

SUMMARY
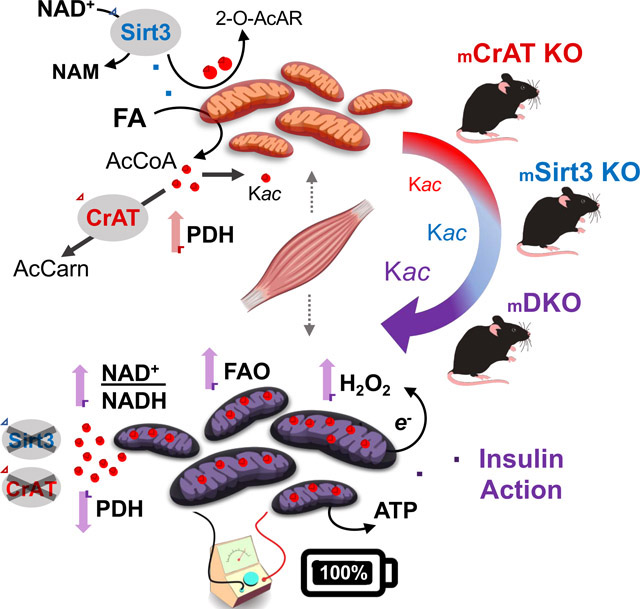
This study sought to examine the functional significance of mitochondrial protein acetylation using a double knockout (DKO) mouse model harboring muscle-specific deficits in acetyl CoA buffering and lysine deacetylation, due to genetic ablation of carnitine acetyltransferase and Sirtuin 3, respectively. DKO mice are highly susceptible to extreme hyperacetylation of the mitochondrial proteome and develop a more severe form of diet-induced insulin resistance than either single KO mouse line. However, the functional phenotype of hyperacetylated DKO mitochondria is largely normal. Of the >120 measures of respiratory function assayed, the most consistently observed traits of a markedly heightened acetyl-lysine landscape are enhanced oxygen flux in the context of fatty acid fuel and elevated rates of electron leak. In sum, the findings challenge the notion that lysine acetylation causes broad-ranging damage to mitochondrial quality and performance, and raise the possibility that acetyl-lysine turnover, rather than acetyl-lysine stoichiometry, modulates redox balance and carbon flux.
Graphical Abstract
INTRODUCTION
Acetyl-CoA holds a central position in intermediary metabolism as the two-carbon end product of glucose, fatty acid and amino acid catabolism. In addition to fueling the TCA cycle, acetyl CoA has gained increasing recognition as a substrate for protein acetylation, a posttranslational modification (PTM) that can occur on the epsilon amino group of lysine residues as well as the N-terminal end of a polypeptide (Glozak et al., 2005; Weinert et al., 2014; Weinert et al., 2013). Lysine acetylation is found prominently on mitochondrial proteins (Kim et al., 2006), which are presumed to be especially vulnerable to non-enzymatic acylation due to the high acyl CoA content and slightly basic pH of the matrix (Davies et al., 2016; Paik et al., 1970; Poburko et al., 2011; Wagner and Payne, 2013). Numerous studies have found that the detectable mitochondrial lysine acetylome increases in the context of obesity, due in part to nutrient-induced expansion of the mitochondrial acetyl CoA pool (Davies et al., 2016; Hirschey et al., 2011; Romanick et al., 2018; Zamora-Mendoza et al., 2018). Relative abundance of these PTMs in tissues such as skeletal muscle, heart and liver has been shown to correlate with perturbations in oxidative metabolism and glucose homeostasis (Davies et al., 2016; Hirschey et al., 2011; Jing et al., 2011; Lantier et al., 2018; Lantier et al., 2015). These observations have fueled widespread speculation that lysine acetylation serves as a key mechanism by which carbon surplus disrupts protein function and mitochondrial quality, thereby compromising metabolic and respiratory reserve and increasing organ susceptibility to energetic stress (Baeza et al., 2016). The best evidence to support this theory comes from studies in mice lacking Sirtuin 3 (SIRT3), an NAD+-dependent enzyme that acts as the major deacetylase in the mitochondrial matrix. Whereas mice with deficiency of SIRT3 have modest phenotypes under basal conditions (Fernandez-Marcos et al., 2012; Hebert et al., 2013; Lantier et al., 2015; Lombard et al., 2007; Peterson et al., 2018), they tend to show increased susceptibility to a variety of metabolic insults, including diet-induced glucose intolerance (Hirschey et al., 2011) and muscle insulin resistance (Lantier et al., 2015), supporting a link between protein deacetylation and bioenergetic resilience (Elkhwanky and Hakkola, 2018; McDonnell et al., 2015; van de Ven et al., 2017). Nonetheless, direct evidence demonstrating that protein acetylation per se actually contributes to mitochondrial and/or metabolic dysfunction in animal models of disease remains surprisingly sparse. Moreover, questions pertaining to the functional relevance of mitochondrial lysine acetylation have emerged from studies showing that most of these PTMs occur at occupancy rates of less than 1% (Nakayasu et al., 2014; Weinert et al., 2014, 2015).
The current study sought to test the hypothesis that wide-ranging hyperacetylation of the mitochondrial proteome disrupts mitochondrial bioenergetics and insulin action in skeletal muscle. To this end, we developed a mouse model with profound susceptibility to nutrient-induced hyperacetylation of mitochondrial proteins by coupling muscle-specific deficiency of SIRT3 with ablation of carnitine acetyltransferase (CrAT), an enzyme that buffers the mitochondrial acetyl group pool by interconverting acetyl CoA and acetylcarnitine (Beenakkers and Klingenberg, 1964). Deletion of CrAT promotes the “on” reaction (i.e. acetylation) (Davies et al., 2016), whereas Sirt3 deficiency blocks the “off” reaction (deacetylation). Therefore, concomitant ablation of both enzymes was predicted to act additively or perhaps synergistically, resulting in a uniquely robust expansion of the acetylproteome in muscle tissues. The double-knockout (DKO) model was combined with a recently developed platform for comprehensive mitochondrial diagnostics (Fisher-Wellman et al., 2018) to query the impact of protein hyperacetylation on a broad range of respiratory and energy fluxes. We found that DKO mice did in fact develop a more severe form of diet-induced insulin resistance, which corresponded with elevated rates of mitochondrial hydrogen peroxide (H2O2) production. Notably however, the overall respiratory and bioenergetic phenotypes of hyperacetylated mitochondria were surprisingly negative, contrary to the idea that acetyl PTMs disrupt mitochondrial quality and respiratory performance. The findings offer unexpected insights into the roles of SIRT3 and lysine acetylation in regulating metabolic function, and led us to propose an alternative model that positions SIRT3-mediated acetyl-lysine turnover, rather than acetyl-lysine stoichiometry, as a mechanism that modulates muscle fuel selection to defend whole body glucose control.
RESULTS
Combined deficiency of CrAT and Sirt3 leads to dramatic increases in the mitochondrial acetylproteome.
The goal of this study was to examine changes in muscle energy metabolism and respiratory function in response to graded increases in the acetylation state of the mitochondrial proteome. To this end, we generated mice with muscle-specific deletion of CrAT (CrATM−/−), Sirt3 (Sirt3M−/−), or both CrAT and Sirt3 (double-knockout, DKO) (Figure 1A) and fed the animals either a standard chow (SC) or a 45% high fat (HF) diet (~12 wks unless otherwise noted). Western blot analysis confirmed tissue-specific deletion of CrAT and Sirt3 in both heart and skeletal muscles of DKO mice as compared to CrATfl/fl x Sirt3fl/fl double-floxed controls (DFC) (Figure 1A), whereas mRNA expression of CrAT and Sirt3 was unaffected in the liver and white adipose tissue (Figure S1A and S1B). Preliminary assessments of total acetylation by western blot analysis (Figure 1B) revealed a progressive increase in lysine acetylation (Kac) that ranged from a modest 2.5-fold increase in semi-pure muscle mitochondria from DFC fed a HF diet, to a dramatic 45-fold increase in DKO mitochondria from mice fed the HF diet (relative to SC-fed DFC mice). Remarkably, the effect of ablating both CrAT and SIRT3 on mitochondrial Kac levels was amplified ~10-fold by feeding DKO mice a HF diet.
Figure 1. Combined deficiency of CrAT and Sirt3 leads to dramatic increases in the mitochondrial acetylproteome.

(A) Mouse models of CrAT and/or Sirt3 deficiency. Tissue-specific knockout of protein abundance in skeletal muscle and heart, with no effect on brown adipose tissue (BAT). Memcode staining was used to visualize and quantify protein loading. (B) Acetyl-lysine (Kac) western blot performed using lysates prepared from skeletal muscle mitochondria. (C) Quantification of Kac abundance normalized to VDAC expression. (D–F) Volcano plot depicting relative occupancy (Log2 fold change on x-axis) vs. statistical significance (−log p value on y-axis) of acetyl-peptides identified in quadriceps tissue from: (D) CrATfl/fl and CrATM−/− mice, (E) Sirt3fl/fl and Sirt3M−/− mice, and (F) Double Knockout (DKO) CrAT/Sirt3M−/− and double floxed controls (DFC) CrATfl/fl/Sirt3fl/fl. Colored and grey dots indicate peptides matched to mitochondrial or non-mitochondrial proteins, respectively. (G) Correlation of relative occupancy of specific acetyl-peptides in DKO/DFC compared to Sirt3M−/−/Sirt3fl/fl. (H–J) Histograms comparing the distribution of relative occupancy of mitochondrial and non-mitochondrial acetyl-peptides identified in muscle tissues from (H) CrATfl/fl versus CrATM−/− mice (I) Sirt3fl/fl versus Sirt3M−/− mice, and (J) DKO versus DFC mice. (K) Histogram comparing the distribution of relative changes in mitochondrial acetyl-peptide abundance measured in DKO/DFC versus Sirt3M−/−/Sirt3fl/fl. Data represent mean ±SEM. (A) N=2 per group. (B–C) N=4 per group. (C) data were analyzed by two-way ANOVA. *** represents a significant difference between DKO chow and DKO HF after Tukey post-hoc testing. (D–K) N=3–5 per group. The control (fl/fl) groups had N=5 for plex#1 and N=4 for plex#2. N=3 was used for the CrATM−/−, Sirt3M−/− and DKO groups. A scheme representing the acetylproteomics workflow is depicted in Supplemental Figure 1D and 1E. ***P≤0.001. N represents biological replicates.
To ascertain global changes in Kac as a result of the DKO manipulation as compared to either single gene deletion, isobaric tag-based quantitative nLC-MS/MS acetylproteomics was performed according to the workflow depicted in Figures S1C–E using lysates prepared from quadriceps muscles of mice fed the HF diet. The first acetylproteomic experiment (10-plex #1) directly comparing DKO (Figure S1D) and CrAT deficient muscles to their respective control groups quantified 1,280 acetylpeptides (from 1,508 identified at 1% FDR), with 560 mapping to 159 mitochondrial proteins (Calvo et al., 2016). A second experiment (10-plex #2) comparing the impact of DKO to SIRT3 deficiency (Figure S1E) quantified 845 acetylpeptides (1,283 identified), 486 mapping to 122 mitochondrial proteins. Each genetic deficiency resulted in a clear pattern of increased mitochondrial protein acetylation. The proportion of mitochondrial acetylpeptides with increased abundance based on fold-change alone was 95% (531/560), 76% (369/486) and 98% (478/486 in 10-plex #2) in CrATKO, SIRT3KO and DKO, respectively (Figure 1H–J); or 10% (55/560), 24% (117/486), and 91% (441/486 in 10-plex #2) after correction for multiple hypothesis testing using a 5% quantitative FDR (Benjamini and Hochberg, 1995; Kammers et al., 2015; Phipson et al., 2016; Ritchie et al., 2015). These results (Table S1) are consistent with previous acetylproteomics investigations of CrAT (Davies et al., 2016) and SIRT3 (Dittenhafer-Reed et al., 2015) deficiency in skeletal muscle.
When comparing the magnitude of Kac changes in the three models relative to their appropriate floxed controls, CrAT deletion led to a modest increase in mitochondrial Kac (Figures 1D and 1H), Sirt3 deletion resulted in an intermediate response (Figures 1E and 1I), and the DKO manipulation (Figures 1F and 1J) caused the most robust increase in abundance of a broad range of acetylated peptides belonging to mitochondrial proteins. Thus, as intended, we achieved a gradient of muscle Kac levels where in CrATM−/− <Sirt3M−/−<DKO. Notably, not only did the additional loss of CrAT increase overall mitochondrial Kac in DKO as compared Sirt3M−/− mice (Figures 1G, 1K and S1H), but the responses at the level of individual lysine residues were strongly correlated between the two models (Figure 1G). Of the 117 acetylpeptides with statistically significant (FDR<5%) increases as a result of SIRT3 ablation, all but two increased at this same threshold in the DKO muscles, albeit with an average fold-change three times that observed with SIRT3 deficiency alone. This finding supports the idea that some SIRT3-targeted lysine residues are distinctly vulnerable to non-enzymatic acetylation (Weinert et al., 2015), and therefore react with acetyl CoA in a reproducible, dose-dependent manner. Proteins exhibiting the greatest degree of hyperacetylation in muscles from DKO mice mapped to a broad range of mitochondrial pathways, including: beta-oxidation, the TCA cycle, oxidative phosphorylation and detoxification of reactive oxygen species (ROS) (Table S1). Levels of Kac were similar between DFC compared to Sirt3fl/fl mice (Figures S1F–G). Lastly, aside from the genetically targeted proteins, neither KO model produced appreciable changes in the muscle proteome, as assessed by nLC-MS/MS analysis of the unenriched or “input” fractions (Table S1).
Combined loss of CrAT and Sirt3 Exacerbates Diet-induced Defects in Glucose Homeostasis and Muscle Insulin Action.
We next sought to determine if changes in mitochondrial Kac were accompanied by proportional shifts in whole-body energy homeostasis, including glucose control, energy balance and body composition (Figures 2, S2 and S3). Consistent with previous reports, the whole-body metabolic phenotypes of all three models of hyperacetylation were largely unremarkable when animals were fed SC (Figures 2A–C, S2A–C, and S3A–C). Similar to previous findings (Muoio et al., 2012), CrATM−/− mice fed a HF diet exhibited marked intolerance to an oral glucose challenge (Figure S2C) concurrent with lower levels of plasma insulin (Figure S2D), suggesting loss of CrAT might exacerbate obesity-related pancreatic β-cell dysfunction. Despite robust remodeling of the mitochondrial acetylproteome in Sirt3M−/− mice, glucose tolerance and energy balance was comparable to their respective controls (Figures S3 and S4). Likewise, as compared to the DFC, DKO mice had no discernable metabolic phenotype when fed SC, and showed only a subtle genotype-specific sensitivity to the HF diet, as assessed by blood glucose levels and a modified HOMA-IR index, both of which were slightly elevated (Figures 2B and 2E).
Figure 2. Combined deficiency of CrAT and Sirt3 exacerbates diet-induced perturbations in glucose homeostasis and muscle insulin action.

(A) Body mass prior to oral glucose tolerance test. (B) 5 h fasting blood glucose. (C) Oral glucose tolerance. (D) Plasma insulin during the oral glucose tolerance test. (E) HOMA-IR calculated with values from the 15 min time point. (F) Arterial glucose levels during the insulin clamp (IC). (G) IC glucose infusion rate. (H) Plasma arterial insulin levels during the IC. (I) IC glucose turnover. (J) Muscle glucose uptake during the IC. (K) IC muscle hexokinase index. (L) Average glucose infusion rate (GIR) during the IC steady state for each genotype compared to littermate controls. Data represent mean ±SEM. (A–E) N=7–9 per group. (B) Data were analyzed by two-way ANOVA and * represents a significant difference between HF DFC and HF DKO after Tukey post-hoc testing. (F–K) N=10 per group. (H) Data were analyzed by two-way ANOVA. ** represents a main effect of insulin on arterial insulin levels. (L) N=7–13 per group, data shown in (C–G and I–L) were analyzed by Student’s t-test. *P≤0.05, **P≤0.01, ***P≤0.001. Outliers are represented as open grey circles. N represents biological replicates.
Considering that whole-body glucose tolerance can appear normal despite impaired insulin sensitivity (Best et al., 1996), we proceeded to assess insulin action in all three genetic models using the gold-standard hyperinsulinemic-euglycemic (insulin) clamp method, which was performed only in cohorts of mice fed the HF diet (Figures 2, S2, and S3). Arterial blood glucose values were clamped between 130–160 mg/dL (Figures 2F, S2F, and S3F) and insulin levels were significantly elevated during the insulin clamp (Figures 2H, S2H, and S3H). This test revealed a modest level of insulin resistance in CrATM−/− mice (Figures 2L, S2G), and no detectable perturbations in Sirt3M−/− mice (Figures 2L and S3G). Remarkably however, as compared to their DFC littermates, the DKO line exhibited a severe form of insulin resistance, evidenced by a dramatic reduction in the steady-state glucose infusion rate (GIR) (Figure 2G and 2L). Basal rates of endogenous glucose production (EndoRa) and glucose disappearance (Rd) were similar between DKO and DFC mice (Figure 2I). By contrast, the clamp condition revealed higher EndoRa and lower Rd in the DKO group, indicating defects in both hepatic and peripheral insulin action, respectively. The decreased Rd in DKO mice was accompanied by diminished glucose uptake (Rg) measured in gastrocnemius, TA and soleus muscles, along with a lower hexokinase (HK) index (2DGP/2DG) (Figure 2K), which together reflect a substantial level of muscle insulin resistance. In sum, when considering only the extreme ends of the acetylproteome spectrum (DFC versus DKO mice), the genetic and dietary condition that resulted in the highest degree of Kac also produced the most severe insulin resistance. However, when the analysis was expanded to include all genetic models (CrATM−/−, Sirt3M−/−, and DKO) and nutritional states, relative levels of mitochondrial Kac correlated poorly with alterations in whole-body glucose homeostasis and muscle insulin action.
