

Abstract
AVB‐S6‐500 neutralized growth arrest‐specific 6 (GAS6) protein and effectively inhibited AXL signaling in preclinical cancer models. A target‐mediated drug disposition (TMDD) pharmacokinetic/pharmacodynamic (PK/PD) model was used to select first‐in‐human (FIH) doses for AVB‐S6‐500 based on predicted target (GAS6) suppression in the clinic. The effect of TMDD on AVB‐S6‐500 clearance was incorporated into a standard two‐compartment model, providing parallel linear and nonlinear clearance. Observed AVB‐S6‐500 and GAS6 concentration data in cynomolgus monkeys and relevant interspecies differences were used to predict the PK (serum concentration)/PD (GAS6 suppression) relationship in humans. Human exposure and GAS6 suppression were simulated for the proposed FIH doses of 1, 2.5, 5, and 10 mg/kg. A dose of 1 mg/kg was selected to target GAS6 suppression for 2 weeks in the initial healthy volunteer study. The cynomolgus monkey:human ratios for the highest proposed FIH dose were anticipated to yield more than a 10‐fold margin to the nonclinical no observed adverse event level while maintaining > 90% GAS6 suppression. In human subjects, the first dose (1 mg/kg) model‐projected and clinically observed maximal concentration (Cmax) was within 10% of predicted; repeat dosing at 5 mg/kg was within 1% (Cmax) and 45% (area under the serum concentration‐time curve from time 0 to end of dosing interval) of predicted. Predicted GAS6 suppression duration of 14 days was accurate for the 1 mg/kg dose. A PK/PD model expedited clinical development of AVB‐S6‐500, minimized exposure of patients with cancer to subtherapeutic doses, and rationally guided the optimal dosing in patients.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE ON THE TOPIC?
☑ Availability of serum‐based mechanistic biomarkers that can be used as pharmacodynamic (PD) assays in first‐in‐human (FIH) oncology clinical trials are uncommon. Minimum anticipated biological effect level approaches are recommended by the European Medicines Agency for monoclonal antibodies.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ Can PD and pharmacokinetic (PK) data from a relevant nonhuman species be used to identify FIH dosing?
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ FIH dosing predictions based on PK/PD modeling of nonclinical data in a relevant species were successful for AVB‐S6‐500, which supports the use of similar modeling approaches for fusion protein or monoclonal antibody products. Species scaling approaches based on body surface area assume dose‐proportional exposure over the dose ranges tested and systemic clearance of the drug proportional to the ratio of the species weight to the power of 0.67. However, target‐mediated drug disposition had a notable effect on the PK of AVB‐S6‐500, and systemic clearance was proportional to the ratio of the species weight to the power of 0.75.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑An appropriate biomarker can expedite clinical development of novel drugs, minimize exposure of patients with cancer to subtherapeutic doses, avoid unnecessary tolerability issues related to inability to ascertain efficacious dose, and rationally guide the optimal dosing. This study may establish precedent for future use of mechanistic biomarkers in FIH studies for novel oncology drugs.
State‐of‐the‐art cancer treatments include antibodies and proteins designed to target specific dysregulated signaling pathways identified based on increasing knowledge of the molecular basis of cancer. Protein‐based therapeutics could provide many benefits, including a reduction in drug‐drug interactions with the background standard of care chemotherapeutic agents.1, 2, 3
AXL, a member of the TAM family of receptor tyrosine kinases, is highly expressed in primary tumors and metastases in comparison with normal tissues and plays a role in tumor proliferation and survival, metastasis, and drug resistance.4, 5, 6, 7, 8 AXL has a single specific ligand, growth arrest‐specific 6 (GAS6) protein, which has also been implicated in poor prognosis, metastasis, and drug resistance in several forms of cancer.1, 4 The expression of GAS6 is widespread in many tissues and cells, including immune cells, endothelial cells, vascular smooth muscle cells, bone marrow cells, adipocytes, platelets, and various cancer cells.9 Signaling via the TAM kinase receptors activate different downstream signaling cascades and regulate diverse functions, including cell migration, adhesion, inflammation, cell growth, survival, and other cell type‐specific functions.10, 11, 12 In the cancer cell, activation of the AXL/GAS6 pathway allows the cancer cell to survive under less than ideal conditions and to invade and migrate into healthy tissue. The pathway is also active in the tumor microenvironment where AXL signaling contributes to the immunosuppressive and protumorigenic phenotypes. Of note, there are no currently identified mutations of AXL from human tumor samples known to result in loss of function or gain of functional activity. These characteristics make AXL and GAS6 promising targets for cancer therapeutics.
