

Abstract

Starting from a hit series from a GNF compound library collection and based on a cell-based proliferation assay of Plasmodium falciparum, a novel imidazolopiperazine scaffold was optimized. SAR for this series of compounds is discussed, focusing on optimization of cellular potency against wild-type and drug resistant parasites and improvement of physiochemical and pharmacokinetic properties. The lead compounds in this series showed good potencies in vitro and decent oral exposure levels in vivo. In a Plasmodium berghei mouse infection model, one lead compound lowered the parasitemia level by 99.4% after administration of 100 mg/kg single oral dose and prolonged mice survival by an average of 17.0 days. The lead compounds were also well-tolerated in the preliminary in vitro toxicity studies and represents an interesting lead for drug development.
Introduction
After thousands of years, malaria is still one of the major infectious diseases that affect millions of people, especially those in underdeveloped countries.1 In the absence of effective antimalarial vaccines, low molecular weight antimalarial drugs are important weapons against the disease.2 Quinine, chloroquine, mefloquine, and artemisinin derivatives have played an important role in fighting against malaria. However, widespread drug resistance has made them less effective with artemisinin derivatives as the only exception. Artemisinin based combination therapies (ACT) recommended by WHO will help to delay the emergence of clinical resistance against artemisinins, but reports of increased parasite clearance times have emerged recently.3 Therefore, it is important to discover antimalarials with novel chemical entities that are effective against the multidrug resistant parasite strains. Here we report our continued effort to develop novel antimalarials from a cell-based screening strategy.4
In our quest to discover novel chemotypes that inhibit Plasmodium falciparum, we previously disclosed a more extensive screening campaign on a library of about 2 million compounds using a cell-based proliferation assay of Plasmodium falciparum (strain 3D7).5 Five-thousand of the reconfirmed hits have been subsequently disclosed in a public malaria database (see https://www.ebi.ac.uk/chembldb/index.php), and we analyzed the hit list with the following criteria: (1) good potency, where IC50 values were less than 1 μM against wild type as well as drug resistant strain; (2) good safety index, greater than 20-fold in a six-cell line toxicity panel; (3) easy synthesis and preferably with multiple active hits within the scaffold.
Compounds 1a–c consisting of imidazolopiperazine moiety presented themselves as an attractive hit series based on these criteria (Table 1). Their structures were distinct from the current known antimalarials such as aminoquinolines and endo-peroxides. Compounds 1a–c represented decent starting points in terms of potency in both wild type and drug resistant strain, and the scaffold exhibited a high selectivity index (>20-fold) against Huh7 cell line. Moreover, one of the representative compound, 1a, was highly soluble (>175 μM at pH 6.8) and clean on most commonly drug affected cytochrome P450 isoforms (IC50 values were greater than 10 μM for CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4). Despite the basic amine functionality on R1, compound 1a was a moderate hERG substrate as determined by binding assay (IC50 = 19 μM, 57% inhibition at 10 μM). However, compound 1a exhibited poor plasma oral exposure with Cmax = 320 nM and AUC(0–5h) = 972 (h·nM) in an oral mouse snapshot PK study, indicating the presence of metabolic soft spots within the molecule or overall poor permeability properties.6
Table 1. Structures of Hits from the HTS Screen.

|
P. falciparum strain IC50 a (nM) |
|||||
|---|---|---|---|---|---|
| compd | EBI code | R1 | 3D7 | W2 | cyotoxicity (μM) Huh7 |
| 1a | GNF-Pf-5069 | Gly | 63 [460] | 97 [473] | >10 [>100] |
| 1b | GNF-Pf-5179 | (dl)-Phe | 235 [119] | 271 [122] | >10 [>79] |
| 1c | GNF-Pf-5466 | (dl)-Leu | 116 [30] | 119 [29] | >10 [42] |
Values are the mean of at least two experiments. Data in brackets are for resynthesized powders; otherwise data are from HTS DMSO stock solutions.
The major goal of optimization was to improve potency and remove metabolically vulnerable functionality of the compounds. Figure 1 shows our initial objectives. In line with the three possible metabolic soft spots of compound 1a, we decided to study the three peripheral sections: the amino acid moiety R1, the phenyl moiety R2, and the aniline moiety R3. We hoped changes in these parts would help us deliver a candidate with enhanced potency as well as improved oral exposure.
Figure 1.

Initial hit of imidazolopiperazine series and plans to optimize the three peripheral parts.
Chemistry
The synthesis of imidazolopiperazine compounds such as 5 is outlined in Scheme 1. A Groebke–Blackburn three-component reaction of 2-aminopyrazine with 4-fluorophenyl isocyanide and 4-fluorobenzaldehyde efficiently furnished the skeleton.7 A PtO2 mediated reduction of the pyrazine ring then afforded desired core structure.8 Compound 5 was obtained by a HATU mediated amidation reaction with N-Boc-glycine followed by a TFA mediated deprotection of the amino group. When not commercially available, isocyanides were prepared in a formylation–dehydration sequence from the corresponding anilines.8
Scheme 1. Representative Synthetic Route of Imidazolopiperazines.

Reagents and conditions: (a) HClO4, MeOH, room temp, 48%; (b) PtO2, H2 (balloon), MeOH, room temp, quantitative; (c) N-Boc-glycine, HATU, DIEA, DMF, room temp, 72%; (d) TFA, DCM, room temp, quantitative; (e) TFA, DCM, room temp, quantitative; (f) 1-bromo-3-fluorobenzene, Pd2(dba)3, Xantphos, Cs2CO3, dioxane, 150 °C, 66%.
To efficiently evaluate the R3 section and avoid repeated preparation and usage of notoriously noxious isocyanides, compound 6 was prepared from the same Groebke–Blackburn three-component reaction of 2-aminopyrazine with tert-butyl isocyanide and 4-fluorobenzaldehyde. After removal of the tert-butyl group, the obtained compound 7 was then used as a common intermediate. A palladium mediated amination with different aryl bromides provided various analogues of compound 7.9
Results and Discussion
Structure–Activity Relationship
The potency of the compounds was determined against the 3D7 and W2 multidrug resistant strains using Sybergreen assay previously described in our HTS disclosure (Table 2).5 Each assay plate had mefloquine and artemisinin as standards to ensure the reliability of the assay. We decided to (1) test various natural and unnatural amino acids on R1, (2) add substituents to the right side phenyl ring, and (3) replace the methylenedioxyl moiety with substituted phenyl rings, heterocycles, and aliphatic rings. We began exploring the SAR studies on the initial hit compounds 1a–c by determining the minimal pharmacophore needed for the antiplasmodial activity. On the basis of the preliminary results, it was clear that dearomatized compound 3 provided us with a decent starting point for further SAR exploration on all the three parts of the molecule. Installation of a glycine group gave us compound 5, which was 4-fold more potent than the original hit compound 1a. On the basis of this result, we focused our efforts on evaluating various amino acids. Increasing the chain length of the amino acid moiety led to 8, which had inferior activity. Various monosubstitution on the α position provided potent compounds (compounds 9, 10, 12, and 13) which were less than 100 nM in potency range. The testing of both of the enantiomers of phenylalanine derivatives indicated that the stereochemistry of the amino acid moiety was not important. Removal of chiral center from compound 9 led us to α-methylalanine derivative 11. This compound had a decent activity of 20 nM against the parasites but offered an enormous advantage of blocking a potential metabolic soft spot and removing chirality in the molecule. A number of second and tertiary amines on the amino acid side chain had deleterious effects on the potency (compounds not shown). The future analogues were optimized using glycine or α-methylalanine as the side chain.
Table 2. SAR Study of R1 Moiety.

