

Abstract
Macrophages play a crucial role in the pathogenesis of atherosclerosis, but the molecular mechanisms remain poorly understood. Here we show that microRNA-34a (miR-34a) is a key regulator of macrophage cholesterol efflux and reverse cholesterol transport by modulating ATP-binding cassette transporters ATP-binding cassette subfamily A member 1 (ABCA1) and ATP-binding cassette subfamily G member 1 (ABCG1). miR-34a also regulates M1 and M2 macrophage polarization via liver X receptor α. Furthermore, global loss of miR-34a reduces intestinal cholesterol or fat absorption by inhibiting cytochrome P450 enzymes CYP7A1 and sterol 12α-hydroxylase (CYP8B1). Consistent with these findings, macrophage-selective or global ablation of miR-34a markedly inhibits the development of atherosclerosis. Finally, therapeutic inhibition of miR-34a promotes atherosclerosis regression and reverses diet-induced metabolic disorders. Our studies outline a central role of miR-34a in regulating macrophage cholesterol efflux, inflammation, and atherosclerosis, suggesting that miR-34a is a promising target for treatment of cardiometabolic diseases.
Keywords: atherosclerosis, cholesterol efflux, inflammation, ABCA1, ABCG1, LXR, miR-34a
Graphical Abstract
Atherosclerosis is the leading cause of death in Western countries. Zhang and colleagues show that microRNA-34a (miR-34a) plays a key role in regulating macrophage cholesterol efflux, inflammation, and atherosclerosis. They also show that antagonism of miR-34a promotes regression of atherosclerosis and improves metabolic homeostasis.
Introduction
Atherosclerosis is a chronic inflammatory disease characterized by lipid accumulation in arterial walls and is the major cause of coronary heart disease.1, 2 Macrophages play a key role in the pathogenesis of atherosclerosis by participating in both inflammatory response and foam cell formation. ATP-binding cassette subfamily A member 1 (ABCA1) and ATP-binding cassette subfamily G member 1 (ABCG1) play a key role in promoting macrophage cholesterol efflux,3, 4 which is the first step of reverse cholesterol transport (RCT) that prevents foam cell formation and the development of atherosclerosis. On the other hand, M1 (inflammatory) or M2 (anti-inflammatory) macrophage polarization is also a key event during the progression or regression of atherosclerosis.5 So far, the molecular mechanisms governing macrophage cholesterol efflux and inflammation remain poorly understood.
MicroRNAs (miRNAs) are small non-coding RNA molecules that regulate gene expression by binding to 3′ UTRs of target mRNAs, usually leading to mRNA degradation or translational repression. miR-34a is expressed in macrophages, endothelial cells, smooth muscle cells (SMCs), hepatocytes, adipocytes, etc., and is shown to be induced in the livers of mice with metabolic syndrome or patients with non-alcoholic fatty liver disease (NAFLD).6, 7 Increased hepatic miR-34a expression promotes liver steatosis, whereas inhibition of hepatic miR-34a expression has an opposite effect.6, 7 In addition, adenovirus-mediated overexpression of miR-34a in the liver protects against the development of atherosclerosis because of reduced secretion of apolipoprotein B (ApoB)-containing lipoproteins from the liver.6
Lentivirus-mediated downregulation of miR-34a prevents diet-induced adiposity likely by suppressing adipocyte fibroblast growth factor (FGF) 21 signaling and sirtuin 1 (SIRT 1) function.8 Very recently, Pan et al.9 demonstrate that adipocyte-secreted exosomal miR-34a plays a key role in promoting obesity-induced adipose inflammation. miR-34a is also shown to promote vascular calcification via vascular SMC mineralization by inhibiting SMC proliferation and inducing SMC senescence.10, 11 In addition, miR-34a is reported to induce endothelial cell senescence in part through suppression of SIRT1,12 whereas antagonism of miR-34a inhibits endothelial cell apoptosis and the progression of atherosclerosis in Apoe−/− mice.13
So far, the role of macrophage miR-34a in atherosclerosis is completely unknown. In this report, we demonstrate that miR-34a is a key modulator of macrophage cholesterol efflux and macrophage polarization by coordinated regulation of ABCA1, ABCG1, and liver X receptor α (LXRα). As a result, myeloid- or macrophage-selective ablation of miR-34a inhibits the development of atherosclerosis in Apoe−/− or Ldlr−/− mice. In addition, global loss of miR-34a reduces intestinal cholesterol or fat absorption by inhibiting cholesterol 7α-hydroxylase (CYP7A1) and sterol-12α hydroxylase (CYP8B1). Finally, therapeutic treatment of mice with miR-34a inhibitors improves metabolic homeostasis and promotes regression of atherosclerosis.
Results
miR-34a Regulates ABCA1 and ABCG1 Expression and Cholesterol Efflux in Macrophages
Previous studies show that hepatic miR-34a is induced under the condition of common metabolic stress.6, 7 Interestingly, miR-34a was also significantly induced in atherosclerotic plaques of human patients (Figure S1A) or Apoe−/− mice (Figure S1B). Treatment of RAW267.4 cells with cholesterol, palmitate, oxidized low-density lipoprotein (ox-LDL), tumor necrosis factor α (TNF-α), or interleukin-6 (IL-6) significantly increased miR-34a expression (Figure S1C), which might partly explain the increased miR-34a expression in atherosclerotic plaques. Interestingly, in human atherosclerotic lesions, the mRNA levels of ABCA1, ABCG1, LXRα, arginase 1 (ARG1), and mannose receptor C-type 1 (MRC1) were repressed, whereas TNFα and IL6 were induced (Figure S1D).
To investigate whether miR-34a regulates cholesterol homeostasis in macrophages, we first determined whether miR-34a regulated the expression of ABCA1 or ABCG1, two of the most important cholesterol efflux transporters in macrophages. Treatment of human THP-1 cells with miR-34a mimics inhibited ABCA1 or ABCG1 protein expression by >50%, whereas miR-34a inhibitors had opposite effects (Figure 1A). The changes in protein expression were consistent with those in mRNA levels (Figure S2A). In addition, treatment of mouse RAW267.4 cells or human primary macrophages with miR-34a mimics or inhibitors had similar effects as those observed in THP-1 cells (Figure 1B; Figures S2B and S2C). In agreement with these data, peritoneal macrophages isolated from miR-34a−/− mice had a >2-fold increase in the levels of Abca1 or Abcg1 mRNA (Figure S2D) or proteins (Figure 1C).
Figure 1.

miR-34a Ablation Induces ABCA1 and ABCG1 to Promote Macrophage Reverse Cholesterol Transport
(A and B) Human THP-1 cells (A) or mouse RAW267.4 cells (B) were treated with 75 nM scramble (Scr) mimics, miR-34a mimics, scramble inhibitors (Scr inhb), or miR-34a inhibitors (34a inhb) (n = 4). After 24 h, proteins were isolated for western blot assays (top panels), and protein levels were quantified (bottom panels). (C) Peritoneal macrophages were isolated from chow-fed miR-34a+/+ mice or miR-34a−/− mice (n = 4). Western blotting was performed (top panel), and protein levels were quantified (bottom panel). (D) RAW267.4 cells were infected with Ad-Empty or Ad-miR-34a for 36 h. RNA immunoprecipitation (RIP) was performed using an AGO2 antibody (top panel). Abca1, Abcg1, Lxrα, and Lxrβ mRNA levels were then quantified by quantitative real-time PCR (bottom panel) (n = 4). (E) Plasmids expressing Abca1 (left panel) or Abcg1 (right panel) wild-type or mutant 3′ UTRs were transfected into HepG2 cells together with scramble or miR-34a mimics. After 36 h, relative luciferase units (RLUs) were determined (n = 5). (F and G) Peritoneal macrophages were isolated from chow-fed miR-34a+/+ or miR-34a−/− mice (F), or from chow-fed miR-34a+/+A1G1fl/fl, miR-34a+/+A1G1M−, miR-34a−/−A1G1fl/fl, or miR-34a−/−A1G1M− mice (G). Cholesterol efflux to ApoA-I (left panel) or HDL (right panel) was determined in the presence or absence of acetylated LDL (Ac-LDL; 50 μg/mL) (n = 6). (H–K) Peritoneal macrophages were isolated from miR-34a+/+ or miR-34a−/− mice carrying or lacking both Abca1 and Abcg1, and then incubated with [3H]cholesterol for 48 h, followed by i.p. injection to C57BL/6 mice (H). Plasma levels of 3H-tracers at 24 (left panel) or 48 h (right panel) were analyzed (n = 8) (I). Hepatic 3H-tracer levels at 48 h were quantified (n = 8) (J). Fecal 3H-tracer levels at 24 (left panel) or 48 h (right panel) were determined (n = 8) (K). In (H)–(K), A1G1 refers to ABCA1/ABCG1. See also Figures S1–S3. All of the data are expressed as mean ± SEM. A two-tailed Student’s t test was used for statistical analysis. *p < 0.05, **p < 0.01. Veh, vehicle.
