

Abstract
Hypoxic microenvironments exist in developing embryonic tissues and determine stem cell fate. We previously demonstrated that hypoxic priming plays roles in lineage commitment of embryonic stem cells. In the present study, we found that hypoxia-primed embryoid bodies (Hyp-EBs) efficiently differentiate into the myogenic lineage, resulting in the induction of the myogenic marker MyoD, which was not mediated by hypoxia-inducible factor 1α (HIF1α) or HIF2α, but rather by Sp1 induction and binding to the MyoD promoter. Knockdown of Sp1 in Hyp-EBs abrogated hypoxia-induced MyoD expression and myogenic differentiation. Importantly, in the cardiotoxin-muscle injury mice model, Hyp-EB transplantation facilitated muscle regeneration in vivo, whereas transplantation of Sp1-knockdown Hyp-EBs failed to do. Moreover, we compared microRNA (miRNA) expression profiles between EBs under normoxia versus hypoxia and found that hypoxia-mediated Sp1 induction was mediated by the suppression of miRNA-92a, which directly targeted the 3′ untranslated region (3′ UTR) of Sp1. Further, the inhibitory effect of miRNA-92a on Sp1 in luciferase assay was abolished by a point mutation in specific sequence in the Sp1 3′ UTR that is required for the binding of miRNA-92a. Collectively, these results suggest that hypoxic priming enhances EB commitment to the myogenic lineage through miR-92a/Sp1/MyoD regulatory axis, suggesting a new pathway that promotes myogenic-lineage differentiation.
Keywords: hypoxic microenvironment, myogenic differentiation, microRNA, embryoid bodies, transcription factor
Graphical Abstract
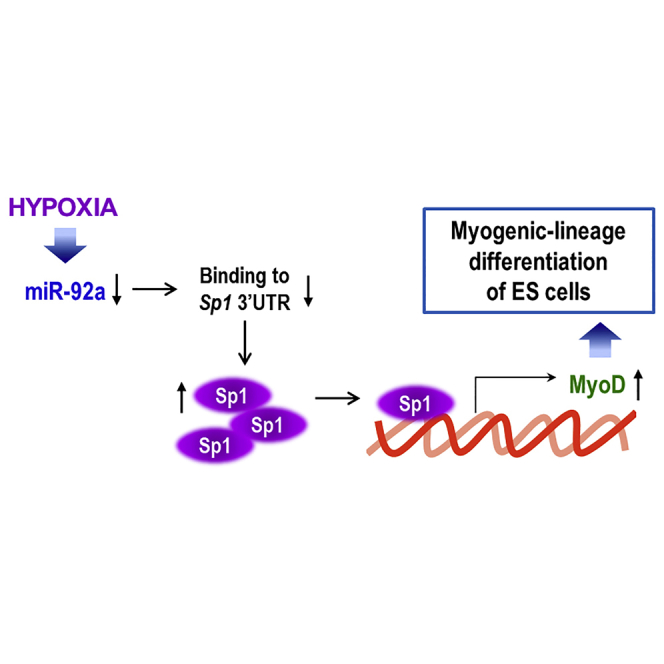
Lee et al. describe the enhanced differentiation of hypoxia-primed embryoid bodies (EBs) into the myogenic lineage. Interestingly, this process was not mediated by HIF1α or HIF2α, but rather by Sp1 induction, via binding to the MyoD promoter. Moreover, the transplantation of hypoxia-primed EBs was found to facilitate muscle regeneration in vivo, and this was also found to depend on Sp1. By analyzing miRNA expression profiles, the authors demonstrate that hypoxia induced Sp1 in EBs via suppression of miRNA-92a that was found to directly target the 3′ UTR of Sp1.
Introduction
Embryonic stem cells (ESCs) possess the capacity for unlimited cell growth and the ability to self-renew and differentiate into all types of mature cells.1 Understanding the regulatory mechanisms underlying the differentiation of ESCs into specific cell types might be useful in manipulating stem cell fate for cell therapy. In cancer biology, hypoxia affects various pathophysiologic processes including cell survival, cell apoptosis, DNA repair, vascular development, and angiogenesis during tumor formation.2, 3, 4, 5 Also in stem cell biology, hypoxia is important because low oxygen gradients exist widely in developing embryonic tissues because of limited oxygen diffusion due to increases in embryo size and dense organ structure formation.6 Hypoxic microenvironments can determine stem cell fate by modulating processes such as proliferation, differentiation, and maintenance.7 We previously reported that hypoxia stimulates ESC differentiation into the vascular lineage via hypoxia-inducible factor 1 (HIF1), and that HDAC6 downregulation by hypoxia potentiates the myogenic differentiation of ESCs.8, 9 Thus, we have been interested in the mechanisms of how the unique microenvironment such as hypoxia regulates stem cell differentiation.
Sp1 belongs to a family of zinc-finger transcription factors involved in cell-cycle regulation, hormonal activation, apoptosis, and angiogenesis.10, 11 It also regulates the expression of genes involved in differentiation and embryonic development.12, 13 This protein is abundant in most cells, but its expression changes during development and varies in different cell types.14 Sp1 binds GC-rich sequences in promoters and, specifically, it recognizes the consensus sequence 5′-GCCCCGCCCCTC-3′ or other related GC-rich sequences.15 Interestingly, Sp1-deficient mouse (Sp1−/−) embryos survive until day 9.5 (E9.5) of gestation but display severely retarded growth with phenotypic abnormalities, and these animals die at approximately E11. This suggests that Sp1 is essential for normal early embryonic development and plays an important role in maintaining differentiated cells.12
Recent advances in small RNA research have suggested that microRNAs (miRNAs) are important regulators of development and differentiation in stem cells.16 miRNAs comprise a large family of noncoding small RNAs of approximately 22 nucleotides (nt) in length that act as negative regulators of gene expression.17, 18 Mature miRNAs recognize their target mRNAs to modulate translational efficiency and/or mRNA degradation by binding complementary sequences within the 3′ untranslated region (3′ UTR).17, 18 Further, many miRNA sequences are often highly conserved in Drosophila, mice, and humans. Moreover, the expression of miRNAs is regulated in tissue-specific and developmental-stage-specific manners19, 20, 21 and one miRNA can target many mRNAs that are involved in various cellular functions such as organ development, differentiation, cancer, and metabolism.16, 22, 23, 24
Muscle loss is the fundamental phenomenon that increases the risk of death in muscle-wasting disorders, chronic disease, or aging.25 Accordingly, ESCs might serve as a good source of muscle cells to treat these conditions;26 therefore, methods to invoke specific myogenic differentiation have been investigated, but these approaches are not yet sufficient. Four myogenic regulatory factors (MRFs), namely MyoD, Myf-5, myogenin, and MRF4, control the specification and the differentiation of the muscle lineage.27 MyoD was the first identified MRF that can convert fibroblasts to skeletal myoblasts.28 The genetically engineered ESCs expressing MyoD efficiently differentiate into the muscle lineage.26
We previously reported that hypoxic exposure during first few days of differentiation efficiently stimulates the ESC differentiation to the meso-endoderm.8 Therefore, we here investigated whether hypoxic priming could enhance the differentiation of ESCs toward the myogenic differentiation among mesoderm lineage, compared to that under normoxic conditions. Indeed, the effects of hypoxia on muscle-lineage differentiation and the underlying mechanisms have not been fully investigated. We also examined the muscle-regenerative ability of hypoxia-primed embryoid bodies (EBs) in a mouse muscle injury model. Particularly, with regard to the mechanism underlying differentiation, we investigated hypoxia-regulated miRNA and the transcription factor Sp1 in addition to the regulatory effect of Sp1 on MyoD expression. Our findings enhance the knowledge of myogenic differentiation and might provide useful insights into the mechanisms of stem cell differentiation.
Results
Hypoxia Induces the Differentiation of Mouse Embryonic Stem Cell (mESC)-Derived EBs to a Myogenic Lineage
We previously reported that hypoxia stimulates stem cell differentiation toward meso-endoderm lineages and especially vascular lineage.8 Because we were also interested in ESC commitment to the myogenic lineage among the mesoderm lineages, we analyzed the expression of myogenic markers (Figure 1). EBs were formed for 3 days, exposed to normoxic (21% oxygen) or hypoxic (1% oxygen) conditions for 16 h, and further cultured under spontaneous differentiation conditions (DMEM/10% FBS) for up to 10 days (Figure 1A). mRNA expression of the pluripotency marker Oct4 was remarkably downregulated upon differentiation and its expression was significantly lower at every time point in response to hypoxic conditions as compared to that under normoxic conditions (Figure 1B). Moreover, myogenic marker genes (MyoD, Myf5) were markedly upregulated in normoxic cells upon differentiation. Interestingly, only the expression of MyoD, a master regulator of myogenesis,29 was significantly increased in hypoxic cells than in normoxic cells (Figure 1B). We next performed immunofluorescence staining for MyoD and myosin heavy chain (MyHC) to confirm the myogenic differentiation potential (Figure 1C). MyoD and MyHC were rarely detectable in normoxic-EBs (Nor-EB), whereas strong staining was observed in hypoxia-primed EBs (Hyp-EB) (Figure 1C), indicating that hypoxia efficiently induces differentiation into the myogenic lineage.
Figure 1.

