

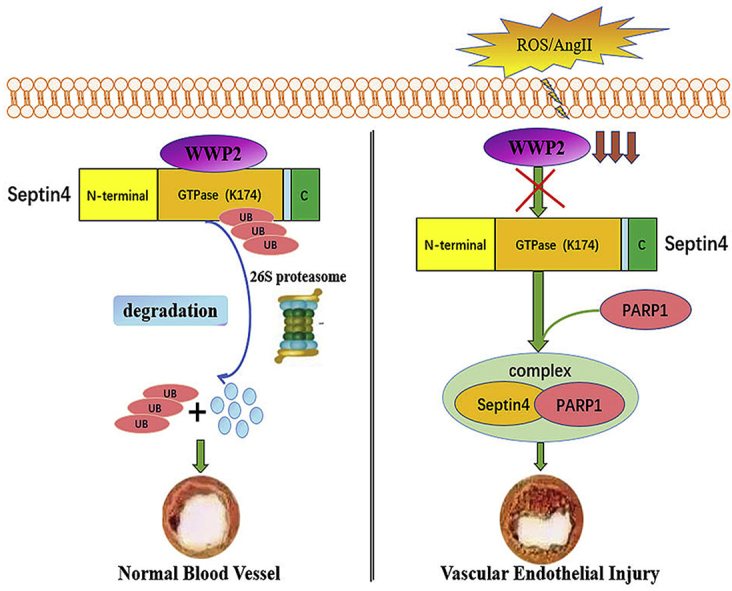
Abstract
Oxidative stress-associated endothelial injury is the initial event and major cause of multiple cardiovascular diseases such as atherosclerosis and hypertensive angiopathy. A protein homeostasis imbalance is a critical cause of endothelial injury, and homologous to E6AP C-terminus (HECT)-type E3 ubiquitin ligases are the core factors controlling protein homeostasis. Although HECT-type E3 ubiquitin ligases are involved in the regulation of cardiac development and diseases, their roles in endothelial injury remain largely unknown. This study aimed to identify which HECT-type E3 ubiquitin ligase is involved in endothelial injury and clarify the mechanisms at molecular, cellular, and organism levels. We revealed a novel role of the HECT-type E3 ubiquitin ligase WWP2 in regulating endothelial injury and vascular remodeling after endothelial injury. Endothelial/myeloid-specific WWP2 knockout in mice significantly aggravated angiotensin II/oxidative stress-induced endothelial injury and vascular remodeling after endothelial injury. The same results were obtained from in vitro experiments. Mechanistically, the endothelial injury factor Septin4 was identified as a novel physiological substrate of WWP2. In addition, WWP2 interacted with the GTPase domain of Septin4, ubiquitinating Septin4-K174 to degrade Septin4 through the ubiquitin-proteasome system, which inhibited the Septin4-PARP1 endothelial damage complex. These results identified the first endothelial injury-associated physiological pathway regulated by HECT-type E3 ubiquitin ligases in vivo as well as a unique proteolytic mechanism through which WWP2 controls endothelial injury and vascular remodeling after endothelial injury. These findings might provide a novel treatment strategy for oxidative stress-associated atherosclerosis and hypertensive vascular diseases.
Keywords: Ubiquitination, Oxidative stress, WWP2, Septin4, Cardiovascular diseases
Abbreviations: HECT, homologous to E6AP C-terminus; ROS, reactive oxygen species; PTM, protein post-translational modification; AngII, angiotensin II; HE, hematoxylin and eosin; HUVECs, Human umbilical vein endothelial cells; DMEM, Dulbecco's modified Eagle medium; FBS, fetal bovine serum; SD, standard deviation; CHX, cycloheximide
Graphical abstract
Highlights
-
•
WWP2 but not WWP1 is involved in oxidative stress vascular endothelial injury.
-
•
Tek Cre+;WWP2FL/FL mice significantly aggravates hypertensive angiopathy.
-
•
Oxidative stress injury factor Septin4 is a novel physiological substrate of WWP2.
-
•
WWP2 degrades Septin4 through the ubiquitin-proteasome system.
-
•
Lysine 174 is of critical importance of Septin4 ubiquitination by WWP2.
1. Introduction
Cardiovascular diseases are one of the major threats to human health, representing the number one killer worldwide. Oxidative stress including accumulation of reactive oxygen species (ROS) plays a critical role as a pathological factor for endothelial injury that constitutes the initial event and major cause of multiple cardiovascular diseases such as atherosclerosis and hypertensive vascular diseases [[1], [2], [3]]. Post-translational modification of proteins represents an important regulatory mechanism in cardiovascular diseases, in which ubiquitination controls protein stabilization and transport as well as protein-protein interactions through a chain-like polyubiquitin reaction [[4], [5], [6], [7], [8]].
E3 ubiquitin ligases mainly include RING-type and HECT-type ligases that specifically recognize downstream substrates and are the core factors in ubiquitination. HECT-type E3 ubiquitin ligases catalyze Lys ubiquitination in many proteins, which is essential for protein homeostasis and cell signal transduction [9,10]. Recent findings have revealed that HECT-type E3 ubiquitin ligases are involved in the regulation of myocardial injury and heart failure [[11], [12], [13]]. However, no report has assessed HECT-type E3 ubiquitin ligases in endothelial injury. Therefore, it is extremely important to evaluate the involvement of HECT-type E3 ubiquitin ligases in endothelial injury and explore the underlying molecular mechanisms, which may provide new strategies for the prevention and treatment of atherosclerosis and hypertensive vascular diseases.
We found that the HECT-type E3 ubiquitin ligase WWP2, a major NEDD4 family member, but not WWP1, another member of the NEDD4 family, was involved in endothelial injury and vascular remodeling after endothelial injury as a novel regulatory factor. Endothelial/myeloid-specific WWP2 knockout in mice significantly aggravated angiotensin II (AngII)-induced endothelial injury and vascular remodeling after endothelial injury, which was consistent with in vitro findings. A new physiological substrate of WWP2 was found while investigating the underlying mechanism, namely the endothelial injury factor Septin4 [14]. We found that WWP2 interacted with the GTPase domain of Septin4 and promoted Septin4 degradation via the ubiquitin-proteasome pathway. We also clarified that Septin4 lysine residue 174 (K174) was the key ubiquitination site modified by WWP2. This inhibited formation of the Septin4-PARP1 endothelial injury complex. The present study revealed the roles of WWP2 and Septin4 at molecular, cellular, and organism levels, and the WWP2-Septin4 axis may be a novel target for the prevention and treatment of atherosclerosis and hypertensive vascular diseases.
2. Materials and methods
2.1. Establishment of endothelial/myeloid-specific WWP2 knockout mice
Conditional endothelial/myeloid WWP2 knockout mice (Tek Cre+;WWP2FL/FL) and Tek Cre-;WWP2FL/FL mice were established by the Shanghai Biomodel Organism Science & Technology Development. Endothelial/myeloid WWP2 knockout mice were confirmed by western blotting (Fig. 1B and C) and detailed Tek Cre+;WWP2FL/FL and Tek Cre-;WWP2FL/FL mouse information is shown in Fig. 1A. All animals were maintained under pathogen-free conditions. Experiments were performed using 8–10-week-old male mice. For NaCl and AngII (A9525, Sigma, USA) infusion models, Tek Cre+;WWP2FL/FL and Tek Cre-;WWP2FL/FL mice were implanted with osmotic minipumps (model 2002; Alzet), according to the manufacturer instructions. Isoflurane inhalation was first used to anesthetize the mice. An incision was made in the middle scapular region, and an osmotic minipump was implanted subcutaneously into the back of the mouse. Mice were infused with NaCl or AngII (1.5 mg/kg/day) for 14 days at 0.5 μL/h. The mice were divided into four groups, Tek Cre-;WWP2FL/FL with NaCl (nine mice), Tek Cre-;WWP2FL/FL with AngII (nine mice), Tek Cre+;WWP2FL/FL with NaCl (nine mice), and Tek Cre+;WWP2FL/FL with AngII (nine mice), with a total of 36 mice. Before sampling, the mice were anesthetized with isoflurane and then sacrificed by neck off. Blood pressure was measured daily by the tail-cuff method. Endothelial/myeloid WWP2 knockout in vivo at the study endpoint was assessed by western blotting. All animal handling complied with animal welfare regulations of China Medical University. The Animal Subject Committee of China Medical University approved the animal study protocol (permission number: 2019001).