Argonaute-2 (AGO2) is one of the main components of the RNA-induced silencing complex that mediates miRNA-directed gene silencing. We performed ribonucleoprotein immunoprecipitation (RNP-IP) using AGO2 antibody/beads and RAW267.4 cell lysates. miR-34a overexpression significantly enriched the association of Abca1, Abcg1, or Lxrα mRNAs, but not Lxrβ mRNA, with the AGO2-containing complex (Figure 1D), suggesting that miR-34a directly regulates Abca1, Abcg1, and Lxrα. In human or mouse ABCA1 or ABCG1 3′ UTRs, there were conserved binding sites for miR-34a (Figure S2E). Treatment with miR-34a mimics significantly inhibited ABCA1 or ABCG1 3′ UTR activity, whereas mutations of the miR-34a binding sites completely abolished the inhibitory effects of miR-34a (Figure 1E). These data indicate that miR-34a directly binds to ABCA1 or ABCG1 3′ UTRs to regulate their expression.
To determine whether miR-34a regulated macrophage cholesterol efflux, we isolated peritoneal macrophages from miR-34a+/+ or miR-34a−/− mice and treated them with either vehicle or acetylated LDL (Ac-LDL). Ac-LDL treatment or miR-34a ablation markedly increased Abca1 or Abcg1 mRNA levels (Figures S2F and S2G). Consistent with the changes in mRNA levels, miR-34a ablation caused a marked increase in cholesterol efflux to ApoA-I or high-density lipoprotein (HDL) (Figure 1F). Interestingly, Ac-LDL treatment increased cholesterol efflux to ApoA-I or HDL in wild-type macrophages, but not in miR-34a−/− macrophages (Figure 1F). One plausible explanation is that either miR-34a ablation or Ac-LDL treatment has increased the macrophage cholesterol efflux capacity to the maximal level via the same mechanism: by inducing ABCA1 and ABCG1 expression.
When peritoneal macrophages were treated with Ac-LDL, miR-34a ablation led to a reduction in cellular levels of total cholesterol and free cholesterol (Figure S3A) but had not much of an effect on cellular cholesterol ester (CE) content (Figure S3A) or cholesterol uptake (p = 0.075; Figure S3B). Nonetheless, we cannot fully rule out the possibility that miR-34a deficiency may prevent cholesterol accumulation in macrophages partly via reducing cholesterol uptake. When miR-34a−/− mice, miR-34aM−/−Apoe−/− mice, miR-34a−/−Apoe−/− mice, and their control mice were fed a Western diet, miR-34a ablation reduced macrophage size (Figures S3C–S3E) and cellular total cholesterol and free cholesterol levels (Figures S3F–S3H). These data are consistent with a role of miR-34a ablation in promoting macrophage cholesterol efflux.
miR-34a Ablation Increases Macrophage Cholesterol Efflux and RCT Dependent on ABCA1 and ABCG1
To investigate whether miR-34a ablation enhances macrophage cholesterol efflux via induction of ABCA1 and ABCG1, we crossed floxed Abca1 and Abcg1 (Abca1fl/flAbcg1fl/fl) mice with miR-34a+/+ mice, miR-34a−/− mice, and/or LysM-Cre mice to generate miR-34a+/+ mice or miR-34a−/− mice carrying or lacking both Abca1 and Abcg1 in macrophages. Consistent with the data of Figure 1F, miR-34a−/− macrophages had increased cholesterol efflux to ApoA-I or HDL (Figure 1G). However, the increases in cholesterol efflux were completely abolished in the absence of Abca1 and Abcg1 (Figure 1G).
To investigate whether ablation of macrophage miR-34a promoted reverse cholesterol transport (RCT) in vivo, we isolated peritoneal macrophages from mice lacking miR-34a, Abca1, or Abcg1 and their control littermates (Figure 1H). These macrophages were loaded with [3H]cholesterol and then injected into C57BL/6 mice (Figure 1H). As expected, miR-34a ablation increased [3H]tracers in the plasma (Figure 1I), liver (Figure 1J), and feces (Figure 1K) at 24 or 48 h. Importantly, these increases were completely abolished when both Abca1 and Abcg1 were abrogated (Figures 1I–1K). Thus, the data of Figures 1G–1K demonstrate that miR-34a ablation stimulates macrophage cholesterol efflux and RCT via induction of ABCA1 and ABCG1.
miR-34a Regulates M1/M2 Macrophage Polarization Partly via LXRα
Inflammation plays a key role in the pathogenesis of atherosclerosis. We therefore investigated the role of miR-34a in macrophage inflammation. miR-34a abrogation prevented lipopolysaccharide (LPS) from inducing inflammatory genes, including Nf-κb, Tnfα, Il6, and Il1β (Figure S4A), suggesting that miR-34a may promote inflammation. In human THP-1 cells, miR-34a mimics induced M1 macrophage markers (TNFα, IL6, IL1β, and CD80) (Figures 2A and 2B) but repressed M2 macrophage markers (ARG1, MRC1, CD163) and LXRα (Figures 2B and 2C). In contrast, miR-34a inhibitors had the opposite effects (Figures 2A–2C). Similar observations were collected when mouse RAW267.4 cells were used; miR-34a mimics induced M1 macrophage markers (Tnfα, Il6, Il1β, and Nos2) but repressed M2 macrophage markers (Arg1, Mrc1, Ym1, Retnla) and Lxrα, whereas miR-34a inhibitors had the opposite effects (Figures 2D–2F). Consistent with these findings, miR-34a−/− macrophages had a marked reduction in M1 macrophage markers and a >2-fold increase in M2 macrophage markers and Lxrα (Figures 2G and 2H). Similar observations were collected when macrophages were treated with Ac-LDL (Figure S4B). Importantly, treatment of human primary macrophages with miR-34a mimics or inhibitors regulated M1 markers (TNF-α, IL-6, IL-1β, CD80), M2 markers (ARG1, MRC1, CD163), or LXRα (Figures S4C and S4D) in a pattern similar to those observed in THP-1 or RAW267.1 cells. In addition, miR-34a−/− mice also had lower levels of plasma TNF-α, IL-6, and IL-1β (Figure S4E), consistent with a change in gene expression in peritoneal macrophages.
Figure 2.

mir-34a Regulates Macrophage Polarization Partly via LXRα
(A–F) THP-1 cells (A–C) or RAW267.4 cells (D–F) were transfected with 75 nM miR-34a mimics, miR-34a inhibitors, or scramble control oligos (n = 4–6). After 24 h, mRNA levels (A, C, D, and F) or protein levels (B and E) were determined. (G and H) Peritoneal macrophages were isolated from chow-fed miR-34a+/+ or miR-34a−/− mice. mRNA (G) and protein (H) levels were determined (n = 3–4). (I and J) Peritoneal macrophages were isolated from chow-fed miR-34a+/+ or miR-34a−/− mice and then infected with lentiviruses expressing shRNA against scramble sequences (Lenti-shScr) or Lxrα (Lenti-shLxrα) for 48 h. mRNA levels were determined (n = 3). (K and L) Peritoneal macrophages were isolated from chow-fed miR-34a+/+ or miR-34a−/− mice and then treated for 24 h with vehicle, LPS (1 μg/mL) plus IFNγ (50 ng/mL) (K), or IL-4 (20 ng/mL) (L) (n = 4–6). The mRNA levels for M1 (K) or M2 (L) macrophages were quantified. See also Figures S4–S6. All of the data are expressed as mean ± SEM. In (A)–(J), a two-tailed Student’s t test was used for statistical analysis. In (K) and (L), a two-way ANOVA test was used for statistical analysis. *p < 0.05, **p < 0.01.
LXRs are nuclear hormone receptors that play a key role in inhibiting macrophage inflammation and promoting macrophage cholesterol efflux.14 Interestingly, miR-34a overexpression or inhibition in peritoneal macrophages, THP-1 cells, or RAW267.4 cells did not affect the expression of Lxrβ (Figures S5A–S5D). The finding that miR-34a regulates LXRα expression suggests that LXRα might be implicated in miR-34a-regulated inflammatory response. To test this hypothesis, we infected miR-34a+/+ macrophages or miR-34a−/− macrophages with lentiviruses expressing shRNA against scramble sequences (Lenti-shScr) or Lxrα (Lenti-shLxrα). As expected, infection with Lxrα shRNA markedly reduced Lxrα expression and its target genes Abca1 and Abcg1 in both genotypes (Figure S5E). In addition, inhibition of Lxrα also induced M1 macrophage markers (Tnfα, Il6, Il1β) (Figure 2I) but repressed M2 macrophage markers (Arg1, Mrc1, Ym1) (Figure 2J) in miR-34a+/+ macrophages. Consistent with the data of Figures 2A–2H, miR-34a ablation repressed M1 macrophage markers but induced M2 macrophage markers in miR-34a−/− macrophages infected with Lenti-shScr; however, miR-34a ablation failed to exert similar effects when Lxrα was inhibited (Figures 2I and 2J). These data demonstrate that LXRα signaling is required, at least in part, for miR-34a to regulate M1 or M2 macrophage polarization. In line with these data, miR-34a overexpression repressed Lxrα expression in peritoneal macrophages (Figure S5A) and also Lxrα 3′ UTR activity in luciferase-reporter assays (Figure S5F). In addition to LXRα, miR-34a also regulated Kruppel-like factor 4 (Klf4) expression in THP-1 cells, RAW267.4 cells, or peritoneal macrophages (Figure S5G). KLF4 is known to play an important role in macrophage polarization15 and is regulated by miR-34a.9 Interestingly, inhibition of Klf4 by small interfering RNA (siRNA) also partially blunted the effects of miR-34a ablation on modulation of M1/M2 macrophage polarization (Figure S5H). Thus, our data show that both LXRα and KLF4 are required for miR-34a ablation to modulate macrophage polarization.
