

Abstract
The taccalonolides are a class of microtubule stabilizers that circumvent clinically relevant forms of drug resistance due to their unique mechanism of microtubule stabilization imparted by the covalent binding of the C-22−–-23 epoxide moiety to tubulin. A taccalonolide (8) with a fluorescein group attached with a linker at C-6 was generated and biochemical and cell-based assays showed that it bound directly to tubulin and stabilized microtubules. This pharmacological probe has allowed, for the first time, a direct visualization of a taccalonolide binding to microtubules, verifying their cellular binding site. This C-6-modified taccalonolide showed potency comparable to the untagged compound in biochemical experiments; however, its potency was lower in cellular assays, presumably due to decreased cellular permeability. These studies provide a valuable tool to facilitate the further understanding of taccalonolide pharmacology and demonstrate that C-6 is a promising site for a linker to be added to this novel class of microtubule stabilizers for targeted drug delivery.
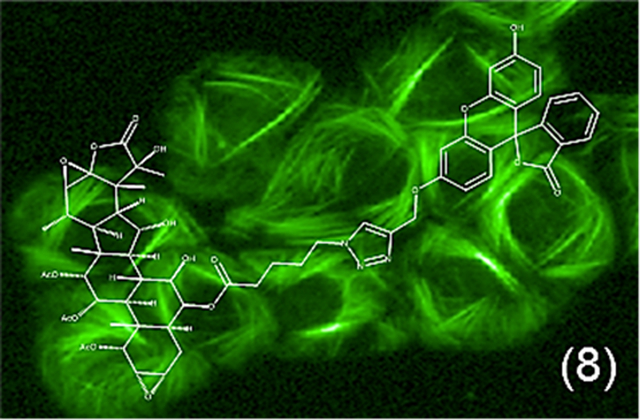
Graphical Abstract
Microtubule stabilizers, such as the taxanes paclitaxel and docetaxel, are some of the most effective drugs used for the treatment of breast cancers. Additionally, taxanes have been approved to treat prostate, pancreatic, non-small-cell lung, and head and neck cancers.1 Although these drugs are effective for many patients, acquired and inherent drug resistance limits their efficacy.2,3 The taccalonolides are a class of microtubule stabilizers that circumvent clinically relevant forms of drug resistance in vitro and in vivo, including drug efflux by P-glycoprotein or MRP7, βIII-tubulin expression, and mutations in the taxane binding site.2,3 The taccalonolides are isolated from the roots and rhizomes of plants of the Tacca species and taccalonolide A is the most abundant natural metabolite. Taccalonolide A is efficiently converted by semi-synthesis to the potent taccalonolides AF and AJ, which contain the C-22–C-23 epoxide that is critical for tubulin binding.4–6 The taccalonolides cause interphase microtubule bundling, which leads to disruption of cellular signaling and trafficking, as well as the formation of multipolar spindles, which lead to mitotic arrest.4,7,8 The ability of the taccalonolides to circumvent multiple forms of drug resistance is likely due to their covalent binding to microtubules.6,9 This covalent binding however, limits biochemical and pharmacokinetic analyses since the taccalonolides cannot be dissociated from tubulin and detected after binding. A functionally tagged taccalonolide (e.g., taccalonolide linked to a fluorescent probe) would allow for more detailed evaluations of the uptake, target binding, and distribution of these compounds in vitro and in vivo.
In vivo antitumor studies demonstrate that taccalonolide AF has a narrow therapeutic window when administered systemically.6 The in vivo activity of taccalonolide AF is hindered by its hydrolysis to taccalonolide AJ, a compound that has no therapeutic window when administered systemically. However, the taccalonolides AF and AJ have potent and highly persistent antitumor efficacy when directly administered to the tumor, suggesting that they could be excellent candidates for targeted drug delivery.10 Antibody-drug conjugates (ADCs) are an emerging class of cancer therapeutics that combine the selectivity of targeted therapy with the cytotoxic efficacy of chemotherapeutics.11,12 Intriguingly, the most effective ADCs currently on the market for the treatment of cancer employ microtubule disrupting agents as their warhead cargo.13 One advantage of taccalonolides AF and AJ, as compared to the other classes of microtubule targeting drugs, is their ability to promote persistent and durable antitumor effects when directly administered to the tumor as a consequence of their covalent binding.10 The targeted delivery of a taccalonolide is a promising strategy for the treatment of taxane-resistant cancers. This strategy would facilitate effective tumor delivery of the drug and be expected to increase the therapeutic window to mitigate toxicity.12,13
Linker optimization has facilitated the production and development of safer and more effective ADCs, which indicates that the linker itself has an important role in the safety and efficacy of ADCs.11 One limitation in creating an ADC includes identification of a potential site for the attachment of a linker that doesn’t compromise the mechanism of action of the chemotherapeutic. For consideration as an ADC, one would first need to identify a site where a linker could be added to the taccalonolide skeleton, without compromising their microtubule binding and stabilizing activities. In the current study, we evaluated the biochemical and cellular effects of a new semi-synthetic derivative of taccalonolide AJ with an ester-linked fluorescein tag at the C-6 position. This tagged taccalonolide maintained microtubule binding and stabilizing activities that allowed it to serve as a functional taccalonolide probe. Additionally, these findings demonstrate that C-6 is a promising site on the taccalonolide backbone to conjugate a linker for the generation of antibody-drug conjugates for targeted therapeutic approaches.14
RESULTS AND DISCUSSION
Chemistry.
