

Summary
Infection with human cytomegalovirus (HCMV) remains a significant cause of morbidity and mortality following hematopoietic stem cell transplant (HSCT), due to various hematologic problems, including myelosuppression. Here we demonstrate that latently expressed HCMV miR-US5-2 down-regulates the transcriptional repressor NGFI-A binding protein (NAB1) to induce myelosuppression of uninfected CD34+ hematopoietic progenitor cells (HPCs) through an increase in TGF-β production. Infection of HPCs with an HCMVΔmiR-US5-2 mutant resulted in decreased TGF-β expression and restoration of myelopoiesis. In contrast, we show that infected HPCs are refractory to TGF-β signaling as another HCMV miRNA, miR-UL22A, down-regulates SMAD3, which is required for maintenance of latency. Our data suggests that latently-expressed viral miRNAs manipulate stem cell homeostasis by inducing secretion of TGF-β, while protecting infected HPCs from TGF-β-mediated effects on viral latency and reactivation. These observations provide a mechanism through which HCMV induces global myelosuppression following HSCT while maintaining lifelong infection in myeloid lineage cells.
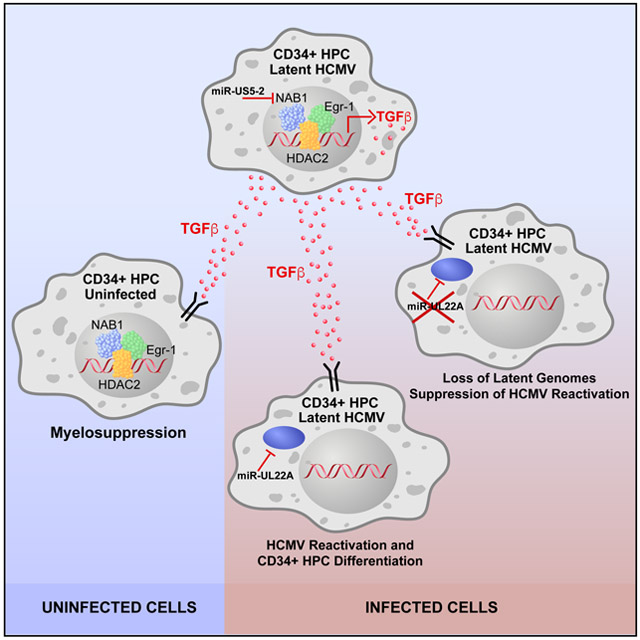
Graphical Abstract
eTOC Blurb
HCMV infection causes significant morbidity and mortality in stem cell transplant recipients due to virus-induced myelosuppression. Hancock et al show that HCMV miRNAs produced during latent infection induces the expression of the myelosuppressive cytokine TGFb while protecting the infected cell from TGFb signaling for efficient viral latency.
Introduction
Human Cytomegalovirus (HCMV) infection of allogeneic hematopoietic stem cell transplant (HSCT) recipients remains a significant cause of morbidity and mortality with up to 80% of patients requiring antiviral therapy (Ljungman et al., 2011; Safdar and Armstrong, 2019). A common clinical manifestation of HCMV disease in these patients is severe pancytopenia, myelosuppression, bone marrow hypoplasia, and delayed engraftment or graft failure (Fries et al., 1993). Over the past 40 years HCMV infection of hematopoietic progenitor cells (HPCs) in the bone marrow has been implicated as the causative agent of the myelosuppression observed in these recipients. While the effects of HCMV infection in the bone marrow are significant, the number of infected hematopoietic progenitor cells is extremely small (Torok-Storb et al., 1992) indicating that factors other than HCMV direct infection of these cells mediates myelosuppression. Despite the clinical relevance in these recipients, surprisingly little is currently known about the mechanism(s) by which HCMV causes these hematologic abnormalities.
Transforming growth factor beta (TGF-β) is a member of a multifunctional cytokine family that regulates a multitude of cellular processes including proliferation, cell survival, differentiation, and migration (Blank and Karlsson, 2015) which are important for regulation of HPC homeostasis (Capron et al., 2010). Several studies have shown that TGF-β is a potent inhibitor of early progenitor cell proliferation (Capron et al., 2010; Sing et al., 1988; Sitnicka et al., 1996) but in contrast can also promote HPC division at low concentrations (Kale, 2004), and more differentiated progenitors are unaffected by TGF-β (Challen et al., 2010). TGF-β regulation of cytokine receptors (McNiece et al., 1992; Sing et al., 1988), cell cycle regulators (Batard et al., 2000; Dao et al., 2002) and apoptosis (Sánchez-Capelo, 2005) are important for progenitor cell self-renewal and quiescence during normal homeostasis (Blank and Karlsson, 2015). Altering TGF-β levels in the bone marrow compartment dramatically alters HPC quiescence and normal hematopoiesis (Bataller et al., 2019).
Expression of TGF-β is regulated by Early Growth Response Gene-1 (EGR1), a zinc finger transcription factor induced by growth factor stimulation of the MEK/ERK pathway (Christy et al., 1988; Lim et al., 1987; Milbrandt, 1987; Sukhatme et al., 1987). EGR1 upregulates TGF-β through two GC-rich EGR1 binding sites within the TGF-β promoter (Kim et al., 1989; Liu et al., 1996; Yoo et al., 1996). EGR1-mediated transcription is negatively regulated by the transcriptional repressor NGFI-A binding protein (NAB1) through direct interaction of sumoylated NAB1 and the R1 domain of EGR1 which brings histone deacetylase 2 (HDAC2) and other chromatin modifiers to EGR1-regulated promoters in order to repress transcription (Cai et al., 2017; García-Gutiérrez et al., 2011; Russo et al., 1995; Swirnoff et al., 1998; Thiel et al., 2000). Mutation of the NAB1 interaction domain of EGR1 results in enhanced expression of TGF-β (Liu et al., 1996). Therefore, NAB1 can modulate TGF-β expression through regulation of EGR1-mediated TGF-β transcription.
TGF-β is secreted from the cell in a latent form that is processed by cellular proteases to produce biologically active TGF-β that can bind to its cognate receptors. Ligand binding results in receptor phosphorylation which recruits and phosphorylates the receptor-associated SMADs (R-SMADs) SMAD2 and/or SMAD3. Phosphorylation of R-SMADs enhances their interaction with the co-SMAD, SMAD4, which results in shuttling of the R-SMAD/SMAD4 complex to the nucleus. SMADs regulate transcription by altering chromatin structure and generally have only weak affinity for the SMAD binding element, therefore other DNA binding proteins are required for selective binding of the SMAD complexes to target enhancer elements (Reviewed in (Massagué et al., 2000)). Thus, the transcriptional outcome of canonical TGF-β signaling depends critically on a SMAD DNA binding cofactor, whose expression and localization are regulated by additional cellular signaling pathways initiated in a context-dependent and cell type-specific manner.
HCMV encodes multiple miRNAs that regulate expression of both cellular and viral genes involved in viral replication, cytokine expression, cell survival, and innate and adaptive immunity (Diggins and Hancock, 2018). HCMV miRNAs miR-US5-1, miR-US5-2 and miR-UL112-3p target multiple cellular genes within the endocytic recycling pathway to allow for efficient virion assembly compartment formation and to limit the release of pro-inflammatory cytokines (Hook et al., 2014). Cytokine production and secretion were also shown to be regulated by HCMV miR-UL148D-1 that targets ACVR1B to limit IL-6 secretion (Lau et al., 2016) and RANTES/CCL5 (Kim et al., 2012). Additionally, HCMV miRNAs miR-US5-1 and miR-UL112-3p were reported to directly target the IKK complex kinases IKKα and IKKβ to inhibit NFκB signaling in order to limit the release of pro-inflammatory cytokines (Hancock et al., 2017). Furthermore, miR-UL112-3p was shown to target the TLR2 transcript to limit signaling through the TLR2-IRAK1 axis and block NFκB activation (Landais et al., 2015). Recently, miR-US22 was shown to directly target EGR1 that regulates proliferation, differentiation and viral reactivation in CD34+ HPCs (Mikell et al., 2019). These examples illustrate how HCMV miRNAs manipulate the cell to facilitate latency and reactivation, as well as shape the antiviral immune response.
In this study, we demonstrate that latent HCMV infection of CD34+ HPCs induces the secretion of TGF-β that is responsible for virus-mediated myelosuppression. We observe that HCMV miR-US5-2 down-regulation of the transcriptional repressor NAB1 was sufficient to induce expression of the cytokine and result in myelosuppression of uninfected CD34+ HPCs. We also observe that cellular signaling induced by TGF-β in the latently infected cell was blocked by viral miR-UL22A targeting of SMAD3 which is critical for latency in CD34+ HPCs. These results provide a mechanism for how latent HCMV infection of a minor population of CD34+ HPCs in the bone marrow of HSCT patients induces global myelosuppression. By inducing the production of TGF-β and simultaneously blocking the negative effects of TGF-β on latently infected cells the virus successfully maintains a lifelong infection in myeloid-lineage cells.
Results
Latent HCMV infection of CD34+ HPCs mediates myelosuppression through secretion of TGF-β.