Fig. 1.

Endothelial/myeloid-specific WWP2 knockout in mice significantly aggravates AngII-induced hypertensive vascular oxidative stress. (A) Establishment scheme of Tek Cre+;WWP2FL/FL and Tek Cre-;WWP2FL/FL mice. (B) Total protein was obtained from blood vessel tissues of Tek Cre+;WWP2FL/FL and Tek Cre-;WWP2FL/FL mice following NaCl (vehicle) or AngII infusions for 2 weeks. Western blot analyses were then performed to assess WWP2 expression levels. (C) Quantification of data is shown as means ± SD (n = 9 mice per group; ***P < 0.001, unpaired Student's t-test; ###P < 0.001, two-way ANOVA with Bonferroni's post-hoc test). (D) Western blot analyses were carried out to assess 3-nitrotyrosine, OGG1, and SOD1 expression levels. (E) Quantification of data is shown as means ± SD (n = 9 mice per group; ***P < 0.001, unpaired Student's t-test; ###P < 0.001, two-way ANOVA with Bonferroni's post-hoc test).
2.2. MicroCT and 3D reconstruction
Micro-computed tomography (microCT-Imaging skycan 1276, Bruker, Germany) was performed at 70 kV (200 μA), acquiring 1237 projections (1520 × 1264) in 6 min 43 s with tubes continuously rotating. Angiograms were obtained in 20 × 20 × 20 μm3 voxels by DataViewer software (Bruker) with correction for ring artefacts. After image reconstruction, data visualization was carried out using NRecon software (Bruker), and CTAn software (Bruker) was employed for further assessment. Upon 3D spine segmentation by interactive delineation of the aorta in 100 and 200 slices (2 and 4 mm, respectively), the artery and vein circumference were assessed by the mean heart tissue brightness following contrast agent injection into an artery and precontrast agent injection set at 100% and 0%, respectively [15].
2.3. Immunohistochemical analysis
Mouse vascular tissues were immersed in 4% paraformaldehyde for 4 h and then transferred to 70% ethanol. Individual lobes of the tissues were placed in processing cassettes, dehydrated through a serial alcohol gradient, and then embedded in paraffin. Before immunostaining, 5 μm-thick vascular tissue sections were dewaxed with xylene, rehydrated through decreasing concentrations of ethanol, washed in PBS, and then stained with hematoxylin and eosin (HE) and a Masson's Trichrome Stain Kit (G1340, Solarbio, China). After staining, the sections were dehydrated through increasing concentrations of ethanol and xylene.
2.4. Cell culture, transfection, and immunoprecipitation
Human umbilical vein endothelial cells (HUVECs) were obtained from Cambrex (China Center for Type Culture Collection, Wuhan, China) and cultured in Dulbecco's modified Eagle's medium (DMEM) (HyClone, Logan, UT, USA) with 10% fetal bovine serum (FBS) (HyClone) at 37 °C in a humidified atmosphere with 5% CO2. The HUVECs were used at passage 4–6 for experiments. Plasmid transfections were carried out using Lipofectamine 3000 (Invitrogen, California, USA), according to the manufacturer's instructions (plasmid/transfection reagent = 1 μg/2.4 μl). For immunoprecipitation, cells were washed twice and lysed with flag lysis buffer (50 mM Tris, 137 mM NaCl, 1 mM EDTA, 10 mM NaF, 0.1 mM Na3VO4, 1% NP-40, 1 mM DTT, and 10% glycerol, pH 7.8) containing fresh protease inhibitors. Cell lysates were incubated with an antibody (antibody/cell lysates = 1 μg/mg) for 3 h and then incubated with 30 μl Protein A/G immunoprecipitation magnetic beads (B23202, Biotool, USA) for 12 h at 4 °C or 30 μl anti-Flag Affinity Gel (B23102, Biotool) for 12 h at 4 °C. Bound complexes were washed with flag lysis buffer and subjected to SDS-PAGE.
2.5. WWP2 knockdown in HUVECs
Control and WWP2 siRNAa were obtained from RIBOBIO (China). WWP2 knockdown was carried out using jetPRIME transfection reagent obtained from PolyPlus (France) (siRNA/transfection reagent = 12.5 pmol/μl). For WWP2 gene targeting, we used three effective target sequences to exclude off-target effects. The efficiency of WWP2 knockdown was verified by western blot analysis. The target sequences were as follows.
WWP2 siRNA-1: GATCTGGGAAATGTGCCTA
WWP2 siRNA-2: GGTGCTTCAGCCAGAACAA
WWP2 siRNA-3: CGGACGTGTCTATTATGTT
2.6. Plasmid construction
Plasmids encoding full-length human WWP2 (China Shanghai, Genechem) were cloned into Flag and HA-tagged destination vectors, and ubiquitin (Genechem) was cloned into a HA-tagged destination vector for immunoprecipitation or immunoblotting. Full-length Septin4 and lysine site-mutated Septin4 (K174R) were cloned into the Flag vector (GeneChem). Four truncated Septin4 plasmids containing different domains were constructed: N-terminal and GTPase domains with a Flag-tag; N-terminal domain alone with a Flag-tag; C-terminal domain alone with a Flag-tag; C-terminal and GTPase domains with a Flag-tag.
2.7. Antibodies and reagents
Antibodies included polyclonal goat anti-Septin4 (1:1000; abcam, USA), polyclonal rabbit anti-WWP2 (1:1000; abcam; 1:500; Proteintech, USA), monoclonal rabbit anti-HA (1:1000; Cell Signaling Technology, USA), monoclonal rabbit anti-Myc (1:1000; Cell Signaling Technology), monoclonal rabbit anti-Flag (1:1000; abcam), polyclonal rabbit anti-caspase-3 (1:1000; Cell Signaling Technology), monoclonal rabbit anti-PARP1 (1:1000; Cell Signaling Technology), polyclonal rabbit anti-collagen I (1:1000; Proteintech), polyclonal rabbit anti-aerfa5-SMA (1:1000; Proteintech), polyclonal rabbit anti-OGG1 (1:500; Proteintech), polyclonal rabbit anti-SOD1 (1:500; Proteintech), monoclonal mouse anti-3-nitrotyrosine (1:1000; abcam), monoclonal mouse anti-β-tubulin (1:2000; abcam), and monoclonal mouse anti-GAPDH (1:2000; abcam). MG132 was reconstituted in DMSO (10 μM), which was obtained from Biotool (USA). CHX was obtained from Biotool and used at 10 μM in DMSO.
2.8. Ubiquitination assay
HUVECs and 293T cells were lysed in 1% SDS buffer (Tris, pH 7.5, 0.5 mM EDTA, and 1 mM DTT) and boiled for 10 min. The cell lysates (500 μl) were incubated with the anti-Septin4 antibody (antibody/cell lysate = 1 μg/mg) for 3 h and then incubated with 30 μl of Protein A/G immunoprecipitation magnetic beads for 12 h at 4 °C or 30 μl of anti-Flag Affinity Gel for 12 h at 4 °C. Analyses of Septin4 or Septin4 (K174R) ubiquitination were performed by anti-HA immunoblotting.