Taccalonolides modified at the C-7 or C-15 positions were found to be susceptible to hydrolysis in aqueous solutions and modification at C-25 resulted in the loss of biological activity.15 During the course of performing those reactions, serendipity led to the identification of reaction products that included modification of the C-6 position. The potential for targeting the semisynthetic modification of C-6 with reasonable regioselectivity led to the hypothesis that it could serve as a novel site for linker installation. A targeted reaction scheme was therefore employed to generate a C-6-taccalonolide-fluorescein probe (Scheme 1). The reduction of taccalonolide B (1) by NaNH3CN resulted in the stereospecific formation of the 6S-OH derivative 2, which was ideally suited for esterification to yield 3 containing an azide linker. The fluorescein (4) derivatives 5 and 6 with alkyne linkers were synthesized as previously described.16 Compounds 3 and 6 were covalently bonded via “click” chemistry17 to generate 7 followed by C-22–C-23 epoxidation to produce 8. The structures of 2, 3, 7, and 8 were confirmed through analysis of their 1D and 2D NMR data together with LC-MS and HRESIMS analysis (Tables 1 – 3, Supporting Information).
Scheme 1.

Synthesis of Compound 8
Table 1.
1H and 13C NMR Data of Compounds 2 and 3 (Methanol-d4)
| No. | 2 | 3 | ||
|---|---|---|---|---|
| 13C | 1H (J) | 13C | 1H (J) | |
| 1 | 75.7 | 4.58, d (5.5) | 75.4 | 4.65, d (5.5) |
| 2 | 51.8 | 3.39, dd (5.5, 4.0) | 51.5 | 3.39, dd (5.5, 3.7) |
| 3 | 54.5 | 3.36, m | 53.9 | 3.31, m |
| 4 | 26.9 | 2.36, m 1.83, m |
26.7 | 1.89, m |
| 5 | 34.7 | 1.71, m | 33.8 | 2.00, m |
| 6 | 73.7 | 3.63, t (2.9) | 76.1 | 5.21, t (2.9) |
| 7 | 75.9 | 3.35, m | 74.2 | 3.60, dd (2.9, 10.0) |
| 8 | 35.7 | 2.12, m | 36.5 | 2.07, m |
| 9 | 42.0 | 2.19, t (11.3) | 42.0 | 2.30, t (11.1) |
| 10 | 38.5 | 38.4 | ||
| 11 | 72.7 | 5.32, dd (11.3, 2.8) | 72.5 | 5.36, dd (11.1, 2.8) |
| 12 | 75.4 | 5.23, d (2.8) | 75.2 | 5.25, d (2.8) |
| 13 | 45.5 | 45.6 | ||
| 14 | 57.8 | 1.97, dd (8.5, 11.0) | 57.6 | 1.99, m |
| 15 | 72.6 | 4.01, dd (8.5, 10.5) | 72.4 | 4.37, dd (7.9, 10.5) |
| 16 | 51.2 | 2.50, dd (10.5, 13.5) | 51.3 | 2.50, m |
| 17 | 49.3 | 1.83, m | 49.2 | 1.83, m |
| 18 | 13.4 | 1.00, s | 13.5 | 1.02, s |
| 19 | 14.5 | 1.03, s | 14.3 | 1.03, s |
| 20 | 20.6 | 0.89, d (7.0) | 20.6 | 0.90, d (7.1) |
| 21 | 32.2 | 2.23, m | 32.2 | 2.23, m |
| 22 | 112.1 | 5.01, d (1.6) | 112.1 | 5.02, d (1.6) |
| 23 | 156.0 | 155.9 | ||
| 24 | 52.0 | 52.0 | ||
| 25 | 80.3 | 80.3 | ||
| 26 | 177.3 | 177.3 | ||
| 27 | 22.2 | 1.63, s | 22.1 | 1.62, s |
| 28 | 25.4 | 1.30, s | 25.4 | 1.29, s |
| 29 | 174.8 | |||
| 30 | 34.8 | 2.52, m | ||
| 31 | 23.4 | 1.75, m | ||
| 32 | 29.4 | 1.68, m | ||
| 33 | 52.2 | 3.33, dd (6.8, 13.0) | ||
| OAc-1 | 171.8 | 171.6 | ||
| 20.7 | 2.09, s | 20.7 | 2.10, s | |
| OAc-11 | 172.3 | 172.3 | ||
| 21.0 | 2.07, s | 21.0 | 2.08, s | |
| OAc-12 | 171.5 | 171.5 | ||
| 21.5 | 1.89, s | 21.5 | 1.90, s | |
Table 3.