Hyperacetylation of Mitochondrial Proteins is Accompanied by Increased Mitochondrial Fat Oxidation in Muscle Fiber Bundles.
To determine if hyperacetylation of the mitochondrial proteome leads to perturbations in mitochondrial respiratory function, we next measured rates of oxygen consumption in permeabilized fiber bundles (PFBs), which retain the acetylation profiles detected in whole tissues (Figure S6). Because our previous work had already characterized the impact of CrAT deficiency on muscle mitochondria (Muoio et al., 2012; Seiler et al., 2015)), the current study focused on comparisons between the Sirt3M−/− and DKO mouse lines, fed either a SC or HF diet. These experiments were performed using malate plus glutamate (G/M), octanoylcarnitine (Oc/M) or palmitoylcarnitine (Pc/M) as substrates, in the absence or presence of 1 mM ADP to stimulate maximal rates of ATP synthesis. Overall, the only evidence linking Kac to diminished respiratory capacity was a small reduction in maximal G/M-supported respiration observed in Sirt3M−/− PFBs after exposure to HF feeding (Figure 3B). Respiration fueled by octanoylcarnitine, a medium chain fatty acid substrate, was unaffected by genotype. By contrast, respiration supported by the long chain fatty acid (Pc/M) increased as a result of Sirt3 deficiency, but only when Sirt3KO mice were the HF diet (Figure 3F). Interestingly, the same outcome was apparent in DKO fiber bundles, regardless of the diet condition (Figures 3E and F). Notably, we found no evidence the CrAT deficiency alone modified respiratory responses when measured in PFBs prepared from mice consuming the HF diet (Figure S6).
Figure 3. Hyperacetylation of mitochondrial proteins is accompanied by increased oxidation of long-chain fatty acid fuel in permeabilized myofibers.

Mitochondrial oxygen consumption (JO2) was assayed in permeabilized fiber bundles from red gastrocnemius muscle from mice fed standard chow (A, C, and E) or a high fat diet (B, D, and F). Glutamate/Malate promotes electron flux through complex I, whereas succinate engages complex II. (G) Scheme depicting 13C-glucose labeling strategy and 13C metabolic flux analysis (MFA) in soleus muscles from DKO and DFC mice fed standard chow. Muscles were incubated with 10 mM [U-13C]glucose + 200 uM palmitate ±100 nM insulin. The citrate labeling pattern provides information on the relative contribution of pyruvate to the acetyl-CoA pool via PDH flux as compared to alternative routes of entry into the tricarboxylic acid cycle (TCAC). When [U-13C]pyruvate enters into the TCAC solely via PDH, the first condensation reaction produces M+2 citrate. By contrast, entry of [U-13C]pyruvate via malic enzyme (ME) or pyruvate carboxylase (PC) results in M+3 malate/oxaloacetate (OAA), which then forms M+3 or M+5 citrate upon condensation with unlabeled or M+2 acetyl-CoA, respectively. After the first spin of the TCAC, malate will be labeled M+2 from PDH flux or M+4 from combined PDH+ME/PC flux (see left text box*). Subsequent turns of the TCAC can produce more highly enriched citrate isotopomers (e.g. M+4, M+6). (H) Average percent 13C enrichment in TCAC metabolites after incubation with [U-13C]glucose without insulin. (I) Average percent 13C enrichment in TCAC metabolites after incubation with [U-13C]glucose plus 100 nM insulin. (J) Citrate mass isotopomer data (MID) from soleus muscles incubated with insulin. (K) The citrate M+5 to aspartate M+3 ratio provides insight into the enrichment of the acetyl-CoA pool because M+5 citrate is produced when M+3 OAA condenses with M+2 acetyl-CoA (see G). Aspartate M+3 was similar between genotypes and was used as a proxy for OAA M+3. Ratios were estimated by normalizing MID to the sum of the labeled isotopomers. Data represent mean ±SEM. (A–F) N=10–13 per group. (H–K) N=8 per group, and were analyzed by Student’s t-test. *P≤0.05, **P≤0.01, ***P≤0.001. N represents biological replicates.
To investigate whether the elevated capacity for Pc/M flux in DKO mitochondria might impact glucose oxidation in intact muscles, mass spectrometry-based metabolic flux analysis was performed using isolated soleus muscles incubated with [U-13C]glucose and unlabeled palmitate, in the presence or absence of insulin (Figure 3G). Muscles were harvested from DFC and DKO mice fed a SC diet to ascertain potential perturbations in glucose trafficking occurring prior to dietary challenge. These experiments revealed a subtle but significant tendency for DKO muscles to amass greater 13C labeling in fumarate and malate under basal conditions (Figures 3H and S6A–H), and more enrichment of succinate upon insulin stimulation (Figure 3I). Also noteworthy, insulin shifted the citrate isotopomer distribution such that the relative abundance of both M+1 and M+3 (# of heavy carbons) was greater in the DKO group, whereas the M+5 and M+6 species were less abundant than the DFC controls (Figure 3J). The ratio of citrate M+5 relative to aspartate M+3 provided an estimate of acetyl-CoA M+2 enrichment (Figure 3K). The lower ratio in DKO muscles is consistent with diminished flux through pyruvate dehydrogenase (PDH). In aggregate, DKO muscles exhibit a modest shift in pyruvate flux, away from PDH entry into the TCA cycle and towards alternative routes leading to synthesis of malate, fumarate and succinate, possibly via the actions of malic enzyme and/or pyruvate carboxylase (Figures 3G and I).
Comprehensive Mitochondrial Diagnostics Using the Creatine Kinase Energetic Clamp Technique.
Because the foregoing metabolic analyses revealed reciprocal shifts in fatty acid and pyruvate flux in DKO muscles, we next sought to gain a more comprehensive and less biased picture of mitochondrial bioenergetics in the context of physiologic and/or genetic perturbations that promote Kac. To this end, we transitioned from the PFB system to isolated mitochondria. Whereas PFBs provide the advantage of retaining the mitochondrial reticulum, technical limitations of this system prohibit measurement of key outcomes such as redox state and membrane potential. These measures were critical to our study because several dehydrogenase (DH) enzymes and proteins involved in the electron transport system (ETS) were among the most prominent targets of lysine acetylation. We therefore leveraged a recently developed mitochondrial diagnostics platform (Figure 4A) in which multiplexed assessment of energy transduction is conducted on freshly isolated mitochondria under a variety of distinct substrate and energetic conditions (Fisher-Wellman et al., 2018). The platform features a modified version of the creatine kinase (CK) energetic clamp technique wherein the extramitochondrial ratio of ATP:ADP, and thus the energy of ATP hydrolysis (ΔGATP), is precisely controlled by a large excess of CK and defined concentrations of creatine, phosphocreatine (PCr) and adenylates, which together confer unlimited capacitance to compensate for deficits in rates of mitochondrial ATP regeneration (Glancy et al., 2008; Messer et al., 2004). Figure 4C depicts the relationship between the primary CK clamp components (ATP, ADP, Cr, PCr) across a span of physiological ATP free energies. As mitochondria are exposed to more PCr, the ATP:ADP ratio increases, resulting in diminished respiratory flux. In simple terms, this technique serves as an in vitro “stress test” that evaluates mitochondrial respiratory responses to a graded energetic challenge. Transitions from a high to low ATP:ADP ratio mimic an increase in energy demand, akin to a transition between rest and exercise. Analysis of the linear relationship between energy demand (ATP:ADP, ΔGATP) and steady-state oxygen flux (JO2) allows for an estimation of respiratory “conductance” (i.e., reciprocal of resistance), such that a steeper slope indicates greater sensitivity and improved kinetics. Importantly, the energetic demands modeled by the CK clamp are comparable to those observed in vivo.
Figure 4. Comprehensive mitochondrial diagnostics using the CK energetic clamp.

(A) Mitochondria isolated from skeletal muscle were used intact for measuring rates of ATP synthesis (JATP); or permeabilized with alamethecin for assaying rates of NADH and NADPH (JNAD(P)H) generation by mitochondrial dehydrogenases in a 96-well format. Complex V activity was assessed in mitochondrial lysates. Rates of oxygen consumption (JO2) and respiratory sensitivity were evaluated using the Oroboros-O2K system paired with the creatine kinase (CK) energetic clamp technique and a buffer containing 20 U/mL CK, 5 mM creatine, and 1.5 – 30 mM phosphocreatine (PCr). Parallel measurements of membrane potential (ΔΨ), redox potential (NAD(P)H/NAD(P)+), and JH2O2 emissions were obtained via spectrofluorometric assays using a QuantaMaster Spectrofluorometer. (B) Mitochondrial energy transduction consists of three main regulatory nodes: 1) matrix dehydrogenases (DH), 2) electron transport system (ETS), and 3) ATP synthesis and transport. (C) Concentrations of Cr, PCr, ATP and ADP during the CK clamp, which is used to titrate the extra-mitochondrial ATP:ADP and permit assessment of respiratory control over a range of ATP free energy states, expressed as kcals/mol. (D) Respiratory conductance (slope of JO2 vs. ΔGATP) of skeletal muscle mitochondria measured in the presence of pyruvate/malate (Pyr/M) compared to succinate/rotenone (S/R). A steeper slope indicates greater sensitivity and improved kinetics. Pyr/M activates the pyruvate DH complex and promotes electron flux through complex I of the ETS. The combination of succinate and rotenone (S/R) promotes electron flux through complex II while inhibiting complex I, which decreases respiratory conductance due to diminished ΔΨ. (E) Respiratory efficiency plot (JO2 vs. ΔΨ). The leftward shift of the S/R plot shows that mitochondria are maintaining a less polarized ΔΨ for any given rate of oxygen consumption (JO2), indicative of diminished respiratory efficiency or a lower P:O ratio. (F) Representative trace of H2O2 emissions (JH2O2) from skeletal muscle mitochondria fueled by PyrM or S/R measured under state 4 conditions, followed by sequential additions of auranofin (AF; thioredoxin (TRX) reductase inhibitor), ATP and phosphocreatine (PCr) to execute the CK clamp, and then CDNB to deplete glutathione (GSH). Disruption of the TRX and GSH matrix scavenging systems permits an estimation of absolute H2O2 production rates.
Both the absolute rates of oxygen consumption and respiratory conductance (slope) depend on energy gradients and fluxes controlled by three principal regulatory nodes: 1) the DH enzymes, 2) the ETS and, 3) ATP synthesis and transport, which together mediate the transfer of energy from that available in carbon substrates to electron potential energy (ΔGredox) – to the proton motive force (PMF, ΔGH+) – to the free energy of ATP hydrolysis (ΔGATP) (Figure 4B). To gain insight into the free energies that drive the transduction process, the foregoing dynamic JO2 assays are combined with parallel assessments of membrane potential (ΔΨm, the primary contributor to the proton motive force, PMF), NAD(P)H/NAD(P)+ redox state and ROS production. The example in Figure 4D depicts the respiratory phenotype of skeletal muscle mitochondria fueled by pyruvate/malate (Pyr/M) as compared to succinate/rotenone (S/R). Due to rotenone inhibition of complex I (CI) and heavy electron flux through complex II, the S/R condition is characterized by a comparatively lower conductance (shallow slope) and less polarized ΔΨm. The resulting leftward shift in the relationship between JO2 and ΔΨm (Figure 4E) indicates that mitochondria are maintaining a lower (less polarized) ΔΨm for any given rate of oxygen consumption, which could reflect a lower P:O ratio (moles of ATP produced per molecule of O2 consumed), although it is important to underscore that the CK clamp assay does not permit direct assessment of ATP turnover (Fisher-Wellman et al., 2018). In vivo, the energy harnessed in the ΔΨm determines the extent to which CV can displace the ATP:ADP ratio from equilibrium. Thus, the ability of mitochondria to maintain ΔΨm and the relationship between ΔΨm and JO2 provide key metrics of energetic stability and respiratory efficiency. Lastly, because H2O2 flux (JH2O2) changes as a function of total electron flux as well as energetic backpressure, rates of electron leak are measured in the context of the CK clamp to better mimic physiologically relevant thermodynamics. The electron leak assays also include compounds added at the beginning and end of the CK clamp to block matrix scavenging systems dependent on thioredoxin and glutathione, respectively, which thereby permits an estimation of maximal/unbuffered JH2O2 (Figure 4F).
A second arm of the assay platform serves to validate and/or further elucidate specific functional perturbations identified within each control node (i.e. DH enzymes and CV-ANT). This is accomplished through direct assessment of maximal ATP synthesis and transport rates (JATP) measured in intact mitochondria exposed to various substrates and saturating ADP concentrations, as well as a 96-well plate assay that evaluates carbon flux though multiple DH enzyme activities (JNAD(P)H) in alamethicin permeabilized mitochondria that retain organization of protein complexes. Finally, complex V activity is measured in mitochondrial lysates. Collectively, the entire suite of biochemical assays provides diagnostic information across wide-ranging pathways of the mitochondrial metabolic network. A more detailed explanation of the rationale and caveats of these assays are provided in (Fisher-Wellman et al., 2018).
DKO Mitochondria Do Not Exhibit Latent Defects in Bioenergetics.
Presuming that the foregoing metabolic phenotypes of DKO PFBs and isolated soleus muscles were attributable to a buildup of acetyl-PTMs throughout the mitochondrial proteome, we reasoned that a more comprehensive assessment of DKO mitochondria might reveal underlying perturbations in bioenergetics. To test this prediction, the diagnostics platform was first applied to DKO mitochondria from mice fed SC using Pc/M, Pyr/M or G/M as respiratory substrates. As described previously (Fisher-Wellman et al., 2018), Pc/M is an especially low flux substrate in skeletal muscle mitochondria. Interestingly, similar to results in the PFBs (Figures 3E and 3F), Pc/M-supported JO2 was greater in DKO as compared to DFC mitochondria (Figure S6I). Notably, despite Kac levels that increased by ~4-fold (on average) in the context of a SC diet (Figures 1B and 1C), the DKO manipulation did not produce detectable deficits in JO2 (Figure S6I), ΔΨm (Figure S6J), NAD(P)H/NAD(P)+ redox potential (Figure S6K), respiratory efficiency (Figure S6L), NAD-dependent enzyme fluxes (Figure S6M), maximal JATP-ANT (Figure S6N), or electron leak (Figures S6O and S6P). The only hint of compromised enzyme activity was observed in the PDH flux assay when L-carnitine was present (Figure S6M), an outcome that probably stems from CrAT deficiency (Muoio et al., 2012).
Chronic HF Feeding Leads to Fuel-specific Shifts in Respiratory Kinetics and Thermodynamics.
Before applying the diagnostics platform to DKO mitochondria exposed to HF feeding, we first sought to characterize the bioenergetics profile associated with diet-induced remodeling of the acetylproteome. Here, muscle mitochondria from DFC mice fed either the SC or HF diets were analyzed under three substrate conditions; Pc/M, Pyr/M and Pc/M+Pyr+L-carnitine, the latter of which was meant to represent a physiological mixture of glucose and fatty acid fuels. Mitochondrial purity was similar between the chow versus HF diet preparations (Figure S7). As anticipated, the HF diet led to a pronounced increase in Pc/M-supported JO2, evidenced by both the absolute flux rates as well as the respiratory conductance (slope) (Figure 5A). By contrast, respiratory conductance in the context of Pyr/M was modestly reduced by the HF diet, possibly due to lipid-induced downregulation of PDH activity (Pehleman et al., 2005). Parallel measurements of membrane potential (Figure 5B) and redox state (Figure 5C) revealed a diet-induced increase in the NAD(P)H percent reduction and a corresponding leftward shift in the respiratory efficiency plot (relationship between JO2 and ΔΨm) when mitochondria were fueled by a mixture of substrates (Figure 5D). This finding is consistent with adaptations that favor greater oxidative flux of fatty acids relative to pyruvate, leading to a lower P:O ratio at any given energy demand. Lastly, the ROS emission assays revealed a striking effect of overnutrition on the JH2O2 profile. In general, the HF diet promoted JH2O2 and/or electron leak (% JH2O2/JO2), regardless of the substrate provided (Figures 5E and 5F). This effect was most pronounced under low-demand (~resting) energetic conditions. In sum, our results show that relatively modest changes in the mitochondrial acetyl-landscape caused by HF feeding were accompanied by discernable shifts in mitochondrial bioenergetics and electron leak.
Figure 5. Chronic high fat feeding alters respiratory kinetics and thermodynamics.

