

Abstract
Endothelin-1 (ET-1) and its two receptors, endothelin receptor A (ETAR) and endothelin receptor B (ETBR) exhibit deregulated overexprerssion in pancreatic ductal adenocarcinoma (PDAC) and pancreatitis. We examined the expression pattern of endothelin (ET) axis components in the murine models of chronic and acute inflammation in the presence or absence of oncogenic K-ras. While the expression of endothelin converting enzyme-1 (ECE-1), ET-1, ETAR and ETBR in the normal pancreas is restricted predominantly to the islet cells, progressive increase of ET receptors in ductal cells and stromal compartment is observed in the KC model (Pdx-1 Cre; K-rasG12D) of PDAC. In the murine pancreas harboring K-rasG12D mutation (KC mice), following acute inflammation induced by cerulein, increased ETAR and ETBR expression is observed in the amylase and CK19 double positive cells that represent cells undergoing pancreatic acinar to ductal metaplasia (ADM). As compared to the wild type (WT) mice, cerulein treatment in KC mice resulted in significantly higher levels of ECE-1, ET-1, ETAR and ETBR, transcripts in the pancreas. Similarly, in response to cigarette smoke-induced chronic inflammation, the expression of ET axis components is significantly upregulated in the pancreas of KC mice as compared to the WT mice. In addition to the expression in the precursor pancreatic intraepithelial neoplasm (PanIN lesions) in cigarette smoke-exposure model and metaplastic ducts in cerulein-treatment model, ETAR and ETBR expression is also observed in infiltrating F4/80 positive macrophages and α-SMA positive fibroblasts and high co-localization was seen in the presence of oncogenic K-ras. In conclusion, both chronic and acute pancreatic inflammation in the presence of oncogenic K-ras contribute to sustained upregulation of ET axis components in the ductal and stromal cells suggesting a potential role of ET axis in the initiation and progression of PDAC.
Keywords: Endothelin, Acinar-ductal metaplasia, PanIN lesions, Oncogenic K-ras, Pancreatic injury, Precancerous lesions, Pancreatitis
Introduction
Cellular plasticity is critical for tissue repair and regeneration. Cellular reprogramming in the exocrine pancreas under benign conditions involves a reversible trans-differentiation sequence of acinar to ductal phenotype during repair and resolution of inflammation [1]. The presence of constitutively active K-ras mutations however, disrupt this sequence and irreversibly maintain the ductal reprogramming leading to the development of pre-neoplastic lesions termed as pancreatic intraepithelial neoplasia (PanIN). The PanIN lesions further undergo a series of histological and morphological changes and progress into malignant pancreatic ductal adenocarcinoma (PDAC). The most commonly used genetically engineered mouse (GEM) model of PDAC generated by knock-in of mutant K-ras allele (G12D) exhibits acinar to ductal metaplasia (ADM) and develops a complete spectrum of PanIN lesions leading to PDAC. This process is characterized by the loss of acinar cell compartment (markers like amylase) and gain of the ductal phenotype, characterized by CK19 expression [2]. The earliest evidence of ADM was provided by studies in transgenic mice overexpressing TGF-α in the pancreas [3]. These animals develop fibrosis and dysplastic lesions accompanied by loss of zymogen granules and acquisition of ductal features [4], implying that maintenance of acinar cell organization is critical to prevent trans-differentiation into ductal architecture [5]. Activation of the mutant K-ras in the cells of the acinar lineage under the control of elastase or Mist1 promoter led to spontaneous low-grade lesion formation in the mouse pancreas. However, progression to high-grade PanIN lesions requires chronic inflammation [6]. Epidemiological studies also suggest that patients with chronic pancreatitis (CP) exhibit an elevated risk of developing PDAC. Indeed, constitutive activation of K-ras in CP patients is associated with malignant transformation of the pancreas [7]. Further, cerulein-induced CP in mice expressing oncogenic K-ras (K-rasG12V) resulted in the development of high grade lesions along with increased infiltration of inflammatory cells [8], atrophy of pancreatic parenchyma, development of metaplastic cells via inhibition of oncogene mediated senescence [9]. Carriere et al. demonstrated that the episodes of cerulein induced pancreatitis favor rapid cancer progression and initiate a cascade of events in mice expressing mutated K-ras in the nestin-positive cell lineage [10], [11]. Interestingly, in acute pancreatitis (AP) model, mutant K-ras favors reprogramming and metaplastic conversion of acinar cells into precancerous lesions in a beta-catenin dependent manner [1].
Components of endothelin axis, which include endothelin-1 (ET-1), endothelin-2 (ET-2), endothelin-3 (ET-3), endothelin converting enzymes (ECEs) along with high affinity G protein-coupled receptors, endothelin A receptor (ETAR) and endothelin B receptor (ETBR), are deregulated in inflammation and cancer. Accumulating evidence suggests that ET-1 plays a significant role in the pathophysiology of pancreatic inflammation. In patients with severe AP, an elevated level of ET-1 correlated with disease severity and inflammation [12]. In addition, significantly elevated circulating levels of ET-1 and its strong expression in the pancreas of CP patients with a history of smoking was observed [13]. In experimental pancreatitis models induced by cerulein or sodium taurocholate a remarkable increase in the serum ET-1 level and damage to pancreatic parenchyma was observed [14], [15], [16]. Importantly, administration of ET-1 excerbates cerulein-induced pancreatic damage and a remarkable change in the pancreas morphology compared to cerulein alone [17]. ET-1 favors acinar cell necrosis, edema, increase in serum amylase and elastase levels, and promotes inflammatory response indicating the role of ET-1 in disease aggravation. Several pancreatic cancer cell lines produce high levels of ET-1 suggesting the possible role of ET axis in PDAC. [18], [19]. A recent study has also demonstrated that ET-1 and ETBR are overexpressed in human PDAC tissues [20] while we have observed the upregulation of ET-1, ETAR and ETBR in tumor cells and various compartments of tumor microenvironment (Gupta et al, unpublished observations).
Given the functional involvement of ET-1 in pancreatic inflammation, expression of ET axis components in PDAC, the role of oncogene-associated inflammation in driving pancreatic neoplastic transformation, and the ability of ET axis to exert pleiotropic effects to promote tumorigenesis, it is possible that ET axis plays a key role in inflammation-driven pancreatic tumorigenesis in the presence of constitutively active oncogenic K-ras mutant. However, the expression of ET axis proteins in premalignant lesions in underlying acute and chronic pancreatic inflammation associated with oncogenic K-ras remains unexplored. We therefore examined the expression pattern of ET axis components in the murine models of chronic and acute inflammation in the presence and absence of oncogenic K-ras. The findings from the current study, demonstrate that cerulein-induced acute inflammatory insult results in the upregulation of ET-1 and ETAR expression, which is subsequently restored to basal levels in mice with wild type K-ras. However, in the presence of mutant K-ras, there is a sustained upregulation of ET axis components with progressive neoplasia. Cigarette smoke exposure, a model of chronic inflammation, in KC mice results in increased expression of ET axis components in the precancerous lesions and pancreatic stroma indicating that oncogenic K-ras mutation results in sustained activation of ET axis in the pancreatic tissues. Overall, the present work suggests a possible role of ET axis in pancreatic inflammation, repair and progression to adenocarcinoma in the presence of oncogenic K-ras.
