

Abstract
In the last decade, drug development has tackled substantial challenges to improve efficiency and facilitate access to innovative medicines. Integrated clinical protocols and the investigation of targeted oncology drugs in healthy volunteers (HVs) have emerged as modalities with an increase in scope and complexity of early clinical studies and first‐in‐human (FIH) studies in particular. However, limited work has been done to explore the impact of these two modalities, alone or in combination, on the scientific value and on the implementation of such articulated studies. We conducted an FIH study in HVs with an oncology targeted drug, an Mnk 1/2 small molecule inhibitor. In this article, we describe results, advantages, and limitations of an integrated clinical protocol with an oncology drug. We further discuss and indicate points to consider when designing and conducting similar scientifically and operationally demanding FIH studies.
Study Highlights.
WHAT IS THE CURRENT KNOWLEDGE OF THE TOPIC?
☑ Integrated clinical protocols and investigation of oncology targeted drugs in healthy volunteers (HVs) have emerged as modalities with an increase of complexity and scope of early clinical studies. However, no formal work has assessed the impact of these two approaches, alone or in combination, on the scientific value and on the execution of early clinical studies.
WHAT QUESTION DID THIS STUDY ADDRESS?
☑ This investigation reports on the simultaneous conduct of an integrated clinical protocol and the investigation of an oncology targeted drug, an Mnk 1/2 small molecule inhibitor, in an HV‐first‐in‐human (FIH) study.
WHAT DOES THIS STUDY ADD TO OUR KNOWLEDGE?
☑ This work provides evidence on the benefits and limitations of conducting integrated clinical protocols to investigate oncology targeted drugs in HV‐FIH studies.
HOW MIGHT THIS CHANGE CLINICAL PHARMACOLOGY OR TRANSLATIONAL SCIENCE?
☑ This experience supports the design and the conduct of innovative, albeit scientifically and operationally demanding, FIH integrated protocol studies.
In the last decade, drug development and clinical trials have entered a phase of accelerated innovation of multiple aspects (e.g., study design, execution, etc.), which is fully unfolding in current times.1, 2, 3, 4 The transformation has been driven by multiple factors1, 3, 5, 6, 7 and has spread across all clinical phases tackling common and specific issues2, 7 of early (i.e., phases I and II/proof of concept) and late (i.e., phases III and IV) stages.2, 4, 7, 8 We report and discuss the use of integrated protocols in first‐in‐human (FIH) studies with healthy volunteers (HVs) and oncology drugs.5, 7
In early clinical development, the deeper understanding of disease biology and genetics (i.e., mechanisms of action) of new compounds has shifted FIH studies toward more demanding designs.5, 7 FIH studies tend now to have more mechanistic objectives related to the efficacy and safety of the new drugs and are commonly referred to as “integrated protocols.”9, 10 A universal definition of “integrated protocol” does not exist, and, depending on the authors, the protocol can also be presented as “umbrella,” “adaptive,” or without any specific characterization. The 2017 European Medicines Agency (EMA) guidelines for risk mitigation in FIH and early clinical trials10, 11 broadly describes the “integrated protocol” as “… a protocol that… combine(s) a number of different study parts…” and “…data generated …(are) used to … initiate a subsequent study part … or… components … sequentially or in overlapping fashion…” For the purpose of this paper, we will use “integrated” protocol as a working definition. Two surveys were conducted recently across European countries to understand the use and features of integrated protocols among various stakeholders (e.g., Pharma, academia, etc.). The two reviews covered about 3,000 phase I and phase I−II clinical studies in the decade of 2004–2014. The evaluation showed that integrated protocols have become common practice, representing about 30% of assessed clinical studies.10, 12 The analysis revealed that integrated protocols mainly incorporate two to three different trial elements. These could be single, multiple dose, or food effect, with other adaptive components (e.g., dose increments, sample size, study population, etc.), but the nature and the modality of combinations could vary broadly.10, 12 Published literature associated integrated protocols with increased complexity of design and approval process and with execution delay. Nevertheless, integrated protocols are perceived as the roadway to explore fundamental scientific questions in a time‐efficient and cost‐effective manner.10, 11, 12
In oncology, the advent of noncytotoxic, targeted drugs with a better safety profile compared with conventional chemotherapy agents has prompted the execution of FIH studies in HVs, rather than in patients as historically done.7, 13, 14, 15, 16 In 2012, Iwamoto et al.17 documented 35 studies in HVs investigating ~30 noncytotoxic oncology drugs. The authors did not mention specific categories or drugs but described single dose escalations, multiple doses, pharmacokinetics (PKs), drug interaction, bioavailability, food effect, or pharmacodynamics (PDs) as the most common study types. In the same year, Gupta et al.18 discussed the investigation of noncytotoxic oncology drugs in HVs. The authors mentioned mainly small molecules (i.e., kinase inhibitors such as bosutinib, LY2584702, and GSK2256098) and, less frequently, large molecules such as monoclonal antibodies (e.g., trastuzumab), quoting the same types of studies as Iwamoto et al.17 Subsequently, other papers reported FIHs or phase I investigations with small molecule targeted anticancer drugs in HVs, affirming the interest of such an option in contemporary early drug development.19, 20 All researchers remarked that the administration of noncytotoxic, targeted compounds, to HVs rather than patients with cancer in early‐phase studies, required additional work to comply with stricter regulatory guidelines.21, 22 Even so, the investigation of noncytotoxic, targeted drugs in HVs was associated with better profiling of key safety, PK, and possibly PD and mechanisms of action properties of the drug and, depending on molecules, with a positive impact on the overall development process.19, 20, 23, 24
The mitogen‐activated protein kinase‐interacting protein kinases 1 and 2 (Mnk 1 and 2 kinases) have been recognized as an important therapeutic target for the treatment of malignancies25, 26 and possibly autoimmune disorders, diabetes, and obesity27, 28 (Figure 1). Activated Mnk 1 and 2 kinases (Mnk 1/2) have been shown to phosphorylate eIF4E in a variety of human cancers25, 26 and to regulate cytokines, chemokines, and growth factors in other diseases.27, 28 Moreover, the inhibition of Mnk 1/2 kinases does not affect normal cell growth and development.29
Figure 1.
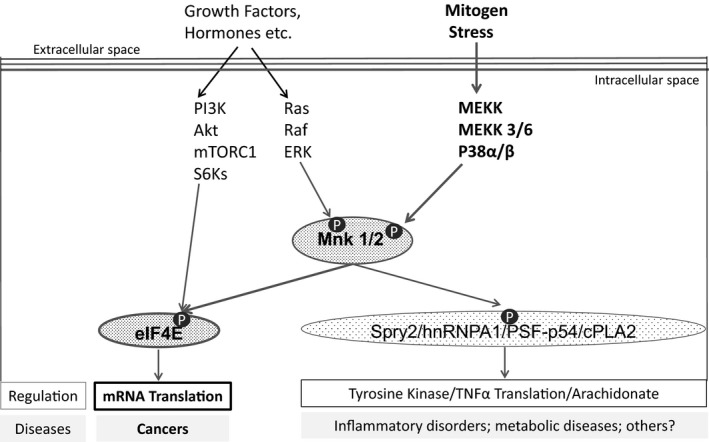
Schematic diagram of Mnk 1/2 signaling. The extracellular signal‐regulated kinase and p38 mitogen‐activated protein kinase pathways phosphorylate and activate Mnk 1/2 kinases. Mnk 1/2 kinases phosphorylate the eukaryotic translation initiation factor 4E, which initiates protein translation and promotes tumor development. Mnk 1/2 kinases affect other less explored targets. The PI3K/Akt/mTOR/S6Ks pathway may also phosphorylate the eIF4E. P, phosphorylate.