Mitochondria were isolated from skeletal muscle of mice fed standard chow (SC) or a 20-week high fat (HF) diet. (A–C) Relationship between (A) JO2, (B) ΔΨ and (C) NAD(P)H/NAD(P)+ redox state versus Gibb’s Energy of ATP hydrolysis (ΔGATP) measured in mitochondria fueled by palmitoylcarnitine+malate (Pc/M), pyruvate+malate (Pyr/M), or Pyr/M+Pc+free carnitine (Carn). (D) Mitochondrial respiratory efficiency represented as JO2 plotted against ΔΨ. (E) Mitochondrial H2O2 emissions (JH2O2). (F) Mitochondrial electron leak, expressed as a percentage of oxygen flux (JH2O2/JO2 x 100 = % Electron Leak). Right triangles represent increasing concentrations of ATP relative to ADP (ATP:ADP) during the CK clamp, resulting in reciprocal changes in energy demand and thus JO2. Data represent mean ± SEM. (A–F) N=6–10 per group. (A–C) Measurements made at submaximal JO2 were analyzed by a two-way ANOVA (* main effect of diet, # diet:ΔGATP interaction, P< 0.05). Maximal JO2 (ΔGATP =−12.95) was analyzed by t-test (‡ P<0.05). (D) Mitochondrial respiratory efficiency was analyzed by ANCOVA using submaximal data to determine whether slopes and intercepts differed as a result of diet (# diet:ΔΨ interaction, P< 0.05, and ¥ indicates these data could not be fit via linear regression). Data in (E and F) were analyzed by Student’s t-test *P≤0.05, **P≤0.01, ***P≤0.001. N represents biological replicates.
The DKO Model Reveals a Paradoxical Disconnect between Mitochondrial Lysine Acetylation and Respiratory Dysfunction.
Considering that exposure of DKO mice to HF feeding caused an exponential rise in mitochondrial Kac (Figure 1), we questioned whether or not the combined loss of Sirt3 and CrAT would amplify or exacerbate the foregoing respiratory phenotype observed in the context of a HF diet (Figure 5). Notably, respiratory rates in the presence of Pc/M were still slightly elevated in DKO versus DFC mitochondria, even after chronic exposure to overnutrition (Figure 6A). A similar outcome was evident when DKO mitochondria were fueled by S/R, another condition (like Pc/M) that drives substantial FAD-linked electron flux. By contrast, G/M-supported JO2 was slightly decreased in DKO mitochondria, but only under the lowest ATP free energy charge when flux approached maximal rates (Figure 6A). Importantly, regardless of the substrate conditions, DKO mitochondria maintained a similar or more polarized ΔΨm (Figure 6B), which argues against potential limitations in DH enzyme flux. It was therefore surprising to find that in the presence of either G/M or mixed substrates, DKO mitochondria were operating at a more oxidized NAD(P)H/NAD(P)+ redox potential (Figures 6C), especially during energetic conditions within a physiological range (i.e. submaximal JO2). Nonetheless, the respiratory efficiency plots were either unchanged or shifted rightward (Figure 6D), suggesting that energy transfer from ΔGredox to ΔΨm was maintained or improved. Further evidence that the DKO manipulation did not compromise flux through NAD-dependent DHs emerged from the enzyme-specific JNADH assays performed in alamethecin permeabilized mitochondria (Figure 6E), which again produced mostly negative results. The one exception was an 8% decline in NADP-linked GDH flux. Likewise, maximal JATP-ANT was similar between DKO and DFC under all substrate conditions tested, except G/M (Figure 6F). Together, these findings suggested that the mechanisms contributing to the lower NAD(P)H/NAD(P)+ charge in DKO mitochondria require intact respiratory-competent mitochondria, and/or might involve repartitioning of carbon flux between DH enzymes involving FAD, NAD and/or NADP-linked electron transfer. In sum, despite pronounced proteome-wide upregulation of Kac, the overall respiratory phenotype of DKO mitochondria exposed to HF feeding was surprisingly modest, as we found no evidence of overt mitochondrial dysfunction. On the contrary, energy transfer from ΔGredox to ΔΨm appeared to be augmented.
Figure 6. The DKO model reveals a paradoxical disconnect between mitochondrial Kac and respiratory dysfunction.

Mitochondria were isolated from skeletal muscles of DFC and DKO mice fed a high fat (HF) diet. (A–C) Relationship between (A) JO2, (B) ΔΨ and (C) NAD(P)H/NAD(P)+ redox state versus Gibb’s Energy of ATP hydrolysis (ΔGATP) measured in mitochondria fueled by palmitoylcarnitine+malate (Pc/M), succinate/rotenone (S/R), pyruvate+malate (Pyr/M), glutamate+malate (G/M), or Pyr/M+Pc+free carnitine (Carn). (D) Mitochondrial respiratory efficiency represented as JO2 plotted against ΔΨ. (E) Rates of maximal mitochondrial dehydrogenase enzyme flux shown as NADH or NADPH production (JNADH or JNADPH) measured in alamethecin permeabilized mitochondria. (F) Maximal ATP efflux (JATP) in intact mitochondria fueled by octanoylcarnitine+malate (Oc/M), S/R, Pyr/M, or G/M. (G) Complex V activity in mitochondrial lysates. Right triangles represent increasing concentrations of ATP relative to ADP (ATP:ADP) during the CK clamp, resulting in reciprocal changes in energy demand and thus JO2. Data represent mean ± SEM. (A–F) N=5–10 per group. (A–C) Measurements made at submaximal JO2 were analyzed by a two-way ANOVA (* main effect of genotype, # genotype:ΔGATP interaction, P< 0.05). Maximal JO2 (ΔGATP =−12.95) was analyzed by T-test (‡ P<0.05). (D) Mitochondrial respiratory efficiency was analyzed by ANCOVA using submaximal data to determine whether slopes and intercepts differed as a result of diet (# diet:ΔΨ interaction, P< 0.05). Data in (E–G) were analyzed by Student’s t-test *P≤0.05, **P≤0.01, ***P≤0.001. N represents biological replicates.
Disruption of Acetyl Group Balance in DKO Mitochondria Exacerbates Diet-induced Mitochondrial H2O2 Emissions and Electron Leak Under Physiologically Relevant Energetic Conditions.
In light of the finding that respiratory efficiency was maintained or enhanced in DKO+HF mitochondria in the context of a lower NAD(P)H/NAD+ redox potential, we posited that the apparent enhancement of electron transfer might impact ROS production. Consistent with this prediction, marked elevations in both JH2O2 and % electron leak were evident in DKO mitochondria during all substrate conditions tested, except S/R (Figures 7A and 7B). Since rotenone inhibits transfer of electrons from iron-sulfur (Fe–S) centers of CI to ubiquinone (Q), the foregoing H2O2 profile pointed to this complex as a likely site of ROS generation. Complex I, a large L-shaped oxidoreductase positioned in the inner mitochondrial membrane, catalyzes the exergonic transfer of two electrons from NADH to Q, thereby forming ubiquinol (QH2). To further examine the interplay between diet and genotype, we measured both the NADH oxidase and Q reductase activities of CI using lysates prepared from DFC and DKO muscles from mice fed either the SC or HF diet (Figures 7C and 7D). Interestingly, we detected a main effect of the HF diet to lower maximal NADH oxidase activity (Figure 7C), and a diet-dependent effect of the DKO genotype, which resulted in a modest 16% decrease in the maximal rate of Q reduction only after HF feeding (Figure 7D). These results suggest that overnutrition dampens NADH-linked electron flux at CI, which might contribute to the diet-related shift in mitochondrial redox charge (Figure 5C). Moreover, since Q reductase activity is thought to be rate-limiting for electron flux from the CI flavin cofactor (FMN) to the Q pool (Ransac et al., 2012; Verkhovskaya et al., 2008), the genotype-specific lowering of this activity might promote electron leak and JH2O2 in DKO mitochondria. It is noteworthy that the acetylation states of 11 distinct CI lysine residues were increased in the DKO+HF condition, including a 40-fold increase in acetylated K42 of the NDUSF3 subunit. We therefore sought to determine if perturbations in maximal Q reductase activity were evident in muscles from SIRT3M−/− mice fed a HF diet, another setting that promoted acetylation of NDUSF3 K42, but to a lesser extent (15-fold change over controls) than the DKO model (Table S1). The results proved negative (not shown), suggesting either that this PTM is inconsequential or the Kac stoichiometry in Sirt3M−/− muscles did not achieve a level that disrupts protein function. In aggregate, the bioenergetics platform identified several diet-induced and substrate-specific perturbations in mitochondrial respiration and energy transduction. The two major phenotypes identified in DKO mitochondria were a small but reproducible elevation in PcM-supported respiration, and pronounced increases in CI-dependent electron leak, which occurred only after mice were fed a HF diet.
Figure 7. Disruption of acetyl-lysine turnover in DKO mitochondria exacerbates diet-induced mitochondrial H2O2 emissions and electron leak under physiologically relevant energetic conditions.

Mitochondria were isolated from skeletal muscles of DFC and DKO mice fed a high fat (HF) diet. (A) H2O2 emissions (JH2O2) and (B) electron leak, expressed as a percentage of oxygen flux (JH2O2/JO2 x 100 = % Electron Leak) were measured in mitochondria fueled by palmitoylcarnitine+malate (Pc/M), succinate/rotenone (S/R), pyruvate+malate (Pyr/M), glutamate/malate (G/M), or Pyr/M+Pc+free carnitine (Carn). Right triangles represent increasing concentrations of ATP relative to ADP (ATP:ADP) during the CK clamp, resulting in reciprocal changes in energy demand and thus JO2. Data represent mean ±SEM. Muscle lysates were used to assay (C) Complex I NADH oxidation and (D) Complex I quinone reduction. (E) Working model. Sirt3 consumes NAD regenerated by Complex I, which raises the local NADH/NAD+ redox charge and thereby inhibits the penultimate step in fatty acid oxidation (FAO). In DKO mice fed a high fat (HF) diet, Complex I is hyperacetylated at K42 of the NDUSF3 subunit, possibly perturbing electron transfer and/or Q reductase activity. The combination of disinhibited FAO and altered redox control promotes electron leak and production of reactive oxygen species (ROS). (A and B) N=5–10 per group, analyzed by Student’s t-test. *P≤0.05, **P≤0.01, ***P≤0.001. (C) N= 8 per group, (D) N=13 per group, data shown in (C and D) were analyzed by Two-way ANOVA with Tukey post-hoc testing, in (C) *** represents a main effect of diet. (D) ** represents a difference between DFC and DKO after Two-way ANOVA with Tukey post-hoc testing. N represents biological replicates.
DISCUSSION
The advent and application of high resolution mass spectrometry instruments able to detect low stoichiometric PTMs has revealed thousands of unique, nutrient-responsive lysine acetylation sites identified throughout the mitochondrial proteome (Hosp, 2017). The widely held presumption that many of these events confer functional consequences stems largely from studies in which site-directed mutagenesis strategies were employed to mimic 0% as compared to near 100% acetyl stoichiometry of specific lysine residues on targeted proteins of interest, typically assayed in cultured cells or as a purified protein. This approach can be problematic and potentially misleading because a growing number of studies have concluded that the vast majority of acetyl PTMs occur at occupancy rates of less than 1% (Nakayasu et al., 2014; Weinert et al., 2014; Weinert et al., 2015). It is therefore important to underscore that a significant change in the relative abundance of a low stoichiometric acetyl modification in response to physiological and/or genetically-engineered perturbations might have little biological impact. By contrast, protein phosphorylation of specific amino acid residues often occurs at a much higher stoichiometries, thereby regulating enzyme activity and/or protein function (Covian and Balaban, 2012) (Humphrey et al., 2015). Nonetheless, the prevailing view in the acylation field suggests that nutritional stress causes a subset of these acetyl-PTMs to surpass a critical stoichiometric threshold, leading to enzyme inhibition, impairment of oxidative metabolism and increased vulnerability to energetic challenges such as starvation, caloric restriction and nutrient surplus (Carrico et al., 2018; Elkhwanky and Hakkola, 2018; van de Ven et al., 2017). The current study sought to investigate this paradigm by engineering a model that would approach or exceed the “worst case scenario” in a stepwise fashion. To this end, a series of transgenic mice harboring muscle-specific defects in mitochondrial acetyl CoA buffering and/or SIRT3 activity were fed standard chow or a calorically enriched diet. According to the acetylproteome analyses, the desired endpoint was achieved. Thus, relative to the SC control groups, the total detectable mitochondrial lysine acetylome increased progressively from a 2.5-fold effect of the HF diet, to a 4-fold impact of the DKO, a 20-fold impact of Sirt3M−/−+HF, and a 45-fold elevation observed in muscles of DKO+HF.
As predicted, the DKO mice were more susceptible to diet-induced insulin resistance than either of the single KO models. However, integration of the whole-body metabolic phenotype with comprehensive mitochondrial bioenergetics produced several unanticipated and enlightening findings. First, the studies failed to establish a clear acetyl-lysine dosing effect wherein the degree of mitochondrial protein acetylation correlated with perturbations in insulin action and/or respiratory performance. Moreover, despite relative upregulation of acetyl-PTM abundance that exceeded a median change of 3.7-fold and a maximum of 216-fold in skeletal muscles from the DKO versus DFC mice, extensive evaluation of mitochondrial bioenergetics failed to identify any overt deficits in respiratory kinetics or efficiency, even after the additional stress of chronic high fat feeding. Moreover, we found no evidence linking protein acetylation to inhibition of NAD-dependent mitochondrial DH enzymes. In fact, contrary to the idea that increased Kac imposes negative feedback on enzymes involved in beta-oxidation (Hirschey et al., 2010; Thapa et al., 2018), the most consistently observed trait of a hyperacetylated proteome was elevated JO2 and enhanced respiratory sensitivity when mitochondria were fueled by a long chain fatty acid substrate. This was evident in Sirt3M−/− mitochondria harvested from mice fed a HF diet and DKO mitochondria regardless of the diet, despite increased abundance (ranging from 1.8 to 142-fold) of 119 acetylpeptides mapping to 20 mitochondrial proteins involved in beta-oxidation (Table S1). Thus, collectively, SIRT3 ablation appeared to disinhibit skeletal muscle fat oxidation.
Among the strongest phenotypes identified in mitochondria exposed to HF feeding were: a) pronounced elevation of Pc/M-supported JO2, b) a more reduced NAD(P)H/NADP+ redox state in the presence of mixed substrates, and c) increased rates of H2O2 production and electron leak. The HF diet also led to a 13% decrease in CI NADH oxidase activity. Intriguingly, as compared to the corresponding control group, DKO+HF mitochondria tended to maintain a more oxidized NAD(P)H/NAD(P)+ redox state but a more polarized ΔΨm, while also producing substantially more H2O2 when fueled by NAD-linked substrates. We also found that HF feeding diminished maximal CI-Q reductase activity in DKO but not DFC muscles. Although the mechanisms underlying diet-induced suppression of CI activity remain uncertain, diminished Q reductase activity in DKO muscles was accompanied by a 40-fold increase in acetylated K42 of the NDUFS3 subunit. Thus, the stoichiometry of this PTM might have reached a level that alters electron flux and escape. The hydrophilic peripheral arm of complex I contains a FMN that accepts two electrons from NADH, followed by a series of 8 Fe-S clusters that mediate single electron transfer to Q (Wirth et al., 2016). NDUFS3 is part of the CI Q-module, which connects the peripheral and membrane-associated arms of the complex and functions as the site of Q reduction to ubiquinol. Binding and release of Q is thought to be rate-limiting for CI-mediated electron transfer; and slower turnover at the Q-module can amplify electron leak at the upstream Fe–S clusters (Lambert and Brand, 2004; Ransac et al., 2012; Verkhovskaya et al., 2008). Another key factor that can contribute to flux limitations and electron leak at CI is a more reduced Q pool (Lambert and Brand, 2004). To this point, it is noteworthy that increased rates of fat catabolism favor Q reduction (Guaras et al., 2016). This is because the first reaction in the beta-oxidation pathway is catalyzed by an FAD-linked DH enzyme that transfers electrons directly to Q via the electron transfer flavoprotein:ubiqionone oxidoreductase (ETFQOR), bypassing CI. In theory, circumstances that promote beta-oxidation while also disrupting Q reductase activity should promote electron leak from CI Fe–S clusters, which is the precise outcome observed in DKO mitochondria. Furthermore, adaptation to a HF diet promotes muscle lipid catabolism, both by supplying fatty acid substrate and upregulating protein abundance of multiple beta-oxidation enzymes, including fatty acyl CoA DHs and ETFQOR. Together, these observations provide a plausible explanation for the elevated rates of H2O2 production and electron leak observed in muscle mitochondria from overfed DKO mice.
Importantly, the H2O2 emission assays described herein were performed in the context of a physiologically relevant ATP:ADP energy charge, suggesting that similar diet/genotype-specific outcomes might occur in vivo. In light of substantial evidence linking states of elevated mitochondrial ROS emission to the development of glucose intolerance and insulin resistance (Anderson et al., 2009; Fisher-Wellman and Neufer, 2012; Patti and Corvera, 2010), we presume that the severity of diet-induced insulin resistance in the DKO mice might be consequent to a shift in mitochondrial redox balance. This could involve regulation of redox-sensitive signaling cascades that target cysteine residues and protein disulfide bonds (Muoio and Neufer, 2012), and/or redox-mediated rerouting of glucose traffic (Madiraju et al., 2018). In the current investigation, results of 13C-MFA in DKO versus DFC soleus muscles from chow fed mice were consistent with a small reduction in PDH flux and a reciprocal shift in pyruvate trafficking via malic enzyme and/or pyruvate carboxylase. Moreover, profound diet-induced insulin resistance in DKO mice was accompanied by an apparent decrease in muscle HK activity. This result aligns with that of a previous study in mice with whole body Sirt3 deficiency (Lantier et al., 2015), and builds evidence that mitochondrial deacetylation plays a role in modulating glycolytic flux.