1H and 13C NMR Data of Compound 8 (Methanol-d4)
| No. | 8 | ||||
|---|---|---|---|---|---|
| 13C | 1H (J) | 13C | 1H (J) | ||
| 1 | 75.3 | 4.61, dd (5.5, 2.0) | 29 | 174.6 | |
| 2 | 51.5 | 3.35, m | 30 | 34.5 | 2.46, t (7.4) |
| 3 | 53.9 | 3.29, m | 31 | 22.9 | 1.62, m |
| 4 | 26.6 | 1.81, m | 32 | 30.5 | 1.98, m |
| 5 | 33.8 | 1.96, m | 33 | 51.0 | 4.46, t (6.9) |
| 6 | 76.1 | 5.16, t (2.5) | 34 | 125.4 | 8.08, s |
| 7 | 74.2 | 3.57, dd (2.5, 10.1) | 35 | 144.6 | |
| 8 | 36.5 | 1.97, m | 36 | 62.8 | 5.24, s |
| 9 | 42.0 | 2.26, t (10.8) | 37 | 161.6 | |
| 10 | 38.4 | 38 | 103.2 | 6.99, d (2.5) | |
| 11 | 72.5 | 5.28, d (10.8) | 39 | 154.0 | |
| 12 | 75.1 | 5.15, m | 40 | 113.3 | |
| 13 | 45.5 | 41 | 130.3 | 6.67, d (8.9) | |
| 14 | 57.1 | 1.94, m | 42 | 113.3 | 6.74, dd (2.5, 8.9) |
| 15 | 72.5 | 4.24, br t (10.0) | 43 | 154.0 | |
| 16 | 47.9 | 2.07, m | 44 | 103.6 | 6.69, d (2.3) |
| 17 | 45.6 | 2.07, m | 45 | 161.6 | |
| 18 | 13.2 | 0.86, d (2.4) | 46 | 113.8 | 6.53, dd (2.3, 8.8) |
| 19 | 14.4 | 0.93, s | 47 | 130.3 | 6.59, d (8.8) |
| 20 | 18.7 | 0.98, dd (2.1, 7.3) | 48 | 111.4 | |
| 21 | 33.8 | 1.53, m | 49 | nd | |
| 22 | 67.3 | 3.27, d (1.8) | 50 | 154.0 | |
| 23 | 92.8 | 51 | 125.4 | 7.20, d (7.7) | |
| 24 | 48.4 | 52 | 136.4 | 7.76, t (7.7) | |
| 25 | 80.1 | 53 | 131.1 | 7.69, t (7.7) | |
| 26 | 176.8 | 54 | 125.9 | 7.80, d (7.7) | |
| 27 | 25.2 | 1.72, s | 55 | 128.2 | |
| 28 | 20.1 | 1.24, d (2.5) | 56 | 171.5 | |
| OAc-1 | 171.6 | ||||
| 20.7 | 2.10, s | ||||
| OAc-11 | 172.2 | ||||
| 21.0 | 2.08, s | ||||
| OAc-12 | 171.5 | ||||
| 21.5 | 1.88, s | ||||
Cellular Effects of the C-6-Fluorescein Taccalonolide.