ETC‐206 is a small molecule, with potent and selective inhibition of the enzymatic activity of Mnk 1/2 kinases, with half‐maximal inhibitory concentration (IC50) of 64 and 86 nM, respectively. ETC‐206 has been shown to inhibit the phosphorylation of eIF4E in vitro and in vivo, on tumor and surrogate tissues (i.e., hair follicles (HFs) and plasma peripheral blood mononuclear cells (PBMCs), and to decrease plasma levels of pro‐inflammatory cytokines, chemokines, and growth factors in vivo.30
ETC‐206 as a single agent was mildly antioproliferative in in vitro studies with no antitumor activity in an in vivo mouse model of chronic myeloid leukemia in blast crisis (CML‐BC). In combination with tyrosine kinase inhibitors, ETC‐206 impeded survival of stem cells derived from patients with CML‐BC or Philadelphia chromosome–positive acute lymphoblastic leukemia and inhibited tumor growth in the mouse model of CML‐BC.30
ETC‐206 did not show any mutagenicity in a bacterial reverse mutation assay, in in vitro induction of chromosome aberrations or in an in vivo micronucleus test in rat. ETC‐206 showed moderate activity on human ether‐a‐go‐go‐related gene channel (IC50 5.24 μM) and phosphodiesterase 3 (IC50 2.25 μM). Dog was shown to be the more sensitive species compared with rat. In a single oral dose–scalation study in dogs (3, 10, and 30 mg/kg/day), ETC‐206 affected cardiovascular (CV) parameters (i.e., blood pressure, heart rate, and cardiac repolarization) at the 30 mg/kg/day. In a 4‐week study in dogs at the same dose levels, ETC‐206 showed no CV effects, but there was an increase in liver enzymes (alanine aminotransferase, alkaline phosphatase, and gamma glutamyltransferase) and bile duct proliferation (minimal to mild) at the medium and high doses. After drug discontinuation, liver enzymes fully recovered, whereas bile duct proliferation showed a tendency to improve. The no observed adverse effect level (NOAEL) was defined in dogs at dose and exposures well below where the CV and liver findings were observed (i.e., 3 mg/kg/day; total mean peak plasma concentration (Cmax) and area under the concentration‐time curve from time of administration up to the time of the last quantifiable concentration (AUC0−last) of 935 ng/mL and 7,230 ng*hour/mL, respectively).
We report on the integrated clinical protocol in HVs with an oral Mnk 1/2 kinase inhibitor (ETC‐206) developed to treat hematological malignancies. We describe advantages, limitations, and points to consider when conducting an integrated FIH clinical protocol with an oncology targeted drug in HVs.
Methods
Subjects population
The study was approved by the Health Science Authority of Singapore (CTA9900394) and by the Centralised Institutional Review Board of the SingHealth Group of Singapore. The study, planned in healthy male and female subjects, recruited only male subjects (see DISCUSSION). The study was conducted according to the Declaration of Helsinki and Good Clinical Practice guidelines (relevant inclusion and exclusion criteria are given in Supplementary Material, Table S1 ). Participants provided informed consent before starting any study‐related procedure.
Study design and treatment
This was a double‐blind, randomized, placebo‐controlled, within‐subject dose‐escalation study of ETC‐206 to assess safety, PKs, PDs, maximum tolerated dose, electrocardiogram (ECG) changes, and food effect. Figure 2 describes study design, treatment, and other key features of the investigation (study procedures are described in Supplementary Material, Table S2 ).
Figure 2.
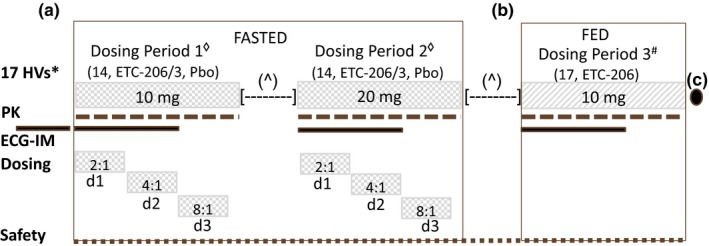
Study design outline, treatments, and key features. (a) Seventeen healthy volunteers were planned to receive single doses of ETC‐206 (n = 14) or placebo (n = 3) under fasted conditions in two consecutive dosing periods (DPs; 10 mg in DP1 and 20 mg in DP2). In each of the two DPs in fasted condition, ETC‐206 and Pbo were given in a staggered manner on day 1, day 2, and day 3 according to the ratio of 2:1, 4:1, 8:1, respectively. (b) In a subsequent DP3#, the entire group of 17 HVs was planned to receive a single dose of 10 mg of ETC‐206 (without Pbo) under fed condition. A minimum of 26 days [—] elapsed between doses for each subject. The next dose was administered after review (^) of safety, tolerability, and pharmacokinetic data from all subjects who received ETC‐206 or Pbo. Subjects were housed for 3 days and monitored ambulatory until day 7 postdose, during each DP. (c) A follow‐up visit was planned on day 14 (3), after the administration of ETC‐206 in the last DP. Electrocardiogram intensive monitoring – standard and triplicate (within 5 minutes) 12‐lead ECGs were performed prior to dosing and at 1, 2, 8, and 24 hours (±10 minutes) after dosing, with the triplicate ECG also being performed at 3, 6, 12, 36, and 48 hours (±10 minutes) after dosing. A 24‐hour continuous ECG recording was conducted on day −1 before DP1, and a 48‐hour continuous ECG recording was conducted on day 1 of each DP, starting prior to drug administration. d1, day 1; d2, day 2; d3, day 3; ECG‐IM, electrocardiogram intensive monitoring; HVs, healthy volunteers; Pbo, placebo; PK, pharmacokinetic.
Safety assessment and stopping criteria
Clinical and laboratory assessments were performed at predose and at multiple time points postdose (during each dosing period) and after the last dosing period. Intensive cardiac monitoring was also conducted during each dosing period (Figure 2). Study safety stopping criteria were implemented (details are provided in Supplementary Material, Table S3 ). Adverse events were evaluated using the National Cancer Institute's Common Terminology Criteria for Adverse Events version 4.03.
PK assessments
Blood samples for ETC‐206 PKs were collected at predose and multiple time points postdose (details are provided in Supplementary Material, Table S4 ).
PD assessments
The levels of relative phosphorylation of eIF4E (p‐eIF4E) in blood (PBMCs), HFs, and skin and the levels of circulating cytokines, chemokines, and growth factors in plasma were assessed at predose and multiple time points postdose (details on methods and time points are provided in Supplementary Material, Table S5 ).
