

Abstract
Expression of programmed death ligand 1 (PD-L1) on the surface of tumor cells and its interaction with programmed cell death protein 1 (PD-1) on tumor-infiltrating lymphocytes suppress anti-tumor immunity. In breast tumors, PD-L1 expression levels are the highest in estrogen receptor-negative, progesterone receptor-negative, and human epidermal growth factor receptor 2-negative (triple-negative) cancers. In this study, we show that a portion of PD-L1 exogenously expressed in several triple-negative breast cancer cell lines, as well as endogenous PD-L1, is proteolytically cleaved by cell surface metalloproteases. The cleavage generates an ~ 37-kDa N-terminal PD-L1 fragment that is released to the media and a C-terminal PD-L1 fragment that remains associated with cells but is efficiently eliminated by lysosomal degradation. We identify ADAM10 and ADAM17, two closely related members of the ADAM family of cell surface metalloproteases, as enzymes mediating PD-L1 cleavage. Notably, treatment of cells with ionomycin, a calcium ionophore and a known activator of ADAM10, or with phorbol 12-myristate 13-acetate, an activator of ADAM17, dramatically increases the release of soluble PD-L1 to the media. We postulate that ADAM10 and/or ADAM17 may contribute to the regulation of the PD-L1/PD-1 pathway and, ultimately, to anti-tumor immunity in triple-negative breast cancer.
Electronic supplementary material
The online version of this article (10.1007/s00262-019-02437-2) contains supplementary material, which is available to authorized users.
Keywords: PD-L1, Breast cancer, Metalloprotease, Immunotherapy, Checkpoint blockade
Introduction
Immune checkpoint blockade represents one of the most promising immunotherapies for the treatment of solid tumors. The therapy involves blocking the interaction between a negative immune checkpoint regulator, such as programmed cell death protein 1 (PD-1) on T cells, with programmed death ligand 1 (PD-L1) expressed in the tumor microenvironment [1]. Since the PD-1/PD-L1 pathway attenuates T cell activity and promotes tumor immune escape, anti-PD-1/PD-L1 therapeutics can stimulate anti-tumor responses, reduce tumor growth, or even cause tumor remission in patients with advanced cancers [1].
In breast cancer, the PD-1/PD-L1 axis is considered a particularly promising therapeutic target in triple-negative tumors (estrogen receptor-negative, progesterone receptor-negative, human epidermal growth factor receptor 2-negative) [2]. Triple-negative breast cancer (TNBC) is the most immunogenic breast cancer subtype, with more prominent immune infiltration and higher expression levels of PD-L1 than other tumor subtypes [3, 4]. Within tumors, PD-L1 is expressed both in tumor cells and in tumor-infiltrating lymphocytes, including CD4+ or CD8+ T cells, and macrophages [3, 4]. Despite the favorable immune characteristics of TNBCs, their response to anti-PD-1/PD-L1 therapy is rather modest and varies significantly among patients [2]. Recently reported objective response rates to anti-PD-1/PD-L1 monotherapies in patients with metastatic TNBCs remained below 25% [5, 6], and response rates to a combination of anti-PD-L1 antibody plus chemotherapy were lower than 60% [7]. To further improve therapeutic outcomes of PD-1/PD-L1 blockade and to help identify biomarkers that can predict patient’s response to anti-PD-1/PD-L1 therapies, a better understanding of all aspects of the PD-1/PD-L1 pathway is needed, including the role of tumor-derived PD-L1 and its regulation in tumor cells.
PD-L1 gene expression is regulated at the transcriptional and post-transcriptional levels [8], and PD-L1 protein is further regulated post-translationally via ubiquitination, glycosylation, palmitoylation, or lysosomal degradation [9–13]. Transmembrane PD-L1, the main protein isoform of PD-L1, resides at the cell surface, but also on the surface of exosomes that are secreted to the extracellular milieu [14, 15]. Soluble forms of PD-L1, containing an intact receptor-binding domain and lacking the transmembrane domain, have also been described, and they result either from an alternative PD-L1 mRNA splicing [16, 17] or from cleavage of the transmembrane PD-L1 protein. Proteolytic cleavage of PD-L1 was described in renal cell carcinoma [18], mesenchymal stromal cells [19, 20], and head and neck squamous cell carcinoma [21], but it has not been investigated in breast cancer.
In this study, we demonstrate unambiguously a proteolytic cleavage of PD-L1 in triple-negative breast cancer cell lines. The cleavage generates a distinct soluble N-terminal PD-L1 fragment, which is detectable by ELISA and immunoblotting, and a C-terminal PD-L1 fragment that remains associated with cells but is efficiently eliminated by lysosomal degradation. We also identify a disintegrin and metalloprotease 10 (ADAM10) and ADAM17, two closely related members of the ADAM family of cell surface metalloproteases [22], as enzymes mediating PD-L1 cleavage. We postulate that ADAM10 and/or ADAM17 may contribute to the regulation of the PD-L1/PD-1 pathway and, ultimately, to anti-tumor immunity in TNBC.
Materials and methods
Reagents and antibodies
Recombinant human IFN-γ was from eBioscience (San Diego, CA), batimastat, aprotinin, pepstatin, leupeptin, matrix metalloprotease (MMP)-9 inhibitor I, GI254023X, CL-82198, bafilomycin A1, monensin, ammonium chloride, and phorbol 12-myristate 13-acetate (PMA) were from MilliporeSigma (Burlington, MA), AEBSF was from Fisher Scientific (Hampton, NH), ionomycin and tumor necrosis factor protease inhibitor 2 (TAPI-2) were from Cayman Chemical (Ann Arbor, MI). Fetal bovine serum (FBS) and horse serum (HS) were from Gibco Thermo Fisher Scientific (Waltham, MA). ON-TARGETplus human ADAM10 small interfering RNAs (siRNAs; J-004503-06 and J-004503-07; siA10#1 and siA10#2, respectively), siGENOME human ADAM17 siRNAs (D-003453-02 and D-003453-03; siA17#1 and siA17#2, respectively), and Dharmafect 4 transfection reagent were from Dharmacon (Lafayette, CO). Human PD-L1 DuoSet ELISA kit was from R&D Systems (Minneapolis, MN). Anti-PD-L1 mAbs, clones E1L3N and E1J2J, anti-ADAM10 pAb #14194, and anti-glyceraldehyde 3-phosphate dehydrogenase (GAPDH) mAb, clone D16H11, were from Cell Signaling Technology (Danvers, MA), anti-ADAM17 pAb was from QED Bioscience (San Diego, CA), anti-Myc tag mAb, clone 9E10, was from Invitrogen (Carlsbad, CA), and anti-FLAG tag mAb (DYKDDDDK) was from GenScript (Piscataway, NJ).
Cell culture
MDA-MB-231, BT549, MCF10A, A549, and DU-145 cells were from the American Tissue Culture Collection (Manassas, VA). SUM149 and SUM159 cell lines were obtained from Asterand (Detroit, MI). MDA-MB-231, DU-145, and A549 cells were cultured in DMEM/F12 medium with 10% FBS and 10 mM HEPES. BT549 cells were grown in RMPI-1640 medium containing 10% FBS and 5 μg/ml insulin. SUM149 and SUM159 cells were cultured in Ham’s F-12 medium supplemented with 5% FBS, 10 mM HEPES, 5 μg/ml insulin, and 1 μg/ml hydrocortisone. MCF10A cells were cultured in DMEM/F-12 supplemented with 5% horse serum, 0.5 µg/ml hydrocortisone, 20 ng/ml human EGF, and 10 µg/ml insulin. Cells were maintained at 37 °C under humidified atmosphere containing 5% CO2. siRNA transfections were performed using 50 nM siRNA (total concentration) and 2 μl Dharmafect 4 (for MDA-MB-231 cells) or Dharmafect 2 reagent (for A549 and DU-145 cells) per well in six-well plates. One day after transfection, cells were transferred to fresh complete media and incubated for additional 24–48 h.
