

Abstract
Background
Loeys-Dietz syndrome (LDS) is a rare connective tissue disorder for which 6 genes in the TGF-β pathway have been identified as causative. With the widespread use of genetic testing, the range of known clinical and genetic profiles has broadened, but these features have not been fully elucidated thus far.
Methods and results
Using gene panel sequencing or whole exome sequencing, we identified 54 unique rare variants in LDS genes in 57 patients with thoracic aneurysms/dissections, including 27 pathogenic mutations (P + LP) and 27 variants of unknown significance (VUSLP + VUS). Genotype-phenotype correlation analysis revealed that carriers with P/LP/ VUSLP variants in TGFBR1/TGFBR2/SMAD3 genes had significantly more severe cardiovascular features (cardiovascular death/dissection) than carriers with VUSs in these 3 genes at an early age and had less favorable event-free survival. Additionally, carriers with VUS in combination with other risk factors, such as hypertension, might be prone to developing an aortic dissection, as indicated by the fact that 5/8 (62.5%) patients with VUSs in our cohort developed aortic dissections in the presence of hypertension, compared with 25.0% (3/12) in the absence of hypertension (p = 0.047).
Conclusions
To date, this was the largest cohort of LDS patients ever reported in China, and the present study expanded the known mutation and phenotypic spectra of LDS, which might help refine our knowledge of LDS.
Keywords: Loeys-Dietz syndrome, Genetic testing, Phenotypic spectrum
Background
Loeys-Dietz syndrome (LDS) is an autosomal dominant connective tissue disorder characterized by cardiovascular deformity (aortic aneurysms and/or dissections, multiple arterial aneurysms and arterial tortuosity) and skeletal problems (arachnodactyly, pectoral deformities, scoliosis and joint laxity) as well as other craniofacial and cutaneous abnormalities, sharing some features with Marfan syndrome (MFS) and differing in other respects [1]. Initially, LDS was generally thought to be more aggressive than MFS, with dissections at younger ages and at smaller arterial diameters, which led to a lower threshold (4.2 cm) for prophylactic surgical intervention by the 2010 American Heart Association (AHA) guideline [2].
Mutations in TGFBR1 and TGFBR2 were discovered in 2005 as the first known causative mutations for LDS [1]. Subsequently, other genes in the TGF-β signaling pathway, including SMAD3 [3], TGFB2 [4, 5], SMAD2 [6] and TGFB3 [7], were also found to be associated with LDS. Subsequently, the definition of LDS expands to all patients who carry a heterozygous pathogenic variant in any of these six genes in combination with the presence of an artery aneurysm/dissection or corresponding systemic manifestations.
The full spectrum of phenotypes and mutations associated with TGFBR1- and TGFBR2-related LDS has been extensively described and well recognized. However, for the more recently identified LDS genes (TGFB2, TGFB3 and SMAD2, SMAD3), the phenotypic and genotypic spectra have not yet been fully elucidated and need further expansion. Current studies reveal that patients with TGFB2-, TGFB3- and SMAD2-related LDS tend to have mild cardiovascular features and that their mutations have lower penetrance than those that cause TGFBR1- and TGFBR2-related LDS [4, 7, 8]. Therefore, further clinical and genetic data on LDS from around the world should be collected and analyzed to define gene-specific vascular treatment guidelines for LDS, rather than treating them all with the same approach. In this study, we identified 54 unique rare variants in LDS genes in aortic aneurysm/dissection patients and summarized the clinical data of these patients, especially their vascular phenotypic data, which could help further refine our knowledge of LDS.
Materials and methods
Patients
More than 900 patients with aortic disease and/or diagnosed or suspected MFS had been referred from the Aortic Surgery Department to the Center for Molecular Diagnosis at Fuwai Hospital and had undergone panel testing involving 15 genes (ACTA2, COL3A1, FBN1, FBN2, MYH11, MYLK, NOTCH1, PRKG1, SKI, SLC2A10, SMAD3, SMAD4, TGFB2, TGFBR1, TGFBR2) since Feb 2014 [9]. Furthermore, more than 200 aortopathy patients were performed whole exome sequencing. From these patients, we included a total of 57 patients in this study, in whom a rare variant in any of the six genes TGFBR1, TGFBR2, SMAD3, TGFB2, SMAD2 and TGFB3 was detected with no other suspected causative mutations.
Variant classification
Variants were analyzed for pathogenicity in line with recommendations from the American College of Medical Genetics (ACMG) and classified into one of 5 categories: benign, likely benign, unknown significance, likely pathogenic or pathogenic [10], with the detailed evidences listed behind. Besides, we additionally defined a subclassification, VUSLP, for internal use (See “Results” section for details).
Statistical analysis
Statistical analyses were performed using SPSS software. Survival curves were estimated using the Kaplan–Meier method and tested by Log Rank tests. Comparisons between continuous variables were made by Student’s t-test. A one-tailed Chi-Square test was used to test if the presence of hypertension facilitated aortic dissections in patients with VUSs in LDS genes. P value below 0.05 was considered as statistically significant.