The IC50 value of 8 was determined to be 2.5 ± 0.1 μM. This represented a 600-fold decrease in cellular potency as compared to taccalonolide AJ, which is the C-22–C-23 epoxidated form of 1.4 Fluorescein was utilized as a control to demonstrate that the probe itself had no activity in the cells. We next evaluated the ability of 8 to stabilize cellular microtubules by performing β-tubulin immunofluorescence using HCC1937 breast cancer cells treated with vehicle or 8 for 24 h. An increase in stabilized, bundled microtubules was observed in cells treated with 5 μM 8 as compared to vehicle controls (Figures 1A and 1B). This demonstrates that 8 was able to enter and stabilize microtubules in intact cells. The fluorescent probe on 8 allowed us, for the first time, to visualize the colocalization of a taccalonolide with the microtubule bundles it induces (Figures 1B and 1C). Importantly, the colocalization of the fluorescein-tagged taccalonolide with microtubules 24 h after addition to the media demonstrated that a taccalonolide linked to a probe through C-6 was stable in aqueous solution for extended time periods. Since 8 induced microtubule bundling and co-localized with β-tubulin by immunofluorescence, we next sought to detect this fluorescent taccalonolide by live cell fluorescence imaging. Within 6 h of treating HCC1937 breast cancer cells with 5 μM 8, we observed bright intracellular staining that was strikingly reminiscent of stabilized microtubules (Figure 2). This finding further demonstrated the ability of this fluorescently tagged taccalonolide to retain microtubule stabilizing activity and supported our hypothesis that microtubules are the primary binding site of the taccalonolides in cells.
Figure 1.

Fluorescein-tagged taccalonolide (8) co-localizes with microtubule bundles. HCC1937 cells were treated with vehicle (A) or 5 μM 8 (B, C) for 24 h. Microtubules were visualized by β-tubulin immunofluorescence (orange) and 8 was visualized directly (green).
Figure 2.

Localization of fluorescence in live HCC1937 cells 6 h after treatment with vehicle or 5 μM 8 (green). Brightfield images are shown for reference.
C-6-Fluorescein Labeled Taccalonolide Directly Binds to and Stabilizes Purified Tubulin.
The direct effects of 8 on tubulin polymerization were evaluated turbidometrically using purified porcine brain tubulin at a final concentration of 20 μM. The concentration-dependent increase in the rate and extent of tubulin polymerization in the presence of 5 – 10 μM taccalonolide AJ is consistent with previously published results.6 At equivalent concentrations, 8 increased tubulin polymerization with similar kinetics and to a similar extent as observed for taccalonolide AJ (Figure 3). Thus, the fluorescein-tagged taccalonolide 8 retained the ability to directly bind and stabilize pure tubulin with comparable potency to the untagged taccalonolide. Based on these observations, we speculated that the relative loss in potency for 8 compared to the unlabeled compound in the cellular assays was due to decreased cellular permeability. Second-generation fluorescently tagged taccalonolides are currently being synthesized with the goal of increasing cellular permeability. The generation of a functional taccalonolide probe will facilitate biochemical, cell biological, and pharmacokinetic studies to foster our understanding of this novel class of microtubule stabilizers. Additionally, the generation of a functional C-6 taccalonolide probe provides a proof- of-principle for utilizing C-6 as a site for conjugation of the taccalonolides for targeted drug delivery.
Figure 3.

Purified tubulin polymerization was measured turbidometrically at a final concentration of 20 μM purified tubulin with 5–10 μM taccalonolides AJ or 8 as compared to vehicle control.
EXPERIMENTAL SECTION
General Experimental Procedures.
1D and 2D NMR spectra were acquired using a 500 or 600 MHz Bruker AVANCE spectrometer (Billerica, MA, USA) using a cryoprobe and CDCl3 as solvent. Mass spectra were measured with an LCMS composed of a Waters Alliance mass spectrometer (Malford, MA, USA) equipped with a 2695 HPLC module, 996 photodiode array detector as well as a Micromass Quattro triple quadrupole mass spectrometer under positive mode ESI conditions. High-resolution electrospray ionization mass spectrometry (HRESIMS) was performed on an Agilent Technologies (Santa Clara, CA, USA) 6224 TOFLC/MS mass spectrometer. TLC was carried out on pre-coated silica gel plates (silica gel 60 F-254, Merck KGaA, Darmstadt, Germany). Spots were visualized by spraying with 20% sulfuric acid in ethanol followed by heating. Preparative HPLC was performed on a Waters 1528 binary pump and 2487 diode array detector system using a Phenomenex (Torrance, CA, USA) Kinetex 5 μm C18 250 × 21 mm and a Phenomenex Luna 5 μm C18 250 mm × 21 mm columns with flow rate of 8 mL/min. Semi-preparative HPLC separations were performed on a Waters 1525 system using a 2998 PDA detector and Gemini 5 μm C18 column (110 Å, 250 mm × 10.0 mm) with a flow rate of 4 mL/min.