Hypoxic Preconditioning Stimulates the Differentiation of mESC-Derived EBs to the Myogenic Lineage
(A) EBs were formed from C57 mESCs by the hanging drop method for 3 days, cultured under normoxic or hypoxic conditions for 16 h, and allowed to attach to a 0.3% gelatin-coated plate in DMEM/10% FBS for 1 day; the medium was then changed to fresh media and cells were further differentiated for up to 10 days. (B) The pluripotency marker Oct4 was significantly downregulated compared to expression under conditions of normoxia, and the myogenic marker MyoD was significantly upregulated in hypoxic cells compared to expression in normoxic cells based on real-time PCR analysis. Graphs show the relative percent change (n = 4); *p < 0.05, **p < 0.01, ***p < 0.001 versus the normoxic group. (C) Immunofluorescence staining for MyoD (red) and MyHC (red, arrows) 10 days after EB reattachment. Nor-EB, normoxic-EBs; Hyp-EB, hypoxia-primed EBs. Nuclear DNA was counterstained with DAPI (blue). Representative confocal microscopic photographs are shown. Magnification, 200×; scale bars, 50 μm.
Hypoxia-Stimulated MyoD Upregulation Is Mediated by Sp1 and Not HIF1 or HIF2
Differentiation into the myogenic lineage was efficiently enhanced by hypoxia; thus, we attempted to determine the mechanism through which this occurs. We focused on MyoD, a master regulator of myogenesis,29 and HIF, a key regulator of hypoxic responses. As shown in Figure 2A, both MyoD and HIF1α proteins were significantly induced in response to hypoxia, whereas Myf5 protein was not changed. To evaluate the effect of HIF1α or HIF2α on MyoD expression, we transfected cells with a HIF1α or HIF2α expression vector under normoxic conditions.8 We expected that MyoD expression would be affected by either HIF1α or HIF2α; however, both protein and mRNA levels were unaffected (Figures 2B and 2C; Figure S1). Moreover, the expression of hypoxia-regulated genes was increased in HIF1α- and HIF2α-overexpressing cells (Figure S1), indicating that the expression plasmids functioned properly.
Figure 2.

MyoD Is Upregulated under Hypoxia, but Its Expression Is Not Stimulated by Either HIF1α or HIF2α
(A) EBs formed for 3 days were cultured under normoxic or hypoxic conditions for 16 h. Western blotting of MyoD, Myf5, and HIF1α (left) was then performed. Quantification of results (right, n = 3; ***p < 0.001 versus the normoxic group). (B and C) Effect of HIF1α or HIF2α overexpression on MyoD and Myf5 in C57 ESCs (B) and E14 ESCs (C). ESCs were transfected with 1 μg pEGFP-HIF1α or pEGFP-HIF2α for 24 h and allowed to form EBs for 3 days under normoxic conditions. Western blotting for HIF1α, HIF2α, MyoD, and Myf5 (left) and quantification (right, n = 3∼4; *p < 0.05, **p < 0.01 versus pMock).
To identify factors that increase MyoD expression in response to hypoxia, we investigated the MyoD promoter region and found candidate regulators such as Sp1 and AP2α (Figure 3A).30, 31 One putative AP2α-binding site and two putative Sp1-binding sites were identified. Sp1 mRNA and protein were upregulated in Hyp-EBs compared to Nor-EBs; however, AP2α mRNA and protein were unaffected (Figures 3B and 3C). We then confirmed the hypoxia-mediated upregulation of Sp1 by immunofluorescence staining, as this protein was strongly detected in EBs grown in hypoxic conditions (Figure 3D). To determine the effect of Sp1 on MyoD expression, we first performed Sp1 knockdown using specific short hairpin RNA (shRNA). Sp1 protein and mRNA were markedly increased under hypoxic conditions, but were significantly decreased after transfection with shSp1 even under hypoxia (Figures 3E and 3F; Figure S2). Interestingly, hypoxia-stimulated MyoD protein and mRNA expression was significantly downregulated in shSp1-EBs (Figures 3E and 3F; Figure S2). These results suggest that MyoD expression depends on Sp1 in response to hypoxic stimuli (Figure 3G).
Figure 3.

MyoD Upregulation by Hypoxia Occurs via Sp1 Stimulation in Hypoxic EBs
(A) The mouse MyoD putative promoter region was found to contain candidate Sp1- and AP2α-binding sites (marked by squares). Nucleotides are numbered relative to the translation start site of MyoD. (B and C) Sp1, but not AP2α, was remarkably increased in hypoxic EBs compared to normoxic EBs (B) Real-time PCR for Sp1 and AP2α mRNA (n = 5, ***p < 0.001). (C) Western blot for Sp1 and AP2α (left). Quantification of western blotting results (right; n = 3, ***p < 0.001). (D) Immunofluorescence staining of Hyp-EBs and Nor-EBs for Sp1 (red) and nuclei (blue) on day 3 after attachment (left). Magnification, 400×; scale bars, 50 μm. Quantitative data for the intensity of Sp1 fluorescence. At least three randomly selected fields were analyzed from three independent experiments (***p < 0.001). (E) Transient transfection of shSp1. ESCs were transfected with 1 μg shMock or shSp1 for 24 h, allowed to form EBs for 3 days under normoxia, and further incubated for 16 h under normoxic or hypoxic conditions. Sp1 protein was markedly reduced after transient transfection of shSp1 compared to that in the non-target shRNA control (shMock). Sp1 knockdown suppressed the increased expression of MyoD protein even under conditions of hypoxia. (F) Quantification of western blotting results for Sp1 and MyoD proteins (n = 3); **p < 0.01, ***p < 0.001 versus Nor-EBs. ##p < 0.01, ###p < 0.001 versus Hyp-shMock-EBs. (G) Sp1 was stimulated by hypoxia and then increased MyoD expression.
Sp1 Increases MyoD Transcription and Activates MyoD Promoter Activity
Because Sp1 knockdown was found to inhibit hypoxia-mediated MyoD upregulation, we tested the ability of Sp1 to stimulate MyoD promoter activity. We first performed chromatin immunoprecipitation (ChIP) assays to examine the binding of Sp1 to the MyoD promoter containing putative Sp1-binding sites (Figure 4A). ChIP analysis shows that Sp1 binding to the MyoD promoter region was increased by hypoxia compared to that under normoxia (Figure 4A). In addition, we examined the effect of hypoxia on the MyoD promoter by performing promoter luciferase assays (data not shown). Reporter gene activity was significantly increased in response to hypoxia in cells transfected with the MyoD-promoter Luc (MyoD-Luc). Next, we further performed promoter luciferase assays to determine the effects of Sp1 overexpression on the MyoD promoter and to identify Sp1-binding sites among two putative regions. C57 ESCs were transfected with Sp1 overexpression vector, and we confirmed that MyoD expression was transcriptionally increased by Sp1 overexpression (Figures 4B and 4C). Figure 4D shows these two sites (red characters, numbered as 1 and 2; 5′-GCTCCGCCCTA-3′ and 5′-CCCCCGCCCC-3′, respectively) and the 6-bp core sequence (5′-CCGCCC-3′) for Sp1 binding are bolded.31
Figure 4.

Sp1 Increased MyoD Expression through Transcriptional Activation of the MyoD Promoter
(A) ChIP analysis shows the Sp1 binding to the MyoD promoter region. Lysates of differentiating C57-ESCs exposed to hypoxia for 8 h were immunoprecipitated with an antibody against Sp1. The precipitated DNA was evaluated by PCR using specific primers for Sp1-binding sites in the MyoD promoter (n = 3) and a representative image is shown. (B and C) MyoD expression in response to Sp1 overexpression. ESCs were transfected with 1 μg pMock or pSp1 for 24 h and allowed to form EBs for 3 days under normoxic conditions. (B) Western blot for MyoD, which was increased by Sp1 overexpression (left). Quantification of western blotting results (right; n = 3, **p < 0.01, ***p < 0.001). (C) Real-time PCR analysis showed that MyoD expression was transcriptionally increased by Sp1 overexpression (n = 4; ***p < 0.001). (D) Segments of the MyoD upstream transcriptional regulatory region containing two putative Sp1-binding sites (red and underlined) located between −91 and −183 bp. (E) Schematic diagram of two Sp1-binding motifs in the full sequence of the MyoD promoter and the promoter deletion mutants (top). Differentiating C57 ESCs cultured in the absence of LIF and feeder cells were co-transfected with various combinations of the 1 μg full-length MyoD promoter, 1 μg MyoD promoter deletion mutants, 0.2 μg pCMV-β-gal, 1 μg pCD-Mock, and 1 μg pCD-Sp1 plasmid for 24 h (bottom, n = 6, ***p < 0.001).
After Sp1 overexpression, luciferase activity was significantly increased in cells transfected with the wild-type (WT)-MyoD promoter region (Figure 4E). Interestingly, cells transfected with the Δ1-mt plasmid, in which the first Sp1-binding site (Sp1-site1) was deleted, showed similar or slightly reduced luciferase activity to that elicited by the WT-MyoD promoter. In contrast, the induction of MyoD reporter activity by Sp1 was significantly inhibited in cells transfected with the Δ2-mt plasmid in which the second Sp1-binding site (Sp1-site2) was deleted. In addition, cells transfected with the Δ1, 2-mt plasmid (both Sp1-binding sites deleted) showed the lowest MyoD reporter activity. From these results, we suggest that induction of MyoD promoter activity by Sp1 depends largely on the Sp1 binding site 2 (5′-CCCCCGCCCC-3′) but weakly on the binding site 1 (Figure 4E).