Finally, miR-34a abrogation also prevented LPS plus IFNγ-induced M1 macrophage polarization (Tnfα, Il6, Il1β) but enhanced IL-4-induced M2 macrophage polarization (Arg1, Mrc1, Ym1) (Figures 2K and 2L). In contrast, miR-34a overexpression had the opposite effects (Figures S6A and S6B). Thus, the data of Figure 2 indicate that miR-34a plays a key role in macrophage polarization via a mechanism involving LXRα.
Macrophage-Selective miR-34a Ablation Protects against the Development of Atherosclerosis
The findings that miR-34a ablation in macrophages leads to increased RCT and M2 macrophage polarization promoted us to hypothesize that macrophage miR-34a abrogation might be sufficient to protect against atherosclerosis. To test this hypothesis, we generated mice lacking both Apoe and miR-34a in macrophages (34aM−Apoe−/−) by crossing miR-34afl/fl mice with LysM-Cre mice and/or Apoe−/− mice. Expression of LysM-Cre reduced miR-34a expression in macrophages by 90% but had no effect on miR-34a expression in other tissues (Figure S7A). Macrophage-selective deletion of miR-34a did not affect plasma triglyceride (TG), total cholesterol, or very low-density lipoprotein (VLDL)/LDL cholesterol (LDL-C) levels, but increased plasma HDL cholesterol (HDL-C) levels (Figures S7B and S7E), the latter of which was consistent with a role of miR-34a ablation in promoting cholesterol efflux to ApoA-I or HDL. When fed a Western diet, macrophage-selective deletion of miR-34a in Apoe−/− mice reduced plasma levels of inflammatory chemokine/cytokines, including MCP-1 (monocyte chemoattractant protein-1), TNF-α, IL-6, and IL-1β (Figures S7F–S7I). Interestingly, macrophage-selective ablation of miR-34a in Apoe−/− mice attenuated atherosclerotic lesions by >50% in both the aortas (Figures 3A and 3B) and aortic roots (Figures 3C and 3D). Indeed, there was a reduction of total cholesterol, free cholesterol, and TG in the aortas of 34aM−Apoe−/− mice (Figure S7J). 34aM−Apoe−/− mice also had increased mRNA levels of Abca1 and Abcg1 but decreased Cd68 mRNA levels in the plaques (Figure 3E). In addition, M1 macrophage markers (Tnfα, Il6, Il1β, Nos2) were repressed, whereas M2 macrophage markers (Arg1, Mrc1) and Lxrα were induced in the plaques of 34aM−Apoe−/− mice (Figure 3F).
Figure 3.

Macrophage- or Myeloid-Selective miR-34a Ablation Protects against Atherosclerosis in Apoe−/− or Ldlr−/− Mice
(A–M) 34afl/flApoe−/− or 34aM−Apoe−/− mice were fed a Western diet for 18 weeks. En face aortas were stained by oil red O (ORO) (A), and the plaque size was quantified (B) (n = 15–17). Aortic roots were sectioned and stained with ORO (C), and the plaque size was determined (D) (n = 14–16). The mRNA levels of genes in the plaques of aortic roots were quantified by quantitative real-time PCR (n = 6) (E and F). The proteins in aortic roots were detected by immunohistochemistry (G and H), and ABCA1 (I) or ABCG1 (J) protein levels in the macrophages of plaques were quantified (n = 5). Negative controls are presented in Figure S8A. Collagens in the aortic roots were stained with picro sirius red (K; top panel), and the collagen content in plaques was quantified (L) (n = 5). In addition, aortic roots were stained with H&E (K; bottom panel), and the necrotic size in the plaques was analyzed (M) (n = 5). (N–Q) Bone marrow from miR-34a+/+ or miR-34a−/− mice was transplanted into irradiated Ldlr−/− mice, which were then fed a Western diet for 16 weeks. En face aortas were stained with ORO (N), and the plaque size was determined (O) (n = 9–12). Aortic roots were also stained by ORO (P), and the plaque size was determined (Q) (n = 9–10). See also Figures S7 and S8. In (G) and (H), arrows point to ABCA1+CD68+ cells (G) or ABCG1+CD68+ cells (H). In (K), the arrow points to the necrotic area. All of the data are expressed as mean ± SEM. A two-tailed Student’s t test was used for statistical analysis. *p < 0.05, **p < 0.01.
Consistent with the change in mRNA levels, we observed increased macrophage ABCA1 and ABCG1 expression (Figures 3G–3J), increased collagen content (Figures 3K and 3L), and reduced CD68 expression (Figure S7K) or necrotic area (Figures 3K and 3M) in the plaques of 34aM−Apoe−/− mice. Macrophage-selective ablation of miR-34a did not affect calcification (Figures S7L and S7M) but increased the content of alpha smooth muscle actin (α-SMA)-positive SMCs (Figures S7N and S7O). These data suggest that macrophage-selective ablation of miR-34a reduces lesion size and increases plaque stability in Apoe−/− mice. In addition, there was not much change in hepatic levels of total cholesterol (Figure S7P) or TG (Figure S7Q), which is consistent with unchanged plasma TG or VLDL/LDL-C levels (Figures S7B–S7E). These latter data suggest that the liver or plasma lipoproteins should have no or little impact on the changes in plaque formation between the two genotypes.
The finding that 34aM−Apoe−/− mice have reduced plasma MCP-1 levels (Figure S7F) led us to determine whether macrophage miR-34a ablation reduced monocyte infiltration to the aorta. By injecting PKH-26-labeled miR-34a+/+ or miR-34a−/− macrophages to 34afl/flApoe−/− or 34aM−Apoe−/− mice, respectively, our data show that miR-34a ablation reduced PKH-26+F4/80+ macrophage content in the aortas of 34aM−Apoe−/− mice by ∼80% (Figure S9A). In contrast, there was not much change in 5-ethynyl-2’-deoxyuridine (EdU)+F4/80+ macrophage content between the two genotypes (Figure S9B). These data indicate that macrophage-selective miR-34a ablation reduces monocyte infiltration but has no effect on local macrophage proliferation.
Finally, transplantation of bone marrow from miR-34a−/− mice to irradiated Ldlr−/− mice also markedly reduced atherosclerotic lesions in both the aortas (Figures 3N and 3O) and aortic roots (Figures 3P and 3Q). Together, our data demonstrate that macrophage-selective miR-34a ablation protects against the development of atherosclerosis by increasing macrophage cholesterol efflux and reducing monocyte infiltration.
Global miR-34a Ablation Ameliorates Dyslipidemia, Atherosclerosis, and Obesity in Apoe−/− or Ldlr−/− Mice
To determine whether global and macrophage-selective loss of miR-34a had similar effects on atherosclerosis, we crossed miR-34a−/− mice with Ldlr−/− mice or Apoe−/− mice. Ablation of miR-34a in Ldlr−/− mice reduced plasma levels of total cholesterol (Figure 4A), TG (Figure 4B), VLDL-cholesterol (Figure 4C), and VLDL-TG (Figure 4D). Consistent with the improved plasma lipid homeostasis, global loss of miR-34a reduced atherosclerotic lesions in both the aortas (Figures 4E and 4F) and aortic roots (Figures 4G and 4H) of Ldlr−/− mice. Similar results were collected when miR-34a was abrogated in Apoe−/− mice (Figures S10A–S10H). Thus, global inactivation of miR-34a improves dyslipidemia and atherosclerosis in both Ldlr−/− mice and Apoe−/− mice.
Figure 4.

Global Loss of miR-34a Improves Hyperlipidemia and Attenuates the Development of Atherosclerosis in Ldlr−/− Mice
(A–H) 34a+/+Ldlr−/− or 34a−/−Ldlr−/− mice were fed a Western diet for 18 weeks. Plasma cholesterol (A) or TG (B) levels were determined (n = 14–15). Plasma cholesterol (C) or TG (D) lipoprotein profiles were analyzed by fast protein liquid chromatography (FPLC). En face aortas were stained by oil red O (ORO) (E), and the plaque size was determined (n = 12) (F). Aortic roots were also stained by ORO (G), and the plaque size was determined (n = 9–10) (H). See also Figures S10 and S11. All of the data are expressed as mean ± SEM. A two-tailed Student’s t test was used for statistical analysis. *p < 0.05, **p < 0.01.
In addition, global loss of miR-34a in Ldlr−/− mice also reduced hepatic TG levels, plasma glucose levels, body fat content, and white adipose tissue amount (Figures S11A–S11C). There was no change in the amount of brown adipose tissue (BAT) or food intake (Figures S11C and S11D). Furthermore, miR-34a inactivation increased oxygen consumption (Figures S11E and S11F), CO2 production (Figures S11G and S11H), and energy expenditure (Figure S11I). There was no change in respiration exchange ratio (RER) (Figure S11J). Finally, global loss of miR-34a significantly induced mRNA levels of genes involved in energy expenditure in BAT, including Pgc1α, Pgc1β, Ucp1, Ucp2, and Ucp3 (Figure S11K). Thus, genetic loss of miR-34a promotes energy expenditure and reduces diet-induced obesity in Ldlr−/− mice.
miR-34a Inactivation Inhibits Lipid Absorption via Inhibiting CYP7A1 and CYP8B1 Expression
The data of Figure 4 and Figure S10 show that global loss of miR-34a in Western diet-fed Ldlr−/− or Apoe−/− mice reduces plasma levels of TG and cholesterol. The improved hyperlipidemia in these mice may also contribute to the athero-protective effect of miR-34a ablation. To determine how global loss of miR-34a improved plasma lipid homeostasis, we determined the effect of miR-34a ablation on lipid absorption. miR-34a−/− mice had reduced cholesterol and fat absorption in the intestine (Figures S12A and S12B), which were associated with unchanged expression of genes involved in lipid absorption in the intestine (Figure S12C).