2.9. Cell viability, apoptosis, and ROS assays
A Cell Counting Kit-8 assay (CCK-8; Dojindo, Kumamoto, Japan) was used to estimate cell viability. In brief, HUVECs were seeded onto 96-well plates (NEST Biotechnology) at 5 × 103 cells/well in DMEM supplemented with 10% FBS and subsequently transfected with control or Flag-WWP2 plasmids using Lipofectamine 3000 (plasmid/transfection reagent = 1 μg/2.4 μl) or control-siRNA or WWP2 siRNAs using jetPRIME (PolyPlus) (siRNA/transfection reagent = 12.5 pmol/μl). After H2O2 exposure, 100 μl CCK-8 solution was added to each well, followed by incubation for 2 h. Cell viability (optical density) was measured at 450 nm by a Bio-Rad microplate reader (Model 680; Bio-Rad Laboratories, Inc., Hercules, CA, USA). An annexin V-fluorescein isothiocyanate (FITC)/PI assay (Beyotime Institute of Biotechnology; cat. no. C1062) was performed to detect apoptosis by flow cytometric analysis, according to the manufacturer's instructions. Briefly, the cells were cultured in 6-well plates (NEST, USA) at 5 × 105 cells/well in DMEM with 10% FBS. Following the various group transfections for 36 h, HUVECs were exposed to H2O2 (500 μmol/l) or PBS for 12 h. The cells were collected and resuspended in 500 μl binding buffer with 5 μL annexin-V and 5 μL PI solutions, followed by incubation for 15 min at room temperature in the dark. Finally, a FACSCalibur flow cytometer was used to analyze the cells. Annexin V-FITC and PI detection were conducted using FL-1 and FL-2 channels, respectively. Intracellular ROS was detected using ROS-mediated conversion of non-fluorescent 2,7-DCFH-DA (1 μl/ml; 10 μM) to fluorescent DCFH, following the various group transfections for 36 h. HUVECs were exposed to H2O2 (500 μmol/l) or PBS for 12 h. Then DCFH was excited at 488 nm and emission was measured at 525 nm by fluorescence microscopy.
2.10. Western blotting
After incubating as described above, to extract proteins, cells were lysed in Co-IP buffer and then centrifuged at 13300 rpm for 20 min at 4 °C. Protein concentrations were determined by the BCA protein assay (Dingguo Changsheng Biotechnology, China) and 50 μg for each sample. After blocking with 5% bovine serum albumin in Tris buffered saline Tween at room temperature for 1 h, membranes were incubated overnight at 4 °C with the corresponding antibody. Data were normalized to the GAPDH or β-Tubulin content of the same sample and analyzed by ImageJ software version 1.46 (National Institutes of Health, USA). At least three blots were used for quantification and quantified data are shown as means ± standard deviation (SD).
2.11. Statistical analysis
Data are presented as means ± SD. Homogeneity of variance was evaluated by the F-test (group pair) or Brown-Forsythe test (multiple groups). The Shapiro-Wilk test was used to assess data normality. Student's and Welch's t-tests were employed to assess data of group pairs with normal and skewed distributions, respectively (two groups). One-way and two-way ANOVAs were performed to compare groups for single and two factors, respectively. P-values were adjusted for multiple comparisons when applicable. All data were analyzed by SPSS 22.0 (SPSS, USA). P < 0.05 was considered as statistically significant.
3. Results
3.1. Endothelial/myeloid WWP2 knockout in mice significantly aggravates AngII-induced hypertensive vascular oxidative stress
To investigate the roles of WWP-E3-ubiquitinated ligases in AngII-induced hypertensive endothelial injury and vascular remodeling after endothelial injury, AngII was used to induce hypertensive angiopathy in C57B6/L mice in vivo. Upon hypertensive angiopathy in mice, the levels of WWP2, but not those of WWP1, were decreased significantly (Supplementary Fig. S1). These results demonstrated that WWP2, but not WWP1, may be involved in hypertensive endothelial injury and vascular remodeling after endothelial injury in vivo.
To further assess the role of WWP2 in hypertensive endothelial injury and vascular remodeling after endothelial injury in vivo, endothelial/myeloid-specific WWP2 knockout mice (Tek Cre+;WWP2FL/FL) were generated. Tek Cre+;WWP2FL/FL and Tek Cre-;WWP2FL/FL mice were subjected to NaCl or AngII infusion via osmotic minipumps for 2 weeks. Then, WWP2 protein expression was examined by western blotting, confirming establishment of Tek Cre+;WWP2FL/FL mice with scrambled Tek Cre-;WWP2FL/FL mice used as controls (Fig. 1A–C).
Hypertensive vascular oxidative stress can lead to injury of endothelial cells, which is crucial for hypertensive angiopathy and atherosclerosis. Therefore, we determined the role of WWP2 in hypertensive vascular oxidative stress induced by AngII. Vascular oxidative stress markers, including 3-nitrotyrosine, OGG1, and SOD1, were detected by western blotting. Compared with Tek Cre-;WWP2FL/FL mice, Tek Cre+;WWP2FL/FL mice had markedly elevated oxidative stress markers 3-nitrotyrosine and OGG1 (Fig. 1D, E) and a significantly decreased antioxidant marker in SOD1 (Fig. 1D and E). These data suggested that WWP2 was involved in the prevention of hypertensive vascular oxidative stress as a protective factor in mice.
3.2. Endothelial/myeloid-specific WWP2 knockout in mice significantly aggravates AngII-induced hypertensive angiopathy
To quantitatively assess hypertensive vascular remodeling after endothelial injury, contrast-enhanced microCT was employed for animal imaging. The 3D micromorphological properties of blood vessels were visualized in Tek Cre+;WWP2FL/FL and Tek Cre-;WWP2FL/FL mice administered AngII for 2 weeks by a longitudinal microCT scan series. The blood vessel perimeter was simultaneously quantified with a contrast solution containing iodine as described previously [20]. Via 2D transversal, sagittal and coronal microCT images, and 3D reconstruction, in vivo microCT allowed characterization of the blood vessel perimeter with a 20 μm voxel size as the spatial resolution. Selected images and videos revealed that vascular endothelial injury resulted in a larger blood vessel perimeter in comparison with the vehicle control group. Accordingly, blood vessel perimeters were markedly greater in mice administered AngII for 2 weeks compared with control mice (P < 0.05) (Fig. 2), confirming that vascular remodeling paralleled the blood vessel perimeter increase. The vascular cross-sections and morphological features of mice in each group were examined (Fig. 2A), and three-dimensional structures were reconstructed (Fig. 2B). Next, vascular circumferences were measured. Comparison between AngII-treated Tek Cre+;WWP2FL/FL and Tek Cre-;WWP2FL/FL mice showed that WWP2 knockout significantly aggravated hypertensive vascular remodeling in mice (Fig. 2C and D). Furthermore, endothelial/myeloid WWP2 knockout further aggravated the increased diastolic blood pressure induced by AngII in mice (Fig. 2E). These results revealed that WWP2 was involved in preventing hypertensive angiopathy as a protective factor in mice.
Fig. 2.