Materials and methods
Cerulein treatment and smoke exposure in vivo
For the studies described herein, we used pancreatic tissues from mice expressing wild type or mutant K-ras(G12D) that were treated with either cerulein or exposed to cigarette smoke for our recently described studies [21], [22]. For cerulein treatment 6 week old KC and WT mice were given 8 hourly intraperitoneal injections (75 µg/kg body weight) on two alternate days and mice were euthanized at 2, 7 and 21 days post-treatment. Saline-treated mice were used as controls (denoted as day 0 in the data) [21]. The KC and WT mice were either sham treated (sham) or exposed to cigarette smoke (smoked) for 20 weeks, 3 h twice a day (150 mg total suspended particles/m3) starting at 10 weeks of age. This exposure regimen resulted in circulating cotinine levels of 125.0 ± 15.6 ng/ml [22].
RNA isolation and real-time quantitative reverse transcriptase (qRT) PCR
Total RNA was isolated from mice pancreas using mirVanaTM isolation kit (Ambion, Foster city, CA, USA). Genomic DNA contamination was removed by DNase treatment using RNeasy mini kit (Qiagen) and integrity of the isolated RNA was measured and processed for reverse transcription. The isolated RNA was converted to cDNA after hybridization with oligo dT using Superscript II Reverse Transcriptase (Invitrogen). The cDNA was used to profile the expression of indicated genes using gene specific primers (Supplementary Table I) normalized to internal control GAPDH by quantitative real time PCR analysis (LightCycler 480 Instrument II, Roche). The relative expression was calculated using delta-delta method using Real Time PCR.
Immunohistochemistry in mice tissues
Slides were baked overnight at 56 °C and deparaffinized with xylene followed by rehydration with decreasing concentration of ethanol. The peroxidase activity was quenched by 3% H2O2 followed by antigens retrieval using 0.01 M preheated citrate buffer (pH 6, 95 °C). After cooling at room temperature (RT), tissue sections were blocked using 2.5% horse serum (ImmPress Universal antibody kit; Vector Laboratories, Burlingame, California, USA) for 2 h and incubated overnight at 4 °C with respective primary antibodies diluted in PBS: anti-ETAR, 1:1000, Abcam, ab117521; anti-ETBR, 1:2000, Abcam, ab117529; anti-ET-1, 1:2000, Abcam, ab117521; anti ECE-1, 1:750, Abcam, ab189843. The slides were washed with PBST (4 washes for 10 min each) and incubated with secondary antibody (ImmPRESS® Universal antibody kit; Vector Laboratories, Burlingame, California, USA) for 30 min at RT. The slides were washed with PBST (4 washes for 10 min each), developed using 3-3′ diaminobenzidine solution (DAB substrate kit, Vector Laboratories) and counter stained with Gill’s hematoxylin for 5 min (Vector Laboratories). Tissues were dehydrated, air dried and mounted with Permount solution (Fischer Scientific, Pittsburg, Pennsylvania, USA). A pathologist at UNMC (Dr. Talmon) using a Nikon light Microscope scored all stained slides and images of the particular area were taken. The staining intensity for ECE-1, ETAR, ETBR, and ET-1 was graded on a scale of 0–3, 0 being negative and 3 strongly positive. The proportion positive of cell for each of the molecule in a given specimen was scored between 1–4 indicating 0–25% for intensity 1, 26–50% for intensity 2, 51–75% for intensity 3 and 76–100% for intensity 4. A composite score was then calculated by multiplying the staining intensity and proportion positivity in a range between 0–12.
Immunofluorescence
The tissues were baked overnight at 56 °C and dewaxed using xylene by washing for 10 min each. After deparaffinization tissues were rehydrated using decreasing concentrations of ethanol and washed with PBS. The tissues were fixed in 100% methanol for 30 min followed by antigen retrieval in 0.01 M preheated citrate buffer (pH 6, 95 °C) in microwave for 15 min. The tissues were blocked with 2.5% horse serum and incubated with primary antibodies overnight diluted in PBS: anti-ETAR, 1:300, Alomone Labs, AER-001; anti-ETAR, 1:300, Santacruz, sc-135902; anti-ETBR, 1:300, Alomone labs, AER-002; anti-ETBR, 1:300, Santacruz, sc-21196; anti-CK19, 1:200, TROMA III, anti-F4/80, 1:80, e-Biosciences, 14-4801-82; anti-α-SMA, 1:200, Abcam, ab7817; anti-amylase 1:300, Sigma. After overnight incubation the slides were washed with PBS thrice for 10 min and incubated for 30 min at room temperature with secondary antibody (FITC conjugated anti-rat, Texas Red conjugated anti-rabbit and FITC conjugated anti-mouse, Alexa 647 conjugated anti-mouse and Alexa 647 conjugated anti-rabbit). The slides were washed thrice with PBS for 10 min, mounted in DAPI containing Vectashield mounting solution and analyzed using Zeiss (Carl Zeiss Microimaging, Thornwood, NY) confocal laser-scanning microscope. The representative images were captured digitally using the 800 LSM software.
Statistical analysis
Student t-test was used to determine the statistical significance between the control and treated group (cerulein and smoking) and p value less than 0.05 was considered statistically significant. Error bars were determined on the basis of standard error values. To measure co-localization using confocal microscopy the images were assessed using ImageJ (National Institute of Health, Bethesda, MD) with Mander’s co-localization using JaCoP plug-in.
Results
Expression of ECE-1, ET-1, ETAR and ETBR in murine pancreas in the presence of activated K-ras
We first compared the expression of ET axis components at the transcript level in the pancreatic tissues harvested from mice harboring K-rasG12D mutation (KC) and their age-matched littermate controls (WT). The levels of ECE-1, ET-1, ETAR and ETBR transcripts were comparable in the pancreas of 10 week old WT and KC mice. However, there was a significantly higher expression of ET axis components in the pancreas of 30, 40 and 50 weeks old KC mice as compared to that of WT controls (Supplementary Fig. 1). In the control pancreas from 50 week old WT mice, a robust expression of ECE-1, ET-1, ETAR, and ETBR was observed in the islets and a relatively weak reactivity was detected in the acinar compartment (Fig. 1A). Importantly, the pancreatic ducts in the WT animals exhibited undetectable expression of ET-1, ETAR and ETBR. In the presence of oncogenic K-ras, the expression ECE-1, ET-1, ETAR, and ETBR progressively increased in the ductal cells from the precursor PanIN lesions at 30 week of age to dysplastic cancerous lesions observed at 40–50 week of age (Fig. 1A). The composite scores for the staining of ET axis components in the neoplastic ducts in KC mice were significantly higher as compared to the staining in the normal ducts in 50 week old WT mice (p < 0.05) (Fig. 1B). In addition to the ductal cells, the expression of ET axis components was detected in the surrounding stromal cells.
Fig. 1.


Expression profile of endothelin axis components in Pdx1-Cre; KrasG12D (KC) murine pancreatic cancer model. (A) Immunohistochemistry analysis of ECE-1, ET-1, ETAR and ETBR in the pancreas of KC mice at various stages of progression. The top panel represents staining pattern in the normal pancreas (50 week old WT mice) and the first column shows histological progression of the neoplastic lesions by Hematoxylin and Eosin (H&E) staining. The islets are marked by arrows and the ducts are marked with boxes and enlarged in insets to show the expression pattern in normal and neoplastic ducts. (B) Composite scores of ECE-1, ET-1, ETAR and ETBR staining in the ductal cells in the 50 week old WT mice (control) and KC mice representing various stages of progression. (n = 3 for ET-1, ETAR and ETBR; n = 4 for ECE-1; p < 0.05 for WT vs KC mice for all stages).