Bile acids are known to promote intestinal fat absorption.16 CYP7A1 and sterol 12α-hydroxylase (CYP8B1) are two rate-limiting enzymes in the classic pathway of bile acid biosynthesis. A previous report shows that miR-34a inhibits FGF15/19 signaling to upregulate CYP7A1 and CYP8B1 expression by targeting the membrane co-receptor β-Klotho of FGF15/19.7 Indeed, overexpression of miR-34a in the liver induced CYP7A1 and CYP8B1 expression (Figures 5A and 5B). Interestingly, miR-34a overexpression repressed the expression of scavenger receptor group B type 1 (SR-BI) (Figures 5A and 5B), the HDL receptor in the liver. Luciferase reporter assays showed that miR-34a mimics inhibited the 3′ UTR activity of SR-BI (Figure S12D). In contrast, miR-34a−/− mice had increased hepatic SR-BI expression but reduced CYP7A1 and CYP8B1 expression (Figures 5C and 5D). Similar observations were seen when C57BL/6 mice were treated with miR-34a inhibitors (Figures S12E and S12F). In addition, miR-34a−/− mice have increased mRNA levels of Abcg5 and Abcg8 (Figure S12G), two transporters that form heterodimers to efflux free cholesterol from hepatocytes to the bile.17 Consistent with the changes in hepatic CYP7A1 and CYP8B1 expression, overexpression of hepatic miR-34a increased the bile acid pool size (Figure 5E), whereas miR-34a−/− mice had a reduced bile acid pool size (Figure 5F). There was not much change in bile acid composition (Figure S12H), which was likely because CYP7A1 and CYP8B1 were regulated in a similar pattern.
Figure 5.

Global Loss of miR-34a Inhibits Lipid Absorption via Inhibiting Hepatic CYP7A1 and CYP8B1 Expression
(A and B) C57BL/6 mice were injected i.v. with Ad-Empty or Ad-miR-34a. After 7 days, hepatic proteins were used for immunoblotting assays (A), and protein levels were quantified (B) (n = 6). (C and D) Hepatic proteins were isolated from chow-fed miR-34a+/+ or miR-34a−/− mice (n = 6). Western blotting assays were performed (C), and hepatic protein levels were quantified (D). (E and F) Bile acid levels in mice infected with Ad-Empty or Ad-miR-34a (E) or in chow-fed miR-34a+/+ or miR-34a−/− mice (F) (n = 7–8). (G–I) miR-34a+/+ or miR-34a−/− mice were fed a Western diet for 18 weeks and then injected i.v. with Ad-Empty (Null) or Ad-CYP7A1 plus Ad-CYP8B1 (7A+8B). After 7 days, hepatic proteins were used for immunoblotting assays (G), and intestinal cholesterol (H) or fat (I) absorption was determined (n = 7–8). See also Figure S12. All of the data are expressed as mean ± SEM. In (A)–(F), a two-tailed Student’s t test was used for statistical analysis. In (H) and (I), a two-way ANOVA test was used for statistical analysis. *p < 0.05, **p < 0.01.
To determine whether global loss of miR-34a reduces lipid absorption via repressing hepatic CYP7A1 and CYP8B1 expression, we normalized hepatic CYP7A1 and CYP8B1 expression in miR-34a−/− mice (Figure 5G). Upon Western diet feeding, miR-34a−/− mice had reduced plasma levels of cholesterol (Figure S12I) and TG (Figure S12J), which were normalized after hepatic CYP7A1 and CYP8B1 levels were recovered (Figures S12I and S12J). In addition, recapitulation of hepatic CYP7A1 and CYP8B1 expression in miR-34a−/− mice to the levels of the control group also normalized intestinal cholesterol absorption (Figure 5H) and intestinal fat absorption (Figure 5I). Thus, miR-34a inactivation improves systemic lipid homeostasis by inhibiting CYP7A1 and CYP8B1 expression.
Therapeutic Inhibition of miR-34a Reverses Metabolic Disorders and Atherosclerosis
The data of Figures 1, 2, 3, 4, and 5 indicate that genetic loss of miR-34a has beneficial metabolic effects. To investigate whether pharmacological inhibition of miR-34a had any beneficial effects, we fed Ldlr−/− mice a Western diet for 14 weeks to induce atherosclerosis and metabolic disorders. These mice were then switched to a normal chow diet, followed by intraperitoneal (i.p.) injection with PBS, locked nucleic acid (LNA) against scramble sequences (LNA-Scr), or miR-34a (LNA-miR-34a) once a week for 5 weeks (Figure 6A). Compared with LNA-Scr or PBS treatment, LNA-miR-34a significantly reduced plasma levels of cholesterol, TG, glucose, and alanine aminotransferease (ALT) with aspartate aminotransferase (AST) levels tending to decrease (Figure 6B). The reduction in plasma cholesterol levels may be because of reduced cholesterol absorption and increased hepatic SR-BI expression (see Figure 5).
Figure 6.

Pharmacological Inhibition of miR-34a Ameliorates Metabolic Disorders and Promotes Regression of Atherosclerosis
(A–T) Ldlr−/− mice were fed a Western diet for 12 weeks and then switched to a standard chow diet. The mice were i.p. injected with PBS, scramble locked nucleic acid (LNA-Scr), or LNA-miR-34a (LNA-34a) once a week for 5 weeks. Echo-MRI and CLAMS were performed at week 4 or 5, respectively. The experimental procedures are illustrated in (A). Plasma levels of cholesterol, triglycerides (TG), glucose, ALT, and AST were analyzed (B) (n = 8). Fat content at day 0 (left panel) or day 28 (right panel) was analyzed by Echo-MRI (C) (n = 7–8). Oxygen consumption (D) or energy expenditure (EE) (E) was determined by CLAMS (n = 7–8). Plasma cholesterol (left) or TG (right) lipoprotein profiles were determined by FPLC (F). En face aortas were stained by oil red O (ORO) (G), and the plaque size was determined (H) (n = 9–10). Aortic roots were also stained by ORO (I), and the plaque size was analyzed (J) (n = 9–10). The mRNA levels of genes in the plaques of aortic roots were determined by quantitative real-time PCR (K) (n = 7). The proteins in aortic roots were immunostained with primary antibodies against ABCA1, ABCG1, or CD68 (L and M), and ABCA1+CD68+ (N) or ABCG1+CD68+ (O) areas in the plaques were quantified (n = 5). Collagens in aortic roots were stained with picro sirius red (P), and the collagen content in the plaques was quantified (Q) (n = 5). In addition, hepatic TG (R) or cholesterol levels (S) were analyzed (n = 8). Livers were stained with picro sirius red (left panel) or H&E (right panel) (T). In (L) and (M), arrows point to cells positive for ABCA1 and CD68 (L) or ABCG1 and CD68 (M). In (T), arrows point to fibrosis. See also Figure S13. All of the data are expressed as mean ± SEM. A two-tailed Student’s t test was used for statistical analysis. *p < 0.05, **p < 0.01.
We also determined the effect of miR-34a inhibition on obesity. At the baseline, there was no change in body fat content among groups (Figure 6C, left panel). After 4-week treatment with LNA-miR-34a, body fat content was remarkably reduced by >50% (Figure 6C, right panel). Consistent with this latter finding, LNA-miR-34a treatment increased oxygen consumption (Figure 6D) and energy expenditure (Figure 6E) during the day or night.
LNA-miR-34a also decreased plasma levels of VLDL-cholesterol and VLDL-TG (Figure 6F). In line with these findings, LNA-miR-34a reduced atherosclerotic lesions by >50% in both the aortas (Figures 6G and 6H) and aortic roots (Figures 6I and 6J). In addition, LNA-miR-34a induced Abca1, Abcg1, Lxrα, and M2 macrophage marker mRNA levels but reduced miR-34a, Cd68, Tnfα, Il6, and Nos2 mRNA levels in atherosclerotic lesions (Figure 6K; Figure S13A). In agreement with the change in mRNA levels, LNA-miR-34a also increased macrophage ABCA1 and ABCG1 protein expression (Figures 6L–6O) and collagen content (Figures 6P and 6Q), but reduced CD68 expression (Figure S13B) and necrosis (Figures S13C and S13D) in the plaques of aortic roots. In addition, LNA-miR-34a treatment did not affect calcification (Figures S13E and S13F) but increased the content of α-SMA-positive SMCs (Figures S13G and S13H) in the plaques of aortic roots. These data suggest that LNA-miR-34a may increase macrophage cholesterol efflux and plaque stability, and decrease macrophage infiltration by inducing M2 macrophage polarization.
Finally, LNA-miR-34a also significantly reduced hepatic accumulation of TG (Figure 6R) or cholesterol (Figure 6S) and improved liver fibrosis and histology (Figure 6T), indicating that LNA-miR-34a reverses NAFLD. Taken together, the data of Figure 6 demonstrate that pharmacological inhibition of miR-34a has a beneficial effect on reversal of diet-induced metabolic disorders and atherosclerosis.
