

Abstract
Diabetes mellitus accelerates vascular calcification (VC) and increases the risk of end-stage renal disease (ESRD). Nevertheless, the impact of VC in renal disease progression in type 2 diabetes mellitus (T2DM) is poorly understood. We addressed the effect of VC and mechanisms involved in renal dysfunction in a murine model of insulin resistance and obesity (ob/ob), comparing with their healthy littermates (C57BL/6). We analyzed VC and renal function in both mouse strains after challenging them with Vitamin D3 (VitD3). Although VitD3 similarly increased serum calcium and induced bone disease in both strains, 24-hour urine volume and creatinine pronouncedly decreased only in ob/ob mice. Moreover, ob/ob increased urinary albumin/creatinine ratio (ACR), indicating kidney dysfunction. In parallel, ob/ob developed extensive intrarenal VC after VitD3. Coincidently with increased intrarenal vascular mineralization, our results demonstrated that Bone Morphogenetic Protein-2 (BMP-2) was highly expressed in these arteries exclusively in ob/ob. These data depict a greater susceptibility of ob/ob mice to develop renal disease after VitD3 in comparison to paired C57BL/6. In conclusion, this study unfolds novel mechanisms of progressive renal dysfunction in diabetes mellitus (DM) after VitD3 in vivo associated with increased intrarenal VC and highlights possible harmful effects of long-term supplementation of VitD3 in this population.
Subject terms: Calcification, Diabetes complications, Type 2 diabetes, Obesity, End-stage renal disease
Introduction
Vascular calcification (VC) is a pathological condition which causes loss of arterial elasticity and augments vascular stiffening associated with increased cardiac work1,2. This contributes to several cardiovascular diseases such as systemic arterial hypertension, coronary artery and cerebrovascular disease, congestive heart failure, and end-stage renal disease (ESRD)3. Previously thought as a process of physiological aging, VC is currently known as an active and complex process, characterized by increased calcifying signaling agonists that overcome inhibitory mediators and resembles skeleton ossification1,2.
Diabetes mellitus is a huge public health problem in the world. It is estimated that diabetic population will reach 430 million people in 2030. Incidence of ESRD is up to 10 times as high in adults with diabetes as those without4. ESRD attributable to diabetes is 12–55%4. In the last decades, numerous advances have been incorporated to the treatment of ESRD. However, the incidence of renal replacement therapy and mortality rates in this population remains high5,6.
Epidemiological studies describe ESRD and VC as independent cardiovascular risk factors5. However, ESRD patients are usually identified with VC7,8, and ESRD together with diabetes mellitus accelerates VC progression7–9. Studies that examined VC in the kidney are scarce and the impact of VC influencing renal disease progression in diabetes mellitus is poorly understood.
Active 1,25-dihydroxyvitamin D3 is implicated in prevention and treatment of hyperparathyroidism in dialysis patients. In addition, there is accumulating evidence that VitD3 has a beneficial role in decreasing proteinuria10, the hallmark of diabetic nephropathy and other chronic kidney diseases. VitD3 may primarily suppress renin-angiotensin-aldosterone system, reduce the inflammatory response, apoptosis, fibrosis, and oxidative stress, and mitigate podocyte cytoskeleton damage10,11. Furthermore, nonskeletal benefits of Vitamin D supplementation was motivated by reports associating low levels of VitD3 and development of pathological conditions such as metabolic syndrome, ESRD, and type 2 diabetes mellitus12. Paradoxically, inappropriate doses of vitamin D, mainly associated with clinical malpractice, can trigger acute kidney injury13, which ultimately lead to permanent kidney damage due to fibrosis, and exaggerated VC (aorta) in a model of monogenic obesity and insulin resistance14,15. To the best of our knowledge, the renal function of individuals subjected to high doses of VitD3 was not previously assessed. Furthermore, no large clinical trials were conducted to investigate harmful effects of Vitamin D supplementation, especially in diabetes mellitus, ESRD and elderly patients. From this perspective, the aim of this study was to address the effect of VitD3-induced intrarenal VC and respective mechanisms involved in renal dysfunction in a murine model of insulin resistance and obesity (ob/ob), comparing to paired healthy littermates (C57BL/6). Accordingly, based on a previous study that demonstrated increased aortic calcification in ob/ob mice after excessive VitD3 supplementation14, we examined the contribution of renal arteries calcification in CKD progression both in ob/ob and in C57BL/6 mice.
Results
Diabetic ob/ob mice developed Vitamin D3-induced kidney dysfunction
Both saline-treated C57BL/6 and ob/ob mice presented similar serum creatinine levels (0.35 ± 0.10 mg/dL versus 0.22 ± 0.04 mg/dL, respectively), Fig. 1A. After VitD3 protocol, serum creatinine increased 92% (0.42 ± 0.03 mg/dL) and 43% (0.50 ± 0.05 mg/dL) in ob/ob and C57BL/6 mice respectively (Fig. 1A) versus paired controls. Enhanced creatinine levels in both strains indicate kidney dysfunction induced by hypervitaminosis D. In parallel, saline-treated ob/ob mice showed lower creatinine clearance than saline-injected C57BL/6 mice (53.5 ± 4.9 µL/min vs. 89.1 ± 13.7 µL/min), Fig. 1C. After VitD3 protocol, creatinine clearance decreased in both strains, more pronouncedly in ob/ob (7.61 ± 1.69 µL/min) in comparison to C57BL/6 mice (28.74 ± 7.01 µL/min), Fig. 1C.
Figure 1.

Renal function analysis of obese insulin-resistant mice (ob/ob) and their littermates (C57BL/6) subjected to Vitamin D3 protocol. (A) Serum creatinine concentration (n = 5–6); (B) 24-hour urine volume quantification (n = 6–8); (C) Creatinine clearance levels evaluation (n = 5–7) and (D) albumin/creatinine ratio determination (n = 5–7) from mice treated (+) with Vitamin D3 or injected with saline (−). α, β, γ, δ = P < 0.05, in comparison to untreated (−) C57BL/6, treated (+) C57BL/6, untreated (−) ob/ob, and treated (+) ob/ob respectively.
To further investigate the effects of VitD3 protocol in renal dysfunction, we assessed 24h-urine volume output, albuminuria and calculated ACR to estimate glomerular damage, which also depicts progressive renal disease in diabetes mellitus16. As expected, the diabetic model showed increased 24h-urine volume in comparison to C57BL/6 (1.49 ± 0.06 mL vs. 1.14 ± 0.17 mL) at baseline. After 21 days of VitD3 treatment, both ob/ob and C57BL/6 strains presented decreased 24h-urine volume. This was more pronounced in ob/ob (0.31 ± 0.11 mL) than in C57BL/6 mice (0.74 ± 0.21 mL), Fig. 1B.
Albumin creatinine ratio was similar both in saline-treated C57BL/6 and in ob/ob mice (106.9 ± 19.1 µg/mg and 340.9 ± 45.4 µg/mg, respectively). ACR from VitD3-treated and from saline-treated C57BL/6 mice were not statistically different (585.1 ± 194.7 µg/mg and 106.9 ± 19.1 µg/mg, respectively), Fig. 1D. Interestingly, ACR from ob/ob VitD3-treated mice increased in comparison to paired saline-treated ob/ob mice (1369.9 ± 218.0 µg/mg vs. 340.9 ± 45.4 µg/mg) and to C57BL/6 VitD3-treated mice (585.1 ± 194.7 µg/mg), Fig. 1D.