|
P. falciparum strain IC50 a (nM) |
|||
|---|---|---|---|
| compd | R1 | 3D7 | W2 |
| 2 | >10 | 5520 | |
| 3 | H | 200 | 175 |
| 5 | Gly | 20 | 23 |
| 8 | β-Ala | 70 | 75 |
| 9 | (l)-Ala | 90 | 64 |
| 10 | (l)-Val | 30 | 24 |
| 11 | α-Me-Ala | 20 | 25 |
| 12 | (l)-Phe | 110 | 121 |
| 13 | (d)-Phe | 70 | 59 |
| mefloquine | 12 | 8 | |
| pyrimethamine | 29 | >10000 | |
| artemisnin | 12 | 14 | |
Values are the mean of at least two experiments.
The study of the R2 group revealed that this portion of the molecule was not amenable to broad changes (Table 3). Compounds 5, 14, and 15 indicated that only small size substituents such as fluorine or hydrogen were tolerated. A comparison of compounds 5, 18, and 19 indicated that para and meta substitutions were tolerated but not ortho substitution. 3,4-Difluoro substitution (compound 20) did not provide additional boost in potency, and hence, it was inferred that 4-fluoro substitution provided the best atom economy for further SAR optimization. Attempts to replace the phenyl ring with a cyclohexyl group led to total loss in potency (compound not shown).
Table 3. SAR Study of R2 Moiety.
|
P. falciparum strain IC50 a (nM) |
||||
|---|---|---|---|---|
| compd | R1 | R2 | 3D7 | W2 |
| 14 | α-Me-Ala | Ph | 200 | 168 |
| 15 | Gly | 4Cl-Ph | 660 | 437 |
| 16 | Gly | 4OMe-Ph | 2270 | 1702 |
| 17 | Gly | 4Me-Ph | 3140 | 3360 |
| 18 | Gly | 3F-Ph | 10 | 30 |
| 19 | α-Me-Ala | 2F-Ph | 1390 | 1284 |
| 20b | α-Me-Ala | 3F,4F-Ph | 60 | 50 |
Values are the mean of at least two experiments.
Compound with 4-methylphenyl as R3.
After optimizing the R1 and R2 groups, we focused our attention on the aniline portion of the molecule (Table 4). One general trend was that compounds with glycine as R1 were usually more potent than their counterparts with dimethylglycine as R1. The potencies for compounds 21 and 22 indicated that aliphatic rings and heterocyclic rings were not tolerated. A number of different substituents (compounds 23–28) at either meta or para position gave active compounds. With the same substituents, 3,4-disubstitution was preferred over 2,4-disubsitution and 3,5-disubstitution patterns (compounds 33 and 34). The most potent compound was 31, which exhibited an EC50 of 3 nM on 3D7 and of 4 nM on W2.
Table 4. SAR Study of R3 Moiety.
| IC50 a (nM) |
|||||
|---|---|---|---|---|---|
| compd | R1 | R2 | R3 | 3D7 | W2 |
| 21 | α-Me-Ala | 4F-Ph | cyclohex- | 9610 | 5980 |
| 22 | α-Me-Ala | 4F-Ph | 3-pyridine | 1413 | 3320 |
| 23 | Gly | 4F-Ph | 3F-Ph | 50 | 34 |
| 24 | α-Me-Ala | 3F-Ph | 4F-Ph | 50 | 71 |
| 25 | Gly | 4F-Ph | 4Me-Ph | 10 | 13 |
| 26 | α-Me-Ala | 4F-Ph | 4Me-Ph | 20 | 24 |
| 27 | Gly | 4F-Ph | 4Cl-Ph | 10 | 9 |
| 28 | α-Me-Ala | 4F-Ph | 4Cl-Ph | 30 | 24 |
| 29 | Gly | 4F-Ph | 3F,4F-Ph | 30 | 23 |
| 30 | α-Me-Ala | 4F-Ph | 3F,4F-Ph | 44 | 36 |
| 31 | Gly | 4F-Ph | 3F,4Cl-Ph | 3 | 4 |
| 32 | α-Me-Ala | 4F-Ph | 3F,4Cl-Ph | 40 | 52 |
| 33 | α-Me-Ala | 4F-Ph | 2F,4F-Ph | 110 | 90 |
| 34 | α-Me-Ala | 4F-Ph | 3F,5F-Ph | 220 | 244 |
Values are the mean of at least two experiments.
ADMET and Pharmacokinetic Properties
Along with the extensive SAR study to optimize the potency of this series, some of the more potent analogues in different categories were evaluated for their in vitro ADME and in vivo pharmacokinetic properties. In general, this series of compounds had very good Lipinski’s rule-of-five compliance and most of the analogues were highly soluble (>175 μM at pH 6.8).
Table 5 summarizes the in vitro ADMET profiles of compounds with good in vitro potency. In general, metabolic stability of the compounds was good to moderate across three species, with rat being the most sensitive species and thus exhibiting the highest hepatic extraction ratio (ER). One interesting finding was that analogues with glycine as R1 and analogues with dimethylglycine as R1 behaved quite differently in terms of hERG activity. Although the glycine analogues offered a safety advantage in terms of low affinity to hERG channel, it was negated by low permeability, as indicated by PAMPA and Caco-2 assays. In general, this also led to lower bioavailability as discussed below. While the dimethylglycines indicated some efflux due to high B–A values, this generally did not lead to lower bioavailability, potentially indicating a saturation of efflux mechanism (such as p-glycoproteins) at the doses used in the oral PK studies.
Table 5. In Vitro ADME of Selected Analoguesa.
| hepatic microsome metabolic stability (extraction ratio) |
|||||||
|---|---|---|---|---|---|---|---|
| compd | PAMPA (% absorbed) | Caco-2 A–B (cm/s × 106) | Caco-2 B–A (cm/s × 106) | mouse | rat | human | hERG (Q-patch) IC50 (μM) |
| 5 | 52 | 0.70 | 2.55 | 0.47 | nt | <0.21 | nt |
| 11 | 81 | 3.05 | 8.38 | 0.51 | 0.63 | 0.52 | 5.6 |
| 18 | low | 0.56 | 2.32 | 0.31 | nt | nt | >30 |
| 24 | 92 | 1.07 | 2.6 | 0.46 | nt | nt | 5.7 |
| 25 | 52 | 1.63 | 3.58 | 0.17 | nt | nt | 11.1 |
| 26 | 96 | 0.96 | 1.66 | 0.19 | 0.87 | 0.54 | 1.5 |
| 27 | 52 | 0.44 | 1.25 | 0.17 | 0.63 | 0.55 | nt |
| 28 | 89 | 1.46 | 1.86 | 0.17 | 0.43 | 0.54 | 2.3 |
| 29 | low | 0.99 | 2.99 | 0.21 | 0.38 | 0.21 | nt |
| 30 | 86 | 0.14 | 1.03 | 0.17 | 0.12 | 0.39 | 4.8 |
| 31 | low | 0.75 | 0.96 | 0.57 | 0.6 | 0.37 | 21 |
| 32 | 94 | 0.84 | 0.87 | 0.76 | 0.67 | 0.48 | 5.6 |
nt = not tested.
The in vivo pharmacokinetic properties were studied in noninfected male balb/C or female CD-1 mice. The overall trend for the oral exposure was in line with the predictions from the permeability assays. Four pairs of head to head comparison (entry 11/5, entry 18/24, entry 25/26, and entry 31/32) of the oral arm Cmax, AUC, and bioavailability revealed that dimethylglycine analogues generally had low hepatic in vivo clearance which resulted in higher AUCs as well as making the compounds more orally available (Table 6). Upon comparison of the pharmacokinetic profiles (Figure 2) for both compounds, it was observed that compound 32 has much higher exposure in the oral arm whereas the intravenous kinetics were comparable for both molecules (graph for compound 31 not shown for clarity). Some of the tested compounds exhibited high volume of distribution (>10 L/kg) and high plasma clearance (>90 (mL/min)/kg) in mice. On the basis of data from in vitro mouse blood to plasma partitioning of structural analogues with blood to plasma ratio of >1, these compounds might be partitioned into red blood cells leading to overestimation of blood clearance and extent of tissue distribution.
Table 6. In Vivo Pharmacokinetic Profile of Selected Analogues after Oral Dose at 30 mg/kg and iv Dose at 5 mg/kg in Micea.
| intravenous PK parameters |
oral PK parameters |
||||||||
|---|---|---|---|---|---|---|---|---|---|
| compd | Vd (L/kg) | CL (mL/min/kg) | T1/2 (h) | AUC(inf) (nM·h) | Cmax (nM) | Tmax (h) | T1/2 (h) | AUC(inf) (nM·h) | F (%) |
| 5 | 12.3 | 188.9 | 2.7 | 1072 | 234 | 2.0 | 4.2 | 965 | 15 |
| 11 | 23.7 | 60.0 | 6.2 | 3442 | 1803 | 1.67 | 9.0 | 17894 | 87 |
| 18 | 21.6 | 192.3 | 1.8 | 1232 | 417 | 2.0 | 1.0 | 1330 | 18 |
| 24 | 12.8 | 134.2 | 2.3 | 1319 | 778 | 0.50 | 2.3 | 3719 | 47 |
| 25 | 39.7 | 222.2 | 3.6 | 976 | 343 | 2.0 | 4.8 | 2343 | 40 |
| 26 | 41.1 | 42.0 | 13.7 | 5253 | 1007 | 2.0 | 15.5 | 17964 | 57 |
| 28 | 40.5 | 65.2 | 8.7 | 2985 | 749 | 1.00 | 37.6 | 28301 | 158 |
| 30 | 14.0 | 27.7 | 7.7 | 7003 | 1940 | 1.67 | 7.7 | 31758 | 76 |
| 31 | 1.5 | 10.3 | 3.8 | 20582 | 790 | 2.0 | 3.6 | 6175 | 5 |
| 32 | 1.7 | 5.8 | 7.8 | 25464 | 3319 | 2.0 | 9.1 | 36668 | 24 |
Intravenous dosing was at 5 mg/kg. Compounds 11 and 30 were dosed at 20 mg/kg. PK values were normalized to 30 mg/kg for comparison. For compounds 11 and 30: Species/strain, Balb/c mice. Formulation: 2.5 mg/mL in PEG300/D5W, 3:1, solution. All of the other compounds were dosed at 30 mg/kg po; strain, CD-1 female mice. Solution formulation with citrate buffer 50 mM (pH 4.5) for iv route and citrate buffer 100 mM (pH 3.5) for po route for compounds 5, 18, 24, 25, 26, and 28. Solution formulation for compounds 31 and 32: Ethanol/PEG400/10% vitamin ETPGS (10:30:60) was used.
Figure 2.
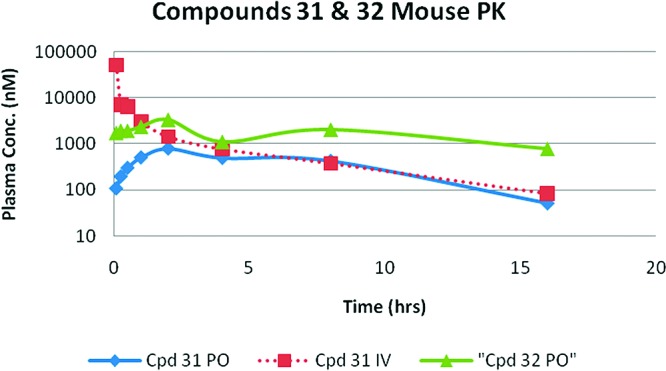
Graphical representation of po and iv kinetics for compounds 31 and 32.
In Vivo Efficacy
The in vivo antimalarial activities of the optimized compounds were evaluated using a Plasmodium berghei mouse model.10 In this test, groups of three P. berghei infected mice were treated orally with compound starting 1 day after infection. The key readouts were the percentage of parasitemia reduction on day 3 postinfection (day 4 postinfection in the case of a 3-day treatment schedule) compared to the untreated mice and the prolongation of survival of the infected mice.
Table 7 summarized the in vivo efficacy test results of selected compounds compared with the standard antimalarials chloroquine (CQ) and artesunate (AS). At 1 × 100 mg/kg dose level, most of the compounds could reduce the parasitemia for more than 99% and prolong mice survival for more than 10 days. Interestingly, for the three head-to-head comparison pairs (entry 25/26, entry 31/32, and entry 18/24), the glycine analogues were less efficacious at 1 × 100 and 1 × 30 mg/kg dose levels despite their better potencies. It was likely that the better oral exposure of the dimethylglycine analogues is responsible for this outcome. Among all the tested compounds, compounds 11 and 30 stood out as the most efficacious compounds at 1 × 100 and 1 × 30 mg/kg dose levels. They both showed parasitemia reduction activities and survival prolongation comparable those of CQ and AS. When the 1 × 30 mg/kg and 3 × 30 mg/kg dose levels are compared, both compounds 11 and 30 demonstrated improved efficacy.
Table 7. In Vivo Efficacy of Selected Analogues on P. berghei Rodent Malaria Model after Different Dose Levelsa.
| 1 × 30 mg/kg |
1 × 100 mg/kg |
3 × 30 mg/kg |
||||
|---|---|---|---|---|---|---|
| compd | parasitemia reduction (%) | survival (day) | parasitemia reduction (%) | survival (day) | parasitemia reduction (%) | survival (day) |
| 11 | 99.3 | 7.7 | 99.6 | 13.3 | 99.9 | 14.0 |
| 18 | 86 | 6.0 | 99.7 | 10.0 | ||
| 24 | 83 | 7.0 | 99.7 | 12.7 | ||
| 25 | 83 | 6.3 | 99.6 | 10.3 | ||
| 26 | 94 | 7.3 | 99.6 | 13 | ||
| 28 | 98 | 6.0 | 99.5 | 13.0 | ||
| 30b | 99.4 | 7.7 | 99.4 | 17.0 | 99.8 | 17.7 |
| 31 | 77 | 6.7 | 99.6 | 9.0 | ||
| 32 | 93 | 7.0 | 99.8 | 11.0 | ||
| CQ | 99.7 | 8.7 | >99.9 | 12 | 98.6 | 18.8 |
| AS | 89 | 7.2 | 97 | 6.7 | 98 | 7.2 |
Activity = average parasitemia reduction. Survival = average lifespan after infection; 6–7 days for untreated mice; 7% Tween 80/3% ethanol formulation.
75% PEG300/25%D5W formulation.
In Vitro Toxicology
Because of their good in vitro potency and in vivo efficacy results, compounds 11 and 30 were evaluated for their toxicity profile. Both compounds did not inhibit cytochromes P450 isoforms (IC50 values were greater than 8.9 μM for CYP1A2, CYP2C19, CYP2C9, CYP2D6, and CYP3A4). The observed cytotoxicity TC50 values (293T, Ba/F3, CHO, HEp2, HeLa, Huh7) were all greater than 12 μM, which translates to a good selectivity index (SI > 100). Compound 11 was well tolerated in the maximum tolerable dose test in rats in vivo with no gross effects at doses up to 500 mg/kg. Compound 30 was negative in the mini-Ames and micronucleus tests. The only safety concern for these two lead compounds were their hERG liability. Although compounds 11 and 30 exhibited IC50 of 5.6 and 4.8 μM, respectively, there was a toxicity index of 100-fold when compared to the cellular potency against the parasites.
Summary
A series of imidazolopiperazines was selected as one of the good hits from a high throughput screen of two million library compounds against a cell based parasite proliferation assay. After extensive optimization, the most potent compounds achieved single digit nanomolar range. The lead compounds showed good physicochemical and pharmacokinetic properties as well as safety profile. In the rodent P. berghei model, compounds 11 and 30 were as efficacious as the known antimalarials at 1 × 100 and 1 × 30 mg/kg dose levels.
The mechanism of actions for the imidazolopiperazine series is not clear. The tested compounds were active against 15 Plasmodium falciparum parasite strains with no potency shift, which proved their effectiveness against the existing multidrug resistant parasite stains. In a time when artemisinin based therapy is the last defense line for multidrug resistant malaria parasite strains, a novel effective chemical entity with distinct structure and mechanism of action will help to offer a new layer of protection. The continued optimization of this series will be reported in due course.
Experimental Section
Biology. Maintenance of Plasmodium falciparum Cultures
Plasmodium falciparum cultures are grown in O+ RBCs using culturing medium: RPMI 1640 medium (no phenol red) with l-glutamine and containing 0.05 mg/mL gentamicin, 0.014 mg/mL hypoxanthine, 38.4 mM Hepes, 0.20% sodium bicarbonate, 0.20% glucose, 3.4 nM NaOH, 5% human serum, and 1.25% albumax. By use of traditional P. falciparum protocols published previously, 25 mL cultures are maintained in 75 cm2 flasks (Fisher catalog no. 10-126-41) at 5% hematocrit and parasitemia ranging between 1% and 10%.11 Once parasitemia reaches 8–10%, cultures are diluted to 1% or higher depending on needs. Maintenance requires daily medium changes and fresh blood every 2 weeks. Cultures are gassed for approximately 30 s to 1 min using a blood–gas mixture to maintain a gas composition of 96% nitrogen, 3% carbon dioxide, and 1% oxygen and incubated at 37 °C. On day 1 of the assay, percent parasitemia is determined by obtaining a 1 μL blood smear. The smear is fixed onto the slide by placing in methanol for 30 s and staining in 10% Geimsa stain, and the percent of parasitized erythrocytes vs uninfected erythrocytes is determined by microscopy with a light microscope.
Antimalarial Proliferation Inhibition Assay: 384-Well Plate Format