Expression of ET axis during acute pancreatic inflammation
To analyze the changes in the ET axis components in response to acute pancreatic inflammation in the presence and absence of oncogenic K-ras, we examined pancreatic tissues from 6-8 week old KC and WT mice treated with cerulein, a cholecystokinin analogue that aggravates secretion of pancreatic enzymes, promotes inflammation, and induces ADM [23]. At day 0, both the WT and KC mice exhibited acinar specific expression for ETAR and ETBR, as evident by their co-localization with amylase (Fig. 2A & B). On day 2-post cerulein treatment in KC mice ETAR and ETBR expression was observed in the metaplastic ducts undergoing ADM, characterized by co-expression of amylase and CK19. The expression of ET receptors was more pronounced in CK19 positive ducts with increased dysplasia at day 7 and day 21 post cerulein administration. In contrast, the WT mice showed recovery of pancreatic parenchyma and co-expression of amylase with ETAR and ETBR (Fig. 2A & B). There was a marked difference in the kinetics and magnitude of changes in the transcripts of ET axis components in the pancreas of cerulein-treated WT and KC mice. In the injury phase (day 2), the levels of ET axis components were not significantly different in the WT and KC mice. In the recovery phase (days 7 and 21) the levels of ECE-1 transcripts were 2.4 and 4.4 fold higher (p < 0.001) in the pancreas of KC mice than WT mice (Fig. 3A). The levels of ET-1 transcripts were 8.1 fold higher in the pancreas of KC mice at day 7 (p < 0.05); however, the levels decreased on day 21 and were not significantly different from WT mice (Fig. 3B). In the pancreas of KC mice, the levels of ET receptors were significantly higher than WT mice at day 7 (ETAR- 8.3 fold, p < 0.05; ETBR-10.5 fold. p < 0.005) following cerulein treatment (Fig. 3C & D). However, at day 21 while the levels of ETAR transcripts remained elevated in KC mice as compared to WT mice (10.1 fold; p < 0.005), the levels of ETBR transcripts declined, but remained higher than WT mice (3.6 fold; p < 0.05) (Fig. 3C & D). No difference in the expression of ET-2 and ET-3 transcripts was observed in KC mice compared to WT mice following cerulein treatment (Supplementary Fig. 2).
Fig. 2.


Alterations in the endothelin receptors during cerulein-induced ADM in KC and WT mice. Triple color immunofluorescence was performed for studying the expression of ETAR (A) and ETBR (B) in the acinar and ductal cells. The ET-receptors are stained with purple fluorophore. Amylase (red) was used as a marker of acinar cells, while CK19 (green) was used as a marker for ductal cells. Representative images of the stained sections of the pancreas of WT and KC mice at 0, 2, 7, 21 days post-treatment with cerulein. (Scale bar = 20 µm;).
Fig. 3.

Alterations in the expression of ET axis components (ECE-1, ET-1, ETAR and ETBR) in the pancreas during acute inflammation and recovery in the presence of WT or oncogenic K-ras. Expression of ET axis components was analyzed by Real Time Quantitative RT-PCR (qRT PCR) on the mRNA isolated from the pancreas of WT and KC mice treated with cerulein at 0, 2, 7 and 21 days post treatment. Histograms show fold change in the levels of ECE-1 (A), ET-1 (B), ETAR (C) and ETBR (D) transcripts in the KC mice comparison to WT mice following cerulein treatment. (n = 3 per time point; * p < 0.05, ** p < 0.005, *** p < 0.001, ns = not significant).
Expression of ET axis during chronic pancreatic inflammation
Chronic inflammation from exposure to cigarette smoke induced alterations in the expression of ET axis components in the presence of activating K-ras mutation. There was a seven- fold increase in the ET-1 transcript levels in the cigarette smoke exposed K-ras mutant mice compared to sham control (p = 0.04) (Fig. 4A). Similarly, the levels of ETAR transcripts were 4.5-fold higher (p = 0.03) (Fig. 4B) and ETBR transcripts 5-fold higher (p = 0.01) (Fig. 4C) in smoke-exposed KC mice as compared to sham controls. In the wild type mice, there was a slight increase in the levels of ET-1 and ETBR transcripts upon smoke exposure, but the difference was not statistically significant. Further, the levels of ECE-1 transcript were elevated upon smoke exposure in both WT and KC mice; however, the differences were not statistically significant as compared to their respective sham controls (Fig. 4D). IHC analysis of the mice pancreas also indicated elevated expression of ET axis components in response to cigarette smoke exposure. In the pancreas of both sham and smoke exposed WT mice, expression levels of ECE-1, ET-1 and both ET receptors were associated with the acinar compartment along with prominent staining in islet cells (Fig. 4E). Interestingly, smoke exposure in the KC mice resulted in an increased ECE-1 expression in the pancreatic ducts. While the staining of ET-1, ETAR and ETBR in the ductal cells in KC mice was much intense than that of WT mice, no difference was observed in their ductal expression in the sham and smoke-exposed mice. These observations were further corroborated by the co-expression of these ET axis components with epithelial marker CK19 (Supplementary Fig. 3). In addition to the ductal cells, expression of ET axis components was also observed in the stroma and infiltrating immune cells. Due to the presence of more preneoplastic lesions and increased stroma, the overall composite score of ET axis components was higher in the pancreas of KC mice as compared to WT mice and the overall scores further increased in smoke exposed KC mice (Fig. 4F). In KC mice, the composite scores of ET-1, ETAR, and ETBR were significantly higher (p < 0.05) in the smoke exposed mice compared to sham control, while that of ECE-1 was higher but not statistically significant.
Fig. 4.


Alterations in the expression of ET axis components (ECE-1, ET-1, ETAR and ETBR) in the pancreas of sham or cigarette smoke exposed WT and KC mice. qRT PCR was performed on the mRNA isolated from the pancreas of the WT or KC mice that were exposed to cigarette smoke or sham treated. The histograms represent fold change in the transcripts of ET axis components, ECE-1 (A), ET-1 (B), ETAR (C) and ETBR (D) transcripts in smoke exposed mice as compared to sham treatment. Immunohistochemistry analysis of the ET axis components in the pancreas of WT and KC mice that were either sham treated or exposed to cigarette smoke (E). Islets are marked with arrows and ducts are marked in boxes and zoomed in insets. The first column shows H & E stained section to demonstrate histological changes due to mutant K-ras or smoke exposure. Box plots (F) represent the composite scores for ET axis components. (n = 3 each group for RT-PCR and n = 6 each group for IHC; * p < 0.05, ns = non-significant).