High dose of Vitamin D3 induced similar bone disease in both ob/ob and C57BL/6 mice
VitD3 treatment increased ≈70% trabecular volume (BV/TV) in both ob/ob and C57BL/6, Table 1. Moreover, both strains augmented trabecular thickness (Tb.Th); this response was 20% less in ob/ob compared to C57BL/6. Both trabecular number (Tb.N) and trabecular separation (Tb.Sp) did not significantly change after VitD3 administration in C57BL/6 and ob/ob mice, Table 1. Static parameters of bone formation showed that VitD3-treated mice increased osteoid surface (OS/BS) in both strains (9 times and 8 times increase in C57BL/6 and in ob/ob, respectively). Similarly, VitD3 enhanced osteoid volume (OV/BV) in both strains (≈50 times for C57BL/6 and ≈35 times for ob/ob), Table 1. Femurs also presented osteoid thickness modification (O.Th) after hypervitaminosis D. Specifically, C57BL/6 and diabetic ob/ob mice increased (25% and 50%, respectively) osteoid thickness in comparison to paired controls. Osteoblastic surface (Ob.S/BS) enhanced after VitD3 in both strains, especially in ob/ob mice (≈6 times and. ≈3 times in ob/ob and C57BL/6 mice respectively), Table 1. Finally, bone resorption parameters demonstrated that osteoclast surface (Oc.S/BS) from ob/ob control femurs was approximately 50% inferior in comparison to C57BL/6 controls. VitD3 reduced Oc.S/BS value (C57BL/6 = 27% and ob/ob = 16%). Eroded surface (ES/BS) from control ob/ob mice was 40% less than in saline-injected C57BL/6 animals. ES/BS decreased after VitD3 in both strains (C57BL/6 = 19% and ob/ob = 14%), Table 1.
Table 1.
Femur’s Histomorphometric parameters from C57BL/6 and from ob/ob mice subjected to Vitamin D3 (+) or to saline (−).
| Parameter | C57BL/6 | ob/ob | ||
|---|---|---|---|---|
| − | + | − | + | |
| BV/TV (%) | 14.95 ± 1.27 | 25.80 ± 0.93a | 10.36 ± 1.53b | 17.81 ± 1.84b |
| Tb.Th (µm) | 31.42 ± 1.46 | 57.37 ± 2.34a | 31.03 ± 0.20b | 45.61 ± 2.93a,b,c |
| Tb.N (n°/mm) | 4.71 ± 0.19 | 4.53 ± 0.24 | 3.33 ± 0.47 | 3.86 ± 0.26 |
| Tb.Sp (µm) | 182.4 ± 10.2 | 166.3 ± 10.1 | 274.9 ± 43.6b | 220.6 ± 21.7 |
| OS/BS (%) | 9.13 ± 1.66 | 81.63 ± 3.77a | 10.59 ± 2.52b | 78.47 ± 5.00a,c |
| OV/BV (%) | 0.82 ± 0.18 | 41.54 ± 0.86a | 1.22 ± 0.62b | 42.32 ± 2.87a,c |
| O.Th (µm) | 1.37 ± 0.12 | 15.66 ± 0.76a | 1.74 ± 0.50b | 13.10 ± 0.90a,c |
| Ob.S/BS (%) | 7.38 ± 1.25 | 22.77 ± 2.09a | 4.84 ± 2.28b | 29.56 ± 4.38a,c |
| Oc.S/BS (%) | 1.72 ± 0.14 | 0.46 ± 0.19a | 0.79 ± 0.52a | 0.12 ± 0.05a |
| ES/BS (%) | 7.21 ± 0.62 | 1.40 ± 0.39a | 2.84 ± 0.66a | 0.40 ± 0.16a,c |
aP < 0.05 in comparison to C57BL/6 control.
bP < 0.05 in comparison to C57BL/6 Vitamin D3-treated.
cP < 0.05 in comparison to ob/ob control.
High dose of Vitamin D3 induced similar damage of bone tissue in both ob/ob and in C57BL/6 mice, as demonstrated by static bone formation parameters and bone resorption data from these animals n = 3–6 (animals injected with saline (−)); n = 6–7 (for animals treated with VitD3 (+)). Increased osteoid matrix and resorption parameters characterize a mixed bone disease.
Vitamin D3 induced intrarenal artery calcification in diabetic mice, but not in paired C57BL/6 littermates
Ex-vivo kidney analysis exhibited high intensity Osteosense-derived fluorescence in VitD3-treated ob/ob mice, but not in VitD3-treated-C57BL/6 nor in paired saline-injected mice, thus demonstrating greater calcification (Fig. 2A). In addition, confocal fluorescence microscopy coincidently showed high-intensity Osteosense 680 EX signal in medial layer of intrarenal arteries from ob/ob VitD3-treated mice, but not in paired VitD3-treated C57BL/6 mice (Fig. 2B).
Figure 2.

Vitamin D3 increased calcification in obese insulin-resistant (ob/ob) mice but not in their littermates (C57BL/6) - Mice were subjected to Vitamin D3 treatment (Vitamin D3) or saline administration (control). 24 h before euthanasia, Osteosense 680 EX was injected i.p., kidneys were isolated and analyzed. (A) Kidneys were imaged ex vivo as described in methods section. Right: fluorescence intensity scale, photon/s. Only kidneys from Vitamin D3-treated ob/ob mice exhibited high fluorescence (average mean and standard error values are indicated as a picture insert), n = 3. Bars = 5 mm. (B) Histological slides of the kidneys were analyzed in a confocal fluorescence microscope. Osteosense 680 EX is depicted in red and nuclei Hoechst-stained are shown in blue. Arrows depict high Osteosense 680 EX signal staining calcification of medial layer of intrarenal arteries from ob/ob mice. Mean Fluorescence intensity normalized by C57BL/6 control: 1.0; 1.2; 1.1 and 2.8 in C57BL/6 control; ob/ob control; C57BL/6 Vitamin D3 and ob/ob Vitamin D3 respectively, n = 2. Bars = 50 µm.
We further confirmed these findings using histochemical techniques. Diabetic ob/ob mice showed increased Alizarin Red S and Von Kossa staining in intrarenal arteries after VitD3 treatment compared to the paired C57BL/6 (Fig. 3A). Negligible or no calcification was identified in intrarenal arteries from saline-treated ob/ob and C57BL/6 mice (Fig. 3A). Quantification of Alizarin Red S staining confirmed these findings: 8717 ± 1.71 µm2 vs. 416 ± 1.71 µm2 in VitD3-treated vs. saline-treated ob/ob mice; 1479 ± 1.66 µm2 vs. 0 ± 1.66 µm2 in VitD3-treated vs. saline-treated C57BL/6 respectively (Fig. 3B). Accordingly, quantification of Von Kossa staining showed: 1582 ± 380.7 µm2 vs. 0 ± 380.7 µm2 in VitD3-treated ob/ob and saline-treated ob/ob mice; 975.9 ± 380.7 µm2 vs. 0.0 ± 380.7 µm2 in VitD3-treated C57BL/6 and saline-treated C57BL/6 mice respectively (Fig. 3C). In conclusion, these data showed greater calcification response of intrarenal arteries from ob/ob than from paired C57BL/6 after VitD3 protocol.