P. falciparum strains to be used for screening are prepared with screening medium (culturing medium without human serum but supplemented with 0.5% Albumax II) and fresh erythrocytes. An amount of 20 μL of screening medium is dispensed via the MicroFlo (BioTek) into 384-well, black clear-bottom assay plates (μClear GNF custom plates by Griener Bio-One). An amount of 50 nL of compounds is transferred using a PlateMate Plus (Matrix) or the GNF Systems PinTool into the assay plates containing screening medium to a final maximum concentration of 10 μM in a 12-point dose response (1/2 log serial dilution). On the basis of the measured parasitemia levels, each parasite strain is diluted to yield 0.5% parasitemia, and 50% hematocrit blood (uninfected erythrocytes) is added to a concentration of 4.17%. This diluted culture is used in dispensing the remaining 60% of the assay volume (30 μL) into the prewarmed assay plates via the MicroFlo liquid dispenser. The final parasitemia is 0.3% and the final hematocrit is 2.5% in a total volume of 50 μL. Assay plates are incubated at 37 °C for 72 h in a gas chamber filled with low oxygen blood gas. After the 72 h of incubation (equivalent to one to two cycles during the blood stage) a prepared mixture of the lysis buffer (5 mM EDTA, 1.6% Triton X-100, 20 mM Tris-HCl, 0.16% saponin) in water and SYBR Green detection (0.1%) reagent is dispensed at 10 μL per well using the MicroFlo. Cultures are incubated for an additional 24 h at 25 °C before measuring fluorescence intensity using the Envision plate reader (Perkin-Elmer) with a 505 dichroic mirror. Excitation and emission filters are 485 and 530 nm, respectively. Data are normalized based on maximum fluorescence signals for DMSO treated wells (no inhibition by compound) and the minimum fluorescence signals for wells containing the highest concentration of inhibitor control compounds, for pyrimethamine, at a final concentration of 10 μM. Data are analyzed on a plate-by-plate basis and compared to reference compounds that are always included on every plate, typically artemisinin, mefloquine, and pyrimethamine. EC50 values are obtained using a custom curve fitting model, and a standard logistic regression model is applied for curve fitting. The quality of the assay run is assessed by the performance of the reference compounds where the EC50 must be within 3-fold of the standard reference values for the assay plate to pass requisite data quality control needs. Additionally, all compounds are typically assayed at a minimum in duplicate (independent assay plates) and EC50 values ideally must not vary by more than 2-fold between plates.
Metabolic Stability
The metabolic stability of drug candidates is determined in human, mouse, and rat liver microsomes using the compound depletion approach, quantified by LC/MS/MS. The assay measures the rate and extent of metabolism of chemical compounds by measuring the disappearance of the parent compound. The assay determines the compound’s in vitro half-life (T1/2) and hepatic extraction ratios (ER) and predicts metabolic clearance in human, rat, and mouse species.12
Pampa Assay
Permeation experiments were carried out in 96-well microtiter filter plates. The compound concentration was determined by UV absorption measured with a SpectraMax190 microplate spectrophotometer (Molecular Devices Corporation, Sunnyvale, CA, U.S.) at absorption wavelengths between 260 and 290 nm.13
Caco-2 Assay
The Caco-2 assay was carried out in a 96-well format, and compound concentrations in each chamber are measured by LC/MS. The assay offers apparent permeability data of test compounds from the apical to the basolateral chambers [Papp(A–B)] and from the basolateral to the apical chambers [Papp(B–A)]. The results can be used to predict the in vivo oral absorption (using Papp(A–B)) and the transport mechanism of the test compounds across GI membrane (ratio Papp(B–A)/Papp(A–B) and log P lipophilicity data).14
hERG (Q-Patch) Assay
The assay utilizes electrophysiology measurements on the electric current passing through hERG channel that is heterologously expressed in a stable CHO cell line. Channels are open by a hERG-specific voltage protocol, and the compound effects are directly characterized by their block of the hERG current. The assay is performed on an automated platform, Q-Patch. All compounds are tested by four-point dose response curves, and IC50 values (between 0.2 and 30 μM) are measured.15
In Vivo Pharmacokinetics Studies in Mice
NITD’s Animal Care and Use Committee (IACUC), registered with Agri-Food and Veterinary Authority (AVA), Government of Singapore, approved all animal experimental protocols. Mice were allowed for acclimatization before initiation of pharmacokinetic (PK) experiments. Feed and water were given ad libitum.
Compounds 5, 18, 24, 25, 26, 28, 31, and 32 were formulated at 3 mg/mL for a dose of 30 mg/kg given orally (po) and at 1 mg/mL for a dose of 5 mg/kg given intravenously (iv). Citrate buffer 100 mM, pH 3.5, was used to formulate compounds 5, 18, 24, 25, 26, and 28 prior to po administration, while citrate buffer 50 mM, pH 4.5, was used for iv administration. Both were clear solutions. For compounds 31 and 32, the solution formulation for both po and iv dosing was ethanol/PEG400/10% vitamin ETPGS (10:30:60). Blood samples were collected at 0.08, 0.25, 0.5, 1, 2, 4, 8, 16, and 24 h following po dosing. Sampling was identical following iv dosing except that the first time point was 0.02 h. Groups of three mice were used for each time point. Blood was centrifuged at 13 000 rpm for 7 min at 4 °C and plasma harvested and stored at −20 °C until analysis.
PK studies for compounds 11 and 30 were conducted at GNF. In-life studies were carried out under protocols approved by the Animal Care and Use Committee (IACUC) of GNF. Compounds 11 and 30 were formulated at 2.5 mg/mL in 75% PEG300 and 25% D5W (5% dextrose in distilled water) solution and were filtered using a 0.45 μm syringe filter. The filtered solutions were dosed intravenously via the lateral tail vein at 5 mg/kg with a dosing volume of 2 mL/kg to male Balb/c mice (n = 3 per group). The filtered solutions were also administered orally at 20 mg/kg (with a dosing volume of 8 mL/kg) to another groups of mice (n = 3 per group). Six blood samples of 50 μL each were collected serially from each animal via retro-orbital sinus up to 24 h after dosing. Blood samples were collected into heparin microtainer and centrifuged, and the plasma was separated and frozen until analysis. Plasma samples were analyzed by LC/MS/MS.
Extraction and LC/MS Analysis
Plasma samples were extracted with acetonitrile/methanol/acetic acid (90:9.8:0.2) for analytes and internal standard (analogue from the same series) using a 8 to 1 extractant to plasma ratio. Analyte quantitation was performed by LC/MS/MS. Liquid chromatography was performed using an Agilent 1100 HPLC system (Santa Clara, CA), with the Agilent Zorbax XDB phenyl (3.5 μ, 4.6 mm × 75 mm) column at an oven temperature of 45 °C, coupled with a QTRAP4000 triple quadruple mass spectrometer (Applied Biosystems, Foster City, CA). Instrument control and data acquisition were performed using the Applied Biosystems software Analyst, version 1.4.2. The mobile phases used were (A) water–acetic acid (99.8:0.2, v/v) and (B) acetonitrile–acetic acid (99.8:0.2, v/v), using a gradient, with flow rate of 1.0 mL/min, and run time of 5 min. Compound detection on the mass spectrometer was performed in electrospray positive ionization mode and using multiple reaction monitoring (MRM) for specificity (transitions: compound 5 384.2/101.2, compound 18 384.3/101.2, compound 24 412.3/327.2, compound 25 380.4/118.2, compound 26 408.5/280.3, compound 28 428.2/343.2, compound 31 418.3/101.4, compound 32 446.3/361.2,) together with their optimized MS parameters. The lower limit of quantification ranged between 0.5 and 23 ng/mL for all the compounds studied.
For PK studies of compounds 11 and 30 in mice, plasma samples (20 μL) were extracted with a solution of acetonitrile/methanol (3:1) containing internal standard. The samples were vortexed, then centrifuged with an Eppendorf centrifuge 5810R (Eppendorf, Hamburg, Germany) at a setting of 4000 rpm for 10 min at room temperature. The supernatant was transferred to a clean 96-well analysis plate for LC/MS/MS analysis. An aliquot (10 μL) was injected onto a Phenomenex C18 guard column (4 mm × 2 mm) followed by a Zorbax SB-C8 analytical column (2.1 mm × 50 mm, 5 μm, Agilent Technologies Inc., Palo Alto, CA, U.S.). Separation was carried out using a gradient elution method with a mobile phase consisting of 0.05% formic acid in water (solvent A) and 0.05% formic acid in acetonitrile (solvent B). The flow rate was 600 μL/min. The HPLC system, consisting of Agilent 1100 series binary pump (Agilent Technologies Inc.), Agilent 1100 series microvacuum degasser (Agilent Technologies Inc.), HTC PAL CTC analytics autosampler (LEAP Technologies, Carborro, NC), and VICI two-position actuator (Valco Int., Huston, Texas, U.S.), was interfaced to a MDS SCIEX API-4000 triple quadruple mass spectrometer (MDS Inc., Toronto, Canada). Mass spectral analyses were carried out using electrospray source in the positive ion mode and using multiple reaction monitoring (MRM) for quantification. MRM transitions of 412.2/327.2 and 430.1/304.0 were used for compounds 11 and 30, respectively.
Pharmacokinetic Analysis
The mean value from the three animals at each time point was plotted against time to give plasma concentration–time profile. Pharmacokinetic parameters were determined using Watson LIMS, version 7.2 (Thermo Electron Corporation, PA, U.S.), by noncompartmental analysis. Pharmacokinetic parameters for compounds 11 and 30 were calculated by noncompartmental regression analysis using an in-house fitting program developed at GNF. The oral bioavailability (F) was calculated as the ratio between the area under the curve (AUC) following oral administration and the AUC following intravenous administration corrected for dose (F = [(AUCpo)(doseiv)]/[(AUCiv)(dosepo)].
In Vivo Antimalarial Activity
All in vivo efficacy studies were approved by the veterinary authorities of the Canton Basel-Stadt. In vivo antimalarial activity was usually assessed for groups of three female NMRI mice (20–22 g) intravenously infected on day zero with 2 × 107 erythrocytes parasitized with P. berghei GFP ANKA malaria strain (PbGFPCON donation from AP Waters and CJ Janse, Leiden University, The Netherlands).10a,16 Untreated control mice die typically between day 6 and day 7 after infection. Experimental compounds were formulated in 7% Tween 80/3% ethanol or 75% PEG300/25% D5W as indicated. Compounds were administered orally in a volume of 10 mL/kg either as a single dose (24 h post infection) or as three consecutive daily doses (24, 48, and 72 h after infection). With the single dose regimen parasitemia was determined (72 h after infection). For the triple dose regimen (96 h after infection), standard flow cytometry techniques were used.16 Activity was calculated as the difference between the mean percent parasitemia for the control and treated groups expressed as a percentage of the control group. The survival time in days was also recorded up to 30 days after infection. A compound was considered curative if the animal survived to day 30 after infection with no detectable parasites.
Chemistry
Unless otherwise noted, materials were obtained from commercial suppliers and were used without purification. Removal of solvent under reduced pressure refers to distillation using Büchi rotary evaporator attached to a vacuum pump (∼3 mmHg). Products obtained as solids or high boiling oils were dried under vacuum (∼1 mmHg).
Purification of compounds by high pressure liquid chromatography was achieved using a Waters autopurification system consisting of a 2767 autosampler/fraction collector, a 2525 binary gradient module, a 2487 UV detector, and a ZQ mass spectrometer. Compounds were purified using flow rate of 30 mL/min with a 50 mm × 20 mm i.d. Ultra 120 5 μm C18Q column (Peeke Scientific, Novato, CA). A 7.5 min linear gradient from 10% solvent A (acetonitrile with 0.035% trifluoroacetic acid) in solvent B (water with 0.05% trifluoroacetic acid) to 90% A was used, followed by a 2.5 min hold at 90% A. 1H NMR spectra were recorded on Bruker XWIN-NMR (400 or 600 MHz) spectrometer. Proton resonances are reported in parts per million (ppm) downfield from tetramethylsilane (TMS). 1H NMR data are reported as multiplicity (s, singlet; d, doublet; t, triplet; q, quartet; quint, quintuplet; sept septuplet; dd, doublet of doublets; dt, doublet of triplets; bs, broad singlet), number of protons, and coupling constant in hertz. For spectra obtained in CDCl3, DMSO-d6, CD3OD, the residual protons (7.27, 2.50, and 3.31 ppm, respectively) were used as the reference.
Analytical thin-layer chromatography (TLC) was performed on commercial silica plates (Merck 60-F 254, 0.25 mm thickness). Compounds were visualized by UV light (254 nm). Flash chromatography was performed either by CombiFlash (separation system Sg. 100c, ISCO) or using silica gel (Merck Kieselgel 60, 230–400 mesh). Elemental analyses were carried out by Midwest Microlabs LLC, Indianapolis, IN.
Purity of Compounds
All compounds were analyzed with a Waters ZQ 2000 LC/MS system, which used an Agilent binary pump and photodiode array detector (PDA) along with a Sedere 75 evaporative light scattering detector (ELSD). Samples were injected from 96-well or 384-well plates with a Leap Technologies HTS Pal autosampler. The mobile phases used were (A) H2O + 0.05% TFA and (B) ACN + 0.035% TFA. A gradient HPLC method with flow of 1.0 mL/min started at 5% B, with a hold of 0.1 min before a linear increase to 95% B at 2.70 min. At 2.71 min the B mobile phase was increased to 100% and held until 2.98 min. Total run time was 3.0 min. The column used was a Waters Atlantis dC18 2.1 mm × 30 mm, 3 μm. The mass spectrometer was operated in positive mode, with a spray voltage of 3.2 kV and cone voltage of 30 V. The source and desolvation temperatures were 130 and 400 °C, respectively, with 600 L/h of nitrogen desolvation flow. The progress of reactions and the purity of products were measured using 254 and 220 nm wavelengths and electrospray ionization (ESI) positive mode. Mass spectra were obtained in ESI positive mode. All the reported compounds were found to be >95% when analyzed at the 254 nm wavelength.
Compound 1a: 2-Amino-1-(3-(benzo[d][1,3]dioxol-5-ylamino)-2-phenyl-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)ethanone
The structure of 1a is as follows:

