

Familial hypercholesterolemia (FH) is a common yet underdiagnosed autosomal dominant disorder that affects ≈1 in 220 individuals globally.1 FH is characterized by lifelong elevation of low‐density lipoprotein cholesterol (LDL‐C) and if untreated leads to early‐onset atherosclerosis and increased risk of cardiovascular events. Affected men and women who are untreated have a 30% to 50% risk of a fatal or nonfatal cardiac event by ages 50 and 60 years, respectively.1
The most common causes of FH are pathogenic variants of the LDL receptor (LDL‐R) gene, which are responsible for 85% to 90% of genetically confirmed FH. Pathogenic variants of the apolipoprotein (ApoB) gene, resulting in decreased binding of LDL to the LDL‐R, or gain‐of‐function mutations in the gene for proprotein convertase subtilisin/kexin 9 (PCSK9), resulting in increased destruction of LDL‐R, are responsible for 5% to 15% and 1% of cases of FH, respectively.2 Autosomal recessive FH, caused by homozygous mutations in the LDL‐R adaptor protein‐1, is associated with a mild homozygous FH (HoFH) phenotype and is beyond the scope of this review.3
With the exception of HoFH, FH is generally a silent disease. HoFH typically presents with pathognomonic physical findings in childhood, including xanthelasmas, tendon xanthomas, and corneal arcus. By contrast, in the Spanish Familial Hypercholesterolemia Cohort study, xanthomas and corneal arcus were present in <15% and 30% of patients with heterozygous FH (HeFH), respectively.4 However, the prevalence of these findings increases with age in untreated individuals.
Elevated total cholesterol (240 mg/dL [6.2 mmol/L]) is found in up to 28.5 million (11.7%) of Americans over the age of 20 years.5 Coronary artery disease (CAD) and myocardial infarction (MI) are also very common cardiovascular conditions, with more than 1 million Americans estimated to have had an MI in 2018.6 Individuals with genetically confirmed FH account for only a small percentage of these cardiac events. In the US National Heart Lung and Blood Institute's Exome Sequencing Project, only 2% of cases of premature MI in men <50 years and women <60 years of age were found to have a genetic defect in the LDL‐R.7 However, as many as 20% of MIs in younger men (<45 years of age) have been attributed to FH.8 Given the broad range of causes of hypercholesterolemia and early‐onset CAD, it is not surprising that FH is not always in the differential diagnosis for healthcare professionals when confronted with a patient presenting with early CAD. This represents a missed opportunity for the screening of family members (cascade screening) and the early initiation of potentially lifesaving therapies.
It is crucial to consider the diagnosis of FH in children with LDL‐C persistently >160 mg/dL (4.1 mmol/L), adults with LDL‐C >190 mg/dL (4.9 mmol/L) (especially if there is a family history of early‐onset CAD), and in all patients with early CAD. Given that FH is an autosomal dominant disorder, an individual who is heterozygous for FH has a 50% chance of passing the gene to his or her children. Homozygous individuals who have identical mutations in both alleles, compound heterozygotes who inherited different mutations in both alleles of the same gene, or double heterozygotes who have mutations in 2 different genes, will have offspring who are all obligate heterozygotes, assuming their partner does not have FH.9
This review will focus on HeFH and aims to highlight the differences between FH, which is present from birth, and hyperlipidemia secondary to suboptimal diet and lifestyle or other causes of acquired hyperlipidemia that develop later in life. Diagnostic tools and potential treatments for individuals with FH will also be discussed.
Diagnosis
FH is significantly underdiagnosed and undertreated, particularly in children.10 The burden of early diagnosis of FH rests on primary care providers, who have the unique potential to help improve the detection and management of FH.11 Universal lipid screening in children between the ages of 9 to 11 years, as recommended by the National Heart, Lung, and Blood Institute, American Academy of Pediatrics, American Heart Association, National Lipid Association, and American College of Cardiology, has the potential to substantially improve case finding.12
Earlier lipid screening at the age of 2 years is recommended if a child has a strong family history of early onset CAD (males, aged <55 years; females, aged <65 years) in a parent, grandparent, aunt, uncle, or sibling; a parent with a total cholesterol >240 mg/dL (6.2 mmol/L); or if the child has underlying cardiac risk factors, such as diabetes mellitus or obesity. Screening between the ages of 9 to 11 years enables the best discernment between those with and without FH and avoids confounding lipid changes secondary to LDL‐C reduction, known to transiently occur during puberty. Survey data suggest that fewer than one third of practicing pediatricians have adopted these recommendations.13 Other obstacles to early diagnosis include the limited public awareness of FH as a disease entity, its inherited nature, and the potential cardiovascular consequences if left untreated. Data from the FH Foundation's CASCADE (Cascade Screening for Awareness and Detection) FH registry demonstrated that the diagnosis of FH occurred at a mean age of 50 years, by which time more than one third of the patients with FH had already experienced an atherosclerotic cardiovascular disease (ASCVD) event.14 This underscores the need for much earlier universal screening, diagnosis, and treatment of FH.
FH is clinically diagnosed on the basis of a weighted combination of physical findings, personal or family history of hypercholesterolemia, early‐onset ASCVD, and the concentration of circulating LDL‐C.15 Extensor tendon xanthomas (typically Achilles, subpatellar, and hand extensor tendons) with extremely elevated LDL‐C levels are considered specific for FH.16 Severe extensor tendon xanthomas have histological features that resemble foam cell formation and lipid accumulation in atherosclerotic plaques (Figure 1). Hence, there is a modest correlation between the extent of tendon xanthomas and the extent of coronary atherosclerosis.17 However, as previously noted in children and young adults, a family history of CAD and elevated LDL‐C levels are often the only findings. The fasting LDL‐C concentration is incorporated in all diagnostic criteria and has been the backbone of FH diagnosis; however, nonfasting lipid testing may be used for initial screening. Blood sampling for the diagnosis of FH should preferably be avoided during acute illnesses, or while taking medications known to increase LDL‐C such as cyclosporine, amiodarone, hydrochlorothiazide, and chlorthalidone.18 Chronic illnesses that elevate LDL‐C, such as hypothyroidism and liver or renal impairment, should be ruled out.19, 20
Figure 1.
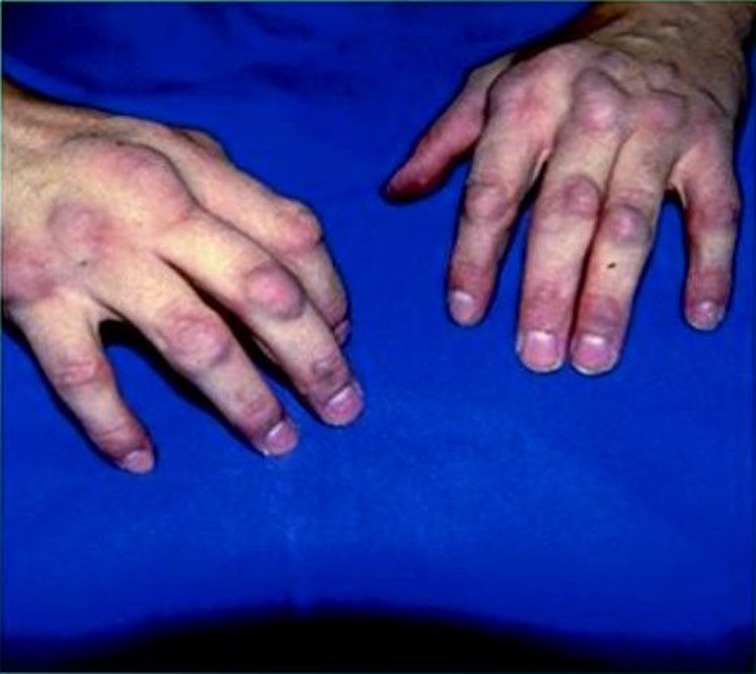
Severe extensor tendon xanthomas. Reprinted with permission of Dr Patrick M. Moriarty.
Although the diagnosis of FH can be made on the basis of clinical features, genetic testing may offer additional insight regarding cardiac risk and diagnosis.1 Recent data from 7 case–control and 5 prospective cohort studies of >26 000 individuals suggest that at any given LDL‐C level, having an identified FH mutation is associated with significantly higher cardiac risk than an individual with the same LDL‐C but no apparent pathogenic FH mutation.21 In this study, individuals with an LDL‐C level ≥190 mg/dL (4.9 mmol/L) and no pathogenic FH mutation were at a 6‐fold higher risk of CAD than the reference group with an LDL‐C ≤130 mg/dL. However, individuals with an LDL‐C level ≥190 mg/dL (4.9 mmol/L) and a pathogenic FH mutation were at a 22‐fold higher risk than the reference group,21 possibly reflecting greater atherogenicity of lifelong LDL‐C elevation in FH compared with LDL‐C elevation acquired later in life.
Although there are no internationally agreed‐upon criteria for the diagnosis of FH, useful diagnostic criteria have been developed. The main diagnostic tools used for FH include the US Make Early Diagnosis to Prevent Early Death (MEDPED) criteria (Table 1),22 the UK Simon Broome system (UK FH Register criteria; Table 2),23 the Dutch Lipid Clinic Network criteria (Table 3),24 and the National Lipid Association expert panel recommendations (Table 4).25, 26 The Dutch Lipid Clinic Network, which uses a scoring system to predict the likelihood of an index patient having FH, and the Simon Broome criteria are the only tools that incorporate genetic test results into their algorithm. In the Simon Broome criteria, a positive genetic test is sufficient for a definitive diagnosis of FH, while in the Dutch Lipid Clinic Network criteria, a positive genetic test should be accompanied by an additional measure (eg, elevated LDL‐C levels) to fulfill the definite diagnosis criteria. Although the existing diagnostic tools differ from each other, both in structure and the cut‐off values of the LDL‐C level necessary for diagnosis, their predictive values are comparable.25 Among these diagnostic strategies, the Dutch Lipid Clinic Network criteria provide the most detailed assessment of the likelihood of a diagnosis of HeFH and are sometimes favored by insurance providers for the purpose of denial or approval of treatment with PCSK9 inhibitors among patients with FH. More recently, the American Heart Association proposed a simple set of criteria; however, these criteria have not yet been widely accepted or implemented (Figure 2).27 Table 5 highlights some of the differences between these diagnostic criteria. The Familial Hypercholesterolemia Foundation, an organization dedicated to improving the awareness, diagnosis, and management of FH, has developed a mobile application28 that can assist both patients and providers with diagnosing FH. A special flow chart to detect FH in children has been developed and tested by the European Atherosclerosis Society consensus panel.29
Table 1.
US MEDPED Criteria for FH Diagnosis22
| FH Is Diagnosed if Total Cholesterol Exceeds These Cut‐Off Points in mg/dL (mmol/L)a | ||||
|---|---|---|---|---|
| Age, y | First‐Degree Relative With FH | Second‐Degree Relative With FH | Third‐Degree Relative With FH | General Population |
| <20 | 220 mg/dL (5.7 mmol/L) | 230 mg/dL (5.9 mmol/L) | 240 mg/dL (6.2 mmol/L) | 270 mg/dL (7.0 mmol/L) |
| 20–29 | 240 mg/dL (6.2 mmol/L) | 250 mg/dL (6.5 mmol/L) | 260 mg/dL (6.7 mmol/L) | 290 mg/dL (7.5 mmol/L) |
| 30–39 | 270 mg/dL (7.0 mmol/L) | 280 mg/dL (7.2 mmol/L) | 290 mg/dL (7.5 mmol/L) | 340 mg/dL (8.8 mmol/L) |
| ≥40 | 290 mg/dL (7.5 mmol/L) | 300 mg/dL (7.8 mmol/L) | 310 mg/dL (8.0 mmol/L) | 360 mg/dL (9.3 mmol/L) |
FH indicates familial hypercholesterolemia; MEDPED, Make Early Diagnosis to Prevent Early Death.
The total cholesterol cut‐off points for FH are dependent upon the confirmed cases of FH in the family. If FH is not diagnosed in the family, then the cut‐off point for diagnosis is as per “general population.”
Table 2.
