

Abstract
Exome and deep sequencing of cells treated with a panel of lentiviral guide RNA demonstrate that both on- and off-target editing proceed in a time-dependent manner. Thus, methods to temporally control Cas9 activity would be beneficial. To address this need, we describe a “self-inactivating CRISPR (SiC)” system consisting of a single guide RNA that deactivates the Streptococcus pyogenes Cas9 nuclease in a doxycycline-dependent manner. This enables defined, temporal control of Cas9 activity in any cell type and also in vivo. Results show that SiC may enable a reduction in off-target editing, with less effect on on-target editing rates. This tool facilitates diverse applications including (1) the timed regulation of genetic knockouts in hard-to-transfect cells using lentivirus, including human leukocytes for the identification of glycogenes regulating leukocyte-endothelial cell adhesion; (2) genome-wide lentiviral sgRNA (single guide RNA) library applications where Cas9 activity is ablated after allowing pre-determined editing times. Thus, stable knockout cell pools are created for functional screens; and (3) temporal control of Cas9-mediated editing of myeloid and lymphoid cells in vivo, both in mouse peripheral blood and bone marrow. Overall, SiC enables temporal control of gene editing and may be applied in diverse application including studies that aim to reduce off-target genome editing.
Keywords: CRISPR, Cas9, genome editing, hematopoietic stem cells, genome-wide screen, lentivirus, doxycycline, off target editing, next generation sequencing, exome sequencing
Graphical Abstract
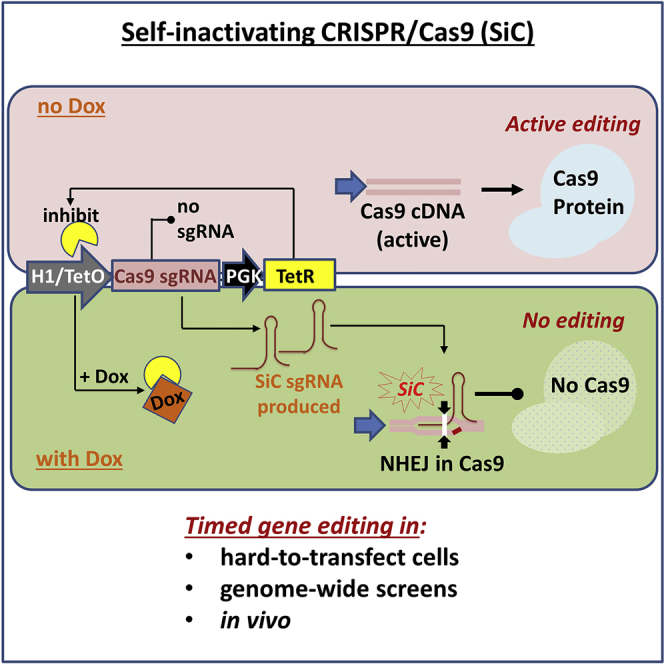
There is a need to control Cas9 activity in a defined time window in order to regulate the efficiency of on-target and off-target CRISPR-Cas9-based editing. This manuscript describes a lentiviral system called “self-inactivating CRISPR (SiC)” that enables such regulation in a doxycycline-dependent manner, both in vitro and in vivo.
Introduction
The CRISPR-Cas9 system is widely used for genome editing due to its simplicity, versatility, and efficiency. Among the endonucleases used in such applications, the Streptococcus pyogenes Cas9 (“Cas9”) is common as it efficiently induces site-directed double stranded breaks to enable either site-specific indel (insertion-deletion) formation during non-homologous end joining (NHEJ) or nucleotide insertion during homology-dependent repair (HDR). Although efficient, a number of studies suggest that CRISPR-Cas9 applications can have unintended off-target effects (OTEs).1, 2 Such OTEs are particularly unacceptable in the context of human clinical applications. Because Cas9 tolerates a few base mismatches, the OTEs can also accumulate in forward genetic screen applications where sgRNA (single guide RNA) and Cas9 are stably integrated into the chromosome.
To improve editing specificity, new software tools have been developed to control sgRNA guanine-cytosine (GC) content, optimize/minimize base (mis)match scores for guide sequences at the 5′ or 3′ ends, or apply additional weighting criteria.3, 4, 5, 6, 7, 8 Additionally, Cas9 has been engineered by (1) designing Cas9 nickase (Cas9n) with single active endonuclease domains in duplex to create paired offset edits;9 (2) fusing Fok1 nuclease with inactive Cas9 so that editing is dimerization-dependent requiring the paired juxtapositioning of sgRNA partners;10, 11 (3) rationally engineering CRISPR-Cas9 complexes so that the homology-based interaction of CRISPR-Cas9 complex with target DNA is more stringent and less tolerant of mismatches;12, 13 (4) designing Cas9 variants with enhanced specificity;14, 15, 16, 17 or (5) splitting Cas9 into two polypeptides for post-translational assembly.18 Finally, a variety of approaches have been developed to limit Cas9 or sgRNA activity in a defined time window using 4-hydroxytamoxifen (4-HT)-based induction of Cas9 activity,19 intein-based nuclease regulation,20 small-molecule-controlled Streptococcus pyogenes Cas9 activity or stability,21, 22, 23 destabilized Cas9,24, 25 light application,26, 27 doxycycline (Dox)-dependent control of Cas9,28, 29, 30, 31 pulsed sgRNA application,30, 32, 33 or in vitro assembled Cas9-sgRNA ribonucleoprotein complexes (RNPs).34 Although these methods are designed to reduce OTEs, none of them are absolute, and complete absence of OTEs is difficult to guarantee. Further, many of these engineering approaches have drawbacks in that their design reduces native Cas9 editing efficiency, limits the number of genome sequences that can be targeted by the nuclease, or are not feasible for library screening applications.
This manuscript aims to improve the specificity and efficiency of genome editing in instances where the stable integration of Cas9 is a necessary part of the protocol, for example in applications involving library/genetic screens;35, 36, 37, 38 those using lentivirus on hard-to-transfect cells like peripheral blood and bone-marrow-derived leukocytes, endothelial cells, neurons, rare cell types, and other primary cells;33, 39, 40 and in vivo applications where Cas9 is part of the host.41 In such usages, it would be attractive to turn off the nuclease activity using Dox or similar drugs after providing a predefined time window for genome editing. With the goal of developing such a system, we describe here a “self-inactivating CRISPR (SiC)” system, which contains an efficient sgRNA that targets and knocks out the Cas9 gene on-demand. The SiC vector is tightly regulated by Dox with no detectable leakage or OTEs. Its function may be delivered on an independent plasmid or in the context of vectors carrying entire sgRNA libraries. The utility of this system to perform genome editing in a time-dependent manner is illustrated for studies involving (1) hard-to-transfect cells in the context of leukocyte-endothelial cell adhesion biology, (2) creation of stable knockout cell pools in sgRNA library screen applications, and (3) in vivo mouse studies that involve leukocytes in peripheral blood and bone marrow.
Results
Self-Inactivating CRISPR (SiC) to Knock Out Cas9-Mediated Editing
A lentiviral system was developed where the S. pyogenes Cas9 gene could be self-edited upon Dox addition and its activity knocked out (Figure 1). The system consists of two multicistronic vectors (Figure 1A; Table S1A): (1) SiC-V1 derived from lentiCRISPR v2,35 which carries Cas9, a dTomato reporter and an sgRNA targeting any gene of interest; and (2) SiC-V2, with a Tet-repressor, Cerulean reporter, and tetracycline/Dox-inducible sgRNA targeting Cas9.
Figure 1.