Statistical analysis
Safety analyses were conducted with descriptive statistics (i.e., number of subjects, mean, SD, median, minimum, and maximum) for continuous variables and frequency and percentage for discrete variables (SAS version 9.4; SAS Institute, Cary, NC). PK analyses were conducted using noncompartmental methods (WinNonlin Professional version 6.4; Pharsight, Mountain View, CA). ECG analysis was based on the central tendency of ECG parameter changes from baseline. A categorical analysis was used for outliers. A morphological analysis was conducted for ECG waveform interpretation. The relationships between ETC‐206 concentrations and changes from baseline of ECG‐corrected QT Fridericia's formula (QTcF) were analyzed with a linear mixed effect modeling approach (Central Laboratory ERT, Philadelphia, PA; details are provided in Supplementary Material, Table S6 ). The exposure–response analysis including the calculation of the partial AUC between 0 and 4 hours postdose and the correlation between the AUC of ETC‐206 concentrations and the AUC of p‐eIF4E levels in PBMCs was conducted using SAS version 9.4.
Results
Subject population
From November 2016 to June 2017, a total of 24 HVs were recruited at the SingHealth Investigational Medicine Unit in Singapore. Figure 2 provides study details on planned subjects’ enrollment, dosing periods, oral study drug administration, single dose levels, and fasted or fed conditions. Table 1 describes the subjects’ main baseline demographics characteristics, study drug dose levels, and administration conditions.
Table 1.
Main baseline subjects’ demographics characteristics, study drug dose level, and administration conditions
| Parameters |
Dosing period 1 10 mg FASTED |
Dosing period 2 20 mg FASTED |
Dosing period 3 10 mg FED |
|---|---|---|---|
| Enrolled/dosed | 24/23 | ||
| Male/female, % | 100/0 | ||
| Mean age, years (range) | 36 (23–54) | ||
| Ethnicity: Chinese/Malay (%) | (87/13) | ||
| Dosed (ETC‐206/Pbo) | 17 + 2e (14 + 2e/3) | 17 (14/3) | 11 (7 + 2e/2d) |
| Withdrawals | 6a | 6c | 0 |
| Replacements | 0 | 6b | 0 |
Pbo, placebo.
The panel shows that, in total, 24 Chinese and Malay healthy volunteers participated in the study; 23 subjects (sbjs) received at least one dose of the study drug (ETC‐206 or placebo (Pbo)) and were included for analysis and discussion, unless differently stated. Of these 23 sbjs, 16 sbjs received ETC‐206, 10 mg single doses in fasted condition; 14 sbjs received ETC‐206, 20 mg single doses in fasted condition; 9 sbjs received ETC‐206, 10 mg single doses in fed condition, and 8 sbjs received Pbo, single doses (3 in each fasted and 2 in fed condition). Sbjs allocation over dosing periods (DPs) was the following. DP1: 17 sbjs received 10 mg single dose study drug (14 ETC‐206 and 3 Pbo); 6 of these sbjsa (5 ETC‐206 and 1 Pbo) withdrew (for nonsafety reasons), did not receive the 20 mg study drug in DP2, and were replaced by other 6 sbjsb. DP2: 17 sbjs received 20 mg SD study drug (14 ETC‐206 and 3 Pbo); 6 of these sbjsc withdrew (4 due to prespecified sbj withdrawal criteria; 2 for non‐safety reasons), did not participate in DP3 and were not replaced. DP3: 11 sbjs remaining from DP2 received 10 mg SD, in fed condition, to assess the food effect. Among these 11 sbjs, 9 sbjs received the planned ETC‐206 and 2 sbjsd received unplanned Pbo; of the 9 sbjs that received ETC‐206, 2 sbjse received first the 10 mg single doses of ETC‐206 in fed condition and then the 10 mg single doses of ETC‐206 in fasted condition. Overall, only 7 sbjs received ETC‐206 as 10 mg single doses in both fasted and fed conditions, and were counted to evaluate food effect.
General safety
Table 2 summarizes main safety and tolerability findings for each ETC‐206 dose level, administration conditions, and placebo assignment (details are described in Supplementary Material, Table S7 ).
Table 2.
Main safety and tolerability findings for each ETC‐206 dose level, administration conditions, and Pbo assignment
| Parameters | ETC‐206 10 mg FASTED (N = 16) | ETC‐206 20 mg FASTED (N = 14) | ETC‐206 10 mg FED (N = 9) | Pbo [entire study] (N = 8) |
|---|---|---|---|---|
| SAEs: seizure (%) | 1 (6)a | 0 | 0 | 0 |
| AEs grade 3 CTCAE: ↑CK,a % | 0 | 1 (7)b | 0 | 0 |
| AEs grade 1–2 CTCAE, %c , h | 9 (56) | 11 (79) | 9 (100) | 5 (62) |
| Gastrointestinal disorders,d% | 4 (25) | 3 (21) | 2 (22) | 0 |
| Diarrhea,e % | 3 (19) | 3 (21)g | 2 (22)f | 0 |
| Investigations, %i | 5 (31) | 1 (7) | 1 (11) | 0 |
| ↓WBC (%) | 3 (19) | 0 | 1 (11) | 0 |
| ↑ALT (%) | 1 (6) | 0 | 0 | 0 |
| ↑CK (%) | 1 (6) | 0 | 0 | 0 |
AEs, adverse events; ALT, alanine aminotransferase; CK, creatine kinase; CTCAE, Common Terminology Criteria for Adverse Events; Pbo, placebo; SAE, serious adverse events; WBC, white blood cell.
The panel shows that one SAEa not drug‐related (seizure) was reported in one subject with ETC‐206 at 10 mg dose level. One grade 3 AEb not drug‐related (CK increase) was reported in one subject with ETC‐206 at 20 mg dose level. AEs, grade 1–2,c were reported across all doses and conditions with the exception of gastrointestinal disordersd that were not reported with subjects receiving placebo (Pbo). Diarrheae (grade 1–2) was reported in 19%, 21%, and 22% of subjects receiving ETC‐206 at the dose of 10 and 20 mg in fasted condition and 10 mg in fed condition, respectively, but not in subjects receiving Pbo. About 50% of diarrhea cases were reported as at least possibly drug‐related (as per clinical judgment of the treating physician), with one grade 2 case at the dose of 10 mg in fed condition definitely relatedf to ETC‐206 administration, and with another grade 2 case at the dose of 20 mg in fasted condition, not relatedg. A decrease of white blood cells was observed in 19% of subjects at the dose of 10 mg in fasted condition and in 11% of subjects at the dose of 10 mg in fed condition. ALT and CK increases were observed, each in 6% of subjects, at the dose of 10 mg in fasted condition. Not‐clinically‐significant, out‐of‐the‐reference‐range laboratory values were reported with all doses and conditions except with Pbo (not shown). hAEs observed in >15% of subjects (and not related to local procedures) are reported; iinvestigation alterations observed in >5% of subjects are reported.