Discussion
In this work, we demonstrate that miR-34a plays a key role in regulating macrophage cholesterol efflux and inflammation, reverse cholesterol transport, and atherosclerosis by coordinating regulation of some genes critically involved in the pathogenesis of atherosclerosis, including ABCA1, ABCG1, LXRα, SR-BI, CYP7A1, and CYP8B1 (Figure 7). As a result, genetic ablation of miR-34a or therapeutic inhibition of miR-34a markedly prevents the development of atherosclerosis or regresses atherosclerosis. Importantly, inhibition of miR-34a also improves dyslipidemia, obesity, and NAFLD. Thus, our current work suggests that miR-34a may be a promising target for treatment of atherosclerosis and other common metabolic disorders.
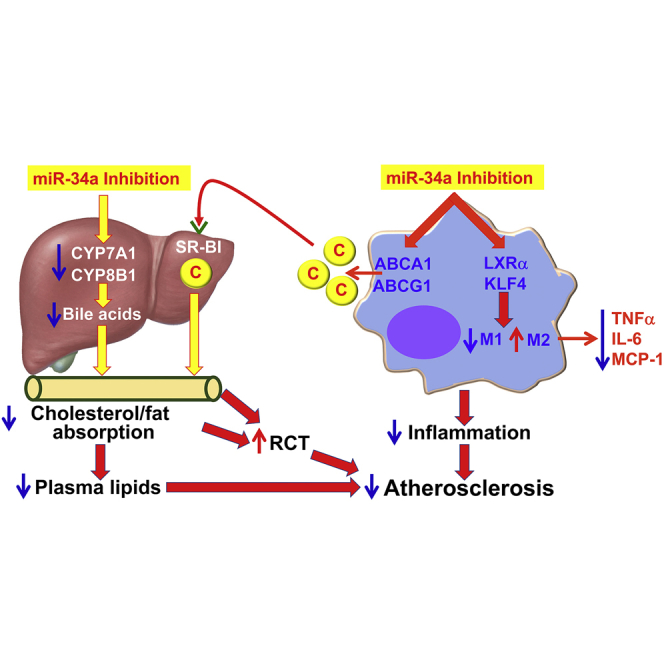
Figure 7.

A Model for Macrophage miR-34a to Regulate Atherogenesis
Inhibition of macrophage miR-34a induces ABCA1 and ABCG1 expression to promote macrophage cholesterol efflux. HDL carries the effluxed free cholesterol to the liver by binding to SR-BI. In the liver, free cholesterol is secreted to the bile and then to the intestine. miR-34a inhibition also suppresses hepatic CYP7A1 and CYP8B1 expression, resulting in a reduction in bile acid synthesis in the liver, as well as cholesterol and fat absorption in the intestine. As a result, reverse cholesterol transport (RCT) is increased and plasma lipids (cholesterol and triglycerides) are decreased. On the other hand, miR-34a inhibition also induces LXRα and KLF4 to promote M2 macrophage polarization, thus reducing macrophage inflammation. By reducing macrophage inflammation and inducing macrophage cholesterol efflux and RCT, inhibition of miR-34a can both prevent and regress atherosclerosis. C, cholesterol.
ABCA1 and ABCG1 are two of the well-characterized and most important cholesterol transporters in macrophages that prevent macrophages from cholesterol overload. ABCA1 protein level is significantly reduced in human atherosclerotic lesions,18 and ABCG1 mRNA is markedly reduced in monocytes of humans with metabolic syndrome.19 These observations are consistent with our findings that miR-34a expression is induced, whereas ABCA1 and ABCG1 are repressed in atherosclerotic lesions, and that miR-34a inhibits ABCA1 and ABCG1 expression. Both LXR activation and miR-33 inhibition are shown to upregulate ABCA1 and ABCG1 expression in macrophages.14, 20 However, LXR activation also induces liver steatosis,21, 22 whereas miR-33 inhibition or ablation is reported to have inconsistent effects on atherosclerosis or metabolic homeostasis.23 Thus, it is becoming necessary and important to identify novel regulator(s) of ABCA1 and ABCG1 that do not have significant side effect(s). The identification of miR-34a as a novel regulator of ABCA1 and ABCG1 appears very attractive because miR-34a inhibition is shown to display many beneficial metabolic effects. Interestingly, our data show that LXRα, but not LXRβ, is a downstream target of miR-34a. Although LXRs are known to regulate ABCA1 and ABCG1 expression, our data show that miR-34a may directly bind to ABCA1 and ABCG1 3′ UTRs to inhibit their expression. Thus, miR-34a regulates ABCA1 and ABCG1 expression likely by directly binding to ABCA1 and ABCG1 3′ UTRs (a direct mechanism) and/or LXRα 3′ UTR (an indirect mechanism). Importantly, our data show that the induction of ABCA1 and ABCG1 is required for miR-34a ablation to increase macrophage cholesterol efflux and reverse cholesterol transport (RCT). Consistent with a critical role of LXRs in regulating macrophage inflammation,24 our data also indicate that LXRα is required for miR-34a ablation to regulate M1/M2 macrophage polarization. Interestingly, our data show that KLF4 is also required for miR-34a to regulate M1/M2 macrophage polarization. It will be interesting to investigate whether any interaction between LXRα and KLF4 is involved in miR-34a-mediated regulation of M1/M2 macrophage polarization.
RCT initiates from macrophage cholesterol efflux. The liver also plays a critical role in RCT. Inhibition of miR-34a not only promotes macrophage cholesterol transport via induction of ABCA1 and ABCG1, but also induces SR-BI in the liver. Increased hepatic expression of SR-BI promotes macrophage RCT and inhibits atherosclerosis, whereas loss of SR-BI has the opposite effects.25 In addition, miR-34a ablation inhibits intestinal cholesterol absorption by suppressing CYP7A1 and CYP8B1 expression and bile acid synthesis. Cholesterol absorption is an important component of RCT because about ∼50% of the cholesterol in the intestine is absorbed or reabsorbed. Bile acids help cholesterol absorption in the intestine. Therefore, miR-34a inhibition promotes RCT via coordinated regulation of macrophage cholesterol efflux (ABCA1, ABCG1), cholesterol uptake in the liver (SR-BI), and cholesterol absorption in the intestine (via hepatic CYP7A1, CYP8B1) (Figure 7).
Increased ApoB-containing lipoproteins in the plasma promote the development of atherosclerosis. Loss of miR-34a lowers plasma ApoB-containing lipoprotein levels by reducing cholesterol and fat absorption in the intestine. These effects are abolished when hepatic CYP7A1 and CYP8B1 levels are normalized in miR-34a−/− mice, supporting a critical role of bile acids in regulating lipid absorption and plasma lipid homeostasis. Previous studies show that miR-34a upregulates CYP7A1 and CYP8B1 expression by suppressing β-Klotho, a co-receptor for FGFR4, therefore attenuating the inhibitory effect of FGF15/19.7 Nonetheless, we cannot rule out other mechanism(s) that may also be involved.
Our studies have suggested that a reduction in bile acid synthesis may have beneficial effects on metabolic regulation. Consistent with our observations, CYP8B1 ablation or inhibition is shown to protect against NAFLD, obesity, and atherosclerosis by mechanisms that may involve reduced fat and cholesterol absorption, reduced ApoB-containing lipoprotein secretion, and altered gut microbial composition.26, 27, 28, 29, 30 Cyp7a1−/− mice also have a marked reduction in cholesterol absorption.31 Loss of Cyp7a1 in the Apoe*3-Leiden background is shown to reduce VLDL secretion.32 These data suggest that inhibition of bile acid synthesis may be sufficient to protect against hyperlipidemia and atherosclerosis.
We have previously shown that acute overexpression of hepatic miR-34a by adenoviruses markedly reduced hepatic expression of hepatocyte nuclear factor 4α (HNFα), resulting in a reduction in VLDL secretion and atherosclerosis in Ldlr−/− mice.6 Because adenovirus-mediated overexpression of miR-34a leads to acute changes in the liver, it remains unknown whether acute or chronic inhibition/ablation of hepatic miR-34a alone has the opposite effects on atherosclerosis. In the current study, we investigate the role of macrophage-selective or global ablation of miR-34a in atherosclerosis, which is a chronic treatment and is not liver focused. Although global ablation of miR-34a improves hyperlipidemia in Apoe−/− or Ldlr−/− mice, the contribution of the improved hyperlipidemia to atherosclerosis in these mice may be limited because macrophage-selective miR-34a ablation or global loss of miR-34a inhibits atherosclerosis to a similar extent (∼50%) in Apoe−/− mice. Thus, our loss-of-function data suggest that miR-34a in macrophages may have an overwhelming effect on regulating atherosclerosis compared with that in the liver or hepatocytes.
Atherosclerosis is often associated with dyslipidemia, obesity, and NAFLD. Our data show that genetic inactivation or pharmacological inhibition of miR-34a induces energy expenditure and reduces obesity, which are associated with increased expression of genes involved in energy metabolism in BAT. The study by Fu et al.8 suggests that miR-34a inhibition reduces obesity by improving adipocyte FGF21 signaling and SIRT1 function. NAFLD is one of the most common liver diseases worldwide and is an independent risk factor for atherosclerosis.33, 34 NAFLD includes simple steatosis and non-alcoholic steatohepatitis (NASH). Our data demonstrate that miR-34a inhibition or ablation reduces diet-induced liver steatosis and fibrosis, as well as plasma ALT levels. The improved liver phenotype may result from an inhibition of CYP7A1 and CYP8B1 and subsequent reduction in cholesterol and fat absorption in the intestine.