Figure 3.

Histochemical analysis and quantification of intrarenal arteries calcification from obese insulin-resistant ob/ob mice and their littermates C57BL/6 subjected to Vitamin D3 protocol. (A) Mice were subjected to Vitamin D3 treatment (Vitamin D3) or saline administration (Control) and tissue secctions were stained with Alizarin Red S and Von Kossa. Both Alizarin Red S (left panels) and Von Kossa (right panels) staining demonstrated increased calcification of intrarenal arteries from VitD3-treated ob/ob in comparison to paired C57BL/6 mice. (B,C) Quantification of stained areas for Alizarin Red S (B) and Von Kossa (C) staining, in µm2, from mice treated with Vitamin D3 (+) or injected with saline (−). α, β, γ, δ = P < 0.05, in comparison to saline (−) C57BL/6, VitD3-treated (+) C57BL/6, saline (−) ob/ob, and VitD3-treated (+) ob/ob, respectively. n = 6 for all groups.
BMP-2 is highly expressed on calcified intrarenal artery from Vitamin D3-treated diabetic mice
As we consistently demonstrated increased VC induced by VitD3 in diabetic mice (Figs. 2 and 3), we evaluated BMP-2 expression, which has a pivotal role in smooth muscle cells osteochondrogenic dedifferentiation and ectopic calcification17,18. We found that BMP-2 was highly expressed in intrarenal arteries exclusively from ob/ob mice subjected to VitD3 protocol (Fig. 4), but not in paired VitD3-injected C57BL/6 samples.
Figure 4.
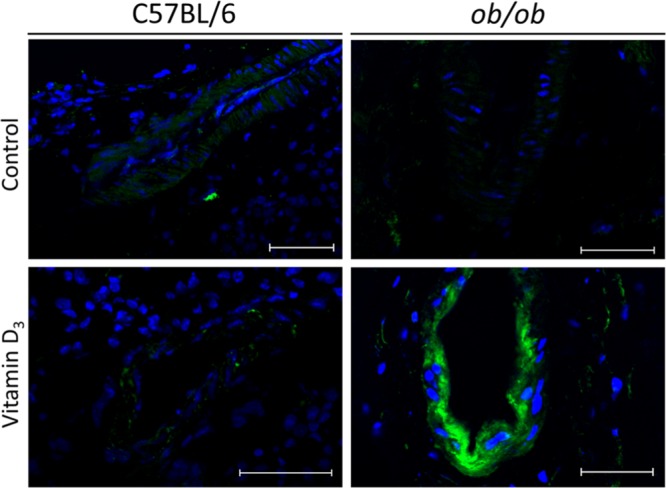
Immunofluorescence analysis of BMP-2 expression in intrarenal arteries from obese insulin-resistant ob/ob mice and their littermates C57BL/6 subjected to Vitamin D3 protocol. Mice were subjected to Vitamin D3 treatment (Vitamin D3) or injected with saline (control). Kidney sections were incubated with anti-BMP-2, followed by secondary AlexaFluor 488-conjugated antibody incubation and analyzed by confocal fluorescence microscopy. Increased BMP-2 expression (green) is observed in calcified intrarenal arteries from Vitamin D3–treated ob/ob mice. On contrary, paired C57BL/6 mice exhibited low BMP-2 expression. Nuclei were stained with Hoechst 33342 (blue). Mean fluorescence intensity and standard error of samples are: 5.32 ± 5.32; 65.95 ± 58.36; 87.08 ± 40.03 and 235.80 ± 21.09 in C57BL/6 control; C57BL/6 Vitamin D3; ob/ob control and ob/ob Vitamin D3 respectively. P < 0.05, ob/ob Vitamin D3 versus all groups, n = 3–4. Bars = 50 µm.
Severe mesangial expansion associated with acute tubular necrosis in VitD3-treated ob/ob mice
In diabetic ob/ob mice, mesangial expansion was more severe when compared to wild type mice, Fig. 5A. Of note, VitD3 accelerated extracellular matrix deposition in mesangial compartment of C57BL/6, demonstrated by statistically similar values of fractional mesangial area in VitD3-treated C57BL/6 compared to VitD3-treated ob/ob mice, Fig. 5B. Both VitD3-treated C57BL/6 and VitD3-treated ob/ob mice’s fractional mesangial area did not show statistical difference when compared to paired saline-treated C57BL/6 and ob/ob mice respectively, Fig. 5B. These findings may suggest a possible effect of inappropriate high-dose of VitD3 in regulating glomerular damage in C57BL/6 mice, since VitD3 promoted increased matrix deposition in mesangial compartment that was able to reach ob/ob levels. However, in pre-existing diabetic nephropathy settings, in which several deleterious pathways have already been activated, VitD3 did not promote additional damage to the mesangial compartment. In parallel, we showed increased glomerular area in ob/ob mice. There was no additive effect of VitD3 in ob/ob mice, but VitD3 was able to equalize glomerular area in C57BL/6 and in ob/ob mice, Fig. 5C.
Figure 5.

Mesangial and histological assessment of the kidneys showing severe mesangial expansion in ob/ob mice, and the effect of Vitamin D3 in glomerular damage and in acute tubular necrosis. (A) PAS staining of glomeruli from C57BL/6 and from ob/ob mice after Vitamin D3 stimulation (+) or after saline injection (−). Bars = 20 µm. (B) VitD3 promoted increased matrix deposition in mesangial compartment of C57BL/6 mice that was able to reach ob/ob levels (no statistical difference among VitD3-treated C57BL/6 and VitD3-treated ob/ob mice). (C) Increased glomerular area in ob/ob mice. There was no additive effect of Vitamin D3 in ob/ob mice, but VitD3 was able to equalize glomerular area in C57BL/6 and in ob/ob mice. α, β, γ, δ = P < 0.05, in comparison to saline-treated C57BL/6 (n = 9), VitD3-treated C57BL/6 (n = 7), saline-treated ob/ob (n = 7), and VitD3-treated ob/ob mice (n = 8), respectively. (D) Vitamin D3 induced acute tubular necrosis (ATN) both in C57BL/6 and in ob/ob mice, as shown by flattening of the renal tubular cells due to tubular dilation (asterisks), loss of brush border (arrows), and degenerative changes characterized by diffuse denudation of the renal cells, presence of necrotic cells, and cellular debris. Insert depicts degenerative changes in a distal tubule. Bars = 20 µm. For saline-injected mice n = 4–6, and for Vitamin D3–treated mice, n = 5–8.
Importantly, VitD3 also induced acute kidney injury, e.g., ATN both in ob/ob and in wild type mice. Cortical and corticomedular tubules showed widespread degenerative changes with luminal dilation and loss of brush border, as well as tubular atrophy, Fig. 5D. These findings may explain the observation of higher serum creatinine levels and lower creatinine clearance and diuresis volume in VitD3-treated animals. Importantly, increased mesangial area in ob/ob mice combined with ATN lesions may explain higher levels of albuminuria, impairing kidney function in these animals.