PKs
Table 3 displays the ETC‐206 PK parameters for each dose level and administration conditions. At 10 mg single dose in fasted condition (see PK section in Discussion), ETC‐206 met predefined PK study stopping criteria (details are described in Supplementary Material, Table S3 ).
Table 3.
Main PK parameters for each ETC‐206 dose level and administration conditions
| Parameters | ETC‐206 10 mg FASTED (N = 16) [SD] | ETC‐206 20 mg FASTED (N = 14) [SD] | ETC‐206 10 mg (FEDa) (N = 9) [SD] |
|---|---|---|---|
| Mean Cmax (ng/mL) | 927 [154] | 1,950 [315] | 733 [78.9] |
| Mean Tmax (hour) | 1.11 [0.37] | 1.00 [0.32] | 4.62 [1.81] |
| Mean t1/2 (hour) | 22.95 [5.63] | 25.76 [4.12] | 27.57 [5.68] |
| Mean AUC0−t (hour*ng/mL) | 21,909 [5,437] | 57,799 [12,335] | 28,746 [6,198] |
| Mean AUC0−inf (hour*ng/mL) | 24,357 [6,434] | 60,279 [12,451] | 30,047 [6,360] |
| CL/F (L/hour) | 0.44 [0.13] | 0.35 [0.07] | 0.35 [0.08] |
| Vd/F (L) | 13.86 [2.16] | 12.52 [1.52] | 13.35 [1.82] |
The panel illustrates that Cmax was reached on average 1 hour postdose in fasted condition and in about 5 hours in the fed condition, with a mean t½ ~23–28 hour. AUC0−t increased in direct proportion with the dose from 10 to 20 mg single doses in fasted condition. The CL/F was estimated at about 0.4 L/hour for the two dose levels, and the Vd/F was limited to about 13 L. Food administration delayed Tmax (3.0 hour), reduced Cmax (20%), and increased AUC0−inf (25%). No formal conclusion on food effect could be drawn due to the small sample size.
AUC0−inf, area under the concentration‐time curve from zero to infinity; AUC0−t, area under concentration‐time profiles; CL/F, apparent clearance; Cmax, peak plasma concentration; PK, pharmacokinetic; t1/2, terminal half‐life; Tmax, time of maximum plasma concentration; Vd/F, apparent volume of distribution.
a N = 9 (2 healthy volunteers (HVs) received placebo). Only 7 HVs received 10 mg in both FASTED and FED conditions.
Cardiac safety
Figure 3 displays the relationships between ETC‐206 plasma concentration and changes from baseline of ECG‐QTcF (concentration‐effect modeling analysis), for each study drug dose level (ETC‐206 and placebo) and administration conditions. Mean changes of ECG parameters from baseline, time averaged, and placebo corrected for each ETC‐206 dose level and administration conditions are shown in Supplementary Material, Table S8 .
Figure 3.
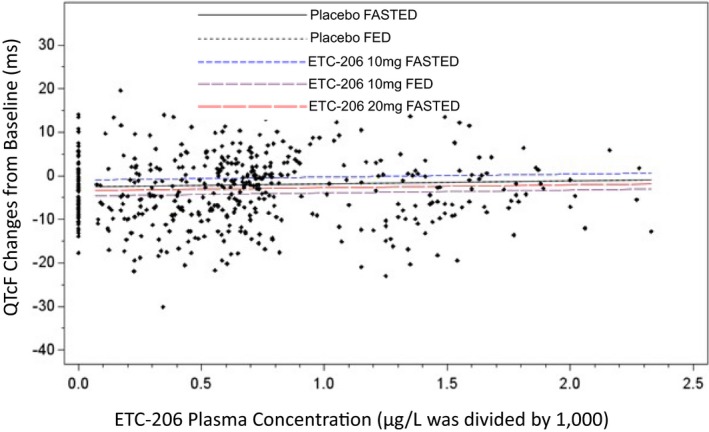
Electrocardiogram‐corrected QTcF changes from baseline vs. ETC‐206 plasma concentration. The mixed effects regression model was used for estimates. Prediction lines are based on model estimates. The chart shows that there is no relationship between ETC‐206 plasma concentrations (at 10 and 20 mg single doses in fasted condition or 10 mg single doses in fed condition) and changes from baseline of QTcF, as shown by the flat slope (P = 0.7) of each dose and condition. Similar findings were for electrocardiogram PR interval, QRS interval, and heart rate (not shown). QTcF, QT Fridericia's formula.
PDs
Figure 4 shows (a) the relative p‐eIF4E mean time course levels in PBMCs (±SD), for each ETC‐206 dose level and administration conditions, together with (b) the correlation of ETC‐206 partial AUC (0–4 hour) with p‐eIF4E partial AUC (0–4 hour) and 95% confidence and prediction limits.
Figure 4.
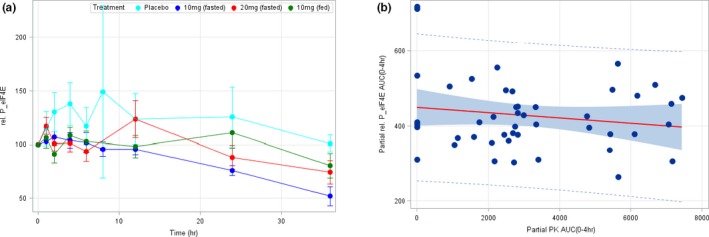
Relative phosphorylation of eIF4E (p‐eIF4E) mean time course levels in peripheral blood mononuclear cells (PBMCs); correlation of ETC‐206 partial area under curve (AUC) with p‐eIF4E partial AUC. (a) Mean relative p‐eIF4E levels in PBMCs (± SD) for placebo and at the doses of 10 mg (fasted), 20 mg (fasted), and 10 mg (fed). (b) Correlation between the partial AUC (0–4 hour) of the PK concentration‐time curve and the partial AUC (0–4 hour) of the relative p‐eIF4E levels in PBMCs (P = 0.00566). The solid red line represents the regression line, the shaded area represents the 95% confidence limits, and the dotted lines identify the 95% prediction limits. AUC, area under the concentration‐time curve; PK, pharmacokinetic.