Reduction of Taccalonolide B (1).
Taccalonolide B (1, 1 eq., 28 mg) was mixed with sodium cyanoborohydride (10 eq., 27 mg) in ethanol (8 mL) containing 5% acetic acid. The mixture was stirred overnight at room temperature, and the resultant product was concentrated under a vacuum. The residue was purified by semipreparative HPLC using a Phenomenex Gemini 5 μm C18 250 mm × 10 mm column eluted with 50% CH3CN/H2O (with 0.1% formic acid) to afford compound 2 (23 mg, 82% yield). Compound 2: white powder; 1H and 13C NMR data, see Table 1; HRESIMS m/z 685.2833 [M + Na]+ (calcd for C33H44O13Na, 685.2831).
Esterification of Compound 2.
5-Azidopentanoic acid (100 μL) was stirred vigorously in 500 μL oxalyl chloride at room temperature for 1 h. The excess oxalyl chloride was removed in vacuo to afford pure 5-azidopentanoic chloride (85 mg). To a dichloromethane solution (2 mL) of compound 2 (1 eq., 23 mg), 5-azidopentanoyl chloride (1 eq., 5.5 μL), triethylamine (100 μL), and 4-DMAP (catalytic amount) were added. The mixture was stirred overnight at room temperature, and the resultant product was concentrated under a vacuum. The residue was resuspended and stirred in 2 mL chloroform at room temperature for 48 h. The solvent was removed in vacuo and the product purified by semipreparative HPLC using a Phenomenex Gemini 5 μm C18 250 mm × 10 mm column eluted with 65% CH3CN/H2O to afford compound 3 (7.2 mg, 26% yield). Compound 3: white powder; 1H and 13C NMR data, see Table 1; HRESIMS m/z 810.3421 [M + Na]+ (calcd for C39H53N3O14Na, 810.3420).
Addition of Fluorescein.
Fluorescein (4, 2 g, 1 eq.) was suspended in absolute DMF (9 mL) before K2CO3 (2.3 g, 2.8 eq.) and propargyl bromide (80% in toluene, 1.82 mL, 4 eq.) were added to the solution at room temperature. The reaction mixture was stirred at 60 °C for 2 h. Afterwards, the reaction mixture was diluted with H2O (220 mL) and the precipitate was collected to yield compound 5 (1.3 g, 52% yield). Compound 5: orange powder; 1H NMR (400 MHz, CDCl3) δ 8.27 (1H, d, J = 7.9 Hz), 7.76 (1H, t, J = 7.9 Hz), 7.68 (1H, t, J = 7.9 Hz), 7.32 (1H, d, J = 7.9 Hz), 7.06 (1H, d, J = 2.3 Hz), 6.90 (1H, d, J = 9.7 Hz), 6.85 (1H, d, J = 9.7 Hz), 6.80 (1H, dd, J = 9.7, 2.5 Hz), 6.54 (1H, dd, J = 9.7, 2.5 Hz), 6.45 (1H, d, J = 2.3 Hz), 4.79 (2H, d, J = 2.2 Hz), 4.62 (1H, dd, J = 2.4, 15.6 Hz), 4.56 (1H, dd, J = 2.4, 15.6 Hz), 2.61 (1H, t, J = 2.2 Hz), 2.32 (1H, t, J = 2.4 Hz); 13C NMR (100 MHz, CDCl3) δ 185.9, 164.6, 161.9, 159.1, 154.2, 149.7, 134.7, 133.2, 131.5, 130.7, 130.4, 130.2, 129.9, 129.8, 129.1, 118.3, 115.6, 113.9, 105.9, 101.7, 77.3, 77.0, 76.5, 75.5, 56.5, 52.9. Compound 5 (150 mg, 1 eq.) was dissolved in THF (6.5 mL) and LiOH∙H2O (79 mg, 5 eq.) in H2O (2 mL) was added at room temperature. After 2 h, the reaction mixture was diluted with H2O (10 mL) and acidified with 2 M HCl (pH 2). The aqueous phase was extracted with EtOAc (3 × 10 mL) and the combined organic phases were evaporated to dryness. The