

Abstract
Amino acid deprivation is a strategy that malignancies utilize to blunt anti-tumor T-cell immune responses. It has been proposed that amino acid insufficiency in T-cells is detected by GCN2 kinase, which through phosphorylation of EIF2α, shuts down global protein synthesis leading to T-cell arrest. The role of this amino acid stress sensor in the context of malignant brain tumors has not yet been studied, and may elucidate important insights into the mechanisms of T-cell survival in this harsh environment. Using animal models of glioblastoma and animals with deficiency in GCN2, we explored the importance of this pathway in T-cell function within brain tumors. Our results show that GCN2 deficiency limited CD8+ T-cell activation and expression of cytotoxic markers in two separate murine models of glioblastoma in vivo. Importantly, adoptive transfer of antigen-specific T-cells from GCN2 KO mice did not control tumor burden as well as wild-type CD8+ T-cells. Our in vitro and in vivo data demonstrated that reduction in amino acid availability caused GCN2 deficient CD8+ T-cells to become rapidly necrotic. Mechanistically, reduced CD8+ T-cell activation and necrosis was due to a disruption in TCR signaling, as we observed reductions in PKCθ and phoshpo-PKCθ on CD8+ T-cells from GCN2 KO mice in the absence of tryptophan. Validating these observations, treatment of wild-type CD8+ T-cells with a downstream inhibitor of GCN2 activation also triggered necrosis of CD8+ T-cells in the absence of tryptophan. In conclusion, our data demonstrate the vital importance of intact GCN2 signaling on CD8+ T-cell function and survival in glioblastoma.
Electronic supplementary material
The online version of this article (10.1007/s00262-019-02441-6) contains supplementary material, which is available to authorized users.
Keywords: GCN2, CD8+ T-cell, Glioma, ISRIB
Introduction
Malignant glioma is a profoundly aggressive tumor of the central nervous system (CNS), which is almost uniformly lethal despite aggressive therapeutic intervention. Despite intensive efforts, both median and long-term survival of glioma patients have not sufficiently improved to achieve long-term survival. Even though the recent surge in immunotherapy has dramatically changed the treatment options for solid tumors [2, 3], immunotherapy for glioma is still rather ineffective [4]. Part of the reason may be because immunosuppression in glioma is considered to be severe, and quantifiably more potent at inducing T-cell exhaustion than most solid tumors [5].
One particularly relevant pathway of immune suppression in glioma is nutrient deprivation, which includes glucose, oxygen, and amino acid availability [6]. Of these limiting factors, amino acid availability is profoundly important for proper immune cell function [7]. Depletion of essential amino acids is a well-recognized modality which immunosuppressive cells use to dampen T-cell dependent immune responses in inflammation [8], tissue rejection [9], and malignancy [10]. Mechanistically, amino acid deprivation is sensed by the canonical stress senor GCN2 (general control nonderepressible 2). This sensor functions by detecting uncharged tRNAs [11], and transmits signals by phosphorylating EIF2α (eukaryotic translation initiation factor 2α) [12], which shuts down global protein translation and upregulates genes that assist in the handling of endoplasmic reticular (ER) stress [13, 14]. This is termed the integrated stress response (ISR), as most forms of cellular stress converge on this same signaling pathway to respond to stress [15].
In the context of glioma, IDO (indolamine 2,3 dioxygenase) mediated tryptophan depletion is the most well-studied mechanism of intratumoral amino acid depletion [16, 17]. These enzymes can be expressed by the tumor [18], infiltrating myeloid-derived suppressor cells (MDSCs) [19], and both can act in concert to deplete tryptophan from the tumor microenvironment (TME). However, as amino acid sensing occurs via the detection of any uncharged tRNA, deficiency of any amino acids may trigger the ISR in glioma. In other solid tumors, arginine is similarly consumed by both immunosuppressive myeloid cells [20] and tumor cells [21, 22]. This has potent inhibitory effects on T-cell function, which can be overcome with arginine pre-treatment [23]. Other amino acids, such as glutamine and asparagine [24, 25], are shown to be limiting in solid tumors.
While substantial work has highlighted why amino acid depletion occurs in tumors, how T-cells respond to this stress remains unclear. The most prominent study demonstrates that GCN2 mediates T-cell anergy and proliferative arrest under conditions of tryptophan depletion [26]. This study suggested that the GCN2 response to amino acid starvation prevents T-cell functionality and that inhibition of GCN2 may reinvigorate T-cells in tumors. However, another study suggests GCN2 is required for CD8+ T-cells proliferative fitness and migration, independent of amino acid sensing [27]. In the CNS, T-cells deficient in GCN2 promote experimental autoimmune encephalomyelitis (EAE) pathology, which suggests that GCN2 dampens T-cell activity [28]. The inconsistent results of these studies suggest GCN2 has pleiotropic effects within the T-cell compartment that require further elucidation.
It is important to dissect the importance of GCN2 on T-cell function in cancer, as studies suggest inhibition of GCN2 (or its downstream pathway) is important for immunotherapeutic intervention in cancer [29, 30]. Perhaps most importantly, a recent study showed that blocking the stress sensing pathway inhibited programmed death-ligand 1 (PD-L1) expression on tumors, which provides a rationale for its use in the clinic as an adjuvant to immunotherapy [30].
Therefore, the goal of this study was to examine the role of GCN2 in adaptive immune function in glioma, and how its deficiency regulates anti-tumor immune responses. Surprisingly, our results show that GCN2 is essential for proper CD8+ T-cell function in brain tumors. Knockout (KO) of GCN2 leads to impaired CD8+ T-cell activation and function in brain tumors, and its inhibition has multiple, deleterious effects on T-cell immunity. We show that polyclonal (and antigen-specific) GCN2 KO CD8+ T-cells have reduced activation and expression of inflammatory cytokines in two separate orthotopic brain tumor models. Furthermore, there was a significant reduction of intratumoral CD8+ T-cells, which was due to their reduced survival in tumors and not migration. While culturing wild-type (WT) CD8+ T-cells in amino acid limiting media stops their proliferation, in GCN2 KO mice, these cells rapidly become necrotic. Western blot analysis revealed a reduction in T-cell receptor (TCR) signaling under amino acid deprivation that is the cause of T-cell necrosis in the absence of GCN2. This was recapitulated using a down-stream inhibitor of GCN2 signaling [integrated stress response inhibitor (ISRIB)], as ISRIB potentiated CD8+ T-cell necrosis in the absence of tryptophan. Our results show that GCN2 is needed for CD8+ T-cell survival in nutrient-poor glioma and that inhibition of this stress sensing pathway has damaging effects on CD8+ T-cells. Therefore, caution must be used when targeting the ISR for cancer therapy in the future.
Materials and methods
Cell lines and culture conditions
CT2A, GL-261 and GL-261 OVA (which stably expresses whole ovalbumin—as generated in [31]) murine syngeneic glioma cell lines were cultured in DMEM (Corning) with 10% fetal bovine serum (FBS, HyClone), 100 U/ml penicillin, and 100 mg/ml streptomycin (Corning), and incubated at 37°C in 5% CO2.
Mouse models and tumor implantation
To study the absence of GCN2 specifically on CD8+ T-cells, we crossed GCN2 KO mice with OT-1 mice and generated GCN2 KO OT-1 mice. Mice were injected with 2 × 105 (GL-261 or CT2A) or 4 × 105 GL-261 OVA cells in 2.5 μl phosphate-buffered saline (PBS, Corning), using a stereotactic frame, at a 3.5 mm depth, in accordance to NIH guidelines.