Results
Among all of the aortopathy patients, a total of 54 unique rare variants in LDS genes were identified in 57 separate patients. Of these variants, 27 were pathogenic or likely pathogenic (summarized in Table 1), mostly in TGFBR1 and TGFBR2 genes (10 in TGFBR1, 10 in TGFBR2), and 24 variants remained unknown significance (summarized in Table 2). Specifically, there were 3 variants which should be classified as VUS on account of lack of evidence according to ACMG criteria. However, they were highly suspected as causative in the light of clinical information and family history. Therefore, we additionally defined a subclassification for these variants, VUSLP, for internal use (Table 1). The variants which could meet the tier “Likely Pathogenic” in ACMG criterion with one more supporting evidence, or those which could be assumed to be de novo according to the family history, were classified as VUSLP. For instance, patient AD1181’s mother suffered a sudden cardiac death at 30, therefore we could not collect her sample to perform the gene testing. However, there was a high probability that she carried the same mutation with her daughter AD1181, which was a de novo mutation because her parents and two sisters were all healthy and did not carry the mutation (Fig. 1).
Table 1.
Definite pathogenic and highly suspected variants in LDS genes detected in our cohort
| Patient ID | Gene | Transcript | Nucleotide change | Amino acid change | MAF in ExAC |
MAF in gnomAD | Domain | Source | Pathogenicity | Evidence | Note |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AD1413 | TGFBR1 | NM_004612 | c.614 T > C | p.Ile205Thr | . | . | Pkinase_Tyr | Maternal | LP | PM2, PP3, PS2$ | |
| AD623–1 | TGFBR1 | NM_004612 | c.644G > C | p.Arg215Pro | . | . | De novo | LP | PS2, PM2, PP3 | ||
| AD808 | TGFBR1 | NM_004612 | c.664G > A | p.Gly222Arg | . | 0.0000289 | Pkinase_Tyr | LP | PM2, PP1_Strong, PP3 | ||
| AD264 | TGFBR1 | NM_004612 | c.683_685del | p.228del | . | . | De novo | LP | PS2, PM2, PS4_Supporting, PM4 | a | |
| AD692–1 | TGFBR1 | NM_004612 | c.702_704del | p.235del | . | . | De novo | LP | PS2, PM2, PM4 | ||
| AD453 | TGFBR1 | NM_004612 | c.722C > T | p.Ser241Leu | . | . | NA | LP | PM2, PS4_Supporting, PS2 | ||
| AD371 | TGFBR1 | NM_004612 | c.934G > A | p.Gly312Ser | 0.00000942 | 0.00000398 | NA | LP | PP3, PM2, PS4_Supporting, PP1_Strong | a | |
| AD641–1 | TGFBR1 | NM_004612 | c.997G > A | p.Asp333Asn | . | . | De novo | LP | PS2, PM2, PP3 | ||
| AD78 | TGFBR1 | NM_004612 | c.1459C > T | p.Arg487Trp | . | . | NA | P | PS4, PM2, PM5, PP1_Strong, PP3 | a | |
| AD703–1 | TGFBR1 | NM_004612 | c.1459C > T | p.Arg487Trp | . | . | Maternal | P | PS4, PM2, PM5, PP1_Strong, PP3 | ||
| AD1346 | TGFBR1 | NM_004612 | c.1459C > T | p.Arg487Trp | . | . | NA | P | PS4, PM2, PM5, PP1_Strong, PP3 | ||
| AD1362 | TGFBR1 | NM_004612 | c.1460G > A | p.Arg487Gln | . | . | Paternal | P | PS2, PS3_Supporting, PS4_Moderate, PM2, PP3 | ||
| AD1804 | TGFBR2 | NM_003242 | c.95-2A > G | 0.0000293 | 0.0006 | Paternal | LP | PVS1, PM2 | |||
| AD257 | TGFBR2 | NM_003242 | c.1067G > C | p.Arg356Pro | . | . | Pkinase_Tyr |
Paternal (Mosaic) |
P | PS2_Very Strong, PS4_Moderate, PM2, PP3 | ab |
| AD22 | TGFBR2 | NM_003242 | c.1139 T > G | p.Leu380Arg | . | . | De novo | LP | PS2, PM2, PP3 | a | |
| AD888 | TGFBR2 | NM_003242 | c.1275G > C | p.Met425Ile | . | . | Pkinase_Tyr | De novo | LP | PS2, PM2, PP3 | |