Endothelial/myeloid WWP2 knockout in mice significantly aggravates AngII-induced hypertensive angiopathy. (A) Aortic blood vessel detection by microCT after administration of a blood pool contrast solution containing iodine (eXIA 160XL), which allowed a spatial resolution of 20 μm voxels in 2D cross-sectional images of Tek Cre-;WWP2FL/FL mice, delineating from the heart precontrast agent injection (0%) to the aorta boundaries in 100 slices (I) and Tek Cre-;WWP2FL/FL mice in 200 slices (II), AngII-treated Tek Cre-;WWP2FL/FL mice in 100 slices (III) and AngII-treated Tek Cre-;WWP2FL/FL mice in 200 slices (IV), Tek Cre+;WWP2FL/FL mice in 100 slices (V) and Tek Cre+;WWP2FL/FL mice in 200 slices (VI), AngII-treated Tek Cre+;WWP2FL/FL mice in 100 slices (VII) and AngII-treated Tek Cre+;WWP2FL/FL mice in 200 slices (VIII). (B) Plane and selected 3D reconstruction images were used to analyze the vascular thickness in Tek Cre-;WWP2FL/FL mice (I-II), AngII-treated Tek Cre-;WWP2FL/FL mice (III-IV), Tek Cre+;WWP2FL/FL mice (V-VI), and AngII-treated Tek Cre+;WWP2FL/FL mice (VII-VIII). (C) Artery circumference and (D) vein circumference in Tek Cre-;WWP2FL/FL and Tek Cre+;WWP2FL/FL mice were determined by in vivo microCT. Data are means ± SD (n = 9 mice per group; **P < 0.01, *P < 0.05, unpaired Student's t-test; #P < 0.05, two-way ANOVA with Bonferroni's post-hoc test). (E) Diastolic blood pressure and (F) systolic blood pressure were measured in Tek Cre+;WWP2FL/FL and Tek Cre-;WWP2FL/FL mice. Data are means ± SD (n = 9 mice per group; ***P < 0.001, unpaired Student's t-test; #P < 0.05, two-way ANOVA with Bonferroni's post-hoc test).
3.3. Endothelial/myeloid-specific WWP2 knockout in mice significantly aggravates hypertensive vascular thickening and fibrosis
Hypertensive endothelial injury induces vascular thickening and fibrosis. Therefore, we assessed the role of WWP2 in vascular thickening and fibrosis induced by Ang II. H&E staining was employed to compare changes in the vessel wall thickness between AngII-treated Tek Cre+;WWP2FL/FL and Tek Cre-;WWP2FL/FL mice. The results showed that WWP2 knockout significantly enhanced vessel wall thickness (Fig. 3A and B). Subsequently, Masson staining was carried out to compare changes in the degree of fibrosis between AngII-treated Tek Cre+;WWP2FL/FL and Tek Cre-;WWP2FL/FL mice. The results suggested that WWP2 knockdown aggravated fibrosis significantly (Fig. 3C and D).
Fig. 3.

Endothelial/myeloid WWP2 knockout in mice aggravates AngII-induced vascular thickening and fibrosis. (A) H&E and (C) Masson's trichrome staining were carried out to assess vascular thickening and fibrosis of vascular tissue specimens from Tek Cre+;WWP2FL/FL and Tek Cre-;WWP2FL/FL mice following NaCl (vehicle) or AngII infusions for 2 weeks. Data (B and D) are means ± SD (n = 9 mice per group; ***P < 0.001, unpaired Student's t-test; ##P < 0.01, two-way ANOVA with Bonferroni's post-hoc test). (E) Western blot analyses were carried out to assess α-SMA and col-1 expression levels. (F) Quantification of immunoblot results are shown as means ± SD (n = 9 mice per group; ***P < 0.001, unpaired Student's t-test; ###P < 0.001, two-way ANOVA with Bonferroni's post-hoc test). (G) Western blot analyses were performed to assess Septin4, cleaved PARP1, and cleaved caspase-3 expression levels. (H) Quantification of immunoblot results are shown as means ± SD (n = 9 mice per group; ***P < 0.001, unpaired Student's t-test; ###P < 0.001, ##P < 0.01, two-way ANOVA with Bonferroni's post-hoc test).
In addition, we assessed whether vascular thickening and fibrosis were accompanied by expression changes of injury-related proteins, including Septin4, cleaved PARP1, and cleaved caspase-3, as well as fibrosis-associated proteins including α-SMA and col-1. Compared with Tek Cre-;WWP2FL/FL mice, Tek Cre+;WWP2FL/FL mice had markedly elevated fibrosis makers α-SMA and col-1 (Fig. 3E and F) and damage-related proteins Septin4, cleaved PARP1 and cleaved caspase-3 (Fig. 3G and H). The above data suggested that WWP2 was involved in the prevention of hypertensive vascular wall thickening and fibrosis as a protective factor in mice.
Taken together, the above data confirmed that WWP2 endothelial/myeloid knockout in mice significantly aggravated AngII-induced vascular ROS production (Fig. 1), vascular remodeling (Fig. 2), vascular wall thickening (Fig. 3), and vascular fibrosis (Fig. 3) after vascular injury, and increased the systolic blood pressure level (Fig. 2).
3.4. WWP2 interacts with Septin4 via its GTPase domain
To explore the mechanisms of WWP2 in endothelial injury, we assessed the interaction between WWP2 and Septin4 that promotes endothelial injury by interacting with PARP1 [14]. First, co-immunoprecipitation of endogenous proteins was used to confirm the interaction between endothelial injury-associated protein Septin4 and WWP2 (Fig. 4A). Next, HA-tagged WWP2 and Flag-Septin4 were used to confirm the interaction between Septin4 and WWP2 by co-immunoprecipitation exogenously (Fig. 4B).
Fig. 4.

WWP2 interacts with Septin4 through the Septin4-GTPase domain. Co-immunoprecipitation was performed to assess the interaction between WWP2 and Septin4. (A) Endogenous Septin4 coimmunoprecipitated with endogenous WWP2, (B) as did HA-tagged WWP2 and Flag-Septin4. This interaction between WWP2 and Septin4 was detected by co-immunoprecipitation with the addition of 500 μmol/L H2O2 (C) and 1 × 10−6 mol/L AngII (D). Co-immunoprecipitation was performed to assess the interaction between WWP2 and the domains of Septin4. (E) Schematic representation of the functional domains of Septin4 and (F) the corresponding Flag-Septin4 fusion constructs used to assess binding of WWP2.
Considering the interaction of WWP2 with Septin4, whether AngII/oxidative stress affects the WWP2-Septin4 interaction was investigated further. Endothelial cells (HUVECs) were incubated in the presence or absence of AngII/H2O2 to induce endothelial injury. Co-immunoprecipitation results showed that AngII/H2O2 increased binding between Septin4 and WWP2 in HUVECs (Fig. 4C and D). The above findings suggested that WWP2 was involved in endothelial injury by interacting with Septin4.
To further clarify which domain of Septin4 interacted with WWP2 and determine modification sites of Septin4 by WWP2, Septin4 was divided into three functional domains, namely N-terminal, C-terminal, and GTPase domains (Fig. 4E). We then performed co-immunoprecipitation to analyze the interaction between endogenous WWP2 and full-length Flag-tagged Septin4 or various Flag-Septin4 truncated mutants. The results showed that WWP2 mainly bound to the GTPase domain of Septin4 (Fig. 4F).
3.5. WWP2 participates in degradation of Septin4 by the proteasome-dependent pathway
To clarify whether WWP2 affects the expression of Septin4, the role of WWP2 in Septin4 stability was examined further. WWP2 was exogenously overexpressed in a gradient. With increasing expression of WWP2, the expression of Septin4 showed a gradual decrease (Fig. 5A and B). Then, siRNA-WWP2 was transfected exogenously to examine Septin4 expression. To prevent off-target effects, three WWP2-siRNA sequences were designed. After HUVEC transfection with control-siRNA, WWP2-siRNA-1, WWP2-siRNA-2 or WWP2-siRNA-3, the knockdown efficiencies of the three WWP2-siRNA sequences were examined by western blotting. The results showed that the three sequences significantly reduced WWP2 expression, but markedly increased Septin4 levels (Fig. 5C and D). Therefore, WWP2 reduces Septin4 protein levels.
