

Abstract
The epigenome is a collection of chemical compounds that attach to and overlay the DNA sequence to direct gene expression. Epigenetic marks do not alter DNA sequence but instead allow or silence gene activity and the subsequent production of proteins that guide the growth and development of an organism, direct and maintain cell identity, and allow for the production of primordial germ cells (PGCs; ova and spermatozoa). The three main epigenetic marks are (1) histone modification, (2) DNA methylation, and (3) noncoding RNA, and each works in a different way to regulate gene expression. This article reviews these concepts and discusses their role in normal functions such as X-chromosome inactivation, epigenetic reprogramming during embryonic development and PGC production, and the clinical example of the imprinting disorders Angelman and Prader–Willi syndromes.
Keywords: epigenomics, histone code, chromatin, DNA methylation, X-chromosome inactivation
The study of genetics focuses on the sequence of DNA itself: its structure, faithful replication of its code, and the effects of mutations. Epigenetics focuses on the non-DNA sequence-related regulatory mechanisms that restrict or allow genes to be transcribed, which affects the production of proteins and, ultimately, cellular activity. Essentially, the epigenome consists of a series of chemical marks that are laid on top of the DNA sequence to “tell the DNA what to do” (National Human Genome Research Institute, 2016). The term epigenetics is credited to Waddington (1942), who described “a causal relationship between genes and their products.” Nanney (1958) later expanded the term to include a “library” of mechanisms that directed gene expression and, importantly, proposed that patterns of epigenetic markings along the genome were passed down through cell divisions to daughter cells along with DNA sequence.
Over time, concepts related to how epigenetic changes were mitotically (i.e., passed along during somatic cell replication to maintain a differentiated cell line, Holliday, 2006) and meiotically heritable (i.e., maintained during meiosis and able to be passed to offspring, Trerotola, Relli, Simeone, & Alberti, 2015) evolved, leading to a contemporary definition of epigenetics as “mechanisms of heredity which do not involve modifications of DNA sequence and are reversible in nature” (Kovalchuk & Kovalchuk, 2012, p. 1). The purpose of this article is to acquaint the reader with the mechanisms underlying epigenetic regulation. The body of the literature focused on associations between epigenetic mechanisms and disease is expanding rapidly across clinical specialties; nurses with a foundational understanding of concepts such as chromatin remodeling, histone modification, DNA methylation, and noncoding RNA will be better equipped to translate new findings into their research and practice.
In the absence of disease, normal epigenetic regulatory mechanisms perform several major functions. First, they direct cell differentiation and maintain a specific cell identity during subsequent replications, ensuring that all generations of somatic cells in a particular line express the correct genes associated with that cell’s structure and function (e.g., mature liver cells continue to give rise to normally functioning liver cells; Trerotola et al., 2015). Second, during meiosis, the removal of epigenetic marks that direct mature cellular differentiation assists in the process of producing totipotent primordial germ cells (PGCs; i.e., the creation of ova and spermatozoa from somatic cells; Allis & Jenuwein, 2016; Kim, Samaranayake, & Pradhan, 2009). In this article, we discuss the mechanisms underlying these epigenetic changes in the genome. It is not the scope of this article to discuss the impact of environmental exposures on epigenetic marks and health or cancer-related epigenetics.
Chromatin
To gain an understanding of epigenetic mechanisms, it is essential to first understand the role of chromatin in gene transcription. To review, RNA transcribes the DNA code in the cell nucleus through messenger RNA (mRNA), which then leaves the nucleus to begin the process of translation of the DNA code to make protein (Dorman, Schmella, & Wesmiller, 2016). The DNA double-helix structure is too large to fit in the cell nucleus; thus, it is compacted into a structure called chromatin (Felsenfeld, 2014). Chromatin, constructed from DNA and proteins (i.e., histones), is the scaffolding for the entire genome, containing the heritable material within the eukaryotic cell (Dawson & Kouzarides, 2012). Chromatin exists in two forms, euchromatin and heterochromatin. Euchromatin is less compacted, allowing easier access for the molecules needed for transcription. Heterochromatin is tightly compacted and is not as easily transcribed (Dawson & Kouzarides, 2012). The epigenetic mechanisms we discuss in this article are associated with changes in the compaction of chromatin that alter its accessibility for transcription.
Epigenetic Mechanisms
The three main epigenetic mechanisms are (1) histone modification, (2) DNA methylation, and (3) noncoding RNA. Rather than changing the DNA sequence, these mechanisms act on top of that sequence, to change gene transcription, thereby altering gene expression (Gayon, 2016).