| AD1181 | TGFBR2 | NM_003242 | c.1363 T > C | p.Trp455Arg | . | . | Pkinase_Tyr | VUSLP | PM2, PP3 | Assumed de novo in AD1181’s mother | |
| AD536 | TGFBR2 | NM_003242 | c.1449dupT | p.Cys483fs | . | . | Pkinase_Tyr | NA | LP | PVS1, PM2 | |
| AD617–1 | TGFBR2 | NM_003242 | 1517delA | p.Asn506fs | . | . | Pkinase_Tyr | NA | P | PVS1, PM2, PP1 | |
| AD1784 | TGFBR2 | NM_003242 | c.1525-1G > C | . | . | NA | LP | PVS1, PM2 | |||
| AD153 | TGFBR2 | NM_003242 | c.1538 T > C | p.Val513Ala | . | . | Pkinase_Tyr | De novo | LP | PS2, PM2 | a |
| AD682–1 | TGFBR2 | NM_003242 | c.1582C > T | p.Arg528Cys | . | . | De novo | P | PS2, PP3, PM2, PS4_Moderate, PS3_Supporting, PM5 | ||
| AD497 | TGFBR2 | NM_003242 | c.1609C > T | p.Arg537Cys | . | . | NA | P | PS2, PS3_Moderate, PS4_Moderate, PM2,PP3, PP1_Strong | a | |
| AD1550 | SMAD3 | NM_005902 | c.233_234insGG | p.Ser78fs | . | . | NA | LP | PVS1, PM2 | ||
| AD1736 | SMAD3 | NM_005902 | c.365_366insGAATCCCTACCAC | p.Val122fs | . | . | Paternal | LP | PVS1, PM2 | ||
| AD1061 | SMAD3 | NM_005902 | c.1041delG | p.Glu347fs | . | . | NA | LP | PVS1, PM2 | ||
| AD792 | SMAD3 | NM_005902 | c.1118G > A | p.Arg373His | . | . | VUSLP | PM2, PP3, PS3_Supporting, PS4_Supporting | |||
| AD1297 | SMAD3 | NM_005902 | c.1247C > T | p.Ser416Phe | . | . | De novo | LP | PS2, PM2, PP3 | ||
| AD535 | SMAD2 | NM_005901 | c.593dupA | p.His198fs | . | . | De novo | LP | PS2, PM2 | ||
| AD802 | TGFB2 | NM_003238 | c.905G > A | p.Arg302His | . | . | TGF_beta | Paternal | VUSLP | PM2, PM5, PP3 | |
| AD1065 | TGFB3 | NM_003239 | c.605_623del | p.Phe202fs | . | . | Maternal | LP | PVS1, PM2 | ||
| AD631–1 | TGFB3 | NM_003239 | c.646 + 2 T > G | . | . | Paternal | LP | PVS1, PM2 |
Note: NA not available; MAF in ExAC was the maximal allele frequency from the public version (20160423), and MAF in gnomAD was the maximal allele frequency from gnomAD v2.1.1; P, pathogenic; LP, likely pathogenic; VUS, variant of uncertain significance; a, reported in our previous article [11]; bThis variant was previously classified as VUS, and then upgraded into pathogenic after the father was confirmed to carry a mosaic mutation in the same site; $ This variant was confirmed to be de novo in patient AD1413’s mother
Table 2.
Variants of unknown significance in LDS genes detected in our cohort
| Patient ID | Gene | Transcript | Nucleotide change | Amino acid change | MAF in ExAC | MAF in gnomAD | Domain | Source | Pathogenicity | Evidence | Note |
|---|---|---|---|---|---|---|---|---|---|---|---|
| AD1039 | TGFBR1 | NM_004612 | c.341C > G | p.Thr114Ser | . | . | VUS | PM2, BP4 | |||
| AD1248 | TGFBR1 | NM_004612 | c.439A > G | p.Ile147Val | 0.0000221 | 0.000098 | Pkinase_Tyr | VUS | BP4 | ||
| AD589 | TGFBR1 | NM_004612 | c.605C > T | p.Ala202Val | . | . | VUS | PM2, PP3 | |||
| AD823 | TGFBR1 | NM_004612 | c.767A > G | p.His256Arg | . | . | VUS | PM2, PP3 | |||
| AD1802 | TGFBR1 | NM_004612 | c.782G > C | p.Gly261Ala | . | . | VUS | PM2, PP3 | |||
| AD183 | TGFBR1 | NM_004612 | c.929C > T | p.Ala310Val | 0.0000221 | 0.0006 | VUS | PP3 | |||
| AD436 | TGFBR1 | NM_004612 | c.935G > T | p.Gly312Val | . | . | VUS | PM2, PP3 | |||
| AD1158 | TGFBR1 | NM_004612 | c.1054 T > G | p.Leu352Val | . | . | VUS | PM2, PP3 | |||
| AD1753 | TGFBR2 | NM_003242 | c.81C > A | p.His27Gln | . | . | NA | VUS | PM2, BP4 | ||
| AD1432 | TGFBR2 | NM_003242 | c.467G > T | p.Ser156Ile | . | . | VUS | PM2, BP4 | |||
| AD1348 | TGFBR2 | NM_003242 | c.578G > A | p.Arg193Gln | 0.000011 | 0.0000544 | TGF_beta | Paternal | VUS | PM2 | |