Collagen I and III deposition in tubule-interstitial area, as demonstrated by Picro Sirius Red staining, was mild in all groups, but slightly increased in VitD3-treated ob/ob mice, Supplemental Fig. I.
Discussion
There is no doubt that the association of DM, VC and ESRD increases morbidity and mortality19. In order to investigate kidney-specific VC mechanisms that may implicate in diabetes mellitus-related kidney dysfunction and pathophysiology, we studied the effect of high dose VitD3 i.p. injection using an experimental model that mimics T2DM and insulin resistance, the ob/ob mouse. Recently, we demonstrated that VitD3 increased expansive vascular remodeling associated with accelerated VC in ob/ob mice, which occurred by the convergence of increased oxidative stress, matrix metalloproteinase activation and absence of VDR downregulation in the vascular wall from these animals14. Conversely, VitD3 and its analogous have been reported as renoprotective, resulting in an attenuation of proteinuria, inflammation, glomerulosclerosis and interstitial fibrosis and an improvement of glomerular ratio20,21. Reciprocal benefits include renin-angiotensin-aldosterone system regulation, anti-inflammatory, anti-oxidative stress and anti-apoptosis properties, podocyte protection, autophagy activation, immunomodulatory effects, hepatocyte growth factor induction, mitochondrial function regulation, and tubular epithelium preservation, via epithelial-mesenchymal transition blockade20–22. Vitamin D3 prescription has been increasing and its inappropriate supplementation may be associated with toxicity and hypercalcemia23. Recently, investigators reported an interesting ESRD murine model24, using phosphorus-rich diet, but it requires twice more time (6 weeks) to develop compared to our model, besides not describing intrarenal arteries calcification. In our study we demonstrated that: 1) VitD3 additionally impacted in diabetic mouse model’s renal function and early markers of glomerular filtration damage, by decreasing creatinine clearance, 24 h urine volume output and by increasing albumin/creatinine ratio in ob/ob in comparison to paired C57BL/6 mice; 2) renal dysfunction occurred in parallel to exaggerated effect of VitD3 in increasing intrarenal VC in ob/ob mice vs. C57BL/6 mice; 3) Increased BMP-2 expression in the vascular wall of calcified intra-renal arteries from ob/ob mice, but not from C57BL/6 animals after VitD3 protocol; 4) increased mesangial expansion in ob/ob mice, without an additive effect of VitD3, associated with severe acute tubular necrosis in VitD3-treated ob/ob mice, contributing to higher levels of albuminuria, and decreased kidney function.
Hypercalcemia, due to increased intestinal and renal calcium resorption is an expected effect of VitD325,26 supplementation at high serum levels27. Hypercalcemia has been extensively studied in this context, since this is a mechanism involved in VC due to high calcium x phosphorus product14,28. To further understand increased VC response of ob/ob mice after our protocol, we performed serum biochemical analysis and assessed bone histomorphometric parameters. We found a significant, but similar increase in serum calcium concentration (≈60% increase), both in ob/ob and in C57BL/6 mice after VitD3 administration when compared to saline-injected mice14. Moreover, both VitD3 and kidney dysfunction-induced effect on bone tissue damage, observed in our study, are demonstrated by increased osteoid matrix and resorption parameters, which characterizes a mixed bone disease29. Bone disease, as shown by parameters of bone resorption (e.g. osteoblastic surface, resorption surface, and diminished number of osteoclasts) was similar in both strains. This implies the reproducibility of the effect of VitD3 in bone/calcium metabolism both in ob/ob and in C57BL/6 mice, conceiving the idea that augmented VC in ob/ob mice translates an individual response from the obese insulin resistance model. A limitation of our study is that we were not able to precisely determine absolute values of serum 25-hidroxyvitamin D both in C57BL/6 and in ob/ob mice after VitD3 stimulation. In this setting, we found that 25-hidroxyvitamin D serum concentration increased to above 100 ng/mL (the limit of the standard curve used, data not shown) in both strains after excessive VitD3 supplementation. In fact, although we used less VitD3 in ob/ob mice versus C57BL/6 mice, these animals showed increased calcification of intrarenal arteries associated with greater kidney dysfunction in comparison to C57BL/6 mice.
VC, which is a condition without specific medical treatment, positively associates with coronary artery disease and cardiovascular events, especially in patients with diabetes mellitus30. Interestingly, now we showed an association of augmented intra-renal arteries calcification and renal dysfunction in ob/ob mice, demonstrated by positive correlation between intrarenal arteries calcification and increased albumin/creatinine ratio, and decreased creatinine clearance. This experiment suggests a direct relationship between arterial calcification and decreased renal function induced by hypervitaminosis D. Furthermore, we demonstrated that high-dose VitD3 stimulation promoted a paradoxical increase in mesangial area in wild type mice. Thus, our work unveils an important effect of VitD3 on mesangial compartment in a toxic dose-dependent manner. We postulate that this effect may include VDR aberrant down-regulation in that compartment and probably in podocytes as well. In ob/ob mice, VitD3 treatment was not implicated in an additive effect in mesangial compartment expansion and in glomerular area augmentation, which may be explained at least by the fact that animals were not treated with insulin and hyperglycemia may continuously aggravated mesangial expansion and abrogated VitD3-mediated renoprotective effects. Furthermore, VitD3 toxicity may induce renal hemodynamic dysfunction, which can be complicated by ATN. Tubular damage could also be explained by diabetes-induced lysosomal dysfunction in proximal tubules31. Therefore, as a surrogate marker of diabetic kidney disease progression, the augmentation in exocytosis-mediated urinary megalin excretion creates a vicious cycle of tubular damage and may also contribute to higher values of albuminuria found in VitD3-treated ob/ob mice. Other possible mechanisms include acute hypercalcemia induced by VitD3 toxicity, which may lead to acute kidney injury by decreasing extracellular fluid volume due to anorexia, nausea, vomiting, and decreased ability to concentrate urine, besides a direct renal vasoconstriction effect, as observed in VitD3-treated animals from our protocol, and in vitamin D intoxication in humans13. VitD3 effects are mediated by VDR. Previously, we demonstrated that VDR downregulation was abrogated in ob/ob mice after VitD3 stimulation, which corroborated to increased VC (aorta) in this mouse model14,15. In the kidneys, VDR is mainly expressed in proximal and distal tubular epithelial cells, podocytes, macula densa of the juxtaglomerular apparatus, and collecting duct epithelial cells32. However, VDR expression is low in glomerular mesangial cells. Despite the low expression of VDR in mesangial compartment, VitD3 may reduce mesangial cell proliferation induced by hyperglycemia in diabetic rat via mTOR pathway modulation and decreasing glomerular volume33. In addition, VitD3 suppressed Monocyte Chemoattractant Protein-1 (MCP-1) expression in mesangial cells by blocking Nuclear factor kappa B (NF-κB) activation, which indicates that vitamin D may protect the kidney by reducing macrophage infiltration34. Moreover, VitD3 per se was able to increase VDR expression at mRNA and protein levels in mesangial cells cultured with either low or high glucose34.