Histone Modification
Histones are negatively charged proteins that associate with the positively charged DNA double-helix structure in the cell nucleus to compact the DNA, so it can fit into the eukaryotic cell nucleus (Felsenfeld, 2014). The first compaction step is the formation of nucleosomes. A nucleosome is formed when eight histone proteins are wrapped with approximately two turns of the DNA double-helix structure (Deichmann, 2016). The histones act like a spool, wrapping the length of DNA double-helix structure around them like a thread to organize and decrease the overall length of the DNA through the formation of a nucleosome. The nucleosomes then wrap into a spiral called a solenoid, continuing to compact the DNA and forming chromatin (Dorman et al., 2016). The chromatin structure looks like “beads on a string,” with the beads being the nucleosomes (Figure 1). Epigenetic modifications of histones change the covalent bonds between and within nucleosomes to alter the chromatin structure (i.e., positioning the nucleosome “beads” farther apart, creating euchromatin, or closer, as with heterochromatin; Duarte, 2013).
Figure 1.
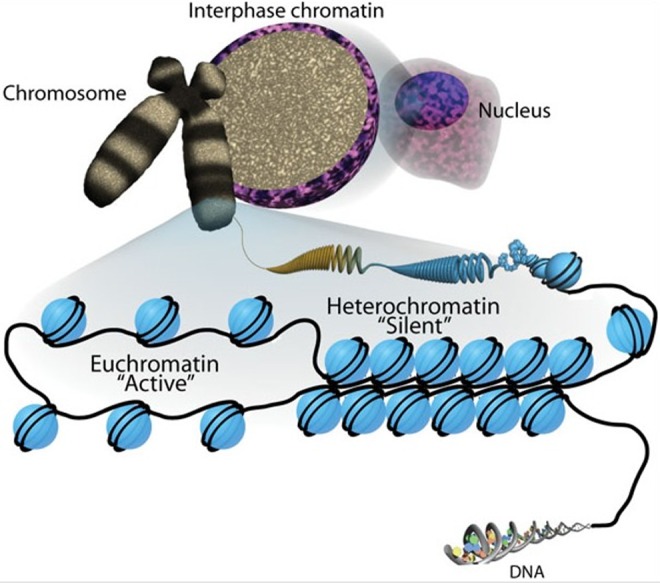
Chromatin. DNA coils around histones to form nucleosomes (blue circles wrapped with two coils). Euchromatin is “active” since it is less compacted and available for transcription. Heterochromatin is “silent” since it is tightly compacted and not accessible for transcription. Note how the nucleosomes appear as “beads on a string.” This figure is reproduced from an open-access article (Sha & Boyer, 2009) distributed under the terms of the Creative Commons Attribution License (CC BY 3.0), which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited. http://creativecommons.org/licenses/by/3.0/
Histones have protruding N-terminal “tails” that extend from the nucleosome and contain modifiable amino acids that help determine how tightly DNA is compacted into chromatin (Dorman et al., 2016; Felsenfeld, 2014). Histone modifications most commonly occur through acetylation, methylation, or phosphorylation of the amino acids on the histone tails (Felsenfeld, 2014; Rodenhiser & Mann, 2006; Figure 2). Depending on the type of modification, the bonds between histones and DNA may weaken or strengthen, thus altering the structure of the chromatin. For example, with acetylation, the positive charge on the histone is removed, which decreases the interaction of the histone tail with negatively charged phosphate groups of the DNA (Rothbart & Strahl, 2014). With less interaction between the positive and negative charges, the chromatin becomes more open and available for gene transcription. When histone modification occurs, the chromatin is altered, but the DNA sequence is not modified, rather it is the effect on the packaging of the DNA that alters gene expression (Figure 2).
Figure 2.

(A) Schematic of epigenetic modifications. Strands of DNA are wrapped around histone octamers, forming nucleosomes. These nucleosomes are organized into chromatin, the building blocks of a chromosome. Reversible and site-specific histone modifications occur at multiple sites through acetylation, methylation, and phosphorylation. DNA methylation occurs at five-position of cytosine residues in a reaction catalyzed by DNA methyltransferases. Together, these modifications provide a unique epigenetic signature that regulates chromatin organization and gene expression. (B) Schematic of the reversible changes in chromatin organization that influence gene expression. Genes are expressed (switched on) when the chromatin is open (active), and they are inactivated (switched off) when the chromatin is condensed (silent). White circles = unmethylated cytosines (these are the circles in the “transcription possible” diagram); red circles = methylated cytosines (these are the circles in the “transcription impeded” diagram). Reprinted, with permission, from Rodenhiser and Mann, (2006), under license from Access Copyright. Further reproduction, distribution, or transmission is prohibited except as otherwise permitted by law.