residue was purified by semipreparative HPLC using a Phenomenex Gemini 5 μm C18 250 mm × 10 mm column eluted with 50% CH3CN/H2O (with 0.1% formic acid) to afford compound 6 (100 mg, 74% yield). Compound 6: orange powder; 1H NMR (400 MHz, CDCl3) δ 8.01 (1H, d, J = 7.6 Hz), 7.67 (1H, t, J = 7.6 Hz), 7.62 (1H, t, J = 7.6 Hz), 7.16 (1H, d, J = 7.6 Hz), 6.85 (1H, d, J = 1.9 Hz), 6.69 (1H, d, J = 8.6 Hz), 6.68 (1H, s), 6.66 (1H, d, J = 1.9, 8.6 Hz), 6.57 (1H, d, J = 8.6 Hz), 6.52 (1H, d, J = 8.6 Hz), 4.71 (2H, d, J = 2.2 Hz), 2.56 (1H, t, J = 2.2 Hz); 13C NMR (100 MHz, CDCl3) δ 170.3, 159.3, 158.2, 153.2, 152.6, 152.5, 135.3, 129.9, 129.4, 129.3, 126.8, 125.2, 124.2, 112.6, 112.2, 112.1, 111.1, 103.3, 102.2, 84.2, 78.0, 76.3, 56.2.
Reaction of 3 and 6.
Compounds 3 (7 mg, 1 eq.) and 6 (7 mg, 2 eq.) were dissolved in a mixture of 1 mL t-BuOH and 1 mL H2O followed by the addition of CuSO4·5H2O (2 mg, 1 eq.) and sodium ascorbate (3.5 mg, 2 eq.). The mixture was stirred overnight at room temperature and then evaporated to dryness. The residue was purified by semipreparative HPLC using a Phenomenex Gemini 5 μm C18 250 mm × 10 mm column eluted with 65% CH3CN/H2O to afford compound 7 (9.8 mg, 95% yield). Compound 7: orange powder; 1H and 13C NMR data, see Table 2; HRESIMS m/z 1180.4268 [M + Na]+ (calcd for C62H67N3O19Na, 1180.4261).
Table 2.
1H and 13C NMR Data of Compound 7 (Methanol-d4)
| No. | 7 | ||||
|---|---|---|---|---|---|
| 13C | 1H (J) | 13C | 1H (J) | ||
| 1 | 75.3 | 4.62, dd (5.6, 2.1) | 29 | 174.6 | |
| 2 | 51.5 | 3.35, m | 30 | 34.6 | 2.47, m |
| 3 | 53.9 | 3.29, m | 31 | 23.0 | 1.63, m |
| 4 | 26.6 | 1.81, m | 32 | 30.5 | 2.00, m |
| 5 | 33.8 | 1.96, m | 33 | 51.0 | 4.46, t (6.9) |
| 6 | 76.1 | 5.17, t (2.5) | 34 | 125.4 | 8.08, s |
| 7 | 74.1 | 3.57, dd (2.5, 10.1) | 35 | 144.6 | |
| 8 | 36.5 | 2.00, m | 36 | 62.8 | 5.23, s |
| 9 | 42.0 | 2.27, t (10.8) | 37 | 161.5 | |
| 10 | 38.3 | 38 | 103.2 | 6.97, d (2.5) | |
| 11 | 72.4 | 5.32, d (10.8) | 39 | 154.0 | |
| 12 | 75.2 | 5.23, m | 40 | 113.3 | |
| 13 | 45.5 | 41 | 130.2 | 6.67, d (8.9) | |
| 14 | 57.7 | 1.95, m | 42 | 113.2 | 6.73, dd (2.5, 8.9) |
| 15 | 72.6 | 4.33, ddd (2.4, 7.8, 10.4) | 43 | 154.0 | |
| 16 | 51.3 | 2.50, m | 44 | 103.6 | 6.69, d (2.3) |
| 17 | 49.2 | 1.82, m | 45 | 161.5 | |
| 18 | 13.6 | 0.98, s | 46 | 113.8 | 6.53, dd (2.3, 8.8) |
| 19 | 14.4 | 0.95, d (2.9) | 47 | 130.2 | 6.57, d (8.8) |
| 20 | 20.6 | 0.87, dd (2.1, 7.1) | 48 | 111.2 | |
| 21 | 32.2 | 2.18, m | 49 | nd | |
| 22 | 112.1 | 5.00, dd (1.6, 4.5) | 50 | 154.0 | |
| 23 | 155.9 | 51 | 125.4 | 7.19, d (7.7) | |
| 24 | 51.9 | 52 | 136.6 | 7.76, t (7.7) | |
| 25 | 80.3 | 53 | 131.2 | 7.69, t (7.7) | |
| 26 | 177.3 | 54 | 125.9 | 7.99, d (7.7) | |
| 27 | 22.1 | 1.61, s | 55 | 128.2 | |
| 28 | 25.4 | 1.27, d (4.2) | 56 | 171.4 | |
| OAc-1 | 171.6 | ||||
| 20.7 | 2.09, s | ||||
| OAc-11 | 172.3 | ||||
| 21.0 | 2.07, s | ||||
| OAc-12 | 171.5 | ||||
| 21.5 | 1.88, d (1.8) | ||||
Epoxidation of 7.