Simon Broome Criteria for the Diagnosis of FH (UK FH Registers Criteria)23
| Criteria | Possibility |
|---|---|
|
In adults: TC >7.5 mmol/L (290.0 mg/dL) (or when available, LDL‐C >4.9 mmol/L [189.5 mg.dL]) In pediatric patients: TC >6.7 mmol/L (259.1 mg/dL), or LDL‐C >4 mmol/L (154.7 mg/dL), AND |
Definite |
| Tendon xanthoma in the patient or first/second‐degree relative, OR alternatively: | |
| Presence of LDL‐R, ApoB, or PCSK9 mutation | |
|
In adults: TC >7.5 mmol/L (290.0 mg/dL) (or when available, LDL‐C >4.9 mmol/L [189.5 mg.dL]) In pediatric patients: TC >6.7 mmol/L (259.1 mg/dL), or LDL‐C >4 mmol/L (154.7 mg/dL), AND |
Possible |
| Family history of MI <50 y old in second‐degree relative or <60 y old in first‐degree relative OR alternatively | |
| Family history of TC >7.5 mmol/L (290.0 mg/dL) in a first‐ or second‐degree relative. |
To convert LDL‐C in mmol/L to mg/dL, multiply by 38.67. ApoB indicates apolipoprotein B; FH, familial hypercholesterolemia; LDL, low‐density lipoprotein; LDL‐R, low‐density lipoprotein receptor; MI, myocardial infarction; PCSK9, proprotein convertase subtilisin/kexin type 9; TC, total cholesterol.
Table 3.
Dutch Lipid Network Criteria for Diagnosis of FH24
| Criteria | Score |
|---|---|
| Family history | |
| Premature CVD (men <55 y old, women <60 y old) in first‐degree relative, OR | 1 |
| LDL >95th percentile in first‐degree relative AND/OR | 1 |
| Tendon xanthoma and/or arcus cornealis in first‐degree relative, OR | 2 |
| LDL >95th percentile in children <18 y old | 2 |
| Personal history | |
| Premature CAD in patient (men <55 y old, women <60 y old) | 2 |
| Premature cerebral or peripheral vascular disease (men <55 y old, women <60 y old) | 1 |
| Clinical examination | |
| Tendon xanthomas, OR | 6 |
| Corneal arcus younger than 45 y old | 4 |
| LDL | |
| >330 mg/dL (8.5 mmol/L) | 8 |
| 250–329 mg/dL (6.5–8.5 mmol/L) | 5 |
| 190–249 mg/dL (4.9–6.4 mmol/L) | 3 |
| 155–189 mg/dL (4.0–4.9 mmol/L) | 1 |
| Presence of functional LDL‐R mutation (in the LDL‐R, ApoB, or PCSK9 gene) | 8 |
| Diagnosis based on the overall score | |
| Definite | >8 |
| Probable | 6–8 |
| Possible | 3–5 |
| Unlikely | <3 |
ApoB indicates apolipoprotein B; CAD, coronary artery disease; CVD, cardiovascular disease; FH, familial hypercholesterolemia; LDL, low‐density lipoprotein; LDL‐R, low‐density lipoprotein receptor; PCSK9, proprotein convertase subtilisin/kexin type 9.
Table 4.
| Children, Adolescents, Young Adults <20 y Old | Adults ≥20 y Old |
|---|---|
|
LDL‐C ≥160 mg/dL (4.1 mmol/L) Non‐HDL‐C ≥190 mg/dL (4.9 mmol/L) |
LDL‐C ≥190 md/dL (4.9 mmol/L) Non‐HDL ≥220 mg/dL (5.7 mmol/L) |
| At the LDL‐C levels listed below, the probability of FH is ≈80% in the setting of general population screening. These LDL‐C levels should prompt the clinician to strongly consider a diagnosis of FH and obtain further family information: | |
| LDL‐C ≥250 mg/dL (6.5 mmol/L) in a patient aged ≥30 y | |
| LDL‐C >220 mg/dL (5.7 mmol/L) for patients aged 20 to 29 y | |
| LDL‐C ≥190 mg/dL (4.9 mmol/L) in patients aged <20 y | |
FH indicates familial hypercholesterolemia; HDL, high‐density lipoprotein; LDL‐C, low‐density lipoprotein cholesterol; MEDPED, Make Early Diagnosis to Prevent Early Death; NLA, National Lipid Association; PCSK9, proprotein convertase subtilisin/kexin 9.
The NLA expert statement was not intended to be a substitute for the MEDPED, Dutch Lipid Clinic Network (DLCN), or Simon Broome criteria. In addition, the NLA recommends use of MEDPED, DLCN, and Simon Broome criteria for diagnosis of FH.
Figure 2.
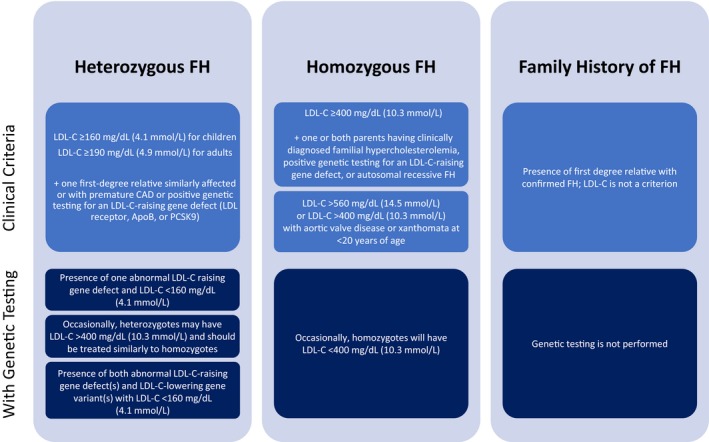
Diagnostic considerations by the American Heart Association.27 ApoB indicates apolipoprotein B; FH, familial hypercholesterolemia; LDL‐C, low‐density lipoprotein cholesterol; PCSK9, proprotein convertase subtulisin/kexin type 9.
Table 5.
| Criteria | MEDPED | DUTCH | SIMON BROOME | NLAa | AHA |
|---|---|---|---|---|---|
| Family history of premature CAD | + | + | + | + | |
| Family history of tendon xanthomas | + | + | |||
| Family history of hypercholesterolemia | + | + | + | + | |
| Patient premature CAD | + | + | |||
| Patient premature PVD | + | ||||
| Tendon xanthomas | + | + | + | ||
| Corneal arcus | + | + | |||
| Elevated LDL‐C | + | + | + | + | + |
| Genetic mutation | + | + | + | + |
AHA indicates American Heart Association; CAD, coronary artery disease; FH, familial hypercholesterolemia; LDL‐C, low‐density lipoprotein cholesterol; MEDPED, Make Early Diagnosis to Prevent Early Death; NLA, National Lipid Association; PVD, peripheral vascular disease.
The NLA recommends the use of MEDPED, Dutch Lipid Clinic Network (DLCN), and Simon Broome criteria for diagnosis of familial hypercholesterolemia.
Genetic Testing
Diagnosis of FH may be confirmed with positive pathogenic genetic testing,25 but cannot be excluded in the absence of a causative mutation. Genetic diagnosis of FH may involve testing for either known pathogenic variants in the genes for LDL‐R, ApoB, and PCSK9 or whole‐gene sequencing.30 Depending on the setting, a substantial percentage of patients with a definitive clinical diagnosis of FH may not have an identifiable mutation, even with next‐generation whole‐gene sequencing.18 In mutation‐negative patients, hypercholesterolemia may be secondary to an unidentified mutation or may be polygenic in nature. However, polygenic hypercholesterolemia would not typically be associated with the autosomal dominant pattern of inheritance seen in many families with a clinical diagnosis of FH. LDL‐C genetic risk scoring, using the weighted sum of the LDL‐C‐raising alleles involved in determining lipid concentrations, can be used as a potential tool to differentiate between patients with polygenic dyslipidemia (but not FH) and patients with FH who test negative using current genetic assays.31 Additionally, 1 expert group has suggested the umbrella of FH to include 2 groups of polygenic individuals whose inheritance pattern suggests an autosomal dominant disorder. They have termed these groups polygenic FH and FH combined with hypertriglyceridemia. Both groups have high polygenic scores and multiple family members with dyslipidemia and CAD. The former group is noted to have LDL‐C >190 mg/dL (4.9 mmol/L) while the latter may have LDL‐C >190 mg/dL or a non‐ HDL‐C >220 mg/dL (5.7 mmol/L) and triglycerides >300 mg/dL (7.8 mmol/L).32 As noted previously, data from Khera and colleagues suggest that cardiovascular risk in these groups with polygenic dyslipidemia, while elevated compared with the general population, is not as high as those individuals with monogenic FH.21
Consultation with a genetic counselor before genetic testing may be beneficial to ensure the patient understands the risks and benefits of genetic testing.
The results of genetic testing should complement clinical and nongenetic laboratory diagnoses. Effective cholesterol‐lowering treatments, which may include statins, ezetimibe, PCSK9 inhibitors, and other agents, are indicated for the treatment of patients with presumed FH regardless of genetic test results.
Risk Prediction in FH
Current evidence suggests that mortality from ASCVD is higher in monogenic cases of FH compared with patients with polygenic hypercholesterolemia. In the Simon Broome Registry, the prevalence of a single pathogenic mutation in those with a definite diagnosis of FH were found to be significantly higher than those labeled as possible FH by the Simon Broome criteria (80% versus 25–30%). By comparing the mortality between these 2 groups, one can clearly show that FH patients with a single pathologic gene mutation are at increased risk of death compared with those with polygenic hypercholesterolemia.33 Humphries et al studied the prevalence of ASCVD in FH patients enrolled in the Simon Broome Registry and assessed the odds of having ASCVD between those without monogenic mutation and those with 3 known pathogenic mutations (LDL‐R, ApoB, or PCSK9 mutations).34 Their results showed that the odds of having ASCVD is higher in monogenic FH. Indeed, each gene conferred different odds of having CAD in affected individuals (odds ratio of 1.8, 3.4, and 19.9, for LDL‐R, ApoB, and PCSK9 mutations, respectively).
Another important aspect in stratifying the risk of atherosclerotic disease in patients with FH is the additive effect of other traditional risk factors of atherosclerosis in these patients. Many studies have found significant variations in the risk of ASCVD in FH patients based on their underlying cardiovascular risks.35, 36 In a longitudinal study of 1900 adult patients with HeFH followed in the CASCADE FH Registry for a mean of 20±11 months, the overall annualized ASCVD event rate was 2.21% (4.57% in patients with prior ASCVD; 0.82% in patients without prior ASCVD). Patient characteristics that were significantly associated with the incidence of ASCVD events were older age at diagnosis of FH, older age at enrollment in the longitudinal registry, male sex, lower HDL‐C, and higher prevalence of hypertension and diabetes mellitus (P<0.001). There were trends for an association among incident ASCVD events and smoking, higher triglycerides, and higher body mass index (P<0.02).37
The International Atherosclerosis Society released an expert consensus in 2016 suggesting risk stratification of FH patients into severe and nonsevere FH categories based on their LDL‐C level and presence of known clinical risk factors for ASCVD.38 International Atherosclerosis Society defines these risk factors as age >40 years without treatment, male sex, smoking, lipoprotein(a) (Lp[a]) >50 mg/dL (75 nmol/L), history of early CAD in first‐degree relatives, body mass index >30 kg/m2, and history of diabetes mellitus or chronic kidney disease. In untreated FH patients at their first presentation, International Atherosclerosis Society defines severe FH as either LDL‐C >400 mg/dL (10.3 mmol/L), or LDL‐C >310 mg/dL (8.0 mmol/L) with 1 high‐risk feature, or LDL‐C >190 mg/dL (4.9 mmol/L) with 2 or more high‐risk features.
FH patients with clinical ASCVD or evidence of advanced subclinical ASCVD are also considered to have severe FH. Markers of subclinical ASCVD are defined as a coronary artery calcium score >100 Agatston units and/or >75th percentile, or computed tomography angiography with at least 1 obstructive lesion (>50%) or multivessel lesions (<50%).
Cascade Screening
Diagnosing FH in a child or parent provides the opportunity for cascade screening, which involves testing all first‐degree relatives for elevated LDL‐C or a known genetic mutation (Figure 4). When first‐degree relatives of the index patient are identified to have FH, they become the locus for further cascade screening of second‐ and third‐degree relatives.18, 39, 40 A number of studies in the primary care setting have successfully implemented strategies of either universal or opportunistic pediatric lipid screening, resulting in the diagnosis of FH in parents and siblings.41, 42 The DECOPIN project also documented significant success in diagnosing FH among children whose parents were diagnosed both clinically and genetically as having definite FH. In fact, cascade screening of relatives has been given a Tier 1 classification by the US Centers for Disease Control and Prevention because of the efficacy with which FH can be diagnosed in this setting.1
Figure 3.
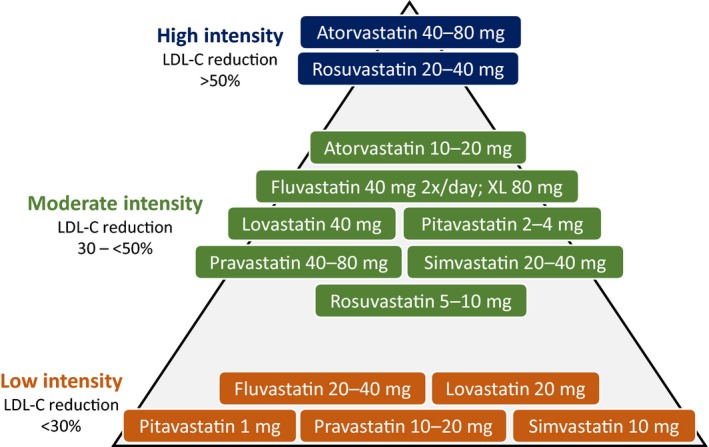
Statin dosing and ACC/AHA classification of intensity. ACC indicates American College of Cardiology; AHA, American Heart Association; LDL‐C, low‐density lipoprotein cholesterol.