In summary, we have identified a novel role of miR-34a in regulating macrophage cholesterol efflux and inflammation, reverse cholesterol transport, and atherosclerosis. Although the current study is focused only on understanding the role of macrophage miR-34a in atherosclerosis, the beneficial effect of miR-34a inhibition on other metabolic disorders, such as obesity and NAFLD, is quite intriguing and important. Thus, inhibition of miR-34a may represent a novel and promising strategy for treatment of atherosclerosis and associated metabolic disorders.
Materials and Methods
Mice and Diets
Mice were housed in a temperature-controlled room under a 12-h light/12-h dark cycle under pathogen-free conditions. 12-week-old male mice were used unless otherwise specified. miR-34afl/fl mice, miR-34a−/− mice, Abca1fl/flAbcg1fl/fl mice, Apoe−/− mice, Ldlr−/− mice, LysM-Cre mice, and C57BL/6J mice were purchased from the Jackson Laboratory and have a C57BL/6 background. These mice were cross-bred to generate miR-34a−/−Apoe−/− mice, miR-34a−/−Ldlr−/− mice, miR-34aM−/−Apoe−/− mice, miR-34a−/−Abca1M−Abcg1M−/− (miR-34a−/−A1G1M−) mice, and respective control littermates. Western diet (42% fat/0.2% cholesterol) was purchased from Envigo (Cat #TD.88137). In general, 10- to 12-week-old male mice were used and randomly allocated. Blinding was performed to prevent bias. Unless otherwise stated, all mice were fasted for 5–6 h prior to euthanization. All of the animal studies were approved by the Institutional Animal Care and Use Committee at Northeast Ohio Medical University and were consistent with the institutional guidelines and NIH Guidelines for the Care and Use of Laboratory Animals.
Plasmids, Human cDNAs, Adenoviruses, and Lentiviruses
ABCA1 and Abcg1 3′ UTR plasmids have been described previously.20 Mutant plasmids containing point mutations in the seed region of the predicted miR-34a binding sites within the 3′ UTRs were generated using a QuickChange Site-Directed Mutagenesis kit (Agilent), and the mutations were confirmed by sequencing. Ad-miR-34a has been described.7 Collection of human carotid endarterectomy specimens was approved by the institutional review board (IRB) at Greenville Health System (South Carolina), and all patients gave written informed consent.35, 36 All procedures were performed in accordance with the NIH guidelines and regulations on human subjects. RNAs isolated from human atherosclerotic lesions or surrounding normal aortas were used for cDNA synthesis.35, 36 Lentiviruses expressing shRNA against Lxrα (LPP-MSH096466-LVRU6GP) or scramble sequences were generated by GeneCopoeia.
Cell Culture and Transfection
THP-1 cells or RAW267.4 cells were purchased from ATCC. Human primary macrophages were purchased from STEMCELL Technologies (Cat #70042). They were transfected with 75 nM miRIDIAN miR-34a mimics, inhibitors, or scramble oligos using DharmaFECT 2 Transfection Reagent (for cell lines) or Viromer Blue transfection reagent (for primary human macrophages; Cat #TT100300; Origene). Mouse peritoneal macrophages were transfected with 75 nM siRNAs against mouse Klf4 or scramble sequences (Cat #SR426926; Origene) using Viromer Blue transfection reagent. Wild-type or mutant 3′ UTRs of ABCA1 or Abcg1 were transfected into HepG2 cells (from ATCC) together with 75 nM miR-34a mimics or scramble mimics. Luciferase activity was determined and normalized to β-galactosidase. Peritoneal macrophages were isolated 4 days after injection of 4% thioglycolate. For macrophage activation experiments, peritoneal macrophages were treated for 24 h with ultrapure LPS (1 μg/mL; Invivogen) plus IFN-γ (50 ng/mL; R&D Systems) for M1 activation, with IL-4 (20 ng/mL; PeproTech) for M2 activation. To inhibit LXR activity, we treated peritoneal macrophages with SR9238 (10 μM) (SML1510; Sigma) or GSK2033 (10 μM) (SML1617; Sigma) for 24 h.
Quantitative Real-Time PCR and Western Blotting
RNA was extracted using TRIzol Reagent (Thermo Fisher), and mRNA levels were quantified by quantitative real-time PCR using SYBR Green (GeneCopoeia) on a 7500 Real Time PCR machine (Applied Biosystems). mRNA levels were normalized to 36B4. miRNAs were extracted using the mirVana miRNA isolation kit, quantified using a TaqMan MicroRNA assay kit (Thermo Fisher), and then normalized to U6. Cell or liver lysates (including total cell lysates or microsomes) were used for western blotting. Antibodies against ABCA1 (NB400-105), ABCG1 (NB400-132), IL-1beta (NB600-633), beta-actin (NB600-501), SR-BI (NB400-101), or calnexin (NB100-1965) were purchased from Novus. Anti-CYP8B1 antibody was purchased from Origene (TA313734). Anti-Tubulin antibody was purchased from Abcam (ab4074). Anti-ARG1 (PA5-29645) or LXRα antibodies (PA1-330) were purchased from Thermo Fisher. Anti-TNF-α antibody (AF-410-NA) was purchased from R&D Systems. CYP7A1 antibody was a gift from Dr. David Russell at UT Southwestern Medical School.
RNP-IP
RAW267.4 cells were infected with Ad-Empty or Ad-miR-34a for 24 h. miRNAs were immunoprecipitated using a mouse Ago2 monoclonal antibody in a MicroRNA Isolation Kit from Wako (292-67301). Ago2-bound Abca1 or Abcg1 mRNA was then quantified using quantitative real-time PCR.
Immunohistochemical Staining and Calcium Staining
Liver or heart was fixed in 10% formalin, embedded in optical cutting temperature compound (OCT), and sectioned. The sections were used for H&E staining, oil red O staining, or picro sirius red staining. For heart sections stained with picro sirius red, polarized light was used for detection of collagen content. For immunohistochemical staining, sections were fixed, washed, and incubated with primary antibodies, followed by incubation with corresponding Alexa Fluor dye-labeled secondary antibodies (Cat #A11012 or #A11006; Thermo Fisher). The nucleus was counterstained with DAPI (Cat #P36935; Thermo Fisher). Anti-CD68 antibody was purchased from Bio-Rad (MCA1957). Anti-α-SMA antibody was purchased from Abcam (Cat #ab5694). Calcium was stained using the Von Kossa Stain kit (Cat #ab150687; Abcam).
Bone Marrow Transplantation
Recipient Ldlr−/− mice were given acidified water (pH 4.5) containing 100 mg/L neomycin (Sigma) and 10 mg/L polymyxin B sulfate (Sigma) 1 week before and 2 weeks after bone marrow transplantation. Ldlr−/− mice were lethally irradiated with 450 rad from a γ-irradiator twice with a 3- to 4-h interval before transplantation. Bone marrow was collected from femurs and tibias of miR-34a+/+ or miR-34a−/− mice by flushing with sterile medium (RPMI 1640, 2% FBS, 5 U/mL heparin, 50 U/mL penicillin, 50 μg/mL streptomycin). Each recipient mouse was injected with about 4–5 × 106 bone marrow cells through intravenous (i.v.) injection. Two weeks later, mice were fed a Western diet for 16 weeks.
VLDL Secretion or Fat or Cholesterol Absorption
VLDL secretion was performed after mice were fasted for 6 h, followed by i.v. injection of 100–200 μL of Triton WR1339 (500 mg/kg). Blood was collected at indicated time points, and TG levels were determined. For fat absorption, mice were fasted for 6 h, followed by i.v. injection of 100–200 μL of Triton WR1339 (500 mg/kg) and then gavage with 200 μL olive oil containing 7 μCi [3H]triolein (Perkin Elmer). Blood was collected at indicated time points, and plasma radioactivity was determined. For cholesterol absorption, a dual isotope ratio method was used as described previously.37
Plasma Biochemistry, Fast Protein Liquid Chromatography, Bile Acid, and Lipid Measurement
Plasma TG, cholesterol, glucose, ALT, and AST levels were measured using Infinity reagents (Thermo Scientific). Bile acids in gallbladder, liver, and intestine were extracted with 95% ethanol overnight, followed by extraction with 80% ethanol for 2 h and methanol/chloroform (2:1) for 2 h at 50°C. Total bile acids were determined using a bile acid assay kit (Diazyme). Plasma lipoproteins were separated by Biologic DuoFlow QuadTec 10 System (Bio-Rad), and TG or cholesterol levels were quantified. HDL or VLDL/LDL cholesterol was measured using an HDL and LDL/VLDL quantification kit (Cat #K613; Biovision). Plasma MCP-1, TNF-α, IL-6, and IL-1β levels were quantified using ELISA kits from PeproTech. Hepatic lipids were extracted in chloroform/methanol (2:1, v/v), and hepatic TG or cholesterol levels were quantified using Infinity reagents (Thermo Scientific). Cellular free cholesterol was measured using a kit from Wako Diagnostics. Macrophage size was measured using a Countless II Automated Cell Counter (Invitrogen).
Body Composition Analysis and Energy Expenditure
Mouse body fat mass was measured by Echo-MRI (EchoMRI). Oxygen consumption, CO2 production, and heat production were determined in a Comprehensive Lab Animal Monitoring System (CLAMS) as detailed previously.38 In brief, mice underwent an acclimation period, and a 24-h measurement of energy expenditure was determined using an eight-chamber system. Each run included two genotypes with four mice per genotype.
