

Abstract
Adult mammalian brains have largely lost neuroregeneration capability except for a few niches. Previous studies have converted glial cells into neurons, but the total number of neurons generated is limited and the therapeutic potential is unclear. Here, we demonstrate that NeuroD1-mediated in situ astrocyte-to-neuron conversion can regenerate a large number of functional new neurons after ischemic injury. Specifically, using NeuroD1 adeno-associated virus (AAV)-based gene therapy, we were able to regenerate one third of the total lost neurons caused by ischemic injury and simultaneously protect another one third of injured neurons, leading to a significant neuronal recovery. RNA sequencing and immunostaining confirmed neuronal recovery after cell conversion at both the mRNA level and protein level. Brain slice recordings found that the astrocyte-converted neurons showed robust action potentials and synaptic responses at 2 months after NeuroD1 expression. Anterograde and retrograde tracing revealed long-range axonal projections from astrocyte-converted neurons to their target regions in a time-dependent manner. Behavioral analyses showed a significant improvement of both motor and cognitive functions after cell conversion. Together, these results demonstrate that in vivo cell conversion technology through NeuroD1-based gene therapy can regenerate a large number of functional new neurons to restore lost neuronal functions after injury.
Keywords: NeuroD1, gene therapy, AAV, ischemic injury, brain repair, astrocyte-to-neuron conversion, motor function, fear conditioning learning
Graphical Abstract
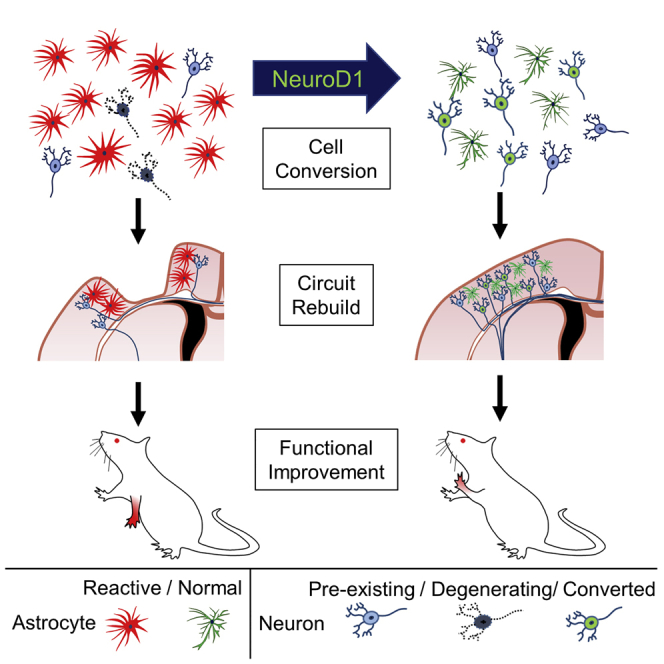
After ischemic brain injury, many neurons die but surviving astrocytes become activated and proliferative. Using NeuroD1 AAV-based gene therapy, Chen and colleagues demonstrate robust neuroregeneration through direct astrocyte-to-neuron conversion and significantly improved functional recovery. This study provides a new paradigm for brain repair using in vivo cell conversion technology.
Introduction
Neuronal loss is a major pathological hallmark of brain injury. Regenerating new neurons to replenish the lost neurons after injury is critical for brain repair. Unfortunately, adult mammalian brains have largely lost neurogenesis capacity, except a few neurogenic niches such as the hippocampus and the subventricular zone (SVZ).1, 2, 3 After ischemic injury, the number of new neurons generated through the internal neurogenesis is typically <1% of total lost neurons in adult mammalian brains.4, 5, 6, 7, 8 Therapeutic approaches have been developed to augment this endogenous neurogenesis such as the use of various neurotrophic factors.9, 10, 11 Alternatively, transplantation of external neural progenitor cells (NPCs) has also been explored as a potential stroke therapy.12, 13, 14 In animal models, transplanted NPCs can survive, proliferate, and regenerate new neurons in stroke areas.15, 16, 17 Even in patients, some clinical trials showed promising results.18 On the other hand, neural stem cells (NSCs) in the SVZ are found to mainly produce reactive astrocytes, not neurons, after migrating to injured cortical areas.19, 20 Engrafting external NSCs for post-stroke treatment also faces serious challenges such as immunorejection, tumorigenesis, and long-term survival.18, 21, 22, 23, 24 Therefore, it is urgent to develop new approaches to regenerate a sufficient number of new neurons in order to achieve long-term functional recovery after brain injury.
We have recently demonstrated a direct conversion of reactive astrocytes into functional neurons by a single transcription factor NeuroD1 in the mouse brain.25 Other groups also reported conversion of glial cells into neurons both in vitro and in vivo26, 27, 28, 29, 30, 31, 32, 33, 34 (reviewed by Li and Chen35). While the in vivo glia-to-neuron conversion approach can regenerate new neurons inside mouse brain and spinal cord, it is unclear whether this technology can generate a sufficient number of new neurons for therapeutic applications. Here, using an engineered adeno-associated virus (AAV) Cre-FLEX system to ectopically express NeuroD1 in reactive astrocytes in an ischemic injury model, we can regenerate 30%–40% of lost neurons in the motor cortex of adult mice. Behavioral tests indicate that NeuroD1-treatment significantly rescues both motor and fear memory deficits after ischemic injury in rodent animals. Together, our studies demonstrate that internal neuroregeneration using in situ cell conversion technology may be an effective approach for functional brain repair after injury.
Results
Astrocyte-to-Neuron Conversion in a Focal Stroke Model Using NeuroD1 AAV-Based Gene Therapy
We have recently demonstrated that NeuroD1 acts as a master transcription factor to directly convert glial cells into functional neurons inside mouse brains.25 However, the total number of neurons generated by retrovirus25 or under non-injury condition36 is somewhat limited. In this study, we used focal ischemic injury as a model system to investigate whether this in situ cell conversion approach can regenerate a sufficient number of functional neurons for brain repair. We initially tried a hypoxia-ischemic stroke model in adult mice but found large variations in the injury areas, from striatum to cortex and hippocampus, making it difficult to compare the effectiveness of in vivo cell conversion among different animals. After some pilot studies, we decided to use a focal stroke model induced by the vasoconstrictive peptide endothelin-1 (ET-1) to produce more consistent local ischemic injury in rodents.37, 38, 39, 40 Importantly, we compared two different ET-1 peptides, one with 21 amino acids (ET-1 [1–21]) and another with 31 amino acids (ET-1 [1–31]). We found that ET-1 (1–31) produced more severe tissue loss than ET-1 (1–21) (Figure S1A). We also found that a mouse strain with FVB genetic background gave more severe stroke injury than the commonly used B6/C57 mice (Figure S1A). Notably, we detected a significant cortical tissue loss over a time course of 2 months when injecting ET-1 (1–31) into the motor cortex of FVB mice (Figures 1A and 1B), establishing a severe focal stroke model with consistent tissue loss in adult mice (5–12 months old).
Figure 1.

NeuroD1-Mediated Astrocyte-to-Neuron Conversion in a Focal Stroke Model
(A) Tissue loss caused by focal ischemic injury. Injection of ET-1 (1–31) into mouse motor cortex led to a gradual tissue loss in 10 weeks. Dashed lines indicate cortical areas. White bar (3 mm) indicates the cortical areas being quantified for tissue loss. Scale bars, 3 mm. (B) Quantification of the remaining cortical tissue (from the midline to 3 mm lateral area, white bar) at 1, 4, and 10 weeks after ischemic injury in the motor cortex (n = 3 for each time point). (C and D) Assessing reactive astrocytes after ischemic injury. Immunostaining of NeuN and GFAP at 5 days (C) and 10 days post stroke (dps) (D) revealed reactive astrocytes at 10 dps. c.c., corpus callosum; ctx, cortex. Scale bars, left panel 200 μm, right panel 40 μm. (E) Retroviruses expressing GFP alone (top row) or NeuroD1-GFP (bottom row), illustrating neuronal conversion by NeuroD1. Viral injection at 10 dps and immunostaining at 17 dpi. Scale bar, 20 μm. (F) Injection of AAV9 expressing GFP alone (hGFAP::GFP, top row) or NeuroD1-GFP (hGFAP::NeuroD1-P2A-GFP, bottom row), illustrating more neurons generated by AAV than retroviruses. Scale bar, 40 μm. (G) Capture of the transitional stage from astrocytes (GFAP) to neurons (NeuN) at early time points of NeuroD1 expression (4 dpi). Injection of AAV9 hGFAP::Cre and CAG::FLEX-NeuroD1-P2A-mCherry resulted in significant NeuroD1 expression in GFAP-labeled astrocytes (top row). Interestingly, some NeuroD1-mCherry labeled cells showed both NeuN and GFAP signal (bottom row), suggesting a transition stage from astrocytes to neurons. Scale bars, upper 40 μm and lower insets 20 μm. (H) Experimental outline for ischemic injury, AAV injection (Cre-FLEX system), and immunostaining analysis. Scale bar, 40 μm. (I) Detection of a large number of NeuroD1-converted neurons using AAV Cre-FLEX system. At 17 dpi, the GFP control group showed many GFAP+ reactive astrocytes (top row, GFAP in purple), whereas the majority of NeuroD1-GFP labeled cells became NeuN-positive (red) neurons (bottom row).