In order to examine the mechanisms of HCMV inhibition of myelopoiesis, hematopoietic colony formation was quantified using myeloid colony forming assays. For these studies, CD34+ HPCs were infected with HCMV strain TB40E-GFP for 48 hours followed by purification of CD34+, GFP+ cells and culture for 14 days in Methocult. Quantitation of hematopoietic colonies indicated that HCMV preferentially suppressed myeloid (CFU-GM and CFU-GEMM) colony formation as compared to erythroid (CFU-E and BFU-E) colony formation (Figure 1A). This myelosuppression was reproducibly observed with HCMV infection of CD34+ HPCs obtained from multiple donors ((data not shown) and from previous studies (Goodrum et al., 2004; Rakusan et al., 1989; Sing and Ruscetti, 1990)). In order to determine if long-term latent HCMV infection was also myelosuppressive to infected HPCs, CD34+ HPCs were infected and sorted as above, then cultured under latency-promoting conditions on stromal cells for 12 days before quantitation of myeloid colony formation. Similar to results obtained with shorter-term infection of CD34+ HPCs, latent HCMV infection suppressed myeloid colony formation indicating that virus-induced myelosuppression was not transient (Figure 1B).
Figure 1. HCMV infection directly suppresses myelopoiesis via upregulation of TGF-β from latently infected HPCs.

CD34+ HPCs were infected with HCMV (TB40E-GFP) for ~42 hrs. Infected cells were sorted for viable, CD34+, and GFP+ HPCs. Uninfected cells were cultured in the same manner and sorted for viable, CD34+ HPCs. A) For analysis of the direct effects of HCMV infection on myeloid differentiation, sorted CD34+ (Mock) or CD34+GFP+ (HCMV) HPCs were plated at 500 cells per dish in Methocult H4434 and cultured for 14 days. Different myeloid (CFU-GM, CFU-GEMM) and erythroid (CFU-E and BFU-E) colonies were counted at 14 days post-plating. B) For analysis of the effects of latent HCMV infection on myeloid differentiation, sorted CD34+ (Mock) or CD34+GFP+ (HCMV) HPCs were plated on stromal cell support for long-term culture to establish HCMV latency. Total cells were counted after 12 days in latency culture (14dpi) and were plated at 1000 cells per dish in Methocult H4434 and cultured for an additional 14 days. Representative experiments are shown. Error bars represent standard deviation for triplicate wells, significance determined by two-way ANOVA with Sidak’s post-test. C) To determine the effect of the HCMV secretome from latently infected CD34+ HPCs, Mock and HCMV-infected HPCs were infected and cultured as in B, then removed and plated in serum-free media for an additional 48hrs prior to collection of cell-free supernatants (secretome samples). CD34+ HPCs pre-treated with or without anti-TGF-β antibody were mixed with diluted (1:5) secretome samples from Mock or HCMV-infected HPCs, plated at 500 cells per dish in Methocult H4434 and cultured for 14 days. Total colonies from a representative experiment are shown. Error bars represent standard deviation for triplicate wells, significance determined by t-test. D) Cell-free secretome samples from latent HCMV-infected CD34+ HPCs or Mock-infected HPCs from long term culture were analyzed for secreted TGF-β by ELISA. Data shown is for one representative experiment out of 5 biological replicates. Additional replicates are summarized in Supplemental Table 1. Error bars represent standard deviation for duplicate wells, significance determined by t-test. See also Figure S1 and Supplemental Table 1.
To determine whether HCMV-induced myelosuppression is due to factors secreted from infected progenitor cells, supernatants from latently infected CD34+ HPCs were examined for myelosuppressive effects on uninfected cells. In these experiments, HCMV-infected CD34+ HPCs were cultured on stromal cell support and supernatants were harvested following establishment of latency. Analysis of CD34+ latently-infected HPCs did not reveal the presence of infectious virus (Supplemental Figure 1).
Supernatants were diluted at a ratio of 1:5 and added to uninfected CD34+ HPCs to quantitate myeloid colony formation. As shown in Figure 1C, supernatants from latently-infected cells (HCMV) were capable of suppressing colony formation compared to supernatants from Mock-infected HPCs cultured under the same conditions. These results indicate that latent HCMV infection of CD34+ HPCs reprograms the cell to produce cytokines that alter hematopoiesis of neighboring uninfected progenitor cells. To identify the HCMV-induced cytokines and growth factors produced by latently infected CD34+ HPCs, HPCs were cultured for an additional two days following the establishment of latency and supernatants were analyzed by Luminex assay and TGF-β ELISA. As shown in Figure 1D and Supplemental Table 1, TGF-β was the only cytokine induced by latent HCMV (~4-fold) in CD34+ HPC supernatants.
TGF-β plays a critical role in regulating the balance between proliferation and differentiation of hematopoietic cells and suppresses hematopoietic colony formation (Blank and Karlsson, 2015). Therefore, to determine if HCMV-induced TGF-β was responsible for myelosuppression, TGF-β neutralizing antibody was added to latent supernatants. In these experiments, 1 μg/ml of anti-TGF-β antibody was added to supernatants obtained from Mock or HCMV-infected CD34+ HPCs at 14 dpi and the supernatants were subsequently added to naïve progenitor cells followed by quantitation of myeloid colony formation. As shown in Figure 1C, addition of anti-TGF-β antibody to supernatants from HCMV-infected cultures restored total myeloid colony formation to levels observed in cells treated with mock-infected supernatants indicating that TGF-β is responsible for viral-mediated myelosuppression in vitro.
HCMV miR-US5-2 targets the transcriptional repressor NAB1 that regulates TGF-β expression.
The ability of latent HCMV infection to induce the secretion of TGF-β in CD34+ HPCs implies that a latently expressed viral gene product regulates this process. Several HCMV-encoded miRNAs are expressed during latency (Mikell et al., 2019). To determine if HCMV miRNAs were capable of inducing secretion of TGF-β, NHDFs were transfected with mimics of latently expressed miRNAs as well as siRNA to TGF-β followed by quantitation of the cytokine in supernatants. As shown in Figure 2A, miR-US5-2 was the only HCMV miRNA that significantly induced secretion of TGF-β from transfected cells. Bioinformatic analysis to identify potential cellular mRNA targets of miR-US5-2 that might regulate TGF-β expression determined that the 3’ UTR of NAB1 encodes two potential miR-US5-2 target sequences (Supplemental Figure 2A). NAB1 is a transcriptional repressor of the transcription factor EGR1 that activates expression of TGF-β (Liu et al., 1996; Yoo et al., 1996). In order to validate NAB1 as a target of miR-US5-2, 293T cells were co-transfected with a miR-US5-2 mimic and luciferase reporters containing either a WT NAB1 3’UTR, or 3’UTRs with mutations of each individual or both potential miR-US5-2 target sites. As shown in Figure 2B, luciferase expression was significantly reduced from the WT NAB1 3’UTR, partially restored by mutation of either of the miR-US5-2 potential target sites and fully restored with mutation of both sequences. Transfection of 293T cells (Figure 2C), Normal Human Dermal Fibroblasts (NHDF) (Figure 2D) or endothelial cells (Figure 2E) with the miR-US5-2 mimic or siRNA to NAB1 resulted in down-regulation of NAB1 protein levels. These data indicate that NAB1 is a valid target of miR-US5-2.
Figure 2. HCMV miR-US5-2 upregulates TGF-β by directly targeting NAB1.

A) NHDFs were transfected with mimics for latently expressed HCMV miRNAs or TGF-β siRNA for 48 hours and secreted TGF-β analyzed by ELISA. Error bars represent standard deviation for triplicate wells, significance determined by one-way ANOVA compared to negative control siRNA. B) HEK293T cells were co-transfected with pSIREN expression vectors containing the WT NAB1 3’ UTR or 3’UTRs containing mutations in the putative miR-US5-2 binding sites and Negative control or miR-US5-2 mimic. Twenty-four hours post-transfection, protein lysates were harvested and examined for luciferase expression. Statistical significance determined by one-way ANOVA. See also Figure S2A. Protein was harvested from 293T (C), Normal Dermal Human Fibroblasts (NHDFs) (D), or human aortic endothelial cells (hAECs) (E) transfected with miR-US5-2 mimic or NAB1 siRNA for 48 hours and analyzed for NAB1 expression via western blot. GAPDH was used as a loading control and fold change in NAB1 expression compared to GAPDH is recorded below.
HCMV miR-US5-2 is sufficient to induce expression of TGF-β and myelosuppression in CD34+ HPCs.
To determine if miR-US5-2 regulated TGF-β expression in hematopoietic cells, pSIREN-GFP vectors expressing either miR-US5-2 or a NAB1 shRNA were transfected into Kasumi-3 cells. Viable, CD34+, GFP+ cells were sorted 24 hours post-transfection, incubated another 24 hours in serum-free media and TGF-β was quantified in cellular supernatants. As shown in Figure 3A, expression of either HCMV miR-US5-2 or an shRNA to NAB1 resulted in a 2-fold increase in secretion of TGF-β. CD34+ HPCs were also transfected with HCMV miR-US5-2 and cultured in HPC maintenance media to determine the effect of the miRNA on hematopoiesis. As shown in Figure 3B and C miR-US5-2 decreased CD34+ HPC expansion. In Figure 3C miR-US5-2 decreased CD34+ HPC proliferation in six different CD34+ HPC donors from 25% to 2-3 fold in comparison to the expression vector control. Quantitation of myeloid colonies from CD34+ HPCs transfected with either miR-US5-2 or a NAB1 shRNA demonstrated a reduction of CFU-GM myeloid colonies as well as total hematopoietic colony formation (Figure 3D and E).
Figure 3. HCMV miR-US5-2 directly targets NAB1 to upregulate TGF-β in HPCs and mediate myelosuppression.