Stable overexpression of PD-L1
Cells were plated in 12-well plates (1x105 cells/well). The next day, 2 ml of fresh media containing human PD-L1 (NM_014143) lentiviral particles (2x105 transducing units/well; OriGene, Rockville, MD) and polybrene (8 μg/ml) were added. Two different PD-L1 clones were used: Myc/FLAG (M/F)-tagged (RC213071L3V) and GFP-tagged (RC213071L4V). The M/F-tagged construct contained the following C-terminal sequence after the PD-L1 insert: TRTRPLEQKLISEEDLAANDILDYKDDDDKVWVGSGATNFSLLKQAGDVEENP, with the Myc and FLAG tags underlined and the P2A peptide sequence italicized. The GFP-tagged construct also contained the C-terminal P2A sequence. P2A is a self-cleaving peptide that separated PD-L1-M/F or PD-L1-GFP and puromycin N-acetyl transferase encoded in the expression vectors. In parallel, cells were incubated with control GFP lentiviral particles (PS100093V). After 24 h, cells were transferred to fresh media, re-plated into six-well plates, and incubated with puromycin for the next 7–10 days. The optimal concentrations of puromycin for each cell line were determined by performing kill curve experiments and were as follows: MDA-MB-231, 0.4 μg/ml; BT549, 0.3 μg/ml; MCF10A, 1 μg/ml; SUM159, 1 μg/ml; SUM149, 0.3 μg/ml; A549, 0.5 μg/ml; and DU-145, 0.5 μg/ml.
PD-L1 ELISA
Cells were incubated in six-well plates for 24–72 h in media with various treatments, as indicated. Conditioned media were centrifuged for 15 min at 21,000×g, and concentrations of soluble PD-L1 (sPD-L1) in the supernatants were measured using human PD-L1 DuoSet ELISA kit (R&D Systems) and a BioTek H1M microplate reader. Cells remaining attached to plates were lysed with lysis buffer, and protein concentrations were determined using the bicinchoninic acid (BCA) assay (Thermo Fisher Scientific). To account for possible differences in cell densities between individual wells, sPD-L1 concentrations measured by ELISA were normalized to equal protein concentration in all cell lysates.
Immunoblotting
Immunoblotting was performed, as described [23]. For signal detection, we used SuperSignal West Pico or West Femto chemiluminescence detection kit (Thermo Fisher Scientific) and Azure c500 digital imaging system.
Results
To determine whether PD-L1 is released from the surface of breast cancer cells, we measured concentrations of sPD-L1 in conditioned media from four different human TNBC cell lines, MDA-MB-231, BT549, SUM159, and SUM149, as well as from the untransformed human mammary cell line MCF10A, using a sandwich ELISA assay employing antibodies specific for the extracellular domain of PD-L1. In parallel, PD-L1 in cell lysates was examined by immunoblotting.
The amount of PD-L1 detected in the lysates of unstimulated cells was the highest in MDA-MB-231 cells and the lowest in SUM149 cells (MDA-MB-231 > SUM159 > BT549 ≈ MCF10A > SUM149) (Fig. 1a). This order of expression corresponded well to PD-L1 mRNA levels determined by RNA sequencing [24, 25] (Gene Expression Omnibus (GEO): GSE73526 and GSE48213; https://www.ncbi.nlm.nih.gov/geo/). Concentrations of sPD-L1 in the conditioned media from unstimulated cells were very low, except for MDA-MB-231 cells, where sPD-L1 reached ~ 100 pg/ml (Fig. 1b). Stimulation of cells for 48 h with 10 ng/ml human IFN-γ dramatically increased cellular levels of PD-L1 (Fig. 1a), consistent with the known role of IFN-γ in the regulation of PD-L1 expression [26]. Importantly, treatment of cells with IFN-γ also significantly increased concentrations of sPD-L1 in the conditioned media from all five cell lines (Fig. 1b).
Fig. 1.
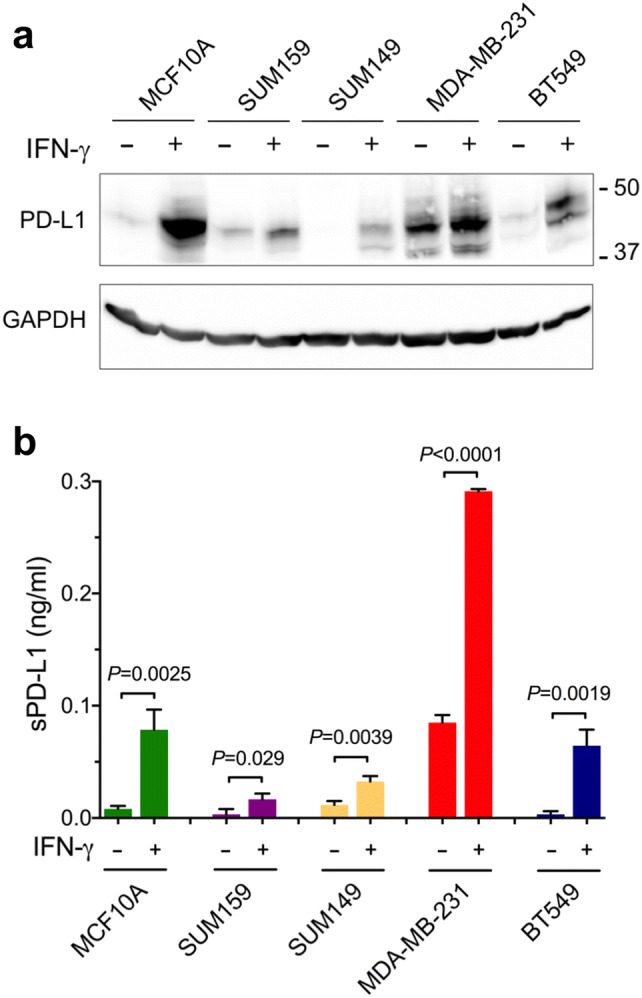
Interferon-γ increases PD-L1 protein levels in cell lysates and in culture media. Human breast cancer cells SUM159, SUM149, MDA-MB-231, BT549, and human non-transformed mammary epithelial cells MCF10A were incubated for 48 h without or with 10 ng/ml human IFN-γ. a Cell lysates were analyzed by Western blotting using anti-PD-L1 antibody (E1L3N). GAPDH is a gel loading control. b Concentrations of sPD-L1 in the media were measured by ELISA. Values are means ± SD from three replicate measurements, normalized to equal protein concentration in all cellular lysates. P values were determined by unpaired two-tailed Student’s t test. Results in a and b are representative of three independent experiments
Human PD-L1 mRNA can be alternatively spliced, generating transcripts that are translated into soluble forms of PD-L1 protein [16, 17]. Similar to the main PD-L1 splice variant, these alternative variants are induced by IFN-γ [16]. To exclude the possibility that increased concentrations of sPD-L1 detected after stimulation with IFN-γ were simply due to an increased expression of a soluble PD-L1 isoform, we established cells with stable constitutive overexpression of transmembrane PD-L1. Recombinant PD-L1 contained a C-terminal GFP tag or a tandem M/F tag. Immunoblotting of cellular lysates showed the presence of several PD-L1 bands, ranging from ~ 70 to ~ 90 kDa for PD-L1-GFP, and from ~ 50 to ~ 70-kDa for PD-L1-M/F (Fig. 2a), which was most likely due to post-translational protein modifications. Importantly, conditioned media from cells with stable overexpression of PD-L1 contained much higher concentrations of sPD-L1 than the parental cells, typically approaching 2–4 ng/ml for MDA-MB-231 cells, 0.4–1 ng/ml for the remaining breast cancer cells, and 0.2–0.75 ng/ml for MCF10A cells after a 2-day incubation period (Fig. 2b). After parallel measurement of PD-L1 concentrations in cell lysates by ELISA, we estimated that sPD-L1 released to the media represented ~ 5–10% of the total PD-L1 protein in breast cancer cells and ~ 1% of PD-L1 in MCF10A cells.