After establishing the ischemic stroke model, we first examined a suitable time window for NeuroD1 viral injection to induce astrocyte-to-neuron conversion in the stroke areas. We performed glial fibrillary acidic protein (GFAP) immunostaining at different time points after stroke to determine when astrocytes became reactive. At 5 days post stroke (dps), we observed a significant loss of neuronal signal NeuN (Figure 1C). The GFAP signal, a commonly used reactive astrocyte marker, was also very low at 5 dps (Figure 1C), suggesting that astrocytes might be injured as well and not activated yet at this early stage. At 10 dps, however, the GFAP signal was significantly upregulated (Figure 1D), indicating that astrocytes had become reactive at this time. We then examined whether these reactive astrocytes induced by ischemic stroke could be converted into neurons. Consistent with our previous report,25 injection of retroviruses expressing NeuroD1 at 10 dps resulted in successful conversion of reactive glial cells into NeuN-positive neurons (Figure 1E). However, the number of neurons was limited due to the fact that retroviruses only expressed NeuroD1 in dividing reactive glial cells. To increase the number of neurons converted from astrocytes, we used AAV to infect both dividing and non-dividing astrocytes in the ischemic injured cortex. AAV has high infection rate and low pathogenicity in humans and has been approved by FDA for clinical trials in the treatment of CNS disorders.41 To infect astrocytes specifically after ischemic injury, we constructed an AAV vector (recombinant serotype AAV9) expressing NeuroD1 under the direct control of a human GFAP promoter (hGFAP::NeuroD1-P2A-GFP). As a control, AAV hGFAP::GFP was found mainly infecting astrocytes but not neurons (Figure 1F, top row). In contrast, AAV hGFAP::NeuroD1-P2A-GFP infected astrocytes gradually turned into NeuN-positive neurons in the ischemic injury areas (Figure 1F, bottom row). Comparing to the NeuroD1 retroviruses (Figure 1E), AAV hGFAP::NeuroD1-P2A-GFP (Figure 1F) generated more neurons. However, we also observed a gradual loss of GFP signal after astrocyte-to-neuron conversion, possibly due to a gradual downregulation of the hGFAP promoter during astrocyte-to-neuron conversion process. The loss of GFP signal after neuronal conversion by hGFAP::NeuroD1-P2A-GFP makes it difficult to distinguish the converted versus non-converted neurons in the viral infected areas. To overcome this problem, we separated the hGFAP promoter and NeuroD1 into two different AAV vectors by using the Cre-FLEX (flip-excision) homologous recombination system.42 One vector contains a human GFAP promoter to drive the expression of Cre recombinase (hGFAP::Cre) in astrocytes. The second vector contains two pairs of heterotypic, antiparallel loxP-type recombination sites flanking an inverted sequence of NeuroD1-P2A-GFP (or NeuroD1-P2A-mCherry) under the control of a strong promoter CAG (FLEX-CAG::NeuroD1-P2A-GFP/mCherry) (Figure S1B). The advantage of this AAV Cre-FLEX system is that the Cre expression is controlled by the hGFAP promoter to maintain the specificity toward astrocytes, while NeuroD1 expression is driven by the strong CAG promoter after Cre-mediated recombination and therefore avoiding being silenced during astrocyte-to-neuron conversion. With this hGFAP::Cre/FLEX-CAG::NeuroD1 system, we found that NeuroD1 was clearly expressed in GFAP+ astrocytes at 4 days post-viral injection (4 dpi, plus 10 dps, total 14 days post stroke) (Figure 1G, top row), which was not detectable if NeuroD1 expression was driven by GFAP promoter (hGFAP::NeuroD1-GFP) for 4 days. Interestingly, a few CAG::NeuroD1-infected astrocytes were captured in a transitional stage, showing co-immunostaining of both NeuN and GFAP (Figure 1G, bottom row), which had never been observed in the control group (hGFAP::Cre/FLEX-CAG::mCherry). After 17 days of NeuroD1 expression in the ischemic injury areas, the majority of CAG::NeuroD1-GFP infected cells were NeuN+ neurons, whereas the control AAV GFP-infected cells remained GFAP+ astrocytes in the injured mouse cortex (Figures 1H and 1I). Note that the CAG::NeuroD1-converted neurons using Cre-FLEX system showed more mature neuronal morphology (Figure 1I, bottom row) comparing to the GFAP::NeuroD1 converted neurons (Figure 1F, bottom row), suggesting that high expression of NeuroD1 under CAG promoter can facilitate neuronal conversion and maturation. Together, these data demonstrate that our engineered hGFAP::Cre/FLEX-CAG::NeuroD1 system can effectively convert astrocytes into neurons in adult mouse cortex after ischemic injury.
Progressive Conversion from Reactive Astrocytes into Neurons in Stroke Areas
We next investigated the time course of NeuroD1-mediated astrocyte-to-neuron conversion in the ischemic injured areas with immunostaining of astrocyte marker GFAP and neuronal marker NeuN (Figure 2). At 4 dpi, both control AAV (hGFAP::Cre + FLEX-CAG::mCherry or GFP) and NeuroD1 AAV (hGFAP::Cre + FLEX-CAG::NeuroD1-mCherry or GFP)-infected cells (red) were mainly GFAP+ astrocytes (green) (Figure 2A’), as guided by hGFAP::Cre AAV. It is worth noting that at 4 dpi about 12% of NeuroD1-infected cells were NeuN positive (Figure 2D). This is surprising but as illustrated in Figure 1G, many of these NeuN+ cells also were GFAP+, representing transitioning stage from astrocytes to neurons rather than off-targeting effect on pre-existing neurons, which should not have GFAP signal. At 7 dpi (Figure 2A’’), while control AAV mCherry-infected cells were still GFAP+ astrocytes (top row, yellow), about half of NeuroD1-mCherry-infected cells had lost GFAP signal (arrowheads). At 17 dpi, control mCherry-infected cells remained GFAP+, but most of the NeuroD1-mCherry-infected cells showed neuronal morphology without GFAP signal (Figure 2A’’’, arrowheads). When NeuN immunostaining was performed at 4 dpi, both control and NeuroD1-infected cells rarely showed NeuN signal (Figure 2B’), consistent with their astrocytic property as shown in Figure 2A’. At 7 dpi, some NeuroD1-infected cells showed clear NeuN signal with immature neuronal morphology (Figure 2B’’, arrowheads). At 17 dpi, the majority of NeuroD1-infected cells became NeuN+ neurons with more mature dendritic morphology (Figure 2B’’’, bottom row, arrowheads), whereas only a few mCherry-infected cells showed NeuN signal (Figure 2B’’’, top row). Quantitative analyses revealed that ∼70% of control mCherry- and GFP-infected cells maintained GFAP+ throughout 4 to 17 dpi (Figure 2C), but more than 70% of NeuroD1-infected cells adopted neuronal identity by 17 dpi (Figure 2D). Importantly, the NeuroD1 AAV-infected astrocytes gradually lost GFAP signal and gradually acquired NeuN signal within 17 days of viral infection, suggesting that the astrocyte-to-neuron conversion process takes ∼2–3 weeks to complete in an injured adult mouse cortex. It is worth mentioning that while AAV can infect both astrocytes and neurons, only ∼10% neurons showed mCherry signal in our control AAV Cre-FLEX system (hGFAP::Cre + FLEX-CAG::mCherry, Figure 1D, 17 dpi), possibly due to low expression of Cre in injured neurons after stroke. Together, these results demonstrate that our AAV NeuroD1 Cre-FLEX system (hGFAP::Cre + FLEX-CAG::NeuroD1-P2A-mCherry) successfully targets NeuroD1 expression in reactive astrocytes after stroke and then triggers a gradual transition from astrocytes into immature neurons and then more mature neurons in 2–3 weeks.
Figure 2.

NeuroD1 Gradually Converts Reactive Astrocytes into Neurons after Stroke
(A’–A’’’) Identification of astrocytes with GFAP immunostaining at 4 (A’), 7 (A’’), or 17 (A’’’) days post viral injection (dpi) in control (mCherry alone, top row) and NeuroD1-mCherry (bottom row) infected areas. Scale bar, 40 μm. (B’–B’’’) Identification of neurons with NeuN immunostaining at 4 (B’), 7 (B’’), or 17 dpi (B’’’). Arrowheads indicate some of the NeuroD1-converted neurons. Scale bar, 40 μm. (C and D) Quantification of GFAP+ cells (C) or NeuN+ cells (D) among all viral infected cells. Note a significant decrease of astrocytes (C) accompanied with a significant increase of neurons (D) in NeuroD1 group. **p < 0.01, ****p < 0.0001. Two-way ANOVA followed by Sidak’s multiple comparison test. n = 3 mice per group. Three images were randomly taken in cortical areas with viral infection. Data are represented as mean ± SEM.
Neuronal Recovery in the Stroke Areas following NeuroD1 AAV Treatment
After demonstrating progressive astrocyte-to-neuron conversion in the stroke areas, we next investigated how many new neurons could be regenerated in an injured mouse cortex after AAV NeuroD1 infection. Figure 3A illustrates the sharp contrast in NeuN staining between control group and NeuroD1-treated group. At 17 dpi (viral injection at 10 days post stroke, total 27 dps), when astrocyte-to-neuron conversion was largely completed, the control group showed a wide injury core (indicated by *) with huge NeuN loss, whereas the NeuroD1 group showed much smaller injury core and thicker cortex than the control group (Figure 3A, left low-power images). The high-power images of Figure 3A (right panels) showed the composition of neurons and astrocytes in the scar border (boxed in low-power images): the control GFP-infected cells were mostly astrocytes (top row), whereas NeuroD1-infected cells were mostly NeuN-positive neurons (bottom row). Importantly, in NeuroD1-converted areas, many GFAP-labeled astrocytes (purple) intermingled with NeuroD1-GFP labeled neurons (Figure 3A, bottom row, right panel), indicating that astrocytes were not depleted after astrocyte-to-neuron conversion. We also noted that besides NeuroD1-GFP labeled neurons (green and yellow in Figure 3A, bottom row), there were many NeuN+ neurons (red, no green) that were not converted by NeuroD1, suggesting a mixture of converted and non-converted neurons intermingled together in the injured areas. This was further confirmed by NeuroD1-immunostaining (Figure 3B, green), showing NeuroD1-converted neurons (yellow) intermingled with non-converted neurons (red only, arrowhead). Quantitative analysis revealed that the neuronal density in the peri-injury core areas in the control group was 27.1 ± 8.1/0.1 mm2 (Figure 3C), whereas in NeuroD1 group, NeuroD1-converted neurons (NeuroD1 and NeuN double positive) reached 39.9 ± 2.4/0.1 mm2, and the non-converted neurons (NeuroD1-negative but NeuN-positive) reached 64.9 ± 7.9/0.1 mm2 (Figure 3C, n = 3 mice per group). Compared to non-stroke cortical neuron density of 141 ± 6.7/0.1 mm2, the total converted plus non-converted neurons in the NeuroD1-converted areas reached 104.8/0.1 mm2, a 74.3% recovery, whereas the control GFP-infected areas only reached 19.2% of non-stroke level (Figure 3C). Notably, the non-converted neurons in the NeuroD1-treated stroke areas more than doubled that in the control GFP-infected stroke areas (Figure 3C, comparing the white bar versus gray bar), suggesting a significant increase of neuronal survival in the injured areas following NeuroD1-mediated astrocyte-to-neuron conversion. The observation of a significant increase of non-converted neurons intermingled with converted neurons suggests that in vivo astrocyte-to-neuron conversion not only regenerates new neurons but also protects injured neurons, which may have important implications in neural repair.
Figure 3.

High Efficiency of Neuroregeneration Achieved by NeuroD1-Mediated Astrocyte-to-Neuron Conversion
(A) Comparison of the NeuN signal in the motor cortex (17 dpi) between the control (top row) and NeuroD1 groups (bottom row). Left panels show the overall NeuN distribution after ischemic injury and viral injection; right panels show enlarged images of the peri-injury core areas. Note that NeuroD1-infected cells (green) were mostly converted into NeuN+ neurons (yellow), but GFAP+ astrocytes (purple) still persisted in the same areas. Scale bars, 500 μm for left panels and 40 μm for right panels. (B) NeuroD1-converted neurons (green and yellow) were intermingled with non-converted neurons (red, arrowheads) in the injury areas. Scale bar, 40 μm. (C) Quantification of total NeuN+ cells in the peri-injury core areas of control group and NeuroD1 group, as well as non-injured cortical areas. Note that the number of non-converted neurons in the NeuroD1 group (white bar) more than doubled the number in the control group, suggesting a neuroprotective effect of NeuroD1 conversion. n = 3 mice in each group. *p < 0.05. Two-way ANOVA followed by Sidak’s multiple comparison tests. Data are represented as mean ± SEM. (D and E) Immunostaining of neuronal dendrite markers SMI32 (D) and MAP2 (E) show much improved neuronal morphology in the NeuroD1 group (bottom row) compared to the control group (top row). Scale bar, 40 μm. (F and G) Immunostaining of axonal marker SMI312 (F), NF200 (G), and axon myelination marker MBP (G) show increased axons and axonal myelination in the NeuroD1 group (bottom row) compared to the control group (top row). Scale bar, 20 μm. (H) RT-PCR analysis revealed a significant increase of neuronal mRNA level including NeuN, Robo2, and Syn1 after NeuroD1 treatment. *p < 0.05. n = 4 mice each group. Unpaired t test. Data are represented as mean ± SEM. (I) Quantification of the total number of NeuN+ cells in the motor cortical areas (500–2,500 μm lateral from the midline). Note a significant increase of the total number of neurons in the NeuroD1 group by 60 dpi. n = 3 mice in each group. *p < 0.05, **p < 0.01. Two-way ANOVA followed by Sidak’s multiple comparison tests. Data are represented as mean ± SEM.