DNA Methylation
DNA methylation is the addition of a methyl group to the DNA base cytosine, forming 5-methylcytosine in a reaction catalyzed by DNA methyltransferases (DNMTs; Schübeler, 2015; Figure 2A). DNA methylation occurs predominantly in cytosine bases that are followed by a guanine base in the promoter region of the gene (i.e., 5′, where gene transcription begins moving down the DNA strand to the 3′ end; Jang, Shin, Lee, & Do, 2017; for a review, see the first article in this primer series, Dorman et al., 2016). The convention has developed of using the CpG label to indicate a potential methylation site in order to differentiate it from the standard notation of the genetic code, where CG would indicate a locus where cytosine is paired with guanine on the DNA double-stranded helix (Jang et al., 2017). Concentrated clusters of unmethylated CpG sites at the 5′ site (i.e., the promoter region) in tissue-specific genes and in “housekeeping” genes are called CpG islands (Inbar-Feigenberg, Choufani, Butcher, Roifman, & Weksberg, 2013; Figure 3). Housekeeping genes are required for basic cell function and, under normal circumstances, maintain a constant level of expression (Eisenberg & Levanon, 2013).
Figure 3.
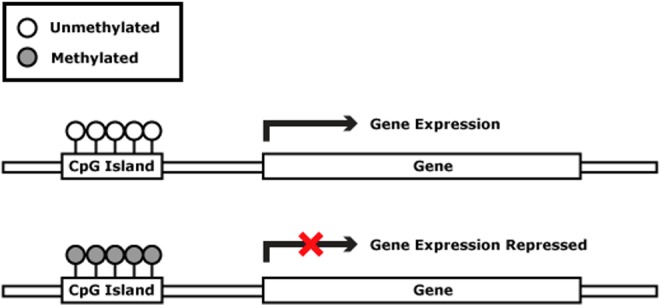
CpG islands on the promoter region of the gene. When these islands are methylated, gene expression is repressed. Reproduced from http://missinglink.ucsf.edu/lm/genes_and_genomes/methylation.html courtesy of Benjamin Huang, MD, University of California San Francisco.
The genes of different cells have different methylation patterns, which contribute to the differences in gene expression (Jang et al., 2017). Transcription factors are proteins that recognize specific DNA sequences to begin transcription of the DNA. The CpG islands are targets for transcription factors that trigger the gene expression process. When unmethylated, the sites are open to gene transcription and expression. When the CpG sites are methylated, however, the transcription factors are unable to bind at the site, resulting in altered gene expression (Rodenhiser & Mann, 2006). During DNA replication, the parent cell’s methylation pattern is copied to the daughter DNA strand, allowing inheritance of the methylation pattern (and thus, the gene’s pattern of expression; Figure 3). DNA methylation, which is maintained by DNMTs, is essential for normal development (Inbar-Feigenberg et al., 2013). Without methylation, essential development processes such as genomic imprinting and X-chromosome inactivation, which we discuss later, would not occur. Abnormal DNA methylation is associated with cancer, autoimmune diseases, and genetic disorders (Rodenhiser & Mann, 2006). Genomewide DNA methylation marks may serve as a biomarker for disease risk and targets for the development of precision interventions (Duarte, 2013).
Noncoding RNA
Researchers have identified many forms of noncoding RNA that contribute to epigenetic regulation of gene expression (Felsenfeld, 2014). Noncoding RNAs are classified as short or long. Short noncoding RNAs (20–25 nucleotide bases) include microRNAs (miRNA) and short interfering RNAs (siRNA; Patil, Zhou, & Rana, 2014). MiRNAs limit gene expression by binding to the 3′-Untranslated region (UTR) sequence of mRNAs. siRNAs bind to specific complementary DNA sequences, silencing target gene expression in a sequence-specific manner (Ozcan, Ozpolat, Coleman, Sood, & Lopez-Berestein, 2015). Long noncoding RNAs increase gene expression by recruiting transcription factors, decrease gene expression by tightening the structure of the chromatin, and change gene expression by altering transcription (Dey, Mueller, & Dutta, 2014). While the role of noncoding RNAs in disease development is not yet fully understood, researchers have reported that their increased expression is associated with fragile X, neurodegenerative diseases, and cancer (Dey et al., 2014; Patil et al., 2014).