Chronic and acute pancreatic inflammation is associated with increased infiltration of ETAR and ETBR expressing macrophages
One of the key events that determines the onset of inflammation and severity of AP is the activation of macrophages [24], [25]. Similar to AP, increased infiltration of macrophages is reported in inflamed sites in both human and mouse CP and macrophages are considered to be critical regulators of inflammation and disease progression [26]. Previous studies have reported increased accumulation of F4/80 positive macrophages in response to smoking in the pancreas of KC mice [27], [22]. Following cerulein-induced acute inflammation, the median number of F4/80 positive macrophage population was not significantly different at day 2 of treatment, however a substantial increase was seen at day 7 (average number of cells/ field = 13) and day 21 (average number of cells/ field = 17) of cerulein treatment in KC mice as compared to WT (Supplementary Fig. 4A). In addition to the acinar compartment, ETAR and ETBR expression was also observed in the infiltrating F4/80 positive macrophages in both WT and KC mice following cerulein treatment (Fig. 5A and C). At days 7 and 21 post-treatment, there was a significantly higher number F4/80-ETAR double positive cells in KC mice as compared to WT. Quantitative analysis of the ETAR expression in F4/80 positive macrophages using Mander’s coefficients indicated a significant increase in the overlapping fractions at day 7 (p = 0.0035) and day 21 (p = 0.0029) of treatment in KC mice as compared to WT mice (Fig. 5B). Similarly, for ETBR significantly increased co-localization with F4/80 was observed as early as day 2 post treatment (p = 0.0001), and remained high at day 7 (p = 0.0005) and day 21 (p = 0.0008) post cerulein trauma in KC mice as compared to WT mice (Fig. 5D). We also extended our studies in the smoking-induced chronic inflammation model using three-color immunofluorescence. Sham control KC mice exhibited increased infiltration of F4/80 positive macrophages in the pancreatic stroma as compared to WT mice and smoke treatment resulted in an increase in both groups (Fig. 6A). For ETAR co-expression with F4/80, the Mander’s overlap coefficient values were significantly higher in sham KC mice as compared to sham or smoke exposed WT mice; however, no significant difference was observed between sham and smoke exposed KC mice (p = 0.06) (Fig. 6B). For ETBR, the Mander’s overlap coefficient values were not significantly different in sham or smoke exposed WT mice, but the values were significantly higher in sham KC mice (p < 0.005) and further increased significantly upon smoke exposure (p < 0.0.05) (Fig. 6C).
Fig. 5.


Increased infiltration of F4/80 positive macrophages is associated with both ETAR and ETBR expression in cerulein induced acute inflammation: Dual color immunofluorescence images of the pancreatic sections stained for ETAR and ETBR (red) and macrophage marker F4/80 (green) following cerulein and saline treatment mice at days 0, 2, 7 and 21. The representative images for ETAR (A) and ETBR (B) are depicted in panels A and C, respectively, with areas showing co-localization of ET-receptors with macrophage markers highlighted in box and zoomed in inset. The degree of overlap between F4/80 positive macrophages and ETAR or ETBR was measured using ImageJ using Manders overlap coefficients (B and D respectively). (** p < 0.005, ns = not significant) (Scale bar = 20 µm; zoom scale bar = 10 µm).
Fig. 6.

Increased infiltration of F4/80 positive macrophages is associated with ETBR expression in smoking induced chronic inflammation: Three color immunofluorescence images of the ETAR (purple), ETBR (red) and F4/80 (green) expression in the pancreas of WT and KC mice that were either sham treated or exposed to cigarette smoke. The representative images are shown in Panel A where the areas of overlap are highlighted in a box and zoomed (inset). The degree of overlap of F4/80 staining with ETAR and ETBR was measured using ImageJ to calculate Manders overlap coefficient (B and C respectively). (* p < 0.05, ** p < 0.005, ns = not significant) (Scale bar = 20 µm; zoom scale bar = 10 µm).
Expression of ETAR and ETBR on pancreatic stellate cells following chronic and acute inflammation
Accumulating evidence indicates that activation of pancreatic stellate cells (PSCs) is pivotal in the development of pancreatitis and pancreatic cancer that results in excessive deposition of extracellular matrix proteins [28], [29]. Studies have demonstrated that smoke exposure leads to the activation of PSCs characterized by the expression of α-SMA in the pancreas of KC mice [22]. The number of activated PSCs following cerulein administration was determined in both WT and KC mice. In comparison to the WT mice, the pancreas of KC mice had a significantly higher number of α-SMA positive cells (p = 0.04) as early as day 2 post cerulein treatment, and their number progressively increased at day 7 and day 21 in KC mice while their number remained unchanged in WT mice. (Supplementary Fig. 4B). At day 7 post-cerulein treatment, the average number of cells/field was 27 in KC and 5 in WT mice (p = 0.02), while the corresponding values at day 21 were 32 and 7 cells/field (p = 0.02). Dual color immunofluorescence analysis revealed limited colocalization of both ETAR and ETBR with α-SMA positive myofibroblasts at day 0 and day 2 post cerulein-treatment in WT and KC mice (Fig. 7a & c). However, in KC mice, there was a significant colocalization of ETAR and ETBR with α-SMA at day 7 and day 21 as compared to WT mice (Fig. 7a & c). The Mander’s overlap coefficient values for both receptors with α-SMA were significantly higher in KC mice as compared to WT mice at day 7 (0.533 vs 0.174 for ETAR; 0.431 vs 0.176 for ETBR) and Day 21 (0.50 vs 0. 213 for ETAR; 0.546 vs 0.206 for ETBR) (Fig. 7b & d) indicating that sustained activation of pancreatic stellate cells in the presence of oncogenic K-ras is associated with the upregulation of ET receptors in this cell type.
Fig. 7.


Analysis of ET-receptor expression on the activated pancreatic fibroblasts/stellate cells in KC and WT mice following cerulein-induced acute inflammation: Dual color immunofluorescence images of the ETAR and ETBR expression (red) with α-SMA (myofibroblast marker) (green) in cerulein and saline treated mice at days 0, 2, 7 and 21 post cerulein administration. Representative images with areas of overlap highlighted in the box and zoomed in the inset (A and C, respectively). The degree of overlap between α-SMA positive fibroblasts and ETAR (B) and ETBR (D) was measured using ImageJ using Manders overlap coefficients. (* p < 0.05, ** p < 0.005, ns = not significant) (Scale bar = 20 µm; zoom scale bar = 10 µm).
We further examined the expression of ET receptors in the PSCs of WT and KC mice that were either sham control or exposed to cigarette smoke. As compared to the KC mice, there were decreased number of α-SMA positive cells in the pancreas of both sham and smoke exposed WT mice, which expressed relatively low levels of ETAR and ETBR (Fig. 8). In sham KC mice, in addition to the prominent expression in neoplastic ducts, both ET receptors were expressed on activated PSCs (Fig. 8A & B). Smoke exposure resulted in increase in the number of activated fibroblasts and there was a further elevation in the levels of both ETAR (Fig. 8A) and ETBR (Fig. 8B). The Mander’s overlap coefficient values of α-SMA with ETAR were significantly higher in smoked KC mice as compared to sham KC (Fig. 8C) while that with ETBR was higher but not statistically significant (Fig. 8D). Importantly, the Mander’s overlap coefficient values for both receptors with α-SMA were significantly higher in sham KC mice than smoke exposed WT mice suggesting that the presence of oncogenic K-ras results in fibroblast activation and upregulation of ET receptors.
Fig. 8.

Analysis of ET-receptors on the activated fibroblasts in the pancreas of WT and KC mice following cigarette smoke exposure. Dual color immunofluorescence images of the ETAR and ETBR staining (red) with α-SMA (myofibroblast marker) staining (green) in pancreatic sections from WT and KC mice that were either sham treated or exposed to cigarette smoke. The representative images are shown in panels A and B (for ETAR and ETBR, respectively) where the areas of overlap are highlighted in a box and zoomed (inset). The degree of overlap of staining of α-SMA with ETAR and ETBR is represented as histograms of Manders overlap coefficients (C and D respectively). (* p < 0.05, ** p < 0.005, ns = not significant (Scale bar = 20 µm; zoom scale bar = 10 µm).