AVB‐S6‐500 (Aravive, Houston, TX) is a fusion of the extracellular domain of AXL, which is the receptor for GAS6, and a human immunoglobulin G1 fragment crystallizable domain. The affinity between endogenous AXL and GAS6 is in the pM range. AVB‐S6‐500 binds GAS6 with low femtomolar range affinity to effectively bind GAS6 and eliminate its ability to stimulate the AXL pathway. The antitumor effect in preclinical studies was strongly associated with the high affinity of the AVB‐S6‐500 protein and suppression of serum GAS6 to below detectable limits.8 In nonclinical studies, AVB‐S6‐500 had the same affinity for cynomolgus monkey GAS6 as human GAS6, indicating that the monkey is a predictive species for human toxicity. The dose that effectively abrogated serum GAS6 levels in monkeys for 1 week is 30‐fold lower than the maximum feasible dose tested, which did not elicit toxicities or adverse effects. In good laboratory practice compliant toxicology studies, AVB‐S6‐500 had a safety profile that made a first‐in‐human (FIH) study in healthy volunteers possible.
Based on the finding that abrogation of serum GAS6 levels related to antitumor effect in preclinical models, a pharmacokinetic/pharmacodynamic (PK/PD) model was used to identify an appropriate starting dose for FIH studies, based on a quantitative understanding of the PK/PD relationship of AVB‐S6‐500 and GAS6 suppression in cynomolgus monkeys, a relevant species.
Methods
To predict human PK/PD at proposed FIH doses and levels, a PK/PD model was developed based on data from AVB‐S6‐500 administration in cynomolgus monkeys then scaled to humans. Predicted values were then compared with PK/PD outcomes in the FIH study.
Nonclinical studies used for PK/PD model development
Observed serum AVB‐S6‐500 and GAS6 concentration data from five nonclinical studies in cynomolgus monkeys covering doses from 0.1150 mg/kg were used to estimate the PK/PD relationship in humans. Procedures involving the care and use of animals were reviewed and approved by the Institutional Animal Care and Use Committee at CiToxLAB Safety and Research Laboratories (Quebec, Canada) and Shin Nippon Biomedical Laboratories (Everett, WA) prior to conduct of each study. During each study, the care and use of animals were conducted in accordance with the principles outlined in the current guidelines published by the Canadian Council on Animal Care and the Guide for the Care and Use of Laboratory Animals and/or 21 Code of Federal Regulations Part 58 Good Laboratory Practice. The study was considered as a “C” category of invasiveness.
Two animals were used per dose level over the range up to 1 mg/kg (0.1, 0.5, and 1 mg/kg); four animals across two studies were administered with 5 mg/kg. Over three studies, 12 animals support the analysis at each dose level over the range of 30100 mg/kg, and 18 animals received 150 mg/kg. In each nonclinical study and across the nonclinical program, the minimum number of animals required to achieve the study objectives was used, based on regulatory requirements, statistical power, and/or availability of historical data. The numbers of animals in each study were within Institutional Animal Care and Use Committee approved guidelines.
PK/PD model development
The choice of model was supported by the results of nonclinical studies, as AVB‐S6‐500 had a PK profile compatible with target‐mediated drug disposition (TMDD) with two parallel elimination paths: typical proteolysis common to all immunoglobulins and a nonlinear saturable pathway related to the target.13, 14 A diagram of the model is shown in Figure 1.
Figure 1.
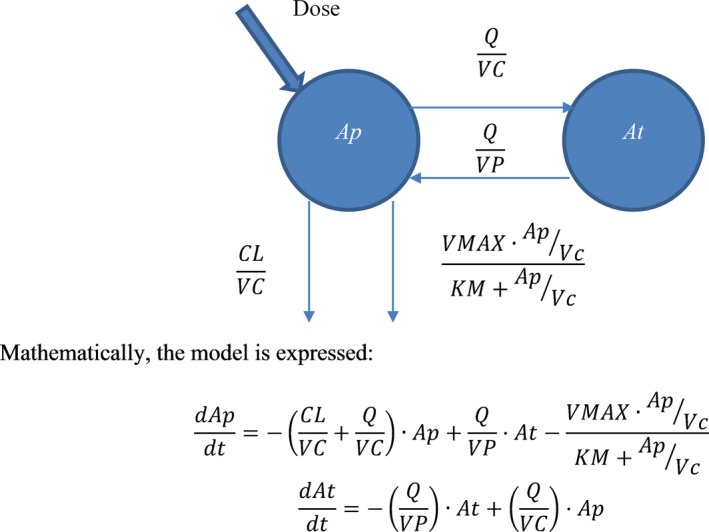
Model diagram. Ap, amount of AVB‐S6‐500 in the plasma compartment; At, amount of AVB‐S6‐500 in the peripheral compartment; CL, systemic clearance; KM, concentration of serum AVB‐S6‐500 at half‐maximal removal by the target; Q, intercompartmental clearance; VC, volume of the central/plasma compartment; Vmax, maximal removal rate of AVB‐S6‐500 from the serum; VP, volume of the peripheral compartment.
The PK/PD analysis set was formed from individual animal AVB‐S6‐500 and GAS6 serum concentrations by sampling time. Individual animal body weight and the actual administered dose were included to accurately capture the effect of individual weight. Data were sourced from Microsoft Excel and Adobe Acrobat format files. AVB‐S6‐500 concentration measurements less than the lower limit of quantification (LLOQ) were flagged as missing (missing dependent variable = 1) for the purposes of fitting. For the purposes of display only, AVB‐S6‐500 concentration measurements and predictions less than the LLOQ are shown as LLOQ/2.