Sp1-Mediated MyoD Expression Is Important for Hypoxic EB Differentiation into the Myogenic Lineage
To confirm the role of Sp1 in regulating myogenic-lineage differentiation, we specifically knocked down its expression (Figure 5). Sp1 protein and mRNA levels were markedly reduced after the transfection of shSp1 (Figure 5A; Figure S3). We then confirmed MyoD expression in various shMock- or shSp1-knockdown clones to preclude interclone variation, and MyoD protein was found to be remarkably decreased in all four Sp1 knockdown ES clones grown under hypoxic conditions (Figure 5A). We then further analyzed the specific myogenic differentiation potential of shSp1-EBs in response to hypoxia, compared to that in shMock-EBs under the same conditions (Figures 5B–5E). The cells were cultured in skeletal muscle induced media (SkIM) for specific muscle differentiation.32, 33 As shown in Figure 5C, the myogenic regulatory factors MyoD and myogenin were remarkably upregulated during the differentiation of Hyp-shMock-EBs compared to control Nor-shMock-EBs (Figure 5C). In contrast, Sp1 knockdown EBs (shSp1) showed impaired myogenic-lineage differentiation even in response to hypoxia (Figure 5C). We next performed immunofluorescence staining for MyoD and MyHC to confirm the involvement of Sp1 in hypoxia-driven muscle-lineage differentiation (Figures 5D and 5E). Strong staining for both markers was observed in Hyp-shMock EBs compared to Nor-shMock EBs. In contrast, MyoD and MyHC were rarely detected in Hyp-shSp1 EBs, indicating that Sp1 is an important regulator of myogenic differentiation in response to hypoxia.
Figure 5.

Knockdown of Sp1 Decreases the Myogenic Differentiation of Hypoxic-EBs
(A) Generation of Sp1-knockdown stable cells. C57 ESCs were transfected with 1 μg shMock or shSp1 for 24 h, and further selected by puromycin treatment. MyoD protein was remarkably decreased in all four Sp1-knockdown hypoxic-ES clones compared to that in all four control clones transfected with shMock. (B) Stable knockdown cells were formed as EBs by the hanging drop method, cultured under normoxic or hypoxic conditions, and plated onto a gelatin-coated plate in DMEM/10% FBS for 1 day. The culture medium was replaced with SkIM and further incubated for up to 20 days. (C) Muscle regulatory factors (MyoD, myogenin) were increased in Hyp-shMock cells compared to those in Nor-shMock cells, and were significantly decreased to a greater extent in Hyp-shSp1 cells compared to expression in Hyp-shMock cells (n = 6); *p < 0.05, **p < 0.01, ***p < 0.001 versus Nor-shMock-EBs and #p < 0.05, ##p < 0.01, ###p < 0.001 versus Hyp-shMock-EBs. (D) Immunofluorescence staining for MyoD (red) or MyHC (red) at day 15 after EB reattachment. Nuclear DNA was counterstained with DAPI (blue). Magnification, 200×; scale bars, 50 μm. (E) Quantitation of the intensity of MyoD and MyHC immunofluorescence (n = 6). ***p < 0.001 versus normoxic shMock-EBs; ###p < 0.001 versus hypoxic shMock-EBs.
Sp1 Knockdown Abrogates Skeletal Muscle Regeneration Induced by Hypoxia-Primed EBs In Vivo
Hyp-EBs showed efficient myogenic-lineage commitment in vitro, and Sp1 was found to mediate hypoxia-primed EB differentiation into the myogenic lineage. We then tested whether hypoxia-primed EBs could undergo myogenic differentiation in vivo, thereby leading to enhanced muscle regeneration, using a mouse model of cardiotoxin (CTX)-induced muscle injury (Figure 6). To monitor the time course of muscle regeneration, we performed rota-rod analysis after CTX muscle injury. We injured the tibialis anterior (TA) muscles of both legs by injecting CTX and then 1 day later transplanted EBs from the three groups including shMock-EBs under normoxia (Nor-shMock EBs), shMock-EBs with hypoxic priming (Hyp-shMock EBs), and hypoxia-primed EBs with Sp1 knockdown (Hyp-shSp1 EBs). We monitored locomotive recovery every week for up to 8 weeks by recording the time until animals fell off the rotating rod (Figures 6A–6C). Consistent with our in vitro results, mice transplanted with Hyp-shMock cells (CTX+Hyp-shMock) showed significantly better motor function than those treated with Nor-shMock cells (CTX+Nor-shMock) from 2 weeks after injury, and this therapeutic effect continued until 8 weeks (Figures 6B and 6C). Interestingly, transplantation of Sp1-knockdown hypoxia-treated EBs (CTX+Hyp-shSp1) resulted in weaker motor function compared to that in animals treated with Hyp-shMock cells (CTX+Hyp-shMock).
Figure 6.

Knockdown of Sp1 in Hypoxia-Primed EBs Diminishes Muscle Differentiation in an In Vivo Mouse Muscle Injury Model
(A) Timetable of rota-rod analysis of mice with skeletal muscle injury. One day before cell transplantation, CTX was injected into the TA muscle (both legs) to induce muscle injury. We then monitored muscle power every week for 8 weeks in five groups: sham group with PBS injection (sham, n = 16), injury only group with CTX injection (CTX only, n = 16), injury and transplantation of normoxic shMock-EBs (CTX+Nor-shMock, n = 15), injury and transplantation of hypoxic shMock-EBs (CTX+Hyp-shMock, n = 16), and injury and transplantation of hypoxic shSp1-EBs (CTX+Hyp-shSp1, n = 16). (B and C) The time required for mice to fall off the rotating rod was measured every week for 8 weeks (B) and at 2 weeks and 6 weeks after cell transplantation (C) as shown; *p < 0.05, **p < 0.01, ***p < 0.001 versus Nor-shMock-EBs and ###p < 0.001 versus Hyp-shMock-EBs. (D) Cross-section of TA muscle was stained with H&E or immunostained for laminin 2α (green)/nuclei (blue) at 8 weeks after transplantation. Transplanted cells were pre-labeled with DiI (red). Most DiI fluorescence-positive red cells were regenerating myofibers with a central nucleus. Magnification, 200×; scale bars, 20 μm. Myofibers positive for DiI in the Hyp-shSp1 group were smaller than those in the Hyp-shMock group. (E) Quantification of DiI(+)/Laminin(+) myofibers, which was high in the Hyp-shMock-EB group but decreased in the Hy-shSp1-EB-transplanted TA muscle (n = 30; ***p < 0.001 versus Nor-shMock-EBs and ###p < 0.001 versus Hyp-shMock-EBs). (F) CSA of regenerating myofibers. TA muscle-transplanted cells were collected at 8 weeks after surgery and H&E staining and CSA (μm2) were measured. CSA of the regenerating myofibers in the Hyp-shSp1 group were significantly smaller than those in the Hyp-shMock group (n = 39; **p < 0.01, ***p < 0.001 versus Nor-shMock-EBs and ##p < 0.01, ###p < 0.001 versus Hyp-shMock-EBs).
We then observed the regenerating myofibers with centrally located nuclei that originated from transplanted EBs marked by DiI labeling (red, arrows; Figure 6D). We also performed laminin immunofluorescence (green) because skeletal muscle fibers are surrounded by the basal lamina and its major components are laminin.34 Regenerating myofibers, based on DiI fluorescence, were more frequently found in the Hyp-shMock group than the Nor-shMock group. In contrast, myofibers showing DiI fluorescence were significantly decreased in the Hyp-shSp1 group (Figures 6D and 6E). The cross-sectional areas of regenerated myofibers were then examined and muscle fibers in the Hyp-shMock group were significantly bigger than those in the Nor-shMock group. In contrast, the cross-sectional areas of muscles transplanted with Hyp-shSp1 cells were much smaller than those in the Hyp-shMock group (Figure 6F). These results support the successful engraftment, differentiation into the myogenic lineage, and improved rota-rod performance of the Hyp-shMock group, which was diminished by Sp1 suppression.
Sp1 Is Upregulated by Hypoxia-Mediated miRNA-92a Suppression
As stated, Sp1 was found to be increased by hypoxia (Figure 3), and thus, we investigated the upstream regulators for Sp1, focusing on miRNAs that typically suppress target gene expression.17 We hypothesized the existence of Sp1-suppressive miRNA(s) that would be reduced by hypoxia. To find miRNAs that are downregulated upon differentiation and further downregulated by hypoxia, we analyzed miRNA expression profiles using miRNA microarray comparing the three groups (normoxic ESCs, normoxic EBs, and hypoxic EBs; Figure 7). Among the 388 miRNAs that were significantly changed between normoxic ESs, normoxic EBs, and hypoxic EBs, 18 were sequentially downregulated in the following order: normoxic ESCs > normoxic EBs > hypoxic EBs. To identify miRNAs that target the 3′ UTR of Sp1 (Sp1 3′ UTR), we used the web-based target prediction tools TargetScan, microRNA.org, and miRBase. We selected eight candidate miRNAs that were common among the three tools, namely miR-7a, miR-7b, miR-27a, miR-92a, miR-128, miR-290, miR-335, and miR-466c (Figure 7A). We next confirmed that the expression of these candidate miRNAs was significantly decreased in normoxic EBs compared to that in normoxic ESCs and were further suppressed in hypoxic EBs (Figure 7B). We then tested which of these eight miRNAs regulate Sp1 expression (Figures 7C and 7D). After transfection with each mimic oligomer, only miR-27a and miR-92a remarkably suppressed Sp1 expression, whereas the overexpression of other mimics did not affect levels of Sp1 (Figures 7C and 7D; Figure S4).