A) Kasumi-3 cells were transfected with pSIREN-GFP plasmids expressing miR-US5-2 or an shRNA against NAB1 and cultured for 24hrs. Pure populations of viable, transfected (GFP+) cells were isolated by FACS and recovered overnight. Supernatants were analyzed by TGF-β ELISA following an additional 24hrs in serum-free media. B and C) CD34+ HPCs were transfected as in A for 48hrs. Pure populations of viable, transfected (GFP+) cells were isolated by FACS and plated in SFEMII to support progenitor cell proliferation for 7 days. Total viable cells were manually counted at 2, 5 and 7 days post plating. The average proliferation for one representative experiment is shown in B with error bars representing standard deviation for replicate wells. Primary CD34+ HPCs from 6 independent donors were transfected with pSIREN-GFP-miR-US5-2 and the fold change in proliferation compared to control vector (pSIREN-GFP) shown in C. Two way ANOVA was performed comparing all donors in C, p<0.05. D and E) CD34+ HPCs were transfected and isolated as above. Pure populations were plated at 500 cells per dish in Methocult H4434 in triplicate for 14 days. Specific myeloid colony types are shown from one representative experiment in D. The average total myeloid colonies from independent CD34+ HPC donors are shown in E for miR-US5-2 (N=11) and NAB1 shRNA (N=5). Error bars represent standard deviation for replicate wells (D) or SEM for replicate experiments (E), significance determined by two-way (D) or one-way (E) ANOVA with Tukey post-test.
In order to determine the contribution of miR-US5-2 to induction of TGF-β and myelosuppression in the context of the virus, the miR-US5-2 pre-miRNA was deleted from a TB40E infectious clone (TB40EΔmiR-US5-2) using GalK recombination (Supplemental Figure 2B). The TB40EΔmiR-US5-2 virus exhibits WT replication in NHDF (data not shown) and infection of NHDF with TB40EΔmiR-US5-2 results in increased NAB1 expression compared to WT-infected cells (Figure 4A). TB40EΔmiR-US5-2 infection of CD34+ HPCs resulted in a significant decrease in TGF-β secretion into the supernatant (Figure 4B). Importantly, deletion of miR-US5-2 significantly restored both CD34+ HPC proliferation (Figures 4C and D) as well as myelopoiesis to levels observed in uninfected cells (Figure 4E). Collectively, these data indicate that miR-US5-2 targeting of NAB1 during HCMV infection in CD34+ HPCs is sufficient to induce TGF-β expression and myelosuppression observed during HCMV infection.
Figure 4. Loss of miR-US5-2 directly abrogates HCMV-mediated TGF-β upregulation in HPCs and controls myelosuppression.

A) NHDF were mock infected or infected with WT HCMV or HCMVΔmiR-US5-2 for the indicated times. Protein lysates were harvested and analyzed for NAB1 expression via western blot. GAPDH was used as a loading control and fold change in NAB1 expression compared to GAPDH is recorded below. B) CD34+ HPCs were infected with HCMV or HCMVΔmiR-US5-2 for 48 hours, then sorted and plated for latency as described in Figure 1. Latently infected HPCs were cultured for an additional 48hrs in serum-free media and analyzed by ELISA for TGF-β expression. Error bars represent standard error of the mean from 4 independent experiments with each independent HPC donor represented by unique symbols, significance determined by one-way ANOVA. C and D) CD34+ HPCs were infected with HCMV, HCMVΔmiR-US5-2, or Mock-infected, sorted by FACS and plated for latency as described in Figure 1. Total viable HPCs were counted at 12d post-plating (14dpi) to assess proliferation during the latency period. The average proliferation for one representative experiment is shown in C, and the fold change compared to Mock-infected cells for 10 independent experiments is shown in D. Error bars represent SEM for replicate experiments and significance was determined by one-way ANOVA. E) Pure populations of infected HPCs were sorted and plated at 500 cells per well in Methocult H4434 in triplicate for analysis of myeloid colony formation. Specific myeloid colony types are shown from one representative experiment out of 3 independent experiments. Error bars represent standard deviation for triplicate wells and significance was determined by two-way ANOVA with Tukey post-test. See also Figure S2B.
HCMV miR-UL22A down-regulates SMAD3 to allow viral reactivation in CD34+ HPCs
Although HCMV-induced TGF-β exhibits myelosuppressive effects on uninfected CD34+ HPCs, the effects of the cytokine on latently infected HPCs is unknown. Since TGF-β can negatively affect the growth of other viruses (Campion et al., 2014; Chiu et al., 2012; Choi et al., 2015; Di Bartolo et al., 2008; DiMaio et al., 2014; Fukuda et al., 2002; Fukuda and Longnecker, 2004; Horndasch et al., 2002; Inman and Allday, 2000; Lei et al., 2012; Lo et al., 2010; Mori et al., 2003; Morris et al., 2016; Prokova et al., 2002; Seo et al., 2005; Tomita et al., 2004; Wood et al., 2007), we hypothesized that HCMV protects infected cells from the negative effects of TGF-β signaling by manipulating components of the TGF-β signaling pathway. To test this hypothesis, we mock or HCMV-infected primary CD34+ HPCs for 48 hours, and isolated a pure population of viable, CD34+, GFP+ HPCs. Following serum starvation, HPCs were stimulated for 4 hours with TGF-β and RNA was analyzed by qRT-PCR. Figure 5A shows that while mock infected CD34+ HPCs respond to TGF-β stimulation by inducing expression of the classic TGF-β-responsive transcript SERPINE, WT infected cells did not induce this transcript, indicating a block in the canonical TGF-β signaling pathway. We next tested whether latently-expressed miRNAs could block the canonical TGF-β signaling pathway. 293T cells were transfected with a SMAD luciferase reporter construct along with negative control miRNA, HCMV miRNA mimics or a SMAD3 siRNA. As shown in Figure 5B, both miR-UL22A-3p and miR-UL22A-5p but not miR-UL112-3p, significantly reduced expression from the SMAD reporter construct. Bioinformatic and biochemical analyses indicated that the R-SMAD SMAD3 contains miRNA binding sites for both miR-UL22A-3p and miR-UL22A-5p within the 3’ UTR (Supplemental Figure 2C) suggesting that these miRNAs potentially target the canonical SMAD signaling pathway. We validated each target site for the respective miRNA using 3’ UTR luciferase assays (Figure 5C). Expression of miR-UL22A-3p and miR-UL22A-5p significantly reduced the expression of SMAD3 transcript (Figure 5D) and protein (Figure 5E), as well as reduced SERPINE expression in response to TGF-β (Figure 5F). These data validate miR-UL22A-5p and miR-UL22A-3p miRNA targeting of SMAD3 as well as the ability of these miRNAs to block signaling through the canonical TGFβ pathway.
Figure 5. HCMV miR-UL22A directly targets the SMAD3 transcript to limit signaling through the TGF-β pathway.

A) CD34+ HPCs were mock-infected or infected with WT TB40E for 48 hours followed by cell sorting for viable, CD34+, GFP+ cells. Cells were serum starved, stimulated with 100pg/mL of TGF-β for 4 hours, and RNA isolated and used for qRT-PCR for SERPINE expression normalized to 18S. Experiment was performed in triplicate. Error bars represent the standard deviation of the mean and significance was determined by t-test B) 293T cells were transfected with a SMAD luciferase reporter construct along with negative control miRNA, SMAD3 siRNA or HCMV miRNA mimics for 24 hours. Cells were serum starved, stimulated with 100pg/mL of TGF-β for 4 hours, then harvested and analyzed for luciferase expression. Experiment was performed in triplicate. Error bars represent the standard error of the mean and significance was determined by t-test. C) Either WT or SMAD3 3’UTR constructs containing mutations in the miR-UL22A-5p or -3p target sites were transfected into 293T cells along with negative control or the corresponding HCMV miRNA mimic. Twenty-four hours later cells were harvested and luciferase expression was measured. Experiment was performed in triplicate. Error bars represent the standard error of the mean and significance was determined by t-test. See also Figure S2C. D and E) NHDF were transfected with negative control miRNA, HCMV miR-UL22A-3p and -5p miRNA mimics or SMAD3 siRNA for 48 hours after which RNA (D) or protein (E) were harvested and analyzed for SMAD3 expression. Protein expression is normalized to GAPDH and relative levels are shown below. F) NHDF were transfected as in D and E, then serum starved and treated with 100pg/mL TGF-β for 4 hours after which time RNA was isolated and qRT-PCR was used to assess SERPINE expression as in (A). * p<0.05, **p<0.01.
We next generated a ΔmiR-UL22A mutant virus by removing the pre-miR-UL22A sequence (deleting both the -3p and -5p arms of the miRNA) from the TB40E strain of HCMV as well as a ΔmiR-UL22A/SMAD3 shRNA virus by replacing the miR-UL22A hairpin with an shRNA targeting SMAD3 (Supplemental Figure 2D). Infection of NHDF with the ΔmiR-UL22A mutant virus results in an increase in SMAD3 protein (Figure 6A), indicating that SMAD3 is a target of miR-UL22A during viral infection. Replacement of miR-UL22A with a SMAD3 shRNA reduced SMAD3 protein levels to those observed during WT infection (Figure 6A). Importantly, infection of CD34+ HPCs with ΔmiR-UL22A restored the transcriptional response to TGF-β, as measured by SERPINE transcript levels, to levels similar to mock infected cells, while infection with the ΔmiR-UL22A/SMAD3 shRNA mutant resulted in a decrease in SERPINE levels to what was observed in WT infected cells (Figure 6B). Thus, miR-UL22A targeting of SMAD3 is directly responsible for the block in canonical TGF-β signaling observed during latent infection of CD34+ HPCs.
Figure 6. HCMV miR-UL22A targeting of SMAD3 is essential for latency in CD34+ HPCs.