Diabetes mellitus may activate specific osteochondrogenic signaling involved in vascular smooth muscle cells dedifferentiation into osteoblast-like cells, thus increasing VC progression. To further investigate this specifically in intrarenal arteries, we assessed BMP-2 expression in the kidney. BMP-2 is a protein secreted by smooth muscle vascular cells, endothelial cells and inflammatory macrophages35. BMP-2 binds to its receptor on the plasma membrane, triggers a specific intracellular signaling cascade, activating osteochondrogenesis and VC. Of note, BMP-2 expression is also regulated by the BMP signaling pathway itself, since BMP-2 is a self-regulatory protein36. In diabetic preclinical animal models, VC is governed by increased MSH homeobox 2 (Msx2) and Tumor Necrosis Factor Alpha (TNF-alpha) expression in the middle layer and in the adventitia of vessel wall, finally activating osteochondrogenic transcription program in smooth muscle cells. Previous reports demonstrated that increased macrophage infiltration in the adipose tissue and in the kidney, as well as augmented TNF-alpha expression in animal models of obesity, play a pivotal role in inducing kidney disease37,38. Accordingly, inflammatory cytokines e.g. C-C chemokine ligand 2 (CCL2) initiates inflammation by binding to C-C chemokine receptor 2 (CCR2) to induce diabetic glomerular sclerosis39. Moreover, investigators showed that blocking CCL2/CCR2 signaling pathway can ameliorate renal injury and proteinuria in a mouse model of obesity and insulin resistance37. BMP-2 activation has also been well characterized in aorta from diabetic mice40. In our laboratory, vascular smooth muscle cells isolated from ob/ob mice aortae showed increased calcification in vitro after BMP-2 stimulation with concurrent upregulation of other osteogenic proteins41. Accordingly, we showed augmented baseline MSX2, BMP-2 expression and Smad 1,5 phosphorylation as well as increased BMP-2-induced osteocondrogenic signaling and dedifferentiation in ob/ob vascular smooth muscle cells in comparison to paired C57BL/6 vascular smooth muscle cells41. In the present study, we identified a significant increase in BMP-2 expression in calcified intrarenal arteries of VitD3-treated ob/ob mice, which also demonstrated renal dysfunction. On the contrary, paired VitD3-treated C57BL/6 mice demonstrated that BMP-2 expression was very low in intrarenal arteries. We speculate that VitD3 induces BMP-2 expression, through the activation of its receptor. To assess the role of leptin-deficiency in vascular smooth muscle cells calcification from ob/ob mice, we demonstrated that calcification increased in leptin-incubated ob/ob vascular smooth muscle cells, but not in leptin-incubated C57BL/6 vascular smooth muscle cells (Supplemental Fig. II). On the contrary of C57BL/6 vascular smooth muscle cells, co-incubation of leptin in BMP-2-treated cells further augmented mineralization in ob/ob vascular smooth muscle cells. These data suggest mechanisms of increased susceptibility of ob/ob mice to develop accelerated vascular calcification, independently of leptin supplementation. A putative explanation for the exaggerated response of ob/ob mice could be that CYP27B1 (1-α-hydroxylase) and CYP24A1 (24-hydroxylase), which are enzymes responsible for regulating active VitD3 metabolites, exhibit altered mRNA and activity levels, thus favoring increased local concentration of bioactive VitD3 and consequently potentiating BMP-2 expression in leptin-deficient diabetic mice. Of note, non-hepatic 25-hydroxylases (Cyp2R1 and Cyp27A1), enzymes responsible for the conversion of cholecalciferol into 25-hydroxyvitamin D and for maintaining VDR levels42, may have contributed to changes in VDR expression levels and the phenotype demonstrated in our model, because 25-hydroxylases lack the tight control that exists for 1-hydroxylase and 24-hydroxylase during excessive cholecalciferol supplementation. Altogether, we postulate that augmented intra-renal calcification potentiates progression of renal dysfunction in ob/ob mice. Moreover, a mechanism involved could be worsening of Windkessel effect, because of increased arterial rigidity and decreased vascular elasticity, which impairs tissue perfusion due to VC24,43. In addition, vascular mineralization may act as an adjunct etiology of this process or even in its progression44,45, adding importance to this study. Nonetheless, we did not find glomerular calcification in ob/ob mice, but we cannot rule out the hypothesis that decreased tissue perfusion and increased BMP-2 expression could have impacted in glomerular dysfunction, by altering podocytes number and/or function46–48 and increasing inflammation19,49. A limitation of our study is that we can’t distinguish whether systemic and local VitD3 effects, e.g. increased serum calcium levels, augmented BMP-2 expression and intrarenal arteries calcification influenced renal dysfunction alone or whether this occurred together with hemodynamic imbalance due to vascular mineralization. This needs further clarification by exploring respective individual impact on renal disease progression. In conclusion our results demonstrate that high-dose VitD3 administration in ob/ob mice, but not in C57BL/6, induce increased intrarenal VC associated with kidney dysfunction. These conditions are common in diabetic patients, bringing high morbidity and mortality50,51. Moreover, pathophysiological and molecular mechanisms investigated in this study represent an important contribution both to understand VitD3 biological properties in the kidneys and to clarify specific aspects of renal disease in diabetic patients, which usually present with VC7,8. Our model may be instrumental for the investigation of new therapeutic targets and/or to develop compounds that attenuate renal dysfunction in diabetes mellitus, especially in the context of increased VC.
Materials and Methods
Animals
We used 16 to 20-week-old male homozygous leptin-deficient ob/ob mice (C57BL/6 background) from Jackson Laboratory (Bar Harbor, ME). A total of 30 ob/ob and 30 C57BL/6 littermates were used to perform the experiments. This study was conducted after approval of the protocol #2242-14 by Sociedade Beneficiente Israelita Albert Einstein’s ethics committee and undertook according to guidelines for the care and use of laboratory animals, which conforms to Guide for the Care and Use of Laboratory Animals (NIH Publication, 8th edition). After 21 days, animals were euthanized with 1 mg/kg IM xylazine chlorohydrate (Bayer, São Paulo, Brazil, Cat#1002181) and 100 mg/kg IM ketamine chlorohydrate (Cristália, São Paulo, Brazil, Cat#404800). Of note, not all 60 animals were represented in all experiments, due to technical difficulties as follow: (i) we were not able to draw blood samples from all animals (dehydration, samples with clots, unsuccessful vein puncture); (ii) automated Abbott® i-STAT Clinical Analyzer failed to give a result in some samples (equipment error) and repeated measurement were not possible due to insufficient sample/blood volume; (iii) we did not have as many metabolic cages as the number of animals used during protocol, in order to collect 24h-urine from all animals; (iv) some animals were anuric, so we were not able to determine albuminuria in some samples/animals, (v) we did not use all animals to perform all the experiments.
Vitamin D3 administration protocol
Ob/ob and C57BL/6 male mice were injected with VitD3 6.4 × 104 IU/day and 4.4 × 104 IU/day respectively i.p. for 18 days and another 3 days of sodium chloride 0.9% i.p. This corresponds to a daily dose of 1.46 × 103 IU/g/day administered to C57BL/6 mice and 1.06 × 103IU/g/day administered to ob/ob mice, considering a body weight of 30 g and 60 g respectively. Sodium chloride 0.9% (saline) i.p. only was used for 21 days in controls. Of note, we did not use the same proportional dose (calculated by body weight) in ob/ob and in C57BL/6 mice, because 80% of ob/ob mice died when we used the aforementioned proportional dose, due to Vitamin D intoxication after 7 to 10 days of protocol. Consequently, we reduced the dose in approximately 30% in ob/ob mice to be able to complete the animal protocol.