In accordance with a significant increase in NeuN+ neurons after NeuroD1-treatment, immunostaining of neuronal dendritic markers MAP2 and SMI32 showed strong and clearly aligned dendrites in the NeuroD1 group, compared to a rather weak and disorganized pattern in the GFP control group (Figures 3D and 3E). Similarly, using axonal markers SMI312 and NF200 along with myelination marker MBP, we found more myelinated axons in the NeuroD1 group than the control group (Figures 3F and 3G). Quantitative analyses for the axonal and dendritic markers are shown in Figure S2. To corroborate with the immunostaining results, we further performed qRT-PCR experiments with tissue lysates of the injured cortices. After ischemic injury, the expression level of neuronal genes including Neun, Robo2, and Syn1 was significantly decreased but partially rescued by NeuroD1-treatment (Figure 3H). Moreover, we quantified the total number of NeuN+ neurons within the ischemia-injured motor cortex (Figure 3I, 500 μm to 2,500 μm lateral from midline). In normal motor cortex without stroke, the total number of NeuN+ cells was around 2,614 ± 182 (counted within 500–2,500 μm lateral from the midline). After ischemic injury, the total number of NeuN-positive neurons within the same cortical areas (500–2,500 μm lateral from the midline) dropped to 20% of the no-stroke level (464 ± 80), indicating that 80% of neurons were lost or injured in our severe focal stroke model. The total number of NeuN+ neurons in the control AAV group did not change very much during 60 days post viral injection (due to gradual tissue loss, Figure 4A), but steadily increased in the NeuroD1 group, reaching 63% of the no-stroke level at 60 dpi (Figure 3I; 60 dpi: control group, 569 ± 145; NeuroD1 group, 1,641 ± 284). Such a significant increase in the total number of cortical neurons after NeuroD1-treatment provides potential building blocks for reconstruction of the disrupted neural circuits.
Figure 4.

Regeneration of Cortical Neurons after NeuroD1-Mediated Astrocyte-to-Neuron Conversion
(A) Low magnification images illustrating gradual tissue loss in the GFP control group (top row) and the rescue by NeuroD1 treatment (bottom row). Note layered structures in NeuroD1 group at 60 dpi. Dashed lines delineate the cortical areas. Scale bar, 400 μm. (B) Quantification of the motor cortical areas (from midline to 3 mm lateral) in the control versus NeuroD1 group. *p < 0.05, ***p < 0.001. Two-way ANOVA followed by Sidak’s multiple comparison test. n = 3 mice per group. (C) Serial brain sections from anterior (A) to posterior (P) further illustrating severe tissue injury in the control group. ctx, cortex; c.c., corpus callosum. Red, NeuN. Blue, DAPI. Scale bar, 400 μm. (D and E) Recovery of laminated structure of motor cortex indicated by layer marker Cux1 (D) and Ctip2 (E). Scale bar, 300 μm. (F) Representative images illustrating NeuroD1-converted neurons (NeuroD1-GFP+) expressing cortical marker Tbr1. Scale bar, 40 μm. (G) Quantification of the neuronal markers among NeuroD1-converted neurons in the ischemic injured cortex. Many converted neurons were immunopositive for cortical markers of glutamatergic neurons including Emx1, Tbr1, and Satb2, while only 10% were GABAergic neurons (PV+ and GABA+). n = 3 mice for each group. Data are represented as mean ± SEM.
Cortical Tissue Repair after Astrocyte-to-Neuron Conversion
With a significant number of new neurons generated through astrocyte-to-neuron conversion, we next examined whether these new neurons could integrate into the cortical tissue after ischemic injury. Figure 4A illustrates the mouse motor cortex at different time points following viral injection (plus 10 days post stroke). Over a time course of 2 months, we observed a gradual cortical tissue loss in the control group injected with AAV GFP (Figure 4A, top row), indicating a severe brain injury in our focal stroke model. In contrast, the cortical tissue in NeuroD1-treated group was largely preserved (Figure 4A, bottom row). Quantitative analysis revealed that the mouse motor cortical volume reduced by 70% over a 2-month period in the control group, whereas only 20% of tissue volume reduction was observed in NeuroD1-treated group (Figure 4B). Cortical volume was quantified from three most severely injured cortical sections, from midline between two hemispheres to 3 mm lateral cortical region, which covered the entire stroke area (Figure 4C). It is critical to point out that while the 60 dpi images in Figure 4A illustrate a sharp contrast showing a cavity in the control group versus a repaired cortex in the NeuroD1 group, the difference between the two groups is much smaller at 7 dpi. As shown in Figure 4B, the major difference was due to the continuous tissue loss in the control group, whereas converting reactive astrocytes into neurons significantly prevented tissue loss in the NeuroD1 group.
We next investigated what kinds of neurons were regenerated after astrocyte-to-neuron conversion in the stroke areas. With immunostaining of cortical neuron markers Cux1 and Ctip2, we found that NeuroD1-GFP positive neurons were distributed in both superficial layer (layer II–III) and deep cortical layer (layer V–VI) (Figures 4D and 4E). Furthermore, the majority of NeuroD1-converted neurons in the injured motor cortex were immunopositive for cortical pyramidal neuron markers including Emx1, Tbr1, and Satb2 (Figures 4F and 4G). In contrast, only ∼10% of NeuroD1-converted neurons in the injured cortex were immunopositive for GABAergic neuron markers including parvalbumin and GABA (Figure 4G), similar to our previous report.25 Therefore, ectopic expression of NeuroD1 in the reactive astrocytes after ischemic injury in the motor cortex generated new neurons with similar neuronal identity to cortical neurons.
Functional Characterization of NeuroD1-Converted Neurons
We further investigated whether NeuroD1-converted neurons are electrophysiologically functional by conducting patch clamp recordings. Cortical slice recordings were performed on NeuroD1-converted neurons at 60 dpi (Figure 5A). Injecting depolarizing currents into the NeuroD1-GFP labeled neurons triggered repetitive action potentials in every neuron recorded (GFP+, n = 22) (Figure 5B). Furthermore, we detected robust spontaneous synaptic events, both excitatory and inhibitory, in NeuroD1-converted neurons (GFP+) in the injury sites after 2 months of astrocyte-to-neuron conversion (Figure 5C), suggesting that these neurons have formed synaptic connections with other neurons. Quantitative analysis revealed that synaptic responses (both EPSCs and IPSCs) decreased in the control group (recorded in non-converted neurons) after stroke, but were significantly rescued in the NeuroD1-GFP group (Figure 5D; EPSC frequency: control, 4.3 ± 0.6 Hz, n = 22; NeuroD1, 6.7 ± 0.8 Hz, n = 25; *p = 0.023. IPSC frequency: control, 8.6 ± 1.3 Hz, n = 22; NeuroD1, 14.0 ± 2.0 Hz, n = 25; *p = 0.032. Student’s t test). Together, these data demonstrate that NeuroD1-mediated astrocyte-to-neuron conversion can regenerate functional neurons in the injury sites after stroke.
Figure 5.

Local and Global Connections after Astrocyte-to-Neuron Conversion
(A and B) Brain slice recording on NeuroD1-converted neurons (GFP) detected repetitive action potential firing (60 dpi, n = 22). (C) Representative traces of spontaneous excitatory (sEPSCs) and inhibitory synaptic events (sIPSCs) recorded in NeuroD1-GFP labeled neurons (60 dpi). (D) Quantification of the frequency of both sEPSCs and sIPSCs in cortical slices without injury (white bar), or with ischemic injury (black bar, GFP control; striped bar, NeuroD1 group). Note that NeuroD1 group showed significantly higher frequency of both sEPSCs and sIPSCs than the control group. Neurons in the control group were the surviving neurons after ischemic injury, not labeled by GFP. Amplitude showed no difference between the control group and NeuroD1 group: EPSC, control, 19.3 ± 2.6 pA; NeuroD1, 16.6 ± 1.3 pA; p > 0.05. IPSC, control, 20.6 ± 1.6 pA; NeuroD1, 21.7 ± 2.0 pA; p > 0.05. n = 22 for control group, and n = 25 for NeuroD1 group. Student’s t test. Electrophysiological properties: Input resistance, non-stroke group 133.8 ± 11.9 MΩ, GFP control group 236.8 ± 27.3 MΩ, NeuroD1 group 180.2 ± 23.3 MΩ; Capacitance, non-stroke group 139.2 ± 10.2 pF, GFP control group 108.0 ± 19.4 pF, NeuroD1 group 128.4 ± 8.5 pF; Resting membrane potential, non-stroke group −70.0 ± 1.9 mV, GFP control group −67.4 ± 1.0 mV, NeuroD1 group −68.1 ± 0.9 mV. n = 21 for non-stroke group, n = 28 for GFP control group, and n = 42 for NeuroD1 group. (E) Representative images illustrating distal axonal projections from NeuroD1-converted neurons. Serial sagittal sections (17 dpi), from medial (M, lower left) to lateral (L, upper right), showing converted neurons in the cortex (inset 1), axonal bundles in the striatum (inset 2), thalamus (inset 3), and hypothalamus (inset 4). Scale bars, 1,000 μm for sagittal images and 40 μm for inset images. (F) CTB retrograding tracing experiment (shown in upper panel, see detail in Supplemental Information) indicate the NeuroD1 converted cells could be labeled by CTB dye. Scale bar, 10 μm.
Long-Range Axonal Projections after Astrocyte-to-Neuron Conversion
With the demonstration of synaptic input to the NeuroD1-converted neurons, we next examined where these newly generated neurons would send out their axon projections in an injured environment. Sagittal sections of the NeuroD1-infected brains at 60 dpi (Figure 5E) revealed robust axonal projections from cortex (box 1) to the striatum (box 2), thalamus (box 3), and hypothalamus (box 4). We also observed significant axon projections from cortex to the hippocampus. Interestingly, the hippocampal CA2-CA3 region was often infected directly when injecting AAV into the cortex. To further investigate axonal projection, we injected retrograde tracer CTB (fluorescently labeled peptide) into the thalamus at 40 dpi (Figure 5F). After 7 days of CTB injection, many NeuroD1-converted neurons in the motor cortex were retrograde labeled with strong CTB signal (Figure 5F), suggesting that these newly generated neurons had sent their projections to the thalamic target. How did the newly generated neurons send their axons to distant targets in an injured adult brain? One possibility is to follow the preexisting axon pathways. To test this hypothesis, we first injected AAV9 Syn::GFP into the motor cortex to label cortical neurons with GFP before stroke. As shown in Figure S3A, the Syn::GFP-expressing neurons can send their axons to distant targets. One week after injecting Syn::GFP, ET-1-31 was injected into the motor cortex to induce ischemic injury. Then, AAV9 NeuroD1-mCherry was injected 10 days after ET-1 induced ischemic injury to convert reactive astrocytes into neurons. One week after NeuroD1 infection, sagittal sections of the viral infected brains were immunostained with GFP and mCherry to trace the axon projections of preexisting neurons and newly converted neurons, respectively (Figure S3A). Because only a small number of neurons were converted at the early time point of 7 dpi, we found a consistent weak mCherry signal in the corpus callosum (box 1), striatum (box 2), and thalamic region (box 3) (Figure S3A), which was in sharp contrast to the massive projections observed at 60 dpi (Figure 5E). Nevertheless, the weak mCherry-labeled new axons appeared to follow the GFP-labeled preexisting axon bundles along the cortex-striatum-thalamus-hypothalamus pathway (Figure S3A). It is important to note that after NeuroD1-mediated conversion, the astrocyte-converted neurons may also start to express Syn::GFP and became GFP-positive as well. Figure S3B illustrates an example of 9 months after NeuroD1 viral injection, showing a coronal section of the motor cortex with NeuroD1-converted neurons sending axons to the contralateral cortex through the corpus callosum. Together, these results suggest that NeuroD1-converted neurons can send out long-range axon projections to global brain regions, possibly by following preexisting axonal pathways.