The effect of GAS6 on the clearance of AVB‐S6‐500 was incorporated into the model as a direct‐effect relationship, as no delay (hysteresis) was observed or expected because AVB‐S6‐500 binds GAS6 rapidly. Time‐matched observed AVB‐S6‐500 and GAS6 serum concentrations were analyzed to determine the AVB‐S6‐500:GAS6 (PK/PD) relationship in cynomolgus monkeys. A maximum effect (Emax)‐type model with Hill‐slope coefficient was fit to the observed data where plasma AVB‐S6‐500 and plasma GAS6 were greater than the LLOQ, with the addition of predose data where plasma AVB‐S6‐500 was assumed zero. For the purposes of display only, GAS6 concentration measurements and predictions less than the LLOQ are shown as LLOQ/2.
Supplementary Material provides the model equations used in detail. R software version 3.4.0 was used to process these source files into formats suitable for analysis by R and NONMEM (nonlinear mixed effects modeling) version 7.3.
Model predictions of clinical PK/PD
PK model parameters for systemic clearance (CL), intercompartmental clearance, and maximal nonlinear elimination (Vmax) were scaled to reflect species differences in body weight, centered on 70 kg, using an allometric scaling coefficient of 0.75. PK model parameters for central/plasma volume of distribution (VC) and peripheral volume of distribution were scaled to reflect species differences in body weight, centered on 70 kg, using an allometric scaling coefficient of 1.0.
No scaling was performed to translate nonhuman primate PD to human PD, as binding affinities to the cynomolgus monkey and human targets were equivalent. Model parameters were scaled to reflect species differences in body weight, centered on 70 kg.
Simulations of human exposure and GAS6 suppression were performed for the proposed FIH dose levels of 1, 2.5, 5, and 10 mg/kg. Predicted human exposure was compared with the cynomolgus monkey exposure at the 150 mg/kg dose (no observed adverse event level). The subject weight for these simulations was assumed to be 75 kg, which is heavier than the overall population (70 kg), owing to the likely enrollment of predominantly young men into the FIH study.
FIH study data for model validation
The phase I safety and PK/PD study (NCT03401528) was conducted in 42 healthy volunteers who ranged in age from 22 to 54 years (mean age was 38 years for men and 42 years for women) from January until July 2018 at the Orlando Clinical Research Center, Orlando, Florida.
The FIH protocol was approved by the institutional review board of the Orlando Clinical Research Center and complied with the principles outlined in the Declaration of Helsinki and the International Committee on Harmonisation E6 Good Clinical Practice Guideline. Participants gave written informed consent prior to screening assessments.
Table 1 shows the baseline characteristics for participants receiving AVB‐S6‐500.
Table 1.
Summary of demographic characteristics in the first‐in‐human study
| AVB‐S6‐500 dose levels | |||||
|---|---|---|---|---|---|
| Single dose | Repeat dose (once weekly for 4 weeks) | ||||
| 1 mg/kg (N = 6) | 2.5 mg/kg (N = 6) | 5 mg/kg (N = 6) | 10 mg/kg (N = 6) | 5 mg/kg (N = 7) | |
| Age, years | |||||
| Mean (SD) | 41.2 (7.88) | 35.2 (10.89) | 36.2 (12.77) | 45.2 (5.27) | 40.3 (8.44) |
| Median | 43.0 | 35.0 | 31.0 | 44.5 | 36.0 |
| Min, Max | 27, 48 | 24, 47 | 22, 54 | 38, 53 | 31, 51 |
| Sex, n (%) | |||||
| Male | 4 (66.7) | 3 (50.0) | 4 (66.7) | 5 (83.3) | 6 (85.7) |
| Female | 2 (33.3) | 3 (50.0) | 2 (33.3) | 1 (16.7) | 1 (14.3) |
| Weight, Kg | |||||
| Mean (SD) | 81.40 (8.945) | 70.73 (11.531) | 79.68 (8.868) | 88.58 (10.265) | 82.81 (9.709) |
| Median | 85.15 | 70.40 | 79.40 | 88.85 | 88.00 |
| Min, Max | 67.0, 89.2 | 59.4, 91.2 | 67.2, 91.5 | 74.5, 103.0 | 72.1, 94.5 |
| Race, n (%) | |||||
| White | 0 | 2 (33.3) | 1 (16.7) | 2 (33.3) | 2 (28.6) |
| Black or African American | 6 (100.0) | 4 (66.7) | 4 (66.7) | 3 (50.0) | 4 (57.1) |
| Asian | 0 | 0 | 0 | 0 | 0 |
| American Indian or Alaska Native | 0 | 0 | 0 | 0 | 0 |
| Native Hawaiian or other Pacific Islander | 0 | 0 | 0 | 0 | 0 |
| Other | 0 | 0 | 1 (16.7) | 1 (16.7) | 1 (14.3) |
| Ethnicity, n (%) | |||||
| Hispanic or Latino | 1 (16.7) | 1 (16.7) | 1 (16.7) | 1 (16.7) | 2 (28.6) |
| Not Hispanic or Latino | 5 (83.3) | 5 (83.3) | 5 (83.3) | 5 (83.3) | 5 (71.4) |
A total of 32 participants were enrolled in the single ascending dose portion of the study and randomized to either AVB‐S6‐500 or placebo administered as a single dose via 60‐minute intravenous infusion. Six participants were enrolled per AVB‐S6‐500 dose level (1, 2.5, 5, and 10 mg/kg); for each group, two additional participants were enrolled to receive placebo (eight subjects total). All participants completed the study and were included in PK analyses.