On the one hand, our findings could be viewed as supportive of a narrative that positions protein acetylation as a prominent mechanism of nutrient-induced mitochondrial stress. On the other hand, the weight of evidence strongly suggests that hyperacetylation per se has limited functional impact on most mitochondrial proteins. Thus, the respiratory phenotype of a heavily acetylated mitochondrial proteome was largely unremarkable. Evidence of compromised bioenergetics in DKO mitochondria was limited to elevated electron leak and a modest reduction in maximal CI activity, which occurred only under the most extreme conditions and an attendant 40-fold increase in acetyl-lysine K42 of NDUFS3. Notably, the reporter ion intensities for the identified peptide containing this specific acetyl-lysine site were exceptionally low or absent in DFC muscles, even after prolonged HF feeding, and the activity of the complex was unaffected in Sirt3M−/−+HF muscles. These findings raise doubt about the biological relevance of this specific PTM, especially outside of the DKO model, and extend results of another recent report questioning the role of the sirtuins in protecting mitochondrial proteins against damages caused by hyperacetylation (Fisher-Wellman et al., 2018). The previous study interrogated heart mitochondria derived from several established genetic models of hyperacylation and found minimal impact of the PTMs on baseline bioenergetics. Conclusions of that work were limited, however, because mitochondria were not evaluated after animals and heart tissue had been exposed to some type of metabolic stress. By contrast, the current study evaluated a more severe genetic manipulation that was further challenged by chronic high fat feeding, which together produced an exponential impact on the mitochondrial acetyl-lysine landscape. In aggregate, the results provide compelling evidence that lysine acetylation per se does not lead to broad-ranging deficits in mitochondrial protein quality and function, even under extreme conditions.
The prevailing view in this field holds that NAD+ availability regulates both SIRT3 activity and acetyl-lysine occupancy to directly alter protein function, bioenergetics and insulin action. Based on the current studies, we propose that SIRT3-mediated acetyl-lysine turnover might confer more functional impact than acetyl-lysine stoichiometry. Thus, this NAD+-consuming deacetylase has the potential to alter the charge of the redox “cloud” surrounding a specific protein complex, and might thereby act as a rheostat that modulates enzyme flux. In this case, recruitment of SIRT3 to multiple acetylated lysine residues belonging to the same protein or complex would favor a more reduced NADH/NAD+ redox state in a localized manner (Figure 7E). This model could explain how multiple low stoichiometric acyl PTMs that spread across enzymes and complexes of a specific metabolic pathway contribute to flux control without having a direct impact on protein conformation and function. If true, increased SIRT3 flux would generate a negative feedback signal to impose a brake on carbon catabolism at specific DH enzymes that are especially sensitive to shifts in the NADH/NAD+ redox potential. Interestingly, among the DH enzymes for which forward flux is most thermodynamically unfavorable are the beta-hydroxy acyl CoA DHs that catalyze the penultimate step in the fatty acid beta-oxidation spiral (Martines et al., 2017). Thus, in the setting of surplus lipid substrate and low energy demand, a small shift towards an increased NADH/NAD+ ratio would dampen rates of beta-oxidation, promoting a fuel switch from fatty acid to glucose. This could explain why SIRT3 ablation augmented PC-supported JO2 while also lowing the NAD(P)H/NAD(P)+ redox charge in the context of mixed substrates. In addition to slowing acetyl CoA production when supply exceeds demand, this mechanism of “fine-tuning” NAD-linked DH activities might guard against excessive ROS production by complex I. The proposed working model correctly predicts that the impact of SIRT3 deficiency on carbon flux, redox charge and/or electron leak would be most evident when the null condition is compared to a control group wherein sirtuin flux and acetyl-lysine turnover are elevated, such as obesity and/or high fat feeding–which aligns with the current results. In this scenario, acetylated-lysine residues provide SIRT3 substrates that promote deacetylase flux, which in turn serves to modulate the redox charge surrounding a specific protein. Further vetting of this model now awaits future study.
LIMITATIONS OF STUDY
The overall conclusion that hyperacetylation per se does not compromise protein quality and respiratory function is based largely on results from in vitro assays performed using isolated mitochondria. Thus, it is possible that the respiratory function assays performed in isolated mitochondria do not fully capture and recapitulate mitochondrial performance in vivo. Additionally, the finding that substantial increases in relative lysine acetylation have little functional impact on respiratory function led us to speculate that the stoichiometry of most acetylation sites is very low. We acknowledge however, that the methods used to perform mitochondrial acetyl-proteomics in this study provide information on relative changes in acetyl-lysine abundance, but do not quantify absolute stoichiometry. Lastly, although insulin resistance in DKO mice was accompanied by enhanced mitochondrial capacity for fat oxidation and elevated rates of electron leak, the study did not provide evidence that either or both of these outcomes played a direct role in disrupting insulin action.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Deborah M. Muoio (debbie.muoio@duke.edu). This study did not generate new unique reagents.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
All animal studies were approved by the Duke University or Vanderbilt University Institutional Animal Care and Use Committee and conducted in Assessment and Accreditation of Laboratory Animal Care-associated facilities. Mice were housed in a light (12h light/12h dark) and temperature (22°C) controlled room an d had ad libitum access to food and water throughout all experiments unless noted otherwise. Male mice on a C57BL/6NJ background were used for all experiments. Mice with a skeletal muscle and heart-specific deletion of CrAT (CrATM−/−) and control animals (CrATfl/fl) were generated as previously described (Muoio et al., 2012). Sirt3fl/fl mice were kindly provided by Dr. Matt Hirschey (Martin et al., 2017) and crossed with MCK-Cre mice purchased from Jackson Labs and backcrossed onto a C57BL6/NJ background. Mice with a muscle and heart-specific deletion of CrAT and Sirt3 (DKO) and controls (DFC) were generated by crossing CrATM−/− mice with Sirt3fl/fl mice. Sirt3M−/− mice and controls were generated by crossing Sirt3M−/− out of the DKO mouse line to ensure genetic similarity between mouse lines. Mice were fed either a standard chow (Lab Diet PicoLab Rodent Diet 20 5053) or high fat (HF) diet with additional sucrose (45% kcal as fat and 25% kcal as sucrose, Research Diets D03021303i). Mice fed a HF diet were placed on the diet starting at 12 wks of age and maintained for 12–25 wks until sacrifice. Unless otherwise stated, mice were anesthetized with Nembutal (intraperitoneal injection; 100mg/kgBW) prior to organ removal.
METHOD DETAILS
Disclosure of Information Related to Experimental Design:
Mice were randomly assigned to diet treatments. When possible, treatments and genotypes were assigned blindly to the experimenter by another member of the lab. Groups of mice or series of experiments were conducted several times over a sufficiently long period of time to avoid time- or condition-dependent bias. Statistical outliers were identified and removed from analysis using Grubbs’ test. Outliers are represented in figures as open grey circles. Sample size estimates were based on previous studies using CrAT KO and Sirt3 KO mouse lines, and mitochondrial diagnostic assays described in(Fisher-Wellman et al., 2018; Fisher-Wellman et al., 2019). A minimum sample size of n=6 was applied to all physiological outcome measures, and where possible, power was improved by increasing the sample size to n=8–12.
Chemicals and Reagents:
All chemicals were purchased from Sigma-Aldrich unless otherwise stated. Creatine kinase from rabbit muscle was purchased from Roche Life Science. Tetramethylrhodamine methyl ester (TMRM) and Amplex Ultra Red were purchased from Thermo Fisher Scientific. Potassium NADP+ was purchased from Ark-Pharm. Potassium pyruvate was purchased from Combi-Blocks. [3-3H]glucose and 2[14C]deoxyglucose were purchased from PerkinElmer.
Mitochondrial isolation from skeletal muscle:
Mice were fasted for 2h with access to water. Skeletal muscle (two gastrocnemii and one quadriceps) was excised and immediately placed in ice-cold Buffer A (phosphate buffered saline (PBS) supplemented with 10mM EDTA (pH 7.4)). Mitochondria were isolated using differential centrifugation (Frezza et al., 2007). The buffers for all isolations are as follows: Buffer B – 100mM KCl, 50mM MOPS, 1mM EGTA, 5mM MgSO4 (pH 7.1); and Buffer C – Buffer B supplemented with 2g/L fatty-acid free bovine serum albumin (BSA). Tissues were minced in 1.4ml Buffer A, transferred to a 50-ml conical, and incubated with 10ml of Buffer A supplemented with 0.05% trypsin on ice for 5min with vortexing every 30sec. The skeletal muscle suspension was centrifuged at 200 x g for 5min at 4°C and the supernatant with trypsin was discarded. The tissue pellet was suspended in 10ml Buffer C and homogenized with an ice-chilled Teflon pestle and borosilicate glass vessel and centrifuged at 500 x g for 10min at 4°C. The supernatant was filtered through two layers of gauze and centrifuged at 9,000 x g for 10min at 4°C. Mitochondrial pellets were suspended in 1.4ml Buffer C using an ice-chilled Teflon pestle and centrifuged at 9,000 x g for 10min at 4°C. Pellets were suspended in 1ml Buffer B and centrifuged at 9,000 x g for 5min at 4°C. Buffer B was aspirated from each tube and mitochondrial pellets were suspended in 80–100ul Buffer B. Protein content was determined using the Pierce BCA protein assay and mitochondria were resuspended at a final concentration of 10mg/ml for all functional assays.
Western blot analysis:
Tissues were freeze clamped, powdered under liquid nitrogen, and homogenized in Cell Lytic-M supplemented with protease inhibitor cocktail, phosphatase inhibitor cocktails 2 and 3, and 10mM nicotinamide using an ice-cold Potter-Elvehjem tissue grinder. Samples were centrifuged for 20min at 13,000 x g for 20min at 4°C, the supernatant was transferred into a clean tube, and samples were stored at −80°C. Isolated mitochondria were suspended in Cell Lytic-M supplemented with protease inhibitor cocktail, phosphatase inhibitor cocktails 2 and 3, and 10mM nicotinamide and pellets were disrupted using an ice-cold Teflon pestle. Protein content was determined via the Pierce BCA protein assay. Samples were diluted in Cell Lytic-M and mixed with an appropriate volume of 5x loading buffer. Fifty micrograms of protein per well were loaded onto a 4–15% Criterion TGX Stain Free SDS-PAGE gel (Bio-Rad), transferred to a nitrocellulose membrane via the Bio-Rad TurboBlot Transfer system, and total protein was visualized using the Pierce Reversible Stain MemCode Kit– a rapid and sensitive alternative to Ponceau S stain for protein detection on nitrocellulose membranes after transfer from polyacrylamide gels (ThermoFisher, https://www.thermofisher.com/order/catalog/product/24580). Membranes were blocked in fish gelatin with tris buffered saline (TBS) for 1h at room temperature and incubated with primary antibodies overnight at 4°C. Following primary incubation, membranes were washed in TBS+0.05% Tween, incubated with appropriate secondary antibody (LICOR Biosciences) diluted in fish gelatin in TBS, washed, and imaged using the Odyssey Imager (LICOR Biosciences). Band intensities were determined using Image Studio version 3.1 (LICOR Biosciences) and ImageJ where appropriate. The following primary antibodies and dilutions were used: CrAT (Abcam #ab153699, 1:1000), Sirt3 (Cell Signaling #D22A3, 1:1000), acetyl-lysine (Cell Signaling #9441, 1:1000), and VDAC (Abcam ab15895).
Gene expression:
RNA was isolated from approximately 25mg pulverized heart, liver, or white adipose tissue (WAT) using a Trizol/chloroform extraction method paired with RNeasy Mini spin columns (Qiagen) with on column DNase treatment. RNA concentration was determined using a NanoDrop. cDNA was synthesized using the iScript cDNA Synthesis Kit (Bio-Rad). Quantitative PCR (qPCR) for CrAT and 18S was performed using Taqman Master Mix (CrAT assay Taqman #_Mm00483985 and 18S assay Applied Biosystems # 4319413E). Sirt3 and 18S gene expression was performed using Sybr Green with the following primers: Sirt3 - Forward 5’ AGG TGG AGG AAG CAG TGA GA 3’ and Reverse 5’ GCT TGG GGT TGT TAA AGA AA 3’, 18S - Forward 5’ GTA ACC CGT TGA ACC CCA TT 3’ and Reverse 5’ CCA TCC AAT CGG TAG TAG CG 3’.
Tissue lysis, digestion, and TMT labeling for acetylproteomics:
Approximately 15 mg of powdered quadriceps muscle was resuspended in ice-cold Urea Lysis Buffer (8M urea in 50 mM Tris, pH 8.0, 40 mM NaCl, 2 mM MgCl2 supplemented with 10 mM nicotinamide, 10 μM trichostatin A, and 1x Roche cOmplete ULTRA EDTA-free protease inhibitor mini tablet) and samples were disrupted with a TissueLyzer (Qiagen) for 30 sec at 30 Hz. Samples were removed, frozen in liquid nitrogen, thawed for three freeze-thaw cycles, and further disrupted by sonication with a probe sonicator (three 5 sec bursts, power setting of 3). Samples were centrifuged at 10,000 x g for 10 min at 4°C, protein content was determined via the Pierce BCA protein assay, 500 μg of protein were transferred to a clean tube, and volumes were normalized with Urea Lysis Buffer (2.5 mg/mL). Samples were reduced with 5 mM DTT at 37°C for 30 min, cooled to room temperature, alkylated with 15 mM iodoacetamide for 30min in the dark and unreacted iodoacetamide was quenched by the addition of DTT to 15 mM. Samples were digested with LysC (Wako Chemicals; 1:100 w:w; 5 μg enzyme per 500 μg protein) for 4h at 37°C; the urea diluted to 1.5 M with 50 mM Tris (pH 8.0), 5 mM CaCl2 and digested with trypsin (50:1 w/w, protein:enzyme) overnight at 37°C. Samples were acidified to 0.5% v/v trifluoroacetic acid (TFA) and centrifuged at 10,000 x g for 10 min at room temperature to pellet any undigested material. The supernatant containing soluble peptides was desalted on a 50 mg tC18 SEP-PAK Solid Phase Extraction (SPE) column (Waters) and peptides were eluted once with 500 μL of 25% acetonitrile/0.1% and twice with 500 μL 50% acetonitrile/0.1% TFA. The eluate was frozen on dry ice and dried overnight in a speed vac. All samples were suspended in 100 uL of 200 mM triethylammonium bicarbonate (TEAB, ThermoFisher Scientific), mixed with a 10-plex Tandem Mass Tag (TMT, 0.8 mg in 50 μL 100% acetonitrile, ThermoFisher Scientific), and vortexed for 4h at room temperature. Samples were quenched with 0.8 μL 50% hydroxylamine, vortexed for an additional 15 min at room temperature, combined, frozen, and dried overnight in a speed vac. Samples were suspended in 1 mL 0.5% TFA and desalted on a 100 mg tC18 SEP-PAK SPE column (Waters) as described above. The eluate was vortexed and ~5% was transferred to a separate tube for quantification of unmodified peptides (“input” material) and assessment of TMT labeling efficiency, and the larger portion (95%) was retained for acetylpeptide enrichment. Both portions were frozen on dry ice, dried overnight in a speed vac, and stored at −80°C.
Acetylpeptide enrichment for proteomics:
Acetylpeptide enrichment was performed using the Cell Signaling PTMScan Acetyl-lysine Motif Kit (#13416). Peptide pellets were suspended in 1.4 mL IAP buffer and centrifuged at 10,000 x g for 5 min at 4°C to remove any insoluble material. The supernatant was transferred into a tube containing PBS-washed antibody beads and incubated on a rotator overnight at 4°C. Samples were centrifuged at 2,000 x g for 30 sec to pellet the beads and beads were washed twice with IAP buffer and three times with chilled milli-Q filtered water. After the last wash step, all remaining liquid was carefully removed, and bound peptides were eluted twice with 100 μL of 0.15% TFA. The eluate was acidified to 0.5% TFA and desalted on a 50 mg tC18 SEP-PAK SPE column (Waters) as previously described, the eluate frozen on dry ice, and dried overnight in a speed vac. Samples were resuspended in 12 μL 0.1% formic acid and stored at −80°C.