Self-Inactivating CRISPR-Cas9 System, SiC
(A) Lentiviral vector with two constructs: SiC-V1 and SiC-V2. SgRNA editing Cas9 are numbered G1–G10, along with their site of action (top). Experiment schematic is shown at the bottom. (B) sgRNA G1–G10 were cloned individually into SiC-V2, and transiently transfected into HEK-Cas9dTomato cells, which stably express Cas9 and dTomato. Following 72 h with and without 2.5 μg/mL Dox, the Surveyor assay was performed. Percent of gene editing was quantified using densitometry. “U” indicates untransfected (wild-type) HEK cells. Arrows indicate indels. sgRNA G7 in the Rec1 lobe was most efficient at editing Cas9. Data are representative of 2–4 repeats for each sgRNA. (C) HEK-Cas9dTomato cells were either transiently transfected (left half of gel) or stably transduced (right half) to express SiC-V2-Cas9G7 (SiC-V2 with guide G7). The latter stably transduced cells are called “HEK-Cas9dTomato-Cas9G7. Dox concentration was varied over 72 h prior to Surveyor assay to monitor Cas9 editing. Cas9 editing is strictly Dox-dependent upon lentiviral transduction. Data are representative of 3–4 repeats. (D–F) Time course studies of Cas9 activity performed using “HEK-Cas9dTomato-Cas9G7” cells. dTomato reporter fluorescence measured using cytometry decreased gradually over 10 days in the presence of 1 μg/mL Dox (D). Genome editing rate was independent of Dox concentration in the range tested (0.5–10 μg/mL, E) Microscopy image shows >90% loss of dTomato signal after 10 days of Dox (F). Data are representative of >10 repeats. See also Figures S1 and S2.
A panel of ten sgRNAs targeting different parts of Cas9 was selected in order to identify the most efficient editing sequence (Figure 1A; Table S1B). To this end, a control scramble (Scr) sgRNA, with nearest target having a 4-base mismatch in the human genome, was cloned into SiC-V1 (Tables S2 and S3). This was stably expressed in HEK293T cells using lentivirus to generate “HEK-Cas9dTomato” cells. The ten sgRNAs (G1–G10) were cloned into SiC-V2 and transiently transfected into HEK-Cas9dTomato cells. Dox was added in some cases. Surveyor assay performed at 72 h demonstrated highest editing by sgRNA G7 (“Cas9G7,” ∼30%) targeting the Rec1 domain (Figures 1A and 1B). Cas9G7 has low homology within the human genome, with the closest sequence having a 4-base mismatch (Table S3). Here, some Cas9 editing was noted even in the absence of Dox.
We hypothesized that the Dox-independent Cas9 editing may be due to the nature of transient transfection, since sgRNA expression may occur before sufficient production of Tet repressor protein. This may not occur in lentivirus-transduced cells. Thus, “HEK-Cas9dTomato” cells were transduced with SiC-V2-Cas9G7 virus (SiC-V2 carrying Cas9G7) to generate stable “HEK-Cas9dTomato-Cas9G7” cells (Figure 1C). Here, consistent with the above hypothesis, Dox-independent editing was completely absent upon lentiviral transduction (Figure 1C, left versus right gel at 0 μg/mL Dox concentration). Gene editing upon transduction was efficient, with ∼40% editing being measured using the Surveyor assay within 3 days independent of Dox concentration from 0.5 to 10 μg/mL. Time course studies that monitored dTomato fluorescence as a surrogate reporter of Cas9 activity showed that 17% of the cells were dTomato-negative at day 2 following Dox addition, with this increasing to 31% and 87% at days 4 and 10 (Figure 1D). Such loss of dTomato signal was independent of Dox dosage from 0.5 to 10 μg/mL (Figure 1E). Fluorescence microscopy confirmed these observations since Cas9G7 abolished dTomato fluorescence upon Dox addition, without affecting Cerulean (Figure 1F). The observation that Cas9 editing was independent of Dox concentration in the range from 0.25 to 1 μg/mL was also noted in Figure S1, where >80% Cas9 editing was measured using deep sequencing within 4 days of Dox addition. Here, the extent of Cas9 editing measured using deep sequencing (Figure S1C) correlated with the magnitude of dTomato signal loss (Figure S1A), only both processes occurred at different timescales. Additionally, Cas9 editing was absent when Dox was omitted, and editing percentage was low (<0.5%) at three top computationally predicted Cas9G7 off-target sites (Figure S1D). Finally, some Dox dose response was observed between 1 and 10 ng/mL Dox, in studies that focused on lower drug dosages (Figure S1B).
In a control experiment, dTomato fluorescence was unchanged in the absence of Cas9G7 lentivirus or upon transduction of HEK-Cas9-dTomato cells with Scr sgRNA lentivirus (SiC-V2-Scr, Figures S2A and S2B). Further, CRISPR-Cas9 genome editing was low in the self-inactivated, dTomato-negative cells upon transient transfection of a functional sgRNA targeting human MGAT1, while dTomato-positive cells displayed 30%–40% editing under identical conditions (Figure S2C). Minor MGAT1 editing is noted in this assay because these are an unsorted population and some Cas9/dTomato-positive cells remain. Overall, the Dox-dependent self-editing of Cas9 using SiC abolishes dTomato fluorescence and this correlates with Cas9 nuclease activity. Dox-independent Cas9-deactivation or “vector leakage” was not detected in this system. In order to remain consistent, all remaining studies used 1 μg/mL Dox to deactivate Cas9.
Effect of Timed Cas9 Inactivation on Off- versus On-Target Editing
Exome sequencing-based studies were undertaken in order to determine the time course of on-target and off-target editing in an unbiased manner (Figure 2). To this end, the HEK-Cas9dTomato-Cas9G7 cells were transduced with an equimolar lentivirus pool encoding for five promiscuous sgRNA that target human EMX1, C1GALT1C1, MGAT1, ST3GAL4, and ZSCAN1, along with blue fluorescence protein/BFP reporter and puromycin selection marker (Table S4). Following viral transduction at an MOI of 0.8–1, puromycin selection was initiated at 24 h. Doxycycline was added at various times to knockout Cas9: 18 h prior to transduction, at the time of transduction (day 0), or at day 2. One sample did not receive Dox, and another control sample remained untransduced. As expected, Cas9-related dTomato fluorescence decreased in a Dox-time-dependent manner (Figure 2B). Exome sequencing was performed on genomic DNA isolated on day 15. Although data analysis was initially attempted for the entire human exome, conclusions were difficult to draw as it is complicated to distinguish between relatively rare off-target edits, spontaneous mutations that are independent of the expressed sgRNA, and Illumina sequencing errors. Thus, detailed search for on- and off-target indels was restricted to the region 3–5 bases upstream of the protospacer adjacent motif (PAM) sequence, in the computationally predicted off-target sites (Figure S3; Tables S5–S9). Such analysis revealed that both on- and off-target edits proceed over the entire 15-day time course, with off-target editing being ten times less prevalent even for the most promiscuous guides (Figure 2C). While 1–2 days was sufficient to obtain maximum on-target edits in the case of some genes, others required more time (Figure 2D). Exome sequencing coverage (i.e., number of reads) was insufficient to draw conclusions for EMX1 and MGAT1.
Figure 2.

Time-Dependent On- and Off-Target Editing Assessed Using Exome and Deep Sequencing
(A) HEK-Cas9dTomato-Cas9G7 cells were transduced with a pool of five lentivirus carrying promiscuous sgRNA targeting human EMX1, C1GalT1C1, MGAT1, ST3Gal4, or ZSCAN2. Cells were red due to Cas9dTomato, cyan due to Cas9G7 sgRNA, which was co-expressed with a Cerulean reporter, and blue due to BFP co-expression by the virus carrying the promiscuous sgRNA. 1 μg/mL Dox was added either 18 h prior to viral transduction, at day 0 (0 day) or at day 2 (2 day) in order to self-inactivate Cas9 (A). Dox was absent in one case, and control cells remained untransduced (without Dox). 1 μg/mL puromycin was added to select for transduced cells starting at day 1 (1 day). Genomic DNA was purified at day 15 for exome sequencing (results in C and D). Selected on- and off-target sites were also amplified from genomic DNA, and indels were quantified using deep sequencing (results in E). (B) Cas9 expression (dTomato, red), presence of sgRNA-library (BFP, blue), and Cas9G7 sgRNA (Cerulean, Cyan) expression were monitored at various times using surrogate fluorescence reporters. Note that some decrease in dTomato fluorescence is observed in No Dox sample at day 15 as the cells were cultured to over-confluence, but there is no Dox editing in these cells based on deep-sequencing. (C and D) In exome sequencing runs, data are presented as mean ± SD for indel formation on 3 on-target genes (C1GalT1C1, ST3Gal4, ZSCAN2) and 32 putative off-target genes. Among the 32, 30 are exonic off-target edits and 2 are intronic. Here, both on-target and off-target editing increased if Dox addition was delayed. Low levels of off-target editing (0.27%) and no on-target editing (0%) was detected in the no-virus control sample, and thus the study design contains only low levels of basal noise. In general, off-target editing was ∼10-fold lower compared to on-target (C). (D) On-target editing percent for three genes based on triplicate runs at each time point. Here, whereas some genes were edited at early times, indels accumulated in others over a period of days (D). (E) In deep-sequencing runs, also, on- and off-target editing increased upon delaying Dox addition. Here, “-T” denotes on-target and “-OT” denotes off-target editing. Samples at each time were analyzed in triplicate as noted using red symbols. Samples with lower duration of Dox treatment are indicated using darker red symbols and these appear to the right in the individual plots. Untransduced cells and cells with earlier Dox addition time points appear to the left. A blue line links the mean “%indel values” of each of the samples, starting with “No virus, No Dox,” to earlier Dox to later, No Dox treatment samples. Percent indel was higher in some runs (left panel) compared to others (right panel), with no editing seen in EMX1-OT10. See also Figure S3.