Ten participants were enrolled in the repeat‐dose cohort; seven received 5 mg/kg AVB‐S6‐500, and three received placebo. Two of the original participants (one on active treatment and one on placebo) discontinued for personal reasons and were replaced. Both AVB‐S6‐500 and placebo were administered via 60‐minute intravenous infusion once weekly for 4 weeks. No protocol deviations impacted the quality of the study or the interpretation of data in either part of the FIH study.
In both cohorts, blood samples for PK/PD analysis were drawn and processed into serum predose (within 45 minutes prior to dosing; 0 hour) and at ~ 1, 2, 4, 6, 8, and 24 hours postdose while participants remained in the clinic. Additional prespecified blood samples were obtained at outpatient visits at 72, 120, 168, and 336 hours postdose. Among participants receiving 5 or 10 mg/kg AVB‐S6‐500 and had measurable levels of AVB‐S6‐500 at the last scheduled visit, additional blood samples were requested at 528 and 696 hours post‐treatment.
As expected, the median subject weight, 75.5 kg, was higher than the population typical 70 kg. Thus, the assumed value of 75 kg for the simulations made prior to the study conduct was reasonable.
Model validation
To verify the PK/PD model, the maximum serum concentration (Cmax), area under the serum concentration‐time curve from time 0 to end of dosing interval (repeat dose), and serum GAS6 time to recovery (single dose) were compared with predicted values as an external validation. All other study data are reported elsewhere.
Noncompartmental PK and PD parameters were calculated from concentration profiles calculated relative to the end of AVB‐S6‐500 infusion using a validated version of Phoenix WinNonlin version 7.0 or later. Actual sampling time points, doses, and infusion durations were used in all calculations. No negative actual times were included. Concentration values below the limit of quantitation (BLOQ) were set to 0. No substitutions were made for missing data points. All subjects who received AVB‐S6‐500 and had no major protocol deviations and sufficient AVB‐S6‐500 sample data to assess PK parameters for each individual concentration‐time profile of AVB‐S6‐500 were included.
GAS6 concentration data were plotted against actual time relative to dosing; summary statistics were calculated for individuals and stratified by cohort.
Results
Model based on PK/PD in cynomolgus monkeys
Data suggested that a dose between 0.5 and 1 mg/kg was the breakpoint between partial and full suppression of GAS6 in the cynomolgus monkey. Increased clearance was observed at dose levels from 0.1 to 5 mg/kg relative to the higher doses (up to 150 mg/kg), a hallmark of TMDD. A single dose of AVB‐S6‐500 at 5 mg/kg relative to lower doses provided the best PK/PD profile in cynomolgus monkeys and demonstrated abrogation of serum‐free GAS6 for ~ 1 week postdose. Model fitting diagnostics for the PK and PD models fit to cynomolgus monkey data are available in the Supplemental Materials.
The cynomolgus monkey:human ratios for the highest proposed FIH dose were anticipated to yield >10‐fold margin to the nonclinical no observed adverse event level/maximum feasible dose while maintaining > 90% GAS6 suppression.
Human dose exposure projections
The Vmax of 3.35 mg/day suggests that 46.9 mg (0.67 mg/kg in a 70 kg human) of AVB‐S6‐500 would be removed by target every 14 days, suggesting that doses < 1 mg/kg (70 mg in a 70 kg human) would not maintain target saturation in healthy human volunteers throughout the dosing interval. At low dose levels (0.1, 0.5, and 1 mg/kg; data not shown), variability in the TMDD parameters (Vmax and concentration of AVB‐S6‐500 at half‐maximal removal by the target) generated a large prediction interval, suggesting that exposure is highly influenced by subject‐subject variability. At higher doses, the TMDD pathway was saturated, and the biphasic serum profile common for monoclonal antibodies was recovered (Table 2). These data are also consistent with a smaller effect of subject‐specific variability on TMDD at doses of 5 and 10 mg/kg.
Table 2.