Discussion
ADM is the earliest recognizable morphological event in the pancreas in response to inflammation or oncogene activation and is regarded as a key event during the development of PDAC. It is characterized by the trans-differentiation of amylase-expressing acinar cells into CK19 positive ductal phenotype. Further, the appearance of precursor lesions (PanINs) precedes the formation of K-ras driven PDAC in experimental mouse models [8], [9]. These precursor lesions are derived from the acinar cells undergoing a trans-differentiation to ductal phenotype, an event usually induced by pancreatitis [6], [30].
ET axis plays a significant role in tissue repair and inflammation of various tissues and has been implicated in the pathophysiology of pancreatic inflammation. Previous studies have shown aberrant expression of ET axis components in surgically resected pancreatic cancer patients and its association with tumorigenesis [20]. ET-1, the major ligand of the family, is detected abundantly in pancreatic cancer cell lines as compared to the other two isoforms, ET-2 and ET-3 [18], [19]. In addition to its pathophysiological role in the pancreas, evidence also suggests its crucial role in normal pancreas physiology and pancreatitis. Studies have demonstrated the presence of ETAR and ETBR in the rat pancreas and their differential binding affinities towards the three endothelin ligands [31]. The present study revealed the expression of ET axis components, ECE-1, ET-1, ETAR and ETBR in pancreatic acinar cells and islet cells and their minimal or low levels in the pancreatic ducts of WT mice. In murine model of PDAC, a progressive increase in the expression was observed in the trans-differentiated or neoplastic ductal cells. Metaplasia is regarded as the conversion or transformation of one cell type into another by an abnormal stimulus and is associated with an early phase of tumor development. In the context of ADM, metaplastic ducts are transitional structures and are characterized by the presence of both acinar cell markers such as amylase and ductal cell marker, CK19. Our analysis revealed elevated expression of both ETAR and ETBR in amylase and CK19 double positive metaplastic ducts in the pancreas of KC mice following cerulein-induced injury, suggesting the upregulation of ET axis signaling during ADM.
Pancreatitis is an established risk factor for PDAC and is characterized by acinar cell necrosis, infiltration of inflammatory cell populations, stromal fibrosis and release of insoluble and soluble mediators [32], [33]. Previous study from our lab has revealed that cerulein induced inflammatory damage to the pancreas is followed by decrease in the amylase expression in the KC mice compared to WT mice [21]. Following cerulein treatment, we observed the expression of both ETAR and ETBR in the metaplastic ducts. The pancreas of KC mice exhibited greater dysplasia and higher expression of ET-receptors in CK19 positive ducts following cerulein treatment on day 7 and 21 (Fig. 2). In contrast to the WT mice, a significant increase in ECE-1, ET-1, ETAR and ETBR transcripts was observed following cerulein treatment at day 7 and these levels continued to remain high even at 21 days post-trauma in KC mice (Fig. 3). This data suggests that acute inflammation results in a persistent upregulation of ET axis components in the pancreas in the presence of oncogenic K-ras, and signaling across ET axis potentially contributes to the reprogramming of acinar cells into ductal morphology to favor neoplastic transformation.
The role of ET-1 has been widely studied in the context of AP where it has been shown to promote disease aggravation. In an experimental rat model of pancreatitis, ET-1 was identified as one of the candidate gene associated with pancreatic inflammation [34]. Additionally, in both sodium taurocholate and cerulein induced pancreatitis, exogenous administration of ET-1 damages the pancreatic parenchyma, promotes acinar cell necrosis and increases amylase and elastase levels [35], [17]. ET-1 is considered to be a significant risk factor for AP and elevated levels correlate with disease severity [12]. Similarly, elevated levels of ET-1 are observed in patients with CP and there is a significant positive correlation between ET-1 expression with smoking history [13]. Studies also indicate a significant increase in the plasma ET-1 levels under the influence of tobacco smoking, suggesting a direct effect on endothelium facilitating the ET-1 peptide release [36], [37]. Cigarette smoke is an established risk factor for PDAC. Our recent study revealed that in the presence of oncogenic K-ras, smoking promotes tumor progression by accelerating the transformation and influencing pancreatic microenvironment by activating pancreatic stellate cells and increasing the accumulation of macrophages [22]. We observed that cigarette smoke results in a more pronounced upregulation of the transcripts of ET axis components (ECE-1, ET-1, ETAR and ETBR) in KC mice as compared to WT mice (Fig. 4). These observations further suggest that elevated ET axis signaling contributes to inflammation-associated neoplastic transformation in the presence of oncogenic K-ras.
Pancreatic inflammation elicits macrophage infiltration and secreted cytokines are considered as potent inducers of pancreatitis-initiated acinar cell transformation to ductal progenitor phenotype [38]. These infiltrated macrophages are considered as drivers of ADM and in the presence of oncogenic signaling favor tumor development [39]. A recent study demonstrated that acinar cells harboring mutant K-ras upregulate ICAM-1 expression, which serves as a chemoattractant for macrophages to drive tumor initiation [40], [41]. ET-1 has also been demonstrated to induced polarization of human macrophages [42] and facilitate cross-talk between breast cancer cells and endothelial cells in an integrin dependent manner [43]. In bladder cancer, ET-1/ETAR interaction favors stromal cross-talk and enhances metastatic colonization in the lung by enhancing the migration and infiltration of tumor cells and tumor associated macrophages, respectively [44]. ET-1 has been shown to stimulate chemotaxis of blood monocytes [45] and induce chemo-kinetic migration of peritoneal macrophages in an ETAR dependent manner [46]. We observed that both chronic and acute pancreatic inflammation was associated with the recruitment of activated macrophages in the pancreatic stroma in the presence of oncogenic K-Ras. In KC mice, cerulein treatment resulted in significant upregulation of ETBR on macrophages as early as 2 days post-treatment while ETAR upregulation was observed at day 7 (Fig. 5). Together with the elevated levels of ET-1, it appears that macrophages are recruited to pancreatic tumor microenvironment in the early stages via signaling across ET axis. However, to fully understand and delineate the molecular mechanisms of ET axis mediated macrophage recruitment in KC mice further studies involving macrophage targeted conditional knock-out of ETAR and ETBR need to be undertaken.
In addition to active infiltration of immune cell populations in response to pancreatic injury, activation of PSCs is recognized as central event in development of pancreatitis and PDAC. The PSCs are activated by a variety of soluble and insoluble mediators such as cytokines, growth factors, oxidative stress, ethanol and its metabolites and pancreatitis. Once activated, PSCs transdifferentiate into myofibroblasts like cells and this phenotypic transformation results in fibrosis and extensive deposition of extracellular matrix proteins [47]. The pro-fibrotic role of ET-1 in various pathologies is well documented and is regulated at transcriptional levels by interaction with various transcription factors such as Smad, TGF-β and activator protein-1 (AP-1) [48], [49]. ET-1 induces a pro-fibrogenic response in lung fibroblasts by enhancing the expression of α-SMA and CTGF via JNK-AP1 and TGF-β pathway [50], [51]. Activated PSCs express ET-1, ETAR and ETBR and are ET-1 responsive, suggesting an autocrine and paracrine loop to stimulate contraction and migration of PSCs by inducing phosphorylation of ERK and MLC but not AKT [52]. To stimulate myofibroblast differentiation, ET-1 has also been shown to promote inflammatory reaction in the pancreas by release of pro-inflammatory mediators such as IL-6 and IL-1β [53]. Moreover, Fitzner et al demonstrated that the pro-fibrogenic effect of ET axis is attenuated by dual ETAR and ETBR antagonist bosentan in experimental CP model [54]. Our results suggest that acute and chronic inflammation induced by cerulein and smoking respectively accelerate the desmoplastic reaction in the K-rasG12D mice and the concomitant expression of endothelin receptors on activated PSCs can possibly promote stromal cross-talk by autocrine and paracrine interactions and aid in tumor progression. We also observed a significant increase in the ET-1 transcript levels in precancerous lesions in the K-rasG12D mice, which further suggests a possible cross-talk between neoplastic ductal and activated stellate cells along ET axis during oncogene-associated pancreatic inflammation and neoplastic transformation. We speculate that persistent increase and activation of ETAR and ETBR with increasing dysplasia and activated myofibroblast signaling plays an essential role in maintenance of PSC activation through ET-1 autocrine loops. However, the molecular mechanisms underlying this phenomenon warrant further investigation.