Role of Epigenetics in Normal Functions
Cell Differentiation and Maintenance of Identity
One major function of epigenetic regulation, as we mentioned earlier, is to direct cellular differentiation as an organism develops from a single-cell zygote into a mature adult (Kim et al., 2009). While there are approximately 25,000 genes in the human genome, only a portion of these are universally expressed, such as the housekeeping genes responsible for basic functions (Eisenberg & Levanon, 2013). Differentiated cells use only a fraction of the available genes and in specific combinations that define their particular cell line’s structure and function. These restricted gene expression patterns are in part due to intracellular transcription factors that recognize and direct the expression of specific DNA sequences (Phillips & Hoopes, 2008) and tissue-enriched genes that are highly expressed in specific cell lines (She et al., 2009). Epigenetic marks are another major regulator for maintaining cell identity. For example, red blood cells express the gene HBB to produce hemoglobin protein, which enables oxygen transport (U.S. National Library of Medicine, 2017). The HBB gene is present in the genome of every other cell type in the body at the same location of chromosome 11p15.4, yet under normal conditions, lifelong restriction of transcription through DNA methylation keeps other cell lines from producing this protein (He et al., 2010).
X-Chromosome Inactivation
A dramatic example of epigenetic gene regulation is the complete silencing of a second X chromosome in a cell nucleus to control the “dose” of gene expression possible from the X chromosome in males and females. Barr and Bertram (1949) reported the first evidence of X-chromosome inactivation when they identified what came to be termed a Barr body, a section of densely packaged heterochromatin consistently located at the periphery of the nucleus in cells in females. Later work demonstrated that the Barr body is actually an X chromosome that is so highly condensed that transcription is not possible (Ohno, Kaplan, & Kinosita, 1959). In female embryos, a single X chromosome contains the necessary dose to guide normal development, so one of the two inherited X chromosomes is randomly selected to become heavily methylated in each cell at the gastrulation stage of female embryonic development (approximately the third week postfertilization; Hill, 2017). The chromatin becomes tightly and permanently condensed, resulting in silencing of gene expression from that body (Disteche & Berletch, 2015). This inactivation is then heritable by all somatic daughter cells throughout the rest of the organism’s life. The remaining active X chromosome functions normally, allowing the development of characteristics typically associated with female phenotypic growth and development (Lyon, 1961, 2005).
A visual example of this phenomenon can be observed in calico cats, where the gene for coat color resides on the X chromosome. In female cats, the distinctive variations in coat color arise due to the random inactivation of the dominant ginger color allele on the silenced X chromosome, allowing the recessive black coat color to be expressed by patches of aggregated cells in what is often described as a mosaic pattern. Male cats in the same litter would be expected to have uniformly ginger or black coats due to their inheritance of a single, active X chromosome (LeMieux, 2017).
More recent research in this area reveals that some genes on the silenced X chromosome may escape complete inactivation, or in the case where additional X chromosomes are present, some cells may be unable to inactivate them (Yang et al., 2011). Examples include trisomy X (47, XXX), which may occur in up to 1 in 1,000 female births, and Klinefelter’s syndrome (47, XXY), seen in about 1.5 of 1,000 males (Swerdlow, Higgins, Schoemaker, Wright, & Jacobs, 2005; Swerdlow, Schoemaker, Higgins, Wright, & Jacobs, 2005). Researchers attribute the significantly increased incidence of inflammatory, metabolic, and cardiovascular morbidities in both of these syndromes in part to excessive gene expression activity from the extra X chromosome (Stochholm, Juul, & Gravholt, 2010; Zitzmann et al., 2015).
Epigenetic Reprogramming
While many epigenetic mechanisms work to establish cellular differentiation and maintain cellular identity over the course of an organism’s lifetime, there are periods where methylation marks must be cleared to allow normal function. These periods include early embryonic development, during the production of PGCs (i.e., ova and spermatozoa) and some instances of cellular differentiation in response to stimuli throughout the life span, such as hematopoietic stem cell differentiation into needed blood cell types in response to hemorrhage or infection (Wu & Sun, 2006).