In order to overcome the limitation of exome sequencing, which had low sequence coverage, we attempted to PCR amplify 57 regions of the genomic DNA products from the above study, including the 5 target regions and 52 off-target sites (primers and expected amplicon sequences listed in Figures S10 and S11). All 57 primer pairs were pooled prior to PCR in this study, due to the large number of treatments and time points. A clean PCR product that migrated as a single band was observed for each of the samples. These were multiplexed with barcodes and unique molecular identifiers (UMIs) and sequenced using Illumina MiSeq platform. Upon analysis, 27 of the 57 amplicon products were represented in the sequencing data, with indels being detected in nine of the products (Figure 2E). Here, highly consistent with the previously described exome sequencing data, both on- and off-target editing proceeded in a time-dependent manner. In some cases like ST3Gal4, 25% on-target editing was measured in the first few days and this increased to 45% over 2 weeks. The corresponding off-target (ST3Gal4-OT22) proceeded more slowly and the editing of this site persisted throughout the time course of the study. Here, off-target editing was low at 4%–5% at initial times, but increased to ∼25% over the 2-week time course. In addition to this, we observed that addition of Dox at early times reduced subsequent editing, consistent with the notion that the SiC system provides a facile strategy to regulate genome editing in a drug-dependent manner.
Because the off-target sites in the above study often contained 3+ base mismatches, we decided to analyze previously described promiscuous sgRNA targeting EMX1 and VEGFA that differ from the target sequence by only two base mismatches (Table S2; Figure 3).42 Thus, these guides were stably expressed using SiC-V1 (red) in HEK cells that already express SiC-V2-Cas9G7 (Cerulean). Dox was added to a portion of the cells at 48 h to knock out Cas9. Specific on- and off-target editing efficiency was then quantified on day 13 using Indel Detection by Amplicon Analysis (Figure 3B; primers listed in Table S2). Here, the EMX1 on-target/off-target editing efficiency ratio improved from 0.8 (= 53.3/62.8) when Dox was absent to 1.3 (= 51.6/40.6) upon addition of Dox. This ratio improved from 2.0 to 4.0 when VEGFA was the target. In both cases, Dox had a minor effect on on-target editing as this proceeded within 2 days. Off-target editing, however, required more time and was higher in absence of Dox. Overall, SiC enables the temporal regulation of genome editing, with the potential that such an approach may reduce OTEs.
Figure 3.

SiC Reduces Off-Target Editing
(A) HEK293T cells stably expressing vector SiC-V2 with Cas9-G7 were transduced with SiC-V1 carrying previously established promiscuous sgRNA targeting either EMX1 or VEGFA. Both guides have well-established on-target (T) and off-target (OT) editing sites. 48 h post transduction, the cells were divided into two groups with one group receiving 1 μg/mL Dox for another 13 days. Genomic DNA was prepared from these four types of cells at the end point and also control untransduced HEK293T-Cas9G7cells cultured with Dox. (B) 6-FAM labeled PCR products generated for EMX1 and VEGFA, on-target and off-target sites, on day 15 were resolved using capillary electrophoresis. Vertical red line indicates unedited PCR fragment from control cells (top row). Numbers in individual panels indicate percent editing based on electrophoresis area-under-the-curve calculations. Off-target editing is reduced upon Dox addition (B, middle versus bottom row). Data are representative of duplicate runs.
SiC Regulates Gene Editing in Hard-to-Transfect Human Leukocytes
We tested whether SiC may aid the study of hematopoietic cells, because these hard-to-transfect cells often require lentivirus application for transgene expression. Such studies edited the human α(2,3)sialyltransferase ST3Gal4, a critical regulator of selectin-dependent leukocyte adhesion in the human inflammatory cell adhesion cascade (Figure 4).43 Here, Cerulean-positive promyeloid HL-60 leukocytes stably transduced with SiC-V2-Cas9G7 (Cerulean) were subsequently transduced with SiC-V2-dTomato (red) virus carrying sgRNA targeting either ST3Gal4 or Scr (Figure 4A). Red and Cerulean, dual-positive fluorescent cells expressing both viruses were fluorescence-activated cell sorting (FACS) sorted on day 3, Dox was added to a portion on day 4, and culture was carried out to day 14. All cells remained >95%–98% viable based on LDS-751 live cell staining, and cell apoptosis was absent based on Annexin-V binding.
Figure 4.

ST3Gal4 Knockout in Human Leukocytes Abolishes Selectin-Dependent Leukocyte Rolling
(A) HL-60s were stably transduced with SiC-V2-Cas9G7 virus to create Cerulean-positive cells. These were subsequently transduced with SiC-V1 virus carrying either scramble-sgRNA or ST3Gal4-sgRNA. Following sorting of dTomato+Cerulean+ cells at day 3, 1 μg/mL Dox was added from days 4 to 14. (B) The Surveyor assay monitored ST3Gal4 (top) and Cas9 (bottom) indels. (C) Fluorescence microscopy at day 14 measured dTomato reporter. Dox treatment resulted in Cas9 editing and loss of red fluorescence. (D) Flow cytometry measured cell surface sialyl Lewis-X expression (using mAb HECA-452), and also P-, E-, and L-selectin binding function (treatments same as C). Loss of ST3Gal4 activity reduced sLeX expression and selectin binding (events in bottom-left quadrant of each sub-plot). (E) No Dox cells from (C) were perfused over selectin substrates in microfluidic flow cell at wall shear stress of 2 dynes/cm2. Cells lacking ST3Gal4 displayed minimal interaction with selectin substrates in bright field. Controls used function blocking mAbs against P- (clone G1), E- (P2H3), and L- (DREG-56) selectin. See also Figure S4.
The Surveyor assay demonstrated robust glycogene editing only in cells carrying ST3Gal4-sgRNA but not Scr (Figure 4B, top). Cas9 indels were only observed upon Dox addition and no Dox-independent leakage was observed at any time point, up to 14 days, using the Surveyor assay (Figure 4B, bottom). Most ST3Gal4 on-target editing was complete by day 4. These observations are consistent with microscopy (Figure 4C) and cytometer studies that noted Cas9 editing/dTomato loss only in the Dox-treated samples (Figure S4). In functional studies, 51%–66% of the cells that expressed ST3Gal4-sgRNA displayed both an abrogation of sialyl Lewis-X/sLeX epitope expression as measured using mAb HECA-452 and diminished binding to L-, E-, and P-selectin (bottom half, Figure 4D). The results are consistent with microfluidics-based cell adhesion assays where ST3Gal4 editing resulted in reduced leukocyte attachment to P-, E-, and L-selectin (Figure 4E). Overall, SiC may be applied to abolish Cas9 activity in hard-to-transfect leukocytes, after 3–4 days when on-target editing is largely complete. This approach provides a rapid and simple method to generate genome-edited leukocytic cell lines.
Use of Cas9 Self-Inactivation in Genome-wide Screens
We determined whether SiC can allow temporal control of Cas9 activity in genome-wide CRISPR screens, using two approaches.