Flow cytometry
For in vivo phenotypic analysis, tumor cells from the brain and spleens were prepared as single cells and stained with fixable viability dye APC-eFluor 780 (eBioscience, Invitrogen) for 20 min, then blocked with anti-CD16/32 (Biolegend) for 10 min and then were stained with, anti-CD45-BV510, anti-CD8-BV605, anti-CD4-Pe-Cy7 and anti-CD44-Percp-cy5.5 (Biolegend). For intracellular staining, cells were fixed and permeabilized with Foxp3 staining kit (eBioscience, Invitrogen), then were stained with Foxp3-eFlour450 (eBioscience, Invitrogen). For intracellular cytokine staining, cells were stimulated with Cell Stimulation Cocktail plus protein transport inhibitors (eBioscience, Invitrogen) for 5 h, according to the manufacturer's instructions. For intracellular cytokine staining cells were stained using the same method as aforementioned, with GZMB-Alexa Fluor 647, IFNγ-Alexa Fluor 700 (Biolegend). For in vivo competition assay, isolated CD8+ T-cells from GCN2 KO OT-1 mice were labeled with Cell Trace Violet, and CD8+ T-cells from OT-1 mice were labeled with Cell Trace Far Red, Cell Proliferation Kit (Invitrogen), according to the manufacturer's instructions. For cell death staining in vivo and in vitro, after surface staining cells were washed with Annexin V binding buffer (BD Biosciences), then stained with Annexin V-APC (1:20 dilution), at room temperature, for 13 min. For CCR7 staining, cells were stained with anti-CCR7-APC and were incubated at 37 °C or 30 min.
T-cell isolation and expansion
Splenic T-cells were isolated from Foxp3-IRES-GFP B6 mice, using EasySep T-cell Isolation Kit (STEMCELL), according to manufacturer instructions. CD8+ T-cells were labeled with anti-CD8-APC, and CD4+ T-cells were labeled with anti-CD4-Pe-Cy7. After isolation, these cells were sorted using BD FACS Aria II cell sorter. After sorting different subsets, cells were cultured and expanded using Dynabeads (Invitrogen) per manufacturer’s instructions. After 5 days of expansion in complete RPMI, different subtypes of T-cells (CD8+ T-cells, CD4+ T-cells, and Tregs) were lifted, and cultured in tryptophan added (+ TRP) and tryptophan free (− TRP) media for 24 h. One day after culture, cells were lifted and were lysed in M-PER (Thermo Fisher) buffer for protein estimation and western blot. To test the effects of limiting amino acid on CD8+ T-cell survival: tryptophan, arginine, and lysine free media were supplemented back with 100%, 10% or 1% amount of the respective amino acid (based on RPMI 1640 formulation levels as previously described [27]) and analyzed for apoptosis and necrosis at 24 and 48 h.
Western blots
Cells were lysed in M-PER buffer (Thermo Fisher), protease and phosphatase inhibitors (Thermo Fisher). Western blots were blocked for an hour at room temperature using 5% milk in 10 mM Tris–HCl pH 7.5, 150 mM NaCl containing 0.1% Tween 20 (TBST). Antibodies used in our western blot experiments were: GCN2 (1:1000), CHOP (1:1000), β-actin (1:5000, Cell Signaling), PKCθ, P-PKCθ (1:1000, Santa Cruz), and P-EIF2α (1:20,000, Abcam).
Trans-ISRIB in vitro treatment
T-cells from WT and GCN2 KO mice were isolated as aforementioned and were treated in five different medias, complete RPMI [10% FBS (HyClone), 10 mM HEPES–sodium pyruvate (Sigma), 1 mM sodium pyruvate (Corning), 0.01% 2-ME, 2 mM l-glutamine (Sigma), 100 U/ml penicillin, and 100 μg/ml streptomycin (Corning)], +TRP, -TRP, 200 nM trans-ISRIB +TRP and 200 nM trans-ISRIB (Cayman Chemicals) − TRP, for 24, 48 and 72 h. After each time point, cells were lifted and analyzed via flow cytometry.
LC–MS analysis
For CD8+ T-cell metabolite content, mice were injected with CT2A or GL-261 tumor cells, and single-cell solution from tumors and spleen was obtained as described above. Cells were then labeled with anti-CD8β-biotin, washed, then bound to anti-Biotin microbeads (Miltenyi Biotec, Bergisch Gladbach, Germany). Cells were then positively selected using positive selection via LD columns (Miltentyi), immediately flash-frozen, then metabolites were isolated using an 80% methanol/20% H20 solution. Pellets were nitrogen flushed and analyzed using utilizing HPLC–MS/MS (performed at Metabolomics developing core at Northwestern University). Peak area values were normalized to total ion count, and creatinine levels were within 1% of each sample, indicating even loading [32].
Statistical analysis
For individual comparisons, Student’s t test was performed. Kaplan–Meier curves were generated to determine the relative survival of glioma bearing animals under different courses of treatment, and log-rank tests were performed to address significance between groups. One-way ANOVA was used for comparisons between multiple groups, and Tukey’s post hoc test was performed to obtain p values for individual comparisons. p < 0.05 was considered significant. *p < 0.05; **p < 0.01; ***p < 0.001; ns not significant.
Results
Absence of GCN2 reduces CD8+ T-cell numbers in the brains of tumor-bearing mice
To study the effect of GCN2 absence on T-cell immunity in glioma, we injected C57BL/6 WT and GCN2 KO mice with the syngeneic glioma mouse model, GL-261. After 14 days post-tumor implantation, animals were euthanized, and the T-cell immune compartment was analyzed by flow cytometry. Interestingly, as demonstrated in Fig. 1a, there was a significant reduction in total number and percentage of CD8+ T-cells (p < 0.01 and p < 0.05, respectively) within the tumors. This phenomenon was specific for CD8+ T-cells, as the same reduction did not occur in other T-cell subsets (Fig. 1a). To test if there was an intrinsic defect in T-cell function or proliferation, we isolated CD8+ and CD4+ T-cells in vitro and checked for their proliferation and activation after 72 h of stimulation. While GCN2 deficiency resulted in CD8+ T-cells and CD4+ T-cells that were significantly less activated based on their CD44 mean fluorescence intensity (MFI), there were no dramatic changes in the proliferation of these cells (Supplementary Fig. 1). This coincides with the periphery of tumor-bearing animals as the spleen of all T-cell subsets did not have any significant change in their T-cell population (Fig. 1b). This suggests that the tumor microenvironment is driving the specific reduction in CD8+ T-cells in GCN2 KO animals.
Fig. 1.

Absence of GCN2 reduces CD8+ T-cell numbers in murine brain tumors. C57BL/6 (WT) and GCN2 KO mice were injected with 2 × 105 GL-261 cells (glioma syngeneic cell line) intracranially, and after 14 days were euthanized for flow cytometric analysis. In a flow cytometric analysis of the total number and percentage of the different T-cell subsets infiltrating the brain tumor microenvironment. In b flow cytometric analysis of the total number of the different T-cell subsets from the spleen. Flow cytometry statistics in a and b were calculated, n = 5 per group, representative of two experiments. Unpaired T test analysis was used to calculate significance. p < 0.05*; p < 0.01**; p < 0.001***. Refer to Supplementary Fig. 7a for the gating strategy
To validate this observation, we repeated the same in vivo experiment with a second syngeneic mouse glioma model (CT2A). The CT2A model also showed a significant reduction in total number and percentage of CD8+ T-cells (p < 0.01 and p < 0.0001, respectively) infiltrating the tumor of GCN2 KO mice, and again, this decline was tumor-specific (Supplementary Fig. 2a and b).