Transcriptome-wide Recovery following NeuroD1-Mediated Astrocyte-to-Neuron Conversion in the Ischemic Injured Cortex
To better understand the global changes of the injured cortical areas after NeuroD1-mediated astrocyte-to-neuron conversion, we conducted RNA sequencing (RNA-seq) to compare the transcriptome profile between the control and NeuroD1 groups by extracting mRNA from the ischemic cortical tissues at 17 dpi (Figure 6). Based on the overall genome-wide expression, the dendrogram of sample relations indicates a closer relationship between the NeuroD1-infected tissues and healthy cortical tissues (no stroke) (Figure 6A). Consistently, differential expression analyses revealed a huge difference in gene-expression profile between no stroke healthy tissues and control virus-infected stroke tissues, with a total of 880 differentially expressed genes (DEGs) identified (Figure 6B). In contrast, only 44 DEGs were identified between NeuroD1-infected stroke tissues and no stroke healthy tissues (Figure 6B). The heatmap in Figure 6C illustrates the overview of the relative changes of DEGs among control virus-infected tissues (n = 2 mice), NeuroD1-infected tissues (n = 2 mice), and healthy tissues without stroke (n = 3 mice). The DEGs showed two distinct expression patterns (Figure 6C): (1) compared to no stroke tissues, the control virus-infected tissues showed highly upregulated genes related to immune response and antigen processing (red color in left control columns); (2) in NeuroD1-infected tissues, the cluster of highly expressed genes with low level in control group (top cluster in the NeuroD1 columns) were related to blood circulation, synaptic transmission, and neuropeptide signaling. Overall, the DEG pattern in NeuroD1-infected stroke tissues is much closer to the healthy tissues than the control virus-infected stroke tissues (Figure 6C). Figure 6D illustrates some representative genes among the DEGs encoding neuronal proteins, such as neural transcription factors (Neurod1, Neurod6, Tbr1, and Satb2), typical neuronal markers (Rbfox3 known as Neun, Tubb3 known as Tuj1, and Map2), and channels and receptors (Kcnc2 and Gabbr2), as well as synaptic proteins (Sv2b, Syn1, Syn2, Rimbp2, and Slc17a7). One repetitive pattern observed is that compared to the no stroke healthy tissues (black bars), the control virus-infected stroke tissues (red bars) showed a consistent decrease of neuronal gene expression, but NeuroD1-infected stroke tissues (blue bars) showed a significant recovery of neuronal gene expression. This is consistent with our immunostaining and RT-PCR experimental results. Together, our transcriptome analysis demonstrates that NeuroD1-mediated astrocyte-to-neuron conversion can largely rescue the neuronal gene expression level following ischemic injury.
Figure 6.

Transcriptomic Analysis of the Gene-Expression Profile at 17 dpi
(A) Sample relationship based on global gene-expression profile revealed a closer relation between NeuroD1-infected tissues and healthy tissues without stroke. Control group (n = 2 mice), NeuroD1 group (n = 2 mice), no stroke group (n = 3 mice). (B) Venn diagram shows the number of differentially expressed genes (DEGs) from pairwise comparisons among control, NeuroD1, and no stroke groups. Note that the number of DEGs between NeuroD1-group and no stroke group is rather small. DEGs are defined as at least 50 base mean value (normalized read counts across all the samples using DESeq2 method) with >3-fold change among samples, and adjusted p value < 0.01. (C) Hierarchical clustering of all the 1,058 DEGs and heatmap of the relative expression level of 1,058 DEGs in all the samples. Red indicates high read count level, whereas blue indicates low read count level. Note the similarity of heatmap pattern between NeuroD1 group and no stroke group. (D) RNA-seq read counts of neuronal genes among different samples. NeuroD1 expression was significantly increased in NeuroD1-infected stroke tissues, as expected. Note a consistent pattern of decreased neuronal gene expression level in stroke tissues infected by control viruses (red bars) but a significant recovery in NeuroD1-infected stroke tissues (blue bars).
Functional Rescue of Motor Deficits
With a significant level of neuroregeneration in the injury areas after NeuroD1-mediated astrocyte-to-neuron conversion, we then investigated whether NeuroD1 treatment can rescue motor deficits caused by ischemic injury in the mouse motor cortex. We analyzed mouse forelimb functions using three behavioral tests—food pellet retrieval, grid walking, and cylinder test—at different time points following viral injection (Figure 7A; see Videos S1, S2, and S3 for behavioral tests). These three tests were designed to focus on the fine movement of the mouse forelimbs. With injection of ET-1 at both the motor cortex and the somatosensory cortex on one side of the brain, we were able to induce severe tissue loss (Figures S4B and S4C) and unilateral deficits of forelimb motor functions with limited spontaneous recovery in FVB mice (Figure 7).
Figure 7.

Motor Functional Improvement after NeuroD1-Treatment
(A) Experimental design for mouse forelimb motor functional tests. Behavioral tests were conducted before ischemic injury to obtain baseline control, and then 9 dps, but 1 day before viral injection to assess injury-induced functional deficits. AAV were injected at 10 dps, and behavioral tests were further performed at 20, 30, 50, and 70 dps to assess functional recovery. (B) Pellet retrieval test. NeuroD1 group (magenta) showed accelerated functional recovery compared to the control group (blue). Ischemic injury at the motor cortex severely impaired the food pellet retrieval capability, dropping from 5–6 pellets/5 min pre-stroke down to 1 pellet/5 min at 9 dps. After NeuroD1 treatment, pellet retrieval ability recovered to 4 pellets/5 min by 60 days post infection. Note that for food pellet retrieval test, only the motor cortex contralateral to the dominant side of forelimb was injured and tested. **p < 0.01, ***p < 0.001. Two-way ANOVA followed by Tukey’s multiple comparison test. Data are represented as mean ± SEM. n = 12 mice for ET-1 plus control AAV group; n = 12 mice for ET-1 plus NeuroD1 AAV group; n = 6 mice for ET-1 plus no virus group; and n = 6 mice for PBS control group. (C) Grid walking test. NeuroD1 group showed lower foot fault rate compared to the control group. Ischemic injury of the motor cortex significantly increased the foot fault rate, which was partially rescued by NeuroD1 treatment. **p < 0.01, ***p < 0.001. Two-way ANOVA followed by Tukey’s multiple comparison test. Data are represented as mean ± SEM. n = 9 mice for ET-1 plus control AAV contralateral (injured) side; n = 9 mice for ET-1 plus NeuroD1 AAV contralateral (injured) side; n = 5 mice for ET-1 plus NeuroD1 ipsilateral (non-injured) side; and n = 4 mice for ET-1 plus control ipsilateral (non-injured) side; n = 6 mice for ET-1 plus no virus contralateral group; and n = 6 mice for PBS contralateral group. (D) Cylinder test. NeuroD1-treated mice showed considerable recovery of rising and touching the sidewall with both forelimbs compared to the control group. **p < 0.01, ****p < 0.0001. Two-way ANOVA followed by Tukey’s multiple comparison test. Data are represented as mean ± SEM. n = 9 mice for ET-1 plus control AAV contralateral (injured) side; n = 11 mice for ET-1 plus NeuroD1 AAV contralateral (injured) side; n = 9 mice for ET-1 plus NeuroD1 ipsilateral (non-injured) side; n = 7 mice for ET-1 plus control ipsilateral (non-injured) side; n = 6 mice for ET-1 plus no virus contralateral group, n = 5 mice for PBS contralateral group.
To test for food pellet retrieval, we deprived mice of food before the training and test to increase their motivation for the food pellet. Before ischemic injury, normal animals could be trained to retrieve 5 or 6 pellets on average out of total 8 pellets in 5 min (see Figure S4A for food pellet retrieving device). After ischemic injury, their pellet retrieval capability was severely impaired, dropping to ∼1 pellet on average in 5 min (Figure 7B). Then, the ischemic injured animals with similar motor deficits were assigned into two groups for viral injection: one group injected with GFP viruses alone and the other injected with NeuroD1-GFP viruses. At 10 days after viral injection, there was no significant difference between the two groups, but after 20 days of viral injection, the NeuroD1 group started to show improvement in food pellet retrieval (Figure 7B). By 60 days after viral injection, the NeuroD1 group reached ∼4 pellets in 5 min, whereas the GFP control group could only retrieve <2 pellets in 5 min (Figure 7B; see Video S1). Similarly, for the grid walking test, normal animals before injury had a low rate of foot fault, typically ∼5% of total steps within 5 min, while walking freely on a grid, but the foot fault rate increased to over 10% after ischemic injury (Figure 7C). After viral injection, the NeuroD1 group showed consistent improvement after 20–60 days of treatment, with the foot fault rate decreasing to ∼7%, whereas the GFP control group remained at a high foot fault rate >9% (Figure 7C; see Video S2). We also performed a cylinder test to assess the forelimb function when animals rise and touch the sidewall of a cylinder. Normal animals typically use both forelimbs to touch and push steadily against the sidewall, with a normal touching rate ∼85% (using both forelimbs). After a unilateral ischemic injury in the forelimb motor cortex, the function of the impaired forelimb was significantly weakened and the impaired limb often either failed to touch the sidewall or dragged along the wall after briefly touching (Figure 7D, normal touching rate dropped to ∼35%). After 20–60 days of viral infection, the NeuroD1 group showed a significant recovery in touching the sidewall with the injured forelimb (∼60%), whereas the majority of the GFP control mice still had a weak forelimb affected by the ischemic injury (Figure 7D; see Video S3). For all three tests, injection of PBS as a sham control did not result in any behavioral deficits (black line in Figures 7B–7D), and injection of ET-1 (1–31) without viral injection produced similar deficits as the GFP control viral injection (orange line in Figures 7B–7D). Because stroke was induced unilaterally to impair the contralateral forelimb, we also assessed the non-injured forelimb (dotted lines in Figures 7C and 7D, labeled as Stroke + NeuroD1 Ipsil. and Stroke + Control Ipsil.). As expected, the non-injured forelimb didn’t show functional deficits. Taken together, by using three different motor behavioral tests, we demonstrate that NeuroD1-treatment can rescue motor functional deficits following ischemic injury in the mouse motor cortex.
After behavioral tests, we dissected out brain tissue for immunostaining. As expected, at 60 dpi, we observed an obvious deep hole in the cortical tissue of the control group (Figure S4B, left), but NeuroD1 group showed better preserved cortical tissue (Figure S4B, right). This was further confirmed with NeuN and GFP immunostaining (Figure S4C). Quantitative analysis after behavioral tests (with two sites of ET-1 injection to induce more severe injury) revealed significant reduction of cortical volume in the control group but not NeuroD1 group (Figure S4D). To investigate whether NeuroD1-converted neurons contributed to motor functions, we put NeuroD1-injected stroked mice (4 months post viral injection) into a running wheel for 30 min running and then rested for 1 h in home cage before sacrificing for c-Fos immunostaining (Figure S4E). Clearly, many NeuroD1-GFP labeled neurons also showed activity-dependent c-Fos signal (Figure S4E), suggesting that the NeuroD1-converted neurons are functionally integrated into the motor cortex. It is important to point out that both NeuroD1-converted and non-converted preexisting neurons contribute to functional recovery.