Fig. 5.

WWP2 is involved in proteasome-dependent Septin4 degradation. (A) Overexpression of WWP2 and assessment of Septin4 expression. (B) Relative protein amounts are shown as means ± SD (***P < 0.001, one-way ANOVA with Bonferroni's post-hoc test). (C) Three target sequences of siRNA-WWP2 were transfected to assess the efficiency of WWP2 silencing and Septin4 expression. (D) Relative protein amounts are shown as means ± SD (***P < 0.001, *P < 0.05, unpaired Student's t-test). (E) Silenced WWP2 gene expression was used to assess the effect of WWP2 on Septin4 expression at various times after CHX administration. (F) Relative protein amounts are shown as means ± SD (**P < 0.01, two-way ANOVA with Bonferroni's post-hoc test). (G) Silenced WWP2 gene expression was used to assess the effect of WWP2 on Septin4 expression at various times after MG132 administration. (H) Relative protein amounts are shown as means ± SD (**P < 0.01, two-way ANOVA with Bonferroni's post-hoc test).
To examine whether WWP2 reduced Septin4 protein levels by inhibiting transcription of the Septin4 gene, a time-gradient treatment with transcription inhibitor cycloheximide (CHX) was performed to inhibit Septin4 gene transcription after transfection with control-siRNA or WWP2-siRNAs. Compared with the WWP2-siRNA group, the control-siRNA group displayed markedly reduced Septin4 protein levels (Fig. 5E and F), suggesting that WWP2 did not affect transcription of the Septin4 gene.
Furthermore, to examine whether WWP2 reduced Septin4 protein levels by promoting Septin4 protein degradation via the proteasome pathway, a time-gradient treatment with proteasome inhibitor MG132 was performed to inhibit Septin4 protein degradation after transfection with control-siRNA or WWP2-siRNAs. The data demonstrated that Septin4 protein levels increased over time after MG132 administration in the control-siRNA group. However, Septin4 protein was maintained at higher levels after MG132 administration in the WWP2-siRNA group (Fig. 5G and H). These findings suggested that WWP2 was dependent on the proteasome pathway to decrease Septin4 protein levels.
3.6. WWP2 ubiquitinates Septin4 via the K174 site
To determine whether WWP2-induced Septin4 degradation by the proteasome was dependent on polyubiquitination, HA-tagged WWP2 and Flag-tagged Septin4 were overexpressed in the presence or absence of proteasome inhibitor MG132. An exogenous co-immunoprecipitation assay showed that binding between WWP2 and Septin4 was enhanced in the absence of MG132 (Fig. 6A). Moreover, an endogenous co-immunoprecipitation assay confirmed enhancement of binding between WWP2 and Septin4 under the action of MG132 (Fig. 6B). Furthermore, after exogenous overexpression of HA-tagged WWP2, Flag-tagged Septin4, and HA-UB in the presence of MG132, changes in Septin4 ubiquitination levels were detected by co-immunoprecipitation. The results showed that Septin4 ubiquitination levels were increased significantly after WWP2 overexpression (Fig. 6C). In addition, Flag-tagged Septin4 and HA-UB were overexpressed simultaneously in normal and WWP2 knockdown cells. As a result, Septin4 ubiquitination levels were decreased significantly after knockdown of WWP2 (Fig. 6D). These findings suggested that WWP2-induced Septin4 degradation by the proteasome was dependent on polyubiquitination.
Fig. 6.

Degradation of Septin4 by WWP2 occurs via the ubiquitin-proteasome pathway at Septin4-K174. (A) Exogenous HA-WWP2 co-immunoprecipitated with exogenous Flag-Septin4 after treatment with MG132. (B) Endogenous WWP2 coimmunoprecipitated with endogenous Septin4 after treatment with MG132. (C) Flag-Septin4, HA-WWP2, and HA-Ub were coexpressed with or without MG132 treatment. Flag-Septin4 purification was performed by IP. Septin4 ubiquitination levels were assessed with anti-HA antibodies. (D) Flag-Septin4, siRNA-WWP2, and HA-Ub were coexpressed with or without MG132 treatment. Flag-Septin4 purification was carried out by IP. Septin4 ubiquitination levels were assessed with anti-HA antibodies. (E) HA-WWP2, HA-Ub, and Flag-WT-Septin4 or Flag-K174R-Septin4 were coexpressed and Flag-Septin4 purification was carried out by IP. Septin4 ubiquitination levels were assessed with anti-HA antibodies.
To further clarify the ubiquitination sites of Septin4 by WWP2 and provide the most direct evidence for ubiquitination of Septin4 by WWP2, we screened the ubiquitination sites according to the interaction domain of Septin4 and WWP2. The potential ubiquitination site of Septin4- (K174) was assessed after K174 mutation (K-R). Wildtype Septin4 and K174R-Septin4 were overexpressed exogenously. Co-immunoprecipitation assays confirmed that overexpression of K174R-Septin4 significantly decreased the ubiquitination levels of Septin4 compared with the wildtype group (Fig. 6E). These results confirmed that WWP2 regulated Septin4 polyubiquitination through the K174 site and promoted Septin4 degradation by the proteasome pathway.
3.7. WWP2 inhibits the endothelial injury complex Septin4-PARP1 to reduce endothelial cell injury
Because Septin4 interacts with PARP1 to form the endothelial injury complex [14], we next clarified whether WWP2 degrades Septin4 and reduces the interaction between Septin4 and PARP1, thereby relieving endothelial cell injury. We found that WWP2 overexpression significantly decreased the interaction between Septin4 and PARP1, whereas WWP2 knockdown significantly increased the interaction between Septin4 and PARP1 by endogenous and exogenous co-immunoprecipitation (Fig. 7A and B).
Fig. 7.

WWP2 inhibits the endothelial injury complex Septin4-PARP1 and alleviates endothelial cells injury. (A) HUVECs underwent transfection with negative control or WWP2 siRNAs for 36 h and then an interaction between PARP1 and Septin4 was assessed by co-immunoprecipitation. (B) HUVECs underwent transfection with control-HA-tagged or HA-WWP2, Myc-PARP1 and Flag-Septin4 for 36 h. The interaction between PARP1 and Septin4 was then assessed by co-immunoprecipitation. (C) HUVECs underwent transfection with Flag-control or Flag-WWP2 for 36 h and then treated with 500 μmol/L H2O2 for 12 h. Cleaved PARP1, cleaved caspase-3, and Septin4 were detected by western blotting. (D) Quantitated data are shown as means ± SD (***P < 0.001, unpaired Student's t-test; ##P < 0.01, #P < 0.05, two-way ANOVA with Bonferroni's post-hoc test). (E) HUVECs underwent transfection with negative control or WWP2 siRNAs for 36 h and were then treated with 500 μmol/L H2O2 for 12 h. Cleaved PARP1, cleaved caspase-3, and Septin4 were detected by western blotting. (F) Quantitated data are shown as means ± SD (***P < 0.001, unpaired Student's t-test; ##P < 0.01, #P < 0.05, two-way ANOVA with Bonferroni's post-hoc test).