| AD1156 | TGFBR2 | NM_003242 | c.617C > T | p.Thr206Met | 0.0000377 | 0.0006 | VUS | BP4 | |||
| AD259 | TGFBR2 | NM_003242 | c.830A > G | p.Lys277Arg | . | . | Pkinase_Tyr | VUS | PM2, PP3 | a | |
| AD667 | TGFBR2 | NM_003242 | c.1188 T > G | p.Cys396Trp | . | . | Pkinase_Tyr | VUS | PM2, PP3 | ||
| AD1162 | TGFBR2 | NM_003242 | c.1254G > T | p.Gln418His | . | . | Pkinase_Tyr | Maternal | VUS | PM2, PP3 | |
| AD324 | SMAD3 | NM_005902 | c.5C > T | p.Ser2Leu | . | . | Paternal | VUS | PM2 | ab | |
| AD1250 | SMAD3 | NM_001145103 | c.53G > A | p.Arg18Gln | . | . | VUS | NA | |||
| AD76 | SMAD3 | NM_005902 | c.140_148del | p.47_50del | . | . | VUS | PM2, PM4, BS2 | a | ||
| AD997 | SMAD3 | NM_005902 | c.364G > A | p.Val122Met | . | . | VUS | PM2, PP3 | |||
| AD850 | SMAD3 | NM_005902 | c.773A > T | p.Asp258Val | . | . | VUS | PM2, PP3 | |||
| AD1288 | SMAD3 | NM_005902 | c.1027 T > C | p.Phe343Leu | . | . | VUS | PM2, PP3 | |||
| AD148 | SMAD3 | NM_005902 | c.1027 T > C | p.Phe343Leu | . | . | VUS | PM2, PP3 | |||
| AD1759 | TGFB2 | NM_003238 | c.893G > A | p.Arg298Gln | 0.0000221 | 0.0002 | NA | VUS | NA | ||
| AD1599 | TGFB2 | NM_003238 | c.1239C > G | p.Cys413Trp | . | . | VUS | PM2, PP3 | |||
| AD985 | TGFB3 | NM_003239 | c.352 + 5G > A | . | . | VUS | PM2, PP3 |
Note: NA not available; MAF in ExAC was the maximal allele frequency from the public version (20160423), and MAF in gnomAD was the maximal allele frequency from gnomAD v2.1.1; P, pathogenic; LP, likely pathogenic; VUS, variant of uncertain significance; a, reported in our previous article [11]; bThis variant was previously misclassified as likely pathogenic [11], and now corrected into VUS
Fig. 1.
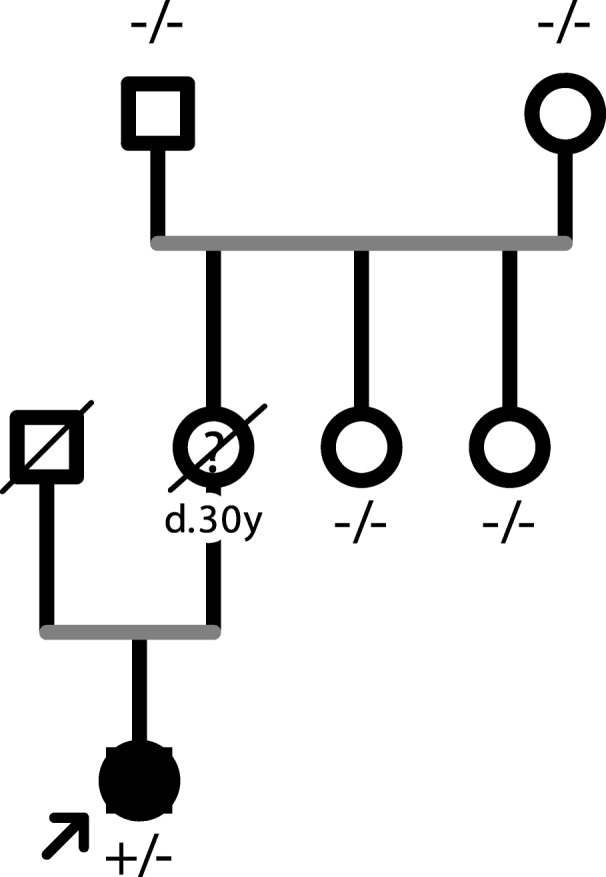
The pedigree of patient AD1181. Black indicated affected while white indicated unaffected.? represented that the person was suspected to have a sudden death due to an aortopathy
The pathogenicity of the variant TGFBR2 c.1067G > C (p. Arg356Pro) initially confused us. This variant was identified in patient AD257 with characteristic signs of LDS, such as descending pseudoaneurysm, bilateral carotid tortuosity, bifid uvula and hypertelorism. This variant had been reported in five individuals with clinical features of Loeys-Dietz syndrome and was found to occur de novo in three of these individuals [12–15]. Furthermore, it was absent from large population studies, and computational prediction tools and conservation analysis suggested that it might impact the protein. All evidence supported that it was a pathogenic mutation. However, we unexpectedly found that the patient’s healthy father also carried the same mutation. Upon a detailed examination of the father’s cardiac structure and arterial tree, there were no apparent abnormalities except for a slight decrease in left ventricular diastolic function. Considering that LDS was a dominant disorder with full penetrance expected at an early age and that the variant was also observed in the patient’s healthy father, this variant was finally downgraded into VUS [11] with conflicting evidence (BS2). When we reanalyzed this case after half a year, we noted that the unequal peak heights suggested probable mosaicism. To test this, we performed deep sequencing (5000×) at this location, and the results showed that the father indeed had a mosaic mutation (Fig. 2), which convincingly explained his lack of LDS symptoms.
Fig. 2.