Tumor-infiltrating CD8+ T-cells in GCN2 KO mice are impaired in both activation status and function
Next, we determined the consequences of GCN2 deficiency on CD8+ T-cells activity/cytotoxic ability in vivo in tumors. We challenged WT and GCN2 KO mice with GL-261, and after 14 days of engraftment, we euthanized the animals and isolated tumor-infiltrating cells for intracellular cytokine staining (Fig. 2a). As demonstrated in Fig. 2a, there is a significant reduction in CD44 MFI (a marker of T-cell activation), and percentages of CD8+ T-cells expressing granzyme B (GZMB, a marker of effector T-cells), (p < 0.001, p < 0.01 respectively), in GCN2 KO tumor-bearing mice. Repeating this experiment with the CT2A tumor model showed the same phenotype (Supplementary Fig. 3).
Fig. 2.

Glioma infiltrating GCN2 KO CD8+ T-cells are dysfunctional. GCN2 KO and WT mice were injected with 2 × 105 GL-261 cells, and after 14 days were euthanized for flow cytometric analysis. In a GL-261 injected tumor-infiltrating lymphocytes were stimulated for 5 h with PMA/Ionomycin, and then stained and analyzed for CD44 mean fluorescence intensity (MFI) and cytokine secretion via flow cytometry. In b, 1 × 106 heat shock killed GL-261 OVA cells were injected intraperitoneally into OT-1 and GCN2 KO OT-1 mice. After 14 days of engraftment, CD8+ T-cells from these mice were isolated and transferred i.v. into RAG1 KO mice previously inoculated with GL-261 OVA. Seven days post transfer, mice were euthanized and analyzed for CD44 MFI and cytokine secretion with flow cytometry. Flow cytometry statistics were calculated, n = 5 per group in a, n = 3 per group in b, representative of two experiments. Unpaired T test analysis was used to calculate significance. p < 0.05*; p < 0.01**; p < 0.001***. Refer to Supplementary Fig. 7b for the gating strategy
Based on our previous findings that CD8+ T-cells were specifically affected by GCN2 deficiency, we determined how this affects antigen-specific CD8+ T-cell function. To test this, we utilized the OT-1 mouse model, in which the TCR of CD8+ T-cells only recognize a specific ovalbumin (OVA) antigen [33], and crossed them to GCN2 KO mice to generate GCN2 KO OT-1 mice. First, we injected control and GCN2 KO OT-1 mice with heat shock killed GL-261 OVA (GL-261 overexpressing the OVA antigen) cells, to educate their TCR with OVA antigen in vivo. After 2 weeks, we isolated CD8+ T-cells and injected them intravenously (i.v.) into Rag1 KO mice previously inoculated intracranially (i.c.) with GL-261 OVA (Fig. 2b). After 7 days, Rag1 KO mice were euthanized, and tumors were analyzed via flow cytometric analysis. In our analysis, we observed that pre-activated CD8+ T-cells from GCN2 KO OT-1 mice were less active and cytotoxic (based on their CD44 and GZMB expression, respectively) (Fig. 2b). Altogether these data confirm that the lack of amino acid sensing enzyme (GCN2) causes functional impairment of antigen-specific CD8+ T-cells in the glioma microenvironment.
GCN2 promotes intratumoral CD8+ T-cell accumulation and promotes survival of animals with glioma
To determine if the absence of GCN2 has a measurable effect on animal survival, we directly challenged WT OT-1 and GCN2 KO OT-1 mice with GL-261 OVA i.c. and analyzed their survival. Interestingly, GCN2 KO OT-1 mice had significantly reduced survival compared to control OT-1 mice (p < 0.05) (Fig. 3a). As a global KO of GCN2 may have effects on other cellular subsets, we injected GL-261 OVA i.c. into Rag1 KO mice, and after 14 days, injected previously activated CD8+ T-cells from either WT or GCN2 KO OT-1 mice. Importantly, this experiment also showed Rag1 KO mice that received WT OT-1 T-cells survived significantly (p < 0.01) longer than the ones that received GCN2 KO OT-1 cells (Fig. 3b). The results of these experiments suggest that GCN2 controls either the recruitment or survival of CD8+ T-cells within glioma.
Fig. 3.

GCN2 promotes antigen-specific CD8+ T-cell responses against glioma. In a, WT OT-1 and GCN2 KO OT-1 mice were challenged directly with 4 × 105 GL-261 OVA and were analyzed for survival. In b, OVA activated CD8+ T-cells from WT OT-1 or GCN2 KO OT-1 mice were transferred i.c. into tumor-bearing Rag1 KO hosts and analyzed for survival. In c, OVA activated CD8+ T-cells from WT OT-1 and GCN2 KO OT-1 mice were isolated and labeled with cell proliferation dye eFluor 450 (GCN2 KO CD8+ T-cells) and cell proliferation dye eFluor 670 (WT OT-1 CD8+ T-cells), and were transferred at a 1:1 ratio into GL-261 OVA tumor-bearing mice either i.c. (left panel) or i.v. (right panel). After 3 days, mice were euthanized for flow cytometric analysis. In a, b, Kaplan–Meier curves were generated from n = 7 mice per group from two independent experiments, and statistical significance calculated using the Log-Rank analysis. In c, flow cytometry statistics calculated from n = 3 mice per condition. Unpaired T test analysis was used to calculate significance. p < 0.05*; p < 0.01**
To address these two possibilities, we performed two different competition assays. Isolated and activated CD8+ T-cells from WT and GCN2 KO OT-1 mice were labeled with two different fluorescent dyes, and were transferred at a 1:1 ratio either intracranially (Fig. 3c, left panel) or intravenously (Fig. 3c, right panel) into GL-261 OVA tumor-bearing hosts. After 3 days, mice were euthanized and analyzed for OT-1 infiltration. Surprisingly, only with intracranial injection was there a significant (p < 0.05) decline in the amount of GCN2 KO OT-1 CD8+ T-cells in the brain. There was no significant change in the infiltration of CD8+ T-cells delivered systemically, suggesting that GCN2 does not regulate T-cell migration to brain tumors. It is important to note that a previous study suggested that the deficiency of GCN2 resulted in an upregulation of C–C chemokine receptor type 7 (CCR7), thus preventing T-cells from leaving the spleen [27].
Converse to this study, we found a downregulation of CCR7 expression in the spleen of mice with little to no detection in the brain (Supplementary Fig. 4). Our data showed CCR7 expression is only expressed in CD44lo cells, which is consistent with the expression of this receptor in memory/naïve cells versus effectors [34]. In summation, these data suggest that in the context of the CNS, the specific reduction in CD8+ T-cells numbers is not due to changes in migratory capabilities.
Considering all these experiments were done with in vitro activated CD8+ T-cells, we decided to demonstrate these findings in the context of in vivo antigen stimulation (Fig. 4). To test this, we performed subcutaneous (s.c) injections of heat shock killed GL-261 OVA cells into WT and GCN2 KO OT-1 mice. After 14 days, in vivo activated CD8+ T-cells from WT or GCN2 KO OT-1 mice were transferred systemically into GL-261 tumor-bearing Rag1 KO mice. The first group of mice was euthanized 7 days after CD8+ T-cell transfer for flow cytometric analysis, and a second group was analyzed for overall survival. Consistent with our previous experiments, Rag1 KO mice that were injected with GCN2 KO OT-1 CD8+ T-cells died significantly earlier (p < 0.05) than the mice that received WT OT-1 CD8+ T-cells (Fig. 4a). Flow cytometric analysis revealed that GCN2 KO OT-1 CD8+ T-cells had reductions in the MFI and percentage of CD127 (precursor memory T-cell marker) and MFI of CD69 (tissue-resident memory T-cell marker) (Fig. 4b, top panels). Interestingly, this reduction was tumor-specific as other organs did not show these changes (Fig. 4b, bottom panels). In conclusion, these data suggest that the lack of amino acid stress-sensing by antigen-specific CD8+ T-cells inhibits their memory capabilities and ultimately results in a lack of animal survival of mice with brain tumors.
Fig. 4.