First, a “SiC-dual-V2” vector was developed. This vector is identical to SiC-V2, only it contains a second constitutively active U6-driven sgRNA downstream of Cerulean (Figure 5A). SgRNA targeting any gene of interest can be cloned into this second site. As an example, in this study, we cloned sgRNA targeting either the Core-1 β3-galactosyltransferase chaperone COSMC or Scr control. Thus, while the second guide (COSMC/Scr) is always active, Cas9 deactivation can be triggered using Dox, enabling gene editing in a defined window. To test this, following transduction of Cas9dTomato+ HL60 cells (red) with SiC-dual-V2 virus targeting COSMC/Scr, Dox was added to deactivate Cas9 at different times (Figure 5B). Flow cytometry analysis on day 9 confirmed that the extent of Cas9 deactivation, measured based on loss of dTomato signal, depended on Dox treatment duration. 65% editing occurring in cells cultured with Dox beginning day 0, 45% if Dox was introduced on day 4, and 0% when Dox was omitted (Figure 5C). Consistent with the notion that on-target Crispr-Cas9 editing occurs rapidly, COSMC editing inferred based on VVA (Vicia villosa agglutinin)-lectin binding40 was 22% (= 16.3% + 5.3%) independent of the time of Dox addition (Figures 5C and 5D). VVA+dTomato− cells lacking both COSMC and Cas9 were noted in Figure 5D (marked by blue arrow). These may be sorted for functional studies because OTEs will not propagate in them. Extending this method, entire sgRNA libraries may be cloned into SiC-dual-V2 at the U6 site for library screening applications in cells constitutively expressing Cas9.
Figure 5.

Use of Cas9G7 in Small- and Genome- Scale CRISPR Screens
(A) SiC-dual-V2 contains two sgRNA, one of which targets Cas9 and other against a target gene. (B) HL-60s were transduced with SiC-V1-Scr virus to create Cas9dTomato cells (red). Titers were adjusted so that 30% of the cells were untransduced (dTomato−), as these serve as internal controls. These cells were then transduced with either Cerulean+ SiC-dual-V2-scramble or SiC-dual-V2-COSMC lentivirus to abolish O-glycan biosynthesis. 1 μg/mL Dox was added on either day 0, 1, 2, 3, or 4 to knockout Cas9. Dox was removed 2 days prior to cytometry analysis on day 9 in order to reduce Dox-induced autofluorescence. U, untransduced cells; -, transduced cells without Dox. (C) On day 9, cells cultured with Dox displayed decreased dTomato signal, indicating Cas9 inactivation (top panels). A portion of the SiC-dual-V2-COSMC transduced cells displayed VVA-FITC binding due to COSMC-knockout (arrow, bottom panel). (D) Dot plot comparing 1 μg/mL Dox treatment for scramble versus COSMC depict a population of COSMC edited cells at day 9, without residual Cas9/dTomato activity (blue arrow). (E) Cas9+ dTomato HL60s were transduced with genome-scale library with ∼90,000 sgRNA in a vector containing BFP and puromycin selection marker. 35% of the cells were BFP-positive corresponding to ∼1 sgRNA/cell (left panel, day 2). BFP+ cells were selected by addition of 1 μg/mL puromycin at day 2 (middle panel, day 4). Cas9G7 sgRNA was electroporated on day 4 to inactivate Cas9. A BFP+ dTomato− population was observed on day 14 (blue arrow). Data in (A)–(D) are representative of three repeats.
Second, a human genome-wide library with ∼91,000 sgRNA members37 was transduced into Cas9-dTomato+ HL-60 s at ∼1/3 MOI (Figure 5E). Puromycin was added at day 2 to remove untransduced cells, followed by electroporation of Cas9G7 sgRNA at day 4 to edit Cas9. 10 days later, a Cas9-dTomato‾BFP+ population is seen (arrow in Figure 5E), corresponding to cells that have members of the genome-wide screen but that lack Cas9-dTomato. Such cells have to be sorted for functional assays, because Cas9 is not deactivated in all cells. Thus, as an alternative to using the Dox-regulated SiC system, we demonstrate that the potent Cas9G7 sgRNA validated above may also be electroporated in order to shut off CRISPR-Cas9 editing in library screening applications.
Regulation of Cas9 Expression In Vivo Using SiC
We applied SiC-V2 to regulate Cas9 expression in mouse hematopoietic stem/progenitor cells (HSPCs) (Figure 6). In one study, lineage-negative (Lin−) bone marrow progenitors were purified from Cas9-GFP mice.41, 44 These cells were transduced using SiC-V2 vector carrying either Cas9G7 sgRNA or Scr. 40%–45% of the transduced Cas9-GFP cells were CFP+ after 2 days of ex vivo culture, at which time Dox was added to a portion (Figure 6A). GFP levels were reduced in ∼25% of the CFP-positive, Cas9G7 carrying cells by 5 days post Dox addition (Figure 6B). Genome editing was absent in the absence of Dox and upon use of Scr sgRNA. GFP-negative cells remained viable for >2 weeks ex vivo.
Figure 6.

SiC Vector Application in Mouse
(A) HSPCs isolated from transgenic Cas9-EGFP mouse bone marrow were transduced with SiC-V2-Scr (control) or SiC-V2-Cas9G7 at ∼40%–45% efficiency (based on CFP signal). 1 μg/mL Dox was added to a portion of the cells on day 2. (B) Cytometry analysis at day 7 shows 25% Cas9 editing (i.e., low GFP signal) upon Dox treatment of SiC-V2-Cas9G7 transduced cells. (C and D) Donor Cas9-EGFP mouse HSPCs (CD45.2+) transduced with SiC-V2-Cas9G7 overnight were transplanted into recipient B6.SJL (CD45.1+) mice. Animals received standard (n = 5) or Dox chow (n = 6). (C) Flow cytometry analysis was performed on peripheral blood cells 1 to 3 weeks post-transplant. Representative dot plot of CD45.2+ CD11b+ peripheral blood cells 1 week post-transplant (left). Percent GFP− CD45.2+ CD11b+ cells at different times in mice fed with standard or Dox chow (right). (D) Bone marrow was analyzed at 4 weeks. Representative dot plot showing appearance of GFP− CD45.2+ CD11b+/Gr-1+ granulocytes upon Dox treatment (left). GFP− CD45.2+ LSK SLAM, CD11b/Gr-1, and B220/CD3 cells in mice fed with standard or Dox chow (right). *p ≤ 0.05; **p ≤ 0.01 (Student’s unpaired, two-tailed t tests). See also Figure S5.
In a second study, Lin− cells from Cas9-GFP donors were transduced with SiC-V2 carrying Cas9G7 overnight, and then transplanted into sub-lethally irradiated syngeneic B6.SJL recipients. Starting 1 day after transplant, mice were fed with standard chow or Dox chow (Figure 6C). Donor and host bone marrow are CD45.2+ and CD45.1+, respectively, and thus CD45.2 mAbs identified the transplanted cells. In serial peripheral blood analysis, mice fed with Dox exhibited a significant reduction in GFP signal in the CD45.2+ CD11b+ population (Figure 6C). GFP signal reduction persisted through the experiment. Four weeks post-transplantation, GFP signal was measured in terminally differentiated and hematopoietic stem cells (HSCs) collected from the bone marrow (Figure 6D; Figure S5). In mice fed with Dox chow, there was a trend toward a decreased GFP signal in the HSC population (defined as Lin−, c-kit+, Sca-1+, CD150+, CD48−).45 Significant reductions in the GFP signal in myeloid (CD11b+/Gr-1+) and lymphoid (B220+/CD3+) bone marrow populations was also observed. Overall, the SiC vectors may be regulated in vivo by supplementing Dox into the mouse diet. This function may be useful in future studies of hematopoietic cell biology.
Discussion
The Cas9 nuclease can tolerate base mismatches. Due to this, the high efficiency of CRISPR-Cas9 editing may be accompanied by promiscuous off-target editing. Both on- and off-target editing proceed in a time-dependent manner, and thus it would be desirable to have a system to dial in the duration of Cas9 activity. We address this issue by developing SiC vectors that efficiently target and self-inactivate Cas9. Such deactivation can be deliberately timed using Dox and can be efficient since >80%–90% of the Cas9 activity is lost upon prolonged Dox treatment. Indeed, while others have proposed complementary methods to reduce Cas9 activity,46, 47, 48, 49 Cas9 inactivation is not timed in these publications because it starts immediately following transfection/transduction. This both limits the amount of Cas9 that is expressed and creates competition between the target sgRNA and Cas9 sgRNA thus limiting the utility of these previous systems to only the most efficient sgRNA.