Model parameter estimates, centered on a 70 kg subject
| Parameter | Estimate (%RSE) | Between‐subject variability (%RSE) |
|---|---|---|
| CL, L/day | 1.10 (7.87) | 29.7 (1.45) |
| VC, L | 2.97 (2.90) | 14.0 (16.1) |
| VP, L | 2.94 (6.29) | 14.3 (15.7) |
| Q, L/day | 2.04 (12.0) | 50.0 (27.4) |
| Vmax, mg/day | 3.35 (3.77) | 98.6 (35.1) |
| KM, ng/mL | 102 (7.01) | 103 (0.874) |
CL, systemic clearance; KM, concentration of serum AVB‐S6‐500 at half‐maximal removal by the target; Q, intercompartmental clearance; RSE, residual standard error; VC, volume of the central/plasma compartment; Vmax, maximal removal rate of AVB‐S6‐500 from the serum; VP, volume of the peripheral compartment.
Based on these simulations, the FIH starting dose of 1 mg/kg was selected to target GAS6 suppression for 2 weeks. Given AVB‐S6‐500 was going to be investigated in oncology clinical trials, a 2‐week dosing interval was considered ideal as it complements the chemotherapy regimens that would be tested in combination.
Comparison of predicted AVB‐S6‐500 concentrations with observed values in human subjects
In healthy human participants, the first dose (1 mg/kg) model‐projected and clinically observed Cmax were within 10%; this pattern was repeated for increasing doses (Table 3). The first dose (1 mg/kg) model‐projected and clinically observed AUC0‐tau were within 30%; prediction accuracy was reduced with increasing doses (Table 3). Figure 2 shows the model‐predicted (typical human subject) and observed AVB‐S6‐500 concentrations.
Table 3.
Ratios of observed and predicted pharmacokinetic parameters in human subjects
| Dose | Dose number | Cmax predicted/observed (predicted:observed) | AUC0‐tau predicted/observed (predicted:observed) |
|---|---|---|---|
| 1 mg/kg | NA | 91.3% (23.2:25.4) | 72.4% (869:1,200) |
| 2.5 mg/kg | NA | 91.4% (58.2:63.7) | 55.9% (2,520:4,510) |
| 5 mg/kg | NA | 96.7% (116:120) | 53.2% (5,290:9,950) |
| 10 mg/kg | NA | 92.5% (233:252) | 44.6% (10,800:24,200) |
| 5 mg/kg q.w. × 4 | 1 | 100% (116:115.6) | 69.8% (5,290:7574.3) |
| 5 mg/kg q.w. × 4 | 4 | 88.1% (134:152.1) | 55.6% (7,060:12687.6) |
AUC0‐tau, area under the serum concentration‐time curve from time 0 to end of dosing interval, µg × hour/mL; Cmax, maximum serum concentration, µg/mL.
Ratios are presented as (predicted/observed) × 100%.
Figure 2.
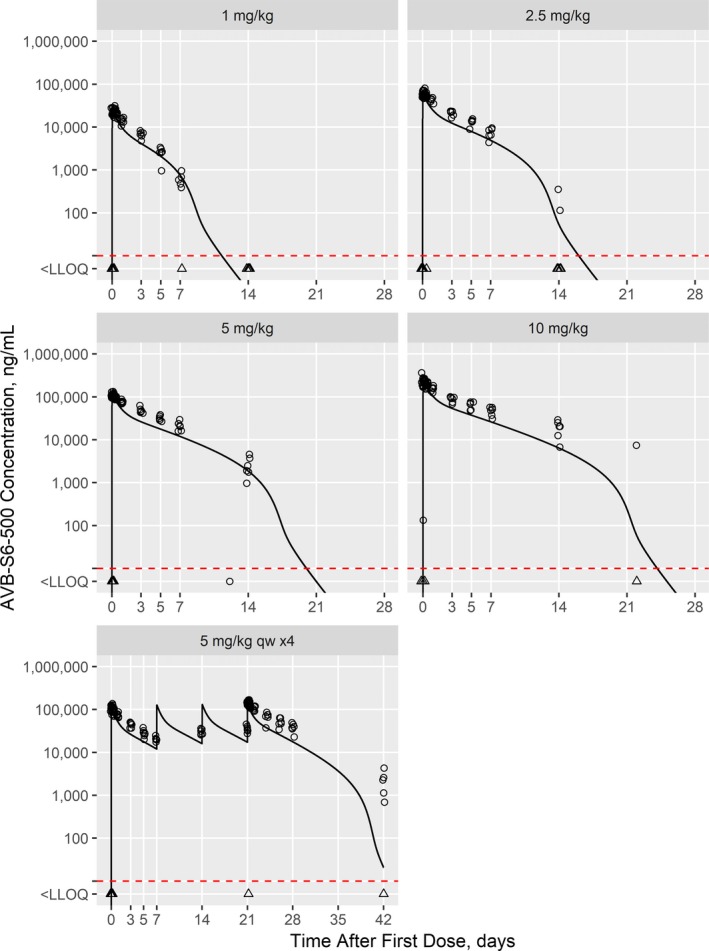
Model‐predicted AVB‐S5‐500 concentrations for the first‐in‐human study with observed concentrations overlaid. Individual observations (points) are shown overlaid with the model‐predicted values (lines) for a typical 75 kg subject. Observations and predictions below the LLOQ (10 ng/mL; dashed red line) are shown at LLOQ/2 and as triangles. LLOQ, lower limit of quantitation.