Processing of the input fraction for proteomics:
The input material was processed using the Pierce High pH Reversed-Phase Peptide Fractionation Kit (ThermoFisher Scientific #84868). Dried input material was dissolved in 300 μL of 0.1% TFA. A 150 μL aliquot was removed, diluted 1:1 with 150 μL 0.1% TFA, and loaded onto a spin column while the remaining 150 μL was stored at −80°C. The eluate was collected for a total of eight fractions per TMT kit. All fractions were frozen on dry ice and dried overnight in a speed vac. Dried samples were re-suspended in 10 μL 0.1% formic acid and peptides were quantified using the Pierce Quantitative Colormetric Peptide Assay (ThermoFisher Scientific #23275). Samples were suspended to a final concentration of 0.1 μg/μL with 0.1% formic acid and stored at −80°C.
nLC-MS/MS for proteomics:
All samples were subjected to nanoLC-MS/MS analysis using a UHPLC system coupled to a Q Exactive Plus Hybrid Quadrupole-Orbitrap mass spectrometer (ThermoFisher Scientific) via a nanoelectrospray ionization source. The default chromatography set-up included an EASY-nLC UPLC system (Thermo) connected to an Acclaim PepMap 100 C18 trapping column (3 um particle size, 75 μm × 20 mm, Thermo) and an Acclaim PepMap RSLC C18 analytical column (2 um particle size, 75 μm × 500 mm column, Thermo). Acetylpeptide samples were analyzed with at least technical duplicate injections of 2 μL and input fractions were subjected to 8 uL (0.8 μg) injections. After trapping at a variable flow rate dictated by max pressure of 500 Bar, each sample underwent analytical separation over a 105 min gradient (flow rate of 300 nL/min) of 5 to 40% solvent B (90% ACN/0.1% FA), with a column temperature of 55°C. MS1 (precursor ions) was performed with default settings of 70,000 resolution, an AGC target of 3x106 ions, and a maximum injection time (IT) of 60 ms. MS2 spectra (product ions) were collected by data-dependent acquisition (DDA) of the top 10 (loop count) most abundant precursor ions with a charge greater than 1 per MS1 scan with dynamic exclusion enabled for a window of 30 sec. Precursor ions were filtered with a 0.7 m/z isolation window and fragmented with a normalized collision energy (NCE) of 30. MS2 scans were performed at 35,000 resolution with an AGC target of 1x105 ions and a maximum IT of 60 ms. For some acetyl runs, 4 μL injections were separated over a 90 min gradient using a nano-Acquity UPLC system (Waters Corporation) connected to a Symmetry C18 trapping column (20 mm x 180 μm, Waters) and a Acquity BEH130 C18 analytical column (1.7 μm particle size, 75 μm x 250 mm, Waters), 140,000 MS1 resolution, and a loop count of 20, 1.2 m/z isolation window, and 60 sec dynamic exclusion for MS2.
Proteomics data analysis:
Data were searched against the UniProt mouse complete proteome database of reviewed (Swiss-Prot) and unreviewed (TrEMBL) proteins, which consisted of 51,434 sequences on the date of download (7/21/2017). Data analysis was performed using Proteome Discoverer 2.2, searching with both Sequest HT and MS Amanda 2.0 with the following default parameters: oxidation (15.995 Da on M) as a variable modification and carbamidomethyl (57.021 Da on C) and TMT10plex (229.163 Da on peptide N-term and K) as fixed modifications, and 2 missed cleavages (full trypsin specificity). TMT labeling efficiency was assessed as a quality control measure by searching for N-terminal TMT as a variable modification—confirming labeling efficiency was ~95% for all samples. All runs with an acetyl-enriched fraction included acetylation (42.011 Da on K) as a variable modification and TMT as a variable modification on K (remaining fixed on peptide N-term). PSMs from each search algorithm were filtered to a 1% FDR using Percolator (Kall et al., 2007) and PTM site localization probabilities were determined using ptmRS (Taus et al., 2011). PSMs were grouped to unique peptides while maintaining a 1% FDR for peptides and a 90% localization threshold for PTMs. Peptides from acetyl and input fractions were grouped to proteins using the rules of strict parsimony and proteins were filtered to 1% FDR using the Protein FDR Validator node of PD2.2. Reporter ion intensities for all PSMs with co-isolation interference below 0.5 (50% of the ion current in the isolation window) and an average S/N >2.5 for reporter ions were summed together at the peptide and protein level, but quantification for each data type (acetyl, input) were kept separate. Peptides shared between protein groups were excluded from protein quantitation calculations.
Proteomics statistical analysis:
Protein and peptide groups tabs from the PDv2.2 results were exported as tab delimited .txt files and analyzed with an in-house Python module based on a workflow previously described in (Fisher-Wellman et al., 2019) with some modifications. Briefly, peptide group reporter intensities for each peptide group in the input material were summed together for each TMT channel, each channel’s sum was divided by the average of all channels’ sums, resulting in channel-specific loading control normalization factors to correct for any deviation from equal protein/peptide input into the ten-sample comparison. Reporter intensities for peptide groups from the acetylpeptide runs and proteins from the input fraction runs were divided by the loading control normalization factors for each TMT channel, respectively. All loading control-normalized quantifications were converted to log2 space. The R-package limma was used to construct linear models for differential abundance of peptides and proteins (Phipson et al., 2016; Ritchie et al., 2015). The eBayes function was used to compute moderated statistical values of differential expression using an empirical Bayesian algorithm (Phipson et al., 2016). All code for the analyses (python and R scripts) can be found online (https://github.com/dmpio/Williams_et_al_2019_Kac_PRX). Protein-level quantification was performed exclusively on Master Proteins–the most statistically significant protein representing a group of parsimonious proteins containing common peptides identified at 1% FDR. Acetylpeptide measurements were calculated alone (abundance) and with normalization to any change in the corresponding Master Protein (relative occupancy) by subtracting Log2 Master Protein values from PTM-containing quantitation values on a sample-specific basis.
Oral glucose tolerance tests:
Mice were singly housed and fasted on Alpha-dri bedding for 5h with access to water. At the start of the test, mice were weighed and a baseline blood sample was obtained from the tail vein for the determination of fasting blood glucose (Bayer Contour Blood Glucose Monitoring System) and plasma insulin prior to an oral gavage of a 1.5g/kg body weight glucose solution (45% glucose diluted in tap water). Subsequent blood glucose samples were obtained via the tail vein at 0, 15, 30, 60, 90, and 120min post-gavage. Blood from the tail vein was collected at 0, 15, 30, and 60min post-gavage using heparinized capillary tubes (Sarstedt Microvette CB 300 LH) and plasma was isolated for the determination of circulating insulin. Plasma insulin was determined using the ALPCO STELLUX Chemi Rodent ELISA kit (ALPCO) per manufacturer’s instructions. A modified version of the homeostatic model assessment of insulin resistance (HOMA-IR) was calculated as [fasting blood glucose (mmol/L)*OGTT 15min insulin (mU/L)]/14.1 (van Dijk et al., 2013).
Body composition and energy balance:
Body weight and composition were measured in fed mice at the Vanderbilt University Mouse Metabolic Phenotyping Center (MMPC) prior to surgical catherization for insulin clamp studies. Body composition was determined using a mq10 nuclear magnetic resonance analyzer (Bruker Optics) and energy balance was measured using indirect calorimetry (Promethion, Sable Systems). For measurements of energy balance, mice were individually housed in metabolic cages with bedding that were identical to their home-cages in a 12h light/dark cycle, temperature and humidity-controlled room for ~1wk. The Promethion system utilizes a pull-mode, negative pressure system with an excurrent flow rate of 2L/min. Cage air is sampled through microperforated stainless steel tubes located at the bottom of the cages to ensure that cage air is sampled uniformly. Water vapor is continuously measured and its dilution effect on O2 and CO2 are mathematically compensated for during data analysis (Lighton, 2008). O2 consumption and CO2 production are measured for each mouse for 30sec every 5min. Room air reference gasses are sampled every 4 cages. The respiratory quotient (RQ) is the ratio of CO2 production over O2 consumption. Energy expenditure is calculated using the Weir equation (Weir, 1949). Ambulatory activity is determined every second with XYZ beams. Data acquisition and raw data processing were coordinated by MetaScreen v2.2.18 and ExpeData v1.7.30. Raw data (Macro13) was uploaded into CalR for data visualization and analysis (https://calrapp.org/, (Mina et al., 2018)). Energy expenditure was analyzed using the Analysis of Covariance (ANCOVA) by regressing energy expenditure (kcal/h) against body weight (Kaiyala, 2014). The significance level for ANCOVA analysis was set a priori to p≤0.05, thus if the group effect was greater than p=0.05, it was determined that the calculation of ANCOVA-adjusted values for energy expenditure was not required. All data were manually inspected and if erroneous values were identified (i.e. energy expenditure ≤ 2 kcal/h) as a consequence of faulty equipment (i.e. poor cage air flow), the subjects were manually excluded using the “Subject exclusion” tab located within CalR.
Hyperinsulinemic-euglycemic (insulin) clamp:
A comprehensive step-by-step description of the insulin clamp surgery, isotope clamp method, and calculations is available from the Vanderbilt MMPC website (https://Vmmpc.org). Insulin clamp studies were performed in chronically catherized, conscious, unstressed, 5h fasted mice at the Vanderbilt University MMPC. Carotid artery and jugular vein catheters were surgically implanted 5–7 days prior to study. Mice were weighed daily and if mice lost more than 10% of their pre-surgery body weight, they were excluded from the study. On the day of study, mice were fasted and [3-3H]glucose was primed and continuously infused into the jugular vein from t= −90 to t=0 (0.04 μCi/min). Prior to the start of the insulin clamp, arterial blood samples were taken at t= −15 and t= −5 for the determination of basal arterial blood glucose, insulin, and [3-3H]glucose turnover. The insulin clamp was started at t=0 with a continuous insulin infusion (4mU/(kg·min)) and a variable infusion of glucose+[3-3H]glucose (0.06 μCi/μL) to maintain euglycemia. The insulin infusion rate was chosen to assess muscle insulin action (Ayala et al., 2006). A mixture of glucose+[3-3H]glucose was employed to minimize alterations in [3-3H]glucose specific activity upon changes in the glucose infusion rate. Heparanized, saline washed erythrocytes were infused to prevent a fall in hematocrit. Arterial glucose was monitored by a hand held glucometer (AccuCheck) every 10min and the glucose infusion rate (GIR) was adjusted to maintain and clamp arterial glucose. Arterial samples for the determination of clamp arterial glucose and [3-3H]glucose turnover were collected at t=80, 90, 100, and 120min and samples for the determination of clamp insulin were collected at t=100 and 120min. At t=120min, a 13μCi bolus of 2[14C]deoxyglucose ([14C]2DG) was administered into the venous line to determine tissue glucose uptake or the glucose metabolic index (Rg). Blood samples were collected at t=122, 125, 135, 145, and 155min to determine [14C]2DG disappearance from the plasma. After the final blood sample was collected, mice were anesthetized and tissues were freeze clamped and stored at −80°C until analysis.
Insulin clamp plasma and tissue analysis:
Plasma samples were deproteinized with barium hydroxide [Ba(OH)2, 0.3N] and zinc sulfate (ZnSO4, 0.3N) and radioactivity of [3-3H]glucose and [14C]2DG was determined by liquid scintillation counting (Packard TRI-CARB 2900TR) with UltimaGold (Packard) as scintillant (Ayala et al., 2007). EndoRa and Rd were calculated using non-steady state equations (Steele et al., 1956). Frozen muscle samples were weighed and homogenized in 0.5% perchloric acid. Muscle homogenates were centrifuged and neutralized with KOH. One portion was counted directly to determine 2[14C]DG (2DG) and 2[14C]DG-6-phosphate (2DGP) and a second portion was deproteinized with Ba(OH)2 (0.3N) and ZnSO4 (0.3N) to remove 2DGP and any tracer incorporation into glycogen prior to scintillation counting for the determination of 2DG radioactivity. 2DGP was calculated by subtracting 2DG values from 2DGP+2DG. Rg calculations reflect 2DGP levels normalized to tissue weight. Plasma insulin was determined by radioimmunoassay (RIA). The hexokinase (HK) index was calculated as tissue 2DGP/2DG.
Preparation of permeabilized fiber bundles:
Mice were fasted for 2h with access to water. Permeabilized fiber bundles were prepared from the red gastrocnemius muscle adapted from previous methods (Perry et al., 2011). A small portion of the muscle was excised (~ 25mg) and transported back to the laboratory in Buffer A (phosphate buffered saline (PBS) supplemented with 10mM EDTA (pH 7.4)). Muscle samples were placed in ice cold Buffer X (7.23mM K2EGTA, 2.77mM CaK2EGTA, 20mM imidazole, 20mM taurine, 6.56mM MgCl2·6H2O, and 50mM MES potassium salt) and trimmed of connective tissue and fat. Small muscle bundles were gently separated along the longitudinal axis with a pair of needle-tipped forceps under magnification. Muscle bundles were then placed in 1.5ml Buffer X containing 30ug/ml saponin and incubated on a rotating platform for 30min at 4°C. Fiber bundles were trans ferred to a new tube containing in 2ml Buffer Z (105mM MES potassium salt, 30mM KCl, 10mM KH2PO4, 5mM MgCl2·6H2O, pH 7.4) supplemented with 0.5mg/ml fatty-acid free BSA 1mM EGTA and incubated for a minimum of 15min or until analysis.
Mitochondrial respiration in permeabilized fiber bundles:
High-resolution respirometry was performed using the OROBOROS Oxygraph-2k (O2K, Oroboros Instruments). Measurements of oxygen consumption were conducted at 37°C in 2ml Buffer Z supplemented with creatine monohydrate (20mM), glucose (5mM), 1mM EGTA, 1U/ml hexokinase (HK), and 10uM blebbistatin. The following substrate combinations were used: Palmitoyl-carnitine/Malate (Pc/M; 20uM/2mM), Octanoyl-carnitine/Malate (Oc/M; 200uM/2mM), and Glutamate/Malate/Succinate (G/M/S; 10/2/10mM). The ADP concentration for all titrations was 1mM. Cytochrome c (10uM) was added at the end of the Pc/M titration to test mitochondrial membrane integrity. At the end of each experiment, fiber bundles were removed from the O2K, washed in double-distilled water to remove salts, placed in a dry, clean tube, frozen at −20°C, and f reeze dried. Data were normalized to mg dry weight and expressed as pmol/sec/mg dry weight.
Ex vivo muscle incubations and 13C-glucose flux analysis.
Soleus muscles were carefully excised from 2 hour fasted anesthetized mice and placed in a low calcium KHB buffer supplemented with 5mM glucose and 0.5mM L-carnitine as previously described (Muoio et al., 2012). Muscles were incubated in a 29°C water bath with shaking and gassing for 20 min. After 20 min, the temperature of the water bath was increased to 37°C and the muscles were incubated for an additional 20 min. After the 40 min incubation period, muscles were transferred to a tracer incubation plate containing 10mM [U-13C]glucose and BSA/fatty acid mixture (200uM palmitate+0.5% BSA) with or without insulin (100nM). The plate was sealed with parafilm and incubated for 90 min with shaking. Once the experiment was complete, the muscles were frozen and stored at −80°C for analysis. Frozen muscles were homogenized in a 1:1:1 mixture of 100% methanol, water mixed with norvaline standard (10ug/ml) and chloroform. The aqueous layer was transferred into a fresh tube and 350ul was dried under nitrogen gas with heating. Twenty-five microliters (ul) of 2wt% methylhydroxylamine in pyridine solution was added and samples were incubated for 90 min at 40°C. Thirty-five microliters (ul) of MTBSTFA + 1% TBDMCS was added and samples were incubated for 30 min at 60°C. The liquid was transferred to an injection vial and injected into the GC-MS according to (Crown et al., 2015).
Mitochondrial respiratory control:
Mice were fasted for 2h with access to water and mitochondria were isolated from skeletal muscle as described above. Steady-state oxygen consumption rates (JO2) ranging from state 2 (i.e. resting or non-phosphorylating) to ~95% of maximal state 3 were sequentially determined using a modified version of the creatine kinase (CK) energetic clamp (Glancy et al., 2008; Glancy et al., 2013; Messer et al., 2004) in the presence of excess CK and known amounts of creatine (Cr), phosphocreatine (PCr), and ATP. The CK reaction is used to couple the conversion of ATP and ADP to PCr and Cr. The buffer for all assays was Buffer Z supplemented with Cr (5mM), PCr (1.5mM), CK (20 U/ml), 2.5mg/ml fatty-acid free BSA, and 1mM EGTA (pH 7.2). At the start of each assay, isolated mitochondria (0.025mg/ml) were added to the assay buffer, followed by the addition of respiratory substrates and ATP (5mM). Next, sequential additions of PCr were added to the chamber to achieve the following final concentrations: 3, 6, 9, 12, 15mM. The purpose of the sequential additions is to alter energy demand or the ATP:ADP and gradually slow JO2 and bring it back down to baseline levels. Under the buffer conditions described above, the free energy of ATP hydrolysis (ΔGATP) after each PCr addition was calculated using the following equation:
where ΔG′°ATP is the standard apparent transformed Gibbs energy (under a specified pH, ionic strength, free magnesium and pressure), R is the gas constant (8.3145 J/kmol) and T is temperature in kelvin (310.15). After each PCr addition, the calculation of both ΔGATP and K′CK were adjusted for changes in buffer strength and free magnesium according to (Golding et al., 1995; Teague et al., 1996) and calculation of ΔGATP at each titration point was performed using a recently developed online tool (https://dmpio.github.io/bioenergetic-calculators/). Respiratory sensitivity, or the ability of mitochondria to respond to a given energy demand (ΔGATP), was calculated by plotting the JO2 against the corresponding ΔGATP. The slope of the linear portion (a linear force-flow relationship) represents the conductance or elasticity of the entire respiratory system under specified substrate conditions. The following substrate conditions were tested: Glutamate/Malate – (G/M; 10/2.5mM), Pyruvate/Malate – (Pyr/M; 5/2.5mM), Palmitoyl-carnitine/Malate – (Pc/M; 20uM/2.5mM), Pyruvate/Palmitoyl-carnitine/Malate/Carnitine (Pyr/Pc/M/Carn; 5mM/20uM/2.5mM/2mM), and Succinate/Rotenone – (S/R; 10/0.005mM)]. Additional information regarding the underlying rationale for each substrate combination is described in detail in (Fisher-Wellman et al., 2018).
