miR-92. CCK-8 and wound healing assays suggested that miR-92 significantly enhanced the proliferation and migration of VSMCs, arrested cells in S-phase in vitro, and accelerated the vascular intimal hyperplasia in the mouse carotid artery injury model in vivo. Together, these results demonstrate that the ability of miR-92 to regulate VSMC viability, including effects on proliferation, migration and the cell cycle, may contribute to vascular injury and artery restenosis.
To further understand the underlying mechanisms associated with the effect of miR-92 on proliferation, migration, and the cell cycle in VSMCs, KLF4 was identified as a potential target of miR-92. As a transcription factor, KLF4 is involved in multiple cellular processes, including inflammatory endothelial activation [37], tumor progression [38], and stem cell biology [39]. It has been reported that KLF4 promotes phenotypic regulation and suppresses proliferation in VSMCs [39,40]. Liu et al [41] reported that treatment with PDGF-BB led to acute upregulation of KLF4 and downregulation of VSMC marker genes, which was blocked by siRNA knockdown of KLF4. Furthermore, KLF4 acts as a potent mediator to activate the tumor suppressor gene cyclin dependent kinase inhibitor 1A (p21) in a p53-dependent manner, thus resulting in reduced proliferation of VSMCs [42]. However, the association between miR-92 and KLF4 has not been fully characterized. In this study, it was revealed that miR-92 could downregulate KLF4 expression at the mRNA and protein expression levels in VSMCs, by directly targeting the 3’-UTR of KLF4, indicating that KLF4 is an important target of miR-92 that could be useful for vascular restenosis prevention and therapy.
Conclusions
In summary, the results reveal that miR-92 promotes VSMC proliferation, migration, and cell cycle in vitro, with smurf1 as a potential functional target. In vivo, a mouse artery injury model was established to validate whether exogenous miR-92 accelerates vascular neointimal hyperplasia. Collectively, these findings offer an appropriate rationale for miR-92 as a potential therapeutic target for the treatment of ASO, vascular injuries and restenosis, and other proliferative vascular disorders.
Acknowledgements
This work was supported by Important Weak Subject Construction Project of Pudong Health and Family Planning Commission of Shanghai (Grant No. PWZbr2017-10), Youth project of Shanghai Pudong New Area Municipal Health Burean (PW2018B-04) and Pudong New Area Science and Technology Development (No: PKJ2018-Y11).
Disclosure of conflict of interest
None.
References
- 1.Go AS, Mozaffarian D, Roger VL, Benjamin EJ, Berry JD, Borden WB, Bravata DM, Dai S, Ford ES, Fox CS, Franco S, Fullerton HJ, Gillespie C, Hailpern SM, Heit JA, Howard VJ, Huffman MD, Kissela BM, Kittner SJ, Lackland DT, Lichtman JH, Lisabeth LD, Magid D, Marcus GM, Marelli A, Matchar DB, McGuire DK, Mohler ER, Moy CS, Mussolino ME, Nichol G, Paynter NP, Schreiner PJ, Sorlie PD, Stein J, Turan TN, Virani SS, Wong ND, Woo D, Turner MB American Heart Association Statistics Committee and Stroke Statistics Subcommittee. Executive summary: heart disease and stroke statistics-2013 update a report from the American heart association. Circulation. 2013;127:143–52. doi: 10.1161/CIR.0b013e318282ab8f. [DOI] [PubMed] [Google Scholar]
- 2.Criqui MH, Vargas V, Denenberg JO, Ho E, Allison M, Langer RD, Gamst A, Bundens WP, Fronek A. Ethnicity and peripheral arterial disease: the san diego population study. Circulation. 2005;112:2703–7. doi: 10.1161/CIRCULATIONAHA.105.546507. [DOI] [PubMed] [Google Scholar]
