

Abstract
Objective
To identify causative mutations in a patient affected by ataxia and spastic paraplegia.
Methods
Whole‐exome sequencing (WES) and whole‐genome sequencing (WGS) were performed using patient's DNA sample. RT‐PCR and cDNA Sanger sequencing were performed on RNA extracted from patient's fibroblasts, as well as western blot.
Results
A novel missense variant in SPG7 (c.2195T> C; p.Leu732Pro) was first found by whole‐exome sequencing (WES), while the second, also unreported, deep intronic variant (c.286 + 853A>G) was identified by whole‐genome sequencing (WGS). RT‐PCR confirmed the in silico predictions showing that this variant activated a cryptic splice site, inducing the inclusion of a pseudoexon into the mRNA sequence, which encoded a premature stop codon. Western blot showed decreased SPG7 levels in patient's fibroblasts.
Interpretation
Identification of a deep intronic variant in SPG7, which could only have been detected by performing WGS, led to a diagnosis in this HSP patient. This case challenges the notion of an autosomal dominant inheritance for SPG7, and illustrates the importance of performing WGS subsequently or alternatively to WES to find additional mutations, especially in patients carrying one variant in a gene causing a predominantly autosomal recessive disease.
Introduction
Hereditary spastic paraplegias (HSPs) are a group of neurodegenerative diseases characterized by progressive loss of upper motor neuron function. The primary pathophysiological feature of the disease is a “dying back type” axonal neuropathy that primarily affects upper motor neurons of the pyramidal tract.1, 2 Collectively, more than 80 genes and at least 76 linkage loci have been associated with HSP.3, 4 Among all of these genes, mutations in SPAST, ATL1, SPG7, and SPG11 remain the most prevalent causes of HSP. Novel sequencing strategies such as whole‐exome sequencing (WES) have accelerated the identification of causative variants associated with HSP, including the identification of novel gene–disease associations,5 although the diagnostic rate reaches only ~ 50% in carefully selected cohorts of patients.6, 7 Whole‐genome sequencing (WGS) can improve the detection of variants, as it provides uniform coverage of exonic regions (especially important to screen regions that are not efficiently probed by WES), the possibility of detecting deep intronic/intergenic variants, and improved calling of structural variants.8 Although WGS has a higher economic cost compared to WES and the interpretation of non‐coding variants remains a challenge, it has already been used successfully to find pathogenic variants in HSP patients and other related neurological conditions (Table S1). In this report, we describe a patient in which a missense heterozygous variant in SPG7 was first found by WES. To find additional mutations, we performed WGS, which identified a second heterozygous deep intronic variant in SPG7 that could not have been detected by WES alone. After functional analysis, we concluded that this second deep intronic variant in SPG7 is responsible for this patient's disease.
Methods
Patients and samples
The proband was recruited and examined at the Neurology department of Donostia University Hospital. Detailed neurological examinations were performed on all members of the family. Blood samples and skin‐derived fibroblast cell lines were obtained using standard methods. Informed consent was obtained from all participants in this study. The research project was approved by the Clinical Research Ethics Committee for Research Ethics Committee of the Bellvitge University Hospital (PR076/14).
Whole‐Exome (WES) and Whole‐Genome Sequencing (WGS)
Genomic DNA was extracted from peripheral blood using standard methods. WES was performed on patient's DNA sample using the SureSelect XT Human All Exon V5 50 Mb kit (Agilent) for DNA capture. Exome and Genome DNA sequencing was performed using the HiSeq 2000 Platform (Illumina) at CNAG (Centre Nacional d'Anàlisi Genòmica, Barcelona). Variants were filtered using RD‐Cat platform (https://rdcat.cnag.crg.eu/) and an in‐house pipeline based on GATK best practice guidelines and prioritization based on accurate patient characterization with HPO terms (Human Phenotype Ontology), interaction networks at physical and functional levels and variant intolerance scores generated by the ExAC and gnomAD consortia. We prioritized variants in HSP genes that had a frequency lower than 0.01 in ExAC (Exome Aggregation Consortium), 1000 genomes, ESP (NHLBI Exome Sequencing Project), and gnomAD control databases. In silico predictors were used to assess the effect of variants on splicing (Human Splicing Finder, MaxEntScan, NNSplice, NetGene2, FSplice, and SpliceAI9). Furthermore, we screened a curated list of HSP (79), spinocerebellar ataxia (49), and spastic ataxia genes (7). Candidate variants were validated and tested for cosegregation in all family members by Sanger sequencing.