Functional Rescue of Cognitive Deficits
We next used a different animal species (rats instead of mice) and a different behavioral task (cognitive rather than motor) to further test the effect of NeuroD1-mediated astrocyte-to-neuron conversion following ischemic injury in the amygdala (Figure 8). It is well established that the associative memory of a conditioned stimulus (tone) and an aversive event (electrical foot shock) is stored in the basolateral nucleus of the amygdala (BLA).43, 44 Lesions of the BLA impair both the acquisition and the subsequent retrieval of an auditory fear memory.45, 46 To assess the NeuroD1 effect on cognitive functions, we injected rats with ET-1 into the BLA to produce an ischemic injury and then submitted to auditory fear conditioning 3 weeks later (Figure 8A). This interval was based on pilot studies showing a partial impairment in fear acquisition combined with partial neuronal injury approximately 21 days after the ET-1 stroke. The 21-day time point also helped to maximize glial scar formation following ET-1 lesion,47 an important factor to consider when using cell conversion strategies. ET-1 has been previously used to induce ischemic lesions in rat models of stroke.37, 47 Accordingly, we found that intra-BLA infusion of ET-1 (1–21) (3 μL/side, 400 pmol) induced a robust lesion in BLA (Figure S5). Behaviorally, rats infused with ET-1 showed a significant reduction in the acquisition of a conditioned freezing response to the tone, in comparison to a saline-infused control group (Figure 8B, day 21). Reduced freezing was also observed during the retrieval test the following day (Figure 8B, day 22), suggesting that the lesion induced by ET-1 impaired the formation of an auditory fear memory. One day later, lesioned rats were separated into two groups receiving infusions of either control virus or NeuroD1 virus (3 μl) into the BLA. After 3 weeks of viral infection, animals were returned to the same box for a fear retrieval test (Figure 8B, day 45). Rats in the control group continued to show reduced freezing levels, as expected. In contrast, freezing in the NeuroD1 group returned to the levels of the saline/saline group, and was significantly higher than the group receiving the control virus infusion (Figure 8B, day 45), suggesting a rescue of the memory deficit by NeuroD1 treatment. There was no effect of NeuroD1 on freezing during the pre-tone period (pre-CS, “x” symbol in Figure 8B), suggesting that NeuroD1 did not induce a general increase in amygdala excitability or non-specific fear. After completion of behavioral tests, we performed immunostaining to confirm viral infection in the BLA. As expected, we found that the majority of NeuroD1-GFP-infected cells were NeuN-positive neurons (Figure 8C). Together, these results suggest that NeuroD1-treatment can rescue the fear memory deficits induced by an ischemic lesion of BLA, possibly through strengthening the residual fear memory via new neurons and connections within the BLA.
Figure 8.

Recovery of Fear Conditioning Memory after NeuroD1 Treatment
(A) Experimental design of fear conditioning test in rats. ET-1 or saline was injected into the BLA, followed by fear conditioning 3 weeks later. Fear memory tests were performed before viral injection and 3 weeks after viral injection to assess the retention of fear memory. Right two panels illustrate the amygdala lesion induced by the infusion of ET-1. Gray areas represent the minimum (dark) and maximum (light) spread of the lesion across different anterior-posterior levels of BLA (−2.12, −2.56, and −2.80 from bregma), condensed in one level for illustration. CeA, central nucleus of the amygdala, op., optical tract. (B) ET-1 lesion reduced freezing during fear conditioning (F(2,31) = 3.98, *p = 0.02, day 21) at both ET-1/Control (blue, *p = 0.021, n = 10) and ET-1/NeuroD1 (magenta, *p = 0.019, n = 14) groups, compared to saline/saline group (black, n = 10). Reduced freezing (F(2,31) = 3.45, *p = 0.044) was also observed on the next day (day 22) in both ET-1/Control (p = 0.030) and ET-1/NeuroD1 (*p = 0.031) groups. Rats were then infused with control virus or NeuroD1 and re-tested 3 weeks later (F(2,31) = 5.86, *p = 0.006, day 45). In the ET-1/NeuroD1 group, freezing returned to the levels of the Saline/Saline group (*p = 0.81), and was significantly higher than ET-1/Control group (*p = 0.004). “x” denotes baseline pre-tone freezing levels. Hab, habituation; Cond, conditioning; pre-CS, pre-conditioned stimulus. One-way ANOVA followed by Duncan’s post hoc test. Data are expressed as mean ± SEM in blocks of two trials. *p < 0.05. (C) After fear conditioning test, immunostaining of rat brain sections confirmed the injection of NeuroD1-GFP viruses into the BLA (green, left panel), and the NeuroD1-infected cells were mostly NeuN-positive neurons (right panels). Scale bar, 1,000 μm.
Discussion
In this study, we demonstrate that in vivo astrocyte-to-neuron conversion mediated through NeuroD1-based gene therapy can efficiently regenerate a large number of functional new neurons in an ischemic injury model and achieve functional rescue of both motor and cognitive deficits in rodent animals. The newly generated neurons intermingled with preexisting neurons and prevented tissue loss in the injury sites. Notably, astrocytes persisted nearby the converted neurons, indicating that astrocytes are not depleted after conversion. Immunostaining together with RNA-seq revealed substantial neuronal recovery after NeuroD1-mediated astrocyte-to-neuron conversion. The astrocyte-converted neurons are electrophysiologically functional and send out long-range axonal projections to the target regions. Both motor function and fear memory tests showed significant improvement after astrocyte-to-neuron conversion. Together, this study demonstrates that in vivo astrocyte-to-neuron conversion can regenerate functional new neurons, form new synaptic circuits, protect injured neurons, and rescue behavioral deficits. This NeuroD1-based gene therapy may open a new avenue for brain repair using internal glial cells to regenerate functional new neurons.
Achieving High Neuroregeneration Efficiency with NeuroD1 AAV-Based Gene Therapy
Many neurological disorders are associated with severe neuronal loss. How to replenish the lost neurons in order to restore the lost brain functions has been proven a very difficult task in the past. We demonstrate here that AAV NeuroD1-based gene therapy can regenerate 400 NeuN+ cells/mm2 in the injured adult mouse cortex, about one third of the cortical neurons (141 ± 6.7 NeuN+ cells/0.1 mm2). Unexpectedly, accompanying astrocyte-to-neuron conversion, we observed a significant neuroprotection of the preexisting mature neurons that would have been lost in the control group. It is therefore the neuroregeneration plus neuroprotection, together with reduction of reactive astrocytes, that results in a significant neural repair. Such induced neuroregeneration efficacy is orders of magnitude higher than the spontaneous adult neurogenesis.4, 5, 6, 7 Generating a large number of functional new neurons in the injury sites may be critical for their survival, because previous studies have reported that the majority of adult newborn neurons failed to survive in injured areas.48 When we used retroviruses to convert only the dividing reactive glial cells into neurons, we also observed a decline of the newly converted neurons within several weeks.25 In this study, we used AAV system to significantly increase the total number of converted neurons. AAV has been extensively used in the CNS for gene expression and circuit mapping due to its high infection rate and low pathogenicity.49, 50 Compared to retroviruses that mainly target dividing cells, AAV has the advantage of infecting both dividing and non-dividing glial cells so that cell conversion is not limited by the number of dividing cells.
On the other hand, because AAV can infect both dividing and non-dividing cells, it becomes critical to target glial cells specifically with glia-specific promoters. Our engineered AAV GFAP::Cre and FLEX-CAG::NeuroD1 system used in this study is trying to balance specificity versus efficacy when developing a suitable viral system as a research tool for long-term tracking of the converted neurons. Because AAV itself is not specific for a particular cell type, we did observe some viral infection of neurons in the control AAV group (GFAP::Cre and FLEX-CAG::mCherry). GFAP::Cre can safeguard the expression of Cre initially in astrocytes, but Cre might “leak” into nearby neurons through direct contacts or exosomes.51 Injured or diseased neurons might also have their glial promoters partially reactivated as reported before,52, 53 resulting in low expression of Cre in infected neurons. Infection by high titer of AAV may lead to higher “leakage,” while low titer will have less “leakage” but the conversion efficiency is also low, creating a dilemma between specificity and efficacy. To solve this problem, it is pivotal to perform side-by-side control experiments such as mCherry versus NeuroD1-mCherry used in this study and examine at different time windows. Different viral systems should also be used to independently verify cell conversion. For example, retroviruses only express target genes in dividing glial cells but not neurons, which will give a clean result as shown in this study (Figure 1E) and our earlier work.25 Another approach is to use glial promoter to directly drive the expression of target genes in glial cells, as shown in Figure 1F, although the conversion efficiency may be affected by the expressed transcription factors. Therefore, it is important to balance the specificity and efficacy when choosing the viral system (retrovirus, AAV, lentivirus) and promoters (general promoters versus cell-specific promoters) for particular type of neural repair.
Unlike classical gene therapy that overexpresses a missing protein to treat genetic defect, we overexpress a neural transcription factor NeuroD1 to change a glial cell into a new neuron. Therefore, our NeuroD1-based in vivo cell conversion technology is a unique approach: a gene therapy-based cell therapy. NeuroD1 is an endogenous neural transcription factor that is not only expressed during early brain development but also in the adult NSCs.54, 55 Even in adult mouse cortex and hippocampus, we can detect a low level of NeuroD1 expression in mature neurons, which triggered us to pick NeuroD1 for in vivo glia conversion in the first place.25 Besides our group, NeuroD1 has also been reported to convert or help the conversion of fibroblast cells or glial cells into neurons by several other groups.36, 56, 57
Functional Integration of NeuroD1-Converted Neurons
Many clinical trials on CNS disorders such as stroke and Alzheimer’s disease have largely failed over the recent years.58, 59 Although different reasons are behind each failed clinical trial, one potential common problem might be the lack of neuroregeneration to support functional recovery. Regenerating a large number of functional new neurons to replace the lost neurons might be the fundamental first step toward neural repair. The high neuroregeneration efficiency presented in this study may solve the long-term problem of how to replenish the lost neurons with sufficient number of functional new neurons. Although adult brains show plasticity and remapping in response to ischemic injury,60 the capacity of such compensation may not be sufficient to overcome the loss of massive number of neurons caused by injury. On the other hand, simply having a massive cluster of new neurons may not be sufficient to rebuild a highly organized neural circuit. The long-range axonal projections from NeuroD1-converted neurons reported here suggest that newly generated neurons might rely upon preexisting axon bundles to reach their target regions. We cannot exclude the possibility that newly converted neurons might send axons to the wrong targets, like that during early brain development. However, we predict that the wrong projections will be eliminated through experience-dependent plasticity if there are no adequate activities to support the connections. Our behavioral tests indicate that NeuroD1-converted neurons contribute toward functional recovery after ischemic stroke. This is consistent with recent reports on in vivo conversion of striatal astrocytes into dopaminergic neurons57 and Muller glia into retinal neurons in adult mice,61, 62 both of which achieved functional improvement as well. Our electrophysiological analysis and c-fos staining further support the notion that NeuroD1-converted neurons integrate into preexisting brain circuits and participate in brain functions. Of course, many more studies are needed to answer precisely how the newly converted neurons establish functional neural circuits with preexisting neurons after injury or disease.