Lipoprotein(a)
Lp[a] consists of an LDL particle to which apolipoprotein(a) is covalently bound. The plasma concentration of Lp(a) is largely genetically determined and is associated with an increased risk of ASCVD when elevated. The results of earlier studies suggested that levels of Lp(a) may be elevated in patients with FH. Among patients with FH, the presence of elevated Lp(a) may be associated with a 2‐fold increase in the risk of ASCVD above the already high risk of ASCVD attributable to FH alone.43 Furthermore, a study including >1000 children with FH whose Lp(a) levels were >30 mg/dL (≈45 nmol/L) demonstrated a 1.45‐times higher incidence (95% CI 0.99–2.13; P=0.05) of having a parent with FH and premature cardiovascular disease (CVD).44 Therefore, Lp(a) levels should be measured in the initial evaluation of patients with FH.45, 44
Treatment
Dietary and lifestyle modifications are the starting points for LDL‐C lowering in patients with FH, but multidrug treatment is often required to achieve adequate LDL‐C levels (Figure 4). In addition, all patients with FH should be counseled on the importance of not smoking or vaping, regular exercise, and maintaining a healthy body weight. ASCVD risk factors and comorbidities, such as hypertension and diabetes mellitus, should be treated.
Figure 4.
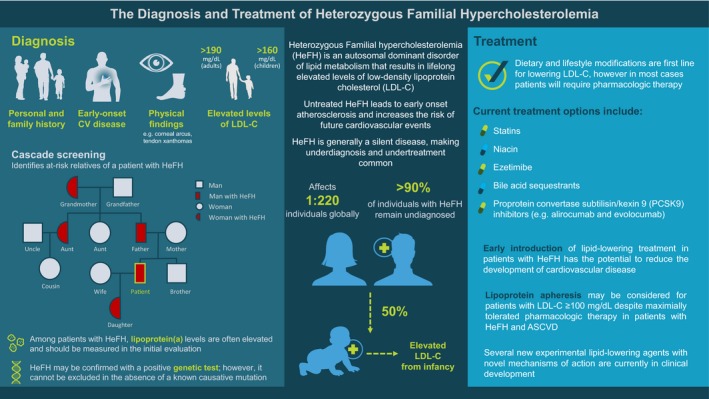
The diagnosis and treatment of heterozygous familial hypercholesterolemia. ASCVD indicates atherosclerotic cardiovascular disease; CV, cardiovascular; HeFH, heterozygous familial hypercholesterolemia; LDL‐C, low‐density lipoprotein cholesterol; PCSK9, proprotein convertase subtilisin/kexin 9. Figure reproduced with permission courtesy of the FH Foundation: https://thefhfoundation.org/family-screening-for-fh-and-the-use-of-genetic-testing. Accessed April 2019.
Pediatric Patients
There are currently no long‐term studies in children with FH that establish a reduction in vascular events in association with early initiation of LDL‐C‐lowering treatment. Results from 1 study demonstrated that children with FH have significantly greater carotid intima‐media thickness than their unaffected siblings, which may be detectable as early as 8 to 10 years of age.46 During longitudinal follow‐up, the difference in carotid intima‐media thickness between FH and non‐FH siblings progressively increased. Results from another study demonstrated small amounts of coronary artery calcification (indicative of advanced plaque) in some adolescents with FH.27 In addition, the results from another study showed that treatment with pravastatin for 2 years in children with FH aged 8 to 18 years resulted in stabilization or possible regression of carotid intima‐media thickness.47 On the basis of these findings, as well as the documented long‐term safety of statins in children over 10 years of follow‐up48 and the Food and Drug Administration (FDA) approval of all 7 statins for the treatment of children with FH, treatment with statins (usually at reduced doses) is recommended beginning between the ages of 8 and 10 years in children with FH. Furthermore, the proven benefits of statins for ASCVD prevention and treatment of FH in adults support their use in this population.49
Until recently there were no long‐term studies in children with FH establishing a reduction in vascular events in association with early initiation of LDL‐lowering therapy. We now have 20‐year follow‐up data on 214 children with genetically confirmed FH documenting that statin treatment during childhood slows the progression of carotid intima‐media thickness. Additionally, childhood statin therapy has now been shown to reduce cardiovascular events and cardiac death assessed through age 39. As compared with their FH affected parents (n = 156), who did not receive statins as children, the incidence of cardiovascular events and death from cardiovascular causes, was much lower in FH patients treated since childhood (1% versus 26% and 0% versus 7%, respectively). These new data affirm the importance of initiating statins in childhood in the setting of FH.50
Therapies for Pediatric Patients with FH
A recently proposed LDL‐C goal in pediatric patients is <110 mg/dL (2.8 mmol/L)51; however, the specific goal in pediatric patients with FH has not yet been determined. LDL‐C goals of either <100 mg/dL (2.6 mmol/L) or <130 mg/dL (3.4 mmol/L) have also been proposed.
Several guidelines recommend the use of statins to lower LDL‐C in children and adolescents who are 8 to 10 years of age or older and have an LDL‐C level that is persistently ≥160 mg/dL (4.1 mmol/L) after 3 to 6 months of lifestyle modification, and a clinical presentation consistent with FH.52 Pravastatin and pitavastatin are approved by the US FDA for children aged 8 years and above,53 whereas the other 5 statins are FDA approved from the age of 10 years for children with FH.54 The cornerstones of European management for HeFH include healthy lifestyle changes and statin treatment from 8 to 10 years of age. Further recommendations include a target LDL‐C level of <130 mg/dL (3.4 mmol/L) if >10 years of age, or ideally a 50% reduction from baseline if 8 to 10 years of age.29
Ezetimibe is approved by the FDA for the treatment of patients with HeFH without age restrictions, but data have not been reviewed for children younger than 10 years of age or premenarchal girls. The 2018 American College of Cardiology/American Heart Association cholesterol guidelines found that the drug provides clinically meaningful LDL‐C reduction without significant safety issues.51 In a prospective, multicenter, placebo‐controlled study including 248 children with FH aged 10 to 17 years, 33 weeks of ezetimibe in combination with simvastatin provided an additional 16% reduction in LDL‐C compared with simvastatin alone. In addition, ezetimibe did not adversely affect growth and development, sexual development, or menstrual cycle length in girls.55
Colesevelam, a bile acid sequestrant, is approved by the FDA for the treatment of boys and postmenarchal girls with HeFH aged 10 to 17 years as monotherapy or in combination with a statin.56 It is modestly effective for lowering the LDL‐C concentration by 13% to 18% and can be useful as adjunctive therapy in some children.
PCSK9 inhibitors, which lead to an increase in available LDL‐R number and a reduction in LDL‐C, have not yet been adequately studied or approved for use in children and adolescents with HeFH; however, clinical trials of alirocumab (NCT02890992) and evolocumab (NCT02392559 and NCT02624869) in this population are ongoing, with results expected soon.
Adult Patients
The 2018 American College of Cardiology/American Heart Association guidelines recommend lipid‐lowering therapies for patients with a baseline LDL‐C level of >190 mg/dL (4.9 mmol/L) or higher and include specific recommendations for patients with FH.51 In patients 30 to 75 years of age with HeFH and an LDL‐C level ≥100 mg/dL (≥2.6 mmol/L) on maximally tolerated statin and ezetimibe therapy, the addition of a PCSK9 inhibitor may be considered. Given that many patients with FH will have documented CAD and will continue to be at very high risk for future ASCVD, the guidelines suggest the use of an LDL‐C threshold of 70 mg/dL (1.8 mmol/L) for the addition of nonstatins to statin therapy.51 Nonstatins include ezetimibe, bile acid sequestrants, and PCSK9 inhibitors. In addition, niacin may have utility for adjunctive LDL‐C lowering in some patients with FH who are unable to achieve LDL‐C goals despite multidrug treatment regimens.57 Lipoprotein apheresis using the Dextran Sulfate Low‐Density Lipoprotein Adsorption system (LA‐15, Kaneka, Osaka, Japan), the only apheresis system available in the United States, may also be considered for select patients who meet the current FDA criteria of LDL‐C >100 mg/dL (2.6 mmol/L) after maximally tolerable therapy in patients with FH and ASCVD.
Therapies for Adults with FH
Various organizations recommend treating patients with FH with high doses of high‐intensity statins, which are capable of lowering LDL‐C by 50% to 60% (Figure 3).58 If high‐dose, high‐intensity statins are not tolerated, the maximally tolerated statin dose should be prescribed. This can include moderate‐intensity statin therapy, which typically lowers LDL‐C by 30% to <50%, or even low‐intensity statins, which lower LDL‐C by <30% (Figure 3). Early introduction of lipid‐lowering treatment in patients with FH appears to reduce the development of coronary events.59 In an observational cohort study of 1950 adults with FH followed for 8.5 years, a 76% reduction in cardiovascular events was reported in statin‐treated patients versus untreated patients.49 Unfortunately, many patients with FH are not identified and treated until later in life, sometimes not until after they have experienced an ASCVD event.60
Ezetimibe targets NPC1L1 (Niemann‐Pick C1‐like protein 1), resulting in inhibition of cholesterol absorption from the intestine. The IMPROVE‐IT (Ezetimibe Added to Statin Therapy after Acute Coronary Syndromes) trial involved a blinded placebo‐controlled treatment of 18 144 recent survivors of acute coronary syndrome with either simvastatin 40 mg daily or simvastatin 40 mg plus ezetimibe 10 mg daily for a median follow‐up of 6 years. The primary end point was a composite of cardiovascular death, nonfatal MI, unstable angina requiring hospitalization, coronary revascularization, and nonfatal stroke. The LDL‐C level was 69.5 mg/dL (1.8 mmol/L) in the simvastatin group and 53.7 mg/dL (1.4 mmol/L) in the simvastatin plus ezetimibe group. At study conclusion, the event rate was 34.7% in the simvastatin‐treated group versus 32.7% in the simvastatin plus ezetimibe group, with an absolute risk reduction of 2.0%. The hazard ratio was 0.936 (CI 0.89–0.99; P=0.016) with a number needed to treat of 50 to prevent 1 event.61
Bile acid sequestrants can be a useful adjunctive therapy for patients who require modest additional LDL‐C lowering after treatment with statins and ezetimibe. Colesevelam is a polymeric water‐absorbing hydrogel that works by binding bile acids in the intestine to form an insoluble complex that is then eliminated from the body in feces.62 In a study conducted in adults with refractory FH, compared with placebo, the addition of colesevelam to a maximally tolerated regimen of a statin plus ezetimibe provided significantly greater reductions in LDL‐C. Similarly, the results of another study demonstrated significantly greater reductions in LDL‐C in pediatric patients with FH receiving colesevelam alone or in combination with statins. In both of these multinational studies, colesevelam was generally well tolerated, with gastrointestinal disorders being the most common treatment‐related adverse event.56 LDL‐C reductions of 14% to 18% can be achieved with colesevelam, with potentially larger reductions of up to 25% to 30% achievable with higher doses of cholestyramine and colestipol.63, 64
Alirocumab and evolocumab are fully human monoclonal antibodies targeted against PCSK9. These agents act to increase available LDL‐R and markedly reduce plasma LDL‐C levels as monotherapy and in combination with statins and ezetimibe. The efficacy and safety of alirocumab in patients with HeFH have been studied in 3 multicenter, randomized, double‐blind, placebo‐controlled, phase III clinical trials: ODYSSEY FH I, ODYSSEY FH II,65 and ODYSSEY HIGH FH.66 All 3 studies included patients who were taking maximally tolerated statin treatment with other lipid‐lowering therapies and who were randomized to 78 weeks of treatment with the primary outcome of percent LDL‐C reduction from baseline assessed at 24 weeks.66, 67
In the ODYSSEY FH I (n=486) and ODYSSEY FH II (n=249) trials, patients were randomly assigned 2:1 to either alirocumab 75 mg subcutaneously every 2 weeks (Q2W) or matching placebo. If the LDL‐C level remained >70 mg/dL (1.8 mmol/L) at 8 weeks, the dose of alirocumab was blindly increased to 150 mg at Week 12. The mean LDL‐C level decreased from 144.7 mg/dL (3.7 mmol/L) at baseline to 71.3 mg/dL (1.8 mmol/L) (−57.9% versus placebo) at Week 24 in patients randomized to alirocumab in ODYSSEY FH I and from 134.6 mg/dL (3.5 mmol/L) to 67.7 mg/dL (1.8 mmol/L) (−51.4% versus placebo) in ODYSSEY FH II (P<0.0001 for both studies). At Week 24, an LDL‐C level <70 mg/dL (1.8 mmol/L) was achieved by 59.8% and 68.2% of patients in ODYSSEY FH I and FH II, respectively. LDL‐C levels achieved at Week 24 were maintained throughout the 78‐week study. Discontinuation because of adverse events was infrequent. In the ODYSSEY HIGH FH trial, 107 patients were randomly assigned 2:1 to either alirocumab 150 mg subcutaneously Q2W or matching placebo. Patients in this study had very high baseline LDL‐C (196.3 mg/dL [5.1 mmol/L] in the alirocumab arm and 201.0 mg/dL [5.2 mmol/L] in the placebo arm). At Week 24, LDL‐C decreased by 45.7% in the alirocumab arm and 6.6% in the placebo arm for a placebo‐controlled difference of 39.1% (P<0.0001). As in the previous studies, LDL‐C reduction was maintained throughout the 78‐week study, and 41% of patients achieved the prespecified LDL‐C goals of <100 mg/dL (2.6 mmol/L) for high‐risk patients and <70 mg/dL (1.8 mmol/L) for very high‐risk patients. As in the previous studies, alirocumab was safe and well tolerated.