The SiC system is based on the key observation that sufficient Tet repressor protein is produced prior to Cas9 sgRNA production only when the SiC-V2 vector is applied in lentivirus format, but not upon transient transfection. Here, it is critical to place the Cas9 nuclease and Cas9 sgRNA on different plasmid vectors, because their co-expression on the same plasmid would result in unwanted Cas9 inactivation during virus production in the packaging cells. The development of the two vector system is also advantageous in that the SiC-V2 vector can now be used in combination with almost any freely-available genome-scale sgRNA library from resources like Addgene, for both humans and other species.35 In such studies, Cas9G7 available in SiC-V2 can be activated during the course of genome-wide screens in a timed manner, to create stable cell pools that can be further selected for desired phenotype. Such timed inactivation of Cas9 is important for genome-wide screens because the selection of the desired phenotype can take several weeks.37, 50 The knocking out of Cas9 in such instances also enables the creation of a stable cell pool that can be repeatedly screened for different endpoints and applications.
The current study demonstrates the application of Cas9 self-inactivation in a variety of systems. This includes the following: (1) studies using hard-to-transfect HL60s, where knocking out the human α(2,3) sialyltransferase ST3Gal4 abolished cell adhesion via selectins. While such experiments previously took many months to perform,40 the SiC expression system expedites the process to 1–2 weeks. Extending this approach, it is also possible to create stable cells carrying SiC-V2-Cas9G7, followed by transduction using a pooled CRISPR library for forward genetic screens. (2) In addition to implementing SiC via lentivirus, self-inactivation is also possible by electroporating Cas9G7 sgRNA into cells as illustrated in the library screening study. Here, Cas9 inactivation is only ∼50% efficient even under optimized conditions though it is readily possible to sort for the desired cells using the fluorescence reporter. (3) While Cas9G7 sgRNA can be used for nuclease inactivation ex vivo, SiC-V2 virus was necessary in vivo in the Cas9-EGFP knockin mouse. Here, Cas9 editing was strictly Dox dependent in HSPCs. CD11b myeloid and B220 lymphoid cells were also edited. Further development of the technology is necessary in order to target HSCs and enable serial transplantation. (4) Finally, a SiC-dual V2 vector was developed in order to co-express both the target and Cas9G7 sgRNA in any cell. Cloning entire pooled libraries of sgRNA is possible into this vector in order to create SiC libraries that allow precise and specific genomic screens in any currently available Cas9+ model cell line.
Overall, the manuscript developed tools that enable the strict control of genome editing using CRISPR-Cas9 in a Dox-dependent manner. This temporal regulatory feature is important when using pooled sgRNA for studies, as some of the weaker but specific guides may require longer/variable gene-editing windows. Although this concept is illustrated using Tet-repressor-based feedback regulation of Cas9 sgRNA expression, similar methods may also be developed for transactivator (tTA or rtTA)-based Dox-inducible systems.51, 52 Additionally, although illustrated for Streptococcus pyogenes Cas9 in the current study, specific SiC sgRNAs may also be designed to extend this method to other CRISPR nucleases like cpf1, SaCas9, or engineered Streptocccus pyogenes Cas9 variants designed to reduce OTEs. Finally, in addition to hematopoietic systems described here, SiC may also be applied to control editing in vivo in instances where lentivirus are applied locally in specific organs.53, 54
Materials and Methods
Cell Culture
HEK293T cells were cultured in DMEM with 10% FBS. HL60 cells (ATCC, Manassas, VA, USA) were maintained in Iscove’s modified Dulbecco’s medium (IMDM) with 10% FBS. SiC vectors were generated using standard molecular biology methods, and lentivirus production method was adapted from a prior publication (Supplemental Methods; Tables S1–S3).55 All reagents were from Thermo Fisher or Sigma Chemicals, unless otherwise mentioned.
Application of SiC Virus to Cells
Cells cultured to 50%–60% confluence were transduced with various lentiviruses in the presence of polybrene (Supplemental Methods).55 Dox was used to inactivate Cas9 activity. Although Dox concentration was varied in some initial studies, the drug concentration was set at 1 μg/mL for a majority of investigations.
Surveyor Assay and Indel Detection by Amplicon Analysis
For the surveyor assay, genomic DNA was prepared from cell pellet using the PureLink Genomic DNA mini kit. 250–400 bp regions spanning the editing site were PCR amplified using specific primer pairs (Table S2), denatured, reannealed, and then incubated with the Surveyor nuclease, enhancer, and MgCl2 for 30 min at 42°C (Integrated DNA Technologies). The final product was separated using a 2.5% Metaphor agarose gel (Lonza, Walkersville, MD, USA) made in 0.5X TBE to detect DNA fragments. In the case of the indel detection by Amplicon Analysis (IDAA) method, the above genomic DNA was used as a template and a tri-primer PCR amplification was performed using the following: (1) 6-FAM labeled “universal forward primer,” (2) gene-specific forward primer containing overhang-binding site for the universal forward primer, and (3) template-specific reverse primer as described previously (Table S2).56 Such PCR amplification was performed both for on- and off-target sites. 1 μL of the 6-FAM labeled PCR product formed in this manner was mixed with 0.5 μL of GeneScan-500LIZ size standard (Applied Biosystems/ABI) and 10 μL HiDi Formamide (ABI). Products formed were analysis using a 3130xl Genetic Analyzer (ABI) and Peak Scanner 2 software (ABI).
Exome Sequencing to Analyze On- and Off-Target Editing
Studies were performed using HEK-Cas9dTomato-Cas9G7 cells, which stably express both Cas9-dTomato and an efficient Dox-regulated sgRNA that self-inactivates Cas9 (Cas9G7). Promiscuous sgRNA targeting EMX1, C1GalT1C1, MGAT1, ST3Gal4, and ZSCAN2 were cloned into pKLV2U6sgRNA(BbsI)PGK-Puro-2A-BFP lentiviral vector (Table S4).37 The selected sgRNA are computationally predicted to have several exonic off-target sites using CCTop software (the input sequence for the interrogated gene and parameters for guide selection, as well as details of targets and off-targets being investigated are provided in Tables S5–S9).57 The above plasmids were pooled in equimolar amounts and used to generate lentivirus that were applied to the HEK-Cas9dTomato-Cas9G7 cells at an MOI of 0.8–1. Puromycin selection (1 μg/mL) was initiated 24 h post-transduction. 1 μg/ml Dox was added in some cases at −18 h (before pooled viral transduction), 0 days (at the time of transduction), or 2 days (2 days post-transduction). Dox was absent in other samples and gene editing continued unabated. HEK-Cas9dTomato-Cas9G7 without the pooled sgRNA-library served as control. Flow cytometry was used to continuously monitor Cas9G7 expression based on Cerulean fluorescence, Cas9 protein expression using dTomato signal, and sgRNA-library transduction/selection using BFP reporter. 15 days post-transduction, genomic DNA was purified using PureLink Genomic DNA Mini kit and prepared for exome sequencing using the Nextera Rapid Capture Exome Library Prep kit (Illumina). The quality of the library was confirmed using the Advanced Analytical Fragment Analyzer and Qubit reagents for concentrations. Captured libraries were pooled and quantified using the Kapa Biosystems Universal qPCR kit. Libraries were diluted to 1.8 pM and loaded onto an Illumina NextSeq 500 sequencer (2 × 75 base paired end sequencing, total reads approximately 130 × 106).
During data analysis, whole-exome reads were aligned to the human genome hg19 version using the BWA aligner.58 On-target editing sites and computationally predicted off-target sites were compiled into bed-formatted tables. These were intersected against the whole-exome sequencing results using bedtools.59 Custom scripts were written in Linux command line to analyze CIGAR strings to detect for indels 3–5 bases upstream of the PAM (code available at https://github.com/neel-lab/MolTher2019). For each gene, indel frequency was calculated by dividing total indel counts by number of gene reads. Indels were only counted when target regions for each gene were sequenced >5 times per sample. Average on-target and off-target editing efficiency data were plotted, only for genes that contained measurable indels.