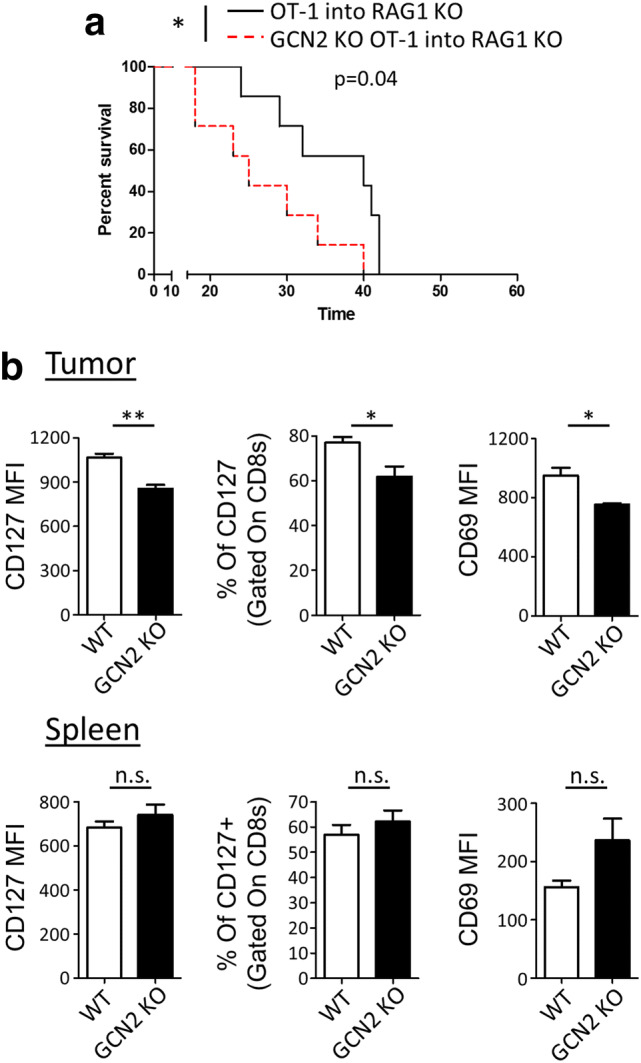
Inhibition of memory and function of in vivo activated CD8+ T-cells with GCN2 deletion. In a, b, 1 × 106 heat shock killed GL-261 OVA cells were injected into OT-1 and GCN2 KO OT-1 mice. After 14 days, CD8+ T-cells from these mice were isolated and transferred i.v. into previously inoculated Rag1 KO mice (4 × 105 GL-261 OVA). The first group of mice was analyzed for survival analysis (a) and the second group of mice was euthanized 7 days post-transfer for flow cytometric analysis (b). In a, the Kaplan–Meier curve was generated from n = 7 per group from two independent experiments, and statistical significance calculated using the Log-Rank analysis. In b, flow cytometry statistics calculated and shown as the total number and percentage positive population, n = 3 per group, representative of two experiments. Unpaired T test analysis was used to calculate significance. p < 0.05*; p < 0.01**. Refer to Supplementary Fig. 7c for the gating strategy
These results above suggest that GCN2 is important for the maintenance of multiple functions of CD8+ T-cells within tumors, suggesting that amino acid sensing is occurring within brain tumors. As previous studies suggest that the tryptophan-catabolic enzyme IDO is important for glioma immunopathology [35], we sought to determine if tryptophan catabolism was contributing to a decreased pool of tryptophan in our glioma models. To test this, we isolated metabolites from the tumor hemisphere of tumor-bearing mice (right hemisphere) and directly compared it to the non-tumor-bearing hemisphere (left hemisphere) (Fig. 5a). While our data show there is considerable IDO activity within tumors, as shown by decreased tryptophan/kynurenine (KYN) ratios in both tumor models (Fig. 5a), the levels of tryptophan were not consistently decreased within tumors (Supplementary Fig. 5a). This supports another study in melanoma, in which they overexpressed another catabolic enzyme TDO (Tryptophan 2,3 dioxygenase), and did not see intratumoral tryptophan levels decrease [36]. However, a limitation of bulk tumor analysis is that it does examine specific subpopulations of cells.
Fig. 5.

Amino acid deficiency causes necrosis in the absence of GCN2 in CD8+ T-cells in murine glioma tumor models. In a, WT mice were injected with GL-261 (top panel) and CT2A (bottom panel) tumor cells, three mice per group. After 14 days of tumor growth, tumor (R. Hemi) and non-tumor (L. Hemi) hemisphere were isolated and measured for TRP/KYN ratios via LC–MS. In b, WT mice were injected with GL-261 (top panel) and CT2A (bottom panel) tumor cells, six mice per group. After 14 days, mice were euthanized and CD8β+ T-cells were isolated from the tumor (brain) and periphery (spleen), and their metabolites were measured using LC–MS. In c, total T-cells were cultured in serial dilutions of three amino acids, and at 24 and 48 h, cells were lifted and stained with annexin-V and fixable viability dye for flow cytometric analysis. Refer to Supplementary Fig. 7d for the gating strategy. In d, WT and GCN2 KO OT-1 CD8+ T-cells were transferred i.v. into previously inoculated with GL-261 OVA, Rag1 KO mice. Seven days post transfer, mice were euthanized, and tumor-infiltrating lymphocytes were stained with annexin-V and fixable viability dye for flow cytometric analysis. Refer to Supplementary Fig. 7e for the gating strategy. In a, the ratio of normalized peak area of TRP to KYN and in b is the normalized number of the peak area of tryptophan, arginine and lysine in the tumor versus non-tumor. In c, Two-way ANOVA followed by Sidak’s multiple comparisons test was used to calculate significance, n = 3 per group. In d, flow cytometry statistics calculated and shown as percentage positive population, n = 3 per group, representative of two experiments. Unpaired T test analysis was used to calculate significance. p < 0.05*; p < 0.01**; p < 0.001***; p < 0.0001****
To overcome this limitation, we performed bead-based isolation and subsequent LC–MS of CD8+ T-cells from the brain and compared them to peripheral CD8+ T-cells with two different glioma tumor models (Fig. 5b). Interestingly we found that in both glioma models, there is a consistent reduction of many proteinogenic amino acids within CD8+ T-cells. (Supplementary Fig. 5b). This suggests that the tumor is outcompeting CD8+ T-cells for multiple resources, not just tryptophan.
Amino-acid depletion and absence of stress sensing cause necrosis of CD8+ T-cells
Considering our data show that there are fewer GCN2 KO CD8+ T-cells in the tumor, and the ones present are less activated, we explored if GCN2 directly influences the survival of CD8+ T-cells under conditions of amino acid stress. As we found that only CD8+ T-cells upregulate GCN2 under amino acid stress (Supplementary Fig. 6a) and that CD8+ T-cells were the only population affected in vivo, we isolated CD8+ T-cells from WT and GCN2 KO mice and activated them under reducing concentrations of three different amino acids: Tryptophan, Arginine, and Lysine (Fig. 5c). Our data indicate that there is a precipitous increase in cell death of GCN2 KO CD8+ T-cells (as compared to WT CD8+ T-cells) when levels of amino acids are reduced (Fig. 5c). The data show that GCN2 deficiency affects the survival of CD8+ T-cells under any depleting amino acid, but may be more sensitive to reductions in levels of tryptophan. After observing this phenomenon in vitro, we decided to explore this effect in vivo. Therefore, we injected WT and GCN2 KO OT-1 CD8+ T-cells into Rag1 KO mice, and after 7 days post transfer, we euthanized the mice and checked for the viability of tumor-infiltrating CD8+ T-cells via flow cytometry. Our flow cytometric analysis revealed that there was a significant increase in both apoptosis and necrosis (p < 0.05) of GCN2 KO OT-1 CD8+ T-cells that infiltrated the tumor as compared to WT OT-1 CD8+ T-cells (Fig. 5d tumor). This effect was tumor specific as there were no significant changes in apoptosis or necrosis of GCN2 KO CD8+ T-cells in the periphery of these tumor-bearing mice (Fig. 5d spleen).