Mosaicism in patient AD257’s family. a, Sanger sequencing indicated an unequal peak height at the mutation site in patient AD257’s father; b, Deep sequencing at the specific location revealed a mosaic mutation in patient AD257’s father
Patient AD535 had positive wrist sign, pectus excavatum, and moderate myopia. He was found to have an aortic root dilation with a diameter of 45 mm and a bicuspid aortic valve (BAV) upon physical examination. A SMAD2 frameshift mutation in the linker region was identified and shown to be de novo; therefore, it was classified as a likely pathogenic mutation. According to the genetic results, the patient was diagnosed with Loeys-Dietz syndrome, and an examination of his whole arterial tree was recommended to check for other arterial aneurysms. To our knowledge, this was the second report of a truncating mutation in the SMAD2 gene and further confirmed haploinsufficiency as its pathogenic mechanism.
Patient AD1162 had an aortic dissection, and also dissections in carotid artery and abdominal aorta. After a detailed investigation, we learnt that the patient had a family history of sudden deaths and retinal detachments (Fig. 3). She had lens subluxation herself. Except that her youngest brother had pectus carinatum and scoliosis, other family members had no obvious skeletal deformities. A 15-gene panel [9] testing revealed that she carried a TGFBR2 mutation (c.1254G > T, p.Gln418His), which was inherited from her mother, who suffered an aortic dissection at age 42. Therefore, she was highly suspected to be LDS. Strangely, it was commonly thought that one of the most distinguishing characteristics between LDS and MFS was that the former rarely included ocular abnormalities such as ectopia lentis or retinal detachments [16]. To exclude a FBN1 large deletion/duplication, multiplex ligation-dependent probe amplification (MLPA) assay was also performed, which was negative (Additional file 1: Figure S1). It suggested that there might be a wider spectrum of LDS than we previously realized.
Fig. 3.
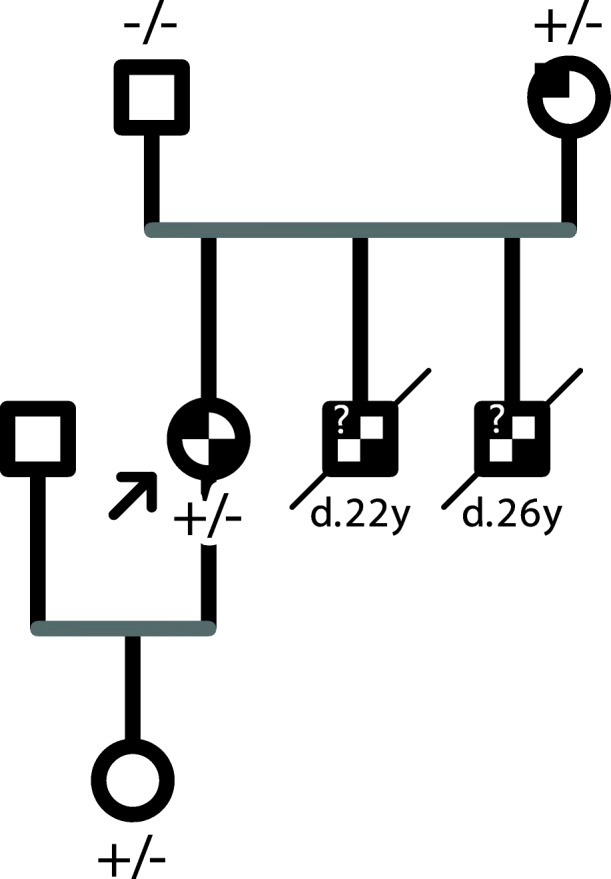
The pedigree of patient AD1162. Black on the top left corner indicated a vascular event, and black on the bottom right corner indicated an ocular event (lens subluxation or retinal detachment).? represented that the person was suspected to have a sudden death due to an aortopathy
To analyze the genotype-phenotype correlation, we divided the patients into two groups according to the variant pathogenicity. Considering TGFBR1/TGFBR2/SMAD3-related LDS often led to a penetrant and severe form of the disease and accounted for the vast majority in our cohort, only patients with these 3 genes were included to perform the analysis. The key cardiovascular information was listed in Table 3. When patients with P/LP/VUSLP variants were set into one group and the others with VUS were set into another, event-free survival was compared and the results showed that patients with a P/LP/VUSLP variant had a significant lower event-free survival rate than those with VUS (p = 0.021 when events defined as aortic dissections or related death; p = 0.025 when events defined as dissections and aortic surgeries and related death) (Fig. 4), indicating that the presence of a pathogenic variant has possible predictive value for disease severity. In addition, the presence of (suspected) pathogenic variants was associated with earlier aortic dissection or surgery than the presence of VUS (29.9 y vs 38.0 y, p = 0.035). Besides, in 20 individuals with VUSs, 8 patients had hypertension while 12 patients were with normal blood pressure, with a dissection rate of 62.5% (5/8) and 25.0% (3/12), separately (p = 0.047) (Additional file 1: Table S1).
Table 3.