Patients successfully completing the aforementioned FH studies were invited to enroll in an open‐label extension program (median total duration of treatment was 2.5 years, including 1.5 years of the parent trial), during which patients were treated with 75 mg of alirocumab Q2W. From Week 12 of the open‐label extension, the dose of alirocumab could be adjusted as per physician's clinical judgment. LDL‐C reductions achieved in the parent study were generally maintained throughout the open‐label extension and alirocumab continued to be well tolerated.68
The effect of evolocumab was evaluated in two 12‐week trials of patients with HeFH: RUTHERFORD69 and RUTHERFORD 2.70 In the RUTHERFORD trial, 167 patients were randomly assigned to evolocumab 350 mg, 420 mg, or placebo every 4 weeks (Q4W) for 12 weeks. Baseline LDL‐C in this study was 156 mg/dL (4.0 mmol/L) while taking maximally tolerated lipid‐lowering therapy (statins and ezetimibe). Patients assigned to the 350 mg dose lowered their LDL‐C by 43%, whereas those assigned to the 420 mg dose lowered their LDL‐C by 55% (P<0.001 for both doses). The placebo group experienced a 1% increase in LDL‐C.69 The RUTHERFORD 2 trial was a multicenter, randomized, double‐blind, placebo‐controlled intervention in 331 patients. Patients were assigned to receive 140 mg of evolocumab Q2W (n=111), 420 mg monthly (n=110), or placebo Q2W (n=55) or monthly (n=55) as a subcutaneous injection. At study conclusion, evolocumab 140 mg Q2W lowered LDL‐C by 59.2%, and evolocumab 420 mg monthly lowered LDL‐C by 61.3% compared with placebo (P<0.0001 for both doses).70 As with the alirocumab studies, patients completing the RUTHERFORD studies were invited to participate in follow‐up long‐term, open‐label, extension studies: OSLER I (52 weeks) and OSLER 2 (48 weeks). Although the duration of open‐label treatment with evolocumab was shorter than the alirocumab studies, the long‐term extension studies confirmed a persistent LDL‐C reduction for up to 64 weeks.71
The efficacy of alirocumab and evolocumab in the secondary prevention of cardiovascular outcomes has also been documented in the ODYSSEY OUTCOMES72 and FOURIER trials,73 respectively (total n=46 488). Although these trials did not specifically recruit patients with FH, it is likely that many patients with FH were included in these studies. The hazard ratio for the primary efficacy end point in each study was 0.85, with nearly identical CIs and highly statistically significant P values obtained in both studies (P<0.001).72, 73 Alirocumab, but not evolocumab, was also shown to be associated with a significant reduction in all‐cause mortality.74 Both agents were safe and well tolerated.
In some circumstances, adults with HeFH and very high LDL‐C following maximally tolerated lipid‐lowering therapy can be considered for lipid or lipoprotein apheresis (formerly known as LDL apheresis). The American Society for Apheresis recently released their 2019 recommendations for the use of apheresis. For the treatment of Lp(a) >50 mg/dL in CVD patients on maximum drug therapy, American Society for Apheresis has assigned a category II, grade 1B classification.75 Category II is defined as a disorder for which apheresis is accepted as second‐line therapy, either as a standalone treatment or in conjunction with other modes of treatment, and a grade 1B recommendation is considered a strong recommendation, with moderate quality evidence, that can apply to most patients in most circumstances without reservation.75 Furthermore, data from observational clinical trials suggest that lipoprotein apheresis may reduce symptoms of angina by improving endothelial function and reduce cardiovascular events when performed regularly.76 The results of other studies demonstrated an acute improvement in coronary microvascular dysfunction after lipoprotein apheresis in patients with FH.77 A long‐term 10‐year study (mean 6 years) evaluated the efficacy and safety of cholesterol‐lowering therapies in conjunction with lipoprotein apheresis in patients with FH with coronary heart disease. This study found that lipoprotein apheresis in combination with cholesterol‐lowering therapy significantly reduced LDL‐C levels compared with those who received cholesterol‐lowering drug therapy alone. In addition, the rate of total coronary events over 10 years of follow‐up was 72% lower in the apheresis group than in the drug therapy alone group (P=0.0088).78 The ODYSSEY ESCAPE study was conducted in 62 patients with HeFH who underwent regular weekly or Q2W lipoprotein apheresis. Study participants were randomly assigned in a 2:1 ratio to receive blinded alirocumab 150 mg (n=41) or placebo (n=21) Q2W subcutaneously for 18 weeks. Alirocumab‐treated individuals demonstrated a 75% reduction in the standardized rate of apheresis treatment compared with placebo‐treated patients (P<0.0001).79 Lipoprotein apheresis has also been shown to reduce Lp(a) levels and CVD events, and is used in some extreme cases specifically for Lp(a) lowering.81
Likewise, the Efficacy and Safety of evolocumab Compared with Continued Lipoprotein Apheresis: Results of a Randomized, Controlled, Open‐Label Study was conducted in 39 patients with pre‐apheresis levels of LDL‐C >100 mg/dL (2.6 mmol/L). Patients were randomly assigned to continue lipoprotein apheresis (n=20) at the same frequency or discontinue lipoprotein apheresis and begin subcutaneous evolocumab 140 mg (n=19) every 2 weeks for 6 weeks. At 6 weeks all participants received open‐label evolocumab 140 mg every 2 weeks for 18 weeks. The primary end point was avoidance of lipoprotein apheresis at week 5 or 6, as a result of achieving an LDL‐C <100 mg/dL (2.6 mmol/L) at week 4 regardless of randomization arm. Significantly more patients in the evolocumab versus the lipoprotein apheresis group met the primary end point of avoiding lipoprotein apheresis (84% versus 10%; treatment difference [95% CI], 74% [45,87]; P<0.0001), suggesting that certain patients may be treated with evolocumab in place of lipoprotein apheresis.81
PCSK9 inhibitors provide an added benefit for statin‐intolerant patients, including patients with FH. The GAUSS‐3 (Goal Achievement After Utilizing an Anti‐PCSK9 Antibody in Statin‐Intolerant Subjects 3) trial was a phase III, multicenter, randomized, double‐blinded study that compared the effectiveness of evolocumab versus ezetimibe in hypercholesterolemic patients (n=511) who were unable to tolerate an effective statin dose.82 Percentage change from baseline in LDL‐C was assessed at Week 24 of treatment with either evolocumab 420 mg monthly or ezetimibe 10 mg daily. Results demonstrated significantly greater mean percentage reductions in LDL‐C levels with evolocumab versus ezetimibe after 24 weeks of treatment (−52.8% versus −16.7% mg/dL, respectively; P<0.001).83 Similarly, the ODYSSEY ALTERNATIVE trial compared alirocumab with ezetimibe in patients at moderate‐to‐high cardiovascular risk with statin intolerance (n=361). Compared with ezetimibe, alirocumab reduced mean LDL‐C levels by 45.0% versus 14.6% (P<0.0001).84
Management of Lipids in Women with FH During Pregnancy
Total cholesterol and LDL‐C increase in all pregnancies by 25% to 50%; however, because women with FH have higher baseline lipids, the absolute increase is more notable in FH pregnancies.85 Compounding this is the recommendation that women discontinue statins, ezetimibe, and PCSK9 inhibitors from the time of attempting to become pregnant through until the completion of breast feeding. Bile acid sequestrants, which are not systemically absorbed, and lipid apheresis are approved during pregnancy.86 Women with FH who have had well‐controlled lipids before pregnancy should not be discouraged from becoming pregnant. Genetic counseling before pregnancy can help couples understand the risk of transmission of FH to their offspring.1
While in general, statins are contraindicated during pregnancy, some experts have recommended their use after organogenesis beginning in the second trimester in the setting of known FH complicated by prevalent ASCVD and in HoFH.87 These experts cited a large cohort study of 886 996 completed pregnancies, which included 1152 (0.13%) pregnancies where a statin was taken, after controlling for pre‐existing conditions including diabetes mellitus. There was no increased risk of organ‐specific malformations found.88 Other data suggest that statins may potentially have a role in the prevention of pre‐eclampsia.89 In fact, 1 group, in conjunction with the FDA, studied the role of pravastatin in 20 high‐risk pregnancies. Beginning between Weeks 12 and 16 of gestation, women were randomized to pravastatin 10 mg or matching placebo. No women in the pravastatin group developed pre‐eclampsia while 4 placebo‐treated women did. Importantly, there was no difference in side effects or congenital anomalies between the groups.90
PCSK9 inhibitors, like other IgG antibodies, are known to cross the placental barrier.91 It is interesting, however, that the package inserts of both alirocumab and evolocumab suggest that this may not occur during the first trimester.92, 93 There are registries devoted to following the pregnancy outcomes of women who become pregnant while on the PCSK9 inhibitors.
More research is needed to determine the best approach to managing lipids in pregnant women with FH. In the absence of randomized clinical trials, all women with FH who become pregnant while on any lipid‐lowering therapy should be closely followed in registries.
Experimental Lipid‐Lowering Therapies in Development
Although options for lipid‐lowering treatment have markedly increased since the first statin came on the market in 1987, there is still a need for new lipid‐lowering agents with novel mechanisms of action. Several such agents are currently in clinical development.
Bempedoic acid is an oral small molecule that inhibits hepatic adenosine triphosphate citrate lyase, leading to inhibition of the production of acetyl coenzyme A, the final common substrate for both fatty acid synthesis and sterol synthesis upstream from 3‐hydroxy‐3‐methylglutaryl–acetyl coenzyme A reductase, which is the target of statins. Bempedoic acid upregulates LDL‐R and leads to LDL‐C reduction. Data from a phase III 52‐week clinical trial (n=2230) that included a small number (n=79) of individuals with FH demonstrated that, when added to maximally tolerated lipid‐lowering therapy (statins [with the exception of simvastatin 40 mg or higher] and ezetimibe), bempedoic acid 180 mg once daily significantly lowered LDL‐C by 18.1% (P<0.001), ApoB by 11.9% (P<0.001), and high‐sensitivity C‐reactive protein by 21.5% (P<0.001), with good safety and tolerability.94 Patients with FH seem to respond similarly to patients without FH. In addition, the ETC‐1002 (Evaluation of Long‐Term Efficacy of Bempedoic Acid) in CLEAR Wisdom (Patients with Hyperlipidemia at High Cardiovascular Risk) trial found that the addition of bempedoic acid to maximally tolerated statins in patients with hypercholesterolemia, including those with FH, significantly reduced LDL‐C levels compared with placebo.95
MGL‐3196 (NCT03038022) is a thyroid hormone receptor‐beta‐selective agonist that has been studied in a phase II,12‐week, double‐blinded, placebo‐controlled trial in 116 patients with HeFH who, despite maximally tolerated statin therapy, had LDL‐C levels >100 mg/dL (2.6 mmol/L). Study subjects initially received a daily dose of 100 mg of MGL‐3196, but some subjects had their dose reduced to 60 mg/d based on pharmacokinetic data obtained at Week 2. LDL‐C was reduced by18.8% versus placebo (P<0.0001). Levels of triglycerides, ApoB, Lp(a), and apolipoprotein CIII were also decreased. MGL‐3196 appeared to be well tolerated. Data from this study were presented as a poster at the European Society of Cardiology (ESC) and have not yet been published.96 However, MGL‐3196 has demonstrated encouraging antidyslipidemic effects in rodent models, reducing non‐HDL‐C and liver triglycerides.97
Inhibitors of angiopoietin‐like 3 are being evaluated in individuals with HeFH, HoFH, and hypertriglyceridemia. Angiopoietin‐like 3 is a protein secreted by the liver that increases triglycerides, LDL‐C, and HDL‐C. Loss‐of‐function variants of the ANGPTL3 gene have been associated with a reduction in triglycerides, LDL‐C, and HDL‐C, in association with reduced risk of CAD. Evinacumab, a fully human monoclonal antibody against angiopoietin‐like 3, was first studied in a single‐group, open‐label, 4‐week study in 9 patients with HoFH (NCT02265952). Study participants were taking multiple lipid‐lowering therapies, including statins, ezetimibe, lomitapide, and PCSK9 inhibitors. Despite multidrug LDL‐C‐lowering treatment, baseline LDL‐C was 376±240 mg/dL (9.7±6.2 mmol/L). Evinacumab substantially decreased LDL‐C by 49±23% (range 25–90%) at Week 4. The absolute LDL‐C reduction was 157±90 mg/dL (4.1±2.3 mmol/L) (range 71–323 mg/dL [1.8–8.4 mmol/L]).98 On the basis of these results, larger trials in both HeFH (NCT03409744) and HoFH (NCT03175367) are under way, with results expected in 2019 and 2022, respectively.