Discussion
The lack of significant independent antiproliferative activity and the absence of potential therapeutic benefit for patients did not justify a single‐agent FIH study of ETC‐206 in a disease population. Instead, ETC‐206's favorable preclinical toxicology profile, and the lack of effect on growth and development of normal cells, warranted an FIH investigation in HVs. We debate below the challenges and the learnings experienced conducting an FIH integrated clinical protocol in HVs to investigate a noncytotoxic, targeted oncology drug.9, 10, 17, 31
The study
The difficulties related to the multiple parts of the integrated protocol were handled through the study logistics, subjects’ management, and a clear sharing of critical procedures. The intensive cardiac monitoring was the most demanding part, mainly due to the multiplicity of activities required to be planned and coordinated to preserve the scientific value of the assessment. Examples were the elimination of the skin punch biopsies from the 10 and 20 mg single dose fasted periods and the strict timing for plasma sampling, to avoid interference with the ECG data recordings32; the ECG data transmission to a dedicated laboratory for ECG‐QTcF analysis; and the novelty of the methodology to the study site and the Clinical Research Organisation (CRO) executing the study. The hurdles associated with the administration of a noncytotoxic oncology drug to HVs were overcome through the implementation of the following broad range of activities. The absence of any genotoxic effect was confirmed with preclinical in vitro and in vivo investigations13, 21 (see nonclinical safety section above). The starting dose of 10 mg single dose in fasted condition required a greater safety factor in HVs (i.e., 1/10 instead of 1/6 of the human equivalent dose estimated according to the NOAEL observed in dogs)11, 21, 22, 33, 34, 35, 36 (see PKs section below). The robustness of the study design was pursued by means of the dose escalation within subject and the blind allocation of placebo to different subjects at each dose level.13, 37 The mitigation of safety risks associated with a new drug investigation was secured with the adherence to tight exclusion criteria for the intensive ECG monitoring32 (see subject population in methods section); the dosing of new subjects in a staggered manner; the implementation of the “sentinel subject” principle9, 11, 13, 36; and the use of specific subjects’ and study safety stopping criteria as recommended by current literature and guidelines9, 11, 13, 36 (Figure 2). The stopping criteria (details are given in Supplementary Material, Table S3 ) initially reflected the preclinical understanding of ETC‐206 but were subsequently amended based on data from the 10 mg single dose in fasted condition to HVs (e.g., high exposure and long half‐life; see PK section below). Although male and female recruitment was planned, only men were recruited. However, as no protocol‐related issues seemed to cause the prevalent male recruitment, the finding did not limit our capacity to investigate both sexes in the following study in patients with hematologic malignancies. Our experience demonstrates that multiple study parts, noncytotoxic oncology drugs, articulated methodological and patient risk issues, and unexpected findings can be successfully managed in an FIH study in HVs.
General safety
General safety (i.e., signs and symptoms and liver, kidney, heart, and hematology laboratory) did not show any serious adverse events related to ETC‐206 (Table 2). The only emerging adverse events possibly related to the study drug were mild to moderate diarrhea (grades 1 and 2), but no apparent link with Cmax was observed. ETC‐206 was safe and well tolerated at 10 and 20 mg single doses in fasted conditions as well as at 10 mg single doses in fed conditions. The strength of these findings was increased by the blind comparison with placebo at 10 and 20 mg single doses in fasted condition. Overall, the risk profile for the administration of ETC‐206 to HVs was similar to that reported with other nononcology drugs (small molecules) administered to HVs in phase I trials36, 38, 39 as well as with oncology, noncytotoxic, targeted small molecules, administered in single and multiple doses (e.g., up to 8 days) to HVs.14, 40, 41, 42, 43 In our study, ETC‐206 was only administered as two single doses; however, the findings indicate that small molecule targeted drugs as kinase inhibitors (i.e., tyrosine, serine, and threonine) or other inhibitors targeting receptors and enzymes can be safely investigated in HVs18, 22, 24 when supported by adequate preclinical evidence.14, 15, 18 The oral availability of ETC‐206, as with most new noncytotoxic compounds, further emphasizes the potential for a shift from patients to HVs studies. It is well recognized that in HV studies the safety assessment is free from bias due to underlying clinical conditions, concurrent or past diseases, medications, etc., and there is no concern on treating subjects at known ineffective doses. Additionally, the execution of a study with 20–24 subjects requires only 4–6 months for an HV study, compared with 18–24 months for a patient study of the same size, with the cost being substantially lower for the HV study.15, 24 Finally, it is also worth mentioning that, unless the compound causes direct DNA damage, regulatory agencies (e.g., the US Food and Drug Administration (FDA) and the EMA) typically allow dosing in HVs.11, 18, 22 Our work highlights that the investigation of noncytotoxic oncology drugs in HVs does not increase the subjects’ level of risk, improves the definition of the drug profile, and is accepted by regulatory agencies.
PKs
The investigation of ETC‐206 in HVs properly characterized the PK key parameters (Table 3). The starting dose of 10 mg single dose in fasted condition was selected with a safety factor of 1 of 10 (instead of 1 of 6 as per oncology patients) of the human equivalent dose estimated according to the NOAEL observed in dogs.22, 33, 34 The total number of doses (i.e., 6) and dose increment levels were also established on the basis of safety findings and NOAEL observed in dogs (see nonclinical safety section above). The incremental doses (i.e., 20, 40, 60, 80, and 100 mg single doses) were generated using a modified Fibonacci's approach,35 as no predictive preclinical PK/PD model was available. After the 10 mg single dose administration in fasted condition, ETC‐206 showed the total mean Cmax and area under the concentration‐time curve from zero to infinity (AUC0−inf) to be similar to (~1:1) and exceeding (~3 times), respectively, the mean total Cmax (935 ng/mL) and mean total AUC0−last (7,230 ng*hour/mL) observed at the NOAEL in dogs, in the absence of any adverse events. As the ETC‐206 exposures met the original PK study stopping criteria agreed with the local regulatory authority (details are described in Supplementary Material, Table S3 ), the protocol was halted and amended accordingly (see amendment section below). Four of the remaining five escalating dose levels (i.e., 20, 40, 60, 80, and 100 mg single doses) and one of the two groups of 17 HVs originally planned were removed. The dose escalation in fasted condition was limited to the dose level of 20 mg administered to the same group of 17 HVs originally dosed at 10 mg single doses. The 20 mg single dose level was maintained according to simulations conducted using the 10 mg single dose exposure in HVs. The predictions indicated that, due to the higher ETC‐206 plasma protein binding in humans than in dogs, at 20 mg single dose the mean Cmax and AUC0−inf concentrations of the unbound fraction of ETC‐206 were still below the corresponding highest unbound mean free fractions Cmax (11.8 ng/mL) and AUC0−last (670 ng*hour/mL) observed at the NOAEL in dogs. ETC‐206 at 20 mg single doses in fasted condition showed dose proportionality and confirmed previous PK predictions were safe and well tolerated. However, as administrations above 20 mg single dose were expected to surpass the limits of unbound Cmax and AUC0−last associated with the NOAEL in dogs, as agreed with the local regulatory authority, no further ETC‐206 dose escalation was attempted. In absence of dose‐limiting toxicity, the 20 mg single dose was then identified as the maximum administered dose rather than the maximum tolerated dose.16, 36, 38 At 10 mg in fed condition, the drug showed a lower and delayed Cmax and higher AUC0−inf in comparison with the fasted condition; although a food effect was elicited, a formal statistical conclusion could not be made (90% confidence interval for Cmax and AUCs were not totally excluded from the boundaries of no effect),33 as the final number of subjects receiving ETC‐206 in both fasted and fed condition (i.e., 7) was smaller than originally planned (i.e., 17). In both fasted and fed conditions, ETC‐206 showed a mean half‐life ranging from ~23 hours for the 10 mg fasted dose to ~28 hours for the 10 mg fed dose, which was substantially longer than observed in preclinical studies. The modeling of PK results in HVs, together with the absence of a formal food effect exclusion, directed the administration of ETC‐206 in fasted condition with an every‐other‐day schedule in the patients’ study, which was planned to follow. Our results, in line with literature,13 prove that the investigation of new drugs in HVs can accurately describe the PK profile and justify the efforts needed to bring new oncology, noncytotoxic, targeted drugs into this paradigm.14, 18