The epoxidation reagent dimethyldioxirane (DMDO) was prepared as an acetone solution using a method that was previously described.4 Compound 7 (8 mg) was dissolved in 0.5 mL and prechilled to −20 °C prior to the addition of 0.5 mL DMDO-acetone solution. The mixture was incubated at −20 °C for 2 h and the solvent removed under a N2 stream. The product was purified by semipreparative HPLC using a Phenomenex Gemini 5 μm C18 250 mm × 10 mm column eluted with 60% CH3CN/H2O to afford compound 8 (7.7 mg, 95% yield). Compound 8: orange powder; 1H and 13C NMR data, see Table 3; HRESIMS m/z 1172.4237 [M - H]¯ (calcd for C62H66N3O20, 1172.4245).
Statement of Purity.
The purity of all compounds was judged by analytical HPLC analysis using the percentage of the integrated signals at UV 205 nm. Different isocratic or gradient solvent systems comprised of CH3OH/H2O or CH3CN/H2O were used to generate the best resolution for each compound. All compounds submitted for bioassay were at least 95% pure as judged by this method.
Biological Assays.
HCC1937 (ATCC CRL-2336™) triple-negative breast cancer cells and HeLa (ATCC CCL-2™) cervical cancer cells were used in this study. Cell lines were validated by STR profiling (Genetica). HCC1937 cells were cultured in RPMI 1640 (Corning) with 10% FBS (Cellgro) and 50 μg/mL gentamicin (Gibco). HeLa cells were grown in BME (Gibco) with 10% FBS, 1X GlutaMax™ Supplement (Gibco), and 50 μg/mL gentamicin.
Antiproliferative Assays.
The antiproliferative and cytotoxic effects of compounds were evaluated using the sulforhodamine B (SRB) assay, as previously described.18,19 Approximately 2,000 HeLa cells were plated per well of a 96-well plate. Cells were treated in triplicate with each concentration of compound or EtOH vehicle control for 48 h in a final volume of 200 μL. Concentration-response curves were generated by non-linear regression analysis using Prism software 7.04 (GraphPad) and the IC50 value of each compound was calculated as the concentration that caused a 50% inhibition of cellular proliferation after the 48 h of drug incubation by comparison to vehicle-treated cells. Three independent experiments were conducted and the values presented as the average ± SEM.
High-Content Live Cell Fluorescence Imaging and Immunofluorescence.
HCC1937 cells were plated in PerkinElmer cell carrier imaging 96-well plates at a density of 8,000 cells/well. Cells were treated with vehicle control or compounds at the indicated final concentration in triplicate for each experiment. For live cell imaging of 8 and its respective controls, cells were washed with phenol red free medium 6 h after treatment and then imaged on the Operetta high-content imager running Harmony software (PerkinElmer). For co-localization experiments, cells were fixed after 24 h of compound treatment and subjected to immunofluorescence for β-tubulin (Sigma T-4026, Invitrogen T-862), while the fluorescein-tagged taccalonolide 8 was directly detected.
Tubulin Polymerization Assay.