RNA splicing analysis
Fibroblast cell lines from the proband, a healthy brother, and an unrelated control were cultured at 37ºC and 5% CO2 in Dulbecco's modified eagle medium (DMEM) supplemented with 10% fetal bovine serum (FBS) and 100U/mL penicillin + 100µl/mL streptomycin. RNA was extracted using the RNeasy Mini Kit (QIAGEN) and cDNA was synthesized using the Superscript IV kit (Life Technologies) following manufacturer's instructions. cDNA was amplified by polymerase chain reaction (PCR) using primers targeting exons 1–4 of SPG7 gene (forward: 5′‐CGGCTTTCAGGCCAACAT‐3′; reverse: 5′‐CGCTCTCGGTACATCTGGTC‐3′) followed by Sanger sequencing.
Western blot
For western blotting analyses, fibroblasts were homogenized in RIPA buffer (150 mmol/L NaCl, 1% Nonidet P40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mmol/L Tris, pH 8.0), sonicated for 2 min at 4ºC, centrifuged, mixed with 4X NuPAGE LDS Sample Buffer (Invitrogen) and heated at 70ºC for 10 min. 60 µg of protein samples was subjected to polyacrylamide gel electrophoresis at 120V in NuPAGE MOPS SDS Running Buffer (Invitrogen) supplemented with 5 mmol/L sodium bisulfite (Ref. 243973, Sigma‐Aldrich). Proteins were transferred to nitrocellulose membranes using iBlot 2 Gel Transfer Device (Invitrogen). After blocking in 5% Bovine Serum Albumin (BSA, Sigma‐Aldrich), 0.05% TBS‐Tween (TBS‐T) for 1 h at room temperature, membranes were incubated with primary antibodies overnight at 4°C. Primary antibodies included Anti‐β‐Actin, mouse (dilution 1/8000, ref:A2228, Sigma‐Aldrich); Anti‐OPA1, mouse (dilution 1/1000, ref: 612607, BD Transduction Laboratories); and Anti‐SPG7, mouse (dilution: 1/500, ref: SC‐514393, Santa Cruz Biotechnology). After incubation with secondary antibodies for 1 h at room temperature, proteins were detected with ChemidocTM Touch Imaging System (BioRad). Bands were quantified with ImageLab (BioRad).
Results
Clinical description
The proband (patient II.1) is a 52‐year‐old man in ongoing follow‐up at the neurology outpatient clinic of Donostia University Hospital due to a 15‐year‐long history of altered gait. Spastic paraplegia with gait ataxia was observed at the first examination in 2005. Mild progressive external ophthalmoplegia (PEO) with ptosis and cerebellar dysarthria were also present. This patient is the first of three sons of a non‐consanguineous couple of Spanish origin. His father had suffered from a paraneoplastic anti‐Hu‐positive cerebellar and polyneuropathic ataxia associated with small‐cell lung cancer and recently died at the age of 73 years. There are no other family members suffering from any disease affecting the central or peripheral nervous system.
A complete diagnostic workup was undertaken to exclude any acquired cause of spastic paraparesis or gait ataxia. Full blood analysis was normal, including absence of acanthocytosis, and negative antineuronal, onconeuronal, and ganglioside antibodies. Only antinuclear antibodies were positive in 1/640 titers for several analyses, but a thorough rheumatology study was unremarkable, including negative autoantibodies and normal salivary gland biopsy. Oxysterol levels ruled out Niemann Pick disease. CSF analysis was also unremarkable. On MRI, there was bilateral frontotemporal and right parietal atrophy with grade 1 subcortical leukoencephalopathy. No T2 hyperintensities were seen in the dentate nuclei. There was concomitant cerebellar atrophy and no signs of myelopathy (Fig. 1A and 1B). Whole‐body muscle‐MRI showed no myopathic changes, and thoracoabdominopelvic CT scan was negative for any mass suggestive of malignancy. There was no evidence of polyneuropathy or myopathy on nerve conduction studies and electromyography. Genetic analysis for different spinocerebellar ataxias (SCA1, 2, 3, 6, 7), DRPLA and Friedreich ataxia were negative. Furthermore, no mutations were found in the mitochondrial DNA.