A) NHDF were infected with WT, ΔmiR-UL22A or ΔmiR-UL22A/SMAD3shRNA and protein was harvested at the indicated timepoints and assessed for SMAD3, HCMV IE2 and GAPDH protein expression. B) CD34+ HPCs were infected with WT, ΔmiR-UL22A and ΔmiR-UL22A/SMAD3 shRNA viruses and analyzed as in Figure 5A to determine SERPINE transcript levels. Experiment was performed in duplicate. Error bars represent the standard error of the mean. Significance was determined by t-test. *p<0.05 C) CD34+ HPCs were infected with HCMV, HCMVΔmiR-UL22A and HCMVΔmiR-UL22A/SMAD3 shRNA and analyzed for latency and reactivation as described in Figure 1. Results from two representative donors are shown. D) HCMV genome copy number in initially sorted (2dpi) and latently-infected HPCs (14dpi) from both two donors was assessed by qPCR for HCMV UL141 and normalized to cellular β-globin. Error bars represent the standard error of the mean of three triplicate wells. See also Figure S2D.
In order to investigate the importance of targeting SMAD3 and blocking the canonical TGF-β signaling pathway during HCMV latency and reactivation in CD34+ HPCs we established latent infections with WT, ΔmiR-UL22A and ΔmiR-UL22A/SMAD3 shRNA viruses. As shown in Figure 6C, the ΔmiR-UL22A virus reactivates with lower frequency compared to WT virus, while the ΔmiR-UL22A/SMAD3 shRNA virus restores the reactivation potential of the ΔmiR-UL22A mutant to WT levels. In order to determine the stage of latency and/or reactivation that is impaired upon infection with the ΔmiR-UL22A mutant virus, viral genomes were quantitated in CD34Δ HPCs at 2 and 14 dpi (Figure 6D). We observed fewer genomes at 14 dpi in ΔmiR-UL22A-infected cells compared to WT while expression of the SMAD3 shRNA in the context of the ΔmiR-UL22A mutant resulted in enhanced viral genomes. Thus, reducing SMAD3 expression and blocking the canonical TGF-β signaling pathway is an essential function of the miR-UL22A miRNAs during latency establishment and/or maintenance in CD34+ HPCs.
Discussion
In this report we demonstrate that latent HCMV infection of CD34+ HPCs induces the expression of TGF-β and inhibits myelopoiesis, which is restored with the addition of a TGF-β neutralizing antibody. We also show that HCMV miR-US5-2 down-regulates NAB1, a repressor of the transcription factor EGR1, and significantly increases expression of TGF-β. Introduction of a mutation into HCMV that blocks expression of HCMV miR-US5-2 inhibits the ability of the virus to induce TGF-β as well as restores CD34+ HPC proliferation and myelopoiesis to levels observed with mock-infected cells. These results provide a potential explanation and a mechanism for the observations in HSCT recipients that despite the presence of very few HCMV-infected cells in the bone marrow HCMV induction of TGF-β results in global myelosuppression.
We also observed that intracellular TGF-β signaling was blocked in infected CD34+ HPCs through HCMV miR-UL22A downregulation of SMAD3 expression. Mutation of miR-UL22A blocked viral reactivation in CD34+ HPCs that was reversed by expression of an shRNA directed against SMAD3. These data indicate that activation of the canonical TGF-β signaling pathway in the infected cell is detrimental for HCMV latency and requires miR-UL22A to block the negative effects of cytokine signaling on the establishment or maintenance of latency and viral reactivation. Thus, HCMV coordinates the expression of two distinct viral miRNAs during latency in order to increase production of TGFβ to manipulate homeostasis of surrounding cells in the bone marrow microenvironment as well as directly protect the infected cell from the negative effects of TGF-β signaling. A model describing the mechanisms of HCMV-mediated myelosuppression through induction of TGF-β and blocking of TGF-β signaling is shown in Figure 7.
Figure 7. HCMV miRNA-mediated regulation of TGF-β production and signaling during viral latency and reactivation in CD34+ HPCs.

Latent infection of CD34+ HPCs results in the expression of HCMV miR-US5-2 which directly targets the TGF-β transcriptional repressor NAB1 resulting in increased production and secretion of TGF-β. Secreted TGF-β acts on neighboring uninfected cells to mediate myelosuppression. Infected cells additionally express miR-UL22A which directly targets SMAD3 to block signaling through the TGF-β pathway which is required for efficient latency establishment and genome maintenance.
TGF-β is one of the most important cytokines regulating CD34+ HPC homeostasis and self-renewal (Blank and Karlsson, 2015). TGF-β can have different effects on cellular functions depending on the differentiation state of the cell and the concentration of the cytokine in the bone marrow microenvironment (Akel et al., 2003; Batard et al., 2000; Capron et al., 2010; Challen et al., 2010; Oh et al., 2000; Sitnicka et al., 1996). While TGF-β produced by stromal and hematopoietic cells is necessary to maintain CD34+ HPC homeostasis (Batard et al., 2000; Zhang et al., 2016), administration of high concentrations of the cytokine to HSCs in vitro is a potent inhibitor of cellular growth and proliferation (Dao et al., 2002; Sing et al., 1988; Sitnicka et al., 1996). While high concentrations of TGF-β negatively affect CD34+ HPC growth and proliferation, physiological concentrations of the cytokine are necessary to maintain stem cell quiescence and self-renewal. The processes involved in the maintenance of the stem cell pool and the signals that promote CD34+ HPC proliferation and differentiation are highly regulated, but incompletely understood (reviewed in (Haas et al., 2018)). As we have shown in this report, altering this balance through HCMV-induced overproduction of TGF-β results not only in myelosuppression but also suppression of HPC proliferation. In contrast to the effects of TGF-β on primitive hematopoietic progenitor cells, more differentiated hematopoietic cells respond differently to the cytokine (Fortunel et al., 2000; Mayani et al., 1995; Oh et al., 2000). Concentrations of TGF-β within the range produced during latent HCMV infection stimulate the proliferation of murine myeloid-biased HSCs, but inhibit lymphoid-biased HSC proliferation (Challen et al., 2010). This situation would promote increased numbers of myeloid lineage cells but decreased numbers of lymphoid cells in the peripheral organs of the host, potentially contributing to an overall immune suppression. Since HCMV preferentially targets myeloid lineage cells for latency and uses these cells a vector to traffic virus throughout the host, induction of TGF-β may provide a better environment for viral persistence.
Since TGF-β is a critical regulator of numerous cellular processes, cytokine expression and release are carefully regulated by the cell. The immediate early transcription factor EGR1 stimulates expression of TGF-β through two GC-rich EGR1 binding sites within the TGF-β promoter (Liu et al., 1996). NAB1 is a transcriptional repressor of EGR1-dependent transcripts and interacts with EGR1 to bring HDAC2 and other chromatin modifiers to the promoter (Cai et al., 2017; García-Gutiérrez et al., 2011; Swirnoff et al., 1998; Thiel et al., 2000). Critically, mutation of the NAB1-interacting domain of EGR1 enhances TGF-β production (Liu et al., 1996), which supports the observations presented here in which downregulation of NAB1 protein by miR-US5-2 enhances TGF-β expression. EGR1 is also a key transcription factor in maintaining the ‘stemness’ of HPCs in the bone marrow by blocking differentiation and promoting self-renewal (Min et al., 2008). Interestingly, we have recently reported that HCMV miR-US22 directly targets the EGR1 transcript (Mikell et al., 2019). miR-US22 is not expressed during latency in CD34+ HPCs, allowing the cells to maintain their stem cell phenotype and produce TGF-β. However, upon reactivation miR-US22 expression is upregulated and plays a key role in reducing EGR1 levels as well as blocking proliferation and enhancing differentiation along the myeloid lineage. Thus, regulated expression of HCMV miRNAs plays key roles in the maintenance of latency and triggering viral reactivation.
Herpesviruses, including Epstein-Barr virus and Kaposi’s sarcoma-associated herpesvirus have been extensively studied for their interactions with the TGF-β signaling pathway and encode proteins and non-coding RNAs or induce cellular miRNAs in order to overcome TGF-β-mediated apoptosis and block expression of growth inhibitory genes (Campion et al., 2014; Chiu et al., 2012; Choi et al., 2015; Di Bartolo et al., 2008; DiMaio et al., 2014; Fukuda et al., 2002; Fukuda and Longnecker, 2004; Horndasch et al., 2002; Inman and Allday, 2000; Lei et al., 2012; Lo et al., 2010; Mori et al., 2003; Morris et al., 2016; Prokova et al., 2002; Seo et al., 2005; Tomita et al., 2004; Wood et al., 2007). In this study we demonstrate that HCMV miR-UL22A reduction of SMAD3 is critical to block TGF-β signaling and plays an essential role in the establishment and/or maintenance of viral latency in CD34+ HPCs. By utilizing a ΔmiR-UL22A mutant virus expressing a SMAD3 shRNA, we are able to conclusively link the lack of viral reactivation observed with the ΔmiR-UL22A mutant virus to the requirement to block the canonical TGF-β signaling pathway. How TGF-β interferes with HCMV latency remains to be determined, but its effects at early stages of latency suggest TGF-β could act on latency establishment or alter the cell such that it is no longer conducive to latency maintenance.
HCMV-induced myelosuppression in HSCT and solid organ transplantation remains a significant problem in these patient populations (Ljungman et al., 2011; Randolph-Habecker et al., 2002). While current clinical practices utilize antiviral drugs that target lytic HCMV infection in transplant patients, the data presented here underlie the need for therapeutics targeting latent infection in order to combat virus-induced myelosuppression. Understanding the mechanisms of how HCMV regulates expression of TGF-β in latently infected CD34+ HPCs to alter hematopoiesis as well as how the virus controls the TGF-β signaling pathway to survive in an environment of enhanced TGF-β expression will be important for development of new therapeutic interventions.