- 3.Setacci C, Castelli P, Chiesa R, Grego F, Simoni GA, Stella A, Galzerano G, Sirignano P, De Donato G, Setacci F. Restenosis: a challenge for vascular surgeon. J Cardiovasc Surg (Torino) 2012;53:735–46. [PubMed] [Google Scholar]
- 4.Kim J, Zhang L, Peppel K, Wu JH, Zidar DA, Brian L, DeWire SM, Exum ST, Lefkowitz RJ, Freedman NJ. Beta-arrestins regulate atherosclerosis and neointimal hyperplasia by controlling smooth muscle cell proliferation and migration. Circ Res. 2008;103:70–79. doi: 10.1161/CIRCRESAHA.108.172338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Inoue T, Node K. Molecular basis of restenosis and novel issues of drug-eluting stents. Circ J. 2009;73:615–21. doi: 10.1253/circj.cj-09-0059. [DOI] [PubMed] [Google Scholar]
- 6.Glass CK, Witztum JL. Atherosclerosis. The road ahead. Cell. 2001;104:503–16. doi: 10.1016/s0092-8674(01)00238-0. [DOI] [PubMed] [Google Scholar]
- 7.Tili E, Michaille JJ, Calin AG. Expression and function of micro RNAs in immune cells during normal or disease state. Int J Med Sci. 2008;5:73–9. doi: 10.7150/ijms.5.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.He L, Hannon GJ. MicroRNAs: small RNAs with a big role in gene regulation. Nat Rev Genet. 2004;5:522–31. doi: 10.1038/nrg1379. [DOI] [PubMed] [Google Scholar]
- 9.Kantharidis P, Wang B, Carew RM, Lan HY. Diabetes complications: the microRNA perspective. Diabetes. 2011;60:1832–1837. doi: 10.2337/db11-0082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trionfini P, Benigni A. MicroRNAs as master regulators of glomerular function in health and disease. J Am Soc Nephrol. 2017;28:1686–1696. doi: 10.1681/ASN.2016101117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Kato M, Natarajan R. MicroRNAs in diabetic nephropathy: functions, biomarkers, and therapeutic targets. Ann N Y Acad Sci. 2015;1353:72–88. doi: 10.1111/nyas.12758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Boucher JM, Peterson SM, Urs S, Zhang C, Liaw L. The miR-143/145 cluster is a novel transcriptional target of Jagged-1/notch signaling in vascular smooth muscle cells. J Biol Chem. 2011;286:28312–21. doi: 10.1074/jbc.M111.221945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wang M, Li W, Chang GQ, Ye CS, Ou JS, Li XX, Liu Y, Cheang TY, Huang XL, Wang SM. MicroRNA-21 regulates vascular smooth muscle cell function via targeting tropomyosin 1 in arteriosclerosis obliterans of lower extremities. Arterioscler Thromb Vasc Biol. 2011;31:2044–53. doi: 10.1161/ATVBAHA.111.229559. [DOI] [PubMed] [Google Scholar]
- 14.Liu X, Cheng Y, Yang J, Xu L, Zhang C. Cell-specific effects of miR-221/222 in vessels: molecular mechanism and therapeutic application. J Mol Cell Cardiol. 2012;52:245–255. doi: 10.1016/j.yjmcc.2011.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rippe C, Blimline M, Magerko KA, Lawson BR, LaRocca TJ, Donato AJ, Seals DR. MicroRNA changes in human arterial endothelial cells with senescence: relation to apoptosis, eNOS and inflammation. Exp Gerontol. 2012;47:45–51. doi: 10.1016/j.exger.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang L, Zhou M, Qin G, Weintraub NL, Tang Y. MiR-92a regulates viability and angiogenesis of endothelial cells under oxidative stress. Biochem Biophys Res Commun. 2014;446:952–958. doi: 10.1016/j.bbrc.2014.03.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bonauer A, Carmona G, Iwasaki M, Mione M, Koyanagi M, Fischer A, Burchfield J, Fox H, Doebele C, Ohtani K, Chavakis E, Potente M, Tjwa M, Urbich C, Zeiher AM, Dimmeler S. MicroRNA-92a controls angiogenesis and functional recovery of ischemic tissues in mice. Science. 2009;324:1710–3. doi: 10.1126/science.1174381. [DOI] [PubMed] [Google Scholar]