Cardiac safety
The intensive cardiac monitoring of ECG parameters did not show clinically significant effects of ETC‐206 at 10 or 20 mg single doses on heart rate, atrioventricular conduction, cardiac depolarization, and repolarization or ECG wave morphology (details are given in Table S8 ). The absence of clinically relevant effects on cardiac repolarization was also indicated by the lack of a significant change of QTcF in the PK/PD model (i.e., flat slope for the relationship; Figure 3) and by the lack of significant categorical outliers for QTcF. Overall, the study showed no evidence for ETC‐206 proarrhythmic risks in the range of doses explored. The results supported the continuation of ETC‐206 investigation in a patient population of hematologic malignancies (in combination with another anticancer drug). It is worth noting that, under given conditions (e.g., justified benefit/risk ratio), published data support the use of drugs potentially affecting QT not only in oncology but also in other diseases.44 The assessment of cardiac safety of new compounds in FIH studies is a matter of innovation in early clinical development. Since the mid‐2000s, the so‐called thorough QT/corrected QT (QTc) study (TQT), assessing cardiac repolarization (ECG/QTc) and arrhythmogenic potential, is mandatory for all new drugs.32 Normally, the TQT study is conducted at an advanced stage of drug development (e.g., after phase II), as that allows the investigation of the effect of a new drug at known therapeutic and supratherapeutic doses.32 However, the TQT study (with placebo and active controls) is resource‐intensive, needs a large number of subjects, is very expensive, and does not always fully address the safety questions.45, 46 Meanwhile, FIH studies, with their broad range of drug doses and exposures investigated, are emerging as an interesting setting to explore the effect of drugs on cardiac repolarization and their arrhythmogenic potential.45, 46 This option has been intensively discussed in the past decade (i.e., sponsors, investigators, and regulatory agencies) with the intent of identifying alternatives45, 46, 47 and possibly waiving TQT studies.48 Our case exemplifies that the accurate investigation of cardiac repolarization (and potential fatal arrhythmias) is feasible even in the complex frame of an FIH integrated protocol with an oncology, noncytotoxic, targeted drug.
PDs
The p‐eIF4E and the regulation of cytokines, chemokines, and growth factors are downstream effects of Mnk 1/2 signaling and are involved in the development of cancers and other diseases25, 26, 27, 28 (see Introduction). Moreover, ETC‐206 was shown to affect levels of p‐eIF4E, cytokine, chemokines, and growth factors in vitro and/or in vivo (see Introduction). For these reasons, the above indicators have been selected as potential PD biomarkers.
The assessment of the effect of SDs of ETC‐206 on relative p‐eIF4E levels showed a large variability (see Figure 4). Nevertheless, a signal of the treatment effect with respect to the placebo response was observed (Figure 4 a) in the initial part of the time course of the relative p‐eIF4E levels in PBMCs (i.e., ≤4 hours postdose). To better assess the magnitude of this signal and the potential relationship with the ETC‐206 plasma levels, the partial area under the ETC‐206 plasma levels (AUC PK (0–4 hour)) and the partial area under the relative p‐eIF4E levels (AUC rel. p‐eIF4E (0–4 hour)) have been computed (Figure 4 b). A statistically significant correlation (P = 0.00566) has been found between these two measures, indicating that as the ETC‐206 plasma levels increase, the relative p‐eIF4E levels decrease. This finding may indicate the presence of an exposure–response relationship between ETC‐206 and a target biomarker and deserves further investigations in coming studies. ETC‐206 did not show any activity on relative p‐eIF4E levels on HFs and skin, or on plasma cytokines, chemokines, and growth factors (i.e., IL‐1β, IL‐2, IL‐4, IL‐6, IL‐8, IFNγ, TNFα; IL‐15, IL‐17A; MCP‐1, and IP‐10).49 Our findings denote that HVs are a valid setting to explore mechanistic hypotheses.
Amendments
Two substantial protocol amendments were made after the start of the clinical study. Neither of these two amendments could be anticipated, and they were not covered by the flexibility of the protocol language already agreed with the local regulatory authority and institutional review board. The first was related to the notable number of subjects with electrolyte (Ca, K, Mg, and Na) values outside the reference range causing a high recruiting failure. That required a rephrase of some exclusion criteria to allow the safe enrollment of subjects with electrolyte values not exceeding grade 1 of Common Terminology Criteria for Adverse Events version 4.03 (relevant inclusion and exclusion criteria are described in Supplementary Material, Table S1 ). The second was related to the high Cmax and AUC0−last observed while dosing ETC‐206 in HVs at 10 mg single doses in fasted condition (see PK section above). That led to the redesign of the study with removal of one group of HVs and four dose levels (40, 60, 80, and 100 mg) initially planned; restriction of the administration of the single dose of 20 mg of ETC‐206 to one group of HVs only (this amendment was also used to insert a later time point (30 hours) for relative p‐eIF4E levels assessment in PBMCs (see Figure 4). The number of amendments was lower than that reported by other authors with integrated phase I protocols10, 12, 50, and they were not attributable to the nature of the study. Furthermore, in contrast with other published data, the integrated nature of our protocol did not cause delays in approval of the study or of subsequent amendments.10, 50 In our case, the main reason for delay was the cited PK result with the related protocol amendment. The finding caused an extension of study timelines triggering the (project) decision to not replace subjects dropped during dosing period 2 (Table 1). Nonetheless, the investigation was completed in <7 months albeit with a smaller sample size and a reduced capacity of detecting food effect. Our experience indicates that an integrated clinical protocol investigating an oncology targeted drug in HVs can be extremely informative and does not necessarily entail a high number of amendments or cause delay (see sections above).
Conclusions
In this paper, we reported on the advantages and limitations of the simultaneous conduct of an integrated clinical protocol and the investigation of oncology targeted drugs in an FIH‐HV study. The multiple components of an integrated protocol, and in particular the intensive cardiac monitoring, required greater efforts in planning and coordination. For example, vein needle punctures and skin biopsy had to be carefully scheduled and/or limited to avoid artifacts in the intensive ECG monitoring. The safe investigation of ETC‐206, an oncology targeted drug, in HVs mandated the conduct of genotoxic studies (not needed for patients with cancer). Moreover, the starting dose in HVs was lower than in patients with cancer due to the need for a higher safety margin with the potential to explore more dose levels to reach clinically relevant concentrations. The use of placebo, stopping criteria, and the “sentinel subject” principle increased the complexity of study preparation and execution. Nevertheless, we learned that in HVs the administered doses of ETC‐206 were safe and well tolerated; the total exposure was higher, and the half‐life was longer than expected from animal studies; QTc or ECG changes were not of concern; PD signals could be detected; and some food effect might exist. The three main limitations of this study were the restriction of the skin punch biopsy only to the fed group, the lack of a formal detection of food effect, and the absence of a repeated dose escalation readout. However, although the first limitation indicates that integrated protocols inevitably reach feasibility limits, the other two were unrelated to the nature of the study. The results of this work guided us to review our understanding of ETC‐206 and to redesign the next study in the patient population and proved that intensive ECG monitoring while preserving scientific integrity is technically feasible. Many factors, compound or project related, can drive the choice of the most appropriate setting (i.e., patients or HV) to conduct an FIH study. Our experience shows that although integrated FIH studies in HVs with oncology drugs are complex and demanding, they can be executed safely and in a time‐ and cost‐efficient manner to obtain critical information. In specific circumstances, they can offer a unique (temporal and scientific) opportunity to interrogate a compound and potentially impact its entire development process.