Figure 7.

Sp1 Suppression by Hypoxia-Responsive MiRNAs
(A) A miRNA array was performed and among 18 miRNAs sequentially downregulated in the order of normoxic-ESCs (Nor-ES) > normoxic-EBs (Nor-EBs) > hypoxic-EBs (Hyp-EBs), and eight candidate miRNAs were selected based on the bioinformatics target prediction tools TargetScan, microRNA.org, and miRBase. (B) The expression of eight candidate miRNAs in Nor-ES, Nor-EBs, and Hyp-EBs was measured by real-time PCR (n = 5; *p < 0.05, **p < 0.01, ***p < 0.001 versus Nor-ES and #p < 0.05, ##p < 0.01, ###p < 0.001 versus Nor-EBs). (C) Differentiating C57 ESCs were transfected with miRNA mimic-oligomer for 2 days under normoxic conditions, and miRNA expression was measured by real-time PCR. Each miRNA was overexpressed in a dose-dependent manner (miR-NC, miRNA-negative control; n = 5, *p < 0.05, **p < 0.01, ***p < 0.001). (D) Sp1 protein was reduced only by miR-27a and miR-92a mimics in C57 ESCs (miR-NC).
Next, we further determined whether forced expression of miR-27a and miR-92a pre-miR miRNA precursors could control Sp1 levels. Both pre-miR27a and pre-miR92a downregulated Sp1 protein, which was consistent with the results of mimic transfections (Figure 8A). Interestingly, a miR-92a precursor (pre-miR-92a) significantly suppressed Sp1 mRNA expression, whereas a miR-27a precursor (pre-miR-27a) had no effect (Figure 8B). From these results, we inferred that miR-27a regulates Sp1 at the protein level, whereas miR-92a regulates Sp1 at the mRNA level. We further examined whether miR-27a or miR-92a could directly target the 3′ UTR of Sp1 by performing luciferase reporter assays (Figures 8C and 8D). The Sp1 3′ UTR is greater than 5 kb, and therefore it was broken up into shorter fragments and cloned into two constructs with overlapping sequence. Fragment A contained two miR-27a-binding sites and one miR-92-binding site, whereas fragment B contained one miR-27a-binding site and two miR-92-binding sites (Figure 8C). After overexpression of pre-miR-27a or pre-miR-92a, only pre-miR-92a significantly suppressed luciferase activity from fragment B of the 3′ UTR of Sp1; however, pre-miR-27a did not affect luciferase with this construct. Luciferase activity from fragment A of the 3′ UTR of Sp1 was not affected by either pre-miR-27a or pre-miR-92a (Figure 8D).
Figure 8.

miR-92a Reduces Sp1 mRNA and Protein by Directly Targeting the 3′UTR of Sp1, whereas miR-27a Suppresses Sp1 Protein but Not mRNA
(A and B) C57 ESCs were transfected with 1 μg pre-miR-27a or pre-miR-92a miR precursors for 2 days under normoxia, and western blotting and real-time PCR were performed. (A) Sp1 western blotting after overexpression of pre-miR-27a or pre-miR-92a miR precursors. Sp1 protein was reduced by both miRNAs (n = 3; **p < 0.01, ***p < 0.001). (B) Suppression of Sp1 mRNA only by pre-miR-92a but not by pre-miR-27a overexpression in C57 cells (n = 4; **p < 0.01, ***p < 0.001). (C) Sequence alignment of putative miR-27a- and miR-92a-targeting sites within the 3′ UTR of Sp1 (5,331 bp). Two luciferase reporter constructs are shown: fragment A, first half of the 3′ UTR of Sp1 (1–2,760 bp); fragment B, last half of the 3′ UTR of Sp1 (2,321–5,331 bp). (D) Luciferase activity reflecting Sp1 expression was suppressed by pre-miR-92a but not by both pre-miR-27a and pre-miR-NC (n = 6; ***p < 0.001). Sp1-3′ UTR-A-Luc, luciferase reporter containing fragment A of the 3′ UTR of the Sp1 gene; Sp1-3′ UTR-B-Luc, luciferase reporter containing fragment B of the 3′ UTR of the Sp1 gene. Differentiating C57 ESCs were co-transfected with various combinations of 1 μg luciferase reporter, 1 μg pre-miR-27a, 1 μg pre-miR-92a, and 1 μg pre-miR negative control, and luciferase activity was measured 24 h later. (E and F) miR-92a was found to directly target the 3′ UTR of the Sp1 gene. (E) Schematic representation of luciferase reporter constructs showing the predicted structures of each base-paired WT (Sp1 3′ UTR-WT) or mutant (Sp1 3′ UTR-mt1, Sp1 3′ UTR-mt2) fragment B of the Sp1 3′ UTR. (F) Luciferase activity showed the reduction of Sp1 expression by pre-miR-92a and that this suppressive activity of pre-miR-92a was abrogated when the first target site (3,737–3,765 bp) of the 3′ UTR was mutated (n = 6; ***p < 0.001, ###p < 0.001). C57 ESCs were co-transfected with various combinations of 1 μg WT, 1 μg mutant Sp1 3′ UTR, 1 μg pre-miR-92a, and 1 μg pre-miR-NC, and luciferase activity was measured 24 h later. (G) Sp1 was increased even under normoxic conditions. C57 mESCs were transfected for 24 h with an antagomiR against miR-92a (50 nM). Hypoxia: 16 h (n = 3; **p < 0.01, ***p < 0.001). (H) Proposed model for stimulation of the myogenic differentiation of mESCs through the miR-92a/Sp1/MyoD axis.
Within the 3′ UTR-fragment B-Luc construct, there were two miR-92a-binding sites including region 1 (transcript position 3,737–3,765) and region 2 (transcript position 4,983–5,011) (Figure 8E). Thus, we investigated whether miR-92a could target either of the two putative target sites within WT or mutated (mt) fragment B sequences of the 3′ UTR of Sp1. Overexpression of pre-miR-92a significantly suppressed luciferase activity from the WT and region 2 mutated versions of this sequence (Figure 8F). In contrast, pre-miR-92a did not affect luciferase activity when region 1 was mutated. Pre-miR negative control (NC pre-miR) also did not affect luciferase activity (Figure 8F). These results indicated that miR-92a directly targets region 1 (transcript position 3,737–3,765) within the 3′ UTR of Sp1. In addition, miR-92a was inhibited under normoxia based on the transfection of an antagomiR against miR-92a, which reproduced the effects of miR-92a reduction under hypoxic conditions, and Sp1 expression was increased even under normoxic conditions (Figure 8G). All these results strongly suggested that Sp1 might be a direct target of miR-92a.
Discussion
The major finding of this study was that hypoxia stimulates myogenic-lineage differentiation and that the transcription factor Sp1 plays an important role in regulating MyoD expression during myogenic differentiation from mESCs under hypoxia. Furthermore, the promoter region of MyoD was found to contain a Sp1-binding site that confers responsiveness to hypoxia. Moreover, miR-92a, which declines in response to hypoxia, directly targets the 3′ UTR of Sp1 to suppress its expression, leading to hypoxia-mediated Sp1 induction. In addition, knockdown of Sp1 abolished hypoxia-induced MyoD expression and myogenic marker expression in differentiating ESCs. Finally, knockdown of Sp1 in ESCs abolished muscle regeneration in vivo.
In our previous report,8 hypoxia was found to induce the differentiation of ESCs toward a meso-endodermal and, further, a vascular lineage. Therefore, we hypothesized that hypoxia might play a role in myogenic-lineage differentiation because this lineage is derived from the mesoderm. In hypoxia-primed EBs, the myogenic regulatory factor MyoD and mature myofiber marker MyHC were significantly increased (Figure 1), whereas Myf5 did not respond to hypoxia. Previously, mice lacking MyoD showed normal muscle development but expressed approximately 4-fold higher levels of Myf5.35 Myf5−/− animals also exhibit normal muscle development.36 However, mice lacking both MyoD and Myf5 demonstrate the complete loss of myoblasts and myofibers,37 indicating that these proteins can compensate for each other but that both are essential for the determination of myogenic precursors during development.
