

Abstract
Inhibition of histone deacetylase 6 (HDAC6) has emerged as a promising therapeutic strategy for the treatment of cancer, chemotherapy-induced peripheral neuropathy, and neurodegenerative disease. The recent X-ray crystal structure determination of HDAC6 enables an understanding of structural features directing affinity and selectivity in the active site. Here, we present the X-ray crystal structures of five HDAC6-inhibitor complexes that illuminate key molecular features of inhibitor linker and capping groups that facilitate and differentiate binding to HDAC6. In particular, aromatic and heteroaromatic linkers nestle within an aromatic cleft defined by F583 and F643, and different aromatic linkers direct the capping group toward shallow pockets defined by the L1 loop, the L2 loop, or somewhere in between these pockets. These results expand our understanding of factors contributing to the selective inhibition of HDAC6, particularly regarding interactions that can be targeted in the region of the L2 pocket.
Graphical Abstract
Introduction
The metal ion-dependent histone deacetylases (HDACs) require a single Zn2+ ion (or possibly Fe2+) to catalyze the hydrolysis of acetyl-l-lysine side chains of protein substrates in vivo or peptide substrates in vitro to yield l-lysine and acetate coproducts.1–5 While these enzymes are named after their first reported substrates,1 thousands of non-histone proteins have since been identified that contain acetyl-l-lysine residues.6 Non-histone proteins, too, undergo strategic acetylation-deacetylation cycles in the regulation of diverse biological functions.
The metal-dependent HDACs consist of the class I isozymes, HDACs 1–3 and 8; the class IIa isozymes, HDACs 4, 5, 7, and 9; the class IIb isozymes, HDACs 6 and 10; and the sole class IV isozyme, HDAC11.7 Despite the common fold and catalytic mechanism shared by the catalytic domain of each HDAC isozyme,5,8–10 the HDACs have varying cellular localizations, expression patterns, client proteins, catalytic activities, and domain assemblies.11 For example, HDAC612,13 is the only isozyme containing two catalytic domains, designated CD1 and CD2.14,15 HDAC6 serves as a tubulin deacetylase in the cell cytosol and helps maintain microtubule acetylation levels that facilitate microtubule dynamics.16 The CD2 domain of HDAC6 is responsible for catalyzing the tubulin deacetylation reaction;17 inhibition of this function suppresses microtubule dynamics and can lead to cell cycle arrest and apoptosis.18 Selective inhibition of HDAC6 CD2 is being explored for cancer chemotherapy, mitigation of chemotherapy-induced peripheral neuropathy, and the treatment of neurodegenerative diseases.19–26
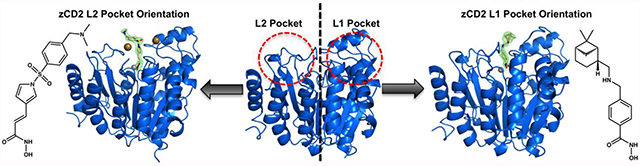
Recent X-ray crystal structure determinations of HDAC6 CD2 from Homo sapiens (human)27 and Danio rerio (zebrafish)27,28 (Figure 1a) have enabled the analysis of enzyme-inhibitor complexes and the characterization of molecular features responsible for affinity and selectivity in the HDAC6 CD2 active site.29–35 Typically, inhibitors consist of a metal-binding group, a linker, and a capping group that makes interactions in the outer active site cleft. Binding interactions of the capping group contribute significantly to affinity and selectivity for HDAC6 CD2. The active site cleft of HDAC6 is slightly wider than the active site clefts of class I HDACs, which confers selectivity for the binding of inhibitors with sterically bulky aromatic linkers; there is an associated entropic advantage that confers selectivity for HDAC6 binding as well.30 The capping groups of these inhibitors generally bind in a pocket defined by the L1 loop flanking the active site. Notably, sterically bulky inhibitors with bifurcated capping groups make interactions in the L1 pocket as well as another shallow pocket defined by the L2 loop and nearby residues G640–N645 (Figure 1b).33 To some degree, the molecular structure of the linker moiety may help steer the capping group toward one pocket or the other.
Figure 1.