STAR Methods
Lead Contact and Materials Availability
All information and unique reagents generated in this study are available from the Lead Contact, Dr. Jay Nelson (nelsonj@ohsu.edu) without restriction.
Experimental Model and Subject Details
Isolation of Primary Hematopoietic Stem/Progenitor Cells from Primary Fetal Liver
CD34+ hematopoietic progenitor cells (HPCs) were isolated from de-identified human fetal liver obtained from Advanced Bioscience Resources using CD34+ magnetic bead separation (Miltenyi) as previously described (Crawford et al., 2017). In brief, single cell suspensions of fetal liver cells were derived by enzymatic and mechanical disruption of the liver tissue followed by purification through a Ficoll gradient. CD34+ HPCs were isolated using the CD34+ HPC MicroBead kit from Miltenyi (Miltenyi Biotec). The purity of isolated CD34+ cells was analyzed using a allophycocyanin (APC)-conjugated monoclonal mouse antibody against CD34 (BioLegend).
Tissue Culture Cells
Normal human dermal fibroblasts (NHDF) and HEK293T cells were obtained from ATCC and cultured in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% heat-inactivated fetal bovine serum (FBS) (HyClone), 100 units/ml penicillin, 100ug/mL streptomycin and 100ug/mL glutamine (ThermoFisher). Human aortic endothelial cells (hAEC) (CC-2535; Lonza) were cultured in EBM-2 basal medium with EGM-2 SingleQuots supplement excluding Heparin (Lonza), as well as 10% FBS, penicillin, and streptomycin. Kasumi-3 cells were obtained from ATCC and maintained in RPMI 1640 with 20% FBS and penicillin, streptomycin, and glutamine. M2-10B4 and S1/S1 stromal cells were obtained from Stem Cell Technologies and maintained in DMEM with 10% fetal bovine serum and penicillin, streptomycin, and glutamine. All cells were maintained at 37°C and 5% CO2.
Method Details
HCMV Constructs
Viruses used in this study include BAC-generated WT TB40/E expressing GFP from the SV40 promoter (Umashankar et al., 2011) and TB40/E mutant viruses lacking the pre-miR-US5-2 sequence, the pre-miR-UL22A sequence or with a SMAD3 shRNA replacing miR-UL22A were generated by galactokinase (galK-)mediated recombination (Warming et al., 2005). Briefly, the galK gene was used to replace the miR-US5-2 pre-miRNA hairpin using homologous recombination (miR-US5-2 galK F: ACGAGAGCGTTCATCGGGGCATGAAGTACGCATTACACAAACTCCATATATTTGTTACGATAGAATACGGAACGGACCTGTTGACAATTAATCATCGGCA, miR-US5-2 galK R: TATGCACAAAAGGTATGTGTGAATGGAAATACATGATGAATGTCATCATCACGCAAAGCAGCCGTGGGAATGGTCTCAGCAAAAGTTCGATTTATTCAAC) or the miR-UL22A hairpin (miR-UL22A galk F: TTTGCTCTAAAAACACCCCGCCTCCGGTCTTTTTTCTTTTGTATTCGGCACGCGAAACACGGTTTCTTCCCATAGCCCTGTTGACAATTAATCATCGGCA, miR-UL22A galK R: ATACGAACCGCTCGAGTCGCGTGTGTTTTGACGGGGCTGGGTGGAGAGGGAAGGTCGCGCCGCCGCAGCATCCCGCATAGCTCAGCAAAAGTTCGATTTA). In the second recombination step, galK is removed using oligos that encompass the regions up- and downstream of the pre-miR-US5-2 sequence (miR-US5-2 F: TACGCATTACACAAACTCCATATATTTGTTACGATAGAATACGGAACGGAACCATTCCCACGGCTGCTTTGCGTGATGATGACATTCATCATGTATTTCC, miR-US5-2 R: GGAAATACATGATGAATGTCATCATCACGCAAAGCAGCCGTGGGAATGGTTCCGTTCCGTATTCTATCGTAACAAATATATGGAGTTTGTGTAATGCGTA) or up and downstream of the pre-miR-UL22A sequence (miR-UL22A oligo F: GTCTTTTTTCTTTTGTATTCGGCACGCGAAACACGGTTTCTTCCCATAGC CTATGCGGGATGCTGCGGCGGCGCGACCTTCCCTCTCCACCCAGCCCCGT, miR-UL22A oligo R: ACGGGGCTGGGTGGAGAGGGAAGGTCGCGCCGCCGCAGCATCCCGCATAGGCTATGGGAAGAAACCGTGTTTCGCGTGCCGAATACAAAAGAAAAAAGAC) or replaced with a SMAD3 shRNA sequence: (TGCTGTTGACAGTGAGCGCTGTGTGAGTTCGCCTTCAATTGAAGCCACAGATGTAATTGAAGGCGAACTCACACACTGCCTACTGCCTCGGA). All virus stocks were propagated and titered on NHDFs.
3’ UTR Luciferase Assays
The NAB1 and SMAD3 3’ UTRs were cloned into the dual luciferase reporter pSiCheck2 (Promega) using the following primer sets: NAB1 F: 5’ GAGCCTGAAGATTCAAGATAG 3’ and NAB1 R: 5’ GTGAGGCACAGAAAATCTTTATTG 3’ and SMAD3 F: 5’ GACCACCAGAGGAGGATGT 3’ and SMAD3 R: 5’ CAGACTGAGCTCCTGGCACAG 3’. Site-directed mutagenesis was performed using the QuikChange PCR method to mutate the potential miR-US5-2 sites within the NAB1 3’ UTR and the potential miR-UL22A sites within the SMAD3 3’ UTR. The primers for mutating miR-US5-2 site 1: Forward: 5’ GATTTAAGGATAATACTCCGAAATAAGCCTTAATAACC 3’ and Reverse: 5’ GGTTATTAAGGCTTATTTCGGAGTATTATCCTTAAATC 3’. The primers for mutating miR-US5-2 site 2: 5’ AGTTCTGCTCTAGTCATAATTGTAA 3’ and Reverse: 5’ TTACAATTATGACTAGAGCAGAACT 3’. The primers for mutating the miR-UL22A 3p site: Forward: 5’ CTAGAGGAGTGGACGTGACTGGATAG 3’ and Reverse: 5’ CTATCCAGTCACGTCCACTCCTCTAG 3’. The primers for mutating the miR-UL22A 5p site: 5’ CTTCCTTGAAGCCACAATTCTTAGTTTTAAAATC 3’ and Reverse: 5’ GATTTTAAAACTAAGAATTGTGGCTTCAAGGAAG 3’. HEK293T cells were seeded into 96 wells plates and transfected with 100ng of pSiCheck2 vector and 100fmol of negative control or HCMV miRNA mimic using Lipofectamine 2000. Twenty-four hours after transfection cells were harvested for luciferase assay using the Dual-Glo Reporter Assay Kit (Promega) according to the manufacturer’s protocol. Luminescence was detected using a Veritas Microplate Luminometer (Turner Biosystems). All experiments were performed at least in triplicate and presented as mean +/− standard deviation.
Luciferase Reporter Assay
293T cells were plated as above and transfected with 100ng of the SMAD Cignal reporter plasmid (Qiagen), 25ng of pRLSV40-Rluc and 100fmol of miRNA mimic or SMAD3 siRNA using Lipofectamine 2000. siRNA targeting NAB1 or SMAD3 were obtained from ThermoFisher Scientific. Cells were incubated overnight and then the media was replaced with serum-free media. After 4 hours of serum starvation 100pg/mL TGFβ was added in serum-free media for an additional 4 hours and the cells were harvested and analyzed as above. Luminescence was detected using a Veritas Microplate Luminometer (Turner Biosystems). All experiments were performed at least in triplicate and presented as mean +/− standard deviation.
Immunoblotting
Protein extracts were run on an 8% SDS-PAGE, transferred to Immobilon-P Transfer Membranes (Milipore Corp., Bedford, MA), and visualized with antibodies specific for NAB1 (sc-81565; Santa Cruz Biotechnology), SMAD3 (ab-40854; Abcam), HCMV IE2 (MAB810; Sigma Aldrich) and GAPDH (ab8245; Abcam). Relative intensity of bands detected by western blotting were quantitated using ImageJ software. Each experiment utilizing western blotting was performed at least in duplicate.
qRT-PCR for cellular transcripts
Total RNA was isolated from transfected or infected cells using the Trizol RNA isolation method. cDNA was prepared using 1000ng of total RNA and random hexamer primers. Samples were incubated at 16°C for 30 minutes, 42°C for 30 minutes and 85°C for 5 minutes. Real-time PCR (Taqman) was used to analyze cDNA levels in transfected or infected samples. An ABI StepOnePlus Real Time PCR machine was used with the following program for 40 cycles: 95°C for 15 sec and 60°C for one minute. Taqman primer and probe sets for SMAD3 (Hs00969210_m1), SERPINE (Hs00167155_m1) and 18S (Hs03928990_g1) were obtained from ThermoFisher Scientific. Relative expression was determined using the ΔΔCt method using 18S as the standard control with error bars representing the standard deviation of at least two experiments.