Gemcabene, a lipid‐modulating agent in phase II development, appears to increase the clearance of very low‐density lipoprotein from plasma, and inhibits cholesterol and triglyceride production in the liver. This results in reduction of very low‐density lipoprotein‐C, LDL‐C, Apo B, triglycerides, and high‐sensitivity C‐reactive protein. Although several phase II studies have been conducted, only 1 small (n=8), nonrandomized, open‐label, 12‐week study included subjects with HoFH and HeFH (NCT02722408). Participants in this study received 300 mg of gemcabene daily for 4 weeks, followed by 600 mg and 900 mg daily for 4 weeks each. As per the company website,99 baseline LDL‐C decreased 39% in patients with HeFH and 15% in patients with HoFH.
Small interfering RNA molecules have recently been used to target hepatic production of PCSK9. Inclisiran (ALN‐PCS) is a long‐acting, synthetic small interfering RNA that is directed against PCSK9 messenger RNA to reduce LDL‐C levels.100 In ORION 1, a phase II multicenter, double‐blind, placebo‐controlled, multiple‐ascending‐dose trial (NCT02597127), inclisiran was administered as a subcutaneous injection in patients at high risk for cardiovascular disease with elevated LDL‐C levels (defined as LDL‐C >70 mg/dL [1.8 mmol/L] in patients with ASCVD and >100 mg/dL [2.6 mmol/L] in patients without ASCVD). Patients were randomized to receive either a single dose of placebo or 200, 300, or 500 mg of inclisiran, or 2 doses (on Day 1 and Day 90) of placebo or 100, 200, or 300 mg of inclisiran. The primary end point was the percentage change of LDL‐C from baseline at 180 days. All doses of inclisiran achieved statistically significant reductions in LDL‐C versus placebo at Day 180. The 2‐dose 300 mg regimen achieved the greatest LDL‐C reduction of 52.6% (P<0.001).101 A number of inclisiran studies in HoFH and HeFH are either completed or ongoing.
Lastly, LIB003 is a recombinant fusion protein consisting of a PCSK9‐binding domain and human serum albumin.102 It is being developed for the reduction of LDL‐C in patients with heterozygous and homozygous FH, CVD, or those at high risk of CVD who require additional LDL‐C reduction.102 By binding to PCSK9 in plasma, LDL‐C is lowered by ≈70% at higher doses, in association with reductions in ApoB and 20% to 30% reductions in Lp(a).102
Ongoing and Future Research
There are still significant gaps in our understanding of optimal diagnostic tools and management methods for individuals with FH. The Familial Hypercholesterolemia Foundation established the ongoing CASCADE‐FH registry to address gaps in knowledge and identify barriers to comprehensive FH screening, identification, and treatment among 40 clinical sites across the United States. Patients with FH are being followed longitudinally to track changes in LDL‐C‐lowering therapy, LDL‐C goal achievement, patient‐reported outcomes, and clinical outcomes.103 Data as of February 2017 demonstrated that <50% of patients were aware of available treatment options and the increased risk of heart disease.104 Results from the CASCADE‐FH registry, and similar registries in Europe and Asia, will help inform optimal screening, diagnosis, and treatment strategies in FH.
Conclusions
FH is a common autosomal dominant disorder that results in markedly elevated LDL‐C levels from birth and causes early‐onset CAD. Early diagnosis provides an opportunity to initiate potentially lifesaving and inexpensive generic pharmacotherapy in childhood. Although multiple guidelines recommend universal screening beginning in childhood, it is estimated that only 10% of the 1.3 million Americans living with FH are aware of their diagnosis. Consequently, FH is often diagnosed in adulthood following a cardiac event. FH is eminently treatable with currently available lipid‐lowering therapies that include statins, ezetimibe, bile acid sequestrants, niacin, and PCSK9 inhibitors, as well as lipoprotein apheresis in more severe cases. However, early initiation of aggressive LDL‐C‐lowering treatment is required to achieve the greatest reduction in ASCVD morbidity and mortality. Additional promising experimental lipid‐lowering agents are in clinical development and may be useful in this endeavor. In the meantime, the prevention of early‐onset ASCVD events and mortality depends on greater awareness of FH among healthcare professionals and patients.
Sources of Funding
Funding for this publication was provided by Sanofi and Regeneron Pharmaceuticals, Inc.
Disclosures
P. Barton Duell reports receiving consultant fees from Akcea, consultant fees from Astra Zeneca, grants and consultant fees from Esperion, grants and consultant fees from Regeneron, grants and consultant fees from Regenxbio, grants and consultant fees from Retrophin, outside the submitted work. Nonfinancial support during the conduct of this study was provided by Sanofi and Regeneron. Seyed Hamed Hosseini Dehkordi reports nonfinancial support from Sanofi and Regeneron Pharmaceuticals Inc., during the conduct of the study. Mary McGowan is a member of the board of the Familial Hypercholesterolemia Foundation and was an employee of Esperion Therapeutics from 2015 to 2018. She has received nonfinancial support during the conduct of this study provided by Sanofi and Regeneron. Patrick Moriarty reports nonfinancial support from Sanofi and Regeneron Pharmaceuticals Inc., during the conduct of the study; grants and personal fees from Amgen, grants and personal fees from Regeneron, grants and personal fees from Kaneka, grants and personal fees from Sanofi, personal fees from Duke, personal fees from Amarin, grants from Ionis, grants from Novartis, grants and personal fees from Renew, grants from FH Foundation, grants from Akcea, grants from Kowa, grants from RegenXBio, personal fees from Esperion, from Ambry Genetics, personal fees from NLA, and personal fees from Academic CME, outside the submitted work.
Acknowledgments
The authors developed the concept, and wrote and approved the publication drafts. Authors received no honoraria related to the development of this publication. Employees of Sanofi and Regeneron Pharmaceuticals, Inc. were permitted to review the manuscript and offer comments. However, the authors were responsible for all content and editorial decisions. Editorial support was provided by Michele Damo, PharmD, of Prime, Knutsford, UK, funded by Sanofi and Regeneron Pharmaceuticals, Inc., according to Good Publication Practice guidelines. Data sharing: There are no data to share in relation to this review article.
J Am Heart Assoc. 2019;8:e013225 DOI: 10.1161/JAHA.119.013225.
References
- 1. Sturm AC, Knowles JW, Gidding SS, Ahmad ZS, Ahmed CD, Ballantyne CM, Baum SJ, Bourbon M, Carrie A, Cuchel M, de Ferranti SD, Defesche JC, Freiberger T, Hershberger RE, Hovingh GK, Karayan L, Kastelein JJP, Kindt I, Lane SR, Leigh SE, Linton MF, Mata P, Neal WA, Nordestgaard BG, Santos RD, Harada‐Shiba M, Sijbrands EJ, Stitziel NO, Yamashita S, Wilemon KA, Ledbetter DH, Rader DJ. Convened by the Familial Hypercholesterolemia Foundation. Clinical Genetic Testing for Familial Hypercholesterolemia: JACC Scientific Expert Panel. J Am Coll Cardiol. 2018;72:662–680. [DOI] [PubMed] [Google Scholar]
- 2. Soutar AK, Naoumova RP. Mechanisms of disease: genetic causes of familial hypercholesterolemia. Nat Clin Pract Cardiovasc Med. 2007;4:214–225. [DOI] [PubMed] [Google Scholar]
- 3. Cuchel M, Bruckert E, Ginsberg HN, Raal FJ, Santos RD, Hegele RA, Kuivenhoven JA, Nordestgaard BG, Descamps OS, Steinhagen‐Thiessen E, Tybjaerg‐Hansen A, Watts GF, Averna M, Boileau C, Boren J, Catapano AL, Defesche JC, Hovingh GK, Humphries SE, Kovanen PT, Masana L, Pajukanta P, Parhofer KG, Ray KK, Stalenhoef AF, Stroes E, Taskinen MR, Wiegman A, Wiklund O, Chapman MJ; European Atherosclerosis Society Consensus Panel on Familial Hypercholesterolaemia . Homozygous familial hypercholesterolaemia: new insights and guidance for clinicians to improve detection and clinical management. A position paper from the Consensus Panel on Familial Hypercholesterolaemia of the European Atherosclerosis Society. Eur Heart J. 2014;35:2146–2157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Mata N, Alonso R, Badimon L, Padro T, Fuentes F, Muniz O, Perez‐Jimenez F, Lopez‐Miranda J, Diaz JL, Vidal JI, Barba A, Piedecausa M, Sanchez JF, Irigoyen L, Guallar E, Ordovas JM, Mata P. Clinical characteristics and evaluation of LDL‐cholesterol treatment of the Spanish Familial Hypercholesterolemia Longitudinal Cohort Study (SAFEHEART). Lipids Health Dis. 2011;10:94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Benjamin EJ, Muntner P, Alonso A, Bittencourt MS, Callaway CW, Carson AP, Chamberlain AM, Chang AR, Cheng S, Das SR, Delling FN, Djousse L, Elkind MSV, Ferguson JF, Fornage M, Jordan LC, Khan SS, Kissela BM, Knutson KL, Kwan TW, Lackland DT, Lewis TT, Lichtman JH, Longenecker CT, Loop MS, Lutsey PL, Martin SS, Matsushita K, Moran AE, Mussolino ME, O'Flaherty M, Pandey A, Perak AM, Rosamond WD, Roth GA, Sampson UKA, Satou GM, Schroeder EB, Shah SH, Spartano NL, Stokes A, Tirschwell DL, Tsao CW, Turakhia MP, VanWagner LB, Wilkins JT, Wong SS, Virani SS; American Heart Association Council on Epidemiology Prevention Statistics Committee, Stroke Statistics Subcommittee . Heart disease and stroke statistics‐2019 update: a report from the American Heart Association. Circulation. 2019;139:e56–e528. [DOI] [PubMed] [Google Scholar]