(a) Ribbon-plot of HDAC6 CD2 from D. rerio (PDB 6PZS). The catalytic Zn2+ ion appears as a gray sphere. (b) Structure of the HDAC6 CD2 complex with the inhibitor RTS-V5, which contains a bifurcated capping group (PDB 6CW8); the molecular surface of the active site illustrates the L1 and L2 pockets. (c) Inhibitors of HDAC6 CD2 (IC50 values are listed below compound numbers): 1, (E)-3-(1-((4-((dimethylamino)methyl)phenyl)sulfonyl)-1H-pyrrol-3-yl)-N-hydroxyacrylamide (Resminostat); 2, 2-(4-bromophenyl)-N-hydroxyoxazole-4-carboxamide; 3, N-(4-(hydroxycarbamoyl)benzyl)-2-(trifluoromethyl)nicotinamide; 4, 4-(((6,6-dimethylbicyclo[3.1.1]heptan-2-yl)amino)methyl)-N-hydroxybenzamide; 5, benzyl ((S)-1-(((S)-1-((4-(hydroxycarbamoyl)-benzyl)amino)-1-oxopropan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate; 6, benzyl (2-((2-((4-(hydroxycarbamoyl)benzyl)amino)-2-oxoethyl)amino)-2-oxoethyl)carbamate; 7, benzyl ((S)-1-(((S)-1-((3-(hydroxycarbamoyl)benzyl)amino)-1-oxopropan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate.
To further explore the role of the linker in steering inhibitor capping groups toward these pockets, and also to determine how the chemical nature of the capping group (i.e., hydrophobicity or hydrophilicity) influences binding in these pockets, we now present five new X-ray crystal structures of zebrafish HDAC6 CD2 complexed with recently-designed inhibitors 1–5 (Figure 1c) determined at resolutions ranging 1.50–2.30 Å. To probe structure-affinity relationships, inhibitors 6 and 7 were also prepared and evaluated but not used for crystallographic studies. Zebrafish HDAC6 CD2 is used for these studies since it yields crystals of far greater quality than the human enzyme; moreover, the active site structures of zebrafish HDAC6 CD2 and human HDAC6 CD2 are essentially identical (with the exception of N530D and N645M substitutions, respectively, at the mouth of the active site).27 These structures provide new insight regarding inhibitor binding interactions in the L1 and L2 pockets, as well as the role of aromatic or heteroaromatic linkers, including benzyl, oxazole, and 3-vinylpyrrole moieties, as they position capping groups in the outer active site cleft.
Results and Discussion
Chemistry.
Newly synthesized compounds 3–7 were prepared as summarized in Schemes 1–5. Detailed synthetic procedures are described in the Experimental Section. The IC50 value of each inhibitor was determined as outlined in the Experimental Section and recorded in Figure 1.
The synthesis of N-(4-(hydroxycarbamoyl)benzyl)-2-(trifluoromethyl)nicotinamide (3) was accomplished as summarized in Scheme 1:
Scheme 1.

The synthesis of 4-(((6,6-dimethylbicyclo[3.1.1]heptan-2-yl)amino)methyl)-N-hydroxybenzamide (4) was achieved in two steps as summarized in Scheme 2:
Scheme 2.

The synthesis of benzyl ((S)-1-(((S)-1-((4-(hydroxycarbamoyl)benzyl)amino)-1-oxopropan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (5) was accomplished as summarized in Scheme 3:
Scheme 3.

The synthesis of benzyl (2-((2-((4-(hydroxycarbamoyl)benzyl)amino)-2-oxoethyl)amino)-2-oxoethyl)-carbamate (6) was accomplished as summarized in Scheme 4:
Scheme 4.

The synthesis of benzyl ((S)-1-(((S)-1-((3-(hydroxycarbamoyl)benzyl)amino)-1-oxopropan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (7) was accomplished using 14 as prepared in the synthesis of 5, outlined below in Scheme 5:
Scheme 5.

X-ray crystal structures of HDAC6 CD2–inhibitor complexes.
The 2.30 Å-resolution crystal structure of the HDAC6 CD2–1 complex contains 2 monomers in the asymmetric unit, and the inhibitor binding mode is identical in each monomer. The inhibitor hydroxamate group chelates the catalytic Zn2+ ion with bidentate coordination geometry (Figure 2). The Zn2+-bound hydroxamate C=O group also accepts a hydrogen bond from Y745, and the Zn2+-bound hydroxamate N–O– group accepts a hydrogen bond from H573. The hydroxamate NH group donates a hydrogen bond to H574.
Figure 2.