Mitochondrial membrane potential (ΔΨ) and NAD(P)H/NAD(P)+ redox:
Fluorescent measurements of mitochondrial membrane potential (ΔΨ) and NAD(P)H/NAD(P)+ redox were determined simultaneously using a QuantaMaster Spectrofluorometer (QM-400; Horiba Scientific). All assays were conducted at 37°C in a 0. 2ml reaction buffer. The buffer for all assays was Buffer Z supplemented with creatine (Cr; 5mM), phosphocreatine (PCr; 1.5mM), creatine kinase (CK; 20U/ml) and TMRM (0.2uM). At the start of each assay, isolated mitochondria (0.1mg/ml) were added to the assay buffer, followed by the addition of respiratory substrates (G/M, Pyr/M, Pc/M, Pyr/Pc/M/Carn, S/R), ATP (5mM), and then sequential additions of PCr to obtain the following final concentrations: 3, 6, 9, 12, 15mM. After the final PCr addition, cyanide (4mM) was added to induce a state of 100% reduction within the NAD(P)H/NAD(P)+ couple and alamethicin (12.5ug/ml) was added to permeabilize the mitochondria. NAD(P)H/NAD(P)+ during the experiment was expressed as a percent reduced according to the following formula: % Reduction = (F-F0%)/(F100%-F0%), where the 0% reduction state is represented as the fluorescent signal (Ex/Em, 340/450nm) recorded in the presence of alamethicin. Mitochondrial membrane potential (ΔΨ) was determined using TMRM by taking the fluorescence ratio of the following excitation/emission parameters [Ex/Em, (572/590nm)/(551/590nm)] as previously described (Scaduto and Grotyohann, 1999). Data were converted from the 572/551nm ratio to millivolts (mV) following a KCl standard curve in the presence of valinomycin (Krumschnabel et al., 2014).
Mitochondrial H2O2 emission (JH2O2) and electron leak in isolated mitochondria:
Rates of mitochondrial H2O2 emissions (JH2O2) were determined fluorometrically (QuantaMaster Spectrofluorometer; Ex:Em 565:600nm) using the Amplex Red (AUR)/horseradish peroxidase (HRP) detection system. All assays were conducted at 37°C in a 0.2ml reaction buffer (Buffe r Z supplemented with creatine (Cr; 5 mM), phosphocreatine (PCr; 1.5 mM), creatine kinase (CK; 20 U/ml), AUR (10uM), HRP (1 U/ml) and superoxide dismutase (20 U/ml). At the start of each assay, isolated mitochondria (0.1mg/ml) were added to the assay buffer, followed by respiratory substrates (G/M, Pyr/M, Pc/M, Pyr/Pc/M/Carn, S/R), auranofin (AF; 0.1uM), ATP (5mM), and then sequential additions of PCr to obtain the following final concentrations: 6 and 15mM. After the final PCr addition, 1-chloro-2,4-dinitrobenzene (CDNB) was added to approximate maximal mitochondrial JH2O2. Electron leak (expressed herein as % electron leak) was calculated by dividing JH2O2 by the corresponding O2 consumption rate (JO2) measured under identical conditions and expressed as a percentage (% Leak = JH2O2/JO2 X 100). Notably, the JH2O2 rates used in the calculation were generated in the presence of auranofin; however, the corresponding JO2 assays did not contain auranofin, as the inhibitor does not impact respiratory conductance (Fisher-Wellman et al., 2018).
Mitochondrial dehydrogenase activity assays (JNAD(P)H):
Rates of mitochondrial NADH and NADPH production (JNAD(P)H) were determined as previously described (Fisher-Wellman et al., 2014). The buffer for all assays (with the exception of aspartate-aminotransferase - GOT2) was Buffer Z, supplemented with alamethicin (0.03mg/mL), rotenone (0.005mM) and NAD+ (2mM) or NADP+ (2mM). For all assays to assess JNADH from pyruvate dehydrogenase (PDH) or alpha-ketoglutarate dehydrogenase (AKGDH), the following cofactors were included: coenzyme A (0.1mM) and thiamine pyrophosphate (0.3mM). Rates of JNADH consumption from GOT2 were determined using the following buffer: HEPES (20mM; pH 8.0), KCl (100mM), KH2PO4 (2.5mM), MgCl2 (2.5mM), glycerol (1%), aspartate (200mM), pyridoxal 5’-phosphate (0.1mM), rotenone (5uM), NADH (0.2mM), and malate dehydrogenase (2U/ml). Prior to the start of the assay, 200ul of assay buffer was loaded into each well of a 96-well plate along with any required cofactors. Isolated mitochondria (10–30ug/well) were added and background rates of NADH and NADPH production were determined via a spectrophotometer (Ex:Em 340:450). The assay was started following the addition of enzymatic substrates and NAD(P)H is determined via auto-fluorescence (Ex:Em 340:450). Data were converted to pmoles of NADH or NADPH/secxmg mitochondria via a NADH and NADPH standard curve and are represented as JNAD(P)H after addition of substrate subtracted from background rates.
Maximal JATP activity:
Rates of mitochondrial ADP-ATP exchange were determined fluorometrically via a magnesium green (MgGr) detection system (Ex/Em 506/530) as described previously (Chinopoulos et al., 2009) with slight modifications. Fluorescence was monitored via a QuantaMaster Spectrofluorometer (Horiba Scientific). Buffer for all assays was an ANT assay buffer – KCl (8mM), K-lactobionate (135mM), NaCl (10mM), HEPES (10mM), KH2PO4 (20mM), EGTA (0.005mM), mannitol (10mM), MgCl2 (1mM), BSA (0.5g/L), pH 7.25. AP5A (50uM) was included in all assays to inhibit ATP production from adenylate kinase. The following substrate conditions were tested: [Octanoyl-carnitine/Malate – (Oct/M; 0.2/2.5mM), Glutamate/Malate – (G/M; 10/2.5mM), Pyruvate/Malate – (Pyr/M; 5/2.5mM), Succinate/Rotenone – (Succ/R; 10/0.005mM)]. Reaction master mixes of 500ul ANT assay buffer, respiratory substrates (Oct/M, G/M, Pyr/M, Succ/R), AP5A (50uM), and MgGr (2uM) were prepared and placed at 37°C to allow for temperature equilibration until use. To begin, 200ul of assay buffer was added to a cuvette followed by the addition of isolated mitochondria (0.05mg/ml for G/M and Pyr/M and 0.075mg/ml for Oct/M and Succ/R) to the assay buffer. The reaction was started by an addition of ADP (250mM) and monitored for 30 seconds. After ADP addition, EDTA (500mM) was added to induce a state of minimum fluorescence followed by an addition of MgCl2 (500mM) to induce a state of maximum fluorescence. For each experiment, the fluorescence signal (i.e. changes in free [Mg2+]) was converted to changes in [ATP] using standard binding equations. The rate of ATP appearance in the extramitochondrial buffer was normalized to mitochondrial protein.
Complex V activity assay:
Mitochondrial lysates (2mg/ml) were prepared from freshly isolated mitochondria in Cell Lytic-M (Sigma-Aldrich). Complex V (CV) activity (pmoles of ATP/sec/mg) was determined by monitoring NADH degradation in the presence of ATP. Of note, NADH oxidation and ATP hydrolysis occur at a 1:1 stoichiometry, thus CV activity can be determined by following the decline in NADH signal over time. In this assay, the following buffer was used: HEPES (20mM; pH 8.0), KCl (100mM), KH2PO4 (2.5mM), MgCl2 (2.5mM), glycerol (1%), lactate dehydrogenase/pyruvate kinase (10U/ml), phosphoenoyl-pyruvate (5mM), rotenone (0.005mM) and NADH (0.2mM). Prior to the start of the assay, 200ul of assay buffer was loaded into individual wells of a 96-well plate followed by mitochondrial lysate (2ug/well). Complex V activity was assessed in the presence and absence of oligomycin (0.005mM) in order to calculate the oligomycin-sensitive rates of ATP hydrolysis. The assay was started following the addition of ATP (5mM) and NADH auto-fluorescence (Ex:Em 376/450nm) was determined.
Complex I activity assay:
Approximately 50mg of pulverized quadriceps tissue was homogenized in mitochondrial isolation media (MIM, 0.3M sucrose, 10mM HEPES, and 1mM EGTA, pH 7.4) with phosphatase and protease inhibitors (1:100) using a protocol adapted from (Barrientos, 2002). Pulverized tissue was diluted 1:20 in ice-cold MIM, transferred into a chilled potter, and homogenized gently using a Teflon/glass homogenizer with 10 controlled strokes. The samples were then centrifuged at 600 x g for 10min at 4C. The supernatant was transferred to a clean tube and a portion was diluted 1:20 for a BCA assay to determine protein content. The samples were diluted to 2ug/ul in hypotonic buffer (25mM K2HPO4, 5.3mM MgCl2, pH 7.2) and stored at −80C until analysis. On the day of analysis, samples were removed from the −80C and subjected to 3 freeze-thaw cycles. Complex I activity (NADH oxidation and Q reduction) was determined in a working reagent containing 50mM Tris, 5mg/ml BSA, 240uM KCN, 4uM Antimycin A, pH=8.0 (Torres et al., 2018). All assays were performed at 340nm following the decrease in absorbance resulting from the oxidation of NADH in 1.5ml cuvettes in a Spectromax M2E spectrophotometer. At the start of the assay, 967ul distilled water, 168ul working reagent, and 200ug of muscle lysates in hypotonic buffer were added to the cuvette. Complex I activity was determined following the oxidation of NADH (0.13mM final) at 340nm for 3min using 1) 50uM oxidized decyl-ubiquinone (DCUox) as the electron acceptor, or 2) following the reduction of DCUox at 280nm in the presence of 0.13mM NADH. Rotenone (4uM) was added at the end of each run to measure rotenone-sensitive NADH-DCU oxidoreductase activity. Rates of complex I activity were normalized to citrate synthase activity. Citrate synthase activity was determined in a 96-well plate using 5ug of muscle lysates diluted in hypotonic buffer from the complex I activity assay. The activity reaction media consisted of 50mM Tris, 1mM EDTA, pH 8. Sample and 180ul of activity reaction media were added to each well and a background reading was obtained at 412nm. The reaction was started upon addition of 10ul of 10mM oxaloacetate and the reaction was monitored at an absorbance of 412nm for approximately 8–10min. Background rates were subtracted from the oxaloacetate-supported rates.
QUANTIFICATION AND STATISTICAL ANALYSIS
Data are presented as means ± SEM. Statistical analyses were preformed using either GraphPad Prism 7.0 (GraphPad Software, San Diego, CA) or JMP 14.0.0 (SAS Institute Inc., Cary, NC) using t-tests one- and two-way ANOVA with Tukey HSD post-hoc testing where warranted. Figures were generated using GraphPad Prism 7.0. The level of significance for all experiments was set at P < 0.05.
For experiments using isolated skeletal muscle mitochondria, statistical analyses were performed using GraphPad Prism 7.0. Submaximal rates of JO2 plotted as a function ΔGATP were used for linear regression analysis using individual replicates, not mean values (* P<0.05). These analyses excluded measurements of maximal JO2 made at the lowest ΔGATP, which represents an extreme, non-physiological energetic condition positioned outside the linear range. Maximal JO2 (ΔGATP =−12.95) was analyzed by unpaired t-test (‡ P<0.05). Measures of ΔΨ and NAD(P)H/NAD(P) redox potential plotted as a function of ΔGATP were analyzed by a one-way ANOVA (diet x ΔGATP or genotype x ΔGATP). These analyses likewise excluded measurements made at the lowest ΔGATP. Mitochondrial respiratory efficiency (JO2 vs ΔΨ) was analyzed by ANCOVA to determine whether slopes and intercepts differed as a result of diet (# diet:ΔΨ interaction, P< 0.05). JNAD(P)H, JATP, JH2O2, electron leak and CV activity data were analyzed by unpaired t-test (* P<0.05). Statistical details can be found in the figure legends including the statistical tests used, exact value of n, what n represents (e.g. number of animals), and dispersion and precision measures (e.g. mean and SEM). Proteomic data were log transformed to produce a normal distribution. A conservative approach was taken in which equal variance was not assumed for unpaired t-tests. Linear regressions were performed on replicates, not mean values, thus reducing chances of Type 1 errors.
DATA AND CODE AVAILABILITY
All raw data for proteomics were submitted to the Proteome Xchange Consortium (Deutsch et al., 2017) via the PRIDE partner repository (Vizcaino et al., 2016) with the dataset identifier PXD014586. All code for the analyses in this published article can be found online (https://github.com/dmpio/Williams_et_al_2019_Kac_PRX).
Supplementary Material
Table S1. TMT-based quantitation of acetylation and protein abundance changes in skeletal muscle, related to Figures 1 and S1. Supplementary Excel file containing proteomic data—both direct output from Proteome Discoverer (Tabs 6 and 7) and summarized information from downstream statistical analysis (Tabs 2–5)—comparing the relative abundance of acetylated peptides and protein abundance, displayed on the following seven spreadsheet tabs:
Tab 1) Key. Includes descriptions of pertinent information fields included in the columns of the subsequent tabs and points to the location (tabs/columns) of key metrics from the study.
Tabs 2–3) Acetylpeptide SUMMARY. Summary of analyzed acetylproteomic data, after normalization of quantitative measurements and statistical analysis across genetic comparisons. Acetylpeptide Relative Abundance is normalized for total peptide amount on each TMT channel in the “input” fraction. Acetylpeptide Relative Occupancy is normalized for any biological changes in the “Master Protein” to which a given acetylated peptide maps (if identified at 1% protein-level FDR and quantified in Tabs 4–5).
Tabs 4–5) Protein SUMMARY. Summary of relative quantitation statistics for all “Master” proteins identified at 1% FDR, which are representatives of parsimonious protein groups containing commonly identified peptides. Protein Relative Abundance is normalized for total peptide amount on each TMT channel.
Tab 6) Peptides _ RAW. Output from Proteome Discoverer 2.2, showing information on all peptides identified at 1% FDR, including both acetylated and unmodified peptides. This non-normalized quantitation was used for downstream analysis.