- 6. Benjamin EJ, Virani SS, Callaway CW, Chamberlain AM, Chang AR, Cheng S, Chiuve SE, Cushman M, Delling FN, Deo R, de Ferranti SD, Ferguson JF, Fornage M, Gillespie C, Isasi CR, Jimenez MC, Jordan LC, Judd SE, Lackland D, Lichtman JH, Lisabeth L, Liu S, Longenecker CT, Lutsey PL, Mackey JS, Matchar DB, Matsushita K, Mussolino ME, Nasir K, O'Flaherty M, Palaniappan LP, Pandey A, Pandey DK, Reeves MJ, Ritchey MD, Rodriguez CJ, Roth GA, Rosamond WD, Sampson UKA, Satou GM, Shah SH, Spartano NL, Tirschwell DL, Tsao CW, Voeks JH, Willey JZ, Wilkins JT, Wu JH, Alger HM, Wong SS, Muntner P; American Heart Association Council on Epidemiology and Prevention Statistics Committee and Stroke Statistics Subcommittee . Heart disease and stroke statistics‐2018 update: a report from the American Heart Association. Circulation. 2018;137:e67–e492. [DOI] [PubMed] [Google Scholar]
- 7. Do R, Stitziel NO, Won HH, Jorgensen AB, Duga S, Angelica Merlini P, Kiezun A, Farrall M, Goel A, Zuk O, Guella I, Asselta R, Lange LA, Peloso GM, Auer PL; NHLBI Exome Sequencing Project , Girelli D, Martinelli N, Farlow DN, DePristo MA, Roberts R, Stewart AF, Saleheen D, Danesh J, Epstein SE, Sivapalaratnam S, Hovingh GK, Kastelein JJ, Samani NJ, Schunkert H, Erdmann J, Shah SH, Kraus WE, Davies R, Nikpay M, Johansen CT, Wang J, Hegele RA, Hechter E, Marz W, Kleber ME, Huang J, Johnson AD, Li M, Burke GL, Gross M, Liu Y, Assimes TL, Heiss G, Lange EM, Folsom AR, Taylor HA, Olivieri O, Hamsten A, Clarke R, Reilly DF, Yin W, Rivas MA, Donnelly P, Rossouw JE, Psaty BM, Herrington DM, Wilson JG, Rich SS, Bamshad MJ, Tracy RP, Cupples LA, Rader DJ, Reilly MP, Spertus JA, Cresci S, Hartiala J, Tang WH, Hazen SL, Allayee H, Reiner AP, Carlson CS, Kooperberg C, Jackson RD, Boerwinkle E, Lander ES, Schwartz SM, Siscovick DS, McPherson R, Tybjaerg‐Hansen A, Abecasis GR, Watkins H, Nickerson DA, Ardissino D, Sunyaev SR, O'Donnell CJ, Altshuler D, Gabriel S, Kathiresan S. Exome sequencing identifies rare LDLR and APOA5 alleles conferring risk for myocardial infarction. Nature. 2015;518:102–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Hopkins PN, Toth PP, Ballantyne CM, Rader DJ; National Lipid Association Expert Panel on Familial Hypercholesterolemia . Familial hypercholesterolemias: prevalence, genetics, diagnosis and screening recommendations from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S9–S17. [DOI] [PubMed] [Google Scholar]
- 9. Hartgers ML, Defesche JC, Langslet G, Hopkins PN, Kastelein JJP, Baccara‐Dinet MT, Seiz W, Hamon S, Banerjee P, Stefanutti C. Alirocumab efficacy in patients with double heterozygous, compound heterozygous, or homozygous familial hypercholesterolemia. J Clin Lipidol. 2018;12:390–396.e398. [DOI] [PubMed] [Google Scholar]
- 10. Nordestgaard BG, Chapman MJ, Humphries SE, Ginsberg HN, Masana L, Descamps OS, Wiklund O, Hegele RA, Raal FJ, Defesche JC, Wiegman A, Santos RD, Watts GF, Parhofer KG, Hovingh GK, Kovanen PT, Boileau C, Averna M, Boren J, Bruckert E, Catapano AL, Kuivenhoven JA, Pajukanta P, Ray K, Stalenhoef AF, Stroes E, Taskinen MR, Tybjaerg‐Hansen A; European Atherosclerosis Society Consensus Panel . Familial hypercholesterolaemia is underdiagnosed and undertreated in the general population: guidance for clinicians to prevent coronary heart disease: consensus statement of the European Atherosclerosis Society. Eur Heart J. 2013;34:3478–3490a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Casula M, Catapano AL, Rossi Bernardi L, Visconti M, Aronica A. Detection of familial hypercholesterolemia in patients from a general practice database. Atheroscler Suppl. 2017;29:25–30. [DOI] [PubMed] [Google Scholar]
- 12. Mihalopoulos NL, Stipelman C, Hemond J, Brown LL, Young PC. Universal lipid screening in 9‐ to 11‐year‐olds before and after 2011 guidelines. Academic Pediatrics. 2018;18:196–199. [DOI] [PubMed] [Google Scholar]
- 13. Gooding HC, Rodday AM, Wong JB, Gillman MW, Lloyd‐Jones DM, Leslie LK, de Ferranti SD. Application of pediatric and adult guidelines for treatment of lipid levels among US adolescents transitioning to young adulthood. JAMA Pediatrics. 2015;169:569–574. [DOI] [PubMed] [Google Scholar]
- 14. Duell PB, Andersen RL, Knickelbine T, Anderson L, Gianos E, O'Brien EC, Kindt I, Shrader P, McCann D, Baum S, Hemphill LC, Ahmed CD, Martin SS, Watson KE, Kullo IJ, Larry JA, Murray M, Fishberg R, Guyton J, Wilemon K, Roe MT, Rader DJ, Ballantyne CM, Underberg JA, Thompson P, Ahmad ZS, Whellan D, Linton MF, Moriarty PM, Shapiro MD, Knowles JW. Abstract 19888: improved longitudinal low density lipoprotein cholesterol goal achievement among familial hypercholesterolemia patients in the CASCADE FH patient registry. Circulation. 2017;136:A19888. [DOI] [PubMed] [Google Scholar]
- 15. Lee SH. Update on familial hypercholesterolemia: diagnosis, cardiovascular risk, and novel therapeutics. Endocrinol Metab (Seoul). 2017;32:36–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bermudez EB, Storey L, Mayo S, Simpson G. An unusual case of multiple tendinous xanthomas involving the extremities and the ears. Case Rep Dermatol. 2015;7:340–344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Patil S, Kharge J, Bagi V, Ramalingam R. Tendon xanthomas as indicators of atherosclerotic burden on coronary arteries. Indian Heart J. 2013;65:491–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Watts GF, Gidding S, Wierzbicki AS, Toth PP, Alonso R, Brown WV, Bruckert E, Defesche J, Lin KK, Livingston M, Mata P, Parhofer KG, Raal FJ, Santos RD, Sijbrands EJ, Simpson WG, Sullivan DR, Susekov AV, Tomlinson B, Wiegman A, Yamashita S, Kastelein JJ; International Familial Hypercholesterolemia Foundation . Integrated guidance on the care of familial hypercholesterolaemia from the International FH Foundation. Eur J Prev Cardiol. 2015;22:849–854. [DOI] [PubMed] [Google Scholar]
- 19. Hartgers ML, Ray KK, Hovingh GK. New approaches in detection and treatment of familial hypercholesterolemia. Curr Cardiol Rep. 2015;17:109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hovingh GK, Davidson MH, Kastelein JJ, O'Connor AM. Diagnosis and treatment of familial hypercholesterolaemia. Eur Heart J. 2013;34:962–971. [DOI] [PubMed] [Google Scholar]
- 21. Khera AV, Won HH, Peloso GM, Lawson KS, Bartz TM, Deng X, van Leeuwen EM, Natarajan P, Emdin CA, Bick AG, Morrison AC, Brody JA, Gupta N, Nomura A, Kessler T, Duga S, Bis JC, van Duijn CM, Cupples LA, Psaty B, Rader DJ, Danesh J, Schunkert H, McPherson R, Farrall M, Watkins H, Lander E, Wilson JG, Correa A, Boerwinkle E, Merlini PA, Ardissino D, Saleheen D, Gabriel S, Kathiresan S. Diagnostic yield and clinical utility of sequencing familial hypercholesterolemia genes in patients with severe hypercholesterolemia. J Am Coll Cardiol. 2016;67:2578–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Williams RR, Hunt SC, Schumacher MC, Hegele RA, Leppert MF, Ludwig EH, Hopkins PN. Diagnosing heterozygous familial hypercholesterolemia using new practical criteria validated by molecular genetics. Am J Cardiol. 1993;72:171–176. [DOI] [PubMed] [Google Scholar]
- 23. Scientific Steering Committee on behalf of the Simon Broome Register Group . Risk of fatal coronary heart disease in familial hypercholesterolaemia. BMJ (Clinical research ed.). 1991;303:893–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. World Health Organization . Familial Hypercholesterolaemia (FH): Report of a second WHO consultation. 1998. Available at: whqlibdoc.who.int/hq/1999/WHO_HGN_FH_CONS_99.2.pdf. Accessed March 29, 2017.
- 25. European Association for Cardiovascular Prevention Rehabilitation , Reiner Z, Catapano AL, De Backer G, Graham I, Taskinen MR, Wiklund O, Agewall S, Alegria E, Chapman MJ, Durrington P, Erdine S, Halcox J, Hobbs R, Kjekshus J, Filardi PP, Riccardi G, Storey RF, Wood D, ESC Committee for Practice Guidelines (CPG) 2008‐2010 and 2010‐2012 Committees. ESC/EAS Guidelines for the management of dyslipidaemias: the Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Eur Heart J. 2011;32:1769–1818. [DOI] [PubMed] [Google Scholar]
- 26. Goldberg AC, Hopkins PN, Toth PP, Ballantyne CM, Rader DJ, Robinson JG, Daniels SR, Gidding SS, de Ferranti SD, Ito MK, McGowan MP, Moriarty PM, Cromwell WC, Ross JL, Ziajka PE. Familial hypercholesterolemia: screening, diagnosis and management of pediatric and adult patients: clinical guidance from the National Lipid Association Expert Panel on Familial Hypercholesterolemia. J Clin Lipidol. 2011;5:S1–S8. [DOI] [PubMed] [Google Scholar]
- 27. Gidding SS, Champagne MA, de Ferranti SD, Defesche J, Ito MK, Knowles JW, McCrindle B, Raal F, Rader D, Santos RD, Lopes‐Virella M, Watts GF, Wierzbicki AS; American Heart Association Atherosclerosis, Hypertension, and Obesity in Young Committee of Council on Cardiovascular Disease in Young, Council on Cardiovascular and Stroke Nursing, Council on Functional Genomics and Translational Biology, and Council on Lifestyle and Cardiometabolic Health . The agenda for familial hypercholesterolemia: a scientific statement from the American Heart Association. Circulation. 2015;132:2167–2192. [DOI] [PubMed] [Google Scholar]
- 28. FH Foundation . FH Diagnosis. 2012. Available at: https://itunes.apple.com/us/app/fh-diagnosis/id543676258?mt=8. Accessed March 14, 2019.