CD34+ HPC Colony Formation Assay
Primary CD34+ HPCs were thawed and recovered overnight in stem cell media (Iscove’s Modified Dulbecco’s Medium (IMDM) containing 10% BIT serum replacement (Invitrogen), penicillin/streptomycin and stem cell cytokines (SCF, FLT3L, IL-3, IL-6 (Peprotech)) and infected with HCMV at a MOI of 3 or transfected with 1ug endotoxin-free plasmid stocks using the P3 primary cell kit and 4D AMAXA (Lonza). All treated HPCs were isolated by FACS (BD FACS Aria equipped with 488, 633 and 405 lasers, running FACS DIVA software) for a pure population of viable, CD34+, GFP+ HPCs. At indicated times, viable CD34+ HPCs were plated in Methocult H4434 (Stem Cell Technologies) in 35mm dishes or 6 well plates in triplicate for myeloid colony assays. Total and specific myeloid colonies were enumerated manually at 7 and 14 days using a standard microscope. Experiments were performed at least in duplicate. Neutralization of TGF-β was performed by treating supernatants with 1ug/mL anti-TGF-β antibody (clone 1D11, MAB1835, R&D) prior to combining with CD34+ HPCs and plating for myeloid colony assays as above.
CD34+ HPC Proliferation Assays
Pure populations of sorted HPCs were plated at 2x103 to 104 cells/mL in progenitor cell proliferation media (SFEMII (Stem Cell Technologies) supplemented with penicillin/streptomycin and stem cell cytokines (SCF, FLT3L, IL-3, IL-6)) at 200uL/well in 96 well plates for proliferation assays. Proliferation was assessed at indicated times as described in the Figures 3 and 4 by Trypan Blue exclusion and manual counting.
HCMV HPC Latency Culture
HCMV latency was established in long-term cultures of CD34+ HPCs using methods as previously detailed (Umashankar and Goodrum, 2014). Briefly, CD34+ HPCs were infected with HCMV TB40E-GFP, TB40E-GFPΔmiR-US5-2, TB40E-GFPΔmiR-UL22A or TB40E-GFPΔmiR-UL22A/SMAD3 shRNA at an MOI of 3 for 42 hours prior to FACS in order to obtain a pure population of viable, CD34+, GFP+ HPCs. HPCs were then co-cultured in transwells above monolayers of irradiated M2-10B4 and S1/S1 stromal cells for 12 additional days to establish latency. The frequency of infectious centers was calculated using ELDA (http://bioinf.wehi.edu.au/software/elda/, (Hu and Smyth, 2009)) at three weeks post plating. Two representative experiments are shown in Figure 6.
Quantitative PCR for viral genomes
Viral DNA was isolated from Trizol at 2 and 14 days post-infection as described in Figure 6 using the manufacturer’s two-step RNA/DNA protocol. Primers and a probe recognizing HCMV UL141 were used to quantify HCMV genomes (probe, CGAGGGAGAGCAAGTT; forward primer, 5′-GATGTGGGCCGAGAATTATGA; reverse primer, 5′-ATGGGCCAGGAGTGTGTCA). Viral genomes synthesized during infection in CD34+ HPCs were normalized to the total cell number using human β-globin as a reference (probe, GGACAGATCCCCAAAGGACT; forward primer, 5′-TTAGGGTTGCCCATAACAGC; reverse primer, 5′-TTGGACCCAGAGGTTCTTTG) as previously described (Crawford et al., 2018). Error bars represent the standard deviation of three replicate wells.
TGF-β ELISA and LUMINEX Assays
CD34+ HPCs, NHDF, or Kasumi-3 cells were maintained as described above. For analysis of cytokine-containing supernatant, cells were transferred to serum-free culture media at equivalent densities and cultured for the indicated times as described in Figures 1, 3, and 4 as well as Supplemental Table 1. Quantification of total TGF-β was performed on duplicate samples using the LegendMAX total TGF-β1 ELISA Kit (Biolegend) following the manufacturer’s instructions and absorbance read at a wavelength of 450nm. Samples for multiplex cytokine analysis were obtained the same way and analyzed using the Cytokine Human Magnetic 30-plex panel (ThermoFisher) according to the manufacturer’s instructions and read on a Luminex LX-200.
Quantification and Statistical Analysis
Data are shown as mean +/− standard deviation or standard error of the mean as indicated in the legends of Figures 1-6. Statistical analysis was performed using GraphPad Prism (v6 or v7) for comparisons between experimental groups using unpaired t-test, paired t-test or one- or two-way analysis of variance (ANOVA) with Tukey post-test as indicated.
Data and Software Availability
This manuscript did not generate any unique datasets or code
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-human TGF-beta 1, 2, 3 | R&D Systems | Cat# MAB1835 |
| Mouse monoclonal anti-human NAB1 | Santa Cruz Biotechnology | Cat# sc-81565 |
| Mouse monoclonal anti-human GAPDH | Abcam | Cat# ab8245 |
| Rabbit monoclonal anti-human SMAD3 | Abcam | Cat# ab40854 |
| Mouse monoclonal anti-HCMV IE2 | Sigma Aldrich | Cat# MAB810 |
| Bacterial and Virus Strains | ||
| HCMV BAC: TB40E-GFP | GenBank ref # EF999921.1 | |
| Biological Samples | ||
| Human: primary fetal liver, de-identified, 12-20 wks | Advanced Bioscience Resources | n/a |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Recombinant human TGF-β | Peprotech | Cat# 100-21 |
| Critical Commercial Assays | ||
| P3 primary cell nucleofection kit | Lonza | Cat# V4XP-3032 |
| Cytokine Human Magnetic 30-plex panel | ThermoFisher | Cat# LHC6003MCytokine |
| LegendMAX total TGF-β1 ELISA | Biolegend | Cat# 436707 |
| SMAD3 Taqman Assay | ThermoFisher | Hs00969210_m1 |
| SERPINE Taqman Assay | ThermoFisher | Hs00167155_m1 |
| 18S rRNA Tarman Assay | ThermoFisher | Hs03928990_g1 |
| Deposited Data | ||
| Experimental Models: Cell Lines | ||
| Human: NHDF | ATCC | Cat# PCS-201-012 |
| Human: HEK293T | ATCC | Cat# CRL-11268 |
| Human: Kasumi-3 | ATCC | Cat# CRL-2725 |
| Human: AEC | Lonza | CC-2535 |
| Mouse: M2-10B4 | Stem Cell Technologies | Cat# 00301 |
| Mouse: S1/S1 | Stem Cell Technologies | Cat# 00302 |
| Experimental Models: Organisms/Strains | ||
| Oligonucleotides | ||
| miR-US5-2 galk F primer: ACGAGAGCGTTCATCGGGGCATGAAGTACGCATTACACAAACTCCATATATTTGTTACGATAGAATACGGAACGGACCTGTTGACAATTAATCATCGGCA | This study | n/a |
| miR-US5-2 galk R primer: TATGCACAAAAGGTATGTGTGAATGGAAATACATGATGAATGTCATCATCACGCAAAGCAGCCGTGGGAATGGTCTCAGCAAAAGTTCGATTTATTCAAC | This study | n/a |
| miR-US5-2 F primer: TACGCATTACACAAACTCCATATATTTGTTACGATAGAATACGGAACGGAACCATTCCCACGGCTGCTTTGCGTGATGATGACATTCATCATGTATTTCC | This study | n/a |
| miR-US5-2 R primer: GGAAATACATGATGAATGTCATCATCACGCAAAGCAGCCGTGGGAATGGTTCCGTTCCGTATTCTATCGTAACAAATATATGGAGTTTGTGTAATGCGTA | This study | n/a |
| NAB1 3’ UTR F primer: GAGCCTGAAGATTCAAGATAG | This study | n/a |
| NAB1 3’ UTR R primer: GTGAGGCACAGAAAATCTTTATTG | This study | n/a |
| miR-US5-2 NAB1 binding site 1, F primer: GATTTAAGGATAATACTCCGAAATAAGCCTTAATAACC | This study | n/a |
| miR-US5-2 NAB1 binding site 1, R primer: GGTTATTAAGGCTTATTTCGGAGTATTATCCTTAAATC | This study | n/a |
| miR-US5-2 NAB1 binding site 2, F primer: AGTTCTGCTCTAGTCATAATTGTAA | This study | n/a |
| miR-US5-2 NAB1 binding site 2, R primer: TTACAATTATGACTAGAGCAGAACT | This study | n/a |
| Please see Table S2 for additional oligonucleotide sequences | ||
| Recombinant DNA | ||
| pRL SV40-Rluc | Promega | Cat# E2231 |
| pSiCheck2 | Promega | PR-C8021 |
| pSIREN-GFP | Clontech | Cat# 632455 |
| SMAD Cignal Reporter Assay | Qiagen | Cat# CCS-017G |
| TGF-β siRNA | ThermoFisher | Cat # 4427038 Assay ID: s14054 |
| NAB1 siRNA | ThermoFisher | Cat # 4427037 Assay ID: s9245 |
| SMAD3 siRNA | ThermoFisher | Cat # 4392420 Assay ID: s535082 |
| RISC-free siRNA negative control | ThermoFisher | Cat# D-001220-01-05 |
| Software and Algorithms | ||
| ImageJ | Schneider et al., 2012 | https://imagej.nih.gov/ij/ |
| FACS Diva | BD | |
| Prism (v6 or v7) | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| Other | ||
Highlights:
Latent HCMV infection of stem cells induces the myelosuppressive cytokine TGFβ
HCMV miR-US5-2 targets the transcriptional repressor NAB1 to mediate TGFβ expression
HCMV miR-UL22A down-regulates SMAD3 to block TGFβ signaling in the infected cell
Blocking TGFβ signaling is critical for HCMV latency and genome maintenance
Acknowledgements
This work was supported by grants for the National Institutes of Health NIAID P01 A127335 and R01 A121640 (JAN). We thank the Oregon National Primate Research Center Endocrine Technology Core for assistance running LUMINEX samples.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interests
The authors declare no competing interests.