Deep Sequencing (Next-Generation Sequencing)
In two studies, genomic DNA regions surrounding putative on-/off-target editing sites were PCR amplified, indexed, and analyzed using deep sequencing. In the first case, genomic DNA was isolated from HEK-Cas9dTomato-Cas9G7 cells treated with different concentration of Dox, in a time series. Amplicons were generated for regions surrounding the Cas9 on-target and top three computationally predicted off target sites (list generated using Cas-offinder;60 primers used listed in Table S11). In the second case, genomic DNA isolated from the above exome sequencing study samples was used as template, and selected on- and off-target sites listed in Table S10 were amplified. In both case, primers designed to amplify both on- and off-target regions for each sample were pooled. Thus, each first-step PCR reaction for the first study contained a pool of 4 primer-pairs while the second study contained a mixture of 57 primer-pairs. The length of the genomic region amplified in each case varied from 181 to 265 bp (Table S11), and all amplicon products were appended with Nextera transposase adaptor overhangs, including Read sequences and 6-base unique molecular identifiers (UMIs), i.e., Read1/Fwd primer overhang: 5′-TCGTCGGCAGCGTCAGATGTGTATAAGAGACAG(N)6-3′; Read2/Rev primer overhangs 5′-GTCTCGTGGGCTCGGAGATGTGTATAAGAGACAG(N)6-3′. A second, low-cycle PCR step was then performed on each of these reactions individually to add the P5 and P7 handles, as well as Nextera DNA combinatorial dual indexes, i.e., using Nextera_P5_index_fwd: 5′-AATGATACGGCGACCACCGAGATCTACAC[i5]TCGTCGGCAGCGTC-3′ and Nextera_P7_index_rev 5′-CAAGCAGAAGACGGCATACGAGAT[i7]GTCTCGTGGGCTCGG-3′. All samples were then pooled. Amplicon size and quality was verified using D1000 Screentape, run on Agilent Tapestation 4200. Amplicon concentration was determined using Kapa Biosystems qPCR quantitation kit for Illumina systems. The sample was loaded at 12 pM final concentration with 5% PhiX control library added. Sequencing was performed using a v2 Micro Flow Cell on an Illumina MiSeq sequencer with 150 cycle paired-end sequencing to generate 9.86 million reads.
Following index de-multiplexing, the .fastq data files were analyzed using a custom script written in MATLAB (available at https://github.com/neel-lab/MolTher2019). Here, overlapping regions of forward and reverse reads were merged to generate the original amplicon sequence. Following this, in order to reduce noise due to sequencing errors, indels were counted based on the number of unique UMIs that satisfied two criteria: (1) they contain base insertions/deletions within the putative sgRNA on-target/off-target sequence; and (2) the measured amplicon length differs from the expected product length of unedited amplicons. Similarly, unedited reads were those that did not satisfy both the above criteria. % indel = 100*[number of indel UMIs]/[number of indel UMIs + number of unedited UMIs].
Flow Cytometry
Cells were washed and suspended in HEPES buffer (30 mM HEPES, 10 mM Glucose, 110 mM NaCl, 10 mM KCl, 1 mM MgCl2, pH 7.2). In some cases, during the lectin binding assays, 2 × 106 cells/mL in HEPES buffer with 0.1% human serum albumin (HSA) and 1.5 mM CaCl2 were incubated on ice for 20 min with fluorescent conjugated lectins at concentrations recommended by the manufacturer (Vector Laboratories, Burlingame, CA, USA). Samples were then analyzed using a BD LSRFortessa X-20 flow cytometer. In other cases, for measuring selectin-ligand binding function, 3 μg/mL P-, E-, or L-selectin-human immunoglobulin G1 (IgG1) conjugate (R&D Systems, Minneapolis, MN, USA) was pre-incubated with 10 μg/mL goat-anti-human IgG-PerCP Ab (Jackson ImmunoResearch, West Grove, PA, USA) for 10 min at room temperature in HEPES buffer containing 1.5 mM CaCl2 and 1% goat serum.61 106 cells/mL were then added to this mixture for an additional 10 min at room temperature, in the presence or absence of 0.5 μg/mL HECA-452 efluor660 (eBioscience-Thermo), prior to cytometry analysis. Selectin binding specificity was confirmed in this assay by incubating the selectin-IgG proteins with blocking mAbs against P- (G1, Ancell, Bayport, MN, USA), E- (P2H3, Affymetrix, Santa Clara, CA, USA), or L- (DREG-56, BD PharMingen, San Jose, CA, USA) selectin during the above runs. The data was analyzed using FCS Express 6 Flow Research Edition.
Microfluidics Assay
Selectin-dependent cell adhesion studies under fluid shear were performed as described previously.43
Murine Studies
All mouse protocols were approved by the Institutional Animal Care and Use Committee of the Roswell Park Comprehensive Cancer Center (Buffalo, NY). Lineage-negative (Lin−) bone-marrow progenitor cells were isolated from femur and tibia of male “Cas9-GFP” mice (i.e., Rosa26-floxed STOP-Cas9 knock-in C57BL6/J mice).44 Lin− cells were cultured overnight in Stemspan serum-free media (Stem Cell Technologies) supplemented with 10 ng/mL recombinant mouse stem cell factor (SCF), thrombopoietin (TPO), Flt3 ligand (Flt3L), and interleukin-3 (IL-3) (Peprotech). The following day, Lin− cells were transduced with SiC-V2 lentivirus for 2 h and re-plated in fresh media overnight. In some cases, transduced cells were maintained for an additional 2 weeks in StemSpan media containing the above growth supplements. In other cases, 3 × 105 of the transduced cells were injected intravenously into the tail vein of sub-lethally irradiated (650 cGy) 6- to 8-week-old male B6.SJL-Ptprca Pepcb/BoyJ mice. Mice were fed a standard or 625 mg/kg Dox hyclate diet (Envigo Teklad) starting the day after transplant and were maintained on this diet for the remainder of the experiment.
Mouse peripheral blood and bone marrow analysis used a panel of anti-mouse mAbs from Biolegend (San Diego, CA, USA) as follows: PE-conjugated CD45.2 (clone 104), APC-conjugates CD45.1 (A20), and PE-Cy7-conjugated CD11b (M1/70) for peripheral blood; Alexa Fluor 700-conugated CD45.2, BV785-conjugated CD45.1, BV605-conjugated CD11b/Gr-1 (RB6-8C5), APC-conjugated B220 (RA3-6B2)/CD3 (17A2), APC-eFluor780-conjugated anti-C-kit (2B8), PE-conjugated anti-Sca-1 (D7), PE-Cy7-conjugated anti-CD150 (TC15-12F12.2), and PerCp-eFluor710-conjugated anti-CD48 (93) for bone marrow. For these runs, erythrocytes were lysed using ACK lysis buffer, and mAb staining was performed for 20 min on ice prior to flow cytometry analysis. The data were analyzed using FlowJo v9.7.6 (TreeStar, Ashland, OR, USA).
Availability
All reagents are available from Addgene. Source code is deposited at https://github.com/neel-lab/MolTher2019. Exome data are available from SRA: https://www.ncbi.nlm.nih.gov/bioproject/561932.
Author Contributions
Conceptualization, A.K., S.N.; Methodology, A.K., Y.Z., T.G., G.S., A.B.S., and N.S.; Investigation, A.K., Y.Z., T.G., G.S., N.S., and M.N.; Resources, A.B.S.; Writing, Original Draft, A.K.; Writing, Review & Editing, all authors; Supervision and Funding Acquisition, S.N. and M.N.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
This work was supported by the NIH grants NHLBI HL103411, GM126537, and NCI P30 CA016056 (for use of Roswell Park Cancer Institute’s Genomic Shared Resource). Illumina sequencing was performed at the University at Buffalo Genomics and Bioinformatics core or Roswell Park Genomic Shared Resource.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2019.09.006.