Overall, our studies demonstrate that ET axis components are upregulated in various cellular compartments during benign and oncogene-associated acute and chronic pancreatic inflammation. Given the elevated expression of ET-receptors at the earliest morphologically identifiable stages of neoplastic transformation, and their sustained upregulation in precursor lesions, it is likely that signaling across ET axis contributes to the initiation and progression of PDAC. Selective and dual specificity ET receptors antagonists are approved for human use for treating pulmonary arterial hypertension and have been investigated for treating cancer and fibrosis [55]. It will be of interest to examine the utility of ET-axis antagonists in reversing or preventing the inflammation-driven neoplastic events in the murine models. Further, it is critical to define the role of ET axis in PDAC pathobiology, particularly in the context of desmoplasia and immune cell infiltration to determine if ET axis antagonists can be combined with chemotherapeutic or immunotherapeutic agents.
Conclusions
In conclusion, our study profiled the expression of ET axis during acute and chronic inflammation associated pancreatic tumor progression in presence of oncogenic t K-rasG12D mutation. Under physiological conditions, the expression of ET axis components is restricted to pancreatic acinar and islet cell compartments. However, during inflammation or injury elevated expression is seen in early precancerous lesions and neoplastic cells. The sustained upregulation of ET axis components in the presence of oncogenic K-ras and overexpression in advanced lesions, suggests that signaling along ET axis possibly contributes to reprogramming of acinar cells into metaplastic ductal cells and drives their transformation into neoplastic lesions. The increased expression in preneoplastic lesions is followed by excessive accumulation of ECM proteins and inflammation in the pancreas, indicating further involvement of ET axis in influencing microenvironmental factors during the initiation and progression of PDAC.
Conflict of interest
The authors declare no conflicts of interest.
Acknowledgements
The work and authors were supported, in part, by grants from the National Institutes of Health (U01 CA213862, R01 CA195586, U01 CA200466). The authors thank the invaluable technical support from Kavita Mallya. We are also thankful to James Talaska and Janice Taylor at the Confocal Microscopy Core Facility for their support.
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.neo.2019.11.001.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
Supplementary Fig. 1.
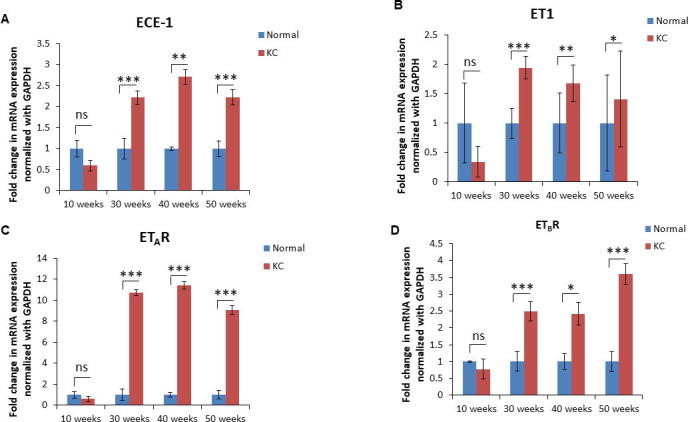
Alterations in the transcripts of ET axis components during the progression on pancreatic cancer in KC mouse model. Real Time PCR analysis showing the fold change in the expression of ECE-1 (A), ET-1 (B), ETAR (C), and ETBR (D) transcripts in the pancreas of KC mice and age-matched littermate controls (WT) at ages representing various stages of progression. (n = 3 per genotype/time point; * p < 0.05, ** p < 0.005, *** p < 0.001).
Supplementary Fig. 2.
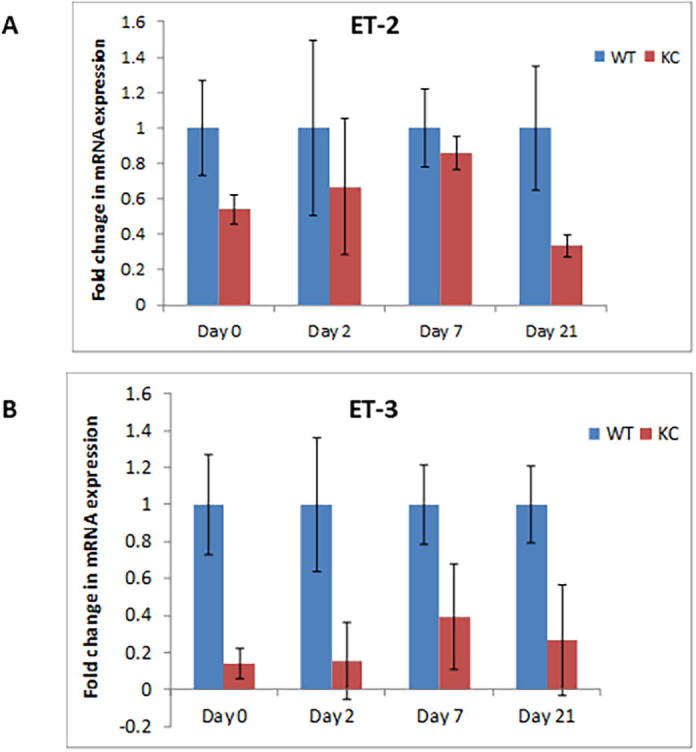
Expression of ET-2 and ET-3 in WT and KC mice following cerulein-induced acute inflammation. Real Time PCR analysis revealed no statistically significant difference in the ET-2 and ET-3 transcripts between WT and KC mice following cerulein treatment. (n = 3 per genotype/time point).
Supplementary Fig. 3.

Expression of ET-1, ETAR and ETBR in spontaneous KC mice model with sham or cigarette smoke exposure: Dual immunofluorescence analysis showing the expression of ET-1, ETAR and ETBR in sham controls and smoke exposed KC mice.
Supplementary Fig. 4.

Increased accumulation of macrophages and fibroblasts following cerulein treatment: Quantitative analysis of the macrophages (A) and fibroblasts (B) in the pancreas of WT and KC mice treated with saline or cerulein. The infiltrating macrophages were identified by positive staining for F4/80, while fibroblasts were identified by positive staining for α-SMA. The histograms represent average count of F4/80 or α-SMA positive cells in five independent fields/tissue section. (n = 3; * p < 0.05, ** p < 0.005, ns = not significant).