Funding
This study was supported by funding from the National Medical Research Council (NMRC), National Research Foundation (NRF), and A*STAR (BMRC), Singapore.
Conflicts of Interest
Vincenzo Teneggi, Veronica Novotny‐Diermayr, Lay Hoon Lee, Maryam Yasin, Kantharaj Ethirajulu, Pauline Yeo, Sylvia Bong Hwa Gan, Stephanie E. Blanchard, Ranjani Nellore, Dhananjay N. Umrani, and Alex Matter are employed at D3, the sponsor of the study. All other authors declared no competing interests for this work.
Author Contributions
V.T. wrote the manuscript. V.T., V.N.‐D., Q.S.L., and A.M. designed the research. V.T., V.N.‐D., D.L.W.T., G.L., Y.C., M.Y., P.Y., K.E., D.N.U., S.E.B., and R.N. performed the research. V.T., Q.S.L., V.N.‐D., R.G., and S.B.H.G. analyzed the data. L.H.L. contributed new reagents/analytical tools.
Supporting information
Table S1. Main General Inclusion/Exclusion Criteria and Specific Exclusion.
Table S2. Study Procedures Overview.
Table S3. Study Stopping Criteria.
Table S4. Methods for Pharmacokinetic Sampling.
Table S5. Methods for Pharmacodynamic Sampling.
Table S6. Statistical Methods.
Table S7. Summary of Treatment‐emergent Adverse Events.
Table S8. Mean changes of ECG parameters from baseline, time averaged, etc.
Acknowledgments
General safety assessments were done by Quest Laboratories, Singapore. The PKs analysis was conducted by PhinC Development, Évry, France. The cardiac safety analysis was conducted by ERT, Philadelphia, PA, USA.
References
- 1. Baird, L.G. et al Accelerated access to innovative medicines for patients in need. Clin. Pharmacol. Ther. 96, 559–571 (2014). [DOI] [PubMed] [Google Scholar]
- 2. Berry, D.A. Emerging innovations in clinical trial design. Clin. Pharmacol. Ther. 99, 82–91 (2016). [DOI] [PubMed] [Google Scholar]
- 3. Leyens, L. & Brand, A. Early patient access to medicines: health technology assessment bodies need to catch up with new marketing authorization methods. Public Health Genom. 19, 187–191 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Eichler, H.G. & Sweeney, F. The evolution of clinical trials: can we address the challenges of the future? Clin. Trials 15, 27–32 (2018). [DOI] [PubMed] [Google Scholar]
- 5. Ivy, S.P. , Siu, L.L. , Garrett‐Mayer, E. & Rubinstein, L. Approaches to phase 1 clinical trial design focused on safety, efficiency, and selected patient populations: a report from the clinical trial design task force of the national cancer institute investigational drug steering committee. Clin. Cancer Res. 16, 1726–1736 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Silva, H. et al A systems approach to enhance clinical research and medicines development. J. Med. Develop. Sci. 1, 59–67 (2015). < 10.18063/jmds.2015.01.004>. [DOI] [Google Scholar]
- 7. Wong, K.M. , Capasso, A. & Eckhardt, S.G. The changing landscape of phase I trials in oncology. Nat. Rev. Clin. Oncol. 13, 106–117 (2016). [DOI] [PubMed] [Google Scholar]
- 8. Dhingra, K. Oncology 2020: a drug development and approval paradigm. Ann. Oncol. 26, 2347–2350 (2015). [DOI] [PubMed] [Google Scholar]
- 9. Ponzano, S. , Blake, K. , Bonelli, M. & Enzmann, H. , on behalf of the European Medicines Agency Committee for Human Medicinal Products “First‐in‐Human Guideline Drafting Group” . Promoting safe early clinical research of novel drug candidates: a European Union regulatory perspective. Clin. Pharmacol. Ther. 103, 564–566 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Erb‐Zohar, K. , Sourgens, H. & Breithaupt‐Groegler, K. Use of integrated clinical trial protocols – a survey in early medicines development. Int. J. Clin. Pharmacol. Ther. 56, 205–211 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. EMEA/CHMP/SWP/28367/07 Rev. 1 . Guideline on strategies to identify and mitigate risks for first‐in‐human and early clinical trials with investigational medicinal products. (2017). [DOI] [PMC free article] [PubMed]
- 12. Fruhner, K. , Hartmann, G. & Sudhop, T. Analysis of integrated clinical trial protocols in early phases of medicinal product development. Eur. J. Clin. Pharmacol. 73, 1565–1577 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Griffin, J. P. & O'Grady, J. Pharmaceutical Medicine, 5th edn., chapter 4, page 168 (Blackwell Publishing, Oxford, UK, 2006). [Google Scholar]
- 14. Kummar, S. , Gutierrez, M. , Doroshow, J.H. & Murgo, A.J. Drug development in oncology: classical cytotoxics and molecularly targeted agents. Br. J. Clin. Pharmacol. 62, 15–26 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Salzberg, M. First‐in‐human phase 1 studies in oncology: the new challenge for investigative sites. Rambam Maimonides Med. J. 3, 1–4 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. EMA/CHMP/205/95 Rev.5 . Committee for Medicinal Products for Human Use (CHMP). Guideline on the evaluation of anticancer medicinal products in man (2017).
- 17. Iwamoto, M. , Iannone, R. & Wagner, J. A. Use of healthy volunteers drives clinical oncology drug development decision making. Clin. Pharmacol. Ther. 92, 571–574 (2012). [DOI] [PubMed] [Google Scholar]
- 18. Gupta, P. , Gupta, V. & Gupta, Y.K. Phase I clinical trials of anticancer drugs in healthy volunteers: need for critical consideration. Indian J. Pharmacol. 44, 540–542 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Xiong, H. et al Studying Navitoclax, a targeted anticancer drug, in healthy volunteers – ethical considerations and risk/benefit assessments and management. Anticancer Res. 34, 3739–3746 (2014). [PubMed] [Google Scholar]
- 20. Yago, M.R. et al The use of betaine HCl to enhance dasatinib absorption in healthy volunteers with rabeprazole‐induced hypochlorhydria. AAPS J. 16, 1358–1365 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. US Food and Drug Administration . Guidance for Industry. M3(R2) Nonclinical Safety Studies for the Conduct of Human Clinical Trials and Marketing Authorization for Pharmaceuticals (2010). [PubMed]
- 22. US Food and Drug Administration . Guidance for Industry. S9 Nonclinical Evaluation for Anticancer Pharmaceuticals (2010).