In the first few hours immediately after fertilization, while the small nuclei from the egg and sperm still exist as separate pronuclear bodies, a critical step involves removal of the epigenetic marks that directed gene expression to support the very specialized functions of the ovum and sperm cells (Seisenberger et al., 2013). This process allows the combination of the contributed DNA and unrestricted expression of the genes necessary for embryonic development. In the first 6 hours postfertilization, the methyl groups within the paternal pronucleus of the sperm cell are rapidly removed via hydroxylation by Ten eleven translocation (TET) proteins (Rasmussen & Helin, 2016). The maternal pronucleus passively demethylates over a longer period; under normal circumstances, DNMT1, a member of the DNMT family of enzymes that maintain methyl group placement, would replace methyl groups along the DNA sequence during each division. During this postfertilization period, DNMT1 is not active, and the lack of replacement of the methyl groups eventually dilutes the prevalence of epigenetic marks, achieving a nadir of methylation levels at the blastocyst stage at about 5–9 days postfertilization. After this stage, global methylation levels begin to rise as the embryo grows and specialized cell lines emerge during development. With the exception of the production of PGCs, the DNA methylation levels in the somatic cells in the growing embryo remain stable over the rest of the life span, maintaining cell-line differentiation (Seisenberger et al., 2013; see Figure 4).
Figure 4.

DNA methylation reprogramming in the mammalian life cycle. In the zygote, DNA from the paternal gamete is rapidly, actively demethylated by TET protein, while passive demethylation of the maternal gamete occurs more slowly through the lack of DNMT1 activity. DPC = days postconception; ICM = inner cell mass; PGC = primordial germ cell; DNMT = DNA methyltransferase. Adapted from Seisenberger et al. (2013), licensed under CC BY 3.0.
Primordial Germ Cell Production and Imprinted Genes
Clearance of epigenetic marks must also occur to create germ cells from mature, differentiated somatic cells, and especially in the case of sperm, chromatin must be tightly compacted during this process. It is important to note that the timing of PGC development differs between the sexes. In postpubescent males, sperm cells are produced throughout the lifetime, but in females, all egg cells are produced and in place in the fetal ovaries at birth, though they do not fully mature until ovulation occurs (Kota & Feil, 2010).
One exception to the reprogramming processes discussed above involves parentally imprinted genes. In normally functioning mammalian cells without DNA mutations, two alleles are present: one inherited from the father and one from the mother. Generally, both alleles are expected to behave in the same way (i.e., either active or silent). X inactivation and gene imprinting are the two normally occurring exceptions, where one allele in a specific gene may be silent and the other active (Barlow & Bartolomei, 2014).
Unlike X inactivation, which occurs randomly in somatic cells, gene imprinting takes place in the germ cells, in a parent-of-origin pattern. This process provides necessary epigenetic variability in genes that are affected by genetic imprinting (also known as differentially methylated regions), most of which are involved in fetal growth and development. Approximately 150 genes in humans are parentally imprinted, but gene expression associated with these marks is very tissue-specific and is generally observed in the placenta and brain (Horsthemke, 2014). Maternal-effect proteins protect these marks during early embryo development, so that they can be preserved in the somatic cell lines. During primordial germ-cell development, if the embryo is female, the maternal parent-of-origin methylation marks will be reset in the oocytes, and if male, they will be reset in spermatozoa according to the paternal parent-of-origin epigenetic contribution (Seisenberger et al., 2013).
Although genetic imprinting is a normal biological process, the resulting silencing of one parental allele may increase the risk of disease should a DNA sequence mutation or other dysfunction affect the remaining allele. A marked difference in phenotype may be observed depending on which parental allele is silenced. For example, dysfunction in a single genetic region, chromosome 15q11.2-q13, may be associated with two different disorders, Angelman or Prader–Willi syndrome. Despite impacting the same chromosomal region, the symptoms associated with each syndrome are quite different (Peters, 2014).
Angelman syndrome is neurodevelopmental disorder related to an inability of the maternal allele to express UBE3A (ubiquitin-protein ligase E3A, located at chromosome 15q11.2, Online Mendelian Inheritance in Man [OMIM], 2017) in brain tissue. The syndrome presents in approximately 1 in 15,000 children and may have one of the four major causes: approximately 70% of Angelman syndrome cases are associated with genetic deletions within the chromosome 15q11-q13 region, about 10% have mutations within the maternal allele of the UBE3A gene, an additional 2–7% result from inheritance of two paternal copies of the gene (also known as uniparental disomy; Clayton-Smith & Laan, 2003; Williams, Driscoll, & Dagli, 2010), and another 3–5% are related to maternal imprinting that results in abnormal silencing of the UBE3A gene in the brain tissue (Sato, 2017). The phenotype of affected children involves severe developmental delay, including speech impairment, difficulty with movement and balance, and behavioral features such as inappropriate laughter and smiling, excitability, and short attention spans (Tan et al., 2011).