These data strongly suggest that GCN2 is required for CD8+ T-cell survival under conditions of amino acid deficiency. This could potentially explain the reason behind the reduction of GCN2 KO CD8+ T-cells in the tumor. To understand this phenomenon from a mechanistic standpoint, we decided to evaluate the TCR signaling status upon activation. Protein kinase C-theta (PKCϴ) is important for T-cell activation and has an important role in TCR signaling, in which deficient activation leads to T-cell apoptosis [37]. Thus, we examined for PKCϴ activation under conditions of amino acid deficiency. Based on this knowledge, we checked protein expression of total PKCϴ and P-PKCϴ on in vitro isolated CD8+ T-cells from WT and GCN2 KO mice, cultured in − TRP and + TRP media for 24 and 48 h. After 24 and 48 h, we lifted the cells and performed western blot analysis. Our results demonstrated that in the absence of TRP and GCN2 deletion, there is a reduction in the expression of PKCϴ and P-PKCϴ in a time-dependent manner (Fig. 6a and Supplementary Fig. 6b). The lack of P-PKCϴ activation in GCN2 KO T-cells is time dependent, as we could detect expression at 24 h after culture, but not after 48 h of culture (Fig. 6a). Furthermore, GCN2 KO T-cells cultured in -TRP media for 72 h did not express PKCϴ and P-PKCϴ at all (Fig. 6b). These data show that by 72 h of amino acid starvation, PKCϴ signaling/activation in GCN2 KO T-cells compared to WT GCN2 T-cells dramatically reduced.
Fig. 6.

EIF2α inhibitor (ISRIB) increases CD8+ T-cell necrosis and deactivation in tryptophan starved conditions. In a, b, CD8+ T-cells were isolated from splenocytes of WT and GCN2 KO mice then cultured in − TRP, + TRP for 24 and 48 h in (a), and − TRP, + TRP or complete RPMI for 72 h in (b). After 24, 48, and 72 h cells were lifted and were lysed for western blot. In c, d, CD8+ T-cells from WT mice were cultured in RPMI, + TRP, − TRP, 200 nM ISRIB + TRP and 200 nM ISRIB − TRP, for 24, 48 and 72 h. At each time-point, cells were lifted and stained for flow cytometric analysis. In c, d, flow cytometry statistics calculated and shown as percentage positive population, n = 3 per group, representative of five experiments. One way ANOVA and Tukey’s post hoc was used to calculate significance. p < 0.05*; p < 0.01**; p < 0.001***. Refer to Supplementary Fig. 7d for the gating strategy
To understand if this phenomenon is relevant under non-transgenic conditions, we treated CD8+ T-cells with the potent and specific inhibitor of the integrated stress response, ISRIB [38]. This compound prevents activation of EIF-2α, thus acting as a pharmacological mimic of GCN2 deficiency. Under conditions of amino acid sufficiency, the compound had no effect of T-cell proliferation or activation (Fig. 6c). However, under conditions of tryptophan deficiency, ISRIB treated CD8+ T-cells became quickly necrotic, with a concomitant decrease in activation status (Fig. 6c, d). Overall, these data suggest that in the context of amino acid deficiency, CD8+ T-cells require stress-sensing via GCN2 to maintain TCR signaling and functionality within brain tumors.
Discussion
The results of this study provide systematic and compelling evidence for the importance of the amino-acid stress sensing pathway in anti-tumor immune responses in glioma. In particular, GCN2 is required for the proliferation, function, and survival of the CD8+ T-cell compartment in brain tumors. As GCN2 is known canonically as an amino acid sensor, we performed experiments that revealed GCN2 acts to prevent apoptosis of CD8+ T-cells under the deprivation of multiple amino acids. However, an important question is whether there is a reduction of amino acids within brain tumors. Our data from bulk metabolomics revealed that amino acid levels are not consistently reduced in tumors, and two other previous microdialysis studies suggest tryptophan [39] (and other amino acids [39, 40]) levels are increased in the interstitium of glioma.
However, a limitation of these types of analyses is that they cannot determine the ability of different subsets of cells to access these pools of amino acids. Perhaps the most illustrative example of this phenomenon is two high impact studies on the deficiency of glucose acquisition by T-cells in solid tumors [41, 42]. While glucose is accessible to the tumor, it outcompetes T-cells for resources. Another layer of complexity is that the expression of specific transporters controls the transport of amino acids. As both T-cells [43] and glioma cells [44, 45] preferentially express large neutral amino acid transporter (LAT-1) to transport essential amino acids, T-cells may be significantly outcompeted for these nutrients.
It is for these manifold reasons that metabolite levels within T-cells in tumors may be more informative. In support of this, our data revealed that CD8+ T-cells had a consistent reduction in most amino acids compared to the periphery. As the deficiency of T-cell infiltration in glioblastoma is more pronounced than any other tumor measured [46, 47], it is not surprising that T-cells are outcompeted for nutrients in the harsh glioma microenvironment. However, this methodology has its limitations (increased processing times), and thus significant future work must be designed to measure amino acid competition at the cellular level in vivo. Despite the complexity of this question, our data undeniably show that GCN2 is required for efficient CD8+ T-cell function in glioblastoma.
The fact that CD8+ T-cells are particularly affected in the brain tumor environment suggests that CD8+ T-cells are particularly reliant on stress sensing in the tumor. This is important because the generation of anti-glioma immune responses relies heavily on functional CD8+ T-cells in both checkpoint blockade [48] and oncolytic adenoviral therapies [49]. As there are distinct metabolic requirements of CD8+ T-cells compared to CD4+ T-cells [50], it makes sense that there are underlying differences in the handling of cellular stress due to metabolic changes in the TME. Nonetheless, this dramatic reduction in CD8+ T-cells was specific within the tumor microenvironment. This suggests that GCN2 has minimal effects on the basal functions of these cells. Indeed, we saw little difference in proliferation/activation of these cells in vitro.
Our competition assays revealed it is not a difference in recruitment that yielded this effect, which is in contrast to a previous study which suggested that GCN2 KO CD8+ T-cells are deficient in CCR7 expression, and are thus retained within the spleens [27]. However, in the context of glioma, the normal recruitment of T-cells to spleens is severely perturbed and thus may mask any effects of splenic retention [51]. Nonetheless, we saw no difference in CCR7 or other chemokine receptor expression, which pointed us to a specific effect within the tumor.
We also observed a reduction in the cytokine production and activation status of GCN2 deficient CD8+ T-cells in the tumor, which occurred in the context of antigen-specific T-cells. Importantly, numerous survival experiments highlight how detrimental GCN2 deficiency is to T-cell function. These results from our adoptive transfer experiments into RAG KO mice bearing tumors further highlight the specific nature and importance of amino acid sensing in CD8+ T-cell function in brain tumors.
This information is at odds with a seminal study on the role of GCN2 in T-cell immunity [26]. This study elegantly showed how IDO depletes tryptophan, which results in T-cell anergy and lack of proliferation. Tryptophan is an essential amino acid, and its deprivation will ultimately shut down the proliferation of any T-cell [52]. This observation has led to the discovery and exploration of both IDO inhibitors for cancer therapy [53] and more recent exploration into GCN2 inhibition [54]. The logic of these studies is that the downstream signaling of EIF2α, such as activating transcription factor 4 (ATF4) and CHOP, is responsible for arresting T-cells. However, this study, and many others on the topic do not address the well-described role of integrated stress response in the maintenance of cell survival under stress [55]. Furthermore, activation of the EIF2α/ATF4 pathway is critical for the initiation of autophagy [56], a metabolic process critical to CD8+ T-cells survival and function [57].