- 23. Dagher, R.N. , Rosario, L.A. , Morse, D.E. & Pazdur, P. Regulatory considerations for early clinical studies of anti‐cancer drugs in healthy volunteers. J. Clin. Oncol. 23, 3101 (2005). [Google Scholar]
- 24. Offman, E . Can small molecule oncology drugs be tested in healthy subjects? <https://www.celerion.com/wp-content/uploads/2016/01/Celerion_-2015-JSCPT_Can-Small-Molecule-Oncology-Drugs-Be-Tested-in-Healthy-Subjects.pdf>.
- 25. Hou, J. , Lam, F. , Proud, C. & Wang, S. Targeting Mnks for cancer therapy. Oncotarget 3, 118–131 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Dreas, A. , Mikulski, M. , Milik, M. , Fabritius, C.H. , Brzózka, K. & Rzymski, T. Mitogen‐activated protein kinase (MAPK) interacting kinases 1 and 2 (MNK1 and MNK2) as targets for cancer therapy: recent progress in the development of MNK inhibitors. Curr. Med. Chem. 24, 3025–3053 (2017). [DOI] [PubMed] [Google Scholar]
- 27. Joshi, S. & Platanias, L.C. Mnk kinase pathway: cellular functions and biological outcomes. World J. Biol. Chem. 5, 321–333 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Moore, C.E. et al MNK1 and MNK2 mediate adverse effects of high‐fat feeding in distinct ways. Sci. Rep. 6, 1–15 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Ueda, T. , Watanabe‐Fukunaga, R. , Fukuyama, H. , Nagata, S. & Fukunaga, R. Mnk2 and Mnk1 are essential for constitutive and inducible phosphorylation of eukaryotic initiation factor 4E but not for cell growth or development. Mol. Cell. Biol. 24, 6539–6549 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Yang, H. et al Optimisation of selective mitogen‐activated protein kinase interacting kinases 1 and 2 (MNK 1/2) inhibitors for the treatment of blast crisis leukaemia. J. Med. Chem. 61, 4348–4369 (2018). [DOI] [PubMed] [Google Scholar]
- 31. US Food and Drug Administration . Clin Trial Gov (2018). <https://clinicaltrials.gov/>. Accessed July 23, 2018).
- 32. ICH E14: The Clinical Evaluation of QT/QTc Interval Prolongation and Proarrhythmic Potential for Non‐antiarrhythmic Drugs. Questions and Answers (R3) (2017).
- 33. US Food and Drug Administration . Guidance Document: Food‐Effect Bioavailability and Fed Bioequivalence Studies (2002).
- 34. EMA/CHMP . Guideline of the Evaluation of Anticancer Medicinal Products in Man. (2006).
- 35. Penel, N. & Kramar, A. What does a modified‐Fibonacci dose‐escalation actually correspond to? BMC Med. Res. Methodol. 12, 103 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Sibille, M. , Patat, A. , Caplain, H. & Donazzolo, Y. A safety grading scale to support dose escalation and define stopping rules for healthy subject first‐entry‐into‐man studies: some points to consider from the French Club Phase I working group. Br. J. Clin. Pharmacol. 70, 736–748 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Agyeman, A.A. & Ofori‐Asenso, R. First in human trials: are there any benefits of including placebo treatments? Drug Dev. Ther. 7, 73–74 (2016). [Google Scholar]
- 38. Sibille, M. , Deigat, N. , Janin, A. , Kirkesseli, S. & Durand, D.V. Adverse events in phase‐I studies: a report in 1015 healthy volunteers. Eur. J. Clin. Pharmacol. 54, 13–20 (1998). [DOI] [PubMed] [Google Scholar]
- 39. Emanuel, E.J. , Bedarida, G. , Macci, K. , Gabler, N.B. , Rid, A. & Wendler, D. Quantifying the risks of non‐oncology phase I research in healthy volunteers: meta‐analysis of phase I studies. BMJ 350, 1–9 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Greenwood, D. , Stratford, I. & Booth, S. A review of first‐in‐human small molecule oncology clinical trials. Int. J. Clin. Trials 3, 77–88 (2016). [Google Scholar]
- 41. Lutfullin, A. , Kuhlmann, J. & Wensing, G. Adverse events in volunteers participating in phase I clinical trials: a single center five year survey in 1,559 subjects. Int. J. Clin. Pharmacol. Ther. 43, 217–226 (2005). [DOI] [PubMed] [Google Scholar]
- 42. Gupta, S. et al Meta‐analysis of the relationship between dose and benefit in phase 1 targeted agent trials. Natl. Cancer Inst. 104, 1860–1866 (2012). [DOI] [PubMed] [Google Scholar]
- 43. US Food and Drug Administration . Guidance document: estimating the maximum safe starting dose in initial clinical trials for therapeutics in adult healthy. Volunteers (2005). [Google Scholar]
- 44. Redfern, W.S. et al Relationships between preclinical cardiac electrophysiology, clinical QT interval prolongation and torsade de pointes for a broad range of drugs: evidence for a provisional safety margin in drug development. Cardiovasc. Res. 58, 32–45 (2003). [DOI] [PubMed] [Google Scholar]
- 45. Darpo, B. et al The IQ‐CSRC prospective clinical phase 1 study: “Can early QT assessment using exposure response analysis replace the thorough QT study?”. Ann. Noninvasive Electrocardiol. 19, 70–81 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Grandi, E. , Morotti, S. , Pueyo, E. & Rodriguez, B. Editorial: safety pharmacology – risk assessment QT interval prolongation and beyond. Front. Physiol. 9, 1–5 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Darpo, B. et al Results from the IQ‐CSRC prospective study support replacement of the thorough QT study by QT assessment in the early clinical phase. Clin. Pharmacol. Ther. 97, 326–335 (2015). [DOI] [PubMed] [Google Scholar]
- 48. Nelson, C.H. et al A quantitative framework to evaluate proarrhythmic risk in a first‐in‐human study to support waiver of a thorough QT study. Clin. Pharmacol. Ther. 98, 630–638 (2015). [DOI] [PubMed] [Google Scholar]
- 49. Teneggi, V. et al First‐in‐Human Phase 1 Study of ETC‐206 an oral MNK 1/2 Kinase Inhibitor in Healthy Volunteers (HVs). (ASCPT, Orlando, FL, 2018). [Google Scholar]
- 50. Getz, K.A. et al The impact of protocol amendments on clinical trial performance and cost. Ther. Innov. Regul. Sci. 50, 436–441 (2016). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Main General Inclusion/Exclusion Criteria and Specific Exclusion.
Table S2. Study Procedures Overview.
Table S3. Study Stopping Criteria.
Table S4. Methods for Pharmacokinetic Sampling.
Table S5. Methods for Pharmacodynamic Sampling.
Table S6. Statistical Methods.
Table S7. Summary of Treatment‐emergent Adverse Events.
Table S8. Mean changes of ECG parameters from baseline, time averaged, etc.