In contrast to the maternal inheritance pattern of Angelman syndrome, Prader–Willi syndrome is associated with a lack of expression of paternally inherited genes in the chromosome 15q11.2-q13 region (Cassidy, Dykens, & Williams, 2000). The incidence and distribution of underlying causes is very similar to those of Angelman syndrome; however, the uniparental disomy and abnormal gene imprinting in Prader–Willi syndrome occurs on the paternal parent-of-origin allele (Irizarry, Miller, Freemark, & Haqq, 2016). The phenotypic presentation includes hypotonia, failure to thrive in infancy and neurocognitive delays from birth to age 2, followed by hyperphagia, rapid weight gain, and continued learning delays into adulthood (Gunay-Aygun, Schwartz, Heeger, O’Riordan, & Cassidy, 2001).
Conclusion
Epigenetic regulation directs the expression of genes to develop and maintain cell identity and to produce totipotent PGCs from mature somatic cells during meiosis. Mechanisms including histone modification, DNA methylation, noncoding RNA, and the degree of compaction of chromatin affect the accessibility of the DNA sequence for the purposes of gene expression. Epigenetic modifications do not change DNA structure but do change function and are integral to the diversity within populations.
We intended for this article to provide a basic overview of these processes, as knowledge continues to rapidly accumulate in the literature and clinical areas regarding the impact of environmental and other disease-related factors on alterations to the epigenome and the resulting health outcomes. A working knowledge of normal epigenetic mechanisms will equip nurses to read and synthesize the growing body of literature describing symptoms and diseases with an epigenetic component.
Footnotes
Authors’ Note: Series Information: The “Primer in Genetics and Genomics” series is a collaboration between Biological Research for Nursing and the International Society of Nurses in Genetics. Sheila A. Alexander, PhD, RN, FCCM, serves as guest editor for the series.
Author Contribution: K. L. Fessele contributed to conception, design, and interpretation; drafted the manuscript; critically revised the manuscript; gave final approval; and agrees to be held accountable for all aspects of work, ensuring integrity and accuracy. F. Wright contributed to conception, design, and interpretation; drafted the manuscript; critically revised the manuscript; gave final approval; and agrees to be held accountable for all aspects of work, ensuring integrity and accuracy.
Declaration of Conflicting Interests: The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding: The author(s) received no financial support for the research, authorship, and/or publication of this article.
References
- Allis C. D., Jenuwein T. (2016). The molecular hallmarks of epigenetic control. Nature Reviews Genetics, 17, 487–500. [DOI] [PubMed] [Google Scholar]
- Barlow D. P., Bartolomei M. S. (2014). Genomic imprinting in mammals. Cold Spring Harbor Perspectives in Biology, 6 doi:10.1101/cshperspect.a018382 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barr M. L., Bertram E. G. (1949). A morphological distinction between neurones of the male and female, and the behaviour of the nucleolar satellite during accelerated nucleoprotein synthesis. Nature, 163, 676. [DOI] [PubMed] [Google Scholar]
- Cassidy S. B., Dykens E., Williams C. A. (2000). Prader-Willi and Angelman syndromes: Sister imprinted disorders. American Journal of Medical Genetics, 97, 136–146. doi:10.1002/1096-8628(200022)97:2<136::AID-AJMG5>3.0.CO;2-V [DOI] [PubMed] [Google Scholar]
- Clayton-Smith J., Laan L. (2003). Angelman syndrome: A review of the clinical and genetic aspects. Journal of Medical Genetics, 40, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson M. A., Kouzarides T. (2012). Cancer epigenetics: From mechanism to therapy. Cell, 150, 12–27. [DOI] [PubMed] [Google Scholar]
- Deichmann U. (2016). Epigenetics: The origins and evolution of a fashionable topic. Developmental Biology, 416, 249–254. [DOI] [PubMed] [Google Scholar]
- Dey B. K., Mueller A. C., Dutta A. (2014). Long non-coding RNAs as emerging regulators of differentiation, development, and disease. Transcription, 5, e944014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Disteche C. M., Berletch J. B. (2015). X-chromosome inactivation and escape. Journal of Genetics, 94, 591–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorman J. S., Schmella M. J., Wesmiller S. W. (2016). Primer in genetics and genomics, Article 1—DNA, genes, and chromosomes. Biological Research for Nursing, 19, 7–17. [DOI] [PubMed] [Google Scholar]