Fig. 2.

Elevated levels of soluble PD-L1 in culture media after stable overexpression of transmembrane PD-L1. PD-L1-GFP, PD-L1-M/F, or GFP alone, were stably overexpressed in MCF10A, SUM159, SUM149, MDA-MB-231, and BT549 cells. a PD-L1 overexpression was confirmed by Western blotting using cell lysates and anti-PD-L1 antibody (E1L3N). Endogenous PD-L1 is not detectable at this exposure time. GAPDH is a gel loading control. b Concentrations of sPD-L1 in conditioned media were measured by ELISA. Values are means ± SD from three replicate measurements, normalized to equal protein concentration in all cell lysates. P value was determined by one-way ANOVA, followed by Tukey’s multiple comparisons test. Results are representative of three independent experiments
Next, we asked whether sPD-L1 represented an authentic cleavage product of transmembrane PD-L1. This question was important because cancer cells often shed large amounts of exosomes [14], and PD-L1-containing exosomes might be detected by ELISA. We focused on three cell lines: MDA-MB-231-PD-L1-M/F, BT549-PD-L1-M/F, and MCF10A-PD-L1-M/F, with high, intermediate, and low secretion of sPD-L1, respectively. After centrifugation of the conditioned media at 120,000×g for 1.5 h, a standard method for sedimentation of exosomes [27], sPD-L1 was detected in the supernatants and not the pellets (Fig. 3a). This result indicated that PD-L1 detected in the conditioned media represented a soluble protein and not exosomal transmembrane PD-L1. Furthermore, incubation of cells in serum-free medium did not diminish the levels of sPD-L1 (Fig. 3b), indicating that cellular proteases, rather than proteolytic enzymes from the serum, played a role in generating sPD-L1.
Fig. 3.

Soluble PD-L1 is generated by a proteolytic cleavage of transmembrane PD-L1. a Conditioned media from MDA-MB-231, BT549, or MCF10A cells with stable overexpression of PD-L1-M/F were centrifuged at 120,000×g for 1.5 h. sPD-L1 in the supernatants (S), pellets (P), and total media prior to centrifugation (T) was assayed by ELISA. Values are means ± SD from three replicate measurements. Results are representative of two independent experiments. b MDA-MB-231, BT549, or MCF10A cells stably overexpressing PD-L1-M/F were incubated for 24 h in media with or without serum. sPD-L1 in the media was assayed by ELISA. Values are means ± SD from three replicate measurements, normalized to equal protein concentration in all cellular lysates. Results in a and b are representative of three independent experiments. c MDA-MB-231 cells with stable overexpression of PD-L1-M/F, PD-L1-GFP, or GFP alone were incubated in serum-free media for 24 h. Conditioned media (70 μl, out of 1 ml of the total volume) or cell lysates (5 μl, out of 500 μl of the total volume) were analyzed by Western blotting using an antibody specific for the extracellular domain of PD-L1 (E1J2J). The N-terminal PD-L1 fragment detected in the media but not the lysates is indicated by a red arrow. GAPDH is a gel loading control for lysate samples. Results are representative of two independent experiments
To unambiguously demonstrate that sPD-L1 was generated by proteolytic cleavage of transmembrane PD-L1, we examined conditioned media from MDA-MB-231 cells overexpressing PD-L1 (which release the highest amounts of sPD-L1) by immunoblotting, using an antibody specific for the extracellular domain of PD-L1 (E1J2J). Cells were incubated in serum-free medium in this experiment to avoid interference from serum albumin in the detection of PD-L1. First, we observed low amounts of full-length PD-L1 in the conditioned media (Fig. 3c), which might be due to a slightly decreased cell viability in the absence of serum. Most importantly, we detected an ~ 37-kDa N-terminal PD-L1 fragment in conditioned media from MDA-MB-231 cells overexpressing PD-L1-M/F or PD-L1-GFP, but not in cell lysates (Fig. 3c). This fragment was not observed in conditioned media from control GFP cells, most likely because the level of endogenous sPD-L1 was below the antibody detection limit. Importantly, while the molecular weights of full-length PD-L1-M/F and PD-L1-GFP were different due to the presence of two different C-terminal tags, the size of the 37-kDa fragment released to the media was the same, indicating that this fragment must have been derived from the recombinant PD-L1-M/F and PD-L1-GFP proteins via the cleavage at the same site within the two constructs.
We next investigated the fate of the C-terminal fragment of PD-L1. If the 37-kDa N-terminal cleavage product is released to the media, a complementary C-terminal PD-L1 fragment should remain associated with cells. Inspection of the lower molecular weight region of the immunoblots obtained with the antibody specific for the cytoplasmic domain of PD-L1 (E1L3N) revealed the presence of a weak ~ 18-kDa band in the lysates of BT549 cells expressing PD-L1-M/F, but not in the lysates of control cells expressing GFP (Fig. 4a). To visualize the 18-kDa fragment in MDA-MB-231-PD-L1-M/F or MCF10A-PD-L1-M/F lysates, cells needed to be incubated for 24 h in the presence of 10 µM chloroquine, a weak base that leads to the inhibition of lysosomal protein degradation. Chloroquine treatment did not have a significant effect on full-length PD-L1-M/F (Fig. 4a). This result suggests that the 18-kDa cleavage product is not stable in MDA-MB-231 or MCF10A cells and is efficiently degraded by lysosomal enzymes. Chloroquine was not required for stabilization of the 18-kDa PD-L1 fragment in BT549 cells (Fig. 4a, see “Discussion”). The same 18-kDa band was also detected with anti-Myc and anti-FLAG antibodies (Fig. 4b), confirming that this band represented the C-terminal fragment of the PD-L1-M/F protein.
Fig. 4.

Detection of the C-terminal proteolytic fragment of PD-L1. a MDA-MB-231, BT549, or MCF10A cells overexpressing PD-L1-M/F or GFP (control) were incubated for 24 h in the absence or presence of 10 μM chloroquine (CQ). Cell lysates were analyzed by Western blotting using an antibody specific for the cytoplasmic (CYT) domain of PD-L1 (E1L3N). The low molecular weight region of the blot (indicated by the box) is also shown after a longer exposure time. b MDA-MB-231 cell lysates from a were analyzed by anti-PD-L1 (E1L3N), anti-Myc, or anti-FLAG antibody. Short and long exposures of the low molecular weight region are shown. CTF, C-terminal fragment. c Parental MDA-MB-231, BT549, or MCF10A cells were incubated for 48 h in the presence or absence of 10 ng/ml IFN-γ, and then for additional 24 h with 10 μM CQ in the presence or absence of IFN-γ, as indicated. Cell lysates were analyzed by Western blotting using the E1L3N anti-PD-L1 antibody. The low molecular weight region of the blot (indicated by the box) is also shown after a longer exposure time. GAPDH is a gel loading control. Results are representative of three (a) or two (b, c) independent experiments
We next used three other drugs to inhibit lysosomal function: ammonium chloride, a weak base, monensin, a Na+/H+ ionophore, or bafilomycin A1, an inhibitor of vacuolar-type H+-ATPase (V-ATPase). As in the case of chloroquine, these treatments increase lysosomal pH and impair lysosomal protein degradation. In MDA-MB-231-PD-L1-M/F or MCF10A-PD-L1-M/F cells, an 18-h treatment with 10 mM NH4Cl or a 4-h treatment with 20 μM monensin or 0.1 μM bafilomycin A1 increased the abundance of the 18-kDa C-terminal PD-L1 fragment (Supplementary Figs. 1–3). In BT549-PD-L1-M/F cells, these treatments did not have a major impact on the C-terminal PD-L1 fragment (Supplementary Figs. 1–3), corroborating the results obtained with chloroquine.