Tab 7) Proteins_ RAW. Output from Proteome Discoverer 2.2, showing information on all proteins to which the identified peptides map, regardless of protein-level FDR or “Master Protein” status. This non-normalized quantitation was used for downstream analysis.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Chemicals, Peptides, and Recombinant Proteins | ||
| Tris Salt of Phosphocreatine | Millipore Sigma | Cat# P1937 CAS# 108321-17-1 |
| Tris Salt of ATP | Millipore Sigma | Cat# A9062 CAS# 102047-34-7 |
| MOPS Free Acid | Millipore Sigma | Cat# M1254 CAS# 1132-61-2 |
| MES Potassium Salt | Millipore Sigma | Cat# M0895 CAS# 39946-25-3 |
| Bovine Serum Albumin (Fatty Acid Free) | Millipore Sigma | Cat# A3803 CAS# 9048-46-6 |
| Potassium Pyruvate | Combi-Blocks | Cat# QA-1116 CAS# 4151-33-1 |
| Potassium NADP+ | Ark-Pharm, Inc | Cat# AK671068 |
| Creatine Kinase from Rabbit Muscle | Millipore Sigma | Cat# 10736988001 |
| Amplex Ultra Red Reagent (AUR) | ThermoFisher | Cat# A36006 |
| CelLytic M | Millipore Sigma | Cat# C2978 |
| Tetramethylrhodamine Methyl Ester (TMRM) | ThermoFisher | Cat# T668 |
| Phosphoenoyl-pyruvate | Millipore Sigma | Cat# 10108294001 CAS# 4265-07-0 |
| Auranofin |
Millipore Sigma | Cat# A6733 |
| Rotenone | Millipore Sigma | Cat# R8875 CAS# 83-79-4 |
| Potassium Cyanide | Millipore Sigma | Cat# 60178 CAS# 151-50-8 |
| Peroxidase from Horseradish (HRP) | Millipore Sigma | Cat# P8375 CAS# 9003-99-0 |
| Superoxide Dismutase (SOD) | Millipore Sigma | Cat# S9697 CAS# 9054-89-1 |
| Glucose-6-phosphate Dehydrogenase (G6PDH) | Millipore Sigma | Cat# G6378 CAS# 9001-40-5 |
| Malate Dehydrogenase (MDH) | Millipore Sigma | Cat# 442610-M CAS# 9001-64-3 |
| Pyruvate Kinase/Lactic Dehydrogenase Enzymes from Rabbit Muscle | Millipore Sigma | Cat# P0294 |
| Hexokinase | Millipore Sigma | Cat# H4502 CAS# 9002-07-7 |
| Trypsin from Porcine Pancreas (Trypsin) | Millipore Sigma | Cat# T4799 CAS# 9001-51-8 |
| Creatine Monohydrate | Millipore Sigma | Cat# C3630 CAS# 6020-87-7 |
| Palmitoyl-L-carnitine | Millipore Sigma | Cat# P1645 CAS# 18877-64-0 |
| Octanoyl-L-carnitine | Millipore Sigma | Cat# 50892 CAS# 25243-95-2 |
| Malic Acid (Malate) | Millipore Sigma | Cat# M1000 CAS# 97-67-6 |
| Glutamic Acid (Glutamate) | Millipore Sigma | Cat# G1501 CAS# 6382-01-0 |
| Succinic Acid (Succinate) | Millipore Sigma | Cat# S3674 CAS# 110-15-6 |
| α-ketoglutaric Acid (AKG) | Millipore Sigma | Cat# K1750 CAS# 328-50-7 |
| Isocitrate | Millipore Sigma | Cat# 58790 CAS# 20226-99-7 |
| Adenosine Diphosphate (ADP) | Millipore Sigma | Cat# A5285 CAS# 72696-48-1 |
| NADH | Millipore Sigma | Cat# N4505 CAS# 104809-32-7 |
| Nicotinamide Adenine Dinucleotide (NAD+) | Millipore Sigma | Cat# N1636 CAS# 53-84-9 |
| L-Aspartic Acid (Aspartate) | Millipore Sigma | Cat# A9256 CAS# 56-84-8 |
| Acetoacetyl-CoA | Millipore Sigma | Cat# A1625 CAS# 1420-36-6 |
| Coenzyme A | Millipore Sigma | Cat# C3019 CAS# 18439-24-2 |
| Acetyl-CoA | Millipore Sigma | Cat# 10101907001 CAS# 32140-51-5 |
| Potassium Chloride | Millipore Sigma | Cat# P5405 CAS# 7447-40-7 |
| Sodium Chloride | Millipore Sigma | Cat# S7653 CAS# 7647-14-5 |
| EDTA | Millipore Sigma | Cat# E0270 CAS# 65501-24-8 |
| EGTA | Millipore Sigma | Cat# E4378 CAS# 67-42-5 |
| Alamethicin | Enzo Life Sciences | Cat# BML-A150-0025 |
| L-Carnitine HCl | Millipore Sigma | Cat# C0283 CAS# 6645-46-1 |
| CDNB | Millipore Sigma | Cat# 237329 CAS# 97-00-7 |
| Nicotinamide | Millipore Sigma | Cat# N3376 CAS# 98-92-0 |
| Antimycin A | Millipore Sigma | Cat# A8674 CAS# 1397-94-0 |
| Oligomycin | Millipore Sigma | Cat# 75351 CAS# 579-13-5 |
| Magnesium Green 5K+ salt | ThermoFisher Scientific | Cat# M3733 CAS# 170516-41-3 |
| Lactobionic Acid | Millipore Sigma | Cat# 153516 CAS# 96-82-2 |
| Glycerol | Millipore Sigma | Cat# G9012 CAS# 56-81-5 |
| Magnesium Chloride Hexahydrate | Millipore Sigma | Cat# M2670 CAS# 7791-18-6 |
| Potassium Dihydrogen Phosphate | Millipore Sigma | Cat# P9791 CAS# 7778-77-0 |
| HEPES | Millipore Sigma | Cat# H3375 CAS# 7365-45-9 |
| AP5A | Millipore Sigma | Cat# D4022 CAS# 4097-04-5 |
| Decylubiquinone | Enzo Life Sciences International | Cat# BML-CM115-0050 |
| D-Glucose (U-13C6, 99%) | Cambridge Isotope Laboratories, Inc | Cat# CLM-1396 CAS#110187-42-3 |
| MOX | Millipore Sigma | Cat# 226904 CAS# 593-56-6 |
| MTBSTFA with 1%TBDMCS | Millipore Sigma | Cat# M-108 CAS# 77377-52-7 |
| Chloroform | Millipore Sigma | Cat# C2432 CAS# 67-66-3 |
| Methanol | Millipore Sigma | Cat# 439193 CAS# 67-56-1 |
| Protease Inhibitor Cocktail | Millipore Sigma | Cat# P8340 |
| Pierce BCA Protein Assay Kit | ThermoFisher Scientific | Cat#23225 |
| Phosphatase Inhibitor Cocktail 2 | Millipore Sigma | Cat# P5726 |
| Phosphatase Inhibitor Cocktail 3 | Millipore Sigma | Cat# P0044 |
| Pierce Reversible Protein Stain Kit for Nitrocellulose Membranes | ThermoFisher Scientific | Cat# 24580 |
| 4–15% Criterion TGX Stain-Free Protein Gel, 18well | Biorad | Cat# 5678084 |
| 10X Tris Glycine SDS Running Buffer | Biorad | Cat# 1610732 |
| 10X Tris Buffered Saline | Biorad | Cat# 1706435 |
| Fish Gelatin | Millipore Sigma | Cat# G7765 |
| Casein | Millipore Sigma | Cat# C0626 |
| Sodium Nitrate | Millipore Sigma | Cat# S8032 |
| Thiamine Pyrophosphate | Millipore Sigma | Cat# C8754 CAS# 154-87-0 |
| Pyridoxal 5′-phosphate | Millipore Sigma | Cat# P9255 CAS# 853645-22-4 |
| Trichostatin A | Millipore Sigma | Cat# T8552 CAS# 58880-19-6 |
| D-(+)-Glucose Solution | Millipore Sigma | Cat# G8769 CAS# 50-99-7 |
| Roche cOmplete ULTRA EDTA-free Protease Inhibitor Mini Tablet | Millipore Sigma | Cat# 05892791001 |
| Roche 1x PhosSTOP Phosphatase Inhibitor Cocktail Tablets | Millipore Sigma | Cat# 04906837001 |
| Barium Hydroxide (0.3N) | Millipore Sigma | Cat# B4059 CAS #17194-00-2 |
| Zinc Sulfate (0.3N) | Millipore Sigma | Cat# Z2976 CAS #7733-02-0 |
| Lysyl Endopeptidase, Mass Spectrometry Grade | Wako Chemicals | Cat# 125-05061 |
| Sequencing Grade Modified Trypsin | Promega | Cat# V5113 |
| tC18 SEP-PAK Solid Phase Extraction Columns (50 mg) | Waters | Cat# WAT054960 |
| tC18 SEP-PAK Solid Phase Extraction Columns (100 mg) | Waters | Cat# WAT036820 |
| Triethylammonium bicarbonate (TEAB) | ThermoFisher | Cat# 90114 |
| TMT10plex™ Isobaric Label Reagent Set (0.8 mg per tag) | ThermoFisher | Cat# 90110 |
| MyTaq Red 2x Mix | Bioline | Cat# BIO-25044 |
| TRIzol Reagent | ThermoFisher | Cat# 15596026 |
| iTaq Universal SYBR Green Supermix | Biorad | Cat# 172-5121 |
| Humulin R U-500 | Lilly USA | |
| [3-3H]glucose | PerkinElmer | Cat# NET331C |
| 2[14C]deoxyglucose | PerkinElmer | Cat# NEC720A |
| UltimaGold Scintillation Fluid | PerkinElmer | Cat# 6013371 |
| Experimental Models: Organisms/Strains | ||
| C57BL/6NJ mice | The Jackson Laboratory | Stock #005304 |
| Skeletal Muscle and Heart Specific Double Knock-Out Mice for CrAT and Sirt3 Genes |
|
|
| Skeletal Muscle and Heart Specific Sirt3 Knock-Out Mice |
|
|
| Software and Algorithms | ||
| Web-based ΔGATP calculator | Fisher-Wellman et al., 2018 | https://dmpio.github.io/bioenergetic-calculators/ck_clamp/ |
| Proteome Discoverer 2.2 | ThermoFisher | N/A |
| Proteomic Statistical Analysis | This Publication* | https://github.com/dmpio/Williams_et_al_2019_Kac_PRX |
| CalR | Mina et al., 2018 |
https://calrapp.org |
| Other | ||
| QuantaMaster Spectrofluorometer | Horiba Scientific | Cat# QM-400 |
| TissueLyser II | Qiagen | Cat# 85300 |
| Oxygraph-2k | Orosboros Instruments | Cat# O2k-Core |
| Promethion Behavioral Phenotyping Mouse System | Sable Systems Int. | N/A |
| Tri-Carb 2900TR Liquid Scintillation Counter | PerkinElmer | N/A |
| Spectromax M2E Spectrophotometer | Molecular Devices | N/A |
| Gas Chromatography Mass Spectrometer | Agilent | Cat# 7890B GC-5977A MSD |
| Bio-Rad Turboblot Transfer System | Biorad | Cat# 1704150EDU |
| Thermo Scientific LTQ Orbitrap XLTM Mass Spectrometer | ThermoScientific | N/A |
| Waters nanoACQUITY Ultra Performance LC (nanoACQUITY UPLC) | ThermoScientific | N/A |
| Deposited Data | ||
| Proteomics Raw Data Files |
This Publication | PRIDE Accession: PXD014586 |
| Oligonucleotides | ||
| Sirt3 RT-qPCR: Fwd: AGGTGGAGGAAGCAGTGAGA | Fernandez-Marcos et al. 2012 | N/A |
| Sirt3 RT-qPCR: Reverse: GCTTGGGGTTGTGAAAGAAA | Fernandez-Marcos et al. 2012 | N/A |
| 18S RT-qPCR: Fwd: GTA ACC CGT TGA ACC CCA | This paper | N/A |
| 18S RT-qPCR: Reverse: CCA TCC AAT CGG TAG TAG CG | This paper | N/A |
| Critical Commercial Assays | ||
| TaqMan Gene Expression Assay on Demand for CrAT | ThermoFisher | Assay ID Mm00483985_m1 |
| Pierce Quantitative Colorimetric Peptide Assay | ThermoFisher | Cat# 23275 |
| PTMScan Acetyl-Lysine Motif [Ac-K] Kit | Cell Signaling Technology | Cat# 13416 |
| Pierce High pH Reversed-Phase Peptide Fractionation Kit | ThermoFisher | Cat# 84868 |
| RNeasy Mini Kit | Qiagen | Cat# 74106 |
| ALPCO STELLUX Chemi Rodent ELISA Kit | ALPCO | Cat# 80-INSMR-CH01 |
| iSCRIPT cDNA Synthesis Kit | Biorad | Cat# 1708840 |
The repository is currently private, so only individuals with a Github account and access to the DMPI repository can access the link at the moment. When the manuscript is accepted for publication, the repository will be made public, with full access to all files at the address: https://github.com/dmpio/Williams_et_al_2019_Kac_PRX
HIGHLIGHTS.
Mitochondria lacking CrAT and Sirt3 are susceptible to extreme protein acetylation
Hyperacetylation is accompanied by disturbances in redox balance and insulin action
Hyperacetylation does not affect mitochondrial respiration and enhances fat oxidation
Sirt3 flux and acetyl-lysine turnover promote a fuel switch from fat to glucose
Williams et al. show that double knockout (DKO) mice harboring muscle-specific deficits in acetyl-CoA buffering and lysine deacetylation are susceptible to extreme mitochondrial hyperacetylation and insulin resistance. However, DKO mitochondria have normal respiratory function and increased fat oxidation. The findings suggest that acetyl-lysine turnover, not stoichiometry, regulates mitochondrial fuel use.
CONTEXT AND SIGNIFICANCE.
Researchers at Duke University in North Carolina sought to understand why cellular engines–known as mitochondria–begin to fail during chronic overnutrition and obesity. One widely accepted theory suggests that surplus carbons derived from dietary fats react with mitochondrial machinery, producing biological debris that disrupts engine performance and compromises metabolic health. Results of the new study challenge this theory by showing that carbon debris does not damage mitochondria in skeletal muscle. Instead, a housekeeping process that cleans the debris draws energy from the engines in a manner that shifts muscle fuel use from fat to sugar, which in turn helps to control blood glucose levels and prevent diabetes. The findings provide new insights into the connection between obesity, type 2 diabetes and mitochondrial health.
ACKNOWLEDGEMENTS
This work was supported by National Institutes of Health, (F32DK105922 (ASW), F30DK1085602 (MTD), F32HL137398 (SBC), F32DK105665 (KFW), R01DK089312 (DMM). The mouse insulin clamp plasma insulin assays were performed at the VUMC Hormone Assay and Analytical Services Core supported by NIH grant DK059637. The insulin clamp plasma and tracer analysis and indirect calorimetry experiments were carried out at the Vanderbilt Mouse Metabolic Phenotyping Center (MMPC), supported by grants DK059637 and 1S10RR028101-01.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
There are no competing interests.