Serum and urine analysis
Blood was collected from the animals before and after the protocol in order to assess serum glucose, urea, creatinine and calcium levels using Abbot® i-STAT Clinical Analyzer and the I-STAT CHEM8+ cartridge (Abbott Laboratories, Illinois, USA, Cat#AB-9P3125). For urine analysis, animals were maintained in a metabolic cage to collect 24-hour urine before and after the protocol. Urine albumin levels were determined by Albumin Mouse ELISA Kit (Abcam, Cat#ab108792). Urine creatinine levels were evaluated by a colorimetric assay Labtest Diagnostics kit (Vista Alegre, Brazil, Cat#10009010034) and quantified by an automatic biochemical analyzer Cobas Mira Plus (Roche, Switzerland).
Bone histomorphometric analysis
After euthanasia, left femurs were dissected, fixed in 70% ethanol, dehydrated, embedded in methyl methacrylate, and sectioned longitudinally with a Policut S microtome (Reichert-Jung, Heidelberg, Germany) in 5 µm-thick sections. Samples were stained with 0.1% toluidine blue (pH 6.4) for histomorphometric analysis which was performed using semiautomatic method with a Labophot-2A microscope (Nikon®), and software Osteomeasure (Osteometrics, Inc, Atlanta, EUA). These histomorphometric parameters are suggested by the American Society of Bone and Mineral Research histomorphometry nomenclature committee52 as follow: bone volume represented as a percentage of tissue volume (BV/TV, %); trabecular thickness (Tb.Th, µm); trabecular separation (Tb.Sp, µm); trabecular number (Tb.N, /mm); osteoid volume as a percentage of bone volume (OV/BV, %); osteoid surface as a percentage of bone surface (OS/BS, %); osteoblast surface as a percentage of bone surface (Ob.S/BS, %); osteoid thickness (O.Th, µm); osteoclast surface as a percentage of bone surface (Oc.S/BS, %); and eroded surface as a percentage of bone surface (ES/BS, %).
Ex vivo calcification assessment of the kidneys using Osteosense 680 EX
24 h before sacrifice, mice were injected with 0.2 µM Osteosense 680 EX (NEV10020EX, Perkin Elmer, USA). After euthanasia, cardiovascular system from mice was perfused with saline, followed by radical nephrectomy. Isolated kidneys were analyzed with a fluorescence detector IVIS ® Lumina LT Series III (PerkinElmer, USA), and fluorescence signals were normalized to estereoradian and ROI of 4.0 cm2 and converted to photon/s53.
Histological quantification of kidneys’ vascular calcification
Kidneys were fixed in 10% phosphate-buffered formalin (pH 7.4), embedded in paraffin and processed for Von Kossa (silver nitrate Sigma, Cat# S1179) and Alizarin Red S (Sigma, Cat#A5533) analysis using a FSX100 microscope (Olympus Life Sciences) and Olympus CellSens software. Kidneys from animals previously labeled with Osteosense 680 EX, were extracted, incubated with sucrose (Sigma, Cat#S9378) 30% overnight, frozen at −80 °C in O.C.T. (optimum cutting temperature) compound (Sakura Finetek, California, USA, VWR Cat# 25608-930) and sections were obtained using a cryostat microtome (Leica Biosystems, Germany). Samples were incubated with Hoechst 33342 (Thermo Fischer Scientific, Cat#H1399) for nuclei staining, and analyzed by Zeiss LSM 710 laser scanning microscope for confocal imaging and the respective software Zen Image (Zeiss®). Osteosense fluorescence quantification was calculated by using the fluorescence intensity divided by vascular area and normalized by C57BL/6 control.
Immunofluorescence analysis of Bone Morphogenetic Protein-2 (BMP-2) expression in the vascular wall
10 µm O.C.T. kidney’s sections were fixed with 4% paraformaldehyde and permeabilized with 0.25% Triton X-100 and PBS for 15 min. Samples were washed, incubated with primary antibody overnight (anti-BMP-2, Abcam, ab:6285) 5 µg/mL, and finally incubated with secondary anti-mouse IgG AlexaFluor 488 (Thermo Fischer Scientific, Cat#A10680) 10 µg/mL for 1 hour, and coverslipped with Hoechst 33342 (Thermo Fischer Scientific, Cat#H1399). Fluorescence image analysis was performed in parallel with controls, using the same settings for all samples in a Zeiss LSM 710 confocal laser scanning microscope and the respective software Zen Image (Zeiss®). BMP-2-derived fluorescence quantification was calculated by using the fluorescence intensity divided by vascular area.
Mesangial and histological assessment of the kidneys
Kidney sections were stained with periodic acid-Schiff (PAS) trichrome staining in each experimental group. Sections were then analyzed by light microscopy (magnification, x400). A quantitative analysis of mesangial expansion was performed. The increase in mesangial matrix was determined by the presence of PAS-positive area in the mesangium, and was expressed in percentage. The glomerular area (μm2) was also traced along the outline of capillary loops using CellSens software (Olympus) in 28–35 randomly selected glomeruli in each animal. Acute tubular necrosis (ATN) was identified by PAS staining, followed by quantification of the following variables: presence of casts, brush border loss, tubular dilation, necrosis, and calcification. Picro Sirius Red staining was assessed by standard light microscopy (magnification, x100).
Statistical analysis
Data are shown as mean ± standard error of the mean (M ± S.E.M). After assessing normality and equal variance, data were analyzed by One-Way ANOVA followed by Tukey test to compare three or more groups, or paired T-test to compare two groups, considering statistically significant if P < 0.05. We used GraphPad Prism 5.0 software (GraphPad Software Inc., La Jolla, CA, USA).
Supplementary information
Acknowledgements
We thank Luciana Cintra (CETEC-HIAE) for her support with the care and maintenance of laboratory animals. This work was supported by Fundação de Amparo à Pesquisa do Estado de São Paulo (FAPESP) 2013/09611-2 and 2016/17961-1; 2013/09652-0 and 2015/25923-0 (scholarships to Carmo and to Pescatore) and Instituto Universidade-Empresa (scholarships to Fessel, Farias-Silva, Andrade and Simplicio-Filho).
Author contributions
Y.E.A. performed animal protocol, histological analysis, analyzed data and wrote the manuscript; M.R.F. assessed immunofluorescence studies, assembled figures, analyzed data and wrote the manuscript; L.S.C. performed animal protocol and Osteosense readings; V.J. processed femurs and assessed bone histomorphometric analysis; E.F.S. performed tissue processing and histochemical techniques; L.A.P. performed animal protocol, BMP-2 immunofluorescence, Osteosense readings and analyzed data; L.F.G. operated fluorescence detector for kidneys’ Osteosense readings and analyzed data; M.C.A. quantified calcification of vascular smooth muscle cells in culture; A.S. assessed mesangial and histological assessment of the kidneys; C.L.P.M. assessed 25-hidroxyvitamin D concentration in plasma; E.B.R. analyzed data, wrote part of introduction and discussion sections; M.L. conceived the study, wrote introduction and discussion sections and revised the manuscript.