References
- Akel S, Petrow-Sadowski C, Laughlin MJ, and Ruscetti FW (2003). Neutralization of autocrine transforming growth factor-beta in human cord blood CD34(+)CD38(−)Lin(−) cells promotes stem-cell-factor-mediated erythropoietin-independent early erythroid progenitor development and reduces terminal differentiation. Stem Cells 21, 557–567. [DOI] [PubMed] [Google Scholar]
- Bataller A, Montalban-Bravo G, Soltysiak KA, and Garcia-Manero G (2019). The role of TGFβ in hematopoiesis and myeloid disorders. Leukemia 33, 1076–1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batard P, Monier MN, Fortunel N, Ducos K, Sansilvestri-Morel P, Phan T, Hatzfeld A, and Hatzfeld JA (2000). TGF-(beta)1 maintains hematopoietic immaturity by a reversible negative control of cell cycle and induces CD34 antigen up-modulation. J Cell Sci 113 ( Pt 3), 383–390. [DOI] [PubMed] [Google Scholar]
- Blank U, and Karlsson S (2015). TGF-beta signaling in the control of hematopoietic stem cells. Blood 125, 3542–3550. [DOI] [PubMed] [Google Scholar]
- Cai L, Tu J, Song L, Gao Z, Li K, Wang Y, Liu Y, Zhong F, Ge R, Qin J, et al. (2017). Proteome-wide Mapping of Endogenous SUMOylation Sites in Mouse Testis. Mol Cell Proteomics 16, 717–727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campion EM, Hakimjavadi R, Loughran ST, Phelan S, Smith SM, D'Souza BN, Tierney RJ, Bell AI, Cahill PA, and Walls D (2014). Repression of the Proapoptotic Cellular BIK/NBK Gene by Epstein-Barr Virus Antagonizes Transforming Growth Factor β1-Induced B-Cell Apoptosis. Journal of virology 88, 5001–5013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Capron C, Lacout C, Lecluse Y, Jalbert V, Chagraoui H, Charrier S, Galy A, Bennaceur-Griscelli A, Cramer-Borde E, and Vainchenker W (2010). A major role of TGF-beta1 in the homing capacities of murine hematopoietic stem cell/progenitors. Blood 116, 1244–1253. [DOI] [PubMed] [Google Scholar]
- Challen GA, Boles NC, Chambers SM, and Goodell MA (2010). Distinct hematopoietic stem cell subtypes are differentially regulated by TGF-beta1. Cell Stem Cell 6, 265–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiu Y-F, Sugden B, Chang P-J, Chen L-W, Lin Y-J, Lan Y-C, Lai C-H, Liou J-Y, Liu S-T, and Hung C-H (2012). Characterization and intracellular trafficking of Epstein-Barr virus BBLF1, a protein involved in virion maturation. Journal of virology 86, 9647–9655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi HS, Jain V, Krueger B, Marshall V, Kim CH, Shisler JL, Whitby D, and Renne R (2015). Kaposi’s Sarcoma-Associated Herpesvirus (KSHV) Induces the Oncogenic miR-17-92 Cluster and Down-Regulates TGF-β Signaling. PLoS pathogens 11, e1005255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christy BA, Lau LF, and Nathans D (1988). A gene activated in mouse 3T3 cells by serum growth factors encodes a protein with "zinc finger" sequences. Proceedings of the National Academy of Sciences of the United States of America 85, 7857–7861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford LB, Kim JH, Collins-McMillen D, Lee BJ, Landais I, Held C, Nelson JA, Yurochko AD, and Caposio P (2018). Human Cytomegalovirus Encodes a Novel FLT3 Receptor Ligand Necessary for Hematopoietic Cell Differentiation and Viral Reactivation. MBio 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford LB, Tempel R, Streblow DN, Kreklywich C, Smith P, Picker LJ, Nelson JA, and Caposio P (2017). Human Cytomegalovirus Induces Cellular and Humoral Virus-specific Immune Responses in Humanized BLT Mice. Sci Rep 7, 937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dao MA, Hwa J, and Nolta JA (2002). Molecular mechanism of transforming growth factor beta-mediated cell-cycle modulation in primary human CD34(+) progenitors. Blood 99, 499–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Bartolo DL, Cannon M, Liu Y-F, Renne R, Chadburn A, Boshoff C, and Cesarman E (2008). KSHV LANA inhibits TGF-β signaling through epigenetic silencing of the TGF-β type II receptor. Blood 111, 4731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diggins NL, and Hancock MH (2018). HCMV miRNA Targets Reveal Important Cellular Pathways for Viral Replication, Latency, and Reactivation. Noncoding RNA 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DiMaio TA, Gutierrez KD, and Lagunoff M (2014). Kaposi"s Sarcoma-Associated Herpesvirus Downregulates Transforming Growth Factor β2 To Promote Enhanced Stability of Capillary-Like Tube Formation. Journal of virology 88, 14301–14309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fortunel NO, Hatzfeld A, and Hatzfeld JA (2000). Transforming growth factor-β: pleiotropic role in the regulation of hematopoiesis. Blood 96, 2022–2036. [PubMed] [Google Scholar]
- Fries BC, Khaira D, Pepe MS, and Torok-Storb B (1993). Declining lymphocyte counts following cytomegalovirus (CMV) infection are associated with fatal CMV disease in bone marrow transplant patients. Exp Hematol 21, 1387–1392. [PubMed] [Google Scholar]
- Fukuda M, Kurosaki W, Yanagihara K, Kuratsune H, and Sairenji T (2002). A Mechanism in Epstein–Barr Virus Oncogenesis: Inhibition of Transforming Growth Factor-β1-mediated Induction of MAPK/p21 by LMP1. Virology 302, 310–320. [DOI] [PubMed] [Google Scholar]
- Fukuda M, and Longnecker R (2004). Latent membrane protein 2A inhibits transforming growth factor-beta 1-induced apoptosis through the phosphatidylinositol 3-kinase/Akt pathway. Journal of virology 78, 1697–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-Gutiérrez P, Juárez-Vicente F, Gallardo-Chamizo F, Charnay P, and García-Domínguez M (2011). The transcription factor Krox20 is an E3 ligase that sumoylates its Nab coregulators. EMBO reports 12, 1018–1023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodrum F, Jordan CT, Terhune SS, High K, and Shenk T (2004). Differential outcomes of human cytomegalovirus infection in primitive hematopoietic cell subpopulations. Blood 104, 687–695. [DOI] [PubMed] [Google Scholar]
- Haas S, Trumpp A, and Milsom MD (2018). Causes and Consequences of Hematopoietic Stem Cell Heterogeneity. Cell Stem Cell 22, 627–638. [DOI] [PubMed] [Google Scholar]
- Hancock MH, Hook LM, Mitchell J, and Nelson JA (2017). Human Cytomegalovirus MicroRNAs miR-US5-1 and miR-UL112-3p Block Proinflammatory Cytokine Production in Response to NF-kappaB-Activating Factors through Direct Downregulation of IKKalpha and IKKbeta. MBio 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hook LM, Grey F, Grabski R, Tirabassi R, Doyle T, Hancock M, Landais I, Jeng S, McWeeney S, Britt W, et al. (2014). Cytomegalovirus miRNAs target secretory pathway genes to facilitate formation of the virion assembly compartment and reduce cytokine secretion. Cell Host Microbe 15, 363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horndasch M, Raschke EE, Bommer G, Schuhmacher M, Dumont E, Kuklik-Roos C, Eick D, and Kempkes B (2002). Epstein-Barr virus antagonizes the antiproliferative activity of transforming growth factor-β but does not abolish its signaling. International Journal of Cancer 101, 442–447. [DOI] [PubMed] [Google Scholar]
- Hu Y, and Smyth GK (2009). ELDA: extreme limiting dilution analysis for comparing depleted and enriched populations in stem cell and other assays. Journal of immunological methods 347, 70–78. [DOI] [PubMed] [Google Scholar]
- Inman GJ, and Allday MJ (2000). Resistance to TGF-β1 correlates with a reduction of TGF-β type II receptor expression in Burkitt’s lymphoma and Epstein–Barr virus-transformed B lymphoblastoid cell lines. Journal of General Virology 81, 1567–1578. [DOI] [PubMed] [Google Scholar]
- Kale V (2004). Differential Activation of MAPK Signaling Pathways by TGF-β1 Forms the Molecular Mechanism Behind Its Dose-Dependent Bidirectional Effects on Hematopoiesis. Stem Cells and Development 13, 27–38. [DOI] [PubMed] [Google Scholar]
- Kim SJ, Jeang KT, Glick AB, Sporn MB, and Roberts AB (1989). Promoter sequences of the human transforming growth factor-beta 1 gene responsive to transforming growth factor-beta 1 autoinduction. Journal of Biological Chemistry 264, 7041–7045. [PubMed] [Google Scholar]