Figure 1.
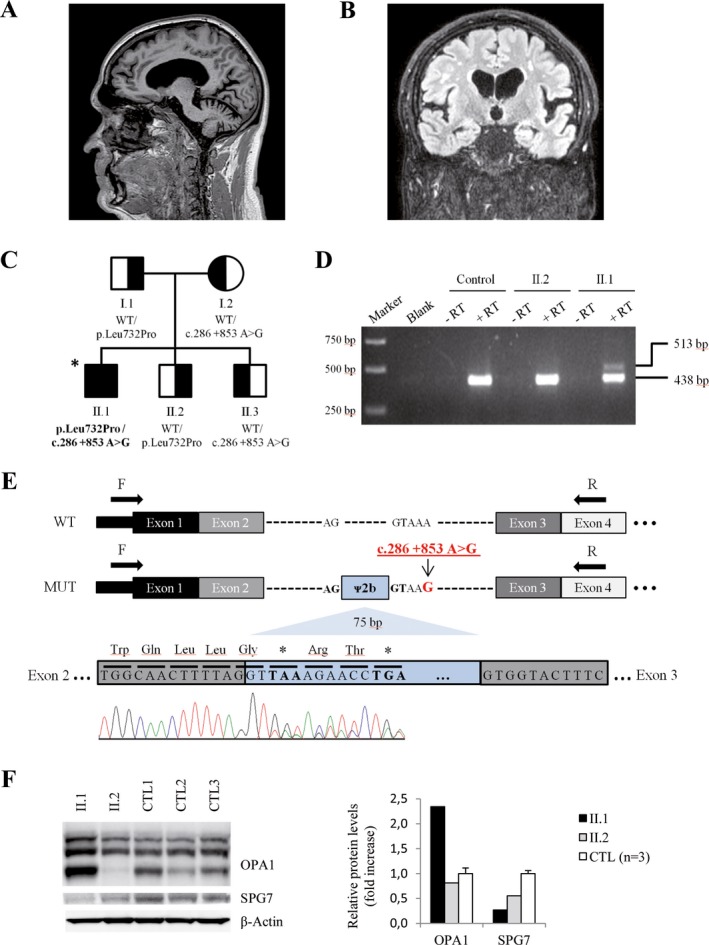
MRI brain images and functional evaluation in fibroblasts from Patient II.1. (A, B) Brain MRI of subject II.1 showing typical cerebellar atrophy of SPG7 disease (sagittal T2w) (A), and bilateral frontotemporal and right parietal atrophy with grade 1 subcortical leukoencephalopathy (coronal FLAIR) (B). (C) Family pedigree and genotype data for the SPG7 variants. Symbols: square, male; circle, female; filled, affected individual; half‐filled, clinically healthy carriers; asterisk, individual sequenced by WES/WGS. Variants found in SPG7 are shown below each symbol. WT: wild‐type. (D) Agarose gel electrophoresis of RT‐PCR products showing an additional fragment (513 bp) in patient II.1. No products were observed in negative RT controls or water controls (blank). A healthy family member (II.2) not carrying the c.286 + 853A>G variant and an unrelated individual were used as healthy controls. (E) Consequence of the c. 286 + 853 A> G variant. WT: structure of wild‐type SPG7 transcript (exons 1–4). MUT: structure of SPG7 transcript generated by the c. 286 + 853 A> G variant. The pseudoexon results from a cryptic donor splice site activation upstream from the c.286 + 853A>G mutation. The in‐frame inclusion of 75 nucleotides of intron 2 (blue, pseudoexon ᴪ2b) translates into premature stop codons at positions 1 and 4 downstream. Arrows indicate primers used for RT‐PCR. cDNA Sanger sequencing shows two populations of mRNA, one corresponding to pseudoexon inclusion and the other to exon 3. (F) Western blot of OPA1 and SPG7 proteins for human fibroblasts of individuals II.1 (patient), II.2 (healthy brother) and controls (n = 3). Right, quantification of western blot bands. Data represented as mean ± SD
The patient's disability has progressed slowly, and at the last visit (May 2019), he is currently able to walk short distances with a walker or cane, requiring a wheelchair for the rest of activities. The Spastic Paraplegia Rating Scale (SPRS) was 15/52 (1,3,2,2,2,2,1,1,1,0,0,0,0), the timed 25‐foot walk test (T25FW) was 15.94 sec, and the 6‐min walk test (6MWT) was 177 meters. No waddling or signs of proximal myopathy were observed. There has been no cognitive or behavioral decline up to this date. He continues to show cerebellar dysarthria. A slight improvement was observed after 1 month of fampridine (Fampyra) (T25FW = 13.73 sec), while other symptomatic treatments failed.