Data availability
The datasets generated during and/or analysed during the current study are available in 1.Liberman, M. Dataset Almeida et al.xlsx. (2019). 10.6084/m9.figshare.7949453.v5.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
is available for this paper at 10.1038/s41598-019-55501-3.
References
- 1.Demer LL, Tintut Y. Vascular calcification: pathobiology of a multifaceted disease. Circulation. 2008;117:2938–2948. doi: 10.1161/CIRCULATIONAHA.107.743161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Sage AP, Tintut Y, Demer LL. Regulatory mechanisms in vascular calcification. Nature reviews. Cardiology. 2010;7:528–536. doi: 10.1038/nrcardio.2010.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liberman M, Pesaro AE, Carmo LS, Serrano CV., Jr. Vascular calcification: pathophysiology and clinical implications. Einstein (Sao Paulo) 2013;11:376–382. doi: 10.1590/s1679-45082013000300021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Roglic Gojka. WHO Global report on diabetes: A summary. International Journal of Noncommunicable Diseases. 2016;1(1):3. doi: 10.4103/2468-8827.184853. [DOI] [Google Scholar]
- 5.Rifkin DE, Ix JH, Wassel CL, Criqui MH, Allison MA. Renal artery calcification and mortality among clinically asymptomatic adults. J. Am. Coll. Cardiol. 2012;60:1079–1085. doi: 10.1016/j.jacc.2012.06.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Finch JL, et al. Phosphate restriction significantly reduces mortality in uremic rats with established vascular calcification. Kidney international. 2013;84:1145–1153. doi: 10.1038/ki.2013.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Paloian NJ, Giachelli CM. A current understanding of vascular calcification in CKD. Am. J. Physiol. Renal Physiol. 2014;307:F891–900. doi: 10.1152/ajprenal.00163.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Palit S, Kendrick J. Vascular calcification in chronic kidney disease: role of disordered mineral metabolism. Curr. Pharm. Des. 2014;20:5829–5833. doi: 10.2174/1381612820666140212194926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hou YC, et al. Role of Vitamin D in Uremic Vascular Calcification. Biomed Res Int. 2017;2017:2803579. doi: 10.1155/2017/2803579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Trohatou O, Tsilibary EF, Charonis A, Iatrou C, Drossopoulou G. Vitamin D3 ameliorates podocyte injury through the nephrin signalling pathway. J. Cell. Mol. Med. 2017;21:2599–2609. doi: 10.1111/jcmm.13180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng X, Cheng J, Shen M. Vitamin D improves diabetic nephropathy in rats by inhibiting renin and relieving oxidative stress. J. Endocrinol. Invest. 2016;39:657–666. doi: 10.1007/s40618-015-0414-4. [DOI] [PubMed] [Google Scholar]
- 12.Al Nozha OM. Vitamin D and extra-skeletal health: causality or consequence. Int J Health Sci (Qassim) 2016;10:443–452. [PMC free article] [PubMed] [Google Scholar]
- 13.Wani M, Wani I, Banday K, Ashraf M. The other side of vitamin D therapy: a case series of acute kidney injury due to malpractice-related vitamin D intoxication. Clin. Nephrol. 2016;86(2016):236–241. doi: 10.5414/CN108904. [DOI] [PubMed] [Google Scholar]
- 14.Carmo LS, et al. Expansive Vascular Remodeling and Increased Vascular Calcification Response to Cholecalciferol in a Murine Model of Obesity and Insulin Resistance. Arteriosclerosis, thrombosis, and vascular biology. 2019;39:200–211. doi: 10.1161/ATVBAHA.118.311880. [DOI] [PubMed] [Google Scholar]
- 15.Barton M. Primum Non Nocere: Why Calcitriol («Vitamin» D) Hormone Therapy Is Not a Magic Bullet. Arteriosclerosis, thrombosis, and vascular biology. 2019;39:117–120. doi: 10.1161/ATVBAHA.118.312105. [DOI] [PubMed] [Google Scholar]
- 16.McFarlane P, Cherney D, Gilbert RE, Senior P. Diabetes Canada Clinical Practice Guidelines Expert, C. Chronic Kidney Disease in Diabetes. Can J Diabetes. 2018;42(Suppl 1):S201–S209. doi: 10.1016/j.jcjd.2017.11.004. [DOI] [PubMed] [Google Scholar]
- 17.Durham AL, Speer MY, Scatena M, Giachelli CM, Shanahan CM. Role of smooth muscle cells in vascular calcification: implications in atherosclerosis and arterial stiffness. Cardiovasc. Res. 2018;114:590–600. doi: 10.1093/cvr/cvy010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Speer MY, et al. Smooth muscle cells give rise to osteochondrogenic precursors and chondrocytes in calcifying arteries. Circulation research. 2009;104:733–741. doi: 10.1161/CIRCRESAHA.108.183053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hudkins KL, et al. BTBR Ob/Ob mutant mice model progressive diabetic nephropathy. Journal of the American Society of Nephrology: JASN. 2010;21:1533–1542. doi: 10.1681/ASN.2009121290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lucisano S, et al. New insights on the role of vitamin D in the progression of renal damage. Kidney Blood Press. Res. 2013;37:667–678. doi: 10.1159/000355747. [DOI] [PubMed] [Google Scholar]
- 21.Yang S, et al. Vitamin D Receptor: A Novel Therapeutic Target for Kidney Diseases. Curr. Med. Chem. 2018;25:3256–3271. doi: 10.2174/0929867325666180214122352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cakici C, et al. Dose-dependent effects of vitamin 1,25(OH)2D3 on oxidative stress and apoptosis. J. Basic Clin. Physiol. Pharmacol. 2018;29:271–279. doi: 10.1515/jbcpp-2017-0121. [DOI] [PubMed] [Google Scholar]
- 23.Dudenkov DV, et al. Changing Incidence of Serum 25-Hydroxyvitamin D Values Above 50 ng/mL: A 10-Year Population-Based Study. Mayo Clin. Proc. 2015;90:577–586. doi: 10.1016/j.mayocp.2015.02.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tani T, Orimo H, Shimizu A, Tsuruoka S. Development of a novel chronic kidney disease mouse model to evaluate the progression of hyperphosphatemia and associated mineral bone disease. Scientific Reports. 2017;7:2233. doi: 10.1038/s41598-017-02351-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Christakos S, Dhawan P, Verstuyf A, Verlinden L, Carmeliet G. Vitamin D: Metabolism, Molecular Mechanism of Action, and Pleiotropic Effects. Physiological reviews. 2016;96:365–408. doi: 10.1152/physrev.00014.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Colussi G, et al. Chronic hypercalcaemia from inactivating mutations of vitamin D 24-hydroxylase (CYP24A1): implications for mineral metabolism changes in chronic renal failure. Nephrology, dialysis, transplantation: official publication of the European Dialysis and Transplant Association - European Renal Association. 2014;29:636–643. doi: 10.1093/ndt/gft460. [DOI] [PubMed] [Google Scholar]
