

Abstract
The combination of congenital bilateral perisylvian syndrome (CBPS) with lower motor neuron dysfunction remains unusual and suggests a potential common genetic insult affecting basic neurodevelopmental processes. Here we identify a putatively pathogenic missense mutation in the MCF2 gene in a boy with CBPS. Using in utero electroporation to genetically manipulate cortical neurons during corticogenesis, we demonstrate that the mouse Mcf2 gene controls the embryonic migration of cortical projection neurons. Strikingly, we find that the CBPS‐associated MCF2 mutation impairs cortical laminar positioning, supporting the hypothesis that alterations in the process of embryonic neuronal migration can lead to rare cases of CBPS.
Introduction
Congenital bilateral perisylvian syndrome (CBPS) is a rare malformation of the cerebral cortex, most often associated with the following clinical signs: pseudobulbar palsy with oromotor apraxia, mild bilateral spastic palsy, cognitive impairment, and epilepsy.1 Despite this well‐reported clinical phenotype, there is often a diagnostic delay, especially when some of the clinical features are missing in the early stages of the disorder. CBPS is considered by certain authors as a peculiar form of cerebral palsy affecting predominantly bulbar muscles, but some patients also display congenital contractures as clinical and neurophysiological signs consistent with an involvement of the lower motor neuron (MN).2, 3, 4 The mechanism underlying this unusual combination of both central and peripheral motor impairment remains unknown, but it most probably involves a common genetic insult affecting basic cellular processes regulating the development of MNs and cortical projection neurons (PNs). During the embryonic period, PNs and MNs are generated in germinal ventricular zones then migrate radially to reach their final anatomical position and interestingly, they share common molecular regulators.5, 6, 7 Here, we identify a missense mutation in the MCF.2 Cell Line Derived Transforming Sequence (MCF2) gene in a boy with CBPS associated to lower MN dysfunction. Using functional in vivo studies in mice, we find that this CBPS‐associated human mutation has a pathogenic effect on the process of embryonic PNs migration in vivo.
Patient, Materials and Methods
Clinical description
The study was approved by the local ethics committee and both patient and parental consent were obtained. The patient, who is a 19‐year‐old boy, originates from Lebanon and was born premature at 31 gestation weeks after a prolonged oligoamnios. Both parents are healthy and not related. The mother has four healthy female siblings. The eldest sister had three children, including one healthy boy. One maternal aunt was born with a spina bifida occulta and carries no neurological sequelae. The patient is a single child. At birth, he exhibited bilateral clubfeet, hip dislocations, and asymmetric paraparesis leading to a presumed diagnosis of congenital arthrogryposis. During infancy and childhood, a severe developmental speech and language impairment was observed along with excessive drooling and feeding problems. Clinical examination of the patient in our institution in late childhood was characterized by a pseudobulbar palsy, an asymmetric lower limb distal paresis with no movement below the knees and severe muscle wasting. Right knee jerk was present but left knee jerk was absent, as were ankle jerks bilaterally. In addition, the patient did not report neither sphincterial dysfunction nor sensory impairment.
MRI study
All MRI data were acquired with a dedicated head coil on a 1.5T machine (Avanto Siemens Erlangen Germany).
Exome sequencing
Exome of the patient was captured using the Agilent SureSelet QXT Human All Exon V5 kit and sequenced on a HiSeq2000 instrument (Illumina). Reads mapping and variant calling were performed using BWA 0.7.13, Picard 2.9.0, GATK HaplotypeCaller 3.7 and annotated with annovar 2017‐07‐17 and UCSC RefSeq (refGene) downloaded on 2018‐08‐10. The variants were searched in various databases including dbSNP151, gnomAD 2.1, ClinVar 2018, and HGMD 2016. Pathogenicity prediction scores were obtained for missense variants using SIFT, PolyPhen2, MutationTaster (MT), CADD.
Mouse in utero electroporation
Animal experiments were conducted according to the Swiss and international guidelines, and approved by the local animal care committee. Embryos from time pregnant embryonic day (E)14.5 CD1 mice were electroporated in the lateral VZ of the dorsal pallium as described previously.8 All constitutive expression of shRNAs and cDNAs was driven by the human U6 promoter, in the PLKO.1 vector for the shRNAs and pUB6/V5‐His A (pUB6) vector for hMCF2 (kind gift from Danny Manor), TOM and GFP. The following shRNAs were electroporated in equal ratios in control and experimental conditions: Mcf2 shRNA (TRCN0000042653, Thermoscientific), and Scramble shRNA (mature sense: CCTAAGGTTAAGTCGCCCTCG, Addgene). The G4A missense mutation was induced in hMCF2 plasmid using site‐directed mutagenesis (InFusion Kit, Takara).