References
- 1.Morris J.P., Cano D.A., Sekine S., Wang S.C., Hebrok M. Beta-catenin blocks Kras-dependent reprogramming of acini into pancreatic cancer precursor lesions in mice. J Clin Invest. 2010;120:508–520. doi: 10.1172/JCI40045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Herreros-Villanueva M., Hijona E., Cosme A., Bujanda L. Mouse models of pancreatic cancer. World J Gastroenterol. 2012;18:1286–1294. doi: 10.3748/wjg.v18.i12.1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wagner M., Luhrs H., Kloppel G., Adler G., Schmid R.M. Malignant transformation of duct-like cells originating from acini in transforming growth factor transgenic mice. Gastroenterology. 1998;115:1254–1262. doi: 10.1016/s0016-5085(98)70098-8. [DOI] [PubMed] [Google Scholar]
- 4.Schmid R.M., Kloppel G., Adler G., Wagner M. Acinar-ductal-carcinoma sequence in transforming growth factor-alpha transgenic mice. Ann N Y Acad Sci. 1999;880:219–230. doi: 10.1111/j.1749-6632.1999.tb09526.x. [DOI] [PubMed] [Google Scholar]
- 5.Shi G., DiRenzo D., Qu C., Barney D., Miley D., Konieczny S.F. Maintenance of acinar cell organization is critical to preventing Kras-induced acinar-ductal metaplasia. Oncogene. 2013;32:1950–1958. doi: 10.1038/onc.2012.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Habbe N., Shi G., Meguid R.A. Spontaneous induction of murine pancreatic intraepithelial neoplasia (mPanIN) by acinar cell targeting of oncogenic Kras in adult mice. Proc Natl Acad Sci U S A. 2008;105:18913–18918. doi: 10.1073/pnas.0810097105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Popovic H.M., Korolija M., Jakic R.J., Pavkovic P., Hadzija M., Kapitanovic S. K-ras and Dpc4 mutations in chronic pancreatitis: case series. Croat Med J. 2007;48:218–224. [PMC free article] [PubMed] [Google Scholar]
- 8.Guerra C., Schuhmacher A.J., Canamero M. Chronic pancreatitis is essential for induction of pancreatic ductal adenocarcinoma by K-Ras oncogenes in adult mice. Cancer Cell. 2007;11:291–302. doi: 10.1016/j.ccr.2007.01.012. [DOI] [PubMed] [Google Scholar]
- 9.Guerra C., Collado M., Navas C. Pancreatitis-induced inflammation contributes to pancreatic cancer by inhibiting oncogene-induced senescence. Cancer Cell. 2011;19:728–739. doi: 10.1016/j.ccr.2011.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Carriere C., Young A.L., Gunn J.R., Longnecker D.S., Korc M. Acute pancreatitis accelerates initiation and progression to pancreatic cancer in mice expressing oncogenic Kras in the nestin cell lineage. PLoS One. 2011;6 doi: 10.1371/journal.pone.0027725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carriere C., Young A.L., Gunn J.R., Longnecker D.S., Korc M. Acute pancreatitis markedly accelerates pancreatic cancer progression in mice expressing oncogenic Kras. Biochem Biophys Res Commun. 2009;382:561–565. doi: 10.1016/j.bbrc.2009.03.068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Milnerowicz S., Milnerowicz H., Nabzdyk S., Jablonowska M., Grabowski K., Tabola R. Plasma endothelin-1 levels in pancreatic inflammations. Adv Clin Exp Med. 2013;22:361–368. [PubMed] [Google Scholar]
- 13.Sliwinska-Mosson M., Milnerowicz S., Nabzdyk S., Kokot I., Nowak M., Milnerowicz H. The effect of smoking on endothelin-1 in patients with chronic pancreatitis. Appl Immunohistochem Mol Morphol. 2015;23:288–296. doi: 10.1097/PAI.0000000000000077. [DOI] [PubMed] [Google Scholar]
- 14.Xiping Z., Ruiping Z., Binyan Y. Protecting effects of a large dose of dexamethasone on spleen injury of rats with severe acute pancreatitis. J Gastroenterol Hepatol. 2010;25:302–308. doi: 10.1111/j.1440-1746.2009.05999.x. [DOI] [PubMed] [Google Scholar]
- 15.Zhang X.P., Xu H.M., Jiang Y.Y. Influence of dexamethasone on mesenteric lymph node of rats with severe acute pancreatitis. World J Gastroenterol. 2008;14:3511–3517. doi: 10.3748/wjg.14.3511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhang X.P., Ye Q., Jiang X.G. Preparation method of an ideal model of multiple organ injury of rat with severe acute pancreatitis. World J Gastroenterol. 2007;13:4566–4573. doi: 10.3748/wjg.v13.i34.4566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu X.H., Kimura T., Ishikawa H. Effect of endothelin-1 on the development of hemorrhagic pancreatitis in rats. Scand J Gastroenterol. 1995;30:276–282. doi: 10.3109/00365529509093277. [DOI] [PubMed] [Google Scholar]
- 18.Kusuhara M., Yamaguchi K., Nagasaki K. Production of endothelin in human cancer cell lines. Cancer Res. 1990;50:3257–3261. [PubMed] [Google Scholar]
- 19.Oikawa T., Kushuhara M., Ishikawa S. Production of endothelin-1 and thrombomodulin by human pancreatic cancer cells. Br J Cancer. 1994;69:1059–1064. doi: 10.1038/bjc.1994.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Cook N., Brais R., Qian W., Hak C.C., Corrie P.G. Endothelin-1 and endothelin B receptor expression in pancreatic adenocarcinoma. J Clin Pathol. 2015;68:309–313. doi: 10.1136/jclinpath-2014-202521. [DOI] [PubMed] [Google Scholar]
- 21.Dey P., Rachagani S., Vaz A.P., Ponnusamy M.P., Batra S.K. PD2/Paf1 depletion in pancreatic acinar cells promotes acinar-to-ductal metaplasia. Oncotarget. 2014;5:4480–4491. doi: 10.18632/oncotarget.2041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kumar S., Torres M.P., Kaur S. Smoking accelerates pancreatic cancer progression by promoting differentiation of MDSCs and inducing HB-EGF expression in macrophages. Oncogene. 2015;34:2052–2060. doi: 10.1038/onc.2014.154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Baer R., Cintas C., Dufresne M. Pancreatic cell plasticity and cancer initiation induced by oncogenic Kras is completely dependent on wild-type PI 3-kinase p110alpha. Genes Dev. 2014;28:2621–2635. doi: 10.1101/gad.249409.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gea-Sorli S., Closa D. Role of macrophages in the progression of acute pancreatitis. World J Gastrointest Pharmacol Ther. 2010;1:107–111. doi: 10.4292/wjgpt.v1.i5.107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Shrivastava P., Bhatia M. Essential role of monocytes and macrophages in the progression of acute pancreatitis. World J Gastroenterol. 2010;16:3995–4002. doi: 10.3748/wjg.v16.i32.3995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Xue J., Sharma V., Hsieh M.H. Alternatively activated macrophages promote pancreatic fibrosis in chronic pancreatitis. Nat Commun. 2015;6:7158. doi: 10.1038/ncomms8158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Edderkaoui M., Xu S., Chheda C. HDAC3 mediates smoking-induced pancreatic cancer. Oncotarget. 2016;7:7747–7760. doi: 10.18632/oncotarget.6820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Apte M.V., Pirola R.C., Wilson J.S. Pancreatic stellate cells: a starring role in normal and diseased pancreas. Front Physiol. 2012;3:344. doi: 10.3389/fphys.2012.00344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Masamune A., Shimosegawa T. Pancreatic stellate cells–multi-functional cells in the pancreas. Pancreatology. 2013;13:102–105. doi: 10.1016/j.pan.2012.12.058. [DOI] [PubMed] [Google Scholar]