Supplemental Information
Supplemental Methods, Tables S1–S4, and Figures S1–S5
References
- 1.Tycko J., Myer V.E., Hsu P.D. Methods for Optimizing CRISPR-Cas9 Genome Editing Specificity. Mol. Cell. 2016;63:355–370. doi: 10.1016/j.molcel.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tsai S.Q., Joung J.K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat. Rev. Genet. 2016;17:300–312. doi: 10.1038/nrg.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Zhou H., Zhou M., Li D., Manthey J., Lioutikova E., Wang H., Zeng X. Whole genome analysis of CRISPR Cas9 sgRNA off-target homologies via an efficient computational algorithm. BMC Genomics. 2017;18(Suppl 9):826. doi: 10.1186/s12864-017-4225-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Hough S.H., Kancleris K., Brody L., Humphryes-Kirilov N., Wolanski J., Dunaway K., Ajetunmobi A., Dillard V. Guide Picker is a comprehensive design tool for visualizing and selecting guides for CRISPR experiments. BMC Bioinformatics. 2017;18:167. doi: 10.1186/s12859-017-1581-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Perez A.R., Pritykin Y., Vidigal J.A., Chhangawala S., Zamparo L., Leslie C.S., Ventura A. GuideScan software for improved single and paired CRISPR guide RNA design. Nat. Biotechnol. 2017;35:347–349. doi: 10.1038/nbt.3804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Allen F., Crepaldi L., Alsinet C., Strong A.J., Kleshchevnikov V., De Angeli P., Páleníková P., Khodak A., Kiselev V., Kosicki M. Predicting the mutations generated by repair of Cas9-induced double-strand breaks. Nat. Biotechnol. 2018;37:64–72. doi: 10.1038/nbt.4317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chakrabarti A.M., Henser-Brownhill T., Monserrat J., Poetsch A.R., Luscombe N.M., Scaffidi P. Target-Specific Precision of CRISPR-Mediated Genome Editing. Mol. Cell. 2019;73:699–713.e6. doi: 10.1016/j.molcel.2018.11.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ran F.A., Hsu P.D., Lin C.Y., Gootenberg J.S., Konermann S., Trevino A.E., Scott D.A., Inoue A., Matoba S., Zhang Y., Zhang F. Double nicking by RNA-guided CRISPR Cas9 for enhanced genome editing specificity. Cell. 2013;154:1380–1389. doi: 10.1016/j.cell.2013.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Tsai S.Q., Wyvekens N., Khayter C., Foden J.A., Thapar V., Reyon D., Goodwin M.J., Aryee M.J., Joung J.K. Dimeric CRISPR RNA-guided FokI nucleases for highly specific genome editing. Nat. Biotechnol. 2014;32:569–576. doi: 10.1038/nbt.2908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guilinger J.P., Thompson D.B., Liu D.R. Fusion of catalytically inactive Cas9 to FokI nuclease improves the specificity of genome modification. Nat. Biotechnol. 2014;32:577–582. doi: 10.1038/nbt.2909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Nishimasu H., Ran F.A., Hsu P.D., Konermann S., Shehata S.I., Dohmae N., Ishitani R., Zhang F., Nureki O. Crystal structure of Cas9 in complex with guide RNA and target DNA. Cell. 2014;156:935–949. doi: 10.1016/j.cell.2014.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Anders C., Bargsten K., Jinek M. Structural Plasticity of PAM Recognition by Engineered Variants of the RNA-Guided Endonuclease Cas9. Mol. Cell. 2016;61:895–902. doi: 10.1016/j.molcel.2016.02.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kleinstiver B.P., Pattanayak V., Prew M.S., Tsai S.Q., Nguyen N.T., Zheng Z., Joung J.K. High-fidelity CRISPR-Cas9 nucleases with no detectable genome-wide off-target effects. Nature. 2016;529:490–495. doi: 10.1038/nature16526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Casini A., Olivieri M., Petris G., Montagna C., Reginato G., Maule G., Lorenzin F., Prandi D., Romanel A., Demichelis F. A highly specific SpCas9 variant is identified by in vivo screening in yeast. Nat. Biotechnol. 2018;36:265–271. doi: 10.1038/nbt.4066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Chen J.S., Dagdas Y.S., Kleinstiver B.P., Welch M.M., Sousa A.A., Harrington L.B., Sternberg S.H., Joung J.K., Yildiz A., Doudna J.A. Enhanced proofreading governs CRISPR-Cas9 targeting accuracy. Nature. 2017;550:407–410. doi: 10.1038/nature24268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zetsche B., Volz S.E., Zhang F. A split-Cas9 architecture for inducible genome editing and transcription modulation. Nat. Biotechnol. 2015;33:139–142. doi: 10.1038/nbt.3149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Liu K.I., Ramli M.N., Woo C.W., Wang Y., Zhao T., Zhang X., Yim G.R., Chong B.Y., Gowher A., Chua M.Z. A chemical-inducible CRISPR-Cas9 system for rapid control of genome editing. Nat. Chem. Biol. 2016;12:980–987. doi: 10.1038/nchembio.2179. [DOI] [PubMed] [Google Scholar]
- 20.Davis K.M., Pattanayak V., Thompson D.B., Zuris J.A., Liu D.R. Small molecule-triggered Cas9 protein with improved genome-editing specificity. Nat. Chem. Biol. 2015;11:316–318. doi: 10.1038/nchembio.1793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maji B., Moore C.L., Zetsche B., Volz S.E., Zhang F., Shoulders M.D., Choudhary A. Multidimensional chemical control of CRISPR-Cas9. Nat. Chem. Biol. 2017;13:9–11. doi: 10.1038/nchembio.2224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Maji B., Gangopadhyay S.A., Lee M., Shi M., Wu P., Heler R., Mok B., Lim D., Siriwardena S.U., Paul B. A High-Throughput Platform to Identify Small-Molecule Inhibitors of CRISPR-Cas9. Cell. 2019;177:1067–1079.e19. doi: 10.1016/j.cell.2019.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Senturk S., Shirole N.H., Nowak D.G., Corbo V., Pal D., Vaughan A., Tuveson D.A., Trotman L.C., Kinney J.B., Sordella R. Rapid and tunable method to temporally control gene editing based on conditional Cas9 stabilization. Nat. Commun. 2017;8:14370. doi: 10.1038/ncomms14370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tu Z., Yang W., Yan S., Yin A., Gao J., Liu X., Zheng Y., Zheng J., Li Z., Yang S. Promoting Cas9 degradation reduces mosaic mutations in non-human primate embryos. Sci. Rep. 2017;7:42081. doi: 10.1038/srep42081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Yang S., Li S., Li X.-J. Shortening the Half-Life of Cas9 Maintains Its Gene Editing Ability and Reduces Neuronal Toxicity. Cell Rep. 2018;25:2653–2659.e3. doi: 10.1016/j.celrep.2018.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nihongaki Y., Kawano F., Nakajima T., Sato M. Photoactivatable CRISPR-Cas9 for optogenetic genome editing. Nat. Biotechnol. 2015;33:755–760. doi: 10.1038/nbt.3245. [DOI] [PubMed] [Google Scholar]
- 27.Polstein L.R., Gersbach C.A. A light-inducible CRISPR-Cas9 system for control of endogenous gene activation. Nat. Chem. Biol. 2015;11:198–200. doi: 10.1038/nchembio.1753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.González F., Zhu Z., Shi Z.-D., Lelli K., Verma N., Li Q.V., Huangfu D. An iCRISPR platform for rapid, multiplexable, and inducible genome editing in human pluripotent stem cells. Cell Stem Cell. 2014;15:215–226. doi: 10.1016/j.stem.2014.05.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Dow L.E., Fisher J., O’Rourke K.P., Muley A., Kastenhuber E.R., Livshits G., Tschaharganeh D.F., Socci N.D., Lowe S.W. Inducible in vivo genome editing with CRISPR-Cas9. Nat. Biotechnol. 2015;33:390–394. doi: 10.1038/nbt.3155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.de Solis C.A., Ho A., Holehonnur R., Ploski J.E. The Development of a Viral Mediated CRISPR/Cas9 System with Doxycycline Dependent gRNA Expression for Inducible In vitro and In vivo Genome Editing. Front. Mol. Neurosci. 2016;9:70. doi: 10.3389/fnmol.2016.00070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang G., Yang L., Grishin D., Rios X., Ye L.Y., Hu Y., Li K., Zhang D., Church G.M., Pu W.T. Efficient, footprint-free human iPSC genome editing by consolidation of Cas9/CRISPR and piggyBac technologies. Nat. Protoc. 2017;12:88–103. doi: 10.1038/nprot.2016.152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Cao J., Wu L., Zhang S.M., Lu M., Cheung W.K.C., Cai W., Gale M., Xu Q., Yan Q. An easy and efficient inducible CRISPR/Cas9 platform with improved specificity for multiple gene targeting. Nucleic Acids Res. 2016;44:e149. doi: 10.1093/nar/gkw660. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Aubrey B.J., Kelly G.L., Kueh A.J., Brennan M.S., O’Connor L., Milla L., Wilcox S., Tai L., Strasser A., Herold M.J. An inducible lentiviral guide RNA platform enables the identification of tumor-essential genes and tumor-promoting mutations in vivo. Cell Rep. 2015;10:1422–1432. doi: 10.1016/j.celrep.2015.02.002. [DOI] [PubMed] [Google Scholar]