Results
MRI and nerve conduction studies
MRI scans of the brain revealed bilateral asymmetric perisylvian polymicrogyria extending to the parietal cortex on the right side. The sylvian sulci also displayed an abnormal configuration bilaterally (Fig. 1A–C). MRI scan of the spinal cord revealed a homogeneous thin spinal cord without the physiological lumbar enlargement (Fig. 1D).
Figure 1.
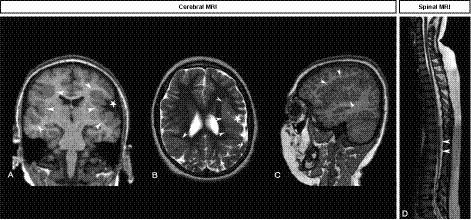
MRI study shows a thin spinal cord and bilateral perisylvian polymicrogyria. (A–C) Brain MRI. Coronal T1‐weighted sequence shows asymmetric bilateral perisylvian polymicrogyria (A), also clearly depicted on axial T2 (B) and sagittal T1‐weighted sequence (C) as a thickened irregular cortex (white arrowheads), with widening of the left sylvian fissure (white star). (D) Spinal MRI. Sagittal T2‐weighted sequence shows a thin dorso‐lumbar spinal cord with no anatomical lumbar bulge (white arrowheads).
Additionally, nerve conduction studies demonstrated a pure motor impairment from L4‐S2 on both sides typical of a lower MN dysfunction.
Exome sequencing
Exome sequencing of the proband and his healthy parents revealed a putatively damaging missense mutation inherited from his mother in the MCF2 gene, located on chromosome Xq27, (NM_005369.5): c.4G>A, p.(Ala2Thr) in exon 1. This variant is absent from the gnomAD database (https://gnomad.broadinstitute.org/) and is predicted as pathogenic by all used algorithms except Polyphen2 which classified it as likely pathogenic. MCF2, also known as DBL, is a member of a large family of guanine nucleotide exchange factors (GEFs) that modulates the activity of Rho GTPases.9, 10 Given that GEFs have been shown to act as key regulators of cellular migration,11 we aimed to test whether the CBPS‐associated MCF2 mutation affected cortical PNs in vivo.
In vivo mouse studies
mMcf2 was found to be expressed in control E17.5 PNs in a previously published RNA sequencing dataset8 and its subcellular cytoplasmic expression in cortical PNs at postnatal day (P)0 was visualized following overexpression of HA‐tagged mMCF2 by in utero electroporation of PNs progenitors at E14.5 in the dorsal pallium (Fig. 2A). A short hairpin RNA (shRNA) targeting mMcf2 induced a 40% knock‐down (KD) of the mRNA expression of mMcf2, as demonstrated by qRT‐PCR of HEK 293T cells cotransfected with pUB6‐mMcf2 and control Scram or mMcf2 targeting shRNA plasmids (Fig. 2B). Using in utero electroporation to genetically manipulate migrating PNs in mice, we next determined whether developmental KD of mMcf2 regulated the migration of PNs. Strikingly, the laminar positioning of PNs was altered by mMcf2 KD. Indeed, ectopic PNs were found in the lower layers of the somatosensory cortex in comparison to control (Scram) PNs, which were mostly located in superficial cortical layers. Interestingly, this phenotype was fully rescued by the overexpression of the WT human ortholog of mMcf2 (hMCF2), resistant to the effect of the mouse‐targeted shRNA, proving the specificity of the mMcf2‐KD‐related migratory defect. In contrast, overexpression of the CBPS‐associated mutated hMCF2 failed to rescue the migratory deficit induced by mMcf2 shRNA, indicating a pathogenic effect of the missense hMCF2 mutation (G4A ‐ p.A2T) on cortical PNs migration (Fig. 2C).
Figure 2.