- Duarte J. D. (2013). Epigenetics primer: Why the clinician should care about epigenetics. Pharmacotherapy, 33, 1362–1368. doi:10.1002/phar.1325 [DOI] [PubMed] [Google Scholar]
- Eisenberg E., Levanon E. Y. (2013). Human housekeeping genes, revisited. Trends in Genetics, 29, 569–574. [DOI] [PubMed] [Google Scholar]
- Felsenfeld G. (2014). A brief history of epigenetics. Cold Spring Harbor Perspectives in Biology, 6, a018200 doi:10.1101/cshperspect.a018200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gayon J. (2016). From Mendel to epigenetics: History of genetics. Comptes Rendus Biologies, 339, 225–230. doi:10.1016/j.crvi.2016.05.009 [DOI] [PubMed] [Google Scholar]
- Gunay-Aygun M., Schwartz S., Heeger S., O’Riordan M. A., Cassidy S. B. (2001). The changing purpose of Prader-Willi syndrome clinical diagnostic criteria and proposed revised criteria. Pediatrics, 108, E92 doi:10.1542/peds.108.5.e92 [DOI] [PubMed] [Google Scholar]
- He Y., Hua Y., Lee J., Liu W., Keep R. F., Wang M. M., Xi G. (2010). Brain alpha- and beta-globin expression after intracerebral hemorrhage. Translational Stroke Research, 1, 48–56. doi:10.1007/s12975-009-0004-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hill M. A. (2017). Gastrulation. Embryology. Retrieved from https://embryology.med.unsw.edu.au/embryology/index.php/Gastrulation
- Holliday R. (2006). Epigenetics: A historical overview. Epigenetics, 1, 76–80. [DOI] [PubMed] [Google Scholar]
- Horsthemke B. (2014). In brief: Genomic imprinting and imprinting diseases. Journal of Pathology, 232, 485–487. [DOI] [PubMed] [Google Scholar]
- Inbar-Feigenberg M., Choufani S., Butcher D. T., Roifman M., Weksberg R. (2013). Basic concepts of epigenetics. Fertility and Sterility, 99, 607–615. doi:10.1016/j.fertnstert.2013.01.117 [DOI] [PubMed] [Google Scholar]
- Irizarry K. A., Miller M., Freemark M., Haqq A. M. (2016). Prader Willi syndrome: Genetics, metabolomics, hormonal function, and new approaches to therapy. Advances in Pediatrics, 63, 47–77. doi:10.1016/j.yapd.2016.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang H. S., Shin W. J., Lee J. E., Do J. T. (2017). CpG and non-CpG methylation in epigenetic gene regulation and brain function. Genes, 8, 148 doi:10.3390/genes8060148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J., Samaranayake M., Pradhan S. (2009). Epigenetic mechanisms in mammals. Cellular and Molecular Life Sciences, 66, 596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kota S. K., Feil R. (2010). Epigenetic transitions in germ cell development and meiosis. Developmental Cell, 19, 675–686. [DOI] [PubMed] [Google Scholar]
- Kovalchuk I., Kovalchuk O. (2012). Epigenetics in health and disease. Upper Saddle River, NJ: FT Press. [Google Scholar]
- LeMieux J. (2017). Calico cats are a walking genetics lesson. Retrieved from the American Council on Science and Health at https://www.acsh.org/news/2016/07/27/calico-cats-are-a-walking-genetics-lesson
- Lyon M. F. (1961). Gene action in the X-chromosome of the mouse (Mus musculus L.). Nature, 190, 372–373. doi:10.1038/190372a0 [DOI] [PubMed] [Google Scholar]
- Lyon M. F. (2005). X chromosome inactivation: No longer all-or-none’. European Journal of Human Genetics: EJHG, 13, 796–797. [DOI] [PubMed] [Google Scholar]
- Nanney D. L. (1958). Epigenetic control systems. Proceedings of the National Academy of Sciences of the United States of America, 44, 712–717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Human Genome Research Institute. (2016). Epigenomics fact sheet. Retrieved from https://www.genome.gov/27532724/
- Ohno S., Kaplan W., Kinosita R. (1959). Formation of the sex chromatin by a single X-chromosome in liver cells of Rattus norvegicus . Experimental Cell Research, 18, 415–418. [DOI] [PubMed] [Google Scholar]
- Online Mendelian Inheritance in Man. (2017). Ubiquitin-protein ligase e3a; ube3a. Retrieved from https://www.omim.org/entry/601623
- Ozcan G., Ozpolat B., Coleman R. L., Sood A. K., Lopez-Berestein G. (2015). Preclinical and clinical development of siRNA-based therapeutics. Advanced Drug Delivery Reviews, 87, 108–119. doi:10.1016/j.addr.2015.01.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patil V. S., Zhou R., Rana T. M. (2014). Gene regulation by non-coding RNAs. Critical Reviews in Biochemistry and Molecular Biology, 49, 16–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters J. (2014). The role of genomic imprinting in biology and disease: An expanding view. Nature Reviews Genetics, 15, 517. [DOI] [PubMed] [Google Scholar]