Finally, we asked whether a similar C-terminal fragment can be detected after the cleavage of endogenous PD-L1. Parental MDA-MB-231, BT549, or MCF10A cells were incubated for 48 h with IFN-γ to boost PD-L1 expression, and for an additional 24 h with chloroquine to stabilize the putative C-terminal cleavage product. As shown in Fig. 4c, a weak 14-kDa band corresponding to the C-terminal PD-L1 fragment was detected in the lysates of IFN-γ- and chloroquine-treated MDA-MB-231 and MCF10A cells. The difference in the molecular weight of this band and the C-terminal PD-L1-M/F fragment, 14 kDa versus 18 kDa, could at least partially be due to the presence of additional 53 residues in the PD-L1-M/F construct (predicted molecular weight of ~ 5.9 kDa, see “Materials and methods”). Similar to BT549-PD-L1-M/F cells, in parental BT549 cells chloroquine did not have a major effect on the stability of the C-terminal PD-L1 fragment.
To determine which enzyme(s) mediate the cleavage of PD-L1, we first incubated cells with inhibitors of different classes of proteases. It was shown previously that high concentrations of ilomastat (GM6001), a broad-spectrum hydroxamate-based metalloprotease inhibitor, partially suppressed the release of sPD-L1 in mouse L-929 cells (connective tissue) [28]. Inhibitors of other classes of proteases have not been tested. Here, MDA-MB-231, BT549, or MCF10A cells overexpressing PD-L1-M/F were treated for 2 days with the following inhibitors: 5 μM batimastat, another broad-spectrum hydroxamate-based metalloprotease inhibitor, 0.1 mM AEBSF or 10 μg/ml aprotinin, two serine protease inhibitors, 10 μg/ml pepstatin, an aspartic protease inhibitor, or 10 μg/ml leupeptin, a cysteine protease inhibitor. In all three cell lines, the release of sPD-L1 was significantly decreased only by batimastat (Fig. 5a–c). These results indicated that PD-L1 cleavage was selectively mediated by the member(s) of the metalloprotease family of enzymes.
Fig. 5.

Cleavage of transmembrane PD-L1 is mediated by ADAM10 and ADAM17. a MDA-MB-231, b BT549, or c MCF10A cells overexpressing PD-L1-M/F were incubated for 48 h in complete media containing 5 μM batimastat (B), 0.1 mM AEBSF (AE), 10 μg/ml aprotinin (Ap), 10 μg/ml pepstatin (P), 10 μg/ml leupeptin (L), or control media without inhibitors (C). d MDA-MB-231, e BT549, or f MCF10A cells overexpressing PD-L1-M/F were incubated for 1 day in serum-free media containing 30 μM GI254023X, 30 μM TAPI-2, 1 μM MMP-9 inhibitor I, 50 μM CL-82198, or control media without inhibitors. In a–f, concentrations of sPD-L1 in the media were measured by ELISA. Values are means ± SD from three replicate measurements, normalized to equal protein concentration in all cellular lysates. P values were determined by unpaired two-tailed Student’s t test. Results are representative of three (a, d–f) or two (b, c) independent experiments. g, h MDA-MB-231-PD-L1-M/F cells were incubated for 1 day in serum-free media containing indicated concentrations of GI254023X (g) or TAPI-2 (h), followed by measuring sPD-L1 in the media by ELISA in triplicates. sPD-L1 concentrations in the absence of inhibitors were set at 100%. IC50 values for each inhibitor were determined after a non-linear regression curve fitting (four-parameter log(inhibitor) vs response) using GraphPad 6.0
Previous reports implicated two matrix metalloproteases, MMP-9 and MMP-13, in the cleavage of PD-L1 in human foreskin fibroblasts and in head and neck cancer cells [19, 21]. Our analysis of the results of global gene expression profiling of breast cancer cells by high-throughput sequencing [24, 25] indicated, however, that the expression of MMP-9 and MMP-13 in MDA-MB-231, BT549, and MCF10A cells is very low (GEO:GSE73526 and GSE48213; https://www.ncbi.nlm.nih.gov/geo/). Since batimastat also inhibits ADAM metalloproteases, in addition to MMPs, and since ADAM10 and ADAM17 are highly expressed in MDA-MB-231, BT549, and MCF10A cells, as well as in SUM159 and SUM149 cells [24, 25], we hypothesized that these two ADAMs may be involved in PD-L1 cleavage in our system. Indeed, treatment of MDA-MB-231, BT549, or MCF10A cells overexpressing PD-L1-M/F with 30 μM GI254023X, a known selective inhibitor of ADAM10 [29], or 30 μM TAPI-2, a selective inhibitor of ADAM17 [30], significantly decreased the release of sPD-L1 to the media (Fig. 5d–f). In contrast, incubation of cells with 1 μM MMP-9 inhibitor I or 50 μM CL-82198, a specific MMP-13 inhibitor, did not block the release of sPD-L1 (Fig. 5d–f). Furthermore, the measured IC50 values for GI254023X (Fig. 5g) and TAPI-2 (Fig. 5h) in MDA-MB-231-PD-L1-M/F cells were 0.20 μM and 0.16 μM, respectively. These submicromolar IC50 values were similar to the IC50 values for GI254023X or TAPI-2 in ADAM10- or ADAM17-mediated cleavage reactions of model substrates in vitro [29, 30], suggesting that these ADAMs might be involved in the cleavage of PD-L1 in intact cells studied here.
To validate our results of pharmacological inhibition of ADAM10 and ADAM17, the two ADAMs were down-regulated using siRNAs. Transfection of two different siRNAs targeting ADAM10 into MDA-MB-231-PD-L1-M/F cells resulted in ~ 70% down-regulation of ADAM10 expression and ~ 60% inhibition of sPD-L1 release to the media (Fig. 6a, b). Transfection of two different siRNAs targeting ADAM17 led to almost complete loss of ADAM17 expression and 40–60% decrease of sPD-L1 in the media (Fig. 6a, b). Combination of all four siRNAs targeting both ADAM10 and ADAM17 resulted in ~ 80% inhibition of the release of sPD-L1 (Fig. 6b). Collectively, these results confirmed that the cleavage of transmembrane PD-L1-M/F in MDA-MB-231 cells was mediated, in a large part, by ADAM10 and ADAM17.
Fig. 6.