- 29. Wiegman A, Gidding SS, Watts GF, Chapman MJ, Ginsberg HN, Cuchel M, Ose L, Averna M, Boileau C, Boren J, Bruckert E, Catapano AL, Defesche JC, Descamps OS, Hegele RA, Hovingh GK, Humphries SE, Kovanen PT, Kuivenhoven JA, Masana L, Nordestgaard BG, Pajukanta P, Parhofer KG, Raal FJ, Ray KK, Santos RD, Stalenhoef AF, Steinhagen‐Thiessen E, Stroes ES, Taskinen MR, Tybjaerg‐Hansen A, Wiklund O. Familial hypercholesterolaemia in children and adolescents: gaining decades of life by optimizing detection and treatment. Eur Heart J. 2015;36:2425–2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Ito MK, Watts GF. Challenges in the diagnosis and treatment of homozygous familial hypercholesterolemia. Drugs. 2015;75:1715–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Futema M, Plagnol V, Li K, Whittall RA, Neil HA, Seed M; Simon Broome Consortium , Bertolini S, Calandra S, Descamps OS, Graham CA, Hegele RA, Karpe F, Durst R, Leitersdorf E, Lench N, Nair DR, Soran H, Van Bockxmeer FM; UK K Consortium , Humphries SE. Whole exome sequencing of familial hypercholesterolaemia patients negative for LDLR/APOB/PCSK9 mutations. J Med Genet. 2014;51:537–544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Masana L, Ibarretxe D, Rodriguez‐Borjabad C, Plana N, Valdivielso P, Pedro‐Botet J, Civeira F, Lopez‐Miranda J, Guijarro C, Mostaza J, Pinto X. Toward a new clinical classification of patients with familial hypercholesterolemia: one perspective from Spain. Atherosclerosis. 2019;287:89–92. [DOI] [PubMed] [Google Scholar]
- 33. Neil HA, Huxley RR, Hawkins MM, Durrington PN, Betteridge DJ, Humphries SE. Comparison of the risk of fatal coronary heart disease in treated xanthomatous and non‐xanthomatous heterozygous familial hypercholesterolaemia: a prospective registry study. Atherosclerosis. 2003;170:73–78. [DOI] [PubMed] [Google Scholar]
- 34. Humphries SE, Whittall RA, Hubbart CS, Maplebeck S, Cooper JA, Soutar AK, Naoumova R, Thompson GR, Seed M, Durrington PN, Miller JP, Betteridge DJ, Neil HA. Genetic causes of familial hypercholesterolaemia in patients in the UK: relation to plasma lipid levels and coronary heart disease risk. J Med Genet. 2006;43:943–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Perez de Isla L, Alonso R, Mata N, Fernandez‐Perez C, Muniz O, Diaz‐Diaz JL, Saltijeral A, Fuentes‐Jimenez F, de Andres R, Zambon D, Piedecausa M, Cepeda JM, Mauri M, Galiana J, Brea A, Sanchez Munoz‐Torrero JF, Padro T, Argueso R, Miramontes‐Gonzalez JP, Badimon L, Santos RD, Watts GF, Mata P. Predicting cardiovascular events in familial hypercholesterolemia: the SAFEHEART registry (Spanish Familial Hypercholesterolemia Cohort Study). Circulation. 2017;135:2133–2144. [DOI] [PubMed] [Google Scholar]
- 36. Villa G, Wong B, Kutikova L, Ray KK, Mata P, Bruckert E. Prediction of cardiovascular risk in patients with familial hypercholesterolaemia. Eur Heart J Qual Care Clin Outcomes. 2017;3:274–280. [DOI] [PubMed] [Google Scholar]
- 37. Duell PB, Gidding SS, Andersen RL, Knickelbine T, Anderson L, Gianos E, Shrader P, Kindt I, O'Brien EC, McCann D, Hemphill LC, Ahmed CD, Martin SS, Larry JA, Ahmad ZS, Kullo IJ, Underberg JA, Guyton J, Thompson P, Wilemon K, Roe MT, Rader DJ, Cuchel M, Linton MF, Shapiro MD, Moriarty PM, Knowles JW. Longitudinal low density lipoprotein cholesterol goal achievement and cardiovascular outcomes among adult patients with familial hypercholesterolemia: the CASCADE FH registry. Atherosclerosis. 2019;289:85–93. [DOI] [PubMed] [Google Scholar]
- 38. Santos RD, Gidding SS, Hegele RA, Cuchel MA, Barter PJ, Watts GF, Baum SJ, Catapano AL, Chapman MJ, Defesche JC, Folco E, Freiberger T, Genest J, Hovingh GK, Harada‐Shiba M, Humphries SE, Jackson AS, Mata P, Moriarty PM, Raal FJ, Al‐Rasadi K, Ray KK, Reiner Z, Sijbrands EJ, Yamashita S. Defining severe familial hypercholesterolaemia and the implications for clinical management: a consensus statement from the International Atherosclerosis Society Severe Familial Hypercholesterolemia Panel. Lancet Diabetes Endocrinol. 2016;4:850–861. [DOI] [PubMed] [Google Scholar]
- 39. Bell DA, Watts GF. Progress in the care of familial hypercholesterolaemia: 2016. Med J Aust. 2016;205:232–236. [DOI] [PubMed] [Google Scholar]
- 40. Umans‐Eckenhausen MA, Defesche JC, Sijbrands EJ, Scheerder RL, Kastelein JJ. Review of first 5 years of screening for familial hypercholesterolaemia in the Netherlands. Lancet. 2001;357:165–168. [DOI] [PubMed] [Google Scholar]
- 41. Ibarretxe D, Rodriguez‐Borjabad C, Feliu A, Bilbao JA, Masana L, Plana N. Detecting familial hypercholesterolemia earlier in life by actively searching for affected children: the DECOPIN project. Atherosclerosis. 2018;278:210–216. [DOI] [PubMed] [Google Scholar]
- 42. McCrindle BW, Gidding SS. What should be the screening strategy for familial hypercholesterolemia? N Engl J Med. 2016;375:1685–1686. [DOI] [PubMed] [Google Scholar]
- 43. Vuorio A, Watts GF, Kovanen PT. Lipoprotein(a) as a risk factor for calcific aortic valvulopathy in heterozygous familial hypercholesterolemia. Atherosclerosis. 2019;281:25–30. [DOI] [PubMed] [Google Scholar]
- 44. Wiegman A, Rodenburg J, de Jongh S, Defesche JC, Bakker HD, Kastelein JJ, Sijbrands EJ. Family history and cardiovascular risk in familial hypercholesterolemia: data in more than 1000 children. Circulation. 2003;107:1473–1478. [DOI] [PubMed] [Google Scholar]
- 45. Langsted A, Kamstrup PR, Benn M, Tybjaerg‐Hansen A, Nordestgaard BG. High lipoprotein(a) as a possible cause of clinical familial hypercholesterolaemia: a prospective cohort study. Lancet Diabetes Endocrinol. 2016;4:577–587. [DOI] [PubMed] [Google Scholar]
- 46. Kusters DM, Wiegman A, Kastelein JJ, Hutten BA. Carotid intima‐media thickness in children with familial hypercholesterolemia. Circ Res. 2014;114:307–310. [DOI] [PubMed] [Google Scholar]
- 47. Wiegman A, Hutten BA, de Groot E, Rodenburg J, Bakker HD, Buller HR, Sijbrands EJ, Kastelein JJ. Efficacy and safety of statin therapy in children with familial hypercholesterolemia: a randomized controlled trial. JAMA. 2004;292:331–337. [DOI] [PubMed] [Google Scholar]
- 48. Kusters DM, Avis HJ, de Groot E, Wijburg FA, Kastelein JJ, Wiegman A, Hutten BA. Ten‐year follow‐up after initiation of statin therapy in children with familial hypercholesterolemia. JAMA. 2014;312:1055–1057. [DOI] [PubMed] [Google Scholar]
- 49. Versmissen J, Oosterveer DM, Yazdanpanah M, Defesche JC, Basart DC, Liem AH, Heeringa J, Witteman JC, Lansberg PJ, Kastelein JJ, Sijbrands EJ. Efficacy of statins in familial hypercholesterolaemia: a long term cohort study. BMJ. 2008;337:a2423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Luirink IK, Wiegman A, Kusters DM, Hof MH, Groothoff JW, de Groot E, Kastelein JJP, Hutten BA. 20‐Year Follow‐up of Statins in Children with Familial Hypercholesterolemia. N Engl J Med. 2019;381:1547‐1556. [DOI] [PubMed] [Google Scholar]
- 51. Grundy SM, Stone NJ, Bailey AL, Beam C, Birtcher KK, Blumenthal RS, Braun LT, de Ferranti S, Faiella‐Tommasino J, Forman DE, Goldberg R, Heidenreich PA, Hlatky MA, Jones DW, Lloyd‐Jones D, Lopez‐Pajares N, Ndumele CE, Orringer CE, Peralta CA, Saseen JJ, Smith SC Jr, Sperling L, Virani SS, Yeboah J. 2018 AHA/ACC/AACVPR/AAPA/ABC/ACPM/ADA/AGS/APhA/ASPC/NLA/PCNA guideline on the management of blood cholesterol: executive summary: a Report of the American College of Cardiology/American Heart Association task force on clinical practice guidelines. J Am Coll Cardiol. 2019;73:3168–3209. [DOI] [PubMed] [Google Scholar]
- 52. Eiland LS, Luttrell PK. Use of statins for dyslipidemia in the pediatric population. J Pediatr Pharmacol Ther. 2010;15:160–172. [PMC free article] [PubMed] [Google Scholar]
- 53. Bristol‐Myers Squibb Company . Highlights of Prescribing Information. PRAVACHOL (pravastatin sodium) Tablets. 2012. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2012/019898s062lbl.pdf. Accessed February 6, 2019.
- 54. Vuorio A, Kuoppala J, Kovanen PT, Humphries SE, Tonstad S, Wiegman A, Drogari E, Ramaswami U. Statins for children with familial hypercholesterolemia. Cochrane Database Syst Rev. 2017;7:Cd006401. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. van der Graaf A, Cuffie‐Jackson C, Vissers MN, Trip MD, Gagne C, Shi G, Veltri E, Avis HJ, Kastelein JJ. Efficacy and safety of coadministration of ezetimibe and simvastatin in adolescents with heterozygous familial hypercholesterolemia. J Am Coll Cardiol. 2008;52:1421–1429. [DOI] [PubMed] [Google Scholar]
- 56. Davidson M. The efficacy of colesevelam HCl in the treatment of heterozygous familial hypercholesterolemia in pediatric and adult patients. Clin Ther. 2013;35:1247–1252. [DOI] [PubMed] [Google Scholar]
- 57. Lambert CT, Sandesara P, Isiadinso I, Gongora MC, Eapen D, Bhatia N, Baer JT, Sperling L. Current treatment of familial hypercholesterolaemia. European Cardiology. 2014;9:76–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Chou RDT, Blazina I, Daeges M, Bougatsos C, Grusing S, Jeanne TL. Statin Use for the Prevention of Cardiovascular Disease in Adults: A Systematic Review for the U.S. Preventive Services Task Force. Available at: https://www.ncbi.nlm.nih.gov/books/NBK396415/pdf/Bookshelf_NBK396415.pdf. Accessed February 6, 2019. [PubMed]
- 59. Perez‐Calahorra S, Laclaustra M, Marco‐Benedi V, Lamiquiz‐Moneo I, Pedro‐Botet J, Plana N, Sanchez‐Hernandez RM, Amor AJ, Almagro F, Fuentes F, Suarez‐Tembra M, Civeira F. Effect of lipid‐lowering treatment in cardiovascular disease prevalence in familial hypercholesterolemia. Atherosclerosis. 2019;284:245–252. [DOI] [PubMed] [Google Scholar]
- 60. deGoma EM, Ahmad ZS, O'Brien EC, Kindt I, Shrader P, Newman CB, Pokharel Y, Baum SJ, Hemphill LC, Hudgins LC, Ahmed CD, Gidding SS, Duffy D, Neal W, Wilemon K, Roe MT, Rader DJ, Ballantyne CM, Linton MF, Duell PB, Shapiro MD, Moriarty PM, Knowles JW. Treatment gaps in adults with heterozygous familial hypercholesterolemia in the United States: data from the CASCADE‐FH Registry. Circ Cardiovasc Genet. 2016;9:240–249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Cannon CP, Blazing MA, Giugliano RP, McCagg A, White JA, Theroux P, Darius H, Lewis BS, Ophuis TO, Jukema JW. Ezetimibe added to statin therapy after acute coronary syndromes. N Engl J Med. 2015;372:2387–2397. [DOI] [PubMed] [Google Scholar]
- 62. Davidson MH, Dicklin MR, Maki KC, Kleinpell RM. Colesevelam hydrochloride: a non‐absorbed, polymeric cholesterol‐lowering agent. Expert Opin Investig Drugs. 2000;9:2663–2671. [DOI] [PubMed] [Google Scholar]
- 63. Shepherd J, Packard CJ, Morgan HG, Third JL, Stewart JM, Lawrie TD. The effects of cholestyramine on high density lipoprotein metabolism. Atherosclerosis. 1979;33:433–444. [DOI] [PubMed] [Google Scholar]
- 64. Superko HR, Greenland P, Manchester RA, Andreadis NA, Schectman G, West NH, Hunninghake D, Haskell WL, Probstfield JL. Effectiveness of low‐dose colestipol therapy in patients with moderate hypercholesterolemia. Am J Cardiol. 1992;70:135–140. [DOI] [PubMed] [Google Scholar]
- 65. Kastelein JJ, Ginsberg HN, Langslet G, Hovingh GK, Ceska R, Dufour R, Blom D, Civeira F, Krempf M, Lorenzato C, Zhao J, Pordy R, Baccara‐Dinet MT, Gipe DA, Geiger MJ, Farnier M. ODYSSEY FH I and FH II: 78 week results with alirocumab treatment in 735 patients with heterozygous familial hypercholesterolaemia. Eur Heart J. 2015;36:2996–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Ginsberg HN, Rader DJ, Raal FJ, Guyton JR, Baccara‐Dinet MT, Lorenzato C, Pordy R, Stroes E. Efficacy and safety of alirocumab in patients with heterozygous familial hypercholesterolemia and LDL‐C of 160 mg/dl or higher. Cardiovasc Drugs Ther. 2016;30:473–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Kastelein JJ, Robinson JG, Farnier M, Krempf M, Langslet G, Lorenzato C, Gipe DA, Baccara‐Dinet MT. Efficacy and safety of alirocumab in patients with heterozygous familial hypercholesterolemia not adequately controlled with current lipid‐lowering therapy: design and rationale of the ODYSSEY FH studies. Cardiovasc Drugs Ther. 2014;28:281–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Farnier M, Hovingh GK, Langslet G, Dufour R, Baccara‐Dinet MT, Din‐Bell C, Manvelian G, Guyton JR. Long‐term safety and efficacy of alirocumab in patients with heterozygous familial hypercholesterolemia: an open‐label extension of the ODYSSEY program. Atherosclerosis. 2018;278:307–314. [DOI] [PubMed] [Google Scholar]