Advantages of In Situ Cell Conversion
Current therapy for acute ischemia stroke is focusing on restoring blood flow through intravenous administration of tissue plasminogen activator (tPA) or mechanical thrombolysis within a narrow time window of 3–4.5 hours following stroke.63, 64 After restoring blood flow, physical rehabilitation of the affected area has been proven to be effective,65 but functional recovery is often limited. One of the most significant advantages of our in vivo cell conversion technology is that we can potentially extend the treatment time window from hours to days and weeks or even months after stroke. This study demonstrates that after 10 days of ischemic injury, reactive astrocytes in the adult mouse cortex can still be efficiently converted into functional neurons with significant tissue repair and behavioral improvement. In our previous work, we demonstrated that reactive astrocytes in 14-month-old 5xFAD mice can still be converted into functional new neurons by NeuroD1.25 Therefore, a broad treatment window might have a major impact on patients who are not able to access immediate treatment after stroke.
Besides a significant increase of treatment time window, our in vivo cell conversion technology also has advantages over the classical cell therapies for stroke treatment. For example, by making use of brain internal glial cells to generate new neurons, the in vivo cell conversion approach is more similar to modulating endogenous adult neurogenesis. However, different from the restricted NSC niches in the adult brain,1, 2, 3 reactive glial cells are widely distributed throughout the CNS. Wherever a neural injury occurs, the local glial cells neighboring to the lost neurons can be used to generate new neurons for replacement, as shown by different groups in different brain and spinal cord regions already.25, 26, 27, 28, 29, 30, 31, 32, 33, 34, 35 Such in situ cell conversion approach is an economical way to generate new neurons for neural repair, with an almost unlimited cell source for regeneration purpose.
Of course, transplantation of external stem cells can also be carried out throughout the CNS via intracranial or intraspinal delivery method, similar to our in vivo cell conversion technology. However, the majority of transplanted cells cannot survive very well in stroke areas and not many functional neurons can be detected after long-term engraftment.18, 24 While many preclinical studies have shown promising functional recovery following stem cell transplantation and some even enter clinical trials, one great challenge is to better understand the precise mechanisms before launching large-scale clinical trials.18, 24 In contrast, our in vivo cell conversion approach shows highly efficient neuroregeneration with long-term survival and functional integration, which may represent the next generation of cell therapy to treat neurological disorders.
Limitations of In Situ Cell Conversion
Like any new technology, in vivo cell conversion approach also has its own limitations and challenges. The precondition for in vivo cell conversion is the presence of reactive glial cells after neural injury. If injury is so severe that even glial cells are lost in a massive way, such as that occurs in middle cerebral artery occlusion (MCAO) model, it might require additional therapies to first reduce cell death in order to preserve some glial cells for conversion purpose. Therefore, in vivo cell conversion technology is complementary to all the acute treatments aiming at neuroprotection immediately after the stroke. The more neurons and glial cells are preserved, the more efficient the cell conversion approach will be. For severe injury that has already resulted in a big tissue loss, such as the empty hole in the mouse cortex at 10 weeks after ET-1 induced ischemic injury (Figure 1A), it may require cell transplantation to fill the hole first before starting a regeneration process. Another challenge is that if an ischemic injury involves multiple brain regions, such as the cortex, striatum, and hippocampus in the MCAO model, it may require different transcription factors to generate different subtypes of neurons in different brain regions for effective repair. Even after successful neuronal conversion, whether the newly generated neurons in different brain regions can form right connections to replace the previously lost connections is also a challenge. Recent studies reported encouraging results that transplanted embryonic neurons or brain organoids in the adult mouse cortex can form wide synaptic connections with their hosts,67, 68 suggesting that newborn neurons are capable of integrating into preexisting brain circuits. Finally, even if rodent animal models show successful neural repair after cell conversion, it is not a guarantee that it will translate into a successful clinical therapy. Non-human primate models may be necessary to further test critical parameters on in vivo cell conversion in order to gain insights into the dosage and time windows for future clinical applications.
In conclusion, our NeuroD1-based gene therapy opens a new path toward efficient in vivo neuroregeneration for brain repair. While many challenges lie ahead, this study provides the proof-of-principle that injured adult mammalian brains can be at least partially repaired through in vivo cell conversion approach.
Materials and Methods
Animals
Mouse
All experiments were performed in the AAALAC-accredited Huck Institute of Life Sciences at The Pennsylvania State University. Animal procedures were performed in accordance with the NIH’s Guide for the Care and Use of Laboratory Animals, and all experimental protocols were approved and overseen by The Pennsylvania State University’s Institutional Animal Care and Use Committee (IACUC). 5- to 12-month-old adult mice were used. Mice were housed in a 12 h light/dark cycle and supplied with sufficient food and water. Both male and female mice were used except in the behavioral experiments in which only male mice were used.
Rat
Male Sprague-Dawley rats (Harlan Laboratories) aging between 3 and 5 months and weighing 300–360 g at the time of the experiment were housed and handled as previously described.69 Rats were maintained on a restricted diet of 18 g per day of standard laboratory rat chow and trained to press a bar for food on a variable interval schedule of reinforcement (VI 60 s) for one week. All experimental procedures in rats were approved by the Institutional Animal Care and Use Committee of the University of Puerto Rico School of Medicine, in compliance with NIH’s Guide for the Care and Use of Laboratory Animals.
Mouse Model of Ischemic Injury and Virus Injection
Wild-type (WT) FVB/NJ and GFAP-GFP transgenic mice were employed for the majority of the experiments described in this study. ET-1 (1–31) was injected into the motor cortex of the adult WT FVB/NJ or GFAPA-GFP transgenic mice (28–40 g, 5–10 months old) to produce focal ischemic injury as previously described.38, 70 Briefly, under anesthesia by intraperitoneal injection of ketamine/xylazine (100 mg/kg ketamine; 12 mg/kg xylazine), mice were placed in a stereotaxic apparatus with the skull and bregma exposed by a midline incision. A small hole of ∼1 mm was drilled in the skull at the coordinates of the forelimb motor cortex (relative to the bregma): +0.2 mm anterior-posterior (AP), ± 1.5 mm medial-lateral. ET-1 (1–31) was dissolved at 2 μg/μL in 1× PBS (OSM ∼320, PH ∼7.3). A total volume of 0.5 μL (1 μg) was injected into each site starting from −1.6 mm dorsal-ventral (DV). The injection was performed by infusion pump throughout 10 min and the injection needle was withdrawn slowly at the speed of 0.1 mm/min. After injection, the needle was kept at 1.1 mm DV for additional 3 min before fully withdrawn. Viral injection followed the similar procedure, except that viral injection was typically performed around 10 days after ET-1 injection through the same hole drilled for ET-1 injection at same depth.
Rat Model of Ischemic Injury and Virus Injection
Rats were anaesthetized with isoflurane (5% for induction, 2.5% for maintenance) and positioned in a stereotaxic frame (Kopf Instruments). Prior to surgery, rats were randomly assigned to receive infusion of either saline or ET-1 into the BLA. A single guide cannula (26 gauge, 9 mm of length, Plastics One) was implanted bilaterally into the BLA (coordinates: anterior-posterior [AP], 2.6 mm from bregma; medial-lateral, 4.9 mm from midline; dorsal-ventral [DV], 8.7 mm from the skull surface). The cannula was fixed to the skull using ortho acrylic cement and four anchoring screws. Immediately following this, an injector extending 1 mm past the tip of the cannula was used to infuse 3 μL/side (400 pmol) of either saline or ET-1 at a rate of 0.15 μL/min. The injector was kept inside the cannula for an additional 10 min to reduce back-flow. The injector was then removed and a stainless-steel obturator (33 gauge) was inserted into the guide cannula to avoid obstruction after infusions were made. Rats were allowed to recover for 3 weeks after surgery before initiating the experiments. This interval was based on pilot studies performed by our group showing a significant impairment in fear acquisition combined with significant neuronal death approximately 21 days after the ET-1 stroke. The 21-day time point also helped to maximize scar glial formation following ET-1 lesion,47 an important factor to consider when using reprogramming strategies.
AAV Vector Construction
The hGFAP promoter was obtained from pDRIVE-hGFAP plasmid (InvivoGen) and inserted into pAAV-MCS (Cell Biolab) between MluI and SacII to replace the cytomegalovirus (CMV) promoter. The Cre gene was obtained by PCR from hGFAP-Cre (Addgene plasmid #40591, gift of Dr. Albee Messing) and inserted into pAAV MCS between EcoRI and Sal1 sites to generate pAAV-hGFAP::Cre vector. To construct pAAV-FLEX-mCherry-P2A-mCherry and pAAV-FLEX-NeuroD1-P2A-mCherry (or pAAV-FLEX-NeuroD1-P2A-GFP) vectors, we obtained the cDNAs coding NeuroD1, mCherry, or GFP by PCR using the retroviral constructs described previously.25 The NeuroD1 gene were fused with P2A-mCherry or P2A-GFP and subcloned into the pAAV-FLEX-GFP vector (Addgene plasmid #28304, gift of Dr. Edward Boyden) between Kpn1 and Xho1 sites. Plasmid constructs were sequenced for verification.
AAV Virus Production
Recombinant AAV9 was produced in 293AAV cells (Cell Biolabs). Briefly, polyethylenimine (PEI, linear, MW 25,000) was used for transfection of triple plasmids: the pAAV expression vector, pAAV9-RC (Cell Biolab) and pHelper (Cell Biolab). 72 h post transfection, cells were scrapped in their medium and centrifuged, frozen, and thawed four times by placing it alternately in dry ice or ethanol and 37°C water bath. AAV crude lysate was purified by centrifugation at 54,000 rpm for 1 h in discontinuous iodixanol gradients with a Beckman SW55Ti rotor. The virus-containing layer was extracted, and viruses were concentrated by Millipore Amicon Ultra Centrifugal Filters. Virus titers were 1.2 × 1012 GC/mL for hGFAP::Cre, hGFAP::NeuroD1-GFP and hGFAP::GFP, 1.4 × 1012 GC/mL for CAG::FLEX-NeuroD1-P2A-GFP and CAG::FLEX-NeuroD1-P2A-mCherry, and 1.6 × 1012 GC/mL for CAG::FLEX-mCherry-P2A-mCherry and CAG::FLEX-GFP-P2A-GFP, determined by QuickTiter AAV Quantitation Kit (Cell Biolabs).
Retrovirus Production
The pCAG-NeuroD1-IRES-GFP and pCAG-GFP were constructed as described previously.25 To package retroviral particles, we transfected gpg helper-free HEK cells with the target plasmid together with vesicular stomatitis virus G protein (VSV-G) vector to produce the retroviruses expressing NeuroD1 or GFP. The titer of retroviral particles was about 107 particles/mL, determined after transduction of HEK cells.
Statistics and Blind Analysis
Statistical analyses were performed using Prism 6 (GraphPad) software. All experiments shown were repeated in at least three animals, and representative data are shown. To determine the significance between groups, we made comparisons using paired two-tailed Student’s t test, or repeated-measurement ANOVA test as indicated. After primarily screening for severe ischemic injury before viral injection, animals were randomly assigned into groups with a matching deficit level. All the images for quantification were taken by one researcher and were quantified by a different researcher who was blind to the animal condition and identity. As mentioned above, mouse behavioral tests were done in a blind fashion. For rat fear conditioning test, freezing was automatically scored using a commercially available video tracking system (Any-Maze, Stoelting).