Cholesterol Efflux and Uptake Assays
Peritoneal macrophages were seeded in a 24-well plate and loaded with 0.5 μCi/mL [3H]cholesterol (PerkinElmer) for 24 h in the presence or absence of Ac-LDL (50 μg/mL). After extensive wash with PBS, cells were incubated in DMEM supplemented with 0.2% fatty acid-free BSA for 4 h. The cells were washed again by PBS and then incubated in fresh DMEM containing 0.2% BSA in the presence or absence of acceptors ApoA-I (15 μg/mL) or HDL (50 μg/mL). Supernatants were collected after 4 h, and the level of [3H]cholesterol was quantified by scintillation counting. Values are expressed as a percentage of total cell [3H]cholesterol content (effluxed [3H]cholesterol + cell-associated [3H]cholesterol). To determine macrophage cholesterol uptake, we treated macrophages with Ac-LDL (50 μg/mL) for a total of 48 h. In the last 4 h, macrophages were treated with PBS or 10 μg/mL Dil-LDL (Cat #J64029; Alfa Aesar). By the end of the study, cells were extensively washed with PBS, and fluorescence activities were measured.
Reverse Cholesterol Transport
Reverse cholesterol transport (RCT) was performed as described previously.39, 40 In brief, peritoneal macrophages were isolated and incubated in DMEM containing 10% FBS containing 5 μCi/mL [3H]cholesterol and 25 μg/mL Ac-LDL for 48 h, followed by wash in PBS and equilibration for 4 h in DMEM supplemented with 0.2% BSA and penicillin/streptomycin. Before injection, cells were pelleted and re-suspended in serum-free DMEM. A total of 0.5 mL of macrophages (∼5 × 106 cells) containing ∼6.5–10 × 106 counts per minute (cpm) was injected i.p. into wild-type mice. Blood and feces were collected at 24 and 48 h. Plasma, hepatic, and fecal 3H-tracers were assayed by scintillation counting, and values are expressed as a percentage of total injected radioactivity.
Atherosclerosis
The aorta, including the ascending arch, thoracic, and abdominal segments, and aortic root were isolated and gently cleaned of the adventitia. Sectioned aortic roots or en face aortas were stained with oil red O, and the atherosclerotic plaque size was determined using the Image-Pro software (Media Cybernetics) as described previously.41
In Vivo Tracing of Recruitment of Circulating Monocytes and Proliferation of Macrophages in Blood Vessels by Flow Cytometry Analysis
Peripheral blood was isolated from miR-34a+/+ or miR-34a−/− mice and incubated with 9 vol of erythrocytes lysis buffer for 10 min on ice containing 0.15 M NH4Cl, 10 mM KHCO3, and 0.1 mM EDTA. Monocytes were isolated with the EasySep mouse monocyte Isolation kit (Cat #19861; STEMCELL Technologies) and labeled with a PKH-26 labeling kit (Cat #PKH26GL; Sigma). Four million labeled cells were injected intravenously into each of the miR-34afl/flApoe−/− or miR-34aM−Apoe−/− mice that had been fed a Western diet for 14 weeks. To measure proliferation of resident macrophages, miR-34afl/flApoe−/− or miR-34aM−Apoe−/− mice that had been fed a Western diet for 14 weeks were injected i.p. with EdU (2 mg/kg/day; Cat #A10044; Thermo Fisher). After 3 days, aortas were carefully dissected out, and fat tissue was removed. Aortas were cut into small pieces and incubated at 37°C for 1 h under slow shaking in an enzyme cocktail containing 4 U/mL Liberase (#5401127001; Roche), 60 U/mL hyaluronidase type I-S (#H3506; Sigma), and 60 U/mL DNase I (#D5025; Sigma) in PBS. The cell suspension was then passed through a 70-μm cell strainer, centrifuged at 600 × g for 5 min at 4°C, and resuspended in flow cytometry buffer (1% BSA/0.1% NaN3).
Macrophages were stained with allophycocyanin (APC) anti-mouse F4/80 (Cat #123116; BioLegend) or its isotope control (BioLegend) on ice for 1 h in the dark. For the proliferation assays, cells were also stained with Click-iT plus Edu Alexa Fluor 488 Flow Cytometry kit (Cat #C10632; Thermo Fisher). After staining, cells were analyzed using a BD Accuri C6 Plus Flow Cytometer (BD Biosciences).
Statistical Analysis
We used GraphPad Prism 8.1.1 (GraphPad, CA, USA) for statistical analysis. Normality and equal variances tests were performed. If data passed both tests, we used unpaired Student’s t test followed by Welch’s corrections. If the sample did not pass normality or equal variance test, statistical analyses were performed using unpaired Mann-Whitney U test. For multiple group comparison, two-way ANOVA was used. All values are expressed as mean ± SEM. Differences were considered statistically significant at p < 0.05. Sample size was based on statistical ANOVA and prior experience with similar studies. All of the measurements were taken from distinct samples. Sample size and statistical methods being used are specified in the figure legends.
Author Contributions
Yanyong Xu, Yang Xu, L.Y., and Y. Zhang conceived and designed the study, and guided the interpretation of the results. Y. Zhang supervised and prepared the manuscript. Yanyong Xu, Yang Xu, and Y. Zhu performed most in vitro and in vivo studies and data analysis. H.S., C.J., and L.Y. performed some studies and data analysis. F.L. performed analysis of bile acid composition. D.F. provided human cDNA samples.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by NIH grants R01DK102619 (to Y. Zhang), R01HL103227 (to Y. Zhang and L.Y.), R01HL142086 (to Y. Zhang), and R01DK118941 (to Y. Zhang).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2019.09.008.
Supplemental Information
References
- 1.Ross R. Atherosclerosis—an inflammatory disease. N. Engl. J. Med. 1999;340:115–126. doi: 10.1056/NEJM199901143400207. [DOI] [PubMed] [Google Scholar]
- 2.Lusis A.J. Atherosclerosis. Nature. 2000;407:233–241. doi: 10.1038/35025203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ye D., Lammers B., Zhao Y., Meurs I., Van Berkel T.J., Van Eck M. ATP-binding cassette transporters A1 and G1, HDL metabolism, cholesterol efflux, and inflammation: important targets for the treatment of atherosclerosis. Curr. Drug Targets. 2011;12:647–660. doi: 10.2174/138945011795378522. [DOI] [PubMed] [Google Scholar]
- 4.Yvan-Charvet L., Wang N., Tall A.R. Role of HDL, ABCA1, and ABCG1 transporters in cholesterol efflux and immune responses. Arterioscler. Thromb. Vasc. Biol. 2010;30:139–143. doi: 10.1161/ATVBAHA.108.179283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Peled M., Fisher E.A. Dynamic Aspects of Macrophage Polarization during Atherosclerosis Progression and Regression. Front. Immunol. 2014;5:579. doi: 10.3389/fimmu.2014.00579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Xu Y., Zalzala M., Xu J., Li Y., Yin L., Zhang Y. A metabolic stress-inducible miR-34a-HNF4α pathway regulates lipid and lipoprotein metabolism. Nat. Commun. 2015;6:7466. doi: 10.1038/ncomms8466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fu T., Choi S.E., Kim D.H., Seok S., Suino-Powell K.M., Xu H.E., Kemper J.K. Aberrantly elevated microRNA-34a in obesity attenuates hepatic responses to FGF19 by targeting a membrane coreceptor β-Klotho. Proc. Natl. Acad. Sci. USA. 2012;109:16137–16142. doi: 10.1073/pnas.1205951109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fu T., Seok S., Choi S., Huang Z., Suino-Powell K., Xu H.E., Kemper B., Kemper J.K. MicroRNA 34a inhibits beige and brown fat formation in obesity in part by suppressing adipocyte fibroblast growth factor 21 signaling and SIRT1 function. Mol. Cell. Biol. 2014;34:4130–4142. doi: 10.1128/MCB.00596-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Pan Y., Hui X., Hoo R.L.C., Ye D., Chan C.Y.C., Feng T., Wang Y., Lam K.S.L., Xu A. Adipocyte-secreted exosomal microRNA-34a inhibits M2 macrophage polarization to promote obesity-induced adipose inflammation. J. Clin. Invest. 2019;129:834–849. doi: 10.1172/JCI123069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Badi I., Mancinelli L., Polizzotto A., Ferri D., Zeni F., Burba I., Milano G., Brambilla F., Saccu C., Bianchi M.E. miR-34a Promotes Vascular Smooth Muscle Cell Calcification by Downregulating SIRT1 (Sirtuin 1) and Axl (AXL Receptor Tyrosine Kinase) Arterioscler. Thromb. Vasc. Biol. 2018;38:2079–2090. doi: 10.1161/ATVBAHA.118.311298. [DOI] [PubMed] [Google Scholar]
- 11.Badi I., Burba I., Ruggeri C., Zeni F., Bertolotti M., Scopece A., Pompilio G., Raucci A. MicroRNA-34a Induces Vascular Smooth Muscle Cells Senescence by SIRT1 Downregulation and Promotes the Expression of Age-Associated Pro-inflammatory Secretory Factors. J. Gerontol. A Biol. Sci. Med. Sci. 2015;70:1304–1311. doi: 10.1093/gerona/glu180. [DOI] [PubMed] [Google Scholar]