Synthesis of 1-2: N-(Benzo[d][1,3]dioxol-5-yl)-2-phenyl-5,6,7,8-tetrahydroimidazo[1,2-a]pyrazin-3-amine

To a stirred solution of isocyanide (158 mg, 1.66 mmol) in 10 mL of MeOH were added benzaldehyde (0.25 mL, 2.49 mmol) and 2-aminopyrazine (281 mg, 1.91 mmol), followed by 1.0 N HClO4 in MeOH (0.17 mL, 0.17 mmol). The reaction mixture was stirred at room temperature for 3 h. The reaction mixture was directly taken to mass-triggered HPLC purification. The collected MeCN/water solution was concentrated and dried on a lyophilizer to give 296 mg of compound 1-2 as a powdery product. 1H NMR (CDCl3, 400 MHz) δ 9.23 (d, J = 1.2 Hz, 1H), 8.03 (d, J = 4.8 Hz, 1H), 7.91–7.94 (m, 3H), 7.26–7.44 (m, 3H), 6.68 (d, J = 8.4 Hz, 1H), 6.32 (br, 1H), 6.25 (d, J = 2.4 Hz, 1H), 6.00 (dd, J = 8.4, 2.4 Hz, 1H), 5.93 (s, 2H).
Synthesis of 1-3: N-(Benzo[d][1,3]dioxol-5-yl)-2-phenyl-5,6,7,8-tetrahydroimidazo[1,2-a]pyrazin-3-amine