Our data suggest that GCN2 preferentially preserves CD8+ T-cell survival under amino-acid stress, which is consistent with the protective role of GCN2 in hepatocytes [58], oligodendrocytes [59] and kidney tissues [60]. The reason for this is that even though phosphorylation of EIF2α shuts down global protein translation, CHOP and ATF4 upregulate a conserved pathway known to alleviate ER stress [13]. The logic is that the deficiency of amino acids will prevent the generation of proper proteins, and thus puts undue stress on the ER. The lack of ability to handle this ER stress results in cell death. Indeed, our data shows that GCN2 deficient T-cells rapidly undergo apoptosis in tryptophan deficient media, while controls merely stop proliferating. Therefore, GCN2 has a dual role in promoting CD8+ T-cell anergy, while also maintaining its ability to starve in nutrient-poor conditions. This is critical information, considering the clinical targeting of EIF2α by the potent and specific inhibitor ISRIB [38] is considered for malignancies. ISRIB has been shown to inhibit aggressive prostate cancer [30], and in another publication has been shown that it regulates PD-L1 expression [61]. While these studies clearly show the benefit of targeting this pathway for cancer inhibition, we must put into context how this inhibition affects the adaptive immunity. The results of this study guide future work and caution in clinical translation.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Acknowledgements
We would like to thank Peng Gao at the Metabolomics Core Facility at Northwestern for performing and analyzing bulk metabolomics included in this study.
Abbreviation
- ATF4
Activating transcription factor 4
- CCR7
C–C Chemokine receptor type 7
- CNS
Central nervous system
- EAE
Experimental autoimmune encephalomyelitis
- EIF2α
Eukaryotic translation initiation factor 2α
- ER
Endoplasmic reticular
- GCN2
General control nonderepressible 2
- GZMB
Granzyme B
- i.c.
Intracranial
- ISRIB
Integrated stress response inhibitor
- ISR
Integrated stress response
- KO
Knockout
- KYN
Kynurenine
- LC-MS
Liquid chromatography–mass spectrometry
- NCI
National Cancer Institute
- PKC-ϴ
Protein kinase C-theta
- + TRP
Tryptophan added
- − TRP
Tryptophan free
- WT
Wild type
Author contributions
AR, JM, and MSL conceived the study. AR, JM, CLC, PZ, DK, and JWK designed and performed the experiments. ALR maintained animal colonies, performed and analyzed the GCN2 backcrosses, and validated all mice used in this study via genotyping. YH and WKP performed all animal surgeries and monitoring of endpoint analyses. KCP, JF, and MT assisted with the construction and provided critical feedback for the manuscript. MSL and JM oversaw the research program and assisted in the preparation of this manuscript. TX performed all statistical analyses and provided feedback regarding animal numbers. All authors have reviewed the manuscript for accuracy and provided feedback during the writing and revision process.
Funding
Financial support comes from an Outstanding Investigator Award from the National Cancer Institute (NCI) to Maciej S Lesniak (R35CA197725) and a grant from the National Institute of Neurological Disorders and Stroke (NINDS) to Maciej S Lesniak (R01NS093903). Jason Miska received a fellowship from the NINDS (1F32NS098737-01A1). This work was supported by the Northwestern University Robert H. Lurie Cancer Center Flow Cytometry Facility Support Grant (NCI CA060553).
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval and ethical standards
The IACUC committee approved all animal work within the Center for Comparative Medicine (CCM) at Northwestern University. Northwestern University has an Animal Welfare Assurance on file with the Office of Laboratory Animal Welfare (A3283-01). Northwestern University conducts its reviews following United States Public Health Service (USPHS) regulations and applicable federal and local laws. The composition of the IACUC meets the requirements of the USPHS policy and the Animal Welfare Act Regulations. The animal protocol number is IS00002459.
Animal source
C57BL/6 (WT), GCN2 KO, Rag1 KO, OT-1, and Foxp3-IRES-GFP C57/B6 mice were purchased directly from The Jackson Laboratory (Bar Harbor, ME).
Cell line authentication
GL-261 was purchased directly from the National Cancer Institute (NCI) Frederick National Tumor Repository Laboratory (Frederick, MD. USA). CT2A was acquired from the Balyasnikova laboratory at Northwestern University, Feinberg School of Medicine (Chicago, IL. USA). All cell lines were used in Northwestern’s Neuro-oncology research. All cell lines are routinely tested for Mycoplasma contamination every 2 months using the Universal Mycoplasma Detection Kit (ATCC® 30-1012 K™). The identity and purity of cell lines were determined using short tandem repeats (STR) profiling performed by the Northwestern sequencing facility.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Aida Rashidi and Jason Miska contributed equally.
References
- 1.Rashidi A, Miska J, Pituch K, Kanojia D, Lopez-Rosas A, Han Y, et al. Gcn2 kinase is essential for adaptive T-cell immunity in glioma. Neuro-Oncology. 2017;19:113. [Google Scholar]
- 2.Wolchok JD, Kluger H, Callahan MK, Postow MA, Rizvi NA, Lesokhin AM, et al. Nivolumab plus ipilimumab in advanced melanoma. N Engl J Med. 2013;369(2):122–133. doi: 10.1056/NEJMoa1302369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Overman MJ, Lonardi S, Wong KYM, Lenz HJ, Gelsomino F, Aglietta M, et al. Durable clinical benefit with nivolumab plus ipilimumab in DNA mismatch repair-deficient/microsatellite instability-high metastatic colorectal cancer. J Clin Oncol. 2018;36(8):773–779. doi: 10.1200/JCO.2017.76.9901. [DOI] [PubMed] [Google Scholar]
- 4.Cloughesy TF, Mochizuki AY, Orpilla JR, Hugo W, Lee AH, Davidson TB, et al. Neoadjuvant anti-PD-1 immunotherapy promotes a survival benefit with intratumoral and systemic immune responses in recurrent glioblastoma. Nat Med. 2019;25(3):477–486. doi: 10.1038/s41591-018-0337-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Woroniecka K, Chongsathidkiet P, Rhodin K, Kemeny H, Dechant C, Farber SH, et al. T-cell exhaustion signatures vary with tumor type and are severe in glioblastoma. Clin Cancer Res. 2018;24(17):4175–4186. doi: 10.1158/1078-0432.CCR-17-1846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Mirzaei R, Sarkar S, Yong VW. T cell exhaustion in glioblastoma: intricacies of immune checkpoints. Trends Immunol. 2017;38(2):104–115. doi: 10.1016/j.it.2016.11.005. [DOI] [PubMed] [Google Scholar]
- 7.Wei J, Raynor J, Nguyen TL, Chi H. Nutrient and metabolic sensing in T cell responses. Front Immunol. 2017;8:247. doi: 10.3389/fimmu.2017.00247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kwidzinski E, Bunse J, Aktas O, Richter D, Mutlu L, Zipp F, et al. Indolamine 2,3-dioxygenase is expressed in the CNS and down-regulates autoimmune inflammation. Faseb J. 2005;19(8):1347. doi: 10.1096/fj.04-3228fje. [DOI] [PubMed] [Google Scholar]
- 9.Munn DH, Zhou M, Attwood JT, Bondarev I, Conway SJ, Marshall B, et al. Prevention of allogeneic fetal rejection by tryptophan catabolism. Science. 1998;281(5380):1191–1193. doi: 10.1126/science.281.5380.1191. [DOI] [PubMed] [Google Scholar]