- 12.Ito T., Yagi S., Yamakuchi M. MicroRNA-34a regulation of endothelial senescence. Biochem. Biophys. Res. Commun. 2010;398:735–740. doi: 10.1016/j.bbrc.2010.07.012. [DOI] [PubMed] [Google Scholar]
- 13.Su G., Sun G., Liu H., Shu L., Liang Z. Downregulation of miR-34a promotes endothelial cell growth and suppresses apoptosis in atherosclerosis by regulating Bcl-2. Heart Vessels. 2018;33:1185–1194. doi: 10.1007/s00380-018-1169-6. [DOI] [PubMed] [Google Scholar]
- 14.Calkin A.C., Tontonoz P. Liver x receptor signaling pathways and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 2010;30:1513–1518. doi: 10.1161/ATVBAHA.109.191197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Liao X., Sharma N., Kapadia F., Zhou G., Lu Y., Hong H., Paruchuri K., Mahabeleshwar G.H., Dalmas E., Venteclef N. Krüppel-like factor 4 regulates macrophage polarization. J. Clin. Invest. 2011;121:2736–2749. doi: 10.1172/JCI45444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Li T., Chiang J.Y. Bile Acid signaling in liver metabolism and diseases. J. Lipids. 2012;2012:754067. doi: 10.1155/2012/754067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wittenburg H., Carey M.C. Biliary cholesterol secretion by the twinned sterol half-transporters ABCG5 and ABCG8. J. Clin. Invest. 2002;110:605–609. doi: 10.1172/JCI16548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Albrecht C., Soumian S., Amey J.S., Sardini A., Higgins C.F., Davies A.H., Gibbs R.G. ABCA1 expression in carotid atherosclerotic plaques. Stroke. 2004;35:2801–2806. doi: 10.1161/01.STR.0000147036.07307.93. [DOI] [PubMed] [Google Scholar]
- 19.Tavoosi Z., Moradi-Sardareh H., Saidijam M., Yadegarazari R., Borzuei S., Soltanian A., Goodarzi M.T. Cholesterol Transporters ABCA1 and ABCG1 Gene Expression in Peripheral Blood Mononuclear Cells in Patients with Metabolic Syndrome. Cholesterol. 2015;2015:682904. doi: 10.1155/2015/682904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Rayner K.J., Suárez Y., Dávalos A., Parathath S., Fitzgerald M.L., Tamehiro N., Fisher E.A., Moore K.J., Fernández-Hernando C. MiR-33 contributes to the regulation of cholesterol homeostasis. Science. 2010;328:1570–1573. doi: 10.1126/science.1189862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Repa J.J., Liang G., Ou J., Bashmakov Y., Lobaccaro J.M., Shimomura I., Shan B., Brown M.S., Goldstein J.L., Mangelsdorf D.J. Regulation of mouse sterol regulatory element-binding protein-1c gene (SREBP-1c) by oxysterol receptors, LXRalpha and LXRbeta. Genes Dev. 2000;14:2819–2830. doi: 10.1101/gad.844900. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schultz J.R., Tu H., Luk A., Repa J.J., Medina J.C., Li L., Schwendner S., Wang S., Thoolen M., Mangelsdorf D.J. Role of LXRs in control of lipogenesis. Genes Dev. 2000;14:2831–2838. doi: 10.1101/gad.850400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Näär A.M. miR-33: a metabolic conundrum. Trends Endocrinol. Metab. 2018;29:667–668. doi: 10.1016/j.tem.2018.04.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Joseph S.B., Castrillo A., Laffitte B.A., Mangelsdorf D.J., Tontonoz P. Reciprocal regulation of inflammation and lipid metabolism by liver X receptors. Nat. Med. 2003;9:213–219. doi: 10.1038/nm820. [DOI] [PubMed] [Google Scholar]
- 25.Zhang Y., Da Silva J.R., Reilly M., Billheimer J.T., Rothblat G.H., Rader D.J. Hepatic expression of scavenger receptor class B type I (SR-BI) is a positive regulator of macrophage reverse cholesterol transport in vivo. J. Clin. Invest. 2005;115:2870–2874. doi: 10.1172/JCI25327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Chevre R., Trigueros-Motos L., Castaño D., Chua T., Corlianò M., Patankar J.V., Sng L., Sim L., Juin T.L., Carissimo G. Therapeutic modulation of the bile acid pool by Cyp8b1 knockdown protects against nonalcoholic fatty liver disease in mice. FASEB J. 2018;32:3792–3802. doi: 10.1096/fj.201701084RR. [DOI] [PubMed] [Google Scholar]
- 27.Patankar J.V., Wong C.K., Morampudi V., Gibson W.T., Vallance B., Ioannou G.N., Hayden M.R. Genetic ablation of Cyp8b1 preserves host metabolic function by repressing steatohepatitis and altering gut microbiota composition. Am. J. Physiol. Endocrinol. Metab. 2018;314:E418–E432. doi: 10.1152/ajpendo.00172.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bertaggia E., Jensen K.K., Castro-Perez J., Xu Y., Di Paolo G., Chan R.B., Wang L., Haeusler R.A. Cyp8b1 ablation prevents Western diet-induced weight gain and hepatic steatosis because of impaired fat absorption. Am. J. Physiol. Endocrinol. Metab. 2017;313:E121–E133. doi: 10.1152/ajpendo.00409.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kaur A., Patankar J.V., de Haan W., Ruddle P., Wijesekara N., Groen A.K., Verchere C.B., Singaraja R.R., Hayden M.R. Loss of Cyp8b1 improves glucose homeostasis by increasing GLP-1. Diabetes. 2015;64:1168–1179. doi: 10.2337/db14-0716. [DOI] [PubMed] [Google Scholar]
- 30.Slätis K., Gåfvels M., Kannisto K., Ovchinnikova O., Paulsson-Berne G., Parini P., Jiang Z.Y., Eggertsen G. Abolished synthesis of cholic acid reduces atherosclerotic development in apolipoprotein E knockout mice. J. Lipid Res. 2010;51:3289–3298. doi: 10.1194/jlr.M009308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Schwarz M., Russell D.W., Dietschy J.M., Turley S.D. Alternate pathways of bile acid synthesis in the cholesterol 7alpha-hydroxylase knockout mouse are not upregulated by either cholesterol or cholestyramine feeding. J. Lipid Res. 2001;42:1594–1603. [PubMed] [Google Scholar]
- 32.Post S.M., Groenendijk M., Solaas K., Rensen P.C., Princen H.M. Cholesterol 7alpha-hydroxylase deficiency in mice on an APOE*3-Leiden background impairs very-low-density lipoprotein production. Arterioscler. Thromb. Vasc. Biol. 2004;24:768–774. doi: 10.1161/01.ATV.0000121572.21122.59. [DOI] [PubMed] [Google Scholar]
- 33.Zhou Y.Y., Zhou X.D., Wu S.J., Fan D.H., Van Poucke S., Chen Y.P., Fu S.W., Zheng M.H. Nonalcoholic fatty liver disease contributes to subclinical atherosclerosis: A systematic review and meta-analysis. Hepatol Commun. 2018;2:376–392. doi: 10.1002/hep4.1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Madan S.A., John F., Pyrsopoulos N., Pitchumoni C.S. Nonalcoholic fatty liver disease and carotid artery atherosclerosis in children and adults: a meta-analysis. Eur. J. Gastroenterol. Hepatol. 2015;27:1237–1248. doi: 10.1097/MEG.0000000000000429. [DOI] [PubMed] [Google Scholar]
- 35.Du F., Yu F., Wang Y., Hui Y., Carnevale K., Fu M., Lu H., Fan D. MicroRNA-155 deficiency results in decreased macrophage inflammation and attenuated atherogenesis in apolipoprotein E-deficient mice. Arterioscler. Thromb. Vasc. Biol. 2014;34:759–767. doi: 10.1161/ATVBAHA.113.302701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Huang X., Li Y., Li X., Fan D., Xin H.B., Fu M. TRIM14 promotes endothelial activation via activating NF-κB signaling pathway. J. Mol. Cell Biol. 2019;2019:mjz040. doi: 10.1093/jmcb/mjz040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turley S.D., Herndon M.W., Dietschy J.M. Reevaluation and application of the dual-isotope plasma ratio method for the measurement of intestinal cholesterol absorption in the hamster. J. Lipid Res. 1994;35:328–339. [PubMed] [Google Scholar]
- 38.Zhang Y., Ge X., Heemstra L.A., Chen W.D., Xu J., Smith J.L., Ma H., Kasim N., Edwards P.A., Novak C.M. Loss of FXR protects against diet-induced obesity and accelerates liver carcinogenesis in ob/ob mice. Mol. Endocrinol. 2012;26:272–280. doi: 10.1210/me.2011-1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Naik S.U., Wang X., Da Silva J.S., Jaye M., Macphee C.H., Reilly M.P., Billheimer J.T., Rothblat G.H., Rader D.J. Pharmacological activation of liver X receptors promotes reverse cholesterol transport in vivo. Circulation. 2006;113:90–97. doi: 10.1161/CIRCULATIONAHA.105.560177. [DOI] [PubMed] [Google Scholar]
- 40.Xu Y., Li F., Zalzala M., Xu J., Gonzalez F.J., Adorini L., Lee Y.K., Yin L., Zhang Y. Farnesoid X receptor activation increases reverse cholesterol transport by modulating bile acid composition and cholesterol absorption in mice. Hepatology. 2016;64:1072–1085. doi: 10.1002/hep.28712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Zhang Y., Wang X., Vales C., Lee F.Y., Lee H., Lusis A.J., Edwards P.A. FXR deficiency causes reduced atherosclerosis in Ldlr-/- mice. Arterioscler. Thromb. Vasc. Biol. 2006;26:2316–2321. doi: 10.1161/01.ATV.0000235697.35431.05. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.