It was found that the upstream regulator that increases MyoD expression under conditions of hypoxia is not either of the well-known hypoxia-responsive transcriptional regulators, HIF1α or HIF2α, but rather, Sp1 (Figures 3 and 4). Sp1 is known to regulate the expression of genes involved in embryonic development and differentiation,12, 13 and its expression level changes during development.14 Several studies have reported its involvement in muscle cell differentiation. Specifically, Sp1 knockdown downregulates gene expression associated with smooth muscle cell differentiation without affecting other cell lineage-related genes in mESCs.38, 39 The amplification of MDM2 in rhabdomyosarcoma cells inhibits MyoD function and inhibits muscle cell differentiation. However, the transfection of an Sp1-overexpression vector restores MyoD activity and C2C12 myoblast differentiation. In that report, MyoD expression was not enhanced by Sp1.40 The human cardiac a-actinin (HCA) promoter contains binding sites for Sp1, serum response factor, and the myogenic basic helix-loop-helix family; moreover, the cooperative DNA binding of these transcriptional activators is required for HCA transcription in C2C12 skeletal myoblast.41 We demonstrated that hypoxia-mediated Sp1 upregulation increases MyoD expression, myogenic differentiation, and muscle regeneration and that Sp1-knockdown EBs could not mediate these effects even after hypoxic priming (Figures 5 and 6). Moreover, cells stained with Sp1 co-localized with MyoD immunofluorescence in reattached hypoxic-EBs (data not shown). Our results thus uncovered a previously unknown pathway, namely the hypoxia-Sp1-MyoD axis, which specifies and potentiates the direction of stem cell differentiation.
We previously reported that hypoxia-induced miR-26a targets HDAC6 and facilitates myogenic differentiation.9 Here, we attempted to identify an upstream regulator that suppresses Sp1 expression and found that the miR-92a directly targets the 3′ UTR of Sp1 and suppresses its expression. Conversely, miR-92a was found to be suppressed by hypoxia (Figures 7 and 8). When we use the strategy of antagomiR-92a, we can mimic the hypoxic condition to enhance the expression of Sp1/MyoD and muscle regeneration as shown in Figure 8G. Thus, based on these data, we can develop new therapeutic agents to suppress miR-92a and to facilitate regeneration of the damaged muscle. There is a muscle-specific miRNA family, the members of which are called myomiRs, which is regulated during muscle differentiation and activity.16, 42 Moreover, hypoxamiRs comprise a specific subset of miRNAs regulated by hypoxia.43 miR-92a and miR-27a are not known as myomiRs, whereas miR-92a was reported to be a hypoxamiR that is downregulated by hypoxia. Hypoxia regulates several fates of hypoxamiR including transcription, maturation, and function.43 For the transcriptional regulation of miRNAs by hypoxia, HIFs play a predominant role. In addition, hypoxia-induced transcription factors such as nuclear factor κB and p53 execute important regulatory functions in hypoxia-driven miRNA transcription.44 Transcription factors such as HIF are upregulated under hypoxic conditions and directly activate the transcription of a subset of hypoxamiRs. In contrast, much less is known about the mechanisms responsible for hypoxia-induced gene repression. At least 10 transcriptional repressors including repressor element-1 silencing transcription factor (REST) have been reported,45 and hypoxia selectively represses some hypoxamiRs through less characterized mechanisms. It is likely that transcriptional repressors participate in the hypoxia-driven repression of hypoxamiRs.
In addition to transcriptional control, hypoxia regulates Drosha and Dicer to mediate hypoxamiR maturation and function. It was reported that Drosha and Dicer mRNA and protein levels were decreased in the lung tissue of rats after exposure to hypoxia. When primary pulmonary fibroblasts are exposed to hypoxia, the expression of Drosha mRNA was decreased.46 It was also reported that some subsets of miRNAs are significantly downregulated in hypoxic human umbilical vein endothelial cells (HUVECs), and that Dicer mRNA and protein levels are decreased in a Von Hippel Lindau (VHL)-dependent manner.47 miR-92a was found to be repressed under hypoxia in our experiments (Figure 7B); this might be possibly through a hypoxia-driven transcriptional repressor, and/or the suppression of miRNA-processing proteins such as Drosha and Dicer. However, further studies focusing on the relative contributions of transcriptional and posttranscriptional events to the regulation of miR-92a by hypoxia will be needed. miR-27a is also known as a hypoxamiR, but its expression pattern varies. The expression of miR-27a was found to be suppressed by hypoxia in our work (Figure 7B) and in hippocampal neurons,48 whereas its expression was reported to increase by hypoxia or HIF1α in pulmonary vascular cells and cancer cells.49, 50 In our current study, miR-27a overexpression reduced Sp1 “protein” level but did not target the 3′ UTR of Sp1 or suppress “mRNA” level (Figures 8A–8D). Further work will be required to clarify these issues.
In conclusion, the hypoxic priming of EBs induced the commitment toward the myogenic lineage through miRNA-92a/Sp1/MyoD axis, which was not only demonstrated in vitro but also confirmed in vivo. We observed enhanced muscle regeneration after the transplantation of hypoxia-primed EBs into a muscle-damaged limb. Accordingly, the hypoxic priming of stem/progenitor cells before transplantation or the modulation of Sp1 expression by pharmacological agents, such as antagomiR-92a, might be plausible new therapeutic strategy to enhance muscle repair in the regenerative medicine.
Materials and Methods
Cell Culture and Differentiation
Undifferentiated E14 mESCs and C57BL/6-background mESCs (C57-mESCs, accession number SCRC-1002; ATCC) were cultured on mitomycin C (Sigma, St. Louis, MO, USA)-treated mouse embryonic fibroblast feeder layers in DMEM (GIBCO, Grand Island, NY, USA) with 20% FBS (HyClone, Fisher Scientific, Pittsburgh, PA, USA), 1% penicillin/streptomycin (GIBCO, Grand Island, NY), 0.1 mM β-mercaptoethanol (BME; Sigma, St. Louis, MO, USA), 1% non-essential amino acids (GIBCO, Grand Island, NY), 2 mM L-glutamine, and 1,000 U/mL leukemia inhibitory factor (LIF; Millipore, Billerica, MA, USA). We considered the C57-ESCs obtained directly from the ATCC as “passage 1,” and performed all experiments in this manuscript with cells between passages 8 and 14. For hypoxic culture conditions, cells were incubated in a Hypoxia Chamber (Forma Scientific, Marietta, OH, USA) under low oxygen tension (1% O2, 5% CO2, and balanced with N2).
EBs were formed by the hanging drop method (one droplet containing 500 cells/20 μL) in the absence of LIF and feeder cells. To induce spontaneous differentiation, we cultured EBs on 0.3% gelatin-coated plates in DMEM/10% FBS. For in vitro myogenic-lineage differentiation, EBs were plated on 0.3% gelatin-coated culture dishes in DMEM/10% FBS for 1 day and further incubated, which was followed by replacement with specific media with some modification, specifically SkIM,32 high-glucose DMEM (GIBCO, Grand Island, NY), 10% FBS (HyClone, Fisher Scientific, Pittsburgh, PA, USA), 5% horse serum (Sigma, St. Louis, MO, USA), 1% penicillin/streptomycin (GIBCO, Grand Island, NY), 1% non-essential amino acids (GIBCO, Grand Island, NY), 0.1 mM BME (Sigma, St. Louis, MO, USA), and recombinant mouse vascular endothelial growth factor (VEGF) (100 ng/mL: R&D Systems, Minneapolis, MN, USA).
Real-Time PCR Analysis
Total RNA was isolated using the QIAshredder and RNeasy mini kit (QIAGEN, Valencia, CA, USA). Up to 1 μg of RNA was converted into cDNA according to the instructions of the PrimeScript 1st strand cDNA Synthesis Kit (Takara, Kyoto, Japan). Real-time PCR was performed using the SYBR Green PCR Master Mix (Roche, Eugene, OR, USA) with specific primers (Table S1). Real-time PCR was performed using an ABI PRISM-7500 sequence detection system (Applied Biosystems, Foster City, CA, USA). 18S rRNA was simultaneously run as a control and used for normalization.
ChIP Assay
mESCs were fixed with 1% formaldehyde for 10 min, which was quenched by adding 125 mM glycine (pH 2.2) for 5 min; the cells were then lysed with ChIP lysis buffer (50 mM Tris-HCl [pH 8.0]), 1 mM EDTA, 0.5% Triton X-100, 0.1% sodium dodecyl chloride, 0.1% sodium deoxyl sulfate, 140 mM NaCl, 1 mM PMSF, and a 1× protease inhibitor cocktail (Roche, Eugene, OR, USA). Lysed samples are sonicated to shear DNA into 500∼1,500-bp fragments using the Bioruptor (Diagenode, Denville, NJ, USA). Anti-Sp1 (Millipore, Billerica, MA, USA) or normal rabbit immunoglobulin G (Cell Signaling Technology, Danvers, MA, USA) were used for immunoprecipitation of the DNA fragments. Protein A/G agarose beads (Abcam, Cambridge, UK) were added to pull down the target-antibody complexes, which were washed four times with ChIP wash buffer (Santa Cruz Biotechnology, Dallas, TX, USA). The crosslinking was reversed by heating at 65°C for 16 h, and DNA was recovered using the QIAquick PCR Purification Kit (QIAGEN, Valencia, CA, USA). The PCR products were resolved on 2% agarose gels stained with ethidium bromide. The PCR primers were designed to amplify a region of the MyoD promoter harboring Sp1-binding sites and the negative binding site, as shown in Table S1.