For additional materials and methods, please see the Supplemental Information.
The datasets generated for this study can be found in GEO: GSE135981.
Author Contributions
G.C. supervised the entire project. G.C. and Y.-C.C. designed most of the experiments, analyzed the data, and wrote the manuscript. Y.-C.C. carried out most of the experiments in mice, quantified the data, and made the figures. N.-X.M. participated in early experiments and performed RNA-seq analysis. Z.-F.P. made viral vectors and provided all the viruses for this study. Z.W. performed brain slice recordings. S.K., E.Y., M.S.C., J.-C.Y., G.L., Y.H., Y.-T.B., and K.L. all contributed to mouse studies including immunostaining and quantification, as well as some behavioral studies. G.J.Q. supervised the rat study on fear conditioning tests, and F.H.D.-M. and A.M.-T. performed the rat experiments and made the figure. G.J.Q. and F.H.D.-M. wrote the part on rat fear conditioning tests.
Conflicts of Interest
G.C. is a co-founder of NeuExcell Therapeutics.
Acknowledgments
This work was supported by grants from the NIH (AG045656) and Alzheimer's Association (ZEN-15-321972) to G.C. It was also supported by the Charles H. Smith Endowment Fund for Brain Repair and the Verne M. Willaman Endowment Fund from the Pennsylvania State University to G.C. The rat fear conditioning test was supported by NIMH grant K99-MH-105549 to F.H.D.-M., NIMH grants R37-MH058883 and P-50-MH-086400 to G.J.Q., and a grant from the University of Puerto Rico President’s Office to G.J.Q. We would like to thank Matthew Keefe and Joseph Gyekis for careful proof reading of our manuscript. We would also like to thank all the Chen lab members for rigorous discussing throughout this project over the past 5 years.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2019.09.003.
Supplemental Information
References
- 1.Ming G.L., Song H. Adult neurogenesis in the mammalian brain: significant answers and significant questions. Neuron. 2011;70:687–702. doi: 10.1016/j.neuron.2011.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Lim D.A., Alvarez-Buylla A. The Adult Ventricular-Subventricular Zone (V-SVZ) and Olfactory Bulb (OB) Neurogenesis. Cold Spring Harb. Perspect. Biol. 2016;8:a018820. doi: 10.1101/cshperspect.a018820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Gonçalves J.T., Schafer S.T., Gage F.H. Adult Neurogenesis in the Hippocampus: From Stem Cells to Behavior. Cell. 2016;167:897–914. doi: 10.1016/j.cell.2016.10.021. [DOI] [PubMed] [Google Scholar]
- 4.Tobin M.K., Bonds J.A., Minshall R.D., Pelligrino D.A., Testai F.D., Lazarov O. Neurogenesis and inflammation after ischemic stroke: what is known and where we go from here. J. Cereb. Blood Flow Metab. 2014;34:1573–1584. doi: 10.1038/jcbfm.2014.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Magnusson J.P., Göritz C., Tatarishvili J., Dias D.O., Smith E.M., Lindvall O., Kokaia Z., Frisén J. A latent neurogenic program in astrocytes regulated by Notch signaling in the mouse. Science. 2014;346:237–241. doi: 10.1126/science.346.6206.237. [DOI] [PubMed] [Google Scholar]
- 6.Sun X., Zhang Q.W., Xu M., Guo J.J., Shen S.W., Wang Y.Q., Sun F.Y. New striatal neurons form projections to substantia nigra in adult rat brain after stroke. Neurobiol. Dis. 2012;45:601–609. doi: 10.1016/j.nbd.2011.09.018. [DOI] [PubMed] [Google Scholar]
- 7.Arvidsson A., Collin T., Kirik D., Kokaia Z., Lindvall O. Neuronal replacement from endogenous precursors in the adult brain after stroke. Nat. Med. 2002;8:963–970. doi: 10.1038/nm747. [DOI] [PubMed] [Google Scholar]
- 8.Parent J.M., Vexler Z.S., Gong C., Derugin N., Ferriero D.M. Rat forebrain neurogenesis and striatal neuron replacement after focal stroke. Ann. Neurol. 2002;52:802–813. doi: 10.1002/ana.10393. [DOI] [PubMed] [Google Scholar]
- 9.Dempsey R.J., Sailor K.A., Bowen K.K., Türeyen K., Vemuganti R. Stroke-induced progenitor cell proliferation in adult spontaneously hypertensive rat brain: effect of exogenous IGF-1 and GDNF. J. Neurochem. 2003;87:586–597. doi: 10.1046/j.1471-4159.2003.02022.x. [DOI] [PubMed] [Google Scholar]
- 10.Keiner S., Witte O.W., Redecker C. Immunocytochemical detection of newly generated neurons in the perilesional area of cortical infarcts after intraventricular application of brain-derived neurotrophic factor. J. Neuropathol. Exp. Neurol. 2009;68:83–93. doi: 10.1097/NEN.0b013e31819308e9. [DOI] [PubMed] [Google Scholar]
- 11.Kobayashi T., Ahlenius H., Thored P., Kobayashi R., Kokaia Z., Lindvall O. Intracerebral infusion of glial cell line-derived neurotrophic factor promotes striatal neurogenesis after stroke in adult rats. Stroke. 2006;37:2361–2367. doi: 10.1161/01.STR.0000236025.44089.e1. [DOI] [PubMed] [Google Scholar]
- 12.Zhang S.C., Wernig M., Duncan I.D., Brüstle O., Thomson J.A. In vitro differentiation of transplantable neural precursors from human embryonic stem cells. Nat. Biotechnol. 2001;19:1129–1133. doi: 10.1038/nbt1201-1129. [DOI] [PubMed] [Google Scholar]
- 13.Reubinoff B.E., Itsykson P., Turetsky T., Pera M.F., Reinhartz E., Itzik A., Ben-Hur T. Neural progenitors from human embryonic stem cells. Nat. Biotechnol. 2001;19:1134–1140. doi: 10.1038/nbt1201-1134. [DOI] [PubMed] [Google Scholar]
- 14.Bliss T.M., Andres R.H., Steinberg G.K. Optimizing the success of cell transplantation therapy for stroke. Neurobiol. Dis. 2010;37:275–283. doi: 10.1016/j.nbd.2009.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kelly S., Bliss T.M., Shah A.K., Sun G.H., Ma M., Foo W.C., Masel J., Yenari M.A., Weissman I.L., Uchida N. Transplanted human fetal neural stem cells survive, migrate, and differentiate in ischemic rat cerebral cortex. Proc. Natl. Acad. Sci. USA. 2004;101:11839–11844. doi: 10.1073/pnas.0404474101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Jin K., Xie L., Mao X., Greenberg M.B., Moore A., Peng B., Greenberg R.B., Greenberg D.A. Effect of human neural precursor cell transplantation on endogenous neurogenesis after focal cerebral ischemia in the rat. Brain Res. 2011;1374:56–62. doi: 10.1016/j.brainres.2010.12.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mine Y., Tatarishvili J., Oki K., Monni E., Kokaia Z., Lindvall O. Grafted human neural stem cells enhance several steps of endogenous neurogenesis and improve behavioral recovery after middle cerebral artery occlusion in rats. Neurobiol. Dis. 2013;52:191–203. doi: 10.1016/j.nbd.2012.12.006. [DOI] [PubMed] [Google Scholar]
- 18.George P.M., Steinberg G.K. Novel Stroke Therapeutics: Unraveling Stroke Pathophysiology and Its Impact on Clinical Treatments. Neuron. 2015;87:297–309. doi: 10.1016/j.neuron.2015.05.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Faiz M., Sachewsky N., Gascón S., Bang K.W., Morshead C.M., Nagy A. Adult Neural Stem Cells from the Subventricular Zone Give Rise to Reactive Astrocytes in the Cortex after Stroke. Cell Stem Cell. 2015;17:624–634. doi: 10.1016/j.stem.2015.08.002. [DOI] [PubMed] [Google Scholar]
- 20.Benner E.J., Luciano D., Jo R., Abdi K., Paez-Gonzalez P., Sheng H., Warner D.S., Liu C., Eroglu C., Kuo C.T. Protective astrogenesis from the SVZ niche after injury is controlled by Notch modulator Thbs4. Nature. 2013;497:369–373. doi: 10.1038/nature12069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lindvall O., Kokaia Z. Stem cells for the treatment of neurological disorders. Nature. 2006;441:1094–1096. doi: 10.1038/nature04960. [DOI] [PubMed] [Google Scholar]
- 22.Bliss T., Guzman R., Daadi M., Steinberg G.K. Cell transplantation therapy for stroke. Stroke. 2007;38(2, Suppl):817–826. doi: 10.1161/01.STR.0000247888.25985.62. [DOI] [PubMed] [Google Scholar]
- 23.Jandial R., Snyder E.Y. A safer stem cell: on guard against cancer. Nat. Med. 2009;15:999–1001. doi: 10.1038/nm0909-999. [DOI] [PubMed] [Google Scholar]
- 24.Goldman S.A. Stem and Progenitor Cell-Based Therapy of the Central Nervous System: Hopes, Hype, and Wishful Thinking. Cell Stem Cell. 2016;18:174–188. doi: 10.1016/j.stem.2016.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Guo Z., Zhang L., Wu Z., Chen Y., Wang F., Chen G. In vivo direct reprogramming of reactive glial cells into functional neurons after brain injury and in an Alzheimer’s disease model. Cell Stem Cell. 2014;14:188–202. doi: 10.1016/j.stem.2013.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Grande A., Sumiyoshi K., López-Juárez A., Howard J., Sakthivel B., Aronow B., Campbell K., Nakafuku M. Environmental impact on direct neuronal reprogramming in vivo in the adult brain. Nat. Commun. 2013;4:2373. doi: 10.1038/ncomms3373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Niu W., Zang T., Zou Y., Fang S., Smith D.K., Bachoo R., Zhang C.L. In vivo reprogramming of astrocytes to neuroblasts in the adult brain. Nat. Cell Biol. 2013;15:1164–1175. doi: 10.1038/ncb2843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Torper O., Pfisterer U., Wolf D.A., Pereira M., Lau S., Jakobsson J., Björklund A., Grealish S., Parmar M. Generation of induced neurons via direct conversion in vivo. Proc. Natl. Acad. Sci. USA. 2013;110:7038–7043. doi: 10.1073/pnas.1303829110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Su Z., Niu W., Liu M.L., Zou Y., Zhang C.L. In vivo conversion of astrocytes to neurons in the injured adult spinal cord. Nat. Commun. 2014;5:3338. doi: 10.1038/ncomms4338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu Y., Miao Q., Yuan J., Han S., Zhang P., Li S., Rao Z., Zhao W., Ye Q., Geng J. Ascl1 Converts Dorsal Midbrain Astrocytes into Functional Neurons In Vivo. J. Neurosci. 2015;35:9336–9355. doi: 10.1523/JNEUROSCI.3975-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Heinrich C., Blum R., Gascón S., Masserdotti G., Tripathi P., Sánchez R., Tiedt S., Schroeder T., Götz M., Berninger B. Directing astroglia from the cerebral cortex into subtype specific functional neurons. PLoS Biol. 2010;8:e1000373. doi: 10.1371/journal.pbio.1000373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Heinrich C., Bergami M., Gascón S., Lepier A., Viganò F., Dimou L., Sutor B., Berninger B., Götz M. Sox2-mediated conversion of NG2 glia into induced neurons in the injured adult cerebral cortex. Stem Cell Reports. 2014;3:1000–1014. doi: 10.1016/j.stemcr.2014.10.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gascón S., Murenu E., Masserdotti G., Ortega F., Russo G.L., Petrik D., Deshpande A., Heinrich C., Karow M., Robertson S.P. Identification and Successful Negotiation of a Metabolic Checkpoint in Direct Neuronal Reprogramming. Cell Stem Cell. 2016;18:396–409. doi: 10.1016/j.stem.2015.12.003. [DOI] [PubMed] [Google Scholar]