Because oxidative stress causes hypertensive endothelial injury, we used H2O2 to simulate HUVECs in vitro. HUVECs were treated with 0, 250, 500, and 750 μmol/L H2O2 for 12 h. Next, we further clarified the role of WWP2 in oxidative stress-induced endothelial injury in vitro. As shown in Supplementary Fig. S2, upon endothelial cell injury, WWP2 expression was decreased significantly, while endothelial cell injury-associated proteins Septin4, cleaved PARP1, and cleaved caspase-3 expression elevated remarkably. In addition, WWP2 overexpression significantly decreased H2O2-induced expression of Septin4, cleaved PARP1, and cleaved caspase-3 (Fig. 7C and D). Conversely, WWP2 knockdown significantly increased the expression of H2O2-induced endothelial cell injury-associated proteins (Fig. 7E and F). These results suggested that WWP2 degraded Septin4 through the ubiquitin-proteasome pathway and inhibited formation of the Septin4-PARP1 complex, thereby counteracting endothelial injury.
3.8. WWP2 maintains endothelial cell viability and suppresses endothelial cell apoptosis and ROS production
As shown in Supplementary Fig. S3, with the increase of the H2O2 concentration, HUVEC viability was decreased significantly, while HUVEC apoptosis and ROS production were increased significantly.
To assess the effects of WWP2 on HUVEC injury, apoptosis, and ROS production induced by H2O2, the cells were transfected with control or Flag-WWP2 plasmids for 36 h and then treated with H2O2 for 12 h. As shown in Fig. 8, overexpression of WWP2 significantly reduced H2O2-associated HUVEC cytotoxicity, apoptosis, and ROS synthesis (Fig. 8A, C, and E). Conversely, the cells underwent transfection with negative control-siRNA or WWP2-siRNAs for 36 h, followed by H2O2 administration for 12 h. As a result, WWP2 knockdown significantly enhanced H2O2-induced HUVEC cytotoxicity, apoptosis, and ROS production (Fig. 8B, D, and F). In addition, WWP2 overexpression significantly decreased oxidative stress markers 3-nitrotyrosine and OGG1, and significantly stabilized antioxidant marker SOD1 (Fig. 8G). Conversely, WWP2 knockdown significantly increased oxidative stress markers 3-nitrotyrosine and OGG1, and significantly decreased antioxidant marker SOD1 (Fig. 8H). These results revealed WWP2 as a novel protective regulator against endothelial injury and vascular remodeling after endothelial injury in vivo and in vitro as well as the molecular mechanism.
Fig. 8.

WWP2 inhibits HUVEC apoptosis and ROS production induced by oxidative stress. HUVECs were transfected with Flag-control or Flag-WWP2 plasmids for 36 h and then treated with H2O2 for 12 h. (A) CCK8 assays were used to assess HUVEC viability. Quantitated data are shown as means ± SD (***P < 0.001, unpaired Student's t-test). (C) Flow cytometry was performed to assess HUVEC apoptosis. Quantitated data are shown as means ± SD (**P < 0.01, two-way ANOVA with Bonferroni's post-hoc test). (E) Immunofluorescence staining of ROS was performed to assess oxidative stress levels in HUVECs. Quantitated data are shown as means ± SD (**P < 0.01, two-way ANOVA with Bonferroni's post-hoc test). (G) 3-Nitrotyrosine, OGG1, and SOD1 were detected by western blotting. Quantitated data are means ± SD (**P < 0.001, *P < 0.05, two-way ANOVA with Bonferroni's post-hoc test). HUVECs underwent transfection with control-siRNA or WWP2-siRNA for 36 h and were then treated with H2O2 for 12 h. (B) CCK8 assays, (D) flow cytometry, (F) immunofluorescence staining of ROS, and (H) western blotting were performed as described above.
4. Discussion
The main findings of this study are that WWP2 interacted with the GTP domain of Septin4, ubiquitinated Septin4-K174, and degraded Septin4 through the ubiquitin-proteasome pathway under physiological conditions, which prevented hypertensive angiopathy. Under oxidative stress or AngII-induced endothelial injury, WWP2 showed significantly decreased expression, losing its ability to bind Septin4-GTP, and then ubiquitination of Septin4-K174 was reduced, and the Septin4-PARP1 apoptosis complex was formed, resulting in hypertensive angiopathy.
The NEDD4 family is a subfamily of HECT-type ligases with three functional domains including N-terminal-C2, WW (double tryptophan), and C-terminal-HECT domains. Previous studies have reported that NEDD4 family members participate in and regulate various biological activities such as membrane protein transport, signal transduction, transcription, and the cell cycle through ubiquitination of various substrates [16,17]. In the present study, the NEDD4 family member WWP2 was involved in endothelial injury and vascular remodeling after endothelial injury as a novel regulatory factor, unlike WWP1 that is a member of the same family. WWP2 was originally identified as a tumor-promoting factor that interacts with and ubiquitinates Smads, which inhibits TGFβ-induced epithelial-mesenchymal transition [18]. WWP2 also interacts with the tumor suppressor PTEN and promotes its degradation via the proteasome pathway, thereby inducing tumorigenesis [19]. In addition, WWP2 maintains stemness of embryonic stem cells by regulating OCT4 [20]. WWP2 is involved in T cell activation, osteoblast differentiation, and bone development by regulating EGR2 ubiquitination and RUNX2 monoubiquitination [21,22]. Furthermore, the WW domain of WWP2 interacts with the CTD domain of the RNA polymerase II functional subunit Rpb1, and WWP2 degrades Rpb1 through the ubiquitin-proteasome pathway, improving the overall efficiency of transcription and ensuring normal transcription [23]. This study showed that WWP2 was involved in endothelial injury as a protective factor, and endothelial/myeloid-specific WWP2 knockout in mice significantly aggravated endothelial cell injury and vascular remodeling after endothelial injury. Mechanistically, the current study demonstrated that the endothelial injury factor Septin4 was a new physiological substrate of WWP2. Moreover, WWP2 ubiquitinated Septin4-K174 by interacting with the GTPase domain of Septin4 and promoted Septin4 degradation by the ubiquitin-proteasome pathway, thereby counteracting endothelial injury and vascular remodeling after endothelial injury.
Septin4 is a mitochondrial protein in eukaryotic cells, which is mainly located at the 17q23 chromosome [24]. It contributes to multiple physiologically relevant processes including cytokinesis, fibrogenesis, cytoskeletal reorganization, vesicle trafficking, and tumor suppression [[25], [26], [27]]. The mitochondrial localization and proapoptotic functions of Septin4 have not been found in other known Septin family members [28,29]. Septin4 does not have the typical IBM structure of most IAP antagonists, but it interacts with the BIR3 domain of XIAP through the specific sequence ARTS-IBM [30,31]. In addition, Septin4 activates cytochrome C and SMAC upstream of the mitochondrial apoptotic pathway to promote apoptosis [32]. Our previous study showed that Septin4 is a major inducer of endothelial cell injury, promoting oxidative stress-associated endothelial cell injury via an interaction with PARP1 [15]. In this study, WWP2 degraded Septin4 through the ubiquitin-proteasome pathway, thereby reducing the Septin4-PARP1 endothelial cell injury complex. This explains the molecular mechanism of WWP2 as a new regulatory factor involved in endothelial injury and vascular remodeling after endothelial injury.