- 18.Liu G, Friggeri A, Yang Y, Park YJ, Tsuruta Y, Abraham E. miR-147, a microRNA that is induced upon toll-like receptor stimulation, regulates murine macrophage inflammatory responses. Proc Natl Acad Sci U S A. 2009;106:15819–15824. doi: 10.1073/pnas.0901216106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kosuge H, Haraguchi G, Koga N, Maejima Y, Suzuki J, Isobe M. Pioglitazone prevents acute and chronic cardiac allograft rejection. Circulation. 2006;113:2613–2622. doi: 10.1161/CIRCULATIONAHA.105.594101. [DOI] [PubMed] [Google Scholar]
- 20.Liu Y, Li W, Ye C, Lin Y, Cheang TY, Wang M, Zhang H, Wang S, Zhang L, Wang S. Gambogic acid induces G0/G1 cell cycle arrest and cell migration inhibition via suppressing PDGF receptor β tyrosine phosphorylation and Rac1 activity in rat aortic smooth muscle cells. J Atheroscler Thromb. 2010;17:901–13. doi: 10.5551/jat.3491. [DOI] [PubMed] [Google Scholar]
- 21.Kamio N, Hirai H, Ashihara E, Tenen DG, Maekawa T, Imanishi J. Use of bicistronic vectors in combination with flow cytometry to screen for effective small interfering RNA target sequences. Biochem Biophys Res Commun. 2010;393:498–503. doi: 10.1016/j.bbrc.2010.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu W, Wang M, Yin H, Yao C, He Q, Yin L, Zhang C, Li W, Chang G, Wang S. MicroRNA-1298 is regulated by DNA methylation and affects vascular smooth muscle cell function by targeting connexin 43. Cardiovasc Res. 2015;107:534–45. doi: 10.1093/cvr/cvv160. [DOI] [PubMed] [Google Scholar]
- 23.Sundaresan M, Yu ZX, Ferrans VJ, Irani K, Finkel T. Requirement for generation of H2O2 for platelet-derived growth factor signal transduction. Science. 1995;270:296–9. doi: 10.1126/science.270.5234.296. [DOI] [PubMed] [Google Scholar]
- 24.Rani UP, Kesavan R, Ganugula R, Avaneesh T, Kumar UP, Reddy GB, Dixit M. Ellagic acid inhibits PDGF-BB-induced vascular smooth muscle cell proliferation and prevents atheroma formation in streptozotocin-induced diabetic rats. J Nutr Biochem. 2013;24:1830–1839. doi: 10.1016/j.jnutbio.2013.04.004. [DOI] [PubMed] [Google Scholar]
- 25.Kim J, Zhang L, Peppel K, Wu JH, Zidar DA, Brian L, DeWire SM, Exum ST, Lefkowitz RJ, Freedman NJ. Beta-arrestins regulate atherosclerosis and neointimal hyperplasia by controlling smooth muscle cell proliferation and migration. Circ Res. 2008;103:70–79. doi: 10.1161/CIRCRESAHA.108.172338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sullivan TR Jr, Karas RH, Aronovitz M, Faller GT, Ziar JP, Smith JJ, O’Donnell TF Jr, Mendelsohn ME. Estrogen inhibits the response-to-injury in a mouse carotid artery model. J Clin Invest. 1995;96:2482–8. doi: 10.1172/JCI118307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chen Z, Wang M, Huang K, He Q, Li H, Chang G. MicroRNA-125b affects vascular smooth muscle cell function by targeting serum response factor. Cell Physiol Biochem. 2018;46:1566–1580. doi: 10.1159/000489203. [DOI] [PubMed] [Google Scholar]
- 28.Li Y, Ouyang M, Shan Z, Ma J, Li J, Yao C, Zhu Z, Zhang L, Chen L, Chang G, Wang S, Wang W. Involvement of microRNA-133a in the development of arteriosclerosis obliterans of the lower extremities via RhoA targeting. J Atheroscler Thromb. 2015;22:424–432. doi: 10.5551/jat.27839. [DOI] [PubMed] [Google Scholar]