- 34.Torper O., Ottosson D.R., Pereira M., Lau S., Cardoso T., Grealish S., Parmar M. In Vivo Reprogramming of Striatal NG2 Glia into Functional Neurons that Integrate into Local Host Circuitry. Cell Rep. 2015;12:474–481. doi: 10.1016/j.celrep.2015.06.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li H., Chen G. In Vivo Reprogramming for CNS Repair: Regenerating Neurons from Endogenous Glial Cells. Neuron. 2016;91:728–738. doi: 10.1016/j.neuron.2016.08.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Brulet R., Matsuda T., Zhang L., Miranda C., Giacca M., Kaspar B.K., Nakashima K., Hsieh J. NEUROD1 Instructs Neuronal Conversion in Non-Reactive Astrocytes. Stem Cell Reports. 2017;8:1506–1515. doi: 10.1016/j.stemcr.2017.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Windle V., Szymanska A., Granter-Button S., White C., Buist R., Peeling J., Corbett D. An analysis of four different methods of producing focal cerebral ischemia with endothelin-1 in the rat. Exp. Neurol. 2006;201:324–334. doi: 10.1016/j.expneurol.2006.04.012. [DOI] [PubMed] [Google Scholar]
- 38.Roome R.B., Bartlett R.F., Jeffers M., Xiong J., Corbett D., Vanderluit J.L. A reproducible Endothelin-1 model of forelimb motor cortex stroke in the mouse. J. Neurosci. Methods. 2014;233:34–44. doi: 10.1016/j.jneumeth.2014.05.014. [DOI] [PubMed] [Google Scholar]
- 39.Hughes P.M., Anthony D.C., Ruddin M., Botham M.S., Rankine E.L., Sablone M., Baumann D., Mir A.K., Perry V.H. Focal lesions in the rat central nervous system induced by endothelin-1. J. Neuropathol. Exp. Neurol. 2003;62:1276–1286. doi: 10.1093/jnen/62.12.1276. [DOI] [PubMed] [Google Scholar]
- 40.Fuxe K., Bjelke B., Andbjer B., Grahn H., Rimondini R., Agnati L.F. Endothelin-1 induced lesions of the frontoparietal cortex of the rat. A possible model of focal cortical ischemia. Neuroreport. 1997;8:2623–2629. doi: 10.1097/00001756-199707280-00040. [DOI] [PubMed] [Google Scholar]
- 41.Ojala D.S., Amara D.P., Schaffer D.V. Adeno-associated virus vectors and neurological gene therapy. Neuroscientist. 2015;21:84–98. doi: 10.1177/1073858414521870. [DOI] [PubMed] [Google Scholar]
- 42.Atasoy D., Aponte Y., Su H.H., Sternson S.M. A FLEX switch targets Channelrhodopsin-2 to multiple cell types for imaging and long-range circuit mapping. J. Neurosci. 2008;28:7025–7030. doi: 10.1523/JNEUROSCI.1954-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Johansen J.P., Cain C.K., Ostroff L.E., LeDoux J.E. Molecular mechanisms of fear learning and memory. Cell. 2011;147:509–524. doi: 10.1016/j.cell.2011.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rogan M.T., Stäubli U.V., LeDoux J.E. Fear conditioning induces associative long-term potentiation in the amygdala. Nature. 1997;390:604–607. doi: 10.1038/37601. [DOI] [PubMed] [Google Scholar]
- 45.Goosens K.A., Maren S. Contextual and auditory fear conditioning are mediated by the lateral, basal, and central amygdaloid nuclei in rats. Learn. Mem. 2001;8:148–155. doi: 10.1101/lm.37601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gale G.D., Anagnostaras S.G., Godsil B.P., Mitchell S., Nozawa T., Sage J.R., Wiltgen B., Fanselow M.S. Role of the basolateral amygdala in the storage of fear memories across the adult lifetime of rats. J. Neurosci. 2004;24:3810–3815. doi: 10.1523/JNEUROSCI.4100-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Abeysinghe H.C., Bokhari L., Dusting G.J., Roulston C.L. Brain remodelling following endothelin-1 induced stroke in conscious rats. PLoS ONE. 2014;9:e97007. doi: 10.1371/journal.pone.0097007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Turnley A.M., Basrai H.S., Christie K.J. Is integration and survival of newborn neurons the bottleneck for effective neural repair by endogenous neural precursor cells? Front. Neurosci. 2014;8:29. doi: 10.3389/fnins.2014.00029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rajasethupathy P., Ferenczi E., Deisseroth K. Targeting Neural Circuits. Cell. 2016;165:524–534. doi: 10.1016/j.cell.2016.03.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chan K.Y., Jang M.J., Yoo B.B., Greenbaum A., Ravi N., Wu W.L., Sánchez-Guardado L., Lois C., Mazmanian S.K., Deverman B.E., Gradinaru V. Engineered AAVs for efficient noninvasive gene delivery to the central and peripheral nervous systems. Nat. Neurosci. 2017;20:1172–1179. doi: 10.1038/nn.4593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Frühbeis C., Fröhlich D., Kuo W.P., Krämer-Albers E.M. Extracellular vesicles as mediators of neuron-glia communication. Front. Cell. Neurosci. 2013;7:182. doi: 10.3389/fncel.2013.00182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hol E.M., Roelofs R.F., Moraal E., Sonnemans M.A., Sluijs J.A., Proper E.A., de Graan P.N., Fischer D.F., van Leeuwen F.W. Neuronal expression of GFAP in patients with Alzheimer pathology and identification of novel GFAP splice forms. Mol. Psychiatry. 2003;8:786–796. doi: 10.1038/sj.mp.4001379. [DOI] [PubMed] [Google Scholar]
- 53.Su M., Hu H., Lee Y., d’Azzo A., Messing A., Brenner M. Expression specificity of GFAP transgenes. Neurochem. Res. 2004;29:2075–2093. doi: 10.1007/s11064-004-6881-1. [DOI] [PubMed] [Google Scholar]
- 54.Kuwabara T., Hsieh J., Muotri A., Yeo G., Warashina M., Lie D.C., Moore L., Nakashima K., Asashima M., Gage F.H. Wnt-mediated activation of NeuroD1 and retro-elements during adult neurogenesis. Nat. Neurosci. 2009;12:1097–1105. doi: 10.1038/nn.2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gao Z., Ure K., Ables J.L., Lagace D.C., Nave K.A., Goebbels S., Eisch A.J., Hsieh J. Neurod1 is essential for the survival and maturation of adult-born neurons. Nat. Neurosci. 2009;12:1090–1092. doi: 10.1038/nn.2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pang Z.P., Yang N., Vierbuchen T., Ostermeier A., Fuentes D.R., Yang T.Q., Citri A., Sebastiano V., Marro S., Südhof T.C., Wernig M. Induction of human neuronal cells by defined transcription factors. Nature. 2011;476:220–223. doi: 10.1038/nature10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Rivetti di Val Cervo P., Romanov R.A., Spigolon G., Masini D., Martín-Montañez E., Toledo E.M., La Manno G., Feyder M., Pifl C., Ng Y.H. Induction of functional dopamine neurons from human astrocytes in vitro and mouse astrocytes in a Parkinson’s disease model. Nat. Biotechnol. 2017;35:444–452. doi: 10.1038/nbt.3835. [DOI] [PubMed] [Google Scholar]
- 58.Turner R.C., Dodson S.C., Rosen C.L., Huber J.D. The science of cerebral ischemia and the quest for neuroprotection: navigating past failure to future success. J. Neurosurg. 2013;118:1072–1085. doi: 10.3171/2012.11.JNS12408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Anderson R.M., Hadjichrysanthou C., Evans S., Wong M.M. Why do so many clinical trials of therapies for Alzheimer’s disease fail? Lancet. 2017;390:2327–2329. doi: 10.1016/S0140-6736(17)32399-1. [DOI] [PubMed] [Google Scholar]
- 60.Winship I.R., Murphy T.H. Remapping the somatosensory cortex after stroke: insight from imaging the synapse to network. Neuroscientist. 2009;15:507–524. doi: 10.1177/1073858409333076. [DOI] [PubMed] [Google Scholar]
- 61.Jorstad N.L., Wilken M.S., Grimes W.N., Wohl S.G., VandenBosch L.S., Yoshimatsu T., Wong R.O., Rieke F., Reh T.A. Stimulation of functional neuronal regeneration from Müller glia in adult mice. Nature. 2017;548:103–107. doi: 10.1038/nature23283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Yao K., Qiu S., Wang Y.V., Park S.J.H., Mohns E.J., Mehta B., Liu X., Chang B., Zenisek D., Crair M.C. Restoration of vision after de novo genesis of rod photoreceptors in mammalian retinas. Nature. 2018;560:484–488. doi: 10.1038/s41586-018-0425-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Hacke W., Kaste M., Bluhmki E., Brozman M., Dávalos A., Guidetti D., Larrue V., Lees K.R., Medeghri Z., Machnig T., ECASS Investigators Thrombolysis with alteplase 3 to 4.5 hours after acute ischemic stroke. N. Engl. J. Med. 2008;359:1317–1329. doi: 10.1056/NEJMoa0804656. [DOI] [PubMed] [Google Scholar]
- 64.National Institute of Neurological Disorders and Stroke rt-PA Stroke Study Group Tissue plasminogen activator for acute ischemic stroke. N. Engl. J. Med. 1995;333:1581–1587. doi: 10.1056/NEJM199512143332401. [DOI] [PubMed] [Google Scholar]
- 65.Veerbeek J.M., van Wegen E., van Peppen R., van der Wees P.J., Hendriks E., Rietberg M., Kwakkel G. What is the evidence for physical therapy poststroke? A systematic review and meta-analysis. PLoS ONE. 2014;9:e87987. doi: 10.1371/journal.pone.0087987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Falkner S., Grade S., Dimou L., Conzelmann K.K., Bonhoeffer T., Götz M., Hübener M. Transplanted embryonic neurons integrate into adult neocortical circuits. Nature. 2016;539:248–253. doi: 10.1038/nature20113. [DOI] [PubMed] [Google Scholar]
- 68.Mansour A.A., Gonçalves J.T., Bloyd C.W., Li H., Fernandes S., Quang D., Johnston S., Parylak S.L., Jin X., Gage F.H. An in vivo model of functional and vascularized human brain organoids. Nat. Biotechnol. 2018;36:432–441. doi: 10.1038/nbt.4127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Quirk G.J., Russo G.K., Barron J.L., Lebron K. The role of ventromedial prefrontal cortex in the recovery of extinguished fear. J. Neurosci. 2000;20:6225–6231. doi: 10.1523/JNEUROSCI.20-16-06225.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Horie N., Maag A.L., Hamilton S.A., Shichinohe H., Bliss T.M., Steinberg G.K. Mouse model of focal cerebral ischemia using endothelin-1. J. Neurosci. Methods. 2008;173:286–290. doi: 10.1016/j.jneumeth.2008.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.