Previous studies have shown that Septin4 expression is mainly affected by transcriptional regulation. However, the present study found that Septin4 was also subjected to post-translational modifications. Indeed, WWP2 was identified as the E3 ubiquitin ligase of Septin4, ubiquitinating Septin4-K174 via an interaction with the GTPase domain of Septin4 that has three domains. The first domain is a disordered N-terminal domain that contains many prolines and regulates protein-protein interactions [[33], [34], [35]]. The second domain, namely the GTPase domain, does not contain the central G4 domain. The proapoptotic activity of Septin4 is decreased by P-loop domain mutation [36,37]. The third domain includes deletion of the C-terminus of the coiled-coil structure, which makes the Septin4 structure more flexible and able to interact with XIAP in different regions to participate in and regulate apoptosis [27]. The Septin4-K174 ubiquitination site was located in the GTPase domain, and Septin4 was regulated by WWP2. Therefore, this study revealed a role of WWP2 in endothelial injury and demonstrated that the WWP2-Septin4 axis participated in endothelial injury and vascular remodeling after endothelial injury as a novel regulatory pathway. The current findings may provide new therapeutic targets for diseases associated with endothelial injury and vascular remodeling after endothelial injury, such as atherosclerosis and hypertensive vascular diseases.
Our study also has some limitations. The Tek-Cre system has substantial non-specificity and might target several immune cell types. Similarly, immune cells have a dramatic effect on endothelial cell functions, especially in the AngII model [38,39]. Therefore, we will use the Cadh5-Cre model for endothelial cell-specific gene deletion to clarify the role of WWP2 in AngII-induced endothelial injury and vascular remodeling after endothelial injury in mice. In addition, further studies will clarify whether other E3 ubiquitin ligases are involved in endothelial injury and vascular remodeling after endothelial injury. Moreover, whether or not WWP2 acts as a protective factor in atherosclerosis and hypertension patients warrants further investigation.
5. Conclusions
WWP2 ubiquitinates the Septin4-K174 site by interacting with the Septin4-GTP domain and degrades Septin4 via the ubiquitin-proteasome pathway. This inhibits formation of the Septin4-PARP1 complex to suppress endothelial injury and vascular remodeling after endothelial injury. These findings provide a theoretical basis to develop new therapeutic drug targets for atherosclerosis and hypertensive vascular diseases.
Declaration of competing interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgments
This study was supported by grants from National Natural Science Foundation of China [Grant No. 81970211 and 81670231] and Postdoctoral Science Funds Project of China [Grant No. 2018M641750] and Natural Science Foundation of Liaoning Provincial Department of Education [Grant No. QN2019002].
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.redox.2019.101419.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Fig. S1.

WWP2 decreases with the occurrence of endothelial injury in mice. (A) Western blot analyses were conducted to assess WWP1 and WWP2 expression levels. (B) Quantification of results is shown as means ± SD (***P < 0.001, unpaired Student's t-test). (C) Cleaved PARP1 and cleaved caspase-3 were increased in the model of hypertensive endothelial injury in mice. Western blot analyses were conducted to assess Septin4, cleaved PARP1, and cleaved caspase-3 expression levels. (D) Quantification of results is shown as means ± SD (***P < 0.001, unpaired Student's t-test). (E) Hypertensive vascular thickening and (F) vascular fibrosis were significantly aggravated after AngII-induced injury in the C57B6/L mouse model in vivo. Quantification of results is shown as means ± SD (***P < 0.001, unpaired Student's t-test). H&E and Masson's trichrome staining were conducted on paraffin-embedded sections of vascular tissues from C57B6/L mice following vehicle infusion (NaCl) or AngII infusion for 2 weeks. n = 9 mice per group.
Fig. S2.

WWP2 decreases gradually with the occurrence of HUVEC injury. (A) Expression of WWP1 and WWP2 in HUVECs was examined after treatment with various concentrations of H2O2 for 12 h by western blotting. (B) Quantification of results is shown as means ± SD (***P < 0.001, one-way ANOVA with Bonferroni's post-hoc test). (C) Expression of Septin4, cleaved PARP1, and cleaved caspase-3 in HUVECs was examined by western blotting. (D) Quantification of results is shown as means ± SD (***P < 0.001, **P < 0.01, *P < 0.05, one-way ANOVA with Bonferroni's post-hoc test).
Fig. S3.

H2O2induces HUVECs apoptosis and ROS production. (A) CCK8 assays were used to assess HUVEC viability with gradient H2O2 treatment. Quantification of results is shown as means ± SD (***P < 0.001, one-way ANOVA with Bonferroni's post-hoc test). (B) Flow cytometry was performed to assess HUVEC apoptosis with gradient H2O2 treatment. Quantification of results is shown as means ± SD (***P < 0.001, one-way ANOVA with Bonferroni's post-hoc test). (C) Immunofluorescence staining of ROS was performed to assess oxidative stress in HUVECs with gradient H2O2 treatment. Quantification of results is shown as means ± SD (***P < 0.001, one-way ANOVA with Bonferroni's post-hoc test).
References
- 1.Karsan A., Harlan J.M. Modulation of endothelial cell apoptosis: mechanisms and pathophysiological roles. J. Atheroscler. Thromb. 1996;3:75–80. doi: 10.5551/jat1994.3.75. [DOI] [PubMed] [Google Scholar]
- 2.Szymanski M.K., Buikema J.H., van Veldhuisen D.J., Koster J., van der Velden J., Hamdani N. Increased cardiovascular risk in rats with primary renal dysfunction; mediating role for vascular endothelial function. Basic Res. Cardiol. 2012;107:242. doi: 10.1007/s00395-011-0242-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li J.M., Shah A.M. Endothelial cell superoxide generation: regulation and relevance for cardiovascular pathophysiology. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2004;287:R1014–R1030. doi: 10.1152/ajpregu.00124.2004. [DOI] [PubMed] [Google Scholar]
- 4.Komander D., Rape M. The ubiquitin code. Annu. Rev. Biochem. 2012;81:203–229. doi: 10.1146/annurev-biochem-060310-170328. [DOI] [PubMed] [Google Scholar]
- 5.Ikeda F., Crosetto N., Dikic I. What determines the specificity and outcomes of ubiquitin signaling? Cell. 2010;143:677–681. doi: 10.1016/j.cell.2010.10.026. [DOI] [PubMed] [Google Scholar]
- 6.Buetow L., Huang D.T. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2016;17:626–642. doi: 10.1038/nrm.2016.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Martins-Marques T., Ribeiro-Rodrigues T., Pereira P., Codogno P., Girao H. Autophagy and ubiquitination in cardiovascular diseases. DNA Cell Biol. 2015;34:243–251. doi: 10.1089/dna.2014.2765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cui Z., Scruggs S.B., Gilda J.E., Ping P., Gomes A.V. Regulation of cardiac proteasomes by ubiquitination, SUMOylation, and beyond. J. Mol. Cell. Cardiol. 2014;71:32–42. doi: 10.1016/j.yjmcc.2013.10.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Buetow L., Huang D.T. Structural insights into the catalysis and regulation of E3 ubiquitin ligases. Nat. Rev. Mol. Cell Biol. 2016;17:626–642. doi: 10.1038/nrm.2016.91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Scheffner M., Kumar S. Mammalian HECT ubiquitin-protein ligases: biological and pathophysiological aspects. Biochim. Biophys. Acta. 2014;1843:61–74. doi: 10.1016/j.bbamcr.2013.03.024. [DOI] [PubMed] [Google Scholar]