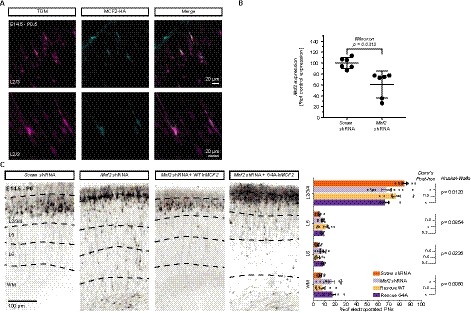
Mouse in vivo study: Mcf2 knock‐down (KD) alters the laminar positioning of projection neurons (PNs) at P0.5 and the G4A missense mutation alters the migratory function of MCF2. (A) The overexpression of hMCF2 by E14.5 electroporation in S1 PNs colabeled with Tomato (TOM) displays a cytoplasmic expression at P0.5. (B) q‐RT‐PCR of mRNA extracts from HEK‐293T cells cotransfected with mMcf2 and Mcf2 or Scramble (Scram) shRNA shows a 40% knock‐down (KD) of the mRNA expression by Mcf2 shRNA, compared to Scram shRNA. Error bars = 95% C.I. (C) shRNA‐mediated Mcf2 KD by in utero electroporation at E14.5 dramatically impairs the laminar positioning of E14.5‐electroporated PNs coexpressing TOM in S1 at P0.5. This phenotype is fully rescued by the overexpression of the shRNA‐resistant hMCF2 but the congenital bilateral perisylvian syndrome (CBPS)‐associated missense mutation G4A prevents any rescue. n = 6–11 brains per condition from ≥3 separate litters. Error bars = 95% CI.
Discussion
In this short report, we have attempted to explore the underlying genetic cause of an unusual but repetitively reported combination of perisylvian polymicrogyria and lower motor neuron dysfunction. These findings have led us to consider a genetic insult affecting basic cellular processes common to the maturation of both PNs and MNs. We have been able to identify an X‐linked recessive inherited CBPS‐associated missense mutation in hMCF2 (G4A ‐ p.A2T). Using cell‐type genetic manipulation in mouse embryos, we observed that mMcf2‐KD impairs corticogenesis by altering the laminar positioning of PNs in vivo. Strikingly, the hMCF2 (G4A ‐ p.A2T) mutation was found to have pathogenic effects on neuronal migration, suggesting that this developmental process could be altered in rare cases of CBPS.
The cell‐specificity of the migratory deficit described in PNs remains to be explored in MNs, but core biological pathways involved in the migration of PNs appear to be shared with MNs.5 In particular, MCF2 is part of the DBL family of RhoGEFs that represents critical regulators of cellular migration.11 RhoGEFs have been involved in regulating neuronal migration through the REELIN pathway12, 13 and the SEMAPHORIN/PLEXIN pathway.14, 15 The molecular underpinnings of the pathological effect of the missense mutation in MCF2 (G4A ‐ p.A2T) are beyond the scope of this study, but based on the structure of MCF2, we hypothesize that it could impact the function of the SEC14 domain of the protein, thus affecting its subcellular localization and its GEF activity.10 Interestingly, another missense mutation in MCF2 has been associated with schizophrenia,16 a complex neurodevelopmental disorder involving risk‐genes regulating neuronal migration.8 Additionally, one missense mutation in MCF2 has been described in a patient displaying undescended testis,17 possibly suggesting a role for MCF2 in cellular migration during organogenesis.
The use of animal models is a powerful tool in order to mechanistically understand neurodevelopmental disorders18 in the context of rare genetic variants associated with complex traits.19 The in vivo model used in this study allowed us to identify an interesting pathophysiological process caused by the acute KD of Mcf2 in the developing mouse. Further studies could take advantage of induced pluripotent stem cells and human organoids to recapitulate human developmental processes. Genomic analyses should be used to search for other rare variants associated with CBPS in a larger sample size to determine whether additional mutations in the MCF2 gene can be identified in rare cases of CBPS with similar lower motor neuron dysfunction. The convergence of additional genetic associations and in vivo/in vitro causal studies would allow to strengthen the pathophysiological mechanism identified in this study.
Author Contributions
AMC designed and performed all the in vivo and in vitro experiments, including all the analyses and wrote the manuscript. AD designed the animal model experiments and wrote the manuscript. JF identified the case and conducted the clinical investigations. MG and SL performed the exome sequencing experiment and analyses. ML analyzed the MRI scans and produced the images for the figure 1.
Conflict of Interest
The authors disclaim no conflict of interest.
Supporting information
Supplementary materials and methods S1
Acknowledgments
The authors would like to thank the patient and his parents, AMC thank Dr Eleanor D’Ersu for proofreading of the translation of the clinical results. This work was supported by the Institute of Genetics and Genomics of Geneva (to AMC and AD) and by the NCCR Synapsy (51NF40‐185897).