The effect of ADAM10 and ADAM17 knockdown on the cleavage of PD-L1. a, b MDA-MB-231-PD-L1-M/F cells were transfected with two different siRNAs targeting ADAM10 or ADAM17, a combination of all four siRNAs, or with control siRNA. Three days after transfection, ADAM10 and ADAM17 expression was analyzed by Western blotting (a), and concentrations of sPD-L1 in the media were measured by ELISA (b). Arrows, the nascent full-length ADAMs; arrowheads, the processed forms; GAPDH is a gel loading control. In b, values are means ± SD from three replicate measurements, normalized to equal protein concentration in all cellular lysates. P values were determined by one-way ANOVA, followed by Dunnett’s multiple comparisons test. c MDA-MB-231-PD-L1-M/F cells were co-transfected with siRNA#1 and #2 targeting ADAM10 or with control siRNA. Two days after transfection, cells were incubated for 45 min in serum-free medium containing 0, 1 μM, or 2.5 μM ionomycin (IO), an activator of ADAM10, followed by ELISA for sPD-L1. d MDA-MB-231-PD-L1-M/F cells were co-transfected with siRNA#1 and #2 targeting ADAM17 or with control siRNA. Two days after transfection, cells were incubated for 2 h in complete medium containing 0, 25 ng/ml, or 50 ng/ml PMA, an activator of ADAM17, followed by measuring sPD-L1 by ELISA. Values are means ± SD from three replicate measurements. In c and d, P values were determined by unpaired two-tailed Student’s t test. Results are representative of three (b) or two (c, d) independent experiments. e Predicted location of the cleavage site in PD-L1. The structure of human PD-L1 (aa 19-232, PDB ID: 3FN3) is shown. The Ig-like V-type domain is required for the interaction with PD-1. The membrane-proximal unstructured region between V225 and H240 most likely contains the metalloprotease cleavage site
ADAM10 and ADAM17 expression is not restricted to breast cancer cells. To determine whether PD-L1 may be cleaved by ADAM10 and/or ADAM17 in other cancer cell types, we overexpressed PD-L1-M/F in DU-145 prostate cancer cells and A549 lung cancer cells (Supplementary Fig. 4a). In both cell lines, we detected increased levels of sPD-L1 in the conditioned media of PD-L1-M/F-expressing cells versus GFP-expressing cells (Supplementary Fig. 4b). ADAM10 or ADAM17 knockdown using siRNAs significantly decreased the concentration of sPD-L1 in the media (Supplementary Fig. 4c, d), indicating that ADAM10- and ADAM17-mediated cleavage of PD-L1-M/F is not restricted to breast cancer cells.
Notably, ADAM10 activity in cell-based assays can be rapidly up-regulated by the calcium ionophore ionomycin, whereas ADAM17 activity can be augmented after stimulation of protein kinase C with phorbol esters [31, 32]. Here, a 45-min incubation of MDA-MB-231-PD-L1-M/F cells with 1 μM or 2.5 μM ionomycin dramatically increased the release of sPD-L1. This effect was dose dependent and was diminished in cells transfected with siRNAs targeting ADAM10 (Fig. 6c). Also, a 2-h treatment of cells with 25 ng/ml or 50 ng/ml PMA augmented the concentration of sPD-L1 in the media in a dose-dependent manner, and this effect was significantly reduced by transfection with siRNAs targeting ADAM17 (Fig. 6d). In sum, these results confirmed the role of ADAM10 and ADAM17 in PD-L1 cleavage and demonstrated that ADAM10- or ADAM17-mediated release of sPD-L1 from the cell surface can be rapidly and potently up-regulated by ionomycin-induced increase of intracellular concentrations of calcium ions or by stimulation of cells with PMA.
Discussion
Our studies provide evidence that human transmembrane PD-L1 is subject to proteolytic cleavage by ADAM10 and ADAM17 in breast cancer cells and in the normal epithelial cell line MCF10A. Importantly, selective up-regulation of ADAM10 activity upon treatment of cells with a calcium ionophore or stimulation of ADAM17 activity via phorbol ester-mediated activation of protein kinase C readily increases the release of sPD-L1. In the tumor microenvironment in vivo, intracellular calcium concentrations or protein kinase C activity may rise in response to a number of extracellular signals. Thus, although PD-L1 cleavage in unstimulated cells in vitro appears low and does not exceed 10% of the total cellular PD-L1, the cleavage in vivo may be considerably augmented by physiological stimuli in the tumor microenvironment.
Inhibition of sPD-L1 release in our experiments by chemical inhibitors or siRNAs targeting ADAM10 and ADAM17 did not exceed ~ 80%. This could be partially due to incomplete inhibition of ADAM activity/expression, in particular, incomplete elimination of ADAM10 by siRNAs (see Fig. 6a). However, we cannot rule out that other mechanisms also contributed to the release of sPD-L1, such as secretion of exosomal PD-L1 or cleavage by other proteases. When cells were incubated in the presence of serum, sedimentation of exosomes did not have a major effect on the measured levels of sPD-L1 in the media. However, after incubation of cells in serum-free media, some full-length PD-L1, in addition to the ~ 37-kDa fragment, was detected in the conditioned media (see Fig. 3), suggesting that some exosomal PD-L1 might have contributed to the pool of PD-L1 in the media, at least under serum-free conditions.
Previous reports linked MMP-9 and MMP-13 to the cleavage of PD-L1 in fibroblasts and in head and neck carcinoma cells [19, 21]. However, these two MMPs are not highly expressed in cells tested here, and thus are unlikely to mediate PD-L1 cleavage. Furthermore, MMP-9 and MMP-13 are secreted enzymes, and their substrates typically include components of the extracellular matrix, rather than transmembrane proteins [33]. ADAM10 and ADAM17 are, in contrast, membrane-bound proteases whose known substrates comprise transmembrane receptors, growth factor or cytokine precursors, and adhesion proteins [34, 35]. Notably, several ADAM10 and/or ADAM17 substrates contain two or more Ig-like domains and a membrane-proximal stalk region, for example, low-affinity IgγFc region receptor III (FcγRIII) [36], lymphocyte activation gene 3 protein (LAG-3) [37], or nectin-4 [38]. PD-L1, with two Ig domains, IgV and IgC, and a short stalk region connecting IgC to the transmembrane domain, is another example of this class of substrates.
ADAM10 or ADAM17-mediated cleavage of FcγRIII, LAG-3, and nectin-4 occurs in their membrane-proximal stalks. Based on our results, we propose that the metalloprotease cleavage site in PD-L1 is also located in the stalk region, between V225 and H240 (Fig. 6e). Cleavage of PD-L1-M/F within this region would generate two products of ~ 24 kDa and ~ 13 kDa (the N-terminal and C-terminal fragments, respectively). Since the extracellular domain of PD-L1 is glycosylated and ~ 12- to 15-kDa larger than the non-glycosylated form [10], the actual size of the N-terminal cleavage product would roughly correspond to the 37-kDa sPD-L1 identified here. The predicted size of the C-terminal cleavage product is smaller than the 18-kDa PD-L1-M/F fragment described here. This discrepancy may be due to abnormal mobility of this fragment in SDS-PAGE or it may be caused by PD-L1 mono-ubiquitination [9]. Since ADAMs do not recognize specific consensus sequences and their cleavage sites are poorly defined, additional studies are needed to determine the exact position of ADAM-mediated cleavage within PD-L1.
The C-terminal PD-L1 cleavage product appeared unstable in MDA-MB-231 and MCF10A cells. This result was somewhat unexpected in the context of recent studies, which identified CMTM6 and CMTM4, two MARVEL domain-containing transmembrane proteins, as critical regulators of PD-L1 stability [12, 13]. Biochemical characterization of CMTM6:PD-L1 complexes using PD-L1/PD-L2 chimeras suggested that the transmembrane domain of PD-L1 is required for the interaction with CMTM6 [13]. The C-terminal PD-L1 fragment observed here comprised the transmembrane domain of PD-L1 but, nevertheless, was significantly less stable than full-length PD-L1 in MDA-MB-231 and MCF10A cells. These results suggest that the presence of the transmembrane domain of PD-L1 is not sufficient for the interaction with CMTM6. This observation may have significant implications when designing methods to disrupt the interaction between PD-L1 and CMTM6, to destabilize PD-L1, and to overcome the immune evasion by tumor cells.