Comparison of observed and predicted GAS6 concentrations in human subjects
In human participants receiving a single dose of 1 mg/kg AVB‐S6‐500, two of six and six of six subjects had quantifiable GAS6 levels at 7 and 14 days after dosing, respectively (Figure 3). The model predicted undetectable and nearly baseline GAS6 at 7 and 14 days after dosing for the typical human subject, and, thus, somewhat overpredicted GAS6 suppression.
Figure 3.
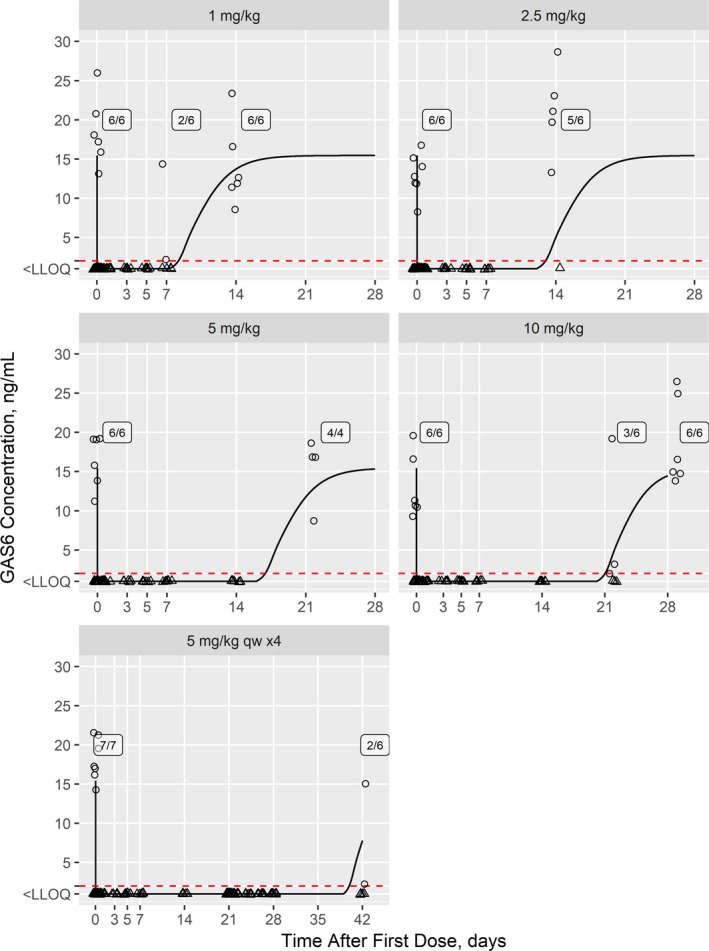
Model‐predicted growth arrest‐specific 6 (GAS6) concentrations for the first‐in‐human study with observed concentrations overlaid. Individual observations (points) are shown overlaid with the model‐predicted values (lines) for a typical 75 kg subject. Observations and predictions below the LLOQ (2 ng/mL; dashed red line) are shown at LLOQ/2 and as triangles. Counts of subjects where GAS6 levels are above the LLOQ are shown. LLOQ, lower limit of quantitation.
For the single dose of 2.5 mg/kg, zero of six and five of six subjects had quantifiable GAS6 levels at 7 and 14 days after dosing. These findings were in agreement with the model, which predicted undetectable and just‐detectable GAS6 at 7 and 14 days after dosing for the typical human subject.
Single doses of 5 and 10 mg/kg suppressed GAS6 levels in all human subjects (six/six) tested at 7 and 14 days. The model predicted undetectable GAS6 for both doses for the typical human subject.
Some participants returned for follow‐up samples at 21 and 28 days in the single‐dose groups of 5 and 10 mg/kg. For the 5 mg/kg cohort, four of four subjects had quantifiable GAS6 levels at 21 days; the model prediction was nearly baseline for the typical subject. For the 10 mg/kg cohort, three of six and six of six subjects had quantifiable GAS6 levels at 21 and 28 days, respectively; the model predicted just‐detectable and nearly baseline levels, respectively, for the typical subject.
Four, weekly infusions of 5 mg/kg AVB‐S6‐500 (repeat‐dose cohort) in healthy subjects resulted in an immediate and sustained maximal reduction in circulating serum GAS6 concentrations to BLOQ. GAS6 was suppressed to BLOQ in all subjects until 504 hours (on day 42) following the final infusion (on day 21), at which point, GAS6 was measurable but below baseline levels in two of six subjects; GAS6 concentrations remained BLOQ in all other (four/six) subjects.