To a stirred solution of compound 1-2 (181 mg, 0.54 mmol) in 10 mL of MeOH was added Pd/C (58 mg, 0.054 mmol). The reaction mixture was evacuated and back-filled with H2. The reaction mixture was stirred at room temperature overnight. The solid was filtered off, and solvent was removed. The 400 MHz proton NMR of the crude product proved it to be 1-3. 1H NMR (MeOH-d4, 400 MHz) δ 7.72–7.4 (m, 2H), 7.38–7.33 (m, 3H), 6.66 (d,, J = 8.29 Hz, 1H), 6.33 (d, J = 2.32 Hz, 1H), 6.14 (dd, J = 8.3, 2.3 Hz, 1H), 5.86 (s, 2H), 4.57 (s, 2H), 4.08 (t, J = 5.7 Hz, 2H), 3.68 (t, J = 5.7 Hz, 2H).
Synthesis of Compound 1a: 2-Amino-1-(3-(benzo[d][1,3]dioxol-5-ylamino)-2-phenyl-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)ethanone
To a stirred solution of N-BOC-glycine (142 mg, 0.81 mmol) in 2 mL of DMF were added HATU (308 mg, 0.81 mmol) and DIEA (0.28 mL, 1.62 mmol). After the mixture was stirred for 10 min, compound 1-3 (180 mg, 0.54 mmol) was added. The reaction mixture was stirred at room temperature for 3 h. Solvent was removed, and the crude product was subjected to MS-triggered HPLC purification. The collected MeCN/water solution was concentrated until no more MeCN was left. The remaining aqueous solution was neutralized with NaHCO3 and extracted with DCM. The organic solution was dried and concentrated. The residue was dissolved in 1:1 MeCN/water solvent and dried on a lyophilizer to give 192 mg of powdery amide coupled product.
A solution of the above obtained amide (265 mg, 0.54 mmol) in 10 mL of 4:1 DCM and TFA was stirred at room temperature for 2 h. Solvent was removed, and the crude product was subjected to MS-triggered HPLC separation. The collected MeCN/water solution was concentrated until no more MeCN was left. The remaining aqueous solution was neutralized with NaHCO3 and extracted with DCM. The organic solution was dried and concentrated. The oil was then dissolved in 1:1 MeCN/water solvent and dried on a lyophilizer to give a powdery product in quantitative yield. 1H NMR (400 MHz, MeOH-d4) δ 7.75 (m, 2H), 7.19 (m, 2H), 7.18 (m, 1H), 6.61 (m, 1H), 6.22 (m, 1H), 6.03 (m, 1H), 5.82 (m, 2H), 4.75 (m, 2H), 3.78–4.05 (m, 6H). M/Z = 392.2 (M + 1).
Compound 1b: 2-Amino-1-(3-(benzo[d][1,3]dioxol-5-ylamino)-2-phenyl-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)-3-phenylpropan-1-one

Compound 1b was synthesized from compound 1-3 by a HATU mediated coupling with 2-(tert-butoxycarbonylamino)-3-phenylpropanoic acid followed by TFA mediated deprotection. 1H NMR (400 MHz, MeOH-d4) δ 7.67 (m, 2H), 7.37–7.20 (m, 8H), 6.65 (m, 1H), 6.25 (m, 1H), 6.07 (td, J = 2.29 Hz, J = 8.14 Hz, 1H), 5.85 (m, 2H), 4.79 (dd, J =5.86 Hz, J = 9.79 Hz, 1H), 4.59–4.19 (m, 2H), 3.57–3.83 (m, 3H), 3.16–2.93 (m, 3H). M/Z = 482.3 (M + 1).
Compound 1c: 2-Amino-1-(3-(benzo[d][1,3]dioxol-5-ylamino)-2-phenyl-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)-4-methylpentan-1-one

Compound 1c was synthesized from compound 1-3 by a HATU mediated coupling with 2-((tert-butoxycarbonyl)amino)-4-methylpentanoic acid followed by TFA mediated deprotection. 1H NMR (400 MHz, MeOH-d4) δ 7.78 (m, 2H), 7.42 (m, 3H), 6.66 (m, 1H), 6.36 (m, 1H), 6.13 (m, 1H), 5.85 (m, 2H), 5.24 (m, 1H), 4.58 (m, 1H), 4.05 (m, 4H), 3.35 (s, 2H), 1.76 (m, 2H), 1.03 (m, 6H). M/Z = 448.2 (M + 1).
Compound 2: N,2-Bis(4-fluorophenyl)imidazo[1,2-a]pyrazin-3-amine

Compound 2 was synthesized by the following way: To a stirred solution of 2-aminopyrazine (683 mg, 7.18 mmol) in 50 mL of MeOH were added 4-fluorobenzaldehyde (1.16 mL, 10.8 mmol) and 1-fluoro-4-isocyanobenzene (1.0 g, 8.25 mmol), followed by 1.0 N HClO4 in MeOH (0.72 mL, 0.72 mmol). The reaction mixture was stirred at room temperature overnight. The reaction mixture was concentrated, and the residue was subjected to flash chromatography purification. The collected organic solution was concentrated to give the compound 2 (1.1 g, 48%) as a yellow oil. 1H NMR (400 MHz, MeOH-d4) δ 8.96 (s, 1H), 7.95 (m, 3H), 7.82 (s, 1H), 7.07 (t, J = 8.84 Hz, 2H), 6.82 (t, J = 8.77 Hz, 2H),6.45 (m, 2H). M/Z = 323.3 (M + 1).
Compound 3: N,2-Bis(4-fluorophenyl)-5,6,7,8-tetrahydroimidazo[1,2-a]pyrazin-3-amine

Compound 3 was synthesized by the following way: To a stirred solution of compound 2 (761 mg, 2.36 mmol) in 10 mL of MeOH was added Pd/C (258 mg, 0.24 mmol). The reaction mixture was evacuated and back-filled with H2. The reaction mixture was stirred at room temperature overnight. The solid was filtered off, and solvent was removed. The product was subjected to mass-triggered HPLC purification. The obtained MeCN/water solution was combined and concentrated to give the final product as a yellow oil after neutralization (quantitative yield). 1H NMR (300 MHz, DMSO-d6) δ 7.81–7.72 (m, 2H), 7.11 (m, 2H), 6.97 (m, 2H), 6.56–6.52 (m, 2H), 3.86 (s, 2H), 3.55 (m, 2H), 3.02 (m, 2H). M/Z = 327.2 (M + 1).
Compound 5: 2-Amino-1-(2-(4-fluorophenyl)-3-(4-fluorophenylamino)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)ethanone

Compound 5 was synthesized from compound 3 by a HATU mediated coupling with N-BOC-glycine followed by TFA mediated deprotection (72% isolated yield) using a protocol for synthesis of compound 1a. 1H NMR (400 MHz, MeOH-d4) δ 7.59–7.56 (m, 2H), 7.12 (t, J = 8.8 Hz, 2H), 6.86 (t, J = 8.8 Hz, 2H), 6.68–6.65 (m, 2H), 5.04–5.00 (m, 2H), 4.08–3.92 (m, 6H). M/Z = 384.2 (M + 1). Elemental analysis (compound + 3HCl): % C, 48.75; % H, 4.50; % N, 14.21 (calculated). % C = 48.86/48.58; % N = 14.11/13.95; % H = 4.69/4.49 (experimental).
Compound 6: N-(tert-butyl)-2-(4-fluorophenyl)imidazo[1,2-a]pyrazin-3-amine

To a stirred solution of 2-aminopyrazine (571 mg, 6.0 mmol) in 10 mL of MeOH were added 4-fluorobenzaldehyde (0.97 mL, 9.0 mmol), 2-isocyano-2-methylpropane (0.78 mL, 6.9 mmol), and 1.0 N HClO4 in MeOH (0.60 mL, 0.60 mmol). The reaction mixture was stirred at room temperature for 3 h. Solvent was removed and the residue was subjected to flash chromatography purification to give compound 6 as white solid. 1H NMR (CDCl3, 400 MHz) δ 8.97 (d, J = 1.6 Hz, 1H), 8.10 (dd, J = 1.6, 4.4 Hz, 1H), 7.90–7.94 (m, 2H), 7.86 (d, J = 4.66 Hz, 1H), 7.13 (t, J = 8.8 Hz, 2H), 1.04 (s, 9H).
Compound 7: 2-(4-fluorophenyl)imidazo[1,2-a]pyrazin-3-amine

A solution of compound 6 (284 mg, 1.0 mmol) in 10 mL of 4:1 DCM and TFA was stirred at room temperature for 2 h. Solvent was removed, and the product was directly used in the next step after neutralization. 1H NMR (300 Hz, CDCl3) δ 8.82 (s, 1H), 8.28 (d, J = 4.8 Hz, 1H), 8.05–8.09 (m, 2H), 7.76 (d, J = 4.5 Hz, 1H), 7.27–7.33 (m, 2H), 5.79 (s, 2H). M/Z = 229 (M + 1).
Compound 8: 3-Amino-1-(2-(4-fluorophenyl)-3-(4-fluorophenylamino)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)propan-1-one

Compound 8 was synthesized from compound 3 by a HATU mediated coupling with 3-(tert-butoxycarbonylamino)propanoic acid followed by TFA mediated deprotection. 1H NMR (CD3OD, 400 MHz) δ 7.61–7.65 (m, 2H), 6.87–6.92 (m, 2H), 6.75–6.80 (m, 2H), 6.45–6.49 (m, 2H), 4.71 (d, J = 4.0 Hz, 2H), 3.82–3.90 (m, 2H), 3.62–3.74 (m, 2H), 2.81–2.84 (m, 2H), 2.51–2.57 (m, 2H). M/Z = 398.4 (M + 1).
Compound 9: (S)-2-Amino-1-(2-(4-fluorophenyl)-3-(4-fluorophenylamino)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)propan-1-one

Compound 9 was synthesized from compound 3 by a HATU mediated coupling with (S)-2-(tert-butoxycarbonylamino)propanoic acid followed by TFA mediated deprotection. 1H NMR (CD3OD, 400 MHz) δ 7.63 (m, 2H), 6.95 (m, 2H), 6.82 (m, 2H), 6.50 (m, 2H), 4.41 (m, 1H), 3.92–3.74 (m, 6H), 1.42 (d, J = 6.95 Hz, 3H). M/Z = 398.2 (M + 1).
Compound 10: (S)-2-Amino-1-(2-(4-fluorophenyl)-3-(4-fluorophenylamino)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)-3-methylbutan-1-one

Compound 10 was prepared from compound 3 by a HATU mediated coupling reaction with (S)-2-(tert-butoxycarbonylamino)-3-methylbutanoic acid followed by a TFA mediated deprotection. 1H NMR (DMSO-d6, 400 MHz) δ 8.15 (bs, 1H), 8.02 (s, 1H), 7.77 (m, 2H), 7.19 (m, 2H), 7.00 (m, 2H), 6.61 (m, 2H), 5.05–4.87 (m, 1H), 4.69–4.41 (m, 1H), 4.23–4.11 (m, 1H), 3.98–3.77 (m, 1H), 3.00 (m, 1H), 2.51 (s, 2H), 2.1 (m, 1H), 0.95 (dd, J = 22, 6.72 Hz, 6H). M/Z = 426 (M + 1)
Compound 11: 2-Amino-1-(2-(4-fluorophenyl)-3-(4-fluorophenylamino)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)-2-methylpropan-1-one