Molecular studies
WES analysis identified a heterozygous missense variant in SPG7 (chr16:89623308T> C hg19, NM_003119.3:c.2195T> C, p.Leu732Pro), which was inherited from his father (Fig. 1C). This variant is completely absent from control databases, is highly conserved in evolution (GERP score: 5.59), is predicted to be deleterious by at least three different predictors (PolyPhen‐2, SIFT, MutationTaster), and is located in the conserved peptidase M41 domain, close to other described missense mutations. Based on these data, we classified the variant as “Likely Pathogenic” according to ACMG/AMP guidelines.10 As the clinical presentation of this patient was compatible with mutations in SPG7, and as we did not find other rare candidate variants in exome data for this patient, we decided to perform WGS on this patient's sample to seek a second variant in SPG7, while using better coverage to rule out other possible disease‐causative genes or novel candidates that could underlie our patient's phenotype.
Indeed, while WGS did not detect candidate variants of interest in other HSP or ataxia genes, it revealed a novel heterozygous variant within intron 2 of the SPG7 gene, which was inherited from the mother (Fig. 1C). This variant (chr16:89577853A> G hg19, NM_003119.3:c.286 + 853A>G) is deep intronic (>100 bp from the splice site), completely absent from control databases, and has not been previously reported as associated with disease. Segregation in two healthy siblings was compatible with pathogenicity of the variant. In silico analysis using various splice site predictors suggested the activation of a cryptic donor site 5 bp upstream from this variant (Table S2). Furthermore, SpliceAI, a very novel tool to predict cryptic splice sites based on deep learning of artificial neural networks,9 additionally predicted the activation of an acceptor site located 80 bp upstream from the c.286 + 853A>G variant at c.286 + 773. This novel acceptor site should function in conjunction with the previous donor site of exon 2 at position c.286 + 1, leading to the inclusion of a cryptic exon from c.286 + 774 to c.286 + 848. The effect appeared specific for the change c.286 + 853A>G, as no donor or acceptor activation events were predicted by SpliceAI for other nucleotide changes, such as c.286 + 853 A> C or A> T. Thus, we set out to validate the prediction and extracted cDNA from the patient's fibroblasts, a healthy sibling, and an additional control. Gel analysis and Sanger sequencing revealed that a 75 bp in‐frame pseudoexon was retained in the cDNA as a consequence of the c.286 + 853A>G variant, matching the SpliceAI prediction (Fig. 1D). This novel mRNA species contains an immediate, premature stop codon at p.(Gly96Gly*1), which is supposed to induce degradation of the transcript by nonsense mediated decay (NMD) (Fig. 1E). No evidence of transcripts containing this pseudoexon was found in the RNA‐seq data of control fibroblasts from the Genotype‐Tissue Expression project (GTEx, https://gtexportal.org). Ensuing this functional validation, the c.286 + 853A>G variant can be classified as “Pathogenic” according to ACMG/AMP guidelines.10 Western blot analyses showed that SPG7 protein was firmly decreased in patient II.1 fibroblasts compared to controls, in concordance with the expected loss‐of‐function effect of the deep intronic variant, while his healthy brother showed a less severe reduction in SPG7 levels, possibly linked to an effect of the p.Leu732Pro variant on protein stability (Fig. 1F). On the other hand, OPA1 protein levels were strongly upregulated in patient II.1, a feature that has also been observed in other SPG7‐mutated patients.11 Thus, protein analysis further confirmed the implication of SPG7 variants on our patient's phenotype.