- Phillips T., Hoopes L. (2008). Transcription factors and transcriptional control in eukaryotic cells. Nature Education, 1, 119. [Google Scholar]
- Rasmussen K. D., Helin K. (2016). Role of TET enzymes in DNA methylation, development, and cancer. Genes & Development, 30, 733–750. doi:10.1101/gad.276568.115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodenhiser D., Mann M. (2006). Epigenetics and human disease: Translating basic biology into clinical applications. CMAJ: Canadian Medical Association Journal, 174, 341–348. doi:10.1503/cmaj.050774 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothbart S. B., Strahl B. D. (2014). Interpreting the language of histone and DNA modifications. Biochimica et Biophysica Acta, 1839, 627–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato M. (2017). Early origin and evolution of the Angelman syndrome ubiquitin ligase gene Ube3a. Frontiers in Cellular Neuroscience, 11, 62 doi:10.3389/fncel.2017.00062 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schübeler D. (2015). Function and information content of DNA methylation. Nature, 517, 321–326. doi:10.1038/nature14192 [DOI] [PubMed] [Google Scholar]
- Seisenberger S., Peat J. R., Hore T. A., Santos F., Dean W., Reik W. (2013). Reprogramming DNA methylation in the mammalian life cycle: Building and breaking epigenetic barriers. Philosophical Transactions of the Royal Society of London. Series B, Biological Sciences, 368, 20110330 doi:10.1098/rstb.2011.0330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sha K, Boyer L. A. (2009). The chromatin signature of pluripotent cells In The Stem Cell Research Community (Ed.), StemBook. Retrieved from http://www.stembook.org/node/585. [PubMed]
- She X., Rohl C. A., Castle J. C., Kulkarni A. V., Johnson J. M., Chen R. (2009). Definition, conservation and epigenetics of housekeeping and tissue-enriched genes. BMC Genomics, 10, 269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stochholm K., Juul S., Gravholt C. H. (2010). Mortality and incidence in women with 47, XXX and variants. American Journal of Medical Genetics Part A, 152, 367–372. [DOI] [PubMed] [Google Scholar]
- Swerdlow A. J., Higgins C. D., Schoemaker M. J., Wright A. F., Jacobs P. A. (2005). Mortality in patients with Klinefelter syndrome in Britain: A cohort study. Journal of Clinical Endocrinology & Metabolism, 90, 6516–6522. [DOI] [PubMed] [Google Scholar]
- Swerdlow A. J., Schoemaker M. J., Higgins C. D., Wright A. F., Jacobs P. A. (2005). Mortality and cancer incidence in women with extra X chromosomes: A cohort study in Britain. Human Genetics, 118, 255–260. [DOI] [PubMed] [Google Scholar]
- Tan W., Bacino C. A., Skinner S. A., Anselm I., Barbieri-Welge R., Bauer-Carlin A.…Glaze D. G. (2011). Angelman syndrome: Mutations influence features in early childhood. American Journal of Medical Genetics Part A, 155, 81–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trerotola M., Relli V., Simeone P., Alberti S. (2015). Epigenetic inheritance and the missing heritability. Human Genomics, 9, 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- U.S. National Library of Medicine. (2017). HBB gene. In Genetics home reference. Retrieved from https://ghr.nlm.nih.gov/gene/HBB
- Waddington C. H. (1942). Canalization of development and the inheritance of acquired characters. Nature, 150, 563–565. [DOI] [PubMed] [Google Scholar]
- Williams C. A., Driscoll D. J., Dagli A. I. (2010). Clinical and genetic aspects of Angelman syndrome. Genetics in Medicine, 12, 385–395. [DOI] [PubMed] [Google Scholar]
- Wu H., Sun Y. E. (2006). Epigenetic regulation of stem cell differentiation. Pediatric Research, 59, 21R–25R. [DOI] [PubMed] [Google Scholar]
- Yang C., Chapman A. G., Kelsey A. D., Minks J., Cotton A. M., Brown C. J. (2011). X-chromosome inactivation: Molecular mechanisms from the human perspective. Human Genetics, 130, 175–185. [DOI] [PubMed] [Google Scholar]
- Zitzmann M., Bongers R., Werler S., Bogdanova N., Wistuba J., Kliesch S.…Tüttelmann F. (2015). Gene expression patterns in relation to the clinical phenotype in Klinefelter syndrome. Journal of Clinical Endocrinology & Metabolism, 100, E518–E523. [DOI] [PubMed] [Google Scholar]