Interestingly, in BT549 cells, the C-terminal PD-L1 fragment appeared more stable and was detectable in the absence of lysosome inhibition (see Fig. 4 and Supplementary Figs. 1–3). According to the Catalogue Of Somatic Mutations In Cancer (COSMIC) database (https://cancer.sanger.ac.uk/cell_lines), BT549 cells carry a homozygous R438C mutation in the VPS18 gene (ENSG00000104142), and this mutation is predicted to be protein-damaging according to the SIFT and PolyPhen prediction tools available at the Ensembl genome browser (http://uswest.ensembl.org). Vacuolar protein sorting-associated protein 18 (VPS18) is a component of the mammalian homotypic fusion and vacuole protein sorting (HOPS) complex required for fusion of endosomes with lysosomes [39]. Thus, it is possible that increased stability of the C-terminal PD-L1 cleavage product generated by ADAM10/17 is a consequence of its impaired delivery to lysosomes, but this hypothesis needs further testing.
An important question remains: what are the biological consequences of PD-L1 cleavage? Specifically, in the context of breast tumors in vivo, does the cleavage weaken or improve anti-tumor immunity? While a soluble extracellular domain of PD-L1 was reported to inhibit T cell responses in some circumstances [20, 40], it is generally believed that a soluble monomeric PD-L1 does not deliver a strong negative regulatory signal through PD-1. This is underscored by a recent discovery of a new secreted PD-L1 variant that has an ability to form homodimers through a Cys residue in its unique C-terminal tail and, unlike a monomeric soluble PD-L1, to inhibit lymphocyte function [17].
Instead, the cleavage of transmembrane PD-L1 may increase anti-tumor immunity, for example by reducing the amount of PD-L1 on tumor cells [19] or by providing sPD-L1 that competes with membrane-bound PD-L1 for PD-1. Furthermore, sPD-L1 may exert an immuno-stimulatory effect through its binding to CD80 (B7-1). While it has been previously established that, in addition to PD-1, PD-L1 also interacts with CD80 [41], recent studies have further demonstrated that PD-L1 and CD80 bind only in cis, on the same cell surface, in a parallel orientation [42–44]. CD80 homodimer, when not bound to PD-L1, interacts in trans with a co-inhibitory receptor CTLA4 or with a co-stimulatory receptor CD28 [45]. Remarkably, PD-L1:CD80 heterotrimer is unable to bind to PD-1 or CTLA4 on the neighboring cell, but binds to and activates CD28 equally well as free CD80 [44]. Thus, it is conceivable that sPD-L1 produced in one location within the tumor, with high local expression of transmembrane PD-L1 and high expression/activity of ADAM10 or ADAM17, diffuses to a different location where it binds to CD80 and switches it from a CTLA4 ligand to a CD28 ligand, effectively promoting T cell activation. Importantly, such a function of sPD-L1 would not be mimicked by exosomal PD-L1, because the exosomal membrane would sterically prevent PD-L1 and CD80 to interact in a parallel orientation.
Finally, within the tumor microenvironment, ADAM10 and ADAM17 are expressed not only in cancer cells but also in tumor-infiltrating immune cells [34]. While we have not tested whether ADAM10 or ADAM17 might cleave PD-L1 in immune cells, the ubiquitous expression of these two ADAMs and regulation of their activities by a number of physiological signals clearly position them as new modulators of the PD-1/PD-L1 immune checkpoint pathway.
Electronic supplementary material
Below is the link to the electronic supplementary material.
Abbreviations
- ADAM
A disintegrin and metalloprotease
- AEBSF
4-(2-Aminoethyl)benzenesulfonyl fluoride hydrochloride
- BCA
Bicinchoninic acid
- FBS
Fetal bovine serum
- GAPDH
Glyceraldehyde 3-phosphate dehydrogenase
- GEO
Gene expression omnibus
- HOPS
Homotypic fusion and vacuole protein sorting
- HS
Horse serum
- FcγRIII
Low-affinity IgγFc region receptor III
- LAG-3
Lymphocyte activation gene 3 protein
- M/F
Myc/FLAG tag
- MMP
Matrix metalloprotease
- PD-1
Programmed cell death protein 1
- PD-L1
Programmed death ligand 1
- PMA
Phorbol 12-myristate 13-acetate
- siRNA
Small interfering RNA
- sPD-L1
Soluble programmed death ligand 1
- COSMIC
The catalogue of somatic mutations in cancer database
- TAPI-2
Tumor necrosis factor protease inhibitor 2
- TNBC
Triple-negative breast cancer
- VPS18
Vacuolar protein sorting-associated protein 18
Author contributions
YR and RW performed the experiments and contributed to writing the manuscript. AZ conceived the study, performed the experiments, and wrote the manuscript. All authors read and approved the final manuscript.
Funding
This work was supported by the National Institutes of Health (US) Grant R01CA172222 and by funds from Kansas State University Johnson Cancer Research Center to Anna Zolkiewska.
Compliance with ethical standards
Conflict of interest
The authors declare that they have no conflict of interest.
Ethical approval and ethical standards
The studies described in this paper were performed using commercially available cell lines. This article does not contain any experiments involving patients or experimental animals. Therefore, no IACUC approval and no informed consent from the donors were required.