Discussion
In this work, we present a case in which WGS was indispensable to identify the causative mutations in an HSP patient. Although various studies in the last years have used WGS to study the etiology of HSP, the variants identified in these works were located near or inside coding regions and could very probably have been detected by WES (Table S1). In contrast, our patient harbored a deep intronic variant inducing the inclusion of a pseudoexon and premature stop sites, which could not have been identified with WES alone, underscoring the usefulness of WGS to increase diagnostic yield. Alternatively, transcriptome sequencing could have been used, as it has recently been proven useful for other genetic diseases.12, 13 Deep intronic mutations are estimated to account for 10% of pathogenic mutations in the general rare genetic disorders population,9, 14 and could underlie a significant number of undiagnosed HSP/ataxia cases as it has been shown in our case,15, 16 although it is still underreported.
This case, in which only one coding variant was detected at first, highlights that the existence of dominant variants in SPG7 should be reexamined. To date, the main argument supporting a dominant inheritance of some variants is the description of four families with patients with two mutations having an affected relative carrying only one mutated allele and confirmed by Sanger sequencing.17, 18, 19 Moreover, at least 10 apparently dominant HSP families have been reported, in which the parents of index patients with two mutations in trans were also affected, although their carrier status was not confirmed by Sanger sequencing.20 However, a full screening of SPG7 in heterozygous affected relatives was not undertaken in any of these works. It is tempting to speculate that some heterozygous patients in these families in fact carry a third pathogenic allele that would have eluded identification in a first screening, thus mimicking a dominant or pseudodominance pattern in the first instance. Indeed, three families with a pseudodominant mode of inheritance have already been described, with three different pathogenic alleles segregating in the family.21, 22 It is worth noting that the p.Ala510Val pathogenic variant is relatively frequent in public control databases (0.25% in ExAC, 0.4% in European population) and was carried in homozygotes by some affected individuals in the three families mentioned. Although the pseudodominance scenario would not reconcile well with the existence of SPG7‐mutated families with three affected generations described in literature, we believe that full gene screening in the apparently heterozygous individuals should be required for claiming the existence of dominant mutations in SPG7 in future cases, including non‐coding regions undetectable by WES. As suggested by this case description, WGS and cDNA/transcriptome sequencing will become key diagnostic tools for uncovering mutations undetectable by WES. In addition, these techniques could also help to assess the pathogenicity of SPG7 variants classified as variants of unknown significance (VUS), while screening for the presence of additional candidate variants in deep intronic regions. This screening will be crucial to better determine the pathogenicity of SPG7 variants and improve current clinical management and genetic counseling for these families.
Finally, the reported case is affected by cerebellar ataxia, a frequent feature in SPG7‐mutated patients (65–90% patients),20, 22, 23 which was clearly predominant over the spastic paraplegia component, and dysarthria (up to 76% of patients in a recent cohort).23 Our patient only harbors one heterozygous loss‐of‐function mutation (the deep intronic cryptic splice variant), which is in line with his predominantly ataxic phenotype, as a recent analysis of 249 patients associated biallelic loss‐of‐function mutations in SPG7 with stronger spasticity phenotypes.20 This patient also had PEO and a high degree of disability, which are less common features in SPG7‐mutated patients. PEO is present in ~ 26% of Spanish patients, although frequency doubles in patients not carrying the common p.Ala510Val mutation in homozygosis.22, 24 Given that SPG7 is one of the most frequent causes of HSP, and also a relatively frequent cause underlying hard‐to‐diagnose ataxias,25 it would be prudent to search for intronic SPG7 variants in patients with ataxia or spastic paraplegia, especially if they have associated PEO, or are already known to harbor one suspicious heterozygous variant in this gene.