- 10.Lob S, Konigsrainer A, Zieker D, Brucher BL, Rammensee HG, Opelz G, et al. IDO1 and IDO2 are expressed in human tumors: levo—but not dextro-1-methyl tryptophan inhibits tryptophan catabolism. Cancer Immunol Immunother. 2009;58(1):153–157. doi: 10.1007/s00262-008-0513-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Dong J, Qiu H, Garcia-Barrio M, Anderson J, Hinnebusch AG. Uncharged tRNA activates GCN2 by displacing the protein kinase moiety from a bipartite tRNA-binding domain. Mol Cell. 2000;6(2):269–279. doi: 10.1016/s1097-2765(00)00028-9. [DOI] [PubMed] [Google Scholar]
- 12.Garcia-Barrio M, Dong J, Ufano S, Hinnebusch AG. Association of GCN1-GCN20 regulatory complex with the N-terminus of eIF2alpha kinase GCN2 is required for GCN2 activation. EMBO J. 2000;19(8):1887–1899. doi: 10.1093/emboj/19.8.1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Fusakio ME, Willy JA, Wang Y, Mirek ET, Al Baghdadi RJ, Adams CM, et al. Transcription factor ATF4 directs basal and stress-induced gene expression in the unfolded protein response and cholesterol metabolism in the liver. Mol Biol Cell. 2016;27(9):1536–1551. doi: 10.1091/mbc.E16-01-0039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Harding HP, Novoa I, Zhang Y, Zeng H, Wek R, Schapira M, et al. Regulated translation initiation controls stress-induced gene expression in mammalian cells. Mol Cell. 2000;6(5):1099–1108. doi: 10.1016/s1097-2765(00)00108-8. [DOI] [PubMed] [Google Scholar]
- 15.Pakos-Zebrucka K, Koryga I, Mnich K, Ljujic M, Samali A, Gorman AM. The integrated stress response. EMBO Rep. 2016;17(10):1374–1395. doi: 10.15252/embr.201642195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wainwright DA, Balyasnikova IV, Chang AL, Ahmed AU, Moon KS, Auffinger B, et al. IDO expression in brain tumors increases the recruitment of regulatory T cells and negatively impacts survival. Clin Cancer Res. 2012;18(22):6110–6121. doi: 10.1158/1078-0432.CCR-12-2130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wainwright DA, Chang AL, Dey M, Balyasnikova IV, Kim C, Tobias AL, et al. Durable therapeutic efficacy utilizing combinatorial blockade against IDO, CTLA-4 and PD-L1 in mice with brain tumors. Clinical Cancer Research. 2014;20:5290–5301. doi: 10.1158/1078-0432.CCR-14-0514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Holmgaard RB, Zamarin D, Li YY, Gasmi B, Munn DH, Allison JP, et al. Tumor-expressed IDO recruits and activates MDSCs in a treg-dependent manner. Cell Rep. 2015;13(2):412–424. doi: 10.1016/j.celrep.2015.08.077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Yu JP, Du WJ, Yan F, Wang Y, Li H, Cao S, et al. Myeloid-derived suppressor cells suppress antitumor immune responses through IDO expression and correlate with lymph node metastasis in patients with breast cancer. J Immunol. 2013;190(7):3783–3797. doi: 10.4049/jimmunol.1201449. [DOI] [PubMed] [Google Scholar]
- 20.Miret JJ, Kirschmeier P, Koyama S, Zhu M, Li YY, Naito Y, et al. Suppression of myeloid cell arginase activity leads to therapeutic response in a NSCLC mouse model by activating anti-tumor immunity. J Immunother Cancer. 2019;7(1):32. doi: 10.1186/s40425-019-0504-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Lind DS. Arginine and cancer. J Nutr. 2004;134(10 Suppl):2837S–2841S. doi: 10.1093/jn/134.10.2837S. [DOI] [PubMed] [Google Scholar]
- 22.Caso G, McNurlan MA, McMillan ND, Eremin O, Garlick PJ. Tumour cell growth in culture: dependence on arginine. Clin Sci (Lond) 2004;107(4):371–379. doi: 10.1042/CS20040096. [DOI] [PubMed] [Google Scholar]
- 23.Geiger R, Rieckmann JC, Wolf T, Basso C, Feng Y, Fuhrer T, et al. l-Arginine modulates T cell metabolism and enhances survival and anti-tumor activity. Cell. 2016;167(3):829–842.e13. doi: 10.1016/j.cell.2016.09.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zhang J, Fan J, Venneti S, Cross JR, Takagi T, Bhinder B, et al. Asparagine plays a critical role in regulating cellular adaptation to glutamine depletion. Mol Cell. 2014;56(2):205–218. doi: 10.1016/j.molcel.2014.08.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pavlova NN, Hui S, Ghergurovich JM, Fan J, Intlekofer AM, White RM, et al. As extracellular glutamine levels decline, asparagine becomes an essential amino acid. Cell Metabolism. 2018;27(2):428. doi: 10.1016/j.cmet.2017.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Munn DH, Sharma MD, Baban B, Harding HP, Zhang Y, Ron D, et al. GCN2 kinase in T cells mediates proliferative arrest and anergy induction in response to indoleamine 2,3-dioxygenase. Immunity. 2005;22(5):633–642. doi: 10.1016/j.immuni.2005.03.013. [DOI] [PubMed] [Google Scholar]
- 27.Van de Velde LA, Guo XJ, Barbaric L, Smith AM, Oguin TH, 3rd, Thomas PG, et al. Stress kinase GCN2 controls the proliferative fitness and trafficking of cytotoxic T cells independent of environmental amino acid sensing. Cell Rep. 2016;17(9):2247–2258. doi: 10.1016/j.celrep.2016.10.079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Keil M, Sonner JK, Lanz TV, Oezen I, Bunse T, Bittner S, et al. General control non-derepressible 2 (GCN2) in T cells controls disease progression of autoimmune neuroinflammation. J Neuroimmunol. 2016;297:117–126. doi: 10.1016/j.jneuroim.2016.05.014. [DOI] [PubMed] [Google Scholar]
- 29.Nakamura A, Nambu T, Ebara S, Hasegawa Y, Toyoshima K, Tsuchiya Y, et al. Inhibition of GCN2 sensitizes ASNS-low cancer cells to asparaginase by disrupting the amino acid response. Proc Natl Acad Sci USA. 2018;115(33):E7776–E7785. doi: 10.1073/pnas.1805523115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nguyen HG, Conn CS, Kye Y, Xue LR, Forester CM, Cowan JE, et al. Development of a stress response therapy targeting aggressive prostate cancer. Sci Trans Med. 2018;10(439):eaar2036. doi: 10.1126/scitranslmed.aar2036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim JW, Miska J, Young JS, Rashidi A, Kane JR, Panek WK, et al. A comparative study of replication-incompetent and -competent adenoviral therapy-mediated immune response in a murine glioma model. Mol Ther Oncolytics. 2017;5:97–104. doi: 10.1016/j.omto.2017.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wu YM, Li L. Sample normalization methods in quantitative metabolomics. J Chromatogr A. 2016;1430:80–95. doi: 10.1016/j.chroma.2015.12.007. [DOI] [PubMed] [Google Scholar]
- 33.Clarke SRM, Barnden M, Kurts C, Carbone FR, Miller JF, Heath WR. Characterization of the ovalbumin-specific TCR transgenic line OT-I: MHC elements for positive and negative selection. Immunol Cell Biol. 2000;78(2):110–117. doi: 10.1046/j.1440-1711.2000.00889.x. [DOI] [PubMed] [Google Scholar]