Main cardiovascular phenotypic information in two subgroups of LDS
| TGFBR1/TGFBR2/SMAD3 | TGFB2/TGFB3/SMAD2 | |||||
|---|---|---|---|---|---|---|
| LP/P | VUSLP | VUS | LP/P | VUSLP | VUS | |
| Numbers | 26 | 2 | 20 | 3 | 1 | 3 |
| Age, years | 29.5 ± 13.3 | 34.0 ± 4.2 | 38.0 ± 11.5 | 20.3 ± 10.4 | 20 | 43.0 ± 8.5 |
| Normal or mild dilation | 3 | 0 | 4 | 3 | 0 | 0 |
| Surgery due to an aortic aneurysm/valve disease | 8 | 0 | 8 | 0 | 1 | 0 |
| Aortic dissection and related death | 15 | 2 | 8 | 0 | 0 | 3 |
Fig. 4.

Kaplan–Meier analysis of event-free survival. Event-free survival was compared in probands with P/LP/VUSLP variants (green curve) versus those with VUS (blue curve). a, Events were defined as aortic dissections and related deaths; b, Events were defined as aortic dissections and related death or aortic surgeries
Discussion
Genetic profiling
TGFBR1 and TGFBR2 are the first genes identified to be associated with LDS. To date, over 200 variants in these genes have been reported in the human gene mutation database (HGMD), most of which (86%) are missense mutations in evolutionarily conserved residues within the serine-threonine kinase (STK) domains. Missense and truncating variants in the STK domain of the TGFBR1 gene induce LDS [1] and multiple self-healing squamous epithelioma (MSSE) [17], presumably through the dominant negative (DN) and haploinsufficiency (HI) effects, respectively. For TGFBR2, there seems to be no difference in TGFBR2-related phenotypic features between individuals with missense mutations and those with truncating mutations [18]. Here, we reported two frameshift mutations and one canonical splicing mutation in the TGFBR2 gene in three separate individuals who had severe vascular events. Patient AD536 suffered a total thoracic and abdominal replacement at his age of 53 and AD1784 suffered an ascending and full aortic arch replacement at 34, while patient AD617 had a positive family history of sudden cardiac death at an early age.
A total of 78 and 8 variants in SMAD3 and SMAD2, respectively, have been reported in LDS patients [8], the majority (63 and 87.5%, respectively) of which are located in the MH2 domain, a highly conserved region responsible for the oligomerization of SMAD2 or SMAD3 with SMAD4 and subsequent Smad-dependent activation of downstream transcription. Other than missense variants, previous reports reveal only one SMAD2 nonsense mutation [19], which is located in the linker region. In this study, we identified the second case of a SMAD2 truncating mutation (c.593dupA), which was also in the linker region. It is very similar to SMAD3 that all pathogenic mutations in linker regions are truncating mutations [8]. For TGFB2 and TGFB3, 44 and 34 mutations have been reported, respectively, mainly distributed in the TGF-β2 cytokine domain, RKKR-motif and latency-associated peptide (LAP). Here, we reported 3 novel TGFB3 mutations and 3 novel TGFB2 mutations.
Cardiovascular phenotypic spectrum
Significant clinical heterogeneity was observed in LDS patients. When first reported as a distinct disease, LDS was described as having more aggressive aortic events than MFS, with a mean age of 26 years at death [1]. A reduced threshold of 42 mm had been proposed for earlier interventions in LDS patients [2]; however, it remained controversial [20, 21]. Current studies revealed that some patients with TGFBR1, TGFBR2 or SMAD3 mutations tended to have an early dissection at a young age or at a relatively small diameter, whereas TGFB2, TGFB3 and SMAD2 carriers often suffered a less severe aortic event [4, 7, 8, 22]. Our data were consistent with previous studies in that all seven individuals with mutations in TGFB2, TGFB3 or SMAD2 had relatively mild aortic events, except that patient AD985, who had the risk factors of extreme hypertension and an intronic mutation predicted to affect normal splicing, suffered an aortic dissection at 32 years of age.
Considering these two clinical forms in LDS, one more severe and penetrant than another, we only involved the severe form when analyzing the genotype-phenotype correlation, so as to avoid the interpretation bias. The results revealed that carriers with P/LP/VUSLP variants have significantly more severe cardiovascular features (aortic dissection and related death) than carriers with VUS, at an early age and less favorable event-free survival. Notably, according to current evidence, many variants (27, 50%) remain VUS and VUSLP. On the one hand, some of these variants may be upgraded to likely pathogenic mutations as further supporting evidence accumulates; on the other hand, these variants may predispose patients to disease in a low-risk or low-penetrance manner and lead to aortic dissection when combined with other risk factors, such as hypertension. This possibility is well supported by our data showing that, when hypertension was present, 5/8 (62.5%) patients with VUS in LDS genes developed aortic dissections, far more than 25.0% (3/12) when hypertension was absent. Based on our current results, carriers with VUSs in LDS genes should receive active control of blood pressure.
Conclusions
In summary, this was the largest cohort of LDS patients ever reported in China, and we expanded the known mutation and phenotypic spectra of LDS. Genetic results not only facilitate an early and accurate diagnosis but also have possible predictive value, which needs to be further investigated because it may influence clinical care.
Supplementary information
Additional file 1. Method S1. Multiplex ligation-dependent probe amplification (MLPA). Figure S1. MLPA assay indicated that there was no FBN1 deletion/duplication in AD1162. Table S1. Main cardiovascular phenotypic information in patients with VUSs in TGFBR1/TGFBR2/SMAD3 genes.