References
- Anderson EJ, Lustig ME, Boyle KE, Woodlief TL, Kane DA, Lin CT, Price JW 3rd, Kang L, Rabinovitch PS, Szeto HH, et al. (2009). Mitochondrial H2O2 emission and cellular redox state link excess fat intake to insulin resistance in both rodents and humans. J. Clin. Invest 119, 573–581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayala JE, Bracy DP, Julien BM, Rottman JN, Fueger PT, and Wasserman DH (2007). Chronic treatment with sildenafil improves energy balance and insulin action in high fat-fed conscious mice. Diabetes 56, 1025–1033. [DOI] [PubMed] [Google Scholar]
- Ayala JE, Bracy DP, McGuinness OP, and Wasserman DH (2006). Considerations in the design of hyperinsulinemic-euglycemic clamps in the conscious mouse. Diabetes 55, 390–397. [DOI] [PubMed] [Google Scholar]
- Barrientos A (2002). In vivo and in organello assessment of OXPHOS activities. Methods 26, 307–316. [DOI] [PubMed] [Google Scholar]
- Beenakkers AM, and Klingenberg M (1964). Carnitine-Coenzyme a Transacetylase in Mitochondria from Various Organs. Biochim. Biophys. Acta 84, 205–207. [DOI] [PubMed] [Google Scholar]
- Benjamini Y, and Hochberg Y (1995). Controlling the False Discovery Rate - a Practical and Powerful Approach to Multiple Testing. J Roy Stat Soc B Met 57, 289–300. [Google Scholar]
- Best JD, Kahn SE, Ader M, Watanabe RM, Ni TC, and Bergman RN (1996). Role of glucose effectiveness in the determination of glucose tolerance. Diabetes Care 19, 1018–1030. [DOI] [PubMed] [Google Scholar]
- Calvo SE, Clauser KR, and Mootha VK (2016). MitoCarta2.0: an updated inventory of mammalian mitochondrial proteins. Nucleic Acids Res 44, D1251–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carrico C, Meyer JG, He W, Gibson BW, and Verdin E (2018). The Mitochondrial Acylome Emerges: Proteomics, Regulation by Sirtuins, and Metabolic and Disease Implications. Cell metabolism 27, 497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chinopoulos C, Vajda S, Csanady L, Mandi M, Mathe K, and Adam-Vizi V (2009). A novel kinetic assay of mitochondrial ATP-ADP exchange rate mediated by the ANT. Biophys. J 96, 2490–2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Covian R, and Balaban RS (2012). Cardiac mitochondrial matrix and respiratory complex protein phosphorylation. American journal of physiology. Heart and circulatory physiology 303, H940–966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crown SB, Marze N, and Antoniewicz MR (2015). Catabolism of Branched Chain Amino Acids Contributes Significantly to Synthesis of Odd-Chain and Even-Chain Fatty Acids in 3T3-L1 Adipocytes. PloS one 10, e0145850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davies MN, Kjalarsdottir L, Thompson JW, Dubois LG, Stevens RD, Ilkayeva OR, Brosnan MJ, Rolph TP, Grimsrud PA, and Muoio DM (2016). The Acetyl Group Buffering Action of Carnitine Acetyltransferase Offsets Macronutrient-Induced Lysine Acetylation of Mitochondrial Proteins. Cell reports 14, 243–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dittenhafer-Reed KE, Richards AL, Fan J, Smallegan MJ, Fotuhi Siahpirani A, Kemmerer ZA, Prolla TA, Roy S, Coon JJ, and Denu JM (2015). SIRT3 mediates multi-tissue coupling for metabolic fuel switching. Cell metabolism 21, 637–646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elkhwanky MS, and Hakkola J (2018). Extranuclear Sirtuins and Metabolic Stress. Antioxidants & redox signaling 28, 662–676. [DOI] [PubMed] [Google Scholar]
- Fernandez-Marcos PJ, Jeninga EH, Canto C, Harach T, de Boer VC, Andreux P, Moullan N, Pirinen E, Yamamoto H, Houten SM, et al. (2012). Muscle or liver-specific Sirt3 deficiency induces hyperacetylation of mitochondrial proteins without affecting global metabolic homeostasis. Scientific reports 2, 425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher-Wellman KH, Davidson MT, Narowski TM, Lin CT, Koves TR, and Muoio DM (2018). Mitochondrial Diagnostics: A Multiplexed Assay Platform for Comprehensive Assessment of Mitochondrial Energy Fluxes. Cell reports 24, 3593–3606 e3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher-Wellman KH, Draper JA, Davidson MT, Williams AS, Narowski TM, Slentz DH, Ilkayeva OR, Stevens RD, Wagner GR, Najjar R, et al. (2019). Respiratory Phenomics across Multiple Models of Protein Hyperacylation in Cardiac Mitochondria Reveals a Marginal Impact on Bioenergetics. Cell reports 26, 1557–1572 e1558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher-Wellman KH, and Neufer PD (2012). Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends in endocrinology and metabolism: TEM 23, 142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fisher-Wellman KH, Weber TM, Cathey BL, Brophy PM, Gilliam LA, Kane CL, Maples JM, Gavin TP, Houmard JA, and Neufer PD (2014). Mitochondrial respiratory capacity and content are normal in young insulin-resistant obese humans. Diabetes 63, 132–141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frezza C, Cipolat S, and Scorrano L (2007). Organelle isolation: functional mitochondria from mouse liver, muscle and cultured fibroblasts. Nature protocols 2, 287–295. [DOI] [PubMed] [Google Scholar]
- Glancy B, Barstow T, and Willis WT (2008). Linear relation between time constant of oxygen uptake kinetics, total creatine, and mitochondrial content in vitro. American journal of physiology. Cell physiology 294, C79–87. [DOI] [PubMed] [Google Scholar]
- Glancy B, Willis WT, Chess DJ, and Balaban RS (2013). Effect of calcium on the oxidative phosphorylation cascade in skeletal muscle mitochondria. Biochemistry (Mosc) 52, 2793–2809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glozak MA, Sengupta N, Zhang X, and Seto E (2005). Acetylation and deacetylation of non-histone proteins. Gene 363, 15–23. [DOI] [PubMed] [Google Scholar]
- Golding EM, Teague WE Jr., and Dobson GP (1995). Adjustment of K’ to varying pH and pMg for the creatine kinase, adenylate kinase and ATP hydrolysis equilibria permitting quantitative bioenergetic assessment. J. Exp. Biol 198, 1775–1782. [DOI] [PubMed] [Google Scholar]
- Guaras A, Perales-Clemente E, Calvo E, Acin-Perez R, Loureiro-Lopez M, Pujol C, Martinez-Carrascoso I, Nunez E, Garcia-Marques F, Rodriguez-Hernandez MA, et al. (2016). The CoQH2/CoQ Ratio Serves as a Sensor of Respiratory Chain Efficiency. Cell reports 15, 197–209. [DOI] [PubMed] [Google Scholar]
- Hebert AS, Dittenhafer-Reed KE, Yu W, Bailey DJ, Selen ES, Boersma MD, Carson JJ, Tonelli M, Balloon AJ, Higbee AJ, et al. (2013). Calorie restriction and SIRT3 trigger global reprogramming of the mitochondrial protein acetylome. Mol. Cell 49, 186–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Goetzman E, Jing E, Schwer B, Lombard DB, Grueter CA, Harris C, Biddinger S, Ilkayeva OR, et al. (2010). SIRT3 regulates mitochondrial fatty-acid oxidation by reversible enzyme deacetylation. Nature 464, 121–125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirschey MD, Shimazu T, Jing E, Grueter CA, Collins AM, Aouizerat B, Stancakova A, Goetzman E, Lam MM, Schwer B, et al. (2011). SIRT3 deficiency and mitochondrial protein hyperacetylation accelerate the development of the metabolic syndrome. Mol. Cell 44, 177–190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Humphrey SJ, James DE, and Mann M (2015). Protein Phosphorylation: A Major Switch Mechanism for Metabolic Regulation. Trends in endocrinology and metabolism: TEM 26, 676–687. [DOI] [PubMed] [Google Scholar]
- Jing E, Emanuelli B, Hirschey MD, Boucher J, Lee KY, Lombard D, Verdin EM, and Kahn CR (2011). Sirtuin-3 (Sirt3) regulates skeletal muscle metabolism and insulin signaling via altered mitochondrial oxidation and reactive oxygen species production. Proc. Natl. Acad. Sci. U. S. A 108, 14608–14613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiyala KJ (2014). Mathematical model for the contribution of individual organs to non-zero y-intercepts in single and multi-compartment linear models of whole-body energy expenditure. PloS one 9, e103301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kall L, Canterbury JD, Weston J, Noble WS, and MacCoss MJ (2007). Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods 4, 923–925. [DOI] [PubMed] [Google Scholar]
- Kammers K, Cole RN, Tiengwe C, and Ruczinski I (2015). Detecting Significant Changes in Protein Abundance. EuPA Open Proteom 7, 11–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SC, Sprung R, Chen Y, Xu Y, Ball H, Pei J, Cheng T, Kho Y, Xiao H, Xiao L, et al. (2006). Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 23, 607–618. [DOI] [PubMed] [Google Scholar]
- Krumschnabel G, Eigentler A, Fasching M, and Gnaiger E (2014). Use of safranin for the assessment of mitochondrial membrane potential by high-resolution respirometry and fluorometry. Methods Enzymol 542, 163–181. [DOI] [PubMed] [Google Scholar]
- Lambert AJ, and Brand MD (2004). Inhibitors of the quinone-binding site allow rapid superoxide production from mitochondrial NADH:ubiquinone oxidoreductase (complex I). J. Biol. Chem 279, 39414–39420. [DOI] [PubMed] [Google Scholar]
- Lantier L, Williams AS, Hughey CC, Bracy DP, James FD, Ansari MA, Gius D, and Wasserman DH (2018). SIRT2 knockout exacerbates insulin resistance in high fat-fed mice. PloS one 13, e0208634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lantier L, Williams AS, Williams IM, Yang KK, Bracy DP, Goelzer M, James FD, Gius D, and Wasserman DH (2015). SIRT3 is crucial for maintaining skeletal muscle insulin action and protects against severe insulin resistance in high fat fed mice. Diabetes [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lighton JRB (2008). Measuring metabolic rates : a manual for scientists (Oxford; New York: Oxford University Press; ). [Google Scholar]
- Lombard DB, Alt FW, Cheng HL, Bunkenborg J, Streeper RS, Mostoslavsky R, Kim J, Yancopoulos G, Valenzuela D, Murphy A, et al. (2007). Mammalian Sir2 homolog SIRT3 regulates global mitochondrial lysine acetylation. Mol. Cell. Biol 27, 8807–8814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madiraju AK, Qiu Y, Perry RJ, Rahimi Y, Zhang XM, Zhang D, Camporez JG, Cline GW, Butrico GM, Kemp BE, et al. (2018). Metformin inhibits gluconeogenesis via a redox-dependent mechanism in vivo. Nat. Med 24, 1384–1394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin AS, Abraham DM, Hershberger KA, Bhatt DP, Mao L, Cui H, Liu J, Liu X, Muehlbauer MJ, Grimsrud PA, et al. (2017). Nicotinamide mononucleotide requires SIRT3 to improve cardiac function and bioenergetics in a Friedreich’s ataxia cardiomyopathy model. JCI Insight 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martines AMF, van Eunen K, Reijngoud DJ, and Bakker BM (2017). The promiscuous enzyme medium-chain 3-keto-acyl-CoA thiolase triggers a vicious cycle in fatty-acid beta-oxidation. PLoS Comput Biol 13, e1005461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell E, Peterson BS, Bomze HM, and Hirschey MD (2015). SIRT3 regulates progression and development of diseases of aging. Trends in endocrinology and metabolism: TEM 26, 486–492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Messer JI, Jackman MR, and Willis WT (2004). Pyruvate and citric acid cycle carbon requirements in isolated skeletal muscle mitochondria. American journal of physiology. Cell physiology 286, C565–572. [DOI] [PubMed] [Google Scholar]
- Mina AI, LeClair RA, LeClair KB, Cohen DE, Lantier L, and Banks AS (2018). CalR: A Web-Based Analysis Tool for Indirect Calorimetry Experiments. Cell metabolism. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muoio DM, and Neufer PD (2012). Lipid-induced mitochondrial stress and insulin action in muscle. Cell metabolism 15, 595–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muoio DM, Noland RC, Kovalik JP, Seiler SE, Davies MN, DeBalsi KL, Ilkayeva OR, Stevens RD, Kheterpal I, Zhang J, et al. (2012). Muscle-specific deletion of carnitine acetyltransferase compromises glucose tolerance and metabolic flexibility. Cell metabolism 15, 764–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayasu ES, Wu S, Sydor MA, Shukla AK, Weitz KK, Moore RJ, Hixson KK, Kim JS, Petyuk VA, Monroe ME, et al. (2014). A method to determine lysine acetylation stoichiometries. Int J Proteomics 2014, 730725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okuda S, Watanabe Y, Moriya Y, Kawano S, Yamamoto T, Matsumoto M, Takami T, Kobayashi D, Araki N, Yoshizawa AC, et al. (2017). jPOSTrepo: an international standard data repository for proteomes. Nucleic Acids Res 45, D1107–D1111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paik WK, Pearson D, Lee HW, and Kim S (1970). Nonenzymatic acetylation of histones with acetyl-CoA. Biochim. Biophys. Acta 213, 513–522. [DOI] [PubMed] [Google Scholar]
- Patti ME, and Corvera S (2010). The role of mitochondria in the pathogenesis of type 2 diabetes. Endocr. Rev 31, 364–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pehleman TL, Peters SJ, Heigenhauser GJ, and Spriet LL (2005). Enzymatic regulation of glucose disposal in human skeletal muscle after a high-fat, low-carbohydrate diet. J Appl Physiol (1985) 98, 100–107. [DOI] [PubMed] [Google Scholar]
- Perry CG, Kane DA, Lin CT, Kozy R, Cathey BL, Lark DS, Kane CL, Brophy PM, Gavin TP, Anderson EJ, et al. (2011). Inhibiting myosin-ATPase reveals a dynamic range of mitochondrial respiratory control in skeletal muscle. Biochem. J 437, 215–222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BS, Campbell JE, Ilkayeva O, Grimsrud PA, Hirschey MD, and Newgard CB (2018). Remodeling of the Acetylproteome by SIRT3 Manipulation Fails to Affect Insulin Secretion or beta Cell Metabolism in the Absence of Overnutrition. Cell reports 24, 209–223 e206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phipson B, Lee S, Majewski IJ, Alexander WS, and Smyth GK (2016). Robust Hyperparameter Estimation Protects against Hypervariable Genes and Improves Power to Detect Differential Expression. Ann Appl Stat 10, 946–963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poburko D, Santo-Domingo J, and Demaurex N (2011). Dynamic regulation of the mitochondrial proton gradient during cytosolic calcium elevations. J. Biol. Chem 286, 11672–11684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ransac S, Heiske M, and Mazat JP (2012). From in silico to in spectro kinetics of respiratory complex I. Biochim. Biophys. Acta 1817, 1958–1969. [DOI] [PubMed] [Google Scholar]
- Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, and Smyth GK (2015). limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romanick SS, Ulrich C, Schlauch K, Hostler A, Payne J, Woolsey R, Quilici D, Feng Y, and Ferguson BS (2018). Obesity-mediated regulation of cardiac protein acetylation: parallel analysis of total and acetylated proteins via TMT-tagged mass spectrometry. Biosci. Rep 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scaduto RC Jr., and Grotyohann LW (1999). Measurement of mitochondrial membrane potential using fluorescent rhodamine derivatives. Biophys. J 76, 469–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seiler SE, Koves TR, Gooding JR, Wong KE, Stevens RD, Ilkayeva OR, Wittmann AH, DeBalsi KL, Davies MN, Lindeboom L, et al. (2015). Carnitine Acetyltransferase Mitigates Metabolic Inertia and Muscle Fatigue during Exercise. Cell metabolism 22, 65–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steele R, Wall JS, De Bodo RC, and Altszuler N (1956). Measurement of size and turnover rate of body glucose pool by the isotope dilution method. Am. J. Physiol 187, 15–24. [DOI] [PubMed] [Google Scholar]
- Taus T, Kocher T, Pichler P, Paschke C, Schmidt A, Henrich C, and Mechtler K (2011). Universal and confident phosphorylation site localization using phosphoRS. J Proteome Res 10, 5354–5362. [DOI] [PubMed] [Google Scholar]
- Teague WE Jr., Golding EM, and Dobson GP (1996). Adjustment of K’ for the creatine kinase, adenylate kinase and ATP hydrolysis equilibria to varying temperature and ionic strength. J. Exp. Biol 199, 509–512. [DOI] [PubMed] [Google Scholar]
- Thapa D, Wu K, Stoner MW, Xie B, Zhang M, Manning JR, Lu Z, Li JH, Chen Y, Gucek M, et al. (2018). The protein acetylase GCN5L1 modulates hepatic fatty acid oxidation activity via acetylation of the mitochondrial beta-oxidation enzyme HADHA. J. Biol. Chem 293, 17676–17684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres MJ, Ryan TE, Lin CT, Zeczycki TN, and Neufer PD (2018). Impact of 17beta-estradiol on complex I kinetics and H2O2 production in liver and skeletal muscle mitochondria. J. Biol. Chem 293, 16889–16898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Ven RAH, Santos D, and Haigis MC (2017). Mitochondrial Sirtuins and Molecular Mechanisms of Aging. Trends Mol Med 23, 320–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Dijk TH, Laskewitz AJ, Grefhorst A, Boer TS, Bloks VW, Kuipers F, Groen AK, and Reijngoud DJ (2013). A novel approach to monitor glucose metabolism using stable isotopically labelled glucose in longitudinal studies in mice. Lab. Anim 47, 79–88. [DOI] [PubMed] [Google Scholar]
- Verkhovskaya ML, Belevich N, Euro L, Wikstrom M, and Verkhovsky MI (2008). Real-time electron transfer in respiratory complex I. Proc. Natl. Acad. Sci. U. S. A 105, 3763–3767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vizcaino JA, Csordas A, del-Toro N, Dianes JA, Griss J, Lavidas I, Mayer G, Perez-Riverol Y, Reisinger F, Ternent T, et al. (2016). 2016 update of the PRIDE database and its related tools. Nucleic Acids Res 44, D447–456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wagner GR, and Payne RM (2013). Widespread and enzyme-independent Nepsilon-acetylation and Nepsilon-succinylation of proteins in the chemical conditions of the mitochondrial matrix. J. Biol. Chem 288, 29036–29045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert BT, Iesmantavicius V, Moustafa T, Scholz C, Wagner SA, Magnes C, Zechner R, and Choudhary C (2014). Acetylation dynamics and stoichiometry in Saccharomyces cerevisiae. Mol Syst Biol 10, 716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinert BT, Iesmantavicius V, Wagner SA, Scholz C, Gummesson B, Beli P, Nystrom T, and Choudhary C (2013). Acetyl-phosphate is a critical determinant of lysine acetylation in E. coli. Mol. Cell 51, 265–272. [DOI] [PubMed] [Google Scholar]
- Weinert BT, Moustafa T, Iesmantavicius V, Zechner R, and Choudhary C (2015). Analysis of acetylation stoichiometry suggests that SIRT3 repairs nonenzymatic acetylation lesions. EMBO J 34, 2620–2632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weir JB (1949). New methods for calculating metabolic rate with special reference to protein metabolism. The Journal of physiology 109, 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wirth C, Brandt U, Hunte C, and Zickermann V (2016). Structure and function of mitochondrial complex I. Biochim. Biophys. Acta 1857, 902–914. [DOI] [PubMed] [Google Scholar]
- Zamora-Mendoza R, Rosas-Vargas H, Ramos-Cervantes MT, Garcia-Zuniga P, Perez-Lorenzana H, Mendoza-Lorenzo P, Perez-Ortiz AC, Estrada-Mena FJ, Miliar-Garcia A, Lara-Padilla E, et al. (2018). Dysregulation of mitochondrial function and biogenesis modulators in adipose tissue of obese children. Int J Obes (Lond) 42, 618–624. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. TMT-based quantitation of acetylation and protein abundance changes in skeletal muscle, related to Figures 1 and S1. Supplementary Excel file containing proteomic data—both direct output from Proteome Discoverer (Tabs 6 and 7) and summarized information from downstream statistical analysis (Tabs 2–5)—comparing the relative abundance of acetylated peptides and protein abundance, displayed on the following seven spreadsheet tabs:
Tab 1) Key. Includes descriptions of pertinent information fields included in the columns of the subsequent tabs and points to the location (tabs/columns) of key metrics from the study.
Tabs 2–3) Acetylpeptide SUMMARY. Summary of analyzed acetylproteomic data, after normalization of quantitative measurements and statistical analysis across genetic comparisons. Acetylpeptide Relative Abundance is normalized for total peptide amount on each TMT channel in the “input” fraction. Acetylpeptide Relative Occupancy is normalized for any biological changes in the “Master Protein” to which a given acetylated peptide maps (if identified at 1% protein-level FDR and quantified in Tabs 4–5).
Tabs 4–5) Protein SUMMARY. Summary of relative quantitation statistics for all “Master” proteins identified at 1% FDR, which are representatives of parsimonious protein groups containing commonly identified peptides. Protein Relative Abundance is normalized for total peptide amount on each TMT channel.
Tab 6) Peptides _ RAW. Output from Proteome Discoverer 2.2, showing information on all peptides identified at 1% FDR, including both acetylated and unmodified peptides. This non-normalized quantitation was used for downstream analysis.
Tab 7) Proteins_ RAW. Output from Proteome Discoverer 2.2, showing information on all proteins to which the identified peptides map, regardless of protein-level FDR or “Master Protein” status. This non-normalized quantitation was used for downstream analysis.
Data Availability Statement
All raw data for proteomics were submitted to the Proteome Xchange Consortium (Deutsch et al., 2017) via the PRIDE partner repository (Vizcaino et al., 2016) with the dataset identifier PXD014586. All code for the analyses in this published article can be found online (https://github.com/dmpio/Williams_et_al_2019_Kac_PRX).