Discussion
A TMDD PK/PD model developed based on data from AVB‐S6‐500 administration in cynomolgus monkeys then scaled to humans was successful in identifying appropriate dosing for the FIH study of AVB‐S6‐500, a novel fusion protein intended for use in cancer treatment. The use of a PK/PD model to guide FIH dosing and the ability to assess the effects of AVB‐S6‐500 on serum GAS6 levels in healthy volunteers will reduce the number of patients with cancer exposed to potentially subtherapeutic doses in clinical trials. Model predictions were made for the typical human subject, meaning between‐subject variability observed in nonhuman primates was not propagated through the simulations. Variability estimates in cynomolgus monkeys for the nonlinear (target) elimination pathway approached 100% relative variability, so the observed data would easily fall within a 90% prediction interval. However, the relationship between variability in purpose‐bred animals and human clinical subjects is dubious.
The AVB‐S6‐500 development program has been facilitated by a high sensitivity biomarker assay for serum GAS6. In preclinical oncology studies, abrogation of serum GAS6 correlated with effects on tumor metastasis,1 confirming serum GAS6 as a relevant serum biomarker to identify a pharmacologically active dose in humans. Model‐predicted GAS6 suppression aligned well with the observed data. The FIH dose of 1 mg/kg was specifically selected to target GAS6 suppression for 2 weeks. The serum GAS6 assay will be used throughout the clinical development program to ensure correct dosing.
The Cmax predictions were within 10% of observed values. At later time points, the model‐predicted concentrations were lower than observations, suggesting that AVB‐S6‐500 clearance was lower than expected. Other efforts in scaling PK in monoclonal antibodies15, 16 encountered difficulties for antibodies exhibiting nonlinear PK; Cmax was consistently overestimated up to 5.3‐fold higher at low concentrations and within 2.3‐fold at high concentrations. Prediction accuracy within 10% for Cmax for AVB‐S6‐500 is a significant improvement compared with these earlier findings,15, 16 Accurate prediction of Cmax only requires that the dominant volume of distribution at the end‐of‐infusion, VC, be accurately predicted. Accurate prediction of AUC, in contrast, require that most (if not all) model parameters be accurately predicted. Linear and nonlinear clearance parameters, CL and Vmax, are particularly important, and how Vmax scales between species is far less well understood.
Species scaling approaches based on body surface area assume dose‐proportional exposure over the dose ranges tested and clearance of the drug proportional to the ratio of the species weight to the power of 0.67. In the case of AVB‐S6‐500, it is likely that neither of these assumptions holds true. TMDD had a notable effect on the PK of AVB‐S6‐500, and systemic clearance was proportional to the ratio of the species weight to the power of 0.75. At higher doses, the TMDD pathway was saturated, and the biphasic serum profile common for monoclonal antibodies was recovered.
For future efforts in this area, an alternative approach to the model estimation step may be considered. The linear model parameters (CL, VC, peripheral volume of distribution, and intercompartmental clearance) are well‐known for antibodies,17 and so fixing these values (or applying them as a Bayesian prior) in the model may improve human prediction accuracy. Nonlinear model parameters (Vmax and concentration of serum AVB‐S6‐500 at half‐maximal removal by the target) specific to an antibody:product pair would be estimated from preclinical data. The correct allometric scaling coefficient for Vmax is unclear and would require multiple species (i.e., a larger weight range than a single, purpose‐bred species) to elucidate. Here, we used 0.75 following prior successes in this area.18
AVB‐S6‐500 has the potential to be an important advance in cancer treatment. AVB‐S6‐500’s mechanism of action, targeting GAS6 to prevent signaling via AXL, makes it suitable for use in combination with products that target specific dysregulated signaling pathways. Such multitarget approaches may have the potential to overcome biological barriers to arresting and reducing cancer proliferation and growth. As a fusion protein, AVB‐S6‐500 is not expected to have drug‐drug interactions with cancer therapies metabolized by CYP450.
The model reported here offers many opportunities for further research. The typical, linear PK parameters for AVB‐S6‐500 were entirely estimated from nonhuman primate data, and then scaled to humans by allometric principles. As values in humans are well‐known in the literature,16, 19 they could be adopted wholesale and scaled to nonhuman primates. Thus, nonhuman primate data would be used to inform the nonlinear portion of the model, and the linear portion would be known from the literature and or fixed with Bayesian priors. Another potential improvement would be to model both drug and target concentrations simultaneously, using more complex TMDD models. In this case, we anticipated that much of the target data would be BLOQ; thus, this approach offers little gain at the expense of much greater complexity.
In summary, FIH dosing predictions based on PK/PD modelling of nonclinical data in a relevant species were successful for AVB‐S6‐500, which supports the use of similar modeling approaches for fusion protein products. These results support the use of similar modeling on relevant biomarkers for products where maximal tolerated dosing is not an appropriate guide point due to a lack of observed toxicity in nonclinical studies.
Funding
All studies referenced herein as well as PK/PD model development and manuscript preparation were funded by Aravive. The work was partially funded by Cancer Prevention & Research Institute of Texas, New Company Product Development Award DP150127 Engineered AXL Decoy Receptor for Treatment of AML & Solid Tumors.
Conflict of Interest
All authors are employees of, or consultants to, Aravive. As employees, L.B., A.G., G.M., R.T., and D.P. have stock options.