Acknowledgements
We thank all subjects who participated in this study.
Abbreviations
- ACMG
American College of Medical Genetics
- AHA
American Heart Association
- DN
Dominant negative
- ESP
Exome Sequencing Project
- HGMD
Human gene mutation database
- LAP
Latency-associated peptide
- LDS
Loeys-Dietz syndrome
- MFS
Marfan syndrome
- MLPA
Multiplex ligation-dependent probe amplification
- MSSE
Multiple self-healing squamous epithelioma
- STK
Serine-threonine kinase
- WES
Whole exome sequencing
Authors’ contributions
HY coordinated the project, carried out data analysis and wrote the manuscript. ML recruited patients and collected clinical information. YM and GZ contributed to accumulation and interpretation of clinical data. YZ performed NGS sequencing experiments and Sanger validation. BL communicated with patients and contributed to follow-up visit. CS was in charge of the clinical evaluation and sample management. ZZ was in charge of the project design and revised the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the grant of CAMS Initiative for Innovative Medicine, China (NO.2016-I2M-1-016) and the National Natural Science Foundation of China (81700419).
Availability of data and materials
The data sets used and analysed during the current study are available from the corresponding author on reasonable request.
Ethics approval and consent to participate
This study was approved by the ethics committee of the institutional review board at Fuwai Hospital (Approval No.:2017–877). All the patients and their related family members accepting the genetic testing assigned a consent form.
Consent for publication
The authors give their consent for publication of this manuscript.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Hang Yang, Yanyun Ma and Mingyao Luo contributed equally to this work.
Contributor Information
Chang Shu, Email: changshu@vip.126.com.
Zhou Zhou, Email: zhouzhou@fuwaihospital.org.
Supplementary information
Supplementary information accompanies this paper at 10.1186/s13023-019-1282-3.
References
- 1.Loeys BL, Chen J, Neptune ER, Judge DP, Podowski M, Holm T, et al. A syndrome of altered cardiovascular, craniofacial, neurocognitive and skeletal development caused by mutations in TGFBR1 or TGFBR2. Nat Genet. 2005;37(3):275–281. doi: 10.1038/ng1511. [DOI] [PubMed] [Google Scholar]
- 2.Hiratzka LF, Bakris GL, Beckman JA, Bersin RM, Carr VF, Casey DE, Jr, et al. 2010 ACCF/AHA/AATS/ACR/ASA/SCA/SCAI/SIR/STS/SVM Guidelines for the diagnosis and management of patients with thoracic aortic disease. A Report of the American College of Cardiology Foundation/American Heart Association Task Force on Practice Guidelines, American Association for Thoracic Surgery, American College of Radiology,American Stroke Association, Society of Cardiovascular Anesthesiologists, Society for Cardiovascular Angiography and Interventions, Society of Interventional Radiology, Society of Thoracic Surgeons,and Society for Vascular Medicine. J Am Coll Cardiol. 2010;55(14):e27–e129. doi: 10.1016/j.jacc.2010.02.015. [DOI] [PubMed] [Google Scholar]
- 3.van de Laar IM, Oldenburg RA, Pals G, Roos-Hesselink JW, de Graaf BM, Verhagen JM, et al. Mutations in SMAD3 cause a syndromic form of aortic aneurysms and dissections with early-onset osteoarthritis. Nat Genet. 2011;43(2):121–126. doi: 10.1038/ng.744. [DOI] [PubMed] [Google Scholar]
- 4.Lindsay ME, Schepers D, Bolar NA, Doyle JJ, Gallo E, Fert-Bober J, et al. Loss-of-function mutations in TGFB2 cause a syndromic presentation of thoracic aortic aneurysm. Nat Genet. 2012;44(8):922–927. doi: 10.1038/ng.2349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Boileau C, Guo DC, Hanna N, Regalado ES, Detaint D, Gong L, et al. TGFB2 mutations cause familial thoracic aortic aneurysms and dissections associated with mild systemic features of Marfan syndrome. Nat Genet. 2012;44(8):916–921. doi: 10.1038/ng.2348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Micha D, Guo DC, Hilhorst-Hofstee Y, van Kooten F, Atmaja D, Overwater E, et al. SMAD2 mutations are associated with arterial aneurysms and dissections. Hum Mutat. 2015;36(12):1145–1149. doi: 10.1002/humu.22854. [DOI] [PubMed] [Google Scholar]