Cell line authentication
MDA-MB-231, BT549, MCF10A, A549, and DU-145 cell lines were from the American Type Culture Collection (Manassas, VA). SUM149 and SUM159 cell lines were obtained from Asterand (Detroit, MI). These cell lines were authenticated by their suppliers using short tandem repeat analysis and have been passaged for fewer than 6 months after culture initiation from an early passage number.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- 1.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–1086. doi: 10.1158/2159-8290.CD-18-0367. [DOI] [PubMed] [Google Scholar]
- 2.Emens LA. Breast cancer immunotherapy: facts and hopes. Clin Cancer Res. 2018;24:511–520. doi: 10.1158/1078-0432.CCR-16-3001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Sobral-Leite M, Van de Vijver K, Michaut M, van der Linden R, Hooijer GKJ, Horlings HM, Severson TM, Mulligan AM, Weerasooriya N, Sanders J, Glas AM, Wehkamp D, Mittempergher L, Kersten K, Cimino-Mathews A, Peters D, Hooijberg E, Broeks A, van de Vijver MJ, Bernards R, Andrulis IL, Kok M, de Visser KE, Schmidt MK. Assessment of PD-L1 expression across breast cancer molecular subtypes, in relation to mutation rate, BRCA1-like status, tumor-infiltrating immune cells and survival. Oncoimmunology. 2018;7:e1509820. doi: 10.1080/2162402X.2018.1509820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Mittendorf EA, Philips AV, Meric-Bernstam F, Qiao N, Wu Y, Harrington S, Su X, Wang Y, Gonzalez-Angulo AM, Akcakanat A, Chawla A, Curran M, Hwu P, Sharma P, Litton JK, Molldrem JJ, Alatrash G. PD-L1 expression in triple-negative breast cancer. Cancer Immunol Res. 2014;2:361–370. doi: 10.1158/2326-6066.CIR-13-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adams S, Loi S, Toppmeyer D, Cescon DW, De Laurentiis M, Nanda R, Winer EP, Mukai H, Tamura K, Armstrong A, Liu MC, Iwata H, Ryvo L, Wimberger P, Rugo HS, Tan AR, Jia L, Ding Y, Karantza V, Schmid P. Pembrolizumab monotherapy for previously untreated, PD-L1-positive, metastatic triple-negative breast cancer: cohort B of the phase 2 KEYNOTE-086 study. Ann Oncol. 2018;30:405–411. doi: 10.1093/annonc/mdy518. [DOI] [PubMed] [Google Scholar]
- 6.Dirix LY, Takacs I, Jerusalem G, Nikolinakos P, Arkenau HT, Forero-Torres A, Boccia R, Lippman ME, Somer R, Smakal M, Emens LA, Hrinczenko B, Edenfield W, Gurtler J, von Heydebreck A, Grote HJ, Chin K, Hamilton EP. Avelumab, an anti-PD-L1 antibody, in patients with locally advanced or metastatic breast cancer: a phase 1b JAVELIN Solid Tumor study. Breast Cancer Res Treat. 2018;167:671–686. doi: 10.1007/s10549-017-4537-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, Dieras V, Hegg R, Im SA, Shaw Wright G, Henschel V, Molinero L, Chui SY, Funke R, Husain A, Winer EP, Loi S, Emens LA, Investigators IMT. Atezolizumab and Nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med. 2018;379:2108–2121. doi: 10.1056/NEJMoa1809615. [DOI] [PubMed] [Google Scholar]
- 8.Zerdes I, Matikas A, Bergh J, Rassidakis GZ, Foukakis T. Genetic, transcriptional and post-translational regulation of the programmed death protein ligand 1 in cancer: biology and clinical correlations. Oncogene. 2018;37:4639–4661. doi: 10.1038/s41388-018-0303-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Horita H, Law A, Hong S, Middleton K. Identifying regulatory posttranslational modifications of PD-L1: a focus on monoubiquitinaton. Neoplasia. 2017;19:346–353. doi: 10.1016/j.neo.2017.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Li CW, Lim SO, Xia W, Lee HH, Chan LC, Kuo CW, Khoo KH, Chang SS, Cha JH, Kim T, Hsu JL, Wu Y, Hsu JM, Yamaguchi H, Ding Q, Wang Y, Yao J, Lee CC, Wu HJ, Sahin AA, Allison JP, Yu D, Hortobagyi GN, Hung MC. Glycosylation and stabilization of programmed death ligand-1 suppresses T-cell activity. Nat Commun. 2016;7:12632. doi: 10.1038/ncomms12632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yang Y, Hsu JM, Sun L, Chan LC, Li CW, Hsu JL, Wei Y, Xia W, Hou J, Qiu Y, Hung MC. Palmitoylation stabilizes PD-L1 to promote breast tumor growth. Cell Res. 2019;29:83–86. doi: 10.1038/s41422-018-0124-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Burr ML, Sparbier CE, Chan YC, Williamson JC, Woods K, Beavis PA, Lam EYN, Henderson MA, Bell CC, Stolzenburg S, Gilan O, Bloor S, Noori T, Morgens DW, Bassik MC, Neeson PJ, Behren A, Darcy PK, Dawson SJ, Voskoboinik I, Trapani JA, Cebon J, Lehner PJ, Dawson MA. CMTM6 maintains the expression of PD-L1 and regulates anti-tumour immunity. Nature. 2017;549:101–105. doi: 10.1038/nature23643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mezzadra R, Sun C, Jae LT, Gomez-Eerland R, de Vries E, Wu W, Logtenberg MEW, Slagter M, Rozeman EA, Hofland I, Broeks A, Horlings HM, Wessels LFA, Blank CU, Xiao Y, Heck AJR, Borst J, Brummelkamp TR, Schumacher TNM. Identification of CMTM6 and CMTM4 as PD-L1 protein regulators. Nature. 2017;549:106–110. doi: 10.1038/nature23669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Whiteside TL. Exosomes and tumor-mediated immune suppression. J Clin Investig. 2016;126:1216–1223. doi: 10.1172/JCI81136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Poggio M, Hu T, Pai CC, Chu B, Belair CD, Chang A, Montabana E, Lang UE, Fu Q, Fong L, Blelloch R. Suppression of exosomal PD-L1 induces systemic anti-tumor immunity and memory. Cell. 2019;177:414–427. doi: 10.1016/j.cell.2019.02.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhou J, Mahoney KM, Giobbie-Hurder A, Zhao F, Lee S, Liao X, Rodig S, Li J, Wu X, Butterfield LH, Piesche M, Manos MP, Eastman LM, Dranoff G, Freeman GJ, Hodi FS. Soluble PD-L1 as a biomarker in malignant melanoma treated with checkpoint blockade. Cancer Immunol Res. 2017;5:480–492. doi: 10.1158/2326-6066.CIR-16-0329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mahoney KM, Shukla SA, Patsoukis N, Chaudhri A, Browne EP, Arazi A, Eisenhaure TM, Pendergraft WF, 3rd, Hua P, Pham HC, Bu X, Zhu B, Hacohen N, Fritsch EF, Boussiotis VA, Wu CJ, Freeman GJ. A secreted PD-L1 splice variant that covalently dimerizes and mediates immunosuppression. Cancer Immunol Immunother. 2018;68:421–432. doi: 10.1007/s00262-018-2282-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Frigola X, Inman BA, Lohse CM, Krco CJ, Cheville JC, Thompson RH, Leibovich B, Blute ML, Dong H, Kwon ED. Identification of a soluble form of B7-H1 that retains immunosuppressive activity and is associated with aggressive renal cell carcinoma. Clin Cancer Res. 2011;17:1915–1923. doi: 10.1158/1078-0432.CCR-10-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dezutter-Dambuyant C, Durand I, Alberti L, Bendriss-Vermare N, Valladeau-Guilemond J, Duc A, Magron A, Morel AP, Sisirak V, Rodriguez C, Cox D, Olive D, Caux C. A novel regulation of PD-1 ligands on mesenchymal stromal cells through MMP-mediated proteolytic cleavage. Oncoimmunology. 2016;5:e1091146. doi: 10.1080/2162402X.2015.1091146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Davies LC, Heldring N, Kadri N, Le Blanc K. Mesenchymal stromal cell secretion of programmed death-1 ligands regulates T cell mediated immunosuppression. Stem Cells. 2017;35:766–776. doi: 10.1002/stem.2509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hira-Miyazawa M, Nakamura H, Hirai M, Kobayashi Y, Kitahara H, Bou-Gharios G, Kawashiri S. Regulation of programmed-death ligand in the human head and neck squamous cell carcinoma microenvironment is mediated through matrix metalloproteinase-mediated proteolytic cleavage. Int J Oncol. 2018;52:379–388. doi: 10.3892/ijo.2017.4221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weber S, Saftig P. Ectodomain shedding and ADAMs in development. Development. 2012;139:3693–3709. doi: 10.1242/dev.076398. [DOI] [PubMed] [Google Scholar]