Funding information
This work was supported by the Institute of Genetics and Genomics of Geneva (to AMC and AD) and by the NCCR Synapsy (51NF40‐185897).
Funding Statement
This work was funded by NCCR Synapsy grant 51NF40‐185897.
References
- 1. Kuzniecky R, Andermann F, Guerrini R, et al. Congenital bilateral perisylvian syndrome: study of 31 patients. Lancet 1993;341:608–612. [DOI] [PubMed] [Google Scholar]
- 2. Clark M, Pitt M, Neville B. Lower motor neuron involvement in perisylvian polymicrogyria. Dev Med Child Neurol 2006;48:842–846. [DOI] [PubMed] [Google Scholar]
- 3. Clark M, Chong WK, Cox T, Neville BG. Congenital perisylvian dysfunction ‐ is it a spectrum? Dev Med Child Neurol 2010;52:33–39. [DOI] [PubMed] [Google Scholar]
- 4. Ravenscroft G, Donato N, Hahn G, et al. Recurrent de novo BICD2 mutation associated with arthrogryposis multiplex congenita and bilateral perisylvian polymicrogyria. Neuromuscul Disord 2016;26:744–748. [DOI] [PubMed] [Google Scholar]
- 5. Cooper JA. Cell biology in neuroscience: mechanisms of cell migration in the nervous system. J Cell Biol 2013;202:725–734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hawthorne A. Repurposing reelin: the new role of radial glia, Reelin and Notch in motor neuron migration. Exp Neurol 2014;256:17–20. [DOI] [PubMed] [Google Scholar]
- 7. Lee H, Kim M, Kim N, et al. Slit and Semaphorin signaling governed by Islet transcription factors positions motor neuron somata within the neural tube. Exp Neurol 2015;269:17–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Molinard‐Chenu A, Dayer A. The candidate schizophrenia risk gene DGCR2 regulates early steps of corticogenesis. Biol Psychiat 2018;83:692–706. [DOI] [PubMed] [Google Scholar]
- 9. Zheng Y. Dbl family guanine nucleotide exchange factors. Trends Biochem Sci 2001;26:724–732. [DOI] [PubMed] [Google Scholar]
- 10. Ognibene M, Vanni C, Blengio F, et al. Identification of a novel mouse Dbl proto‐oncogene splice variant: evidence that SEC14 domain is involved in GEF activity regulation. Gene 2014;537:220–229. [DOI] [PubMed] [Google Scholar]
- 11. Goicoechea SM, Awadia S, Garcia‐Mata R. I’m coming to GEF you: regulation of RhoGEFs during cell migration. Cell Adh Migr 2014;8:535–549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Franco S, Martinez‐Garay I, Gil‐Sanz C, et al. Reelin regulates Cadherin function via Dab1/Rap1 to control neuronal migration and lamination in the neocortex. Neuron 2011;69:482–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Lee GH, D’Arcangelo G. New insights into reelin‐mediated signaling pathways. Front Cell Neurosci 2016;10:122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Azzarelli R, Pacary E, Garg R, et al. An antagonistic interaction between PlexinB2 and Rnd3 controls RhoA activity and cortical neuron migration. Nat Commun 2014;5:3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tamagnone L, Comoglio P, Tamagnone L, Comoglio P. Signalling by semaphorin receptors: cell guidance and beyond. Trends Cell Biol 2000;10:377–383. [DOI] [PubMed] [Google Scholar]
- 16. Piton A, Gauthier J, Hamdan F, et al. Systematic resequencing of X‐chromosome synaptic genes in autism spectrum disorder and schizophrenia. Mol Psychiatr 2011;16:867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Xu W‐H, Zhang C, Zhao W‐M, et al. Mutational analysis of proto‐oncogene Dbl on Xq27 in testicular germ cell tumors reveals a rare SNP in a patient with bilateral undescended testis. World J Urol 2009;27:811. [DOI] [PubMed] [Google Scholar]
- 18. Mitchell KJ, Huang ZJ, Moghaddam B, Sawa A. Following the genes: a framework for animal modeling of psychiatric disorders. BMC Biol 2011;9:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Foo J‐N, Liu J‐J, Tan E‐K. Whole‐genome and whole‐exome sequencing in neurological diseases. Nat Rev Neurol 2012;8:508. [DOI] [PubMed] [Google Scholar]
- 20. Schindelin J, Arganda‐Carreras I, Frise E, et al. Fiji: an open‐source platform for biological‐image analysis. Nat Methods 2012;9:676–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods S1