- 27.Tebben PJ, Singh RJ, Kumar R. Vitamin D-Mediated Hypercalcemia: Mechanisms, Diagnosis, and Treatment. Endocrine reviews. 2016;37:521–547. doi: 10.1210/er.2016-1070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cozzolino M, Dusso AS, Slatopolsky E. Role of calcium-phosphate product and bone-associated proteins on vascular calcification in renal failure. Journal of the American Society of Nephrology: JASN. 2001;12:2511–2516. doi: 10.1681/ASN.V12112511. [DOI] [PubMed] [Google Scholar]
- 29.Moe S, et al. Definition, evaluation, and classification of renal osteodystrophy: a position statement from Kidney Disease: Improving Global Outcomes (KDIGO) Kidney international. 2006;69:1945–1953. doi: 10.1038/sj.ki.5000414. [DOI] [PubMed] [Google Scholar]
- 30.Martin-Timon I, Sevillano-Collantes C, Segura-Galindo A, Del Canizo-Gomez FJ. Type 2 diabetes and cardiovascular disease: Have all risk factors the same strength? World J. Diabetes. 2014;5:444–470. doi: 10.4239/wjd.v5.i4.444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.De S, et al. Exocytosis-Mediated Urinary Full-Length Megalin Excretion Is Linked With the Pathogenesis of Diabetic Nephropathy. Diabetes. 2017;66:1391–1404. doi: 10.2337/db16-1031. [DOI] [PubMed] [Google Scholar]
- 32.Wang Y, Borchert ML, DeLuca HF. Identification of the vitamin D receptor in various cells of the mouse kidney. Kidney international. 2012;81:993–1001. doi: 10.1038/ki.2011.463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Wang H, et al. In vitro and in vivo inhibition of mTOR by 1,25-dihydroxyvitamin D3 to improve early diabetic nephropathy via the DDIT4/TSC2/mTOR pathway. Endocrine. 2016;54:348–359. doi: 10.1007/s12020-016-0999-1. [DOI] [PubMed] [Google Scholar]
- 34.Zhang Z, et al. 1,25-Dihydroxyvitamin D3 targeting of NF-kappaB suppresses high glucose-induced MCP-1 expression in mesangial cells. Kidney international. 2007;72:193–201. doi: 10.1038/sj.ki.5002296. [DOI] [PubMed] [Google Scholar]
- 35.Goumans Marie-José, Zwijsen An, ten Dijke Peter, Bailly Sabine. Bone Morphogenetic Proteins in Vascular Homeostasis and Disease. Cold Spring Harbor Perspectives in Biology. 2017;10(2):a031989. doi: 10.1101/cshperspect.a031989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Katagiri Takenobu, Watabe Tetsuro. Bone Morphogenetic Proteins. Cold Spring Harbor Perspectives in Biology. 2016;8(6):a021899. doi: 10.1101/cshperspect.a021899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Lee SJ, et al. CCR2 knockout ameliorates obesity-induced kidney injury through inhibiting oxidative stress and ER stress. PloS one. 2019;14:e0222352. doi: 10.1371/journal.pone.0222352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Weisberg SP, et al. Obesity is associated with macrophage accumulation in adipose tissue. The Journal of clinical investigation. 2003;112:1796–1808. doi: 10.1172/jci19246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sullivan T, et al. CCR2 antagonist CCX140-B provides renal and glycemic benefits in diabetic transgenic human CCR2 knockin mice. American journal of physiology. Renal physiology. 2013;305:F1288–1297. doi: 10.1152/ajprenal.00316.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Bostrom KI, Jumabay M, Matveyenko A, Nicholas SB, Yao Y. Activation of vascular bone morphogenetic protein signaling in diabetes mellitus. Circ Res. 2011;108:446–457. doi: 10.1161/circresaha.110.236596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Andrade MC, Carmo LS, Farias-Silva E, Liberman M. Msx2 is required for vascular smooth muscle cells osteoblastic differentiation but not calcification in insulin-resistant ob/ob mice. Atherosclerosis. 2017;265:14–21. doi: 10.1016/j.atherosclerosis.2017.07.028. [DOI] [PubMed] [Google Scholar]
- 42.Kagi L, et al. Regulation of vitamin D metabolizing enzymes in murine renal and extrarenal tissues by dietary phosphate, FGF23, and 1,25(OH)2D3. PloS one. 2018;13:e0195427. doi: 10.1371/journal.pone.0195427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Graciolli FG, et al. The complexity of chronic kidney disease-mineral and bone disorder across stages of chronic kidney disease. Kidney international. 2017;91:1436–1446. doi: 10.1016/j.kint.2016.12.029. [DOI] [PubMed] [Google Scholar]
- 44.Vanagt WY, Famaey N, Rega F, Gewillig M. Extreme windkessel effect can cause right heart failure early after truncus repair. Interactive cardiovascular and thoracic surgery. 2012;15:181–182. doi: 10.1093/icvts/ivr133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Georgiadis GS, et al. Upper limb vascular calcification score as a predictor of mortality in diabetic hemodialysis patients. J. Vasc. Surg. 2015;61:1529–1537. doi: 10.1016/j.jvs.2015.01.026. [DOI] [PubMed] [Google Scholar]
- 46.Reiser J, Altintas MM. Podocytes. F1000Res. 2016;5:F1000 Faculty Rev-1114. doi: 10.12688/f1000research.7255.1. [DOI] [Google Scholar]
- 47.Ferland-McCollough D, Slater S, Richard J, Reni C, Mangialardi G. Pericytes, an overlooked player in vascular pathobiology. Pharmacol. Ther. 2017;171:30–42. doi: 10.1016/j.pharmthera.2016.11.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ueda H, et al. Bmp in podocytes is essential for normal glomerular capillary formation. Journal of the American Society of Nephrology: JASN. 2008;19:685–694. doi: 10.1681/ASN.2006090983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pichaiwong W, et al. Reversibility of structural and functional damage in a model of advanced diabetic nephropathy. Journal of the American Society of Nephrology: JASN. 2013;24:1088–1102. doi: 10.1681/ASN.2012050445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ohtake T, Kobayashi S. Impact of vascular calcification on cardiovascular mortality in hemodialysis patients: clinical significance, mechanisms and possible strategies for treatment. Renal Replacement Therapy. 2017;3:13. doi: 10.1186/s41100-017-0094-y. [DOI] [Google Scholar]
- 51.Mizobuchi M, Towler D, Slatopolsky E. Vascular calcification: the killer of patients with chronic kidney disease. Journal of the American Society of Nephrology: JASN. 2009;20:1453–1464. doi: 10.1681/ASN.2008070692. [DOI] [PubMed] [Google Scholar]
- 52.Parfitt AM, et al. Bone histomorphometry: standardization of nomenclature, symbols, and units. Report of the ASBMR Histomorphometry Nomenclature Committee. Journal of bone and mineral research: the official journal of the American Society for Bone and Mineral Research. 1987;2:595–610. doi: 10.1002/jbmr.5650020617. [DOI] [PubMed] [Google Scholar]
- 53.Hjortnaes J, New SE, Aikawa E. Visualizing novel concepts of cardiovascular calcification. Trends in cardiovascular medicine. 2013;23:71–79. doi: 10.1016/j.tcm.2012.09.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The datasets generated during and/or analysed during the current study are available in 1.Liberman, M. Dataset Almeida et al.xlsx. (2019). 10.6084/m9.figshare.7949453.v5.