- 34.Kim S., Kim D., Cho S.W., Kim J., Kim J.S. Highly efficient RNA-guided genome editing in human cells via delivery of purified Cas9 ribonucleoproteins. Genome Res. 2014;24:1012–1019. doi: 10.1101/gr.171322.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Sanjana N.E., Shalem O., Zhang F. Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods. 2014;11:783–784. doi: 10.1038/nmeth.3047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Wang T., Wei J.J., Sabatini D.M., Lander E.S. Genetic screens in human cells using the CRISPR-Cas9 system. Science. 2014;343:80–84. doi: 10.1126/science.1246981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Tzelepis K., Koike-Yusa H., De Braekeleer E., Li Y., Metzakopian E., Dovey O.M., Mupo A., Grinkevich V., Li M., Mazan M. A CRISPR Dropout Screen Identifies Genetic Vulnerabilities and Therapeutic Targets in Acute Myeloid Leukemia. Cell Rep. 2016;17:1193–1205. doi: 10.1016/j.celrep.2016.09.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Han J., Perez J.T., Chen C., Li Y., Benitez A., Kandasamy M., Lee Y., Andrade J., tenOever B., Manicassamy B. Genome-wide CRISPR/Cas9 Screen Identifies Host Factors Essential for Influenza Virus Replication. Cell Rep. 2018;23:596–607. doi: 10.1016/j.celrep.2018.03.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Park R.J., Wang T., Koundakjian D., Hultquist J.F., Lamothe-Molina P., Monel B., Schumann K., Yu H., Krupzcak K.M., Garcia-Beltran W. A genome-wide CRISPR screen identifies a restricted set of HIV host dependency factors. Nat. Genet. 2017;49:193–203. doi: 10.1038/ng.3741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stolfa G., Mondal N., Zhu Y., Yu X., Buffone A., Jr., Neelamegham S. Using CRISPR-Cas9 to quantify the contributions of O-glycans, N-glycans and Glycosphingolipids to human leukocyte-endothelium adhesion. Sci. Rep. 2016;6:30392. doi: 10.1038/srep30392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Platt R.J., Chen S., Zhou Y., Yim M.J., Swiech L., Kempton H.R., Dahlman J.E., Parnas O., Eisenhaure T.M., Jovanovic M. CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell. 2014;159:440–455. doi: 10.1016/j.cell.2014.09.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mondal N., Buffone A., Jr., Stolfa G., Antonopoulos A., Lau J.T.Y., Haslam S.M., Dell A., Neelamegham S. ST3Gal-4 is the primary sialyltransferase regulating the synthesis of E-, P-, and L-selectin ligands on human myeloid leukocytes. Blood. 2015;125:687–696. doi: 10.1182/blood-2014-07-588590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Povinelli B.J., Nemeth M.J. Wnt5a regulates hematopoietic stem cell proliferation and repopulation through the Ryk receptor. Stem Cells. 2014;32:105–115. doi: 10.1002/stem.1513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oguro H., Ding L., Morrison S.J. SLAM family markers resolve functionally distinct subpopulations of hematopoietic stem cells and multipotent progenitors. Cell Stem Cell. 2013;13:102–116. doi: 10.1016/j.stem.2013.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chen Y., Liu X., Zhang Y., Wang H., Ying H., Liu M., Li D., Lui K.O., Ding Q. A self-restricted CRISPR system to reduce off-target effects. Mol. Ther. 2016;24:1508–1510. doi: 10.1038/mt.2016.172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Petris G., Casini A., Montagna C., Lorenzin F., Prandi D., Romanel A., Zasso J., Conti L., Demichelis F., Cereseto A. Hit and go CAS9 delivered through a lentiviral based self-limiting circuit. Nat. Commun. 2017;8:15334. doi: 10.1038/ncomms15334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Moore R., Spinhirne A., Lai M.J., Preisser S., Li Y., Kang T., Bleris L. CRISPR-based self-cleaving mechanism for controllable gene delivery in human cells. Nucleic Acids Res. 2015;43:1297–1303. doi: 10.1093/nar/gku1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Li A., Lee C.M., Hurley A.E., Jarrett K.E., De Giorgi M., Lu W., Balderrama K.S., Doerfler A.M., Deshmukh H., Ray A. A Self-Deleting AAV-CRISPR System for In Vivo Genome Editing. Mol. Ther. Methods Clin. Dev. 2018;12:111–122. doi: 10.1016/j.omtm.2018.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Shalem O., Sanjana N.E., Hartenian E., Shi X., Scott D.A., Mikkelsen T.S., Heckl D., Ebert B.L., Root D.E., Doench J.G. Genome-Scale CRISPR-Cas9 Knockout Screening in Human Cells. Science. 2014;343:84–87. doi: 10.1126/science.1247005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Gossen M., Freundlieb S., Bender G., Müller G., Hillen W., Bujard H. Transcriptional activation by tetracyclines in mammalian cells. Science. 1995;268:1766–1769. doi: 10.1126/science.7792603. [DOI] [PubMed] [Google Scholar]
- 52.Gossen M., Bujard H. Tight control of gene expression in mammalian cells by tetracycline-responsive promoters. Proc. Natl. Acad. Sci. USA. 1992;89:5547–5551. doi: 10.1073/pnas.89.12.5547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Holmgaard A., Askou A.L., Benckendorff J.N.E., Thomsen E.A., Cai Y., Bek T., Mikkelsen J.G., Corydon T.J. In Vivo Knockout of the Vegfa Gene by Lentiviral Delivery of CRISPR/Cas9 in Mouse Retinal Pigment Epithelium Cells. Mol. Ther. Nucleic Acids. 2017;9:89–99. doi: 10.1016/j.omtn.2017.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Fricano-Kugler C.J., Williams M.R., Salinaro J.R., Li M., Luikart B. Designing, Packaging, and Delivery of High Titer CRISPR Retro and Lentiviruses via Stereotaxic Injection. J. Vis. Exp. 2016;(111) doi: 10.3791/53783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Buffone A., Jr., Mondal N., Gupta R., McHugh K.P., Lau J.T.Y., Neelamegham S. Silencing α1,3-fucosyltransferases in human leukocytes reveals a role for FUT9 enzyme during E-selectin-mediated cell adhesion. J. Biol. Chem. 2013;288:1620–1633. doi: 10.1074/jbc.M112.400929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Yang Z., Steentoft C., Hauge C., Hansen L., Thomsen A.L., Niola F., Vester-Christensen M.B., Frödin M., Clausen H., Wandall H.H., Bennett E.P. Fast and sensitive detection of indels induced by precise gene targeting. Nucleic Acids Res. 2015;43:e59. doi: 10.1093/nar/gkv126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Stemmer M., Thumberger T., del Sol Keyer M., Wittbrodt J., Mateo J.L. CCTop: An Intuitive, Flexible and Reliable CRISPR/Cas9 Target Prediction Tool. PLoS One. 2015;10:e0124633. doi: 10.1371/journal.pone.0124633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li H., Durbin R. Fast and accurate long-read alignment with Burrows-Wheeler transform. Bioinformatics. 2010;26:589–595. doi: 10.1093/bioinformatics/btp698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Quinlan A.R., Hall I.M. BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics. 2010;26:841–842. doi: 10.1093/bioinformatics/btq033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Bae S., Park J., Kim J.-S. Cas-OFFinder: a fast and versatile algorithm that searches for potential off-target sites of Cas9 RNA-guided endonucleases. Bioinformatics. 2014;30:1473–1475. doi: 10.1093/bioinformatics/btu048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Beauharnois M.E., Lindquist K.C., Marathe D., Vanderslice P., Xia J., Matta K.L., Neelamegham S. Affinity and kinetics of sialyl Lewis-X and core-2 based oligosaccharides binding to L- and P-selectin. Biochemistry. 2005;44:9507–9519. doi: 10.1021/bi0507130. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Methods, Tables S1–S4, and Figures S1–S5