Polder omit map of the HDAC6 CD2-1 complex (PDB 6PZR; monomer A, contoured at 3.0 σ). Atoms are color-coded as follows: C = light blue (monomer A) or wheat (inhibitor), N = blue, O = red, S = yellow, Zn2+ =gray sphere, and I− = larger sand spheres. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. Locations of the L1 and L2 pockets are indicated.
The 3-vinylpyrrole linker is sandwiched between F583 and F643, making favorable offset aromatic π–π interactions. To achieve this interaction, the π systems of the hydroxamate and the 3-vinylpyrrole moiety of 1 are rotated on average 85° away from planarity and hence not in resonance with each other.
The inhibitor capping group is oriented toward the L2 pocket instead of the L1 pocket. To date, the only other inhibitor with a capping group that binds in the L2 pocket is one containing a bifurcated capping group, such that one substituent binds in the L1 pocket and the other substituent binds in the L2 pocket (Figure 1b).33 Polar residues in the L2 pocket include N530 and N645, as well as S531 near the interface of the L2 and L1 pockets, but these residues do not form hydrogen bonds with the inhibitor capping group.
Unexpectedly, two iodide ions from the crystallization buffer bind in the active site and flank the bound inhibitor (Figure 2). Each iodide ion is quite large (effective ionic radius = 2.20 Å) and makes a number of contacts with the protein and inhibitor. One iodide ion accepts a hydrogen bond from H614 (average I---N separation = 3.7 Å) and makes van der Waals contacts with F643, L712, and the phenylsulfone moiety in the capping group of the inhibitor. The other iodide ion accepts a hydrogen bond from H463 (average I---N separation = 3.7 Å) and makes van der Waals contacts with S531, I532, F533, and F583. These interactions are consistent with the intermolecular interactions of iodide ions classified in a survey of more than 350 iodide binding sites in refined protein structures – negatively charged iodide ions often bind in hydrophobic patches, adjacent to aromatic residues, while also capable of making hydrogen bond interactions.36
The 1.75 Å-resolution crystal structure of the HDAC6 CD2–2 complex contains 2 monomers in the asymmetric unit, and the inhibitor binding mode is identical in each monomer. The inhibitor hydroxamate group chelates the catalytic Zn2+ ion with bidentate coordination geometry in similar fashion to that observed for the binding of 1, making identical metal coordination and hydrogen bond interactions (Figure 3). Unique to 2 is the heterocyclic oxazole linker, which makes favorable offset aromatic π–π interactions with F583 and F643. Because of the geometry and orientation of the 5-membered ring of the oxazole moiety, the bromophenyl capping group is oriented toward solution and does not fully interact with either the L1 or the L2 pocket. Accordingly, a 5-membered heteroaromatic ring linker connected directly to the Zn2+-bound hydroxamate does not appear to be ideal for targeting capping group binding in these pockets.
Figure 3.

Polder omit map of the HDAC6 CD2–2 complex (PDB 6Q0Z; monomer A, contoured at 4.0 σ). Atoms are color-coded as follows: C = light blue (monomer A), dark gray (symmetry mate) or wheat (inhibitor), N = blue, O = red, Br = magenta, and Zn2+ = gray sphere. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. Locations of the L1 and L2 pockets are indicated.
The π systems of the hydroxamate moiety and the oxazole moiety of 2 are not coplanar, and hence not in resonance with each other: the corresponding average N-C-C-C dihedral angle is 50°. However, the oxazole ring and the bromophenyl rings are, on average, within 14° of planarity. Thus, these aromatic rings are in resonance with one another and bind as a nearly rigid unit. The bromine atom of the bromophenyl capping group is oriented toward solvent but does not contact symmetry mate residues T780 or N784.
The 1.50 Å-resolution crystal structure of the HDAC6 CD2–3 complex contains 2 monomers in the asymmetric unit. Unexpectedly, the inhibitor binding mode is different in monomers A and B. Hydroxamate-Zn2+ coordination is monodentate in monomer A: the hydroxamate N–O– group coordinates to Zn2+ and accepts a hydrogen bond from Y745, and the hydroxamate C=O group accepts a hydrogen bond from the Zn2+-bound water molecule, which in turn hydrogen bonds to H573 and H574 (Figure 4a). In monomer B, hydroxamate-Zn2+ coordination is bidentate, with the hydroxamate C=O and N–O– groups chelating the metal ion (Figure 4b).
Figure 4.

(a) Polder omit map of the HDAC6 CD2–3 complex (PDB 6PZO; monomer A, contoured at 3.5 σ). Atoms are color-coded as follows: C = light blue (monomer A), dark gray (symmetry mate) or wheat (inhibitor), N = blue, O = red, F = magenta, Zn2+ =gray sphere, and solvent = small red spheres. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. A red dashed line indicates a possible n→π* interaction between a C-F group and the amide carbonyl. (b) Polder omit map of the HDAC6 CD2–3 complex (PDB 6PZO; monomer B, contoured at 3.5 σ). Atoms are color-coded as follows: C = light blue (monomer B), dark gray (symmetry mate) or wheat (inhibitor), N = blue, O = red, F = magenta, Zn2+ = gray sphere, and solvent = small red spheres. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. Locations of the L1 and L2 pockets are indicated.
In both monomers, the benzyl linker is sandwiched between F583 and F643, making favorable offset aromatic π–π interactions. However, the π systems of the hydroxamate and the benzyl moieties of 3 are oriented differently in each monomer. In monomer A, they are within 8° of planarity. In monomer B, they are rotated 36° away from planarity. These conformational differences may arise from the different Zn2+ coordination modes of the inhibitor in each monomer.
Also different in each monomer is the orientation of the amide connecting the linker to the trifluoromethylpyridine capping group, although the capping group occupies