- 30.Kopp JL, von FG, Mayes E, et al. Identification of Sox9-dependent acinar-to-ductal reprogramming as the principal mechanism for initiation of pancreatic ductal adenocarcinoma. Cancer Cell 2012;22:737–50. [DOI] [PMC free article] [PubMed]
- 31.Hildebrand P., Mrozinski J.E., Jr., Mantey S.A., Patto R.J., Jensen R.T. Pancreatic acini possess endothelin receptors whose internalization is regulated by PLC-activating agents. Am J Physiol. 1993;264:G984–G993. doi: 10.1152/ajpgi.1993.264.5.G984. [DOI] [PubMed] [Google Scholar]
- 32.Flandez M., Cendrowski J., Canamero M. Nr5a2 heterozygosity sensitises to, and cooperates with, inflammation in KRas(G12V)-driven pancreatic tumourigenesis. Gut. 2014;63:647–655. doi: 10.1136/gutjnl-2012-304381. [DOI] [PubMed] [Google Scholar]
- 33.Bai H., Li H., Zhang W. Inhibition of chronic pancreatitis and pancreatic intraepithelial neoplasia (PanIN) by capsaicin in LSL-KrasG12D/Pdx1-Cre mice. Carcinogenesis. 2011;32:1689–1696. doi: 10.1093/carcin/bgr191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Oz H.S., Lu Y., Vera-Portocarrero L.P., Ge P., Silos-Santiago A., Westlund K.N. Gene expression profiling and endothelin in acute experimental pancreatitis. World J Gastroenterol. 2012;18:4257–4269. doi: 10.3748/wjg.v18.i32.4257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Plusczyk T., Bersal B., Westermann S., Menger M., Feifel G. ET-1 induces pancreatitis-like microvascular deterioration and acinar cell injury. J Surg Res. 1999;85:301–310. doi: 10.1006/jsre.1999.5610. [DOI] [PubMed] [Google Scholar]
- 36.Borissova A.M., Tankova T., Kirilov G., Dakovska L., Krivoshiev S. The effect of smoking on peripheral insulin sensitivity and plasma endothelin level. Diabetes Metab. 2004;30:147–152. doi: 10.1016/s1262-3636(07)70100-3. [DOI] [PubMed] [Google Scholar]
- 37.Goerre S., Staehli C., Shaw S., Luscher T.F. Effect of cigarette smoking and nicotine on plasma endothelin-1 levels. J Cardiovasc Pharmacol. 1995;26(Suppl 3):S236–S238. [PubMed] [Google Scholar]
- 38.Liou G.Y., Doppler H., Necela B. Macrophage-secreted cytokines drive pancreatic acinar-to-ductal metaplasia through NF-kappaB and MMPs. J Cell Biol. 2013;202:563–577. doi: 10.1083/jcb.201301001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liou G.Y., Storz P. Inflammatory macrophages in pancreatic acinar cell metaplasia and initiation of pancreatic cancer. Oncoscience. 2015;2:247–251. doi: 10.18632/oncoscience.151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liou G.Y., Doppler H., Necela B. Mutant KRAS-induced expression of ICAM-1 in pancreatic acinar cells causes attraction of macrophages to expedite the formation of precancerous lesions. Cancer Discov. 2015;5:52–63. doi: 10.1158/2159-8290.CD-14-0474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Storz P. The crosstalk between acinar cells with Kras mutations and M1-polarized macrophages leads to initiation of pancreatic precancerous lesions. Oncoimmunology. 2015;4 doi: 10.1080/2162402X.2015.1008794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Soldano S., Pizzorni C., Paolino S. Alternatively activated (M2) macrophage phenotype is inducible by endothelin-1 in cultured human macrophages. PLoS One. 2016;11 doi: 10.1371/journal.pone.0166433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chen C.C., Chen L.L., Hsu Y.T., Liu K.J., Fan C.S., Huang T.S. The endothelin-integrin axis is involved in macrophage-induced breast cancer cell chemotactic interactions with endothelial cells. J Biol Chem. 2014;289:10029–10044. doi: 10.1074/jbc.M113.528406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Said N., Smith S., Sanchez-Carbayo M., Theodorescu D. Tumor endothelin-1 enhances metastatic colonization of the lung in mouse xenograft models of bladder cancer. J Clin Invest. 2011;121:132–147. doi: 10.1172/JCI42912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Achmad T.H., Rao G.S. Chemotaxis of human blood monocytes toward endothelin-1 and the influence of calcium channel blockers. Biochem Biophys Res Commun. 1992;189:994–1000. doi: 10.1016/0006-291x(92)92302-e. [DOI] [PubMed] [Google Scholar]
- 46.Bath P.M., Mayston S.A., Martin J.F. Endothelin and PDGF do not stimulate peripheral blood monocyte chemotaxis, adhesion to endothelium, and superoxide production. Exp Cell Res. 1990;187:339–342. doi: 10.1016/0014-4827(90)90102-g. [DOI] [PubMed] [Google Scholar]
- 47.Masamune A., Watanabe T., Kikuta K., Shimosegawa T. Roles of pancreatic stellate cells in pancreatic inflammation and fibrosis. Clin Gastroenterol Hepatol. 2009;7:S48–S54. doi: 10.1016/j.cgh.2009.07.038. [DOI] [PubMed] [Google Scholar]
- 48.Stow L.R., Jacobs M.E., Wingo C.S., Cain B.D. Endothelin-1 gene regulation. FASEB J. 2011;25:16–28. doi: 10.1096/fj.10-161612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Masaki T. Historical review: endothelin. Trends Pharmacol Sci. 2004;25:219–224. doi: 10.1016/j.tips.2004.02.008. [DOI] [PubMed] [Google Scholar]
- 50.Weng C.M., Yu C.C., Kuo M.L., Chen B.C., Lin C.H. Endothelin-1 induces connective tissue growth factor expression in human lung fibroblasts by ETAR-dependent JNK/AP-1 pathway. Biochem Pharmacol. 2014;88:402–411. doi: 10.1016/j.bcp.2014.01.030. [DOI] [PubMed] [Google Scholar]
- 51.Shi-wen X., Kennedy L., Renzoni E.A. Endothelin is a downstream mediator of profibrotic responses to transforming growth factor beta in human lung fibroblasts. Arthritis Rheum. 2007;56:4189–4194. doi: 10.1002/art.23134. [DOI] [PubMed] [Google Scholar]
- 52.Masamune A., Satoh M., Kikuta K., Suzuki N., Shimosegawa T. Endothelin-1 stimulates contraction and migration of rat pancreatic stellate cells. World J Gastroenterol. 2005;11:6144–6151. doi: 10.3748/wjg.v11.i39.6144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Jonitz A., Fitzner B., Jaster R. Molecular determinants of the profibrogenic effects of endothelin-1 in pancreatic stellate cells. World J Gastroenterol. 2009;15:4143–4149. doi: 10.3748/wjg.15.4143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fitzner B., Brock P., Holzhuter S.A. Synergistic growth inhibitory effects of the dual endothelin-1 receptor antagonist bosentan on pancreatic stellate and cancer cells. Dig Dis Sci. 2009;54:309–320. doi: 10.1007/s10620-008-0366-z. [DOI] [PubMed] [Google Scholar]
- 55.Aubert J.D., Juillerat-Jeanneret L. Endothelin-receptor antagonists beyond pulmonary arterial hypertension: cancer and fibrosis. J Med Chem. 2016;59:8168–8188. doi: 10.1021/acs.jmedchem.5b01781. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.