Western Blot Assays
Cells were harvested and lysed in lysis buffer containing protease inhibitors (Roche, Eugene, OR, USA). Total protein (10∼30 μg) was immunoblotted with specific primary antibodies as follows: anti-Sp1 (Millipore, Billerica, MA, USA), anti-MyoD (Santa Cruz Biotechnology, Dallas, TX, USA), anti-Myf5 (Santa Cruz Biotechnology, Dallas, TX, USA), anti-Pax3 (DSHB, Iowa City, Iowa, USA), anti-HIF1α (Cayman Chemical, Ann Arbor, MI, USA), anti-HIF2α (Novus Biologicals, Littlsanteton, CO, USA), anti-AP2α (Abcam, Cambridge, UK), and anti-α-tubulin (Calbiochem, La Jolla, CA, USA). This was followed by incubation for 1 h with horseradish peroxidase (HRP)-conjugated secondary antibody (Santa Cruz Biotechnology, Dallas, TX, USA). Immunoreactive bands were visualized by enhanced chemiluminescence using the Novex ECL Chemiluminescent Substrate Reagent (Invitrogen Life Technologies, Carlsbad, CA, USA). Quantification of band intensity was performed using ImageJ software (NIH, Bethesda, MD, USA) and normalized to the intensity of α-tubulin.
miRNA Microarray and miRNA Real-Time PCR Analysis
Total RNA from cultures exposed to three experimental conditions (mESCs cultured for 16 h under normoxia, EBs cultured for 16 h under normoxia, EBs cultured for 16 h under hypoxia) was prepared using a Trizol Extraction Kit (Invitrogen, Life Technologies, Carlsbad, CA, USA) according to the manufacturer’s instructions. miRNA microarrays were performed by Genomictree (Daejeon, Korea). In brief, 100 ng of total RNA was labeled and hybridized to miRNA microarrays (Agilent Technologies, Santa Clara, CA, USA) using the mouse miRNA Microarray Kit protocol for use with Agilent miRNA microarrays Version 1.0. Hybridization signals were detected with a DNA microarray scanner G2505B (Agilent Technologies, Santa Clara, CA, USA) and the scanned images were analyzed using Agilent feature extraction software (v9.5.3.1). Data were analyzed using GeneSpring GX 7.3.1 software (Agilent Technologies) and normalized as follows: (1) values below 0.01 were set to 0.01; (2) to make comparisons between one-color expression profiles, each measurement was divided by the 50th percentile of all measurements from the same species. The data presented in this manuscript have been deposited into the NCBI Gene Expression Omnibus and are accessible through GEO: GSE125487 (mouse). For the detection of miRNA levels, single-stranded cDNA was synthesized using the Taqman miRNA RT Kit (Applied Biosystems, Foster City, CA, USA). Real-time PCR of miRNA was performed using TaqMan miRNA assays (Applied Biosystems, Foster City, CA, USA) as follows: miR-7a-5p (assay ID 000268), miR-7b-3p (assay ID 002555), miR-27a-3p (assay ID 000408), miR-92a-3p (assay ID 000430), miR-128-3p (assay ID 002216), miR-290-3p (assay ID 002591), miR-335-3p (assay ID 002185), miR-466c-5p (assay ID 463771_mat), and U6 small nuclear RNA (assay ID 001973) for an internal control. Primer information is outlined in Table S1.
Plasmid Construction, Oligonucleotides, and Transfection
For Sp1 knockdown, we used the MISSION TRC shRNA Target Set (TRCN0000071603) or the control sh-plasmid (MISSION Non-Target shRNA Control SHC002; Sigma, St. Louis, MO, USA). C57 ESCs were transfected with Metafetamin (Biotex Laboratories, Edmonton, Alberta, Canada) and selected by puromycin treatment (10 μg/mL; Sigma, St. Louis, MO, USA) for stable transfection. An Sp1 overexpression vector containing the Sp1 CDS (NM_013672.2) was synthesized by PCR and cloned into pcDNA3.1 (Invitrogen, Carlsbad, CA, USA). Primer information can be found in Table S2.
miRNA mimic oligomers, miRNA precursors, and an miRNA inhibitor were purchased from ThermoFisher (ThermoFisher Scientific, Waltham, MA, USA) as follows: mirVana miRNA mimics: mmu-miR-7a-5p (MC10047), mmu-miR-7b-3p (MC19214), mmu-miR-27a-3p (MC10939), mmu-miR-92a-3p (MC10312), mmu-miR-128-3p (MC11746), mmu-miR-290a-3p (MC12487), mmu-miR-335-3p (MC13018), mmu-miR-466c-5p (MC19405), miRNA mimic negative control (4464058); pre-miR miRNA precursors: pre-miR-27a-3p precursor (PM10939), pre-miR-92a-3p precursor (PM10312), pre-miR miRNA precursor control (AM17110). mirVana miRNA inhibitors: mmu-miR-92a-3p (MH10312), mirVana miRNA inhibitor negative control (4464078). Transfection was performed using Metafetamin (Biotex, Edmonton, Alberta, Canada) according to the manufacturer’s protocol.
Luciferase Assays for MyoD Promoter, Sp1 3′ UTR, and miRNA Target Validation
The promoter region of MyoD, containing putative AP2α- and Sp1-binding sites, was obtained by genomic PCR and cloned into the pGL4 basic luciferase reporter vector (Promega, Madison, WI, USA). Deletion mutants without each Sp1-binding site in the MyoD promoter region were generated using the primer set described in Table S2 with the QuickChange II Site-Directed Mutagenesis System (Staratagene, La Jolla, CA, USA), which was followed by sequence verification. Luciferase assays were performed using the Luciferase Assay System kit (Promega, Madison, WI, USA) with a GloMax luminometer (Promega, Madison, WI, USA). The relative luciferase activity was normalized as relative light units to β-galactosidase activity. The Sp1 3′ UTR was purchased from GeneCopoeia (MmiT030998, Rockville, MD, USA) and subcloned downstream of the Renilla luciferase gene in psiCHECK-2 (Promega, Madison, WI, USA). Because the Sp1 3′ UTR is greater than 5 kb, the UTR was broken up into shorter fragments and cloned into two constructs using overlapping sequences. Site-directed mutagenesis of the miR-92a-binding site in the Sp1 3′ UTR was achieved using QuickChange II Site-Directed Mutagenesis System (Staratagene, La Jolla, CA, USA), which was followed by sequence verification. For reporter assays, C57 ESCs cultured in the absence of LIF and feeder cells were transfected with various combinations of effector plasmids. Luciferase assays were performed using the Dual-Glo luciferase assay system kit (Promega, Madison, WI, USA) with a GloMax luminometer (Promega, Madison, WI, USA). The transfection efficiency was normalized to firefly luciferase activity as an internal control, as the psiCHECK-2 Vector also contains a constitutively expressed firefly luciferase gene. Primer sequences for cloning are described in Table S2.
Cardiotoxin-Muscle Injury Model
All animal experiments were performed with approval from the Institutional Animal Care and Use Committee of Seoul National University Hospital (SNU-170524-2). For the rota-rod test, male C57BL/6 (8 weeks old) mice were anesthetized and 50 μL of CTX (10 μM, Sigma, St. Louis, MO, USA) was injected into both TA muscles of each mouse to induce muscle injury.9, 51 One day later, cells from shMock-EBs/normoxia, shMock-EBs/hypoxia, or shSp1-EBs/hypoxia groups (total cell numbers corresponding to 5 × 104/50 μL PBS) were transplanted into the TA muscle of each group. As a sham control, PBS was injected into the TA muscle. EBs were labeled with DiI Dye (2 μg/mL; Molecular Probes, Eugene, OR, USA) for 30 min before inducing EB formation via the hanging drop method to trace differentiation. The TA muscles were harvested and histological analysis was performed to evaluate differentiation into skeletal muscle cells in injured muscles.
Rota-Rod Test
After the procedures, we evaluated the ability of mice to remain on a rotating rod (Panlab Rota-Rods LE. 8200, Harvard Apparatus, Holliston, MA, USA).51 For this, we measured the time it took the mouse to fall off the rod, which was rotating under continuous acceleration (from 5 to 40 rpm) as a measurement of motor function competence. We performed four trials for each mouse, measured each latency time on the rod, and calculated the average. Mice were allowed to rest for at least 5 min between each trial. For the mouse habituating period, before CTX muscle injury, the mice stayed on a stationary drum for 3 min and then ran on the rotating rod up to three times.
Immunofluorescence Staining
Differentiated cells from EBs on a μ-Dish35mm high (ibidi, Planegg, Germany) were fixed with 4% PFA, blocked with blocking buffer (0.5% goat serum, 0.1% Triton X-100, 1% BSA-PBS), and labeled with specific markers as follows: anti-MyoD (Dako, Burlingame, CA, USA), anti-MyHC (R&D Systems, Minneapolis, MN, USA), and anti-Sp1 (Millipore, Billerica, MA, USA). The samples were then incubated with Alexa 488- or Alexa 555-conjugated (Invitrogen Life Technologies, Carlsbad, CA, USA) secondary antibodies for 2 h in the dark. After cell transplantation, the TA muscle was excised, rinsed with PBS, and frozen in liquid nitrogen to analyze the tissues of CTX-injured mice. Histological tissue sections (4∼8-μm-thick) were prepared from snap-frozen tissue samples, fixed with acetone, blocked in 1% BSA, and incubated with anti-laminin 2α (Cambridge, UK), which was followed by incubation with Alexa 488 (Invitrogen Life Technologies, Carlsbad, CA, USA). The nuclei were stained with DAPI (Thermo Fisher Scientific Waltham, MA, USA) and mounted using fluorescent mounting medium (DAKO, Burlingame, CA, USA). The images of each section were obtained using an LSM710 confocal microscope (Carl Zeiss, Oberkochen, Germany). For the quantification of regenerating myofibers, at least five randomly selected fields from transverse-sectioned slides from four different mice were analyzed.