- 29.Bilder G, Wentz T, Leadley R, Amin D, Byan L, O’Conner B, Needle S, Galczenski H, Bostwick J, Kasiewski C, Myers M, Spada A, Merkel L, Ly C, Persons P, Page K, Perrone M, Dunwiddie C. Restenosis following angioplasty in the swine coronary artery is inhibited by an orally active PDGF-receptor tyrosine kinase inhibitor, RPR101511A. Circulation. 1999;99:3292–3299. doi: 10.1161/01.cir.99.25.3292. [DOI] [PubMed] [Google Scholar]
- 30.Park ES, Kang SI, Yoo KD, Lee MY, Yoo HS, Hong JT, Shin HS, Kim B, Yun YP. Camptothecin inhibits platelet-derived growth factor-BB-induced proliferation of rat aortic vascular smooth muscle cells through inhibition of PI3K/Akt signaling pathway. Exp Cell Res. 2013;319:982–991. doi: 10.1016/j.yexcr.2012.12.024. [DOI] [PubMed] [Google Scholar]
- 31.Sun L, Zhao R, Zhang L, Zhang T, Xin W, Lan X, Huang C, Du G. Salvianolic acid A inhibits PDGF-BB induced vascular smooth muscle cell migration and proliferation while does not constrain endothelial cell proliferation and nitric oxide biosynthesis. Molecules. 2012;17:3333–47. doi: 10.3390/molecules17033333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhan Y, Kim S, Izumi Y, Izumiya Y, Nakao T, Miyazaki H, Iwao H. Role of JNK, p38, and ERK in platelet-derived growth factor-induced vascular proliferation, migration, and gene expression. Arterioscler Thromb Vasc Biol. 2003;23:795–801. doi: 10.1161/01.ATV.0000066132.32063.F2. [DOI] [PubMed] [Google Scholar]
- 33.Heldin CH. Simultaneous induction of stimulatory and inhibitory signals by PDGF. FEBS Lett. 1997;410:17–21. doi: 10.1016/s0014-5793(97)00318-9. [DOI] [PubMed] [Google Scholar]
- 34.Claesson-Welsh L. Platelet-derived growth factor receptor signals. J Biol Chem. 1994;269:32023–32026. [PubMed] [Google Scholar]
- 35.Doran AC, Meller N, Mcnamara CA. Role of smooth muscle cells in the initiation and early progression of atherosclerosis. Arterioscler Thromb Vasc Biol. 2008;28:812–9. doi: 10.1161/ATVBAHA.107.159327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Rudijanto A. The role of vascular smooth muscle cells on the pathogenesis of atherosclerosis. Acta Med Indones. 2007;39:86–93. [PubMed] [Google Scholar]
- 37.Hamik A, Lin Z, Kumar A, Balcells M, Sinha S, Katz J, Feinberg MW, Gerzsten RE, Edelman ER, Jain MK. 44 Kruppel-like factor 4 regulates endothelial inflammation. J Biol Chem. 2007;282:13769–79. doi: 10.1074/jbc.M700078200. [DOI] [PubMed] [Google Scholar]
- 38.Rowland BD, Bernards R, Peeper DS. The KLF4 tumour suppressor is a transcriptional repressor of p53 that acts as a context-dependent oncogene. Nat Cell Biol. 2005;7:1074–82. doi: 10.1038/ncb1314. [DOI] [PubMed] [Google Scholar]
- 39.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouseembryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–676. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 40.Garvey SM, Sinden DS, Schoppee Bortz PD, Wamhoff BR. Cyclosporine up-regulates Kruppel-like factor-4 (KLF4) in vascular smooth muscle cells and drives phenotypic modulation in vivo. J Pharmacol Exp Ther. 2010;333:34–42. doi: 10.1124/jpet.109.163949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Liu Y, Sinha S, McDonald OG, Shang Y, Hoofnagle MH, Owens GK. Kruppel-like factor 4 abrogates myocardin-induced activation of smooth muscle gene expression. J Biol Chem. 2005;280:9719–27. doi: 10.1074/jbc.M412862200. [DOI] [PubMed] [Google Scholar]
- 42.Wassmann S, Wassmann K, Jung A, Velten M, Knuefermann P, Petoumenos V, Becher U, Werner C, Mueller C, Nickenig G. Induction of p53 by GKLF is essential for inhibition of proliferation of vascular smooth muscle cells. J Mol Cell Cardiol. 2007;43:301–7. doi: 10.1016/j.yjmcc.2007.06.001. [DOI] [PubMed] [Google Scholar]