Compound 11 was synthesized from compound 3 by a HATU mediated coupling with 2-(tert-butoxycarbonylamino)-2-methylpropanoic acid followed by TFA mediated deprotection. 1H NMR (MeOH-d4, 400 Hz) δ7.70–7.67 (m, 2H), 7.22 (t, J = 8.8 Hz, 2H), 6.97(t, J = 8.8 Hz,2H), 6.8–6.77 (m, 2H), 5.16 (s, 2H), 4.24 (m, 2H), 4.07 (m, 2H), 1.76 (s, 6H). M/Z = 412.2 (M + 1). Elemental analysis of HCl salt (compound + 2HCl + 0.50 H2O): % C, 53.56; % H, 5.31; % N, 14.19 (calculated). % C = 53.98/53.69; % N = 14.07/13.97; % H = 5.16/4.98 (experimental).
Compound 12: (S)-2-Amino-1-(2-(4-fluorophenyl)-3-(4-fluorophenylamino)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)-3-phenylpropan-1-one

Compound 12 was synthesized from compound 3 by a HATU mediated coupling with (S)-2-(tert-butoxycarbonylamino)-3-phenylpropanoic acid followed by TFA mediated deprotection. 1H NMR (CDCl3, 400 Hz) δ 9.06 (bs), 7.94 (bs), 7.3–6.93 (m, 11H), 6.55 (m, 2H), 5.5–4.62 (m, 2H), 4.28 (m, 1H), 3.68–2.60 (m, 6H). M/Z = 474.4 (M + 1).
Compound 13: (R)-2-Amino-1-(2-(4-fluorophenyl)-3-(4-fluorophenylamino)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)-3-phenylpropan-1-one

Compound 13 was synthesized from compound 3 by a HATU mediated coupling with (R)-2-(tert-butoxycarbonylamino)-3-phenylpropanoic acid followed by TFA mediated deprotection. 1H NMR (400 MHz, MeOH-d4) δ 7.65 (m, 1H), 7.18 (m, 8H), 6.96 (m, 2H), 6.82 (m, 1H), 6.49 (m, 1H), 4.65–4.8 (m, 2H), 4.00–4.49 (m, 2H), 3.38–3.57 (m, 3H), 2.73–3.00 (m, 2H). M/Z = 474.4 (M + 1).
Compound 14: 2-Amino-1-(3-((4-fluorophenyl)amino)-2-phenyl-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)-2-methylpropan-1-one

Compound 14 was synthesized following the same sequence as compound 1 involving an Ugi reaction, reduction using palladium on carbon followed by HATU mediated coupling with N-BOC-dimethylglycine and TFA mediated deprotection. 1H NMR (DMSO-d6, 400 MHz) δ 8.25 (bs, 1H), 8.01 (s, 1H), 7.73 (m, 2H), 7.35 (m, 2H), 7.25 (m, 1H), 7.00 (t, J = 8.04 Hz, 2H), 6.62 (m, 2H), 4.40 (m, 2H), 4.08 (m, 2H), 3.76 (m, 2H), 1.60 (s, 6H). M/Z = 394.4 (M + 1).
Compound 15: 2-Amino-1-(2-(4-chlorophenyl)-3-((4-fluorophenyl)amino)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)ethanone

Compound 15 was synthesized following the same sequence as compound 1 involving an Ugi reaction, reduction using palladium on carbon followed by HATU mediated coupling with N-BOC-glycine and TFA mediated deprotection. 1H NMR (400 MHz, MeOH-d4) δ 7.68 (m, 2H), 7.38 (m, 2H), 6.94 (m, 2H), 6.69 (m, 2H), 5.05–5.00 (m, 2H), 4.12 (s, 2H), 3.83–3.99 (m, 4H). M/Z = 400.2 (M + 1).
Compound 16: 2-Amino-1-(3-((4-fluorophenyl)amino)-2-(4-methoxyphenyl)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)ethanone

Compound 16 was synthesized following the same sequence as compound 1 involving an Ugi reaction, reduction using palladium on carbon followed by HATU mediated coupling with N-Boc-glycine and TFA mediated deprotection. 1H NMR (400 MHz, MeOH-d4) δ 7.58 (d, J = 8.79 Hz, 2H), 6.98 (m, 4H), 6.75 (m, 2H), 5.07–5.00 (m, 2H), 4.14 (s, 2H), 4.02 (m, 4H), 3.18 (s, 3H). M/Z = 396.2 (M + 1).
Compound 17: 2-Amino-1-(3-((4-fluorophenyl)amino)-2-(p-tolyl)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)ethanone

Compound 17 was synthesized following the same sequence as compound 1 involving an Ugi reaction, reduction using palladium on carbon followed by HATU mediated coupling with N-Boc-glycine and TFA mediated deprotection. 1H NMR (400 MHz, MeOH-d4) δ 7.53 (d, J = 8.12 Hz, 2H), 7.17 (m, 2H), 6.94 (m, 2H), 6.72 (m, 2H), 5.07–5.00 (m, 2H), 4.13 (s, 2H), 4.02 (m, 4H), 2.35 (s, 3H). M/Z = 380.4 (M + 1).
Compound 18: 2-Amino-1-(3-((4-fluorophenyl)amino)-2-(3-fluorophenyl)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)ethanone

Compound 18 was synthesized following the same sequence as compound 1 involving an Ugi reaction, reduction using palladium on carbon followed by HATU mediated coupling with N-BOC-glycine and TFA mediated deprotection. 1H NMR (MeOH-d4, 400 Hz) δ 7.55–7.53 (m, 1H), 7.47–7.45 (m, 2H), 7.15 (m, 1H), 6.96–6.94 (m, 2H), 6.84–6.81 (m, 2H), 5.20 (m, 2H), 4.25–4.20 (m, 6H), 3.35 (s, NH). M/Z = 384.2 (M + 1).
Compound 19: 2-Amino-1-(2-(2-fluorophenyl)-3-((4-fluorophenyl)amino)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)-2-methylpropan-1-one

Compound 19 was synthesized following the same sequence as compound 1 involving an Ugi reaction, reduction using palladium on carbon followed by HATU mediated coupling with N-BOC-dimethylglycine and TFA mediated deprotection. 1H NMR (MeOH-d4, 400 Hz) δ 7.41 (m, 1H), 7.28 (m, 1H), 7.08 (m, 2H), 6.79 (m, 2H), 6.56 (m, 2H), 4.93(m, 2H), 4.00 (m, 2H), 3.86 (t, J = 5.17 Hz, 2H), 1.64 (s, 6H). M/Z = 412.2 (M + 1).
Compound 20: 2-Amino-1-(2-(3,4-difluorophenyl)-3-(p-tolylamino)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)-2-methylpropan-1-one

Compound 20 was synthesized following the same sequence as compound 1 involving an Ugi reaction, reduction using palladium on carbon followed by HATU mediated coupling with N-BOC-dimethylglycine and TFA mediated deprotection. 1H NMR (MeOH-d4, 400 Hz) δ 7.53–7.48 (m, 1H), 7.44–7.41 (m, 1H), 7.17 (m, 1H), 6.9 (m, 2H), 6.48 (m, 2H), 4.88 (m, 2H), 4.05 (m, 2H), 3.81 (m, 2H), 2.12 (s, 3H), 1.62 (s, 6H). M/Z = 426.2(M + 1).
Compound 21: 2-Amino-1-(3-(cyclohexylamino)-2-(4-fluorophenyl)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)-2-methylpropan-1-one

Compound 21 was synthesized following the same sequence as compound 1 involving an Ugi reaction, reduction using palladium on carbon followed by HATU mediated coupling with N-BOC-dimethylglycine and TFA mediated deprotection. 1H NMR (MeOH-d4, 400 Hz) δ 7.66 (m, 2H), 7.18 (t, J = 8.74 Hz, 2H), 4.95 (m, 2H), 4.13 (m, 2H), 4.11 (m, 2H), 2.74 (m, 1H), 1.73 (m, 2H), 1.68 (s, 6H), 1.37–1.60 (m, 4H), 1.05–1.15 (m, 4H). M/Z = 400.3 (M + 1).
Compound 22: 2-Amino-1-(2-(4-fluorophenyl)-3-(pyridin-3-ylamino)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)-2-methylpropan-1-one

Compound 22 was synthesized following the same sequence as compound 1 involving an Ugi reaction, reduction using palladium on carbon followed by HATU mediated coupling with N-BOC-dimethylglycine and TFA mediated deprotection. 1H NMR (400 MHz, MeOH-d4) δ 8.15 (m, 1H), 7.7 (m, 3H), 7.57 (m, 2H), 7.1 (t, J = 8.74 Hz, 2H); 4.93 (m, 2H); 4.18 (s, 2H), 3.97 (m, 2H); 1.68 (s, 6H). M/Z = 395.4 (M + 1).
Compound 23: 2-Amino-1-(2-(4-fluorophenyl)-3-(3-fluorophenylamino)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)ethanone
The structure of 23 is as follows:

Synthesis of 23-3: N-(3-Fluorophenyl)-2-(4-fluorophenyl)-5,6,7,8-tetrahydroimidazo[1,2-a] pyrazin-3-amine

To a solution of compound 7 (69 mg, 0.30 mmol) in 6 mL of dioxane were added 1-bromo-3-fluorobenzene (66 μL, 0.60 mmol), Pd2(dba)3 (8 mg, 0.009 mmol), Xantphos (11 mg, 0.018 mmol), and Cs2CO3 (196 mg, 0.60 mmol) at room temperature. The reaction mixture was degassed and stirred at 120 °C under N2 for 5 h. The reaction mixture was cooled to room temperature, and solid was filtered off. The resulting filtrate was concentrated. The residue was subjected to mass-triggered HPLC purification to give a yellow oil (64 mg, 66%) after neutralization. To a stirred solution of the obtained adduct (64 mg, 0.20 mmol) in 5 mL of MeOH was added Pd/C (21 mg, 0.02 mmol). The reaction mixture was evacuated and back-filled with H2. The reaction mixture was stirred at room temperature overnight. The solid was filtered off, and solvent was removed. The residue was subjected to mass-triggered HPLC purification to give a yellow solid (42 mg, 66%).
Synthesis of 23: 2-Amino-1-(2-(4-fluorophenyl)-3-(3-fluorophenylamino)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)ethanone

.Compound 23 was synthesized from compound 23-3 by a HATU mediated coupling with N-Boc-glycine followed by TFA mediated deprotection. 1H NMR (DMSO-d6, 400 MHz) δ 8.24 (bs, 1H), 8.12 (s, 2H), 7.76 (m, 2H), 7.19 (m, 3H), 6.48 (m, 2H), 6.36 (m, 1H), 4.80 (m, 2H), 4.04–3.69 (m, 6H). M/Z = 384.4 (M + 1).
Compound 24: 2-Amino-1-(2-(3-fluorophenyl)-3-(4-fluorophenylamino)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)-2-methylpropan-1-one