Morphometric Analysis Based on H&E Staining
Paraffin sections (4–6-μm-thick) of mouse TA muscles were stained by H&E using standard protocols. The microscopic images were obtained using an Olympus TH4-200 microscope (Olympus, Tokyo, Japan). Myofiber cross-sectional areas (CSA; μm2) were measured using H&E-stained cross-sections with ImageJ software (https://imagej.nih.gov/ij/). For quantification, 8 to 10 randomly selected fields from transverse-sectioned slides from four different mice were analyzed.52
Statistical Analysis
The results are expressed as means ± SEM. Comparisons between two groups were performed using a Student’s t test. p values < 0.05 were considered statistically significant. All analyses were performed using SPSS 17.0 (SPSS, Chicago, IL, USA) or Excel.
Author Contributions
Conceived the project and experimental design: S.-Y.L. Conducted experiments: S.-Y.L., J.Y., J.H.P., W.J.K., and J.L. Assisted with vector construction and experiments: W.J.K., S.-Y.K., I.H., and C.-S.L. Contributed to data analysis: H.K.S. and E.J.L. Performed data analysis, interpretation, and wrote manuscript: S.-Y.L. and J.Y. Contributed to revising manuscript and discussion: H.-S.K. All authors read and approved the final manuscript.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This study is supported by Technology R&D Project “Strategic Center of Cell and Bio Therapy for Heart, Diabetes & Cancer (HI17C2085)” and “Korea Research-Driven Hospital (HI14C1277)” through the Korea Health Industry Development Institute (KHIDI), funded by the Ministry of Health & Welfare, Republic of Korea, and by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (2017R1D1A1B03034649).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2019.08.014.
Supplemental Information
References
- 1.Itskovitz-Eldor J., Schuldiner M., Karsenti D., Eden A., Yanuka O., Amit M., Soreq H., Benvenisty N. Differentiation of human embryonic stem cells into embryoid bodies compromising the three embryonic germ layers. Mol. Med. 2000;6:88–95. [PMC free article] [PubMed] [Google Scholar]
- 2.Lee S.W., Lee Y.M., Bae S.K., Murakami S., Yun Y., Kim K.W. Human hepatitis B virus X protein is a possible mediator of hypoxia-induced angiogenesis in hepatocarcinogenesis. Biochem. Biophys. Res. Commun. 2000;268:456–461. doi: 10.1006/bbrc.2000.2093. [DOI] [PubMed] [Google Scholar]
- 3.Kim M.S., Kwon H.J., Lee Y.M., Baek J.H., Jang J.E., Lee S.W., Moon E.J., Kim H.S., Lee S.K., Chung H.Y. Histone deacetylases induce angiogenesis by negative regulation of tumor suppressor genes. Nat. Med. 2001;7:437–443. doi: 10.1038/86507. [DOI] [PubMed] [Google Scholar]
- 4.Lee S.W., Kim W.J., Choi Y.K., Song H.S., Son M.J., Gelman I.H., Kim Y.J., Kim K.W. SSeCKS regulates angiogenesis and tight junction formation in blood-brain barrier. Nat. Med. 2003;9:900–906. doi: 10.1038/nm889. [DOI] [PubMed] [Google Scholar]
- 5.Semenza G.L. Oxygen sensing, hypoxia-inducible factors, and disease pathophysiology. Annu. Rev. Pathol. 2014;9:47–71. doi: 10.1146/annurev-pathol-012513-104720. [DOI] [PubMed] [Google Scholar]
- 6.Semenza G.L. Regulation of mammalian O2 homeostasis by hypoxia-inducible factor 1. Annu. Rev. Cell Dev. Biol. 1999;15:551–578. doi: 10.1146/annurev.cellbio.15.1.551. [DOI] [PubMed] [Google Scholar]
- 7.Youn S.W., Lee S.W., Lee J., Jeong H.K., Suh J.W., Yoon C.H., Kang H.J., Kim H.Z., Koh G.Y., Oh B.H. COMP-Ang1 stimulates HIF-1α-mediated SDF-1 overexpression and recovers ischemic injury through BM-derived progenitor cell recruitment. Blood. 2011;117:4376–4386. doi: 10.1182/blood-2010-07-295964. [DOI] [PubMed] [Google Scholar]
- 8.Lee S.W., Jeong H.K., Lee J.Y., Yang J., Lee E.J., Kim S.Y., Youn S.W., Lee J., Kim W.J., Kim K.W. Hypoxic priming of mESCs accelerates vascular-lineage differentiation through HIF1-mediated inverse regulation of Oct4 and VEGF. EMBO Mol. Med. 2012;4:924–938. doi: 10.1002/emmm.201101107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Lee S.W., Yang J., Kim S.Y., Jeong H.K., Lee J., Kim W.J., Lee E.J., Kim H.S. MicroRNA-26a induced by hypoxia targets HDAC6 in myogenic differentiation of embryonic stem cells. Nucleic Acids Res. 2015;43:2057–2073. doi: 10.1093/nar/gkv088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cook T., Gebelein B., Urrutia R. Sp1 and its likes: biochemical and functional predictions for a growing family of zinc finger transcription factors. Ann. N Y Acad. Sci. 1999;880:94–102. doi: 10.1111/j.1749-6632.1999.tb09513.x. [DOI] [PubMed] [Google Scholar]
- 11.Wierstra I. Sp1: emerging roles--beyond constitutive activation of TATA-less housekeeping genes. Biochem. Biophys. Res. Commun. 2008;372:1–13. doi: 10.1016/j.bbrc.2008.03.074. [DOI] [PubMed] [Google Scholar]
- 12.Marin M., Karis A., Visser P., Grosveld F., Philipsen S. Transcription factor Sp1 is essential for early embryonic development but dispensable for cell growth and differentiation. Cell. 1997;89:619–628. doi: 10.1016/s0092-8674(00)80243-3. [DOI] [PubMed] [Google Scholar]
- 13.Thomas K., Wu J., Sung D.Y., Thompson W., Powell M., McCarrey J., Gibbs R., Walker W. SP1 transcription factors in male germ cell development and differentiation. Mol. Cell. Endocrinol. 2007;270:1–7. doi: 10.1016/j.mce.2007.03.001. [DOI] [PubMed] [Google Scholar]
- 14.Saffer J.D., Jackson S.P., Annarella M.B. Developmental expression of Sp1 in the mouse. Mol. Cell. Biol. 1991;11:2189–2199. doi: 10.1128/mcb.11.4.2189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Philipsen S., Suske G. A tale of three fingers: the family of mammalian Sp/XKLF transcription factors. Nucleic Acids Res. 1999;27:2991–3000. doi: 10.1093/nar/27.15.2991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Heinrich E.M., Dimmeler S. MicroRNAs and stem cells: control of pluripotency, reprogramming, and lineage commitment. Circ. Res. 2012;110:1014–1022. doi: 10.1161/CIRCRESAHA.111.243394. [DOI] [PubMed] [Google Scholar]
- 17.Bartel D.P. MicroRNAs: genomics, biogenesis, mechanism, and function. Cell. 2004;116:281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 18.Lee H.J. Exceptional stories of microRNAs. Exp. Biol. Med. (Maywood) 2013;238:339–343. doi: 10.1258/ebm.2012.012251. [DOI] [PubMed] [Google Scholar]
- 19.Suh M.R., Lee Y., Kim J.Y., Kim S.K., Moon S.H., Lee J.Y., Cha K.Y., Chung H.M., Yoon H.S., Moon S.Y. Human embryonic stem cells express a unique set of microRNAs. Dev. Biol. 2004;270:488–498. doi: 10.1016/j.ydbio.2004.02.019. [DOI] [PubMed] [Google Scholar]
- 20.Krichevsky A.M., King K.S., Donahue C.P., Khrapko K., Kosik K.S. A microRNA array reveals extensive regulation of microRNAs during brain development. RNA. 2003;9:1274–1281. doi: 10.1261/rna.5980303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lagos-Quintana M., Rauhut R., Yalcin A., Meyer J., Lendeckel W., Tuschl T. Identification of tissue-specific microRNAs from mouse. Curr. Biol. 2002;12:735–739. doi: 10.1016/s0960-9822(02)00809-6. [DOI] [PubMed] [Google Scholar]
- 22.Alvarez-Garcia I., Miska E.A. MicroRNA functions in animal development and human disease. Development. 2005;132:4653–4662. doi: 10.1242/dev.02073. [DOI] [PubMed] [Google Scholar]
- 23.Esquela-Kerscher A., Slack F.J. Oncomirs - microRNAs with a role in cancer. Nat. Rev. Cancer. 2006;6:259–269. doi: 10.1038/nrc1840. [DOI] [PubMed] [Google Scholar]
- 24.Krützfeldt J., Stoffel M. MicroRNAs: a new class of regulatory genes affecting metabolism. Cell Metab. 2006;4:9–12. doi: 10.1016/j.cmet.2006.05.009. [DOI] [PubMed] [Google Scholar]