- 23.Duhachek-Muggy S, Qi Y, Wise R, Alyahya L, Li H, Hodge J, Zolkiewska A. Metalloprotease-disintegrin ADAM12 actively promotes the stem cell-like phenotype in claudin-low breast cancer. Mol Cancer. 2017;16:32. doi: 10.1186/s12943-017-0599-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Marcotte R, Sayad A, Brown KR, Sanchez-Garcia F, Reimand J, Haider M, Virtanen C, Bradner JE, Bader GD, Mills GB, Pe’er D, Moffat J, Neel BG. Functional genomic landscape of human breast cancer drivers, vulnerabilities, and resistance. Cell. 2016;164:293–309. doi: 10.1016/j.cell.2015.11.062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Daemen A, Griffith OL, Heiser LM, Wang NJ, Enache OM, Sanborn Z, Pepin F, Durinck S, Korkola JE, Griffith M, Hur JS, Huh N, Chung J, Cope L, Fackler MJ, Umbricht C, Sukumar S, Seth P, Sukhatme VP, Jakkula LR, Lu Y, Mills GB, Cho RJ, Collisson EA, van’t Veer LJ, Spellman PT, Gray JW. Modeling precision treatment of breast cancer. Genome Biol. 2013;14:R110. doi: 10.1186/gb-2013-14-10-r110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Garcia-Diaz A, Shin DS, Moreno BH, Saco J, Escuin-Ordinas H, Rodriguez GA, Zaretsky JM, Sun L, Hugo W, Wang X, Parisi G, Saus CP, Torrejon DY, Graeber TG, Comin-Anduix B, Hu-Lieskovan S, Damoiseaux R, Lo RS, Ribas A. Interferon receptor signaling pathways regulating PD-L1 and PD-L2 expression. Cell Rep. 2017;19:1189–1201. doi: 10.1016/j.celrep.2017.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xu R, Simpson RJ, Greening DW. A protocol for isolation and proteomic characterization of distinct extracellular vesicle subtypes by sequential centrifugal ultrafiltration. Methods Mol Biol. 2017;1545:91–116. doi: 10.1007/978-1-4939-6728-5_7. [DOI] [PubMed] [Google Scholar]
- 28.Chen Y, Wang Q, Shi B, Xu P, Hu Z, Bai L, Zhang X. Development of a sandwich ELISA for evaluating soluble PD-L1 (CD274) in human sera of different ages as well as supernatants of PD-L1 + cell lines. Cytokine. 2011;56:231–238. doi: 10.1016/j.cyto.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 29.Ludwig A, Hundhausen C, Lambert MH, Broadway N, Andrews RC, Bickett DM, Leesnitzer MA, Becherer JD. Metalloproteinase inhibitors for the disintegrin-like metalloproteinases ADAM10 and ADAM17 that differentially block constitutive and phorbol ester-inducible shedding of cell surface molecules. Comb Chem High Throughput Screen. 2005;8:161–171. doi: 10.2174/1386207053258488. [DOI] [PubMed] [Google Scholar]
- 30.Moss ML, Rasmussen FH. Fluorescent substrates for the proteinases ADAM17, ADAM10, ADAM8, and ADAM12 useful for high-throughput inhibitor screening. Anal Biochem. 2007;366:144–148. doi: 10.1016/j.ab.2007.04.043. [DOI] [PubMed] [Google Scholar]
- 31.Horiuchi K, Le Gall S, Schulte M, Yamaguchi T, Reiss K, Murphy G, Toyama Y, Hartmann D, Saftig P, Blobel CP. Substrate selectivity of epidermal growth factor-receptor ligand sheddases and their regulation by phorbol esters and calcium influx. Mol Biol Cell. 2007;18:176–188. doi: 10.1091/mbc.E06-01-0014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Le Gall SM, Bobe P, Reiss K, Horiuchi K, Niu XD, Lundell D, Gibb DR, Conrad D, Saftig P, Blobel CP. ADAMs 10 and 17 represent differentially regulated components of a general shedding machinery for membrane proteins such as transforming growth factor α, L-selectin, and tumor necrosis factor α. Mol Biol Cell. 2009;20:1785–1794. doi: 10.1091/mbc.E08-11-1135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Vandooren J, Van den Steen PE, Opdenakker G. Biochemistry and molecular biology of gelatinase B or matrix metalloproteinase-9 (MMP-9): the next decade. Crit Rev Biochem Mol Biol. 2013;48:222–272. doi: 10.3109/10409238.2013.770819. [DOI] [PubMed] [Google Scholar]
- 34.Lambrecht BN, Vanderkerken M, Hammad H. The emerging role of ADAM metalloproteinases in immunity. Nat Rev Immunol. 2018;18:745–758. doi: 10.1038/s41577-018-0068-5. [DOI] [PubMed] [Google Scholar]
- 35.Zunke F, Rose-John S. The shedding protease ADAM17: physiology and pathophysiology. Biochim Biophys Acta Mol Cell Res. 2017;1864:2059–2070. doi: 10.1016/j.bbamcr.2017.07.001. [DOI] [PubMed] [Google Scholar]
- 36.Jing Y, Ni Z, Wu J, Higgins L, Markowski TW, Kaufman DS, Walcheck B. Identification of an ADAM17 cleavage region in human CD16 (FcγRIII) and the engineering of a non-cleavable version of the receptor in NK cells. PLoS One. 2015;10:e0121788. doi: 10.1371/journal.pone.0121788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Li N, Wang Y, Forbes K, Vignali KM, Heale BS, Saftig P, Hartmann D, Black RA, Rossi JJ, Blobel CP, Dempsey PJ, Workman CJ, Vignali DA. Metalloproteases regulate T-cell proliferation and effector function via LAG-3. EMBO J. 2007;26:494–504. doi: 10.1038/sj.emboj.7601520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Fabre-Lafay S, Garrido-Urbani S, Reymond N, Goncalves A, Dubreuil P, Lopez M. Nectin-4, a new serological breast cancer marker, is a substrate for tumor necrosis factor-α-converting enzyme (TACE)/ADAM-17. J Biol Chem. 2005;280:19543–19550. doi: 10.1074/jbc.M410943200. [DOI] [PubMed] [Google Scholar]
- 39.Wartosch L, Gunesdogan U, Graham SC, Luzio JP. Recruitment of VPS33A to HOPS by VPS16 is required for lysosome fusion with endosomes and autophagosomes. Traffic. 2015;16:727–742. doi: 10.1111/tra.12283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Liang Z, Tian Y, Cai W, Weng Z, Li Y, Zhang H, Bao Y, Li Y. High-affinity human PD-L1 variants attenuate the suppression of T cell activation. Oncotarget. 2017;8:88360–88375. doi: 10.18632/oncotarget.21729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Butte MJ, Keir ME, Phamduy TB, Sharpe AH, Freeman GJ. Programmed death-1 ligand 1 interacts specifically with the B7-1 costimulatory molecule to inhibit T cell responses. Immunity. 2007;27:111–122. doi: 10.1016/j.immuni.2007.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Chaudhri A, Xiao Y, Klee AN, Wang X, Zhu B, Freeman GJ. PD-L1 binds to B7-1 only in cis on the same cell surface. Cancer Immunol Res. 2018;6:921–929. doi: 10.1158/2326-6066.CIR-17-0316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sugiura D, Maruhashi T, Okazaki IM, Shimizu K, Maeda TK, Takemoto T, Okazaki T. Restriction of PD-1 function by cis-PD-L1/CD80 interactions is required for optimal T cell responses. Science. 2019;364:558–566. doi: 10.1126/science.aav7062. [DOI] [PubMed] [Google Scholar]
- 44.Zhao Y, Harrison DL, Song Y, Ji J, Huang J, Hui E. Antigen-presenting cell-intrinsic PD-1 neutralizes PD-L1 in cis to attenuate PD-1 signaling in T cells. Cell Rep. 2018;24(379–390):e376. doi: 10.1016/j.celrep.2018.06.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Chen L, Flies DB. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat Rev Immunol. 2013;13:227–242. doi: 10.1038/nri3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.