- 11.Razaghi B., Steele S.L., Prykhozhij S.V., Stoyek M.R., Hill J.A., Cooper M.D. hace1 Influences zebrafish cardiac development via ROS-dependent mechanisms. Dev. Dynam. 2018;247:289–303. doi: 10.1002/dvdy.24600. [DOI] [PubMed] [Google Scholar]
- 12.Zhang L., Chen X., Sharma P., Moon M., Sheftel A.D., Dawood F. HACE1-dependent protein degradation provides cardiac protection in response to haemodynamic stress. Nat. Commun. 2014;5:3430. doi: 10.1038/ncomms4430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Otaki Y., Takahashi H., Watanabe T., Funayama A., Netsu S., Honda Y. HECT-type ubiquitin E3 ligase ITCH interacts with thioredoxin-interacting protein and ameliorates reactive oxygen species-induced cardiotoxicity. J. Am. Heart Assoc. 2016;21(1):5. doi: 10.1161/JAHA.115.002485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang N., Zhang Y., Zhao S., Sun Y. Septin4 as a novel binding partner of PARP1 contributes to oxidative stress induced human umbilical vein endothelial cells injure. Biochem. Biophys. Res. Commun. 2018;496:621–627. doi: 10.1016/j.bbrc.2018.01.105. [DOI] [PubMed] [Google Scholar]
- 15.Ehling J., Theek B., Gremse F., Baetke S., Möckel D., Maynard J. Micro-CT imaging of tumor angiogenesis: quantitative measures describing micromorphology and vascularization. Am. J. Pathol. 2014;184:431–441. doi: 10.1016/j.ajpath.2013.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Boase N.A., Kumar S. NEDD4: the founding member of a family of ubiquitin-protein ligases. Gene. 2015;557:113–122. doi: 10.1016/j.gene.2014.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fajner V., Maspero E., Polo S. Targeting HECT-type E3 ligases - insights from catalysis, regulation and inhibitors. FEBS Lett. 2017;591:2636–2647. doi: 10.1002/1873-3468.12775. [DOI] [PubMed] [Google Scholar]
- 18.Soond S.M., Chantry A. Selective targeting of activating and inhibitory Smads by distinct WWP2 ubiquitin ligase isoforms differentially modulates TGFβ signalling and EMT. Oncogene. 2011;30:2451–2462. doi: 10.1038/onc.2010.617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Maddika S., Kavela S., Rani N., Palicharla V.R., Pokorny J.L., Sarkaria J.N. WWP2 is an E3 ubiquitin ligase for PTEN. Nat. Cell Biol. 2011;13:728–733. doi: 10.1038/ncb2240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Xu H., Wang W., Li C., Yu H., Yang A., Wang B. WWP2 promotes degradation of transcription factor OCT4 in human embryonic stem cells. Cell Res. 2009;19:561–573. doi: 10.1038/cr.2009.31. [DOI] [PubMed] [Google Scholar]
- 21.Chen A., Gao B., Zhang J., McEwen T., Ye S.Q., Zhang D. The HECT-type E3 ubiquitin ligase AIP2 inhibits activation-induced T-cell death by catalyzing EGR2 ubiquitination. Mol. Cell. Biol. 2009;29:5348–5356. doi: 10.1128/MCB.00407-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhu W., He X., Hua Y., Li Q., Wang J., Gan X. The E3 ubiquitin ligase WWP2 facilitates RUNX2 protein transactivation in a mono-ubiquitination manner during osteogenic differentiation. J. Biol. Chem. 2017;292:11178–11188. doi: 10.1074/jbc.M116.772277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li H., Zhang Z., Wang B., Zhang J., Zhao Y., Jin Y. Wwp2-mediated ubiquitination of the RNA polymerase II large subunit in mouse embryonic pluripotent stem cells. Mol. Cell. Biol. 2007;27:5296–5305. doi: 10.1128/MCB.01667-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Lin Y.H., Kuo Y.C., Chiang H.S., Kuo P.L. The role of the septin family in spermiogenesis. Spermatogenesis. 2011;1:298–302. doi: 10.4161/spmg.1.4.18326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Neubauer K., Zieger B. The mammalian septin interactome. Front. Cell Dev. Biol. 2017;5:3. doi: 10.3389/fcell.2017.00003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Khairat J.E., Balasubramaniam V., Othman I., Omar A.R., Hassan S.S. Interaction of recombinant Gallus gallus SEPT5 and brain proteins of H5N1-avian influenza virus-infected chickens. Proteomes. 2017;5 doi: 10.3390/proteomes5030023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Østevold K., Meléndez A.V., Lehmann F., Schmidt G., Aktories K., Schwan C. Septin remodeling is essential for the formation of cell membrane protrusions (microtentacles) in detached tumor cells. Oncotarget. 2017;8:76686–76698. doi: 10.18632/oncotarget.20805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mandel-Gutfreund Y., Kosti I., Larisch S. ARTS, the unusual septin: structural and functional aspects. Biol. Chem. 2011;392:783–790. doi: 10.1515/BC.2011.089. [DOI] [PubMed] [Google Scholar]
- 29.García-Fernández M., Kissel H., Brown S., Gorenc T., Schile A.J., Rafii S. Sept4/ARTS is required for stem cell apoptosis and tumor suppression. Genes Dev. 2010;24:2282–2293. doi: 10.1101/gad.1970110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Reingewertz T.H., Shalev D.E., Sukenik S., Blatt O., Rotem-Bamberger S., Lebendiker M. Mechanism of the interaction between the intrinsically disordered C-terminus of the pro-apoptotic ARTS protein and the Bir3 domain of XIAP. PLoS One. 2011;6 doi: 10.1371/journal.pone.0024655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Garrison J.B., Correa R.G., Gerlic M., Yip K.W., Krieg A., Tamble C.M. ARTS and Siah collaborate in a pathway for XIAP degradation. Mol. Cell. 2011;41:107–116. doi: 10.1016/j.molcel.2010.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Edison N., Zuri D., Maniv I., Bornstein B., Lev T., Gottfried Y. The IAP-antagonist ARTS initiates caspase activation upstream of Cytochrome C and SMAC/Diablo. Cell Death Differ. 2012;19:356–368. doi: 10.1038/cdd.2011.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Garcia W., de Araújo A.P., Neto Mde O., Ballestero M.R., Polikarpov I., Tanaka M. Dissection of a human septin: definition and characterization of distinct domains within human SEPT4. Biochemistry. 2006;45:13918–13931. doi: 10.1021/bi061549z. [DOI] [PubMed] [Google Scholar]
- 34.Haynes C., Oldfield C.J., Ji F., Klitgord N., Cusick M.E., Radivojac P. Intrinsic disorder is a common feature of hub proteins from four eukaryotic interactomes. PLoS Comput. Biol. 2006;2:e100. doi: 10.1371/journal.pcbi.0020100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tompa P., Szasz C., Buday L. Structural disorder throws new light on moonlighting. Trends Biochem. Sci. 2005;30:484–489. doi: 10.1016/j.tibs.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 36.Gottfried Y., Rotem A., Lotan R., Steller H., Larisch S. The mitochondrial ARTS protein promotes apoptosis through targeting XIAP. EMBO J. 2004;23:1627–1635. doi: 10.1038/sj.emboj.7600155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hunter A.M., Lacasse E.C., Korneluk R.G. The inhibitors of apoptosis (IAPs) as cancer targets. Apoptosis. 2007;12:1543–1568. doi: 10.1007/s10495-007-0087-3. [DOI] [PubMed] [Google Scholar]
- 38.Guzik T.J., Hoch N.E., Brown K.A., McCann L.A., Rahman A., Dikalov S. Role of the T cell in the genesis of angiotensin II induced hypertension and vascular dysfunction. J. Exp. Med. 2007;204:2449–2460. doi: 10.1084/jem.20070657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wenzel P., Knorr M., Kossmann S., Stratmann J., Hausding M., Schuhmacher S. Lysozyme M-positive monocytes mediate angiotensin II-induced arterial hypertension and vascular dysfunction. Circulation. 2011;124:1370–1381. doi: 10.1161/CIRCULATIONAHA.111.034470. [DOI] [PubMed] [Google Scholar]