Compound 24 was synthesized from compound 23-3 by a HATU mediated coupling with 3-(tert-butoxycarbonylamino)propanoic acid followed by TFA mediated deprotection. 1H NMR (MeOH-d4, 400 Hz) δ 7.50–7.48 (m, 2H), 7.44–7.40 (m, 1H), 7.18 (m, 1H), 6.98 (t, J = 8.8 Hz, 2H), 6.82–6.79 (m, 2H), 5.18 (m, 2H), 4.25 (t, J = 4.8 Hz, 2H), 4.07 (t, J = 4.8 Hz, 2H), 1.76 (s, 6H). M/Z = 412.0 (M + 1). Elemental analysis of HCl salt (compound + 2HCl + 1.3H2O): % C, 52.04; % H, 5.48; % N, 13.79 (calculated). % C = 51.95/52.02; % N = 13.37/13.5; % H = 5.26/5.32 (experimental).
Compound 25: 2-Amino-1-(2-(4-fluorophenyl)-3-(p-tolylamino)-5,6-dihydroimidazo[1,2-a]pyrazine-7(8H)-yl)ethanone

Compound 25 was synthesized following the same sequence as compound 1 involving an Ugi reaction, reduction using palladium on carbon followed by HATU mediated coupling with N-BOC-glycine and TFA mediated deprotection. 1H NMR (400 MHz, MeOH-d4) δ 7.74–7.71 (m, 2H), 7.21–7.17 (m, 2H), 7.03 (t, J = 8.0 Hz, 2H), 6.67 (d, J = 8.4 Hz, 2H), 5.17–5.15 (m, 2H), 4.22–3.98 (m, 6H), 2.22 (s, 3H). M/Z = 380.4 (M + 1). Elemental analysis of HCl salt (compound + 3HCl + 0.1H2O): % C, 51.41; % H, 5.18; % N, 14.27 (calculated). % C = 51.75/51.52; % N = 14.34/14.29; % H = 5.17/5.15 (experimental).
Compound 26: 2-Amino-1-(2-(4-fluorophenyl)-3-(p-tolylamino)-5,6-dihydroimidazo[1,2-a]pyrazine-7(8H)-yl)-2-methylpropan-1-one

Compound 26 was synthesized following the same sequence as compound 1 involving an Ugi reaction, reduction using palladium on carbon followed by HATU mediated coupling with N-BOC-dimethylglycine and TFA mediated deprotection. 1H-NMR (MeOH-d4, 400 MHz) δ 7.72 (m, 2H), 7.18 (t, J = 8.0 Hz, 2H), 7.02 (m, 2H), 6.66 (d, J = 8.35 Hz, 2H), 5.19 (s, 2H), 4.28 (m, 2H), 4.07 (m, 2H), 2.21 (s, 3H), 1.77 (s, 6H). M/Z = 408.2 (M + 1). Elemental analysis of HCl salt (compound + 3HCl + 0.1H2O): % C, 53.26; % H, 5.67; % N, 13.5 (calculated). % C = 53.28/53.18; % N = 13.39/13.32; % H = 5.92/5.81 (experimental).
Compound 27: 2-Amino-1-(3-(4-chlorophenylamino)-2-(4-fluorophenyl)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)ethanone

Compound 27 was synthesized from compound 7 using palladium catalyzed amination, reduction using palladium on carbon followed by HATU mediated coupling with N-BOC-glycine and TFA mediated deprotection. 1H NMR (MeOH-d4, 400 Hz) δ 7.59–7.56 (m, 2H), 7.15–7.09 (m, 4H), 6.68–6.65 (m, 2H), 5.05 (m, 2H), 4.04–3.87 (m, 6H). M/Z = 400.1 (M + 1).
Compound 28: 2-Amino-1-(3-(4-chlorophenylamino)-2-(4-fluorophenyl)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)-2-methylpropan-1-one

Compound 28 was synthesized from compound 7 using palladium catalyzed amination, reduction using palladium on carbon followed by HATU mediated coupling with N-BOC-dimethylglycine and TFA mediated deprotection. 1H NMR (MeOH-d4, 400 Hz) δ 7.70 (m, 2H), 7.21 (m, 4H), 6.8 (m, 2H), 5.19 (m, 2H), 4.26 (m, 2H), 4.0 (m, 2H), 1.77 (s, 6H). M/Z = 428.1 (M + 1). Elemental analysis of HCl salt (compound + 3HCl + H2O): % C, 47.58; % H, 5.08, % N, 12.61 (calculated). % C = 47.46/47.57; % N = 12.48/12.5; % H = 4.72/4.81 (experimental).
Compound 29: 3-Amino-1-(2-(4-fluorophenyl)-3-(4-fluorophenylamino)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)propan-1-one

Compound 29 was synthesized from compound 7 using palladium catalyzed amination, reduction using palladium on carbon followed by HATU mediated coupling with N-BOC-glycine and TFA mediated deprotection. 1H NMR (MeOH-d4, 400 Hz) δ 7.64–7.59 (m, 2H), 6.96–6.89 (m, 3H), 6.38–6.34 (m, 1H), 6.27–6.23 (m, 1H), 4.74–4.63 (m, 2H), 3.94–3.41 (m, 6H). M/Z = 402.1 (M + 1).
Compound 30: 2-Amino-1-(3-(3,4-difluorophenylamino)-2-(4-fluorophenyl)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)-2-methylpropan-1-one

Compound 30 was synthesized following the same sequence as compound 1 involving an Ugi reaction, reduction using palladium on carbon followed by HATU mediated coupling with N-BOC-dimethylglycine and TFA mediated deprotection. 1H NMR (400 MHz, MeOH-d4) δ 7.64–7.70 (m, 2H), 6.95–6.91 (m, 3H), 6.33–6.39 (m, 1H), 6.27–6.22 (m, 1H), 4.97 (s, 2H), 4.17 (m, 2H), 3.73 (t, J = 2.8 Hz, 2H), 1.36 (s, 6H). M/Z = 430.2 (M + 1). Elemental analysis of HCl salt (compound + 2HCl + 0.40H2O): % C, 51.85; % H, 4.91; % N, 13.74 (calculated). % C = 51.73/51.85; % N = 13.5/13.51; % H = 4.87/4.94 (experimental).
Compound 31: 2-Amino-1-(3-(4-chloro-3-fluorophenylamino)-2-(4-fluorophenyl)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)ethanone

Compound 31 was synthesized from compound 7 using palladium catalyzed amination reduction using palladium on carbon followed by HATU coupling using N-Boc-glycine followed by TFA mediated deprotection. 1H NMR (400 MHz, MeOH-d4) δ 7.7 (m, 2H), 7.44–7.40 (m, 3H), 6.71 (m, 1H), 6.64 (m, 1H), 5.16 (s, 2H), 4.18–4.01 (m, 6H). M/Z = 418.0 (M + 1). Elemental analysis of HCl salt (compound + 2HCl + 2H2O): % C, 45.6; % H, 4.59; % N, 13.29 (calculated). % C = 47.46/47.57; % N = 13.17/13.18; % H = 4.04/4.05 (experimental).
Compound 32: 2-Amino-1-(3-(4-chloro-3-fluorophenylamino)-2-(4-fluorophenyl)-5,6-dihydro imidazo[1,2-a]pyrazin-7(8H)-yl)-2-methylpropan-1-one

Compound 32 was synthesized from compound 7 using palladium catalyzed amination, reduction using palladium on carbon followed by HATU mediated coupling with N-BOC-dimethylglycine and TFA mediated deprotection. 1H NMR (MeOH-d4, 400 Hz) δ 7.59–7.56 (m, 2H), 7.2–7.11 (m, 3H), 6.6–6.57 (m, 2H), 5.04 (s, 2H), 4.14 (m, 2H), 3.97 (m, 2H), 1.69 (s, 6H). M/Z = 446.2 (M + 1). Elemental analysis (compound + 3HCl + 0.2H2O): % C,47.28; % H, 4.58; % N, 12.53 (calculated). % C = 47.07/47.19; % N = 12.33/12.34; % H = 4.86/4.82 (experimental).
Compound 33: 2-Amino-1-(3-(2,4-difluorophenylamino)-2-(4-fluorophenyl)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)-2-methylpropan-1-one

Compound 33 was synthesized from compound 7 using palladium catalyzed amination, reduction using palladium on carbon followed by HATU mediated coupling with N-BOC-dimethylglycine and TFA mediated deprotection. 1H NMR (DMSO-d6, 400 MHz) δ 8.24 (bs, 1H), 7.83 (bs, 1H), 7.75 (m, 2H), 7.25 (m, 1H), 7.17 (m, 2H), 6.81 (m, 1H), 6.4 (m, 1H), 4.86 (m, 2H), 4.06 (m, 2H), 3.75 (m, 2H), 1.6 (m, 6H). M/Z = 430.2 (M + 1).
Compound 34: 2-Amino-1-(3-(3,5-difluorophenylamino)-2-(4-fluorophenyl)-5,6-dihydroimidazo[1,2-a]pyrazin-7(8H)-yl)-2-methylpropan-1-one