- 34.Bjorkdahl O, Barber KA, Brett SJ, Daly MG, Plumpton C, Elshourbagy NA, et al. Characterization of CC-chemokine receptor 7 expression on murine T cells in lymphoid tissues. Immunology. 2003;110(2):170–179. doi: 10.1046/j.1365-2567.2003.01727.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ladomersky E, Zhai L, Lenzen A, Lauing KL, Qian J, Scholtens DM, et al. IDO1 inhibition synergizes with radiation and PD-1 blockade to durably increase survival against advanced glioblastoma. Clin Cancer Res. 2018;24(11):2559–2573. doi: 10.1158/1078-0432.CCR-17-3573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sonner JK, Deumelandt K, Ott M, Thome CM, Rauschenbach KJ, Schulz S, et al. The stress kinase GCN2 does not mediate suppression of antitumor T cell responses by tryptophan catabolism in experimental melanomas. Oncoimmunology. 2016;5(12):e1240858. doi: 10.1080/2162402X.2016.1240858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hayashi K, Altman A. Protein kinase C theta (PKCtheta): a key player in T cell life and death. Pharmacol Res. 2007;55(6):537–544. doi: 10.1016/j.phrs.2007.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Sidrauski C, McGeachy AM, Ingolia NT, Walter P. The small molecule ISRIB reverses the effects of eIF2alpha phosphorylation on translation and stress granule assembly. Elife. 2015 doi: 10.7554/eLife.05033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Behrens PF, Langemann H, Strohschein R, Draeger J, Hennig J. Extracellular glutamate and other metabolites in and around RG2 rat glioma: an intracerebral microdialysis study. J Neurooncol. 2000;47(1):11–22. doi: 10.1023/a:1006426917654. [DOI] [PubMed] [Google Scholar]
- 40.Wibom C, Surowiec I, Moren L, Bergstrom P, Johansson M, Antti H, et al. Metabolomic patterns in glioblastoma and changes during radiotherapy: a clinical microdialysis study. J Proteome Res. 2010;9(6):2909–2919. doi: 10.1021/pr901088r. [DOI] [PubMed] [Google Scholar]
- 41.Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. 2015;162(6):1217–1228. doi: 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chang CH, Qiu J, O’Sullivan D, Buck MD, Noguchi T, Curtis JD, et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162(6):1229–1241. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hayashi K, Jutabha P, Endou H, Sagara H, Anzai N. LAT1 is a critical transporter of essential amino acids for immune reactions in activated human T cells. J Immunol. 2013;191(8):4080–4085. doi: 10.4049/jimmunol.1300923. [DOI] [PubMed] [Google Scholar]
- 44.Nawashiro H, Otani N, Uozumi Y, Ooigawa H, Toyooka T, Suzuki T, et al. High expression of L-type amino acid transporter 1 in infiltrating glioma cells. Brain Tumor Pathol. 2005;22(2):89–91. doi: 10.1007/s10014-005-0188-z. [DOI] [PubMed] [Google Scholar]
- 45.Kobayashi K, Ohnishi A, Promsuk J, Shimizu S, Kanai Y, Shiokawa Y, et al. Enhanced tumor growth elicited by L-type amino acid transporter 1 in human malignant glioma cells. Neurosurgery. 2008;62(2):493–503. doi: 10.1227/01.neu.0000316018.51292.19. [DOI] [PubMed] [Google Scholar]
- 46.Thorsson V, Gibbs DL, Brown SD, Wolf D, Bortone DS, Yang THO, et al. The immune landscape of cancer. Immunity. 2018;48(4):812. doi: 10.1016/j.immuni.2018.03.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hendry S, Salgado R, Gevaert T, Russell PA, John T, Thapa B, et al. Assessing tumor-infiltrating lymphocytes in solid tumors: a practical review for pathologists and proposal for a standardized method from the international immuno-oncology biomarkers working group: part 2: TILs in melanoma, gastrointestinal tract carcinomas, non-small cell lung carcinoma and mesothelioma, endometrial and ovarian carcinomas, squamous cell carcinoma of the head and neck, genitourinary carcinomas, and primary brain tumors. Adv Anat Pathol. 2017;24(6):311–335. doi: 10.1097/PAP.0000000000000161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Zeng J, See AP, Phallen J, Jackson CM, Belcaid Z, Ruzevick J, et al. Anti-PD-1 blockade and stereotactic radiation produce long-term survival in mice with intracranial gliomas. Int J Radiat Oncol Biol Phys. 2013;86(2):343–349. doi: 10.1016/j.ijrobp.2012.12.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kleijn A, van den Bossche W, Haefner ES, Belcaid Z, Burghoorn-Maas C, Kloezeman JJ, et al. The sequence of Delta24-RGD and TMZ administration in malignant glioma affects the role of CD8(+)T cell anti-tumor activity. Mol Ther Oncolytics. 2017;5:11–19. doi: 10.1016/j.omto.2017.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Cao Y, Rathmell JC, Macintyre AN. Metabolic reprogramming towards aerobic glycolysis correlates with greater proliferative ability and resistance to metabolic inhibition in CD8 versus CD4 T cells. PLoS One. 2014;9(8):e104104. doi: 10.1371/journal.pone.0104104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chongsathidkiet P, Jackson C, Koyama S, Loebel F, Cui X, Farber SH, et al. Sequestration of T cells in bone marrow in the setting of glioblastoma and other intracranial tumors. Nat Med. 2018;24(9):1459–1468. doi: 10.1038/s41591-018-0135-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Munn DH, Shafizadeh E, Attwood JT, Bondarev I, Pashine A, Mellor AL. Inhibition of T cell proliferation by macrophage tryptophan catabolism. J Exp Med. 1999;189(9):1363–1372. doi: 10.1084/jem.189.9.1363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Prendergast GC, Malachowski WP, DuHadaway JB, Muller AJ. Discovery of IDO1 inhibitors: from bench to bedside. Cancer Res. 2017;77(24):6795–6811. doi: 10.1158/0008-5472.CAN-17-2285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Galezowski M, Sitarz K, Majewska E, Chmielewski S, Michalik K, Masiejczyk M, et al. Development of small molecule selective inhibitors of GCN2 as an immunotherapy aimed at preventing immune escape of tumor cells. Cancer Res. 2017;77:2639. [Google Scholar]
- 55.Wek RC, Jiang HY, Anthony TG. Coping with stress: eIF2 kinases and translational control. Biochem Soc T. 2006;34:7–11. doi: 10.1042/BST20060007. [DOI] [PubMed] [Google Scholar]
- 56.B’chir W, Maurin AC, Carraro V, Averous J, Jousse C, Muranishi Y, et al. The eIF2 alpha/ATF4 pathway is essential for stress-induced autophagy gene expression. Nucleic Acids Res. 2013;41(16):7683–7699. doi: 10.1093/nar/gkt563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Xu XJ, Araki K, Li SZ, Han JH, Ye LL, Tan WG, et al. Autophagy is essential for effector CD8(+) T cell survival and memory formation. Nat Immunol. 2014;15(12):1152–1161. doi: 10.1038/ni.3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Wilson GJ, Bunpo P, Cundiff JK, Wek RC, Anthony TG. The eukaryotic initiation factor 2 kinase GCN2 protects against hepatotoxicity during asparaginase treatment. Am J Physiol Endocrinol Metab. 2013;305(9):E1124–E1133. doi: 10.1152/ajpendo.00080.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.She P, Bunpo P, Cundiff JK, Wek RC, Harris RA, Anthony TG. General control nonderepressible 2 (GCN2) kinase protects oligodendrocytes and white matter during branched-chain amino acid deficiency in mice. J Biol Chem. 2013;288(43):31250–31260. doi: 10.1074/jbc.M113.498469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chaudhary K, Shinde R, Liu HY, Gnana-Prakasam JP, Veeranan-Karmegam R, Huang L, et al. Amino acid metabolism inhibits antibody-driven kidney injury by inducing autophagy. J Immunol. 2015;194(12):5713–5724. doi: 10.4049/jimmunol.1500277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Xu Y, Poggio M, Jin HY, Shi Z, Forester CM, Wang Y, et al. Translation control of the immune checkpoint in cancer and its therapeutic targeting. Nat Med. 2019;25(2):301–311. doi: 10.1038/s41591-018-0321-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.