Author Contribution
AP and CC provided funding to run the study. EV, GFE, ALM, and AP designed and conceptualized the study. EV, AS, GFE, RRM, MZ, LPS, MR, and SF performed the analysis and interpreted the data. EV, GFE, ALM, and AP drafted the manuscript. All authors critically revised the manuscript.
Conflict of Interests
The authors report no disclosures or conflicts of interest relevant to the manuscript.
Supporting information
Table S1. Previous descriptions of HSP patients sequenced by WGS
Table S2. Splice prediction scores obtained for the c.286+853A>G mutation
Acknowledgments
We are indebted to the family members who participated in this study. We thank CERCA Program/Generalitat de Catalunya for institutional support. We also thank Cristina Guilera and Juanjo Martínez for their excellent technical assistance. This study was supported by the Centre for Biomedical Research on Rare Diseases (CIBERER) [ACCI14‐759], the URDCat program (PERIS SLT002/16/00174), the Hesperia Foundation and the Secretariat for Universities and Research of the Ministry of Business and Knowledge of the Government of Catalonia [2017SGR1206] to AP, and Instituto de Salud Carlos III [PI14/00581] (Co‐funded by European Regional Development Fund. ERDF, a way to build Europe) and La Marató de TV3 [345/C/2014] to CC and AP. EV was funded by a grant from the Ministerio de Economia, Industria y Competividad (Juan de la Cierva programme FJCI‐2016‐28811); LPS was funded by a predoctoral grant from the Instituto de Salud Carlos III (PFIS, FI18/00141); SF was funded by the Instituto de Salud Carlos III [Miguel Servet programme CPII16/00016, co‐funded by European Social Fund. ESF investing in your future], and MR was funded by CIBERER.
Edgard Verdura, Agatha Schlüter and Gorka Fernández‐Eulate are Contributed equally.
Adolfo López de Munain and Aurora Pujol are Contributed equally.
Funding Statement
This work was funded by Instituto de Salud Carlos III grants FI18/0041, Miguel Servet programme CPII16/00016, and [PI14/00581]; Fundació la Marató de TV3 grant [345/C/2014]; PERIS (Generalitat de Catalunya) grant URDCat program (SLT002/16/00174); Hesperia Foundation grant ; CIBERER grant [ACCI14‐759]; Secretariat for Universities and Research of the Ministry of Business and Knowledge of the Government of Catalonia grant [2017SGR1206] ; Ministerio de Economia, Industria y Competividad grant Juan de la Cierva programme FJCI‐2016‐28811.
Contributor Information
Adolfo López de Munain, Email: adolfo.lopezdemunainarregui@osakidetza.eus.
Aurora Pujol, Email: apujol@idibell.cat.
References
- 1. DeLuca GC, Ebers GC, Esiri MM. The extent of axonal loss in the long tracts in hereditary spastic paraplegia. Neuropathol Appl Neurobiol 2004;30:576–584. [DOI] [PubMed] [Google Scholar]
- 2. Fink JK. Hereditary spastic paraplegia: clinical principles and genetic advances. Semin Neurol 2014;34:293–305. [DOI] [PubMed] [Google Scholar]
- 3. Lo Giudice T, Lombardi F, Santorelli FM, et al. Hereditary spastic paraplegia: Clinical‐genetic characteristics and evolving molecular mechanisms. Exp Neurol 2014;261:518–539. [DOI] [PubMed] [Google Scholar]
- 4. Parodi L, Coarelli G, Stevanin G, et al. Hereditary ataxias and paraparesias: Clinical and genetic update. Curr Opin Neurol 2018;31:462–471. [DOI] [PubMed] [Google Scholar]
- 5. Novarino G, Fenstermaker AG, Zaki MS, et al. Exome Sequencing links corticospinal motor neuron disease to common neurodegenerative disorders. Science (80‐. ) 2014;343:506–511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Kara E, Tucci A, Manzoni C, et al. Genetic and phenotypic characterization of complex hereditary spastic paraplegia. Brain 2016;139(Pt 7):1904–1918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Schule R, Wiethoff S, Martus P, et al. Hereditary spastic paraplegia: clinicogenetic lessons from 608 patients. Ann Neurol 2016;79(4):646–658. [DOI] [PubMed] [Google Scholar]