- 69. Raal F, Scott R, Somaratne R, Bridges I, Li G, Wasserman SM, Stein EA. Low‐density lipoprotein cholesterol‐lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: the Reduction of LDL‐C with PCSK9 Inhibition in Heterozygous Familial Hypercholesterolemia Disorder (RUTHERFORD) randomized trial. Circulation. 2012;126:2408–2417. [DOI] [PubMed] [Google Scholar]
- 70. Raal FJ, Stein EA, Dufour R, Turner T, Civeira F, Burgess L, Langslet G, Scott R, Olsson AG, Sullivan D, Hovingh GK, Cariou B, Gouni‐Berthold I, Somaratne R, Bridges I, Scott R, Wasserman SM, Gaudet D. PCSK9 inhibition with evolocumab (AMG 145) in heterozygous familial hypercholesterolaemia (RUTHERFORD‐2): a randomised, double‐blind, placebo‐controlled trial. Lancet. 2015;385:331–340. [DOI] [PubMed] [Google Scholar]
- 71. Sabatine MS, Giugliano RP, Wiviott SD, Raal FJ, Blom DJ, Robinson J, Ballantyne CM, Somaratne R, Legg J, Wasserman SM, Scott R, Koren MJ, Stein EA; Open‐Label Study of Long‐Term Evaluation against LDL Cholesterol (OSLER) Investigators . Efficacy and safety of evolocumab in reducing lipids and cardiovascular events. N Engl J Med. 2015;372:1500–1509. [DOI] [PubMed] [Google Scholar]
- 72. Schwartz GG, Steg PG, Szarek M, Bhatt DL, Bittner VA, Diaz R, Edelberg JM, Goodman SG, Hanotin C, Harrington RA, Jukema JW, Lecorps G, Mahaffey KW, Moryusef A, Pordy R, Quintero K, Roe MT, Sasiela WJ, Tamby JF, Tricoci P, White HD, Zeiher AM; ODYSSEY OUTCOMES Committees Investigators . Alirocumab and cardiovascular outcomes after acute coronary syndrome. N Engl J Med. 2018;379:2097–2107. [DOI] [PubMed] [Google Scholar]
- 73. Sabatine MS, Giugliano RP, Keech AC, Honarpour N, Wiviott SD, Murphy SA, Kuder JF, Wang H, Liu T, Wasserman SM, Sever PS, Pedersen TR; FOURIER Steering Committee and Investigators . Evolocumab and clinical outcomes in patients with cardiovascular disease. N Engl J Med. 2017;376:1713–1722. [DOI] [PubMed] [Google Scholar]
- 74. Schwartz GG, Szarek MM, Bhatt DL, Bittner VA, Bregeault M‐F, Dalby AJ, Diaz R, Edelberg JM, Goodman SG, Hanotin C, Harrington RA, Jukema JW, Lecorps G, Mahaffey KW, Moryusef A, Ostadal P, Parkhomenko A, Pordy R, Roe MT, Tricoci P, Vogel R, White HD, Zeiher AM, Steg PG. Alirocumab reduces risk of death after acute coronary syndrome in patients with persistently elevated atherogenic lipoproteins on intensive statin treatment. Circulation. 2018;138:. Abstract:20008. [Google Scholar]
- 75. Padmanabhan A, Connelly‐Smith L, Aqui N, Balogun RA, Klingel R, Meyer E, Pham HP, Schneiderman J, Witt V, Wu Y, Zantek ND, Dunbar NM, Schwartz GEJ. Guidelines on the use of therapeutic apheresis in clinical practice—evidence‐based approach from the writing committee of the American Society for Apheresis: the Eighth Special Issue.. J Clin Apheresis. 2019;34:171–354. [DOI] [PubMed] [Google Scholar]
- 76. Current Julius U. Role of lipoprotein apheresis in the treatment of high‐risk patients. J Cardiovasc Dev Dis. 2018;5:E27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Wu MD, Moccetti F, Brown E, Davidson BP, Atkinson T, Belcik JT, Giraud G, Duell PB, Fazio S, Tavori H, Tsimikas S, Lindner JR. Lipoprotein apheresis acutely reverses coronary microvascular dysfunction in patients with severe hypercholesterolemia. JACC Cardiovasc Imaging. 2019;12:1430–1440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Mabuchi H, Koizumi J, Shimizu M, Kajinami K, Miyamoto S, Ueda K, Takegoshi T. Long‐term efficacy of low‐density lipoprotein apheresis on coronary heart disease in familial hypercholesterolemia. Hokuriku‐FH‐LDL‐Apheresis Study Group. Am J Cardiol. 1998;82:1489–1495. [DOI] [PubMed] [Google Scholar]
- 79. Moriarty PM, Parhofer KG, Babirak SP, Cornier MA, Duell PB, Hohenstein B, Leebmann J, Ramlow W, Schettler V, Simha V, Steinhagen‐Thiessen E, Thompson PD, Vogt A, von Stritzky B, Du Y, Manvelian G. Alirocumab in patients with heterozygous familial hypercholesterolaemia undergoing lipoprotein apheresis: the ODYSSEY ESCAPE trial. Eur Heart J. 2016;37:3588–3595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Moriarty PM, Hemphill L. Lipoprotein apheresis. Cardiol Clin. 2015;33:197–208. [DOI] [PubMed] [Google Scholar]
- 81. Moriarty PM. Efficacy and safety of evolocumab compared with continued lipoprotein apheresis: results of a randomised, controlled, open‐label study. late‐breaking poster P1314. Presented at the ESC Congress 2017.
- 82. Nissen SE, Dent‐Acosta RE, Rosenson RS, Stroes E, Sattar N, Preiss D, Mancini GB, Ballantyne CM, Catapano A, Gouni‐Berthold I, Stein EA, Xue A, Wasserman SM, Scott R, Thompson PD. Comparison of PCSK9 inhibitor evolocumab vs ezetimibe in statin‐intolerant patients: design of the goal achievement after utilizing an anti‐pcsk9 antibody in statin‐intolerant subjects 3 (GAUSS‐3) trial. Clin Cardiol. 2016;39:137–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Nissen SE, Stroes E, Dent‐Acosta RE, Rosenson RS, Lehman SJ, Sattar N, Preiss D, Bruckert E, Ceska R, Lepor N, Ballantyne CM, Gouni‐Berthold I, Elliott M, Brennan DM, Wasserman SM, Somaratne R, Scott R, Stein EA. Efficacy and tolerability of evolocumab vs ezetimibe in patients with muscle‐related statin intolerance: the GAUSS‐3 Randomized Clinical Trial. JAMA. 2016;315:1580–1590. [DOI] [PubMed] [Google Scholar]
- 84. Moriarty PM, Thompson PD, Cannon CP, Guyton JR, Bergeron J, Zieve FJ, Bruckert E, Jacobson TA, Kopecky SL, Baccara‐Dinet MT, Du Y, Pordy R, Gipe DA; Investigators ODYSSEYALTERNATIVE . Efficacy and safety of alirocumab vs ezetimibe in statin‐intolerant patients, with a statin rechallenge arm: the ODYSSEY ALTERNATIVE randomized trial. J Clin Lipidol. 2015;9:758–769. [DOI] [PubMed] [Google Scholar]
- 85. Amundsen AL, Khoury J, Iversen PO, Bergei C, Ose L, Tonstad S, Retterstol K. Marked changes in plasma lipids and lipoproteins during pregnancy in women with familial hypercholesterolemia. Atherosclerosis. 2006;189:451–457. [DOI] [PubMed] [Google Scholar]
- 86. Eapen DJ, Valiani K, Reddy S, Sperling L. Management of familial hypercholesterolemia during pregnancy: case series and discussion. J Clin Lipidol. 2012;6:88–91. [DOI] [PubMed] [Google Scholar]
- 87. Botha TC, Pilcher GJ, Wolmarans K, Blom DJ, Raal FJ. Statins and other lipid‐lowering therapy and pregnancy outcomes in homozygous familial hypercholesterolaemia: a retrospective review of 39 pregnancies. Atherosclerosis. 2018;277:502–507. [DOI] [PubMed] [Google Scholar]
- 88. Bateman BT, Hernandez‐Diaz S, Fischer MA, Seely EW, Ecker JL, Franklin JM, Desai RJ, Allen‐Coleman C, Mogun H, Avorn J, Huybrechts KF. Statins and congenital malformations: cohort study. BMJ. 2015;350:h1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Costantine MM, Cleary K, Kennedy Eunice; Shriver National Institute of Child H, Human Development Obstetric–Fetal Pharmacology Research Units Network . Pravastatin for the prevention of preeclampsia in high‐risk pregnant women. Obstet Gynecol. 2013;121:349–353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Costantine MM, Cleary K, Hebert MF, Ahmed MS, Brown LM, Ren Z, Easterling TR, Haas DM, Haneline LS, Caritis SN, Venkataramanan R, West H, D'Alton M, Hankins G; Eunice Kennedy Shriver National Institute of Child Health and Human Development Obstetric‐Fetal Pharmacology Research Units Network . Safety and pharmacokinetics of pravastatin used for the prevention of preeclampsia in high‐risk pregnant women: a pilot randomized controlled trial. Am J Obstet Gynecol. 2016;214:720.e721–720.e717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91. FDA . Pharmacology/Toxicology BLA Review and Evaluation: Application Number: 125522Orig1s000. 2015. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/nda/2015/125522Orig1s000PharmR.pdf. Accessed August 28, 2019.
- 92. US Food and Drug Administration . REPATHA highlights of prescribing information. 2017. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/125522s014lbl.pdf. Accessed August 28, 2019.
- 93. US Food and Drug Administration . PRALUENT highlights of prescribing information. 2015. Available at: https://www.accessdata.fda.gov/drugsatfda_docs/label/2015/125559Orig1s000lbledt.pdf. Accessed August 28, 2019.
- 94. Ray KK, Bays HE, Catapano AL, Lalwani ND, Bloedon LT, Sterling LR, Robinson PL, Ballantyne CM. Safety and efficacy of bempedoic acid to reduce ldl cholesterol. N Engl J Med. 2019;380:1022–1032. [DOI] [PubMed] [Google Scholar]
- 95. Kumbhani DJ. Efficacy and safety of bempedoic acid added to maximally tolerated statins in patients with hypercholesterolemia and high cardiovascular risk—CLEAR wisdom. 2019. Available at: https://www.acc.org/latest-in-cardiology/clinical-trials/2019/03/16/23/43/clear-wisdom. Accessed March 25, 2019.
- 96. Kastelein JJP, Klausen IC, Hovingh GK, Heggen E, Taub R, Langslet G. LDL cholesterol, apolipoprotein B, lipoprotein(a), apolipoprotein CIII and triglyceride lowering by MGL‐3196, a thyroid hormone beta selective agonist, in a 12 week study in HeFH patients. Eur Heart J. 2018;39(suppl 1):ehy566.P5387. [Google Scholar]
- 97. Jakobsson T, Vedin LL, Parini P. Potential role of thyroid receptor beta agonists in the treatment of hyperlipidemia. Drugs. 2017;77:1613–1621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Gaudet D, Gipe DA, Pordy R, Ahmad Z, Cuchel M, Shah PK, Chyu KY, Sasiela WJ, Chan KC, Brisson D, Khoury E, Banerjee P, Gusarova V, Gromada J, Stahl N, Yancopoulos GD, Hovingh GK. ANGPTL3 inhibition in homozygous familial hypercholesterolemia. N Engl J Med. 2017;377:296–297. [DOI] [PubMed] [Google Scholar]
- 99. Gemphire Therapeutics . Gemphire to Present New COBALT‐1 Clinical Data at the 2017 FH Global Summit. 2017. Available at: http://ir.gemphire.com/phoenix.zhtml?c=254241&p=irol-newsArticle&ID=2302595. Accessed February 7, 2019.
- 100. Kosmas CE, Munoz Estrella A, Sourlas A, Silverio D, Hilario E, Montan PD, Guzman E. Inclisiran: a new promising agent in the management of hypercholesterolemia. Diseases (Basel, Switzerland). 2018;6:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Ray KK, Landmesser U, Leiter LA, Kallend D, Dufour R, Karakas M, Hall T, Troquay RP, Turner T, Visseren FL, Wijngaard P, Wright RS, Kastelein JJ. Inclisiran in patients at high cardiovascular risk with elevated ldl cholesterol. N Engl J Med. 2017;376:1430–1440. [DOI] [PubMed] [Google Scholar]
- 102. Stein EATT, Biernat L, Dimova D, Zhou R, Dai M, Daniels C, Mitchell T. Low density lipoprotein cholesterol reduction and safety with LIB003, an anti‐proprotein convertase subtilsin/kexin type 9 fusion protein (PCSK9): results of a randomized, double‐blind, placebo‐controlled, single ascending dose study. J Am Coll Cardiol. 2019;73:1714. [Google Scholar]
- 103. O'Brien EC, Roe MT, Fraulo ES, Peterson ED, Ballantyne CM, Genest J, Gidding SS, Hammond E, Hemphill LC, Hudgins LC, Kindt I, Moriarty PM, Ross J, Underberg JA, Watson K, Pickhardt D, Rader DJ, Wilemon K, Knowles JW. Rationale and design of the familial hypercholesterolemia foundation CAscade SCreening for Awareness and DEtection of Familial Hypercholesterolemia registry. Am Heart J. 2014;167:342–349.e317. [DOI] [PubMed] [Google Scholar]
- 104. FH Foundation . The Familial Hypercholesterolemia Foundation CASCADE‐FH Registry. 2017. Available at: https://thefhfoundation.org/cascade-fh-registry-clinical. Accessed February 6, 2019.