- Kim Y, Lee S, Kim S, Kim D, Ahn JH, and Ahn K (2012). Human cytomegalovirus clinical strain-specific microRNA miR-UL148D targets the human chemokine RANTES during infection. PLoS pathogens 8, e1002577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Landais I, Pelton C, Streblow D, DeFilippis V, McWeeney S, and Nelson JA (2015). Human Cytomegalovirus miR-UL112-3p Targets TLR2 and Modulates the TLR2/IRAK1/NFkappaB Signaling Pathway. PLoS pathogens 11, e1004881. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau B, Poole E, Krishna B, Sellart I, Wills MR, Murphy E, and Sinclair J (2016). The Expression of Human Cytomegalovirus MicroRNA MiR-UL148D during Latent Infection in Primary Myeloid Cells Inhibits Activin A-triggered Secretion of IL-6. Sci Rep 6, 31205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lei X, Zhu Y, Jones T, Bai Z, Huang Y, and Gao S-J (2012). A Kaposi's sarcoma-associated herpesvirus microRNA and its variants target the transforming growth factor β pathway to promote cell survival. Journal of virology 86, 11698–11711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim RW, Varnum BC, and Herschman HR (1987). Cloning of tetradecanoyl phorbol ester-induced 'primary response' sequences and their expression in density-arrested Swiss 3T3 cells and a TPA non-proliferative variant. Oncogene 1, 263–270. [PubMed] [Google Scholar]
- Liu C, Adamson E, and Mercola D (1996). Transcription factor EGR-1 suppresses the growth and transformation of human HT-1080 fibrosarcoma cells by induction of transforming growth factor beta 1. Proceedings of the National Academy of Sciences 93, 11831–11836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ljungman P, Hakki M, and Boeckh M (2011). Cytomegalovirus in hematopoietic stem cell transplant recipients. Hematol Oncol Clin North Am 25, 151–169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lo AKF, Dawson CW, Lo KW, Yu Y, and Young LS (2010). Upregulation of Id1 by Epstein-Barr Virus-encoded LMP1 confers resistance to TGFβ-mediated growth inhibition. Molecular Cancer 9, 155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Massagué J, Blain SW, and Lo RS (2000). TGFB; Signaling in Growth Control, Cancer, and Heritable Disorders. Cell 103, 295–309. [DOI] [PubMed] [Google Scholar]
- Mayani H, Little MT, Dragowska W, Thornbury G, and Lansdorp PM (1995). Differential effects of the hematopoietic inhibitors MIP-1 alpha, TGF-beta, and TNF-alpha on cytokine-induced proliferation of subpopulations of CD34+ cells purified from cord blood and fetal liver. Experimental Hematology 23, 422–427. [PubMed] [Google Scholar]
- McNiece IK, Bertoncello I, Keller JR, Ruscetti FW, Hartley CA, and Zsebo KM (1992). Transforming growth factor β inhibits the action of stem cell factor on mouse and human hematopoietic progenitors. The International Journal of Cell Cloning 10, 80–86. [DOI] [PubMed] [Google Scholar]
- Mikell I, Crawford LB, Hancock MH, Mitchell J, Buehler J, Goodrum F, and Nelson JA (2019). HCMV miR-US22 down-regulation of EGR-1 regulates CD34+ hematopoietic progenitor cell proliferation and viral reactivation. bioRxiv, 645374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milbrandt J (1987). A nerve growth factor-induced gene encodes a possible transcriptional regulatory factor. Science 238, 797–799. [DOI] [PubMed] [Google Scholar]
- Min IM, Pietramaggiori G, Kim FS, Passegue E, Stevenson KE, and Wagers AJ (2008). The transcription factor EGR1 controls both the proliferation and localization of hematopoietic stem cells. Cell Stem Cell 2, 380–391. [DOI] [PubMed] [Google Scholar]
- Mori N, Morishita M, Tsukazaki T, and Yamamoto N (2003). Repression of Smad-dependent transforming growth factor-β signaling by Epstein-Barr virus latent membrane protein 1 through nuclear factor-κB. International Journal of Cancer 105, 661–668. [DOI] [PubMed] [Google Scholar]
- Morris MA, Dawson CW, Laverick L, Davis AM, Dudman JPR, Raveenthiraraj S, Ahmad Z, Yap L-F, and Young LS (2016). The Epstein-Barr virus encoded LMP1 oncoprotein modulates cell adhesion via regulation of activin A/TGFβ and β1 integrin signalling. Scientific Reports 6, 19533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oh I-H, Lau A, and Eaves CJ (2000). During ontogeny primitive (CD34+CD38−) hematopoietic cells show altered expression of a subset of genes associated with early cytokine and differentiation responses of their adult counterparts. Blood 96, 4160–4168. [PubMed] [Google Scholar]
- Prokova V, Mosialos G, and Kardassis D (2002). Inhibition of Transforming Growth Factor β Signaling and Smad-dependent Activation of Transcription by the Latent Membrane Protein 1 of Epstein-Barr Virus. Journal of Biological Chemistry 277, 9342–9350. [DOI] [PubMed] [Google Scholar]
- Rakusan TA, Juneja HS, and Fleischmann WR Jr. (1989). Inhibition of hemopoietic colony formation by human cytomegalovirus in vitro. J Infect Dis 159, 127–130. [DOI] [PubMed] [Google Scholar]
- Randolph-Habecker J, Iwata M, and Torok-Storb B (2002). Cytomegalovirus mediated myelosuppression. J Clin Virol 25 Suppl 2, S51–56. [DOI] [PubMed] [Google Scholar]
- Russo MW, Sevetson BR, and Milbrandt J (1995). Identification of NAB1, a repressor of NGFI-A- and Krox20-mediated transcription. Proceedings of the National Academy of Sciences 92, 6873–6877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Safdar A, and Armstrong D (2019). Cytomegalovirus In Principles and Practice of Transplant Infectious Diseases, Safdar A, ed. (New York, NY: Springer New York; ), pp. 611–642. [Google Scholar]
- Sánchez-Capelo A (2005). Dual role for TGF-β1 in apoptosis. Cytokine & Growth Factor Reviews 16, 15–34. [DOI] [PubMed] [Google Scholar]
- Seo T, Park J, and Choe J (2005). Kaposi's Sarcoma–Associated Herpesvirus Viral IFN Regulatory Factor 1 Inhibits Transforming Growth Factor-β Signaling. Cancer research 65, 1738–1747. [DOI] [PubMed] [Google Scholar]
- Sing GK, Keller JR, Ellingsworth LR, and Ruscetti FW (1988). Transforming growth factor beta selectively inhibits normal and leukemic human bone marrow cell growth in vitro. Blood 72, 1504–1511. [PubMed] [Google Scholar]
- Sing GK, and Ruscetti FW (1990). Preferential suppression of myelopoiesis in normal human bone marrow cells after in vitro challenge with human cytomegalovirus. Blood 75, 1965–1973. [PubMed] [Google Scholar]
- Sitnicka E, Ruscetti F, Priestley G, Wolf N, and Bartelmez S (1996). Transforming growth factor beta 1 directly and reversibly inhibits the initial cell divisions of long-term repopulating hematopoietic stem cells. Blood 88, 82–88. [PubMed] [Google Scholar]
- Sukhatme VP, Kartha S, Toback FG, Taub R, Hoover RG, and Tsai-Morris CH (1987). A novel early growth response gene rapidly induced by fibroblast, epithelial cell and lymphocyte mitogens. Oncogene research 1, 343–355. [PubMed] [Google Scholar]
- Swirnoff AH, Apel ED, Svaren J, Sevetson BR, Zimonjic DB, Popescu NC, and Milbrandt J (1998). Nab1, a Corepressor of NGFI-A (Egr-1), Contains an Active Transcriptional Repression Domain. Molecular and Cellular Biology 18, 512–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thiel G, Kaufmann K, Magin A, Lietz M, Bach K, and Cramer M (2000). The human transcriptional repressor protein NAB1: expression and biological activity. Biochimica et Biophysica Acta (BBA) - Gene Structure and Expression 1493, 289–301. [DOI] [PubMed] [Google Scholar]
- Tomita M, Choe J, Tsukazaki T, and Mori N (2004). The Kaposi's sarcoma-associated herpesvirus K-bZIP protein represses transforming growth factor β signaling through interaction with CREB-binding protein. Oncogene 23, 8272–8281. [DOI] [PubMed] [Google Scholar]
- Torok-Storb B, Simmons P, Khaira D, Stachel D, and Myerson D (1992). Cytomegalovirus and marrow function. Ann Hematol 64 Suppl, A128–131. [DOI] [PubMed] [Google Scholar]
- Umashankar M, and Goodrum F (2014). Hematopoietic long-term culture (hLTC) for human cytomegalovirus latency and reactivation. Methods Mol Biol 1119, 99–112. [DOI] [PubMed] [Google Scholar]
- Umashankar M, Petrucelli A, Cicchini L, Caposio P, Kreklywich CN, Rak M, Bughio F, Goldman DC, Hamlin KL, Nelson JA, et al. (2011). A novel human cytomegalovirus locus modulates cell type-specific outcomes of infection. PLoS Pathog 7, e1002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warming S, Costantino N, Court DL, Jenkins NA, and Copeland NG (2005). Simple and highly efficient BAC recombineering using galK selection. Nucleic acids research 33, e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood VHJ, O'Neil JD, Wei W, Stewart SE, Dawson CW, and Young LS (2007). Epstein–Barr virus-encoded EBNA1 regulates cellular gene transcription and modulates the STAT1 and TGFβ signaling pathways. Oncogene 26, 4135. [DOI] [PubMed] [Google Scholar]
- Yoo YD, Chiou C-J, Choi KS, Yi Y, Michelson S, Kim S, Hayward GS, and Kim S-J (1996). The IE2 Regulatory Protein of Human Cytomegalovirus Induces Expression of the Human Transforming Growth Factor b1 Gene through an Egr-1 Binding Site. Journal of virology 70, 7062–7070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Kozono DE, O'Connor KW, Vidal-Cardenas S, Rousseau A, Hamilton A, Moreau L, Gaudiano EF, Greenberger J, Bagby G, et al. (2016). TGF-beta Inhibition Rescues Hematopoietic Stem Cell Defects and Bone Marrow Failure in Fanconi Anemia. Cell Stem Cell 18, 668–681. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
This manuscript did not generate any unique datasets or code