- 7.Bertoli-Avella AM, Gillis E, Morisaki H, Verhagen JMA, de Graaf BM, van de Beek G, et al. Mutations in a TGF-beta ligand, TGFB3, cause syndromic aortic aneurysms and dissections. J Am Coll Cardiol. 2015;65(13):1324–1336. doi: 10.1016/j.jacc.2015.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Schepers D, Tortora G, Morisaki H, MacCarrick G, Lindsay M, Liang D, et al. A mutation update on the LDS-associated genes TGFB2/3 and SMAD2/3. Hum Mutat. 2018;39(5):621–634. doi: 10.1002/humu.23407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yang H, Luo M, Fu Y, Cao Y, Yin K, Li W, et al. Genetic testing of 248 Chinese aortopathy patients using a panel assay. Sci Rep. 2016;6:33002. doi: 10.1038/srep33002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Richards S, Aziz N, Bale S, Bick D, Das S, Gastier-Foster J, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet Med. 2015;17(5):405–424. doi: 10.1038/gim.2015.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Luo M, Yang H, Yin K, Chen Q, Zhang J, Fan Y, Zhou Z, Chang Q. Genetic testing of 10 patients with features of Loeys-Dietz syndrome. Clin Chim Acta. 2016;456:144–148. doi: 10.1016/j.cca.2016.02.005. [DOI] [PubMed] [Google Scholar]
- 12.Uike K, Matsushita Y, Sakai Y, Togao O, Nagao M, Ishizaki Y, et al. Systemic vascular phenotypes of Loeys-Dietz syndrome in a child carrying a de novo R381P mutation in TGFBR2: a case report. BMC Res Notes. 2013;6:456. doi: 10.1186/1756-0500-6-456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ben Amor IM, Edouard T, Glorieux FH, Chabot G, Tischkowitz M, Roschger P, Klaushofer K, Rauch F. Low bone mass and high material bone density in two patients with Loeys-Dietz syndrome caused by transforming growth factor beta receptor 2 mutations. J Bone Miner Res. 2012;27(3):713–718. doi: 10.1002/jbmr.1470. [DOI] [PubMed] [Google Scholar]
- 14.Sakai H, Visser R, Ikegawa S, Ito E, Numabe H, Watanabe Y, et al. Comprehensive genetic analysis of relevant four genes in 49 patients with Marfan syndrome or Marfan-related phenotypes. Am J Med Genet A. 2006;140(16):1719–1725. doi: 10.1002/ajmg.a.31353. [DOI] [PubMed] [Google Scholar]
- 15.Ki CS, Jin DK, Chang SH, Kim JE, Kim JW, Park BK, et al. Identification of a novel TGFBR2 gene mutation in a Korean patient with Loeys-Dietz aortic aneurysm syndrome; no mutation in TGFBR2 gene in 30 patients with classic Marfan's syndrome. Clin Genet. 2005;68(6):561–563. doi: 10.1111/j.1399-0004.2005.00535.x. [DOI] [PubMed] [Google Scholar]
- 16.Van Laer L, Dietz H, Loeys B. Loeys-Dietz syndrome. Adv Exp Med Biol. 2014;802:95–105. [DOI] [PubMed]
- 17.Goudie DR, D'Alessandro M, Merriman B, Lee H, Szeverenyi I, Avery S, et al. Multiple self-healing squamous epithelioma is caused by a disease-specific spectrum of mutations in TGFBR1. Nat Genet. 2011;43(4):365–369. doi: 10.1038/ng.780. [DOI] [PubMed] [Google Scholar]
- 18.Stheneur C, Collod-Beroud G, Faivre L, Gouya L, Sultan G, Le Parc JM, et al. Identification of 23 TGFBR2 and 6 TGFBR1 gene mutations and genotype-phenotype investigations in 457 patients with Marfan syndrome type I and II, Loeys-Dietz syndrome and related disorders. Hum Mutat. 2008;29(11):E284–E295. doi: 10.1002/humu.20871. [DOI] [PubMed] [Google Scholar]
- 19.Cannaerts E, Kempers M, Maugeri A, Marcelis C, Gardeitchik T, Richer J, et al. Novel pathogenic SMAD2 variants in five families with arterial aneurysm and dissection: further delineation of the phenotype. J Med Genet. 2019;56(4):220–227. doi: 10.1136/jmedgenet-2018-105304. [DOI] [PubMed] [Google Scholar]
- 20.Attias D, Stheneur C, Roy C, Collod-Beroud G, Detaint D, Faivre L, et al. Comparison of clinical presentations and outcomes between patients with TGFBR2 and FBN1 mutations in Marfan syndrome and related disorders. Circulation. 2009;120(25):2541–2549. doi: 10.1161/CIRCULATIONAHA.109.887042. [DOI] [PubMed] [Google Scholar]
- 21.Schoenhoff FS, Mueller C, Czerny M, Matyas G, Kadner A, Schmidli J, Carrel T. Outcome of aortic surgery in patients with Loeys-Dietz syndrome primarily treated as having Marfan syndrome. Eur J Cardiothorac Surg. 2014;46(3):444–449. doi: 10.1093/ejcts/ezu002. [DOI] [PubMed] [Google Scholar]
- 22.Brownstein AJ, Kostiuk V, Ziganshin BA, Zafar MA, Kuivaniemi H, Body SC, Bale AE, Elefteriades JA. Genes associated with thoracic aortic aneurysm and dissection: 2018 update and clinical implications. Aorta (Stamford) 2018;6(1):13–20. doi: 10.1055/s-0038-1639612. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1. Method S1. Multiplex ligation-dependent probe amplification (MLPA). Figure S1. MLPA assay indicated that there was no FBN1 deletion/duplication in AD1162. Table S1. Main cardiovascular phenotypic information in patients with VUSs in TGFBR1/TGFBR2/SMAD3 genes.
Data Availability Statement
The data sets used and analysed during the current study are available from the corresponding author on reasonable request.