Compound 34 was synthesized from compound 7 using palladium catalyzed amination, reduction using palladium on carbon followed by HATU mediated coupling with N-BOC-dimethylglycine and TFA mediated deprotection. 1H NMR (MeOH-d4, 400 Hz) δ 7.6 (m, 2H), 7.08 (t, J = 8.76 Hz, 2H), 6.25 (m, 3H), 4.98 (s, 2H), 4.11 (t, J = 5.28 Hz, 2H), 3.9 (m, 2H), 1.66 (s, 6H). M/Z = 430.2 (M + 1).
Acknowledgments
This work was supported by a joint grant to Genomics Institute of the Novartis Research Foundation, the Biomedical Primate Research Center, the Swiss Tropical and Public Health Institute, and the Novartis Institute for the Tropical Diseases from the U.K. Wellcome Trust and Medicines for Malaria Venture.
Glossary
Abbreviations Used
- ACT
artemisinin based combination therapy
- WHO
World Health Organization
- MMV
Medicines for Malaria Venture
- GNF
Genomics Institute of the Novartis Research Foundation
- Cmax
the maximum plasma concentration
- IC50
half-maximal effective concentration
- AUC
area under the concentration–time curve
- hERG
human ether-a-go-go-related gene
- PEG
polyethylene glycol
- D5W
5% dextrose in water
Author Contributions
# These authors made equal contribution to this work.
References
- a World Health Organization. http://www.who.int/topics/malaria/en/ (accessed November 27, 2010).; b Annual Report 2009; Medicines for Malaria Ventures (MMV): Geneva, Switzerland, 2009; http://www.mmv.org/sites/default/files/uploads/docs/publications/annual_report_2009_0.pdf. [Google Scholar]; c Milner E.; McCalmont W.; Bhonsle J.; Caridha D.; Carroll D.; Gardner S.; Gerena L.; Gettayacamin M.; Lanteri C.; Luong T.; Melendez V.; Moon J.; Roncal N.; Sousa J.; Tungtaeng A.; Wipf P.; Dow G. Structure–activity relationships amongst 4-position quinoline methanol antimalarials that inhibit the growth of drug sensitive and resistant strains of Plasmodium falciparum. Bioorg. Med. Chem. Lett. 2010, 20 (4), 1347–1351. [DOI] [PubMed] [Google Scholar]; d Milner E.; McCalmont W.; Bhonsle J.; Caridha D.; Cobar J.; Gardner S.; Gerena L.; Goodine D.; Lanteri C.; Melendez V.; Roncal N.; Sousa J.; Wipf P.; Dow G. Anti-malarial activity of a non-piperidine library of next-generation quinoline methanols. Malar. J. 2010, 9 (1), 51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Global Report on Antimalarial Drug Efficacy and Drug Resistance: 2000–2010; World Health Organization: Geneva, Switzerland, 2010; http://whqlibdoc.who.int/publications/2010/9789241500470_eng.pdf. [Google Scholar]; b A research agenda for malaria eradication: drugs. PLoS Med. 2011, 8 ( (1), ), e1000402. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Sadanand S. Malaria: an evaluation of the current state of research on pathogenesis and antimalarial drugs. Yale J. Biol. Med. 2010, 83 (4), 185–191. [PMC free article] [PubMed] [Google Scholar]; d Wu T.; Nagle A. S.; Chatterjee A. K. Road towards new antimalarials: overview of the strategies and their chemical progress. Curr. Med. Chem. 2011, 18 (6), 853–871. [DOI] [PubMed] [Google Scholar]
- Noedl H.; Chanthap L.; Se Y.; Socheat D.; Peou S.; Schaecher K.; Srivichai S.; Teja-Isavadharm P.; Smith B.; Jongsakul K.; Surasri S.; Fukuda M. Artemisinin resistance in Cambodia?. Trop. Med. Int. Health 2007, 12, 69. [Google Scholar]
- a Deng X. M.; Nagle A.; Wu T.; Sakata T.; Henson K.; Chen Z.; Kuhen K.; Plouffe D.; Winzeler E.; Adrian F.; Tuntland T.; Chang J.; Simerson S.; Howard S.; Ek J.; Isbell J.; Tully D. C.; Chatterjee A. K.; Gray N. S. Discovery of novel 1H-imidazol-2-yl-pyrimidine-4,6-diamines as potential antimalarials. Bioorg. Med. Chem. Lett. 2010, 20 (14), 4027–4031. [DOI] [PubMed] [Google Scholar]; b Wu T.; Nagle A.; Sakata T.; Deng X. M.; Chen Z.; Henson K.; Chang J.; Plouffe D.; Tuntland T.; Kuhen K.; Winzeler E.; Gray N.; Chatterjee A. K.. Cell-Based Optimization of Novel Benzamides as Antimalarials. Abstracts of Papers, 236th National Meeting of the American Chemical Society, Philadelphia, PA, Aug 17–21, 2008; American Chemical Society: Washington, DC, 2008; MEDI 339, p 236. [Google Scholar]; c Wu T.; Nagle A.; Sakata T.; Henson K.; Borboa R.; Chen Z.; Kuhen K.; Plouffe D.; Winzeler E.; Adrian F.; Tuntland T.; Chang J.; Simerson S.; Howard S.; Ek J.; Isbell J.; Deng X.; Gray N. S.; Tully D. C.; Chatterjee A. K. Cell-based optimization of novel benzamides as potential antimalarial leads. Bioorg. Med. Chem. Lett. 2009, 19 (24), 6970–6974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plouffe D.; Brinker A.; McNamara C.; Henson K.; Kato N.; Kuhen K.; Nagle A.; Adrian F.; Matzen J. T.; Anderson P.; Nam T. G.; Gray N. S.; Chatterjee A.; Janes J.; Yan S. F.; Trager R.; Caldwell J. S.; Schultz P. G.; Zhou Y.; Winzeler E. A. In silico activity profiling reveals the mechanism of action of antimalarials discovered in a high-throughput screen. Proc. Natl. Acad. Sci. U.S.A. 2008, 105 (26), 9059–9064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu B.; Chang J.; Gordon W. P.; Isbell J.; Zhou Y.; Tuntland T. Snapshot PK: a rapid rodent in vivo preclinical screening approach. Drug Discovery Today 2008, 13 (7–8), 360–367. [DOI] [PubMed] [Google Scholar]
- a Varma R. S.; Kumar D. Microwave-accelerated three-component condensation reaction on clay: solvent-free synthesis of imidazo[1,2-a] annulated pyridines, pyrazines and pyrimidines. Tetrahedron Lett. 1999, 40 (43), 7665–7669. [Google Scholar]; b Bienayme H.; Bouzid K. A new heterocyclic multicomponent reaction for the combinatorial synthesis of fused 3-aminoimidazoles. Angew. Chem., Int. Ed. 1998, 37 (16), 2234–2237. [DOI] [PubMed] [Google Scholar]; c Groebke K.; Weber L.; Mehlin F. Synthesis of imidazo[1,2-a] annulated pyridines, pyrazines and pyrimidines by a novel three-component condensation. Synlett 1998, 6, 661–663. [Google Scholar]
- Kercher T.; Rao C.; Bencsik J. R.; Josey J. A. Diversification of the three-component coupling of 2-aminoheterocycles, aldehydes, and isonitriles: efficient parallel synthesis of a diverse and druglike library of imidazo- and tetrahydroimidazo[1,2-d] heterocycles. J. Comb. Chem. 2007, 9 (6), 1177–1187. [DOI] [PubMed] [Google Scholar]
- Surry D. S.; Buchwald S. L. Biaryl phosphane ligands in palladium-catalyzed amination. Angew. Chem., Int. Ed. 2008, 47 (34), 6338–6361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Fidock D. A.; Rosenthal P. J.; Croft S. L.; Brun R.; Nwaka S. Antimalarial drug discovery: efficacy models for compound screening. Nat. Rev. Drug Discovery 2004, 3 (6), 509–520. [DOI] [PubMed] [Google Scholar]; b Vennerstrom J. L.; Arbe-Barnes S.; Brun R.; Charman S. A.; Chiu F. C.; Chollet J.; Dong Y.; Dorn A.; Hunziker D.; Matile H.; McIntosh K.; Padmanilayam M.; Santo Tomas J.; Scheurer C.; Scorneaux B.; Tang Y.; Urwyler H.; Wittlin S.; Charman W. N. Identification of an antimalarial synthetic trioxolane drug development candidate. Nature 2004, 430 (7002), 900–904. [DOI] [PubMed] [Google Scholar]
- Trager W.; Jensen J. B. Human malaria parasites in continuous culture. Science 1976, 193 (4254), 673–675. [DOI] [PubMed] [Google Scholar]
- a Obach R. S. Prediction of human clearance of twenty-nine drugs from hepatic microsomal intrinsic clearance data: an examination of in vitro half-life approach and nonspecific binding to microsomes. Drug Metab. Dispos. 1999, 27 (11), 1350–1359. [PubMed] [Google Scholar]; b Kalvass J. C.; Tess D. A.; Giragossian C.; Linhares M. C.; Maurer T. S. Influence of microsomal concentration on apparent intrinsic clearance: implications for scaling in vitro data. Drug Metab. Dispos. 2001, 29 (10), 1332–1336. [PubMed] [Google Scholar]; c Chauret N.; Gauthier A.; Nicoll-Griffith D. A. Effect of common organic solvents on in vitro cytochrome P450-mediated metabolic activities in human liver microsomes. Drug Metab. Dispos. 1998, 26 (1), 1–4. [PubMed] [Google Scholar]; d Yan Z.; Caldwell G. W. Metabolic assessment in liver microsomes by co-activating cytochrome P450s and UDP-glycosyltransferases. Eur. J. Drug Metab. Pharmacokinet. 2003, 28 (3), 223–232. [DOI] [PubMed] [Google Scholar]
- a Avdeef A.; Bendels S.; Di L.; Faller B.; Kansy M.; Sugano K.; Yamauchi Y. PAMPA—critical factors for better predictions of absorption. J. Pharm. Sci. 2007, 96 (11), 2893–2909. [DOI] [PubMed] [Google Scholar]; b Avdeef A. The rise of PAMPA. Expert Opin. Drug Metab. Toxicol. 2005, 1 (2), 325–342. [DOI] [PubMed] [Google Scholar]
- Balimane P. V.; Chong S. Cell culture-based models for intestinal permeability: a critique. Drug Discovery Today 2005, 10 (5), 335–343. [DOI] [PubMed] [Google Scholar]
- a Dubin A. E.; Nasser N.; Rohrbacher J.; Hermans A. N.; Marrannes R.; Grantham C.; Van Rossem K.; Cik M.; Chaplan S. R.; Gallacher D.; Xu J.; Guia A.; Byrne N. G.; Mathes C. Identifying modulators of hERG channel activity using the PatchXpress planar patch clamp. J. Biomol. Screening 2005, 10 (2), 168–181. [DOI] [PubMed] [Google Scholar]; b Mathes C. QPatch: the past, present and future of automated patch clamp. Expert Opin. Ther. Targets 2006, 10 (2), 319–327. [DOI] [PubMed] [Google Scholar]; c Zheng W.; Spencer R. H.; Kiss L. High throughput assay technologies for ion channel drug discovery. Assay Drug Dev. Technol. 2004, 2 (5), 543–552. [DOI] [PubMed] [Google Scholar]
- Franke-Fayard B.; Trueman H.; Ramesar J.; Mendoza J.; van der Keur M.; van der Linden R.; Sinden R. E.; Waters A. P.; Janse C. J. A Plasmodium berghei reference line that constitutively expresses GFP at a high level throughout the complete life cycle. Mol. Biochem. Parasitol. 2004, 137 (1), 23–33. [DOI] [PubMed] [Google Scholar]