Tubulin polymerization assays were performed using purified porcine brain tubulin and buffers from Cytoskeleton, Inc. In individual wells of a 96-well plate, 1 μL of each 100x drug stock was incubated with 20 μM porcine brain tubulin in GPEM glycerol buffer (1 mM GTP, 10% glycerol, 80 mM PIPES pH 6.9, 2 mM MgCl2 and 0.5 mM EGTA) in a final volume of 100 μL. Porcine tubulin was prepared on ice to inhibit tubulin polymerization until the assay was initiated while the plate reader was pre-warmed to 37 °C. Tubulin polymerization was measured every minute for an hour by light scattering at 340 nm in a Spectramax plate reader using Softmax software (Molecular Devices). Light scattering was normalized to the initial measurement for each well. Individual experiments were graphed and the experiments replicated at least two times.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported by R01CA121138 from the National Cancer Institute to S.L.M. Support of the UTHSCSA NMR facility and Cancer Center Support grant (CA054174) and CIDD are greatly acknowledged. Dedicated to Dr. Rachel Mata, National Autonomous University of Mexico, Mexico City, Mexico, and Dr. Barbara N. Timmerman, University of Kansas, for their pioneering work on bioactive natural products.
Footnotes
Supporting Information
The Supporting Information containing 1H and 13C NMR data for compounds 2, 3, 5, 6, 7, and 8 is available free of charge on the ACS Publications website.
The authors declare the following competing financial interest(s): A.R.B.O., A.L.R., L.D., R.H.C., and S.L.M. are listed as authors on a pending patent application on the taccalonolides.
REFERENCES
- (1).Ojima I; Lichtenthal B; Lee S; Wang C; Wang X Expert Opin. Ther. Pat 2016, 26, 1–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Tinley TL; Randall-Hlubek DA; Leal RM; Jackson EM; Cessac JW; Quada JC Jr., Hemscheidt TK; Mooberry SL Cancer Res. 2003, 63, 3211–3220. [PubMed] [Google Scholar]
- (3).Risinger AL; Jackson EM; Polin LA; Helms GL; LeBoeuf DA; Joe PA; Hopper-Borge E; Luduena RF; Kruh GD; Mooberry SL Cancer Res. 2008, 68, 8881–8888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Li J; Risinger AL; Peng J; Chen Z; Hu L; Mooberry SL J. Am. Chem. Soc 2011, 133, 19064–19067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Peng J; Risinger AL; Li J; Mooberry SL J. Med. Chem 2014, 57, 6141–6149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Risinger AL; Li J; Bennett MJ; Rohena CC; Peng J; Schriemer DC; Mooberry SL Cancer Res. 2013, 73, 6780–6792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Risinger AL; Riffle SM; Lopus M; Jordan MA; Wilson L; Mooberry SL Mol. Cancer 2014, 13, 41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Rohena CC; Peng J; Johnson TA; Crews P; Mooberry SL Biochem Pharmacol. 2013, 85, 1104–1114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Wang Y; Yu Y; Li GB; Li SA; Wu C; Gigant B; Qin W; Chen H; Wu Y; Chen Q; Yang J Nat. Commun 2017, 8, 15787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Risinger AL; Li J; Du L; Benavides R; Robles AJ; Cichewicz RH; Kuhn JG; Mooberry SL J. Nat. Prod 2017, 80, 409–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Diamantis N; Banerji U Br. J. Cancer 2016, 114, 362–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Tsuchikama K; An Z Protein Cell 2018, 9, 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Klute K; Nackos E; Tasaki S; Nguyen DP; Bander NH; Tagawa ST Onco Targets Ther. 2014, 7, 2227–2236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Beck A; Goetsch L; Dumontet C; Corvaia N Nat. Rev. Drug Discov 2017, 16, 315–337. [DOI] [PubMed] [Google Scholar]
- (15).Ola ARB; Risinger AL; Du L; Zammiello CL; Peng J; Cichewicz RH; Mooberry SL J. Nat. Prod 2018, 81, 579–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Nahrwold M; Weiss C; Bogner T; Mertink F; Conradi J; Sammet B; Palmisano R; Royo Gracia S; Preusse T; Sewald NJ Med. Chem 2013, 56, 1853–1864. [DOI] [PubMed] [Google Scholar]
- (17).Derbre S; Roue G; Poupon E; Susin SA; Hocquemiller R Chembiochem. 2005, 6, 979–982. [DOI] [PubMed] [Google Scholar]
- (18).Tinley TL; Leal RM; Randall-Hlubek DA; Cessac JW; Wilkens LR; Rao PN; Mooberry SL Cancer Res. 2003, 63, 1538–1549. [PubMed] [Google Scholar]
- (19).Skehan P; Storeng R; Scudiero D; Monks A; McMahon J; Vistica D; Warren JT; Bokesch H; Kenney S; Boyd MR J Natl. Cancer Inst 1990, 82, 1107–1112. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.