Author Contributions
L.B., M.D., and G.M. wrote the manuscript. L.B., M.D., A.G., G.M., D.P., and R.T. designed the research. L.B., M.D., A.G., G.M., D.P., and R.T. performed the research. L.B., M.D., A.G., G.M., A.M., D.P., and R.T. analyzed the data.
Supporting information
Supplemental Data. Target‐mediated drug disposition pharmacokinetic/pharmacodynamic model‐informed dose selection for the first‐in‐human study of AVB S6‐500.
Acknowledgments
The authors thank the Cancer Prevention & Research Institute of Texas for the financial support under New Company Product Development Award DP150127 Engineered AXL Decoy Receptor for Treatment of AML & Solid Tumors. Dr. Jing Shang and Mr. Yoshinobu Yokota developed the good laboratory practice analytical methods for AVB‐S6‐500 and serum GAS6 and performing the analyses. Dr. Lisa M. DeTora prepared the initial outline and manuscript drafts for author review. Manuscript preparation was funded by Aravive.
References
- 1. Kariolis, M.S. et al Inhibition of the GAS6/AXL pathway augments the efficacy of chemotherapies. J. Clin. Invest. 127, 183–198 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Fabian, M.A. et al A small molecule‐kinase interaction map for clinical kinase inhibitors. Nat. Biotechnol. 23, 329–336 (2005). [DOI] [PubMed] [Google Scholar]
- 3. Scott, A.M. , Wolchok, J.D. & Old, L.J. Antibody therapy of cancer. Nat. Rev. Cancer. 12, 278–287 (2012). [DOI] [PubMed] [Google Scholar]
- 4. Rankin, E.B. & Giaccia, A.J. The receptor tyrosine kinase AXL in cancer progression. Cancers (Basel) 8, pii: E103 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Rankin, E.B. et al Direct regulation of GAS6/AXL signaling by HIF promotes renal metastasis through SRC and MET. Proc. Natl. Acad. Sci. USA 111, 13373–13378 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Aguilera, T.A. et al Molecular pathways: Oncologic pathways and their role in T‐cell exclusion and immune evasion‐a new role for the AXL receptor tyrosine kinase. Clin. Cancer Res. 23, 2928–2933 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Aguilera, T.A. et al Reprogramming the immunological microenvironment through radiation and targeting AXL. Nat. Commun. 7, 13898 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Kariolis, M.S. et al An engineered Axl 'decoy receptor' effectively silences the GAS6‐AXL signaling axis. Nat. Chem. Biol. 10, 977–983 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Wu, G. et al Molecular insights of Gas6/TAM in cancer development and therapy. Cell Death Dis. 8, e2700 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Goruppi, S. , Ruaro, E. & Schneider, C. Gas6, the ligand of Axl tyrosine kinase receptor, has mitogenic and survival activities for serum starved NIH3T3 fibroblasts. Oncogene 12, 471–480 (1996). [PubMed] [Google Scholar]
- 11. Fridell, Y.W. et al Differential activation of the Ras/extracellular‐signal‐regulated protein kinase pathway is responsible for the biological consequences induced by the Axl receptor tyrosine kinase. Mol. Cell. Biol. 16, 135–145 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. McCloskey, P. et al GAS6 mediates adhesion of cells expressing the receptor tyrosine kinase Axl. J. Biol. Chem. 272, 23285–23291 (1997). [DOI] [PubMed] [Google Scholar]
- 13. Levy, G. Pharmacologic target‐mediated drug disposition. Clin. Pharmacol. Ther. 56, 248–252 (1994). [DOI] [PubMed] [Google Scholar]
- 14. Mager, D.E. & Jusko, W.J. General pharmacokinetic model for drugs exhibiting target‐mediated drug disposition. J. Pharmacokinet. Pharmacodyn. 28, 507–532 (2001). [DOI] [PubMed] [Google Scholar]
- 15. Deng, R. et al Projecting human pharmacokinetics of therapeutic antibodies from nonclinical data. What have we learned? mAbs 3, 61–66 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Dong, J.Q. et al Quantitative prediction of human pharmacokinetics for monoclonal antibodies retrospective analysis of monkey as a single species for first‐in‐human prediction. Clin. Pharmacokinet. 50, 131–142 (2011). [DOI] [PubMed] [Google Scholar]
- 17. Davda, J.P. , Dodds, M.G. , Gibbs, M.A. , Wisdom, W. & Gibbs, J.P. A model‐based meta‐analysis of monoclonal antibody pharmacokinetics to guide optimal first‐in‐human study design. mAbs 6, 1094–1102 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mager, D.E. , Woo, S. & Jusko, W.J. Scaling pharmacodynamics from in vitro and preclinical animal studies to humans. Drug Metab. Pharmacokinet. 24, 16–24 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Dirks, N.L. & Meibohm, B. Population pharmacokinetics of therapeutic monoclonal antibodies. Clin. Pharmacokinet. 49, 633–659 (2010). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Data. Target‐mediated drug disposition pharmacokinetic/pharmacodynamic model‐informed dose selection for the first‐in‐human study of AVB S6‐500.