- 8. Vincent QB, Shang L, Casanova J‐L, et al. Whole‐genome sequencing is more powerful than whole‐exome sequencing for detecting exome variants. Proc Natl Acad Sci 2015;112(17):5473–5478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Jaganathan K, Kyriazopoulou Panagiotopoulou S, McRae JF, et al. Predicting splicing from primary sequence with deep learning. Cell 2019;176:535–548.e24. [DOI] [PubMed] [Google Scholar]
- 10. Richards S, Aziz N, Bale S, et al. Standards and guidelines for the interpretation of sequence variants: a joint consensus recommendation of the American College of medical genetics and genomics and the association for molecular pathology. Genet Med 2015;17(5):405–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Pfeffer G, Gorman GS, Griffin H, et al. Mutations in the SPG7 gene cause chronic progressive external ophthalmoplegia through disordered mitochondrial DNA maintenance. Brain 2014;137(5):1323–1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Kremer LS, Bader DM, Mertes C, et al. Genetic diagnosis of Mendelian disorders via RNA sequencing. Nat Commun 2017;8:15824. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Cummings BB, Marshall JL, Tukiainen T, et al. Improving genetic diagnosis in Mendelian disease with transcriptome sequencing. Sci. Transl. Med. 2017;9:eaal5209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Vaz‐Drago R, Custódio N, Carmo‐Fonseca M. Deep intronic mutations and human disease. Hum Genet 2017;136(9):1093–1111. [DOI] [PubMed] [Google Scholar]
- 15. Minnerop M, Kurzwelly D, Wagner H, et al. Hypomorphic mutations in POLR3A are a frequent cause of sporadic and recessive spastic ataxia. Brain 2017;140(6):1561–1578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Bis‐Brewer DM, Züchner S. Perspectives on the Genomics of HSP Beyond Mendelian Inheritance. Front Neurol 2018;9:958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Klebe S, Depienne C, Gerber S, et al. Spastic paraplegia gene 7 in patients with spasticity and/or optic neuropathy. Brain 2012;135(Pt 10):2980–2993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Sánchez‐Ferrero E, Coto E, Beetz C, et al. SPG7 mutational screening in spastic paraplegia patients supports a dominant effect for some mutations and a pathogenic role for p. A510V. Clin Genet 2013;83:257–262. [DOI] [PubMed] [Google Scholar]
- 19. McDermott CJ, Dayaratne RK, Tomkins J, et al. Paraplegin gene analysis in hereditary spastic paraparesis (HSP) pedigrees in northeast England. Neurology 2001;56:467–471. [DOI] [PubMed] [Google Scholar]
- 20. Coarelli G, Schule R, van de Warrenburg BPC, et al. Loss of paraplegin drives spasticity rather than ataxia in a cohort of 241 patients with SPG7 . Neurology 2019;92:e2679–e2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. van Gassen KLI, van der Heijden CDCC, de Bot ST, et al. Genotype–phenotype correlations in spastic paraplegia type 7: a study in a large Dutch cohort. Brain 2012;135:2994–3004. [DOI] [PubMed] [Google Scholar]
- 22. De la Casa‐Fages B, Fernández‐Eulate G, Gamez J, et al. Parkinsonism and spastic paraplegia type 7: Expanding the spectrum of mitochondrial Parkinsonism. Mov Disord 2019;34:1547–1561. [DOI] [PubMed] [Google Scholar]
- 23. Hewamadduma CA, Hoggard N, O'Malley R, et al. Novel genotype‐phenotype and MRI correlations in a large cohort of patients with SPG7 mutations. Neurol Genet 2018;4:e279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Choquet K, Tétreault M, Yang S, et al. SPG7 mutations explain a significant proportion of French Canadian spastic ataxia cases. Eur J Hum Genet 2016;24:1016–1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Pfeffer G, Pyle A, Griffin H, et al. SPG7 mutations are a common cause of undiagnosed ataxia. Neurology 2015;84:1174–1176. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Previous descriptions of HSP patients sequenced by WGS
Table S2. Splice prediction scores obtained for the c.286+853A>G mutation