- 25.Tedesco F.S., Dellavalle A., Diaz-Manera J., Messina G., Cossu G. Repairing skeletal muscle: regenerative potential of skeletal muscle stem cells. J. Clin. Invest. 2010;120:11–19. doi: 10.1172/JCI40373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ozasa S., Kimura S., Ito K., Ueno H., Ikezawa M., Matsukura M., Yoshioka K., Araki K., Yamamura K.I., Abe K. Efficient conversion of ES cells into myogenic lineage using the gene-inducible system. Biochem. Biophys. Res. Commun. 2007;357:957–963. doi: 10.1016/j.bbrc.2007.04.032. [DOI] [PubMed] [Google Scholar]
- 27.Wang Y.X., Rudnicki M.A. Satellite cells, the engines of muscle repair. Nat. Rev. Mol. Cell Biol. 2011;13:127–133. doi: 10.1038/nrm3265. [DOI] [PubMed] [Google Scholar]
- 28.Tapscott S.J., Davis R.L., Thayer M.J., Cheng P.F., Weintraub H., Lassar A.B. MyoD1: a nuclear phosphoprotein requiring a Myc homology region to convert fibroblasts to myoblasts. Science. 1988;242:405–411. doi: 10.1126/science.3175662. [DOI] [PubMed] [Google Scholar]
- 29.Mokalled M.H., Johnson A.N., Creemers E.E., Olson E.N. MASTR directs MyoD-dependent satellite cell differentiation during skeletal muscle regeneration. Genes Dev. 2012;26:190–202. doi: 10.1101/gad.179663.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Mitchell D.L., DiMario J.X. AP-2 alpha suppresses skeletal myoblast proliferation and represses fibroblast growth factor receptor 1 promoter activity. Exp. Cell Res. 2010;316:194–202. doi: 10.1016/j.yexcr.2009.08.008. [DOI] [PubMed] [Google Scholar]
- 31.Mummaneni P., Yates P., Simpson J., Rose J., Turker M.S. The primary function of a redundant Sp1 binding site in the mouse aprt gene promoter is to block epigenetic gene inactivation. Nucleic Acids Res. 1998;26:5163–5169. doi: 10.1093/nar/26.22.5163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Mizuno Y., Chang H., Umeda K., Niwa A., Iwasa T., Awaya T., Fukada S., Yamamoto H., Yamanaka S., Nakahata T., Heike T. Generation of skeletal muscle stem/progenitor cells from murine induced pluripotent stem cells. FASEB J. 2010;24:2245–2253. doi: 10.1096/fj.09-137174. [DOI] [PubMed] [Google Scholar]
- 33.Chang H., Yoshimoto M., Umeda K., Iwasa T., Mizuno Y., Fukada S., Yamamoto H., Motohashi N., Miyagoe-Suzuki Y., Takeda S. Generation of transplantable, functional satellite-like cells from mouse embryonic stem cells. FASEB J. 2009;23:1907–1919. doi: 10.1096/fj.08-123661. [DOI] [PubMed] [Google Scholar]
- 34.Patton B.L., Connoll A.M., Martin P.T., Cunningham J.M., Mehta S., Pestronk A., Miner J.H., Sanes J.R. Distribution of ten laminin chains in dystrophic and regenerating muscles. Neuromuscul. Disord. 1999;9:423–433. doi: 10.1016/s0960-8966(99)00033-4. [DOI] [PubMed] [Google Scholar]
- 35.Rudnicki M.A., Braun T., Hinuma S., Jaenisch R. Inactivation of MyoD in mice leads to up-regulation of the myogenic HLH gene Myf-5 and results in apparently normal muscle development. Cell. 1992;71:383–390. doi: 10.1016/0092-8674(92)90508-a. [DOI] [PubMed] [Google Scholar]
- 36.Kaul A., Köster M., Neuhaus H., Braun T. Myf-5 revisited: loss of early myotome formation does not lead to a rib phenotype in homozygous Myf-5 mutant mice. Cell. 2000;102:17–19. doi: 10.1016/s0092-8674(00)00006-4. [DOI] [PubMed] [Google Scholar]
- 37.Kassar-Duchossoy L., Gayraud-Morel B., Gomès D., Rocancourt D., Buckingham M., Shinin V., Tajbakhsh S. Mrf4 determines skeletal muscle identity in Myf5:Myod double-mutant mice. Nature. 2004;431:466–471. doi: 10.1038/nature02876. [DOI] [PubMed] [Google Scholar]
- 38.Zhang L., Jin M., Margariti A., Wang G., Luo Z., Zampetaki A., Zeng L., Ye S., Zhu J., Xiao Q. Sp1-dependent activation of HDAC7 is required for platelet-derived growth factor-BB-induced smooth muscle cell differentiation from stem cells. J. Biol. Chem. 2010;285:38463–38472. doi: 10.1074/jbc.M110.153999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Xiao Q., Luo Z., Pepe A.E., Margariti A., Zeng L., Xu Q. Embryonic stem cell differentiation into smooth muscle cells is mediated by Nox4-produced H2O2. Am. J. Physiol. Cell Physiol. 2009;296:C711–C723. doi: 10.1152/ajpcell.00442.2008. [DOI] [PubMed] [Google Scholar]
- 40.Guo C.S., Degnin C., Fiddler T.A., Stauffer D., Thayer M.J. Regulation of MyoD activity and muscle cell differentiation by MDM2, pRb, and Sp1. J. Biol. Chem. 2003;278:22615–22622. doi: 10.1074/jbc.M301943200. [DOI] [PubMed] [Google Scholar]
- 41.Biesiada E., Hamamori Y., Kedes L., Sartorelli V. Myogenic basic helix-loop-helix proteins and Sp1 interact as components of a multiprotein transcriptional complex required for activity of the human cardiac alpha-actin promoter. Mol. Cell. Biol. 1999;19:2577–2584. doi: 10.1128/mcb.19.4.2577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.McCarthy J.J. The MyomiR network in skeletal muscle plasticity. Exerc. Sport Sci. Rev. 2011;39:150–154. doi: 10.1097/JES.0b013e31821c01e1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Nallamshetty S., Chan S.Y., Loscalzo J. Hypoxia: a master regulator of microRNA biogenesis and activity. Free Radic. Biol. Med. 2013;64:20–30. doi: 10.1016/j.freeradbiomed.2013.05.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cummins E.P., Taylor C.T. Hypoxia-responsive transcription factors. Pflugers Arch. 2005;450:363–371. doi: 10.1007/s00424-005-1413-7. [DOI] [PubMed] [Google Scholar]
- 45.Cavadas M.A.S., Cheong A., Taylor C.T. The regulation of transcriptional repression in hypoxia. Exp. Cell Res. 2017;356:173–181. doi: 10.1016/j.yexcr.2017.02.024. [DOI] [PubMed] [Google Scholar]
- 46.Caruso P., MacLean M.R., Khanin R., McClure J., Soon E., Southgate M., MacDonald R.A., Greig J.A., Robertson K.E., Masson R. Dynamic changes in lung microRNA profiles during the development of pulmonary hypertension due to chronic hypoxia and monocrotaline. Arterioscler. Thromb. Vasc. Biol. 2010;30:716–723. doi: 10.1161/ATVBAHA.109.202028. [DOI] [PubMed] [Google Scholar]
- 47.Ho J.J., Metcalf J.L., Yan M.S., Turgeon P.J., Wang J.J., Chalsev M., Petruzziello-Pellegrini T.N., Tsui A.K., He J.Z., Dhamko H. Functional importance of Dicer protein in the adaptive cellular response to hypoxia. J. Biol. Chem. 2012;287:29003–29020. doi: 10.1074/jbc.M112.373365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Cai Q., Wang T., Yang W.J., Fen X. Protective mechanisms of microRNA-27a against oxygen-glucose deprivation-induced injuries in hippocampal neurons. Neural Regen. Res. 2016;11:1285–1292. doi: 10.4103/1673-5374.189194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kang B.Y., Park K.K., Green D.E., Bijli K.M., Searles C.D., Sutliff R.L., Hart C.M. Hypoxia mediates mutual repression between microRNA-27a and PPARγ in the pulmonary vasculature. PLoS ONE. 2013;8:e79503. doi: 10.1371/journal.pone.0079503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Jin F., Yang R., Wei Y., Wang D., Zhu Y., Wang X. HIF-1alpha-induced miR-23a approximately 27a approximately 24 cluster promotes colorectal cancer progression via reprogramming metabolism. Cancer Lett. 2019;440-441:211–222. doi: 10.1016/j.canlet.2018.10.025. [DOI] [PubMed] [Google Scholar]
- 51.Darabi R., Gehlbach K., Bachoo R.M., Kamath S., Osawa M., Kamm K.E., Kyba M., Perlingeiro R.C. Functional skeletal muscle regeneration from differentiating embryonic stem cells. Nat. Med. 2008;14:134–143. doi: 10.1038/nm1705. [DOI] [PubMed] [Google Scholar]
- 52.Liu N., Williams A.H., Maxeiner J.M., Bezprozvannaya S., Shelton J.M., Richardson J.A., Bassel-Duby R., Olson E.N. microRNA-206 promotes skeletal muscle regeneration and delays progression of Duchenne muscular dystrophy in mice. J. Clin. Invest. 2012;122:2054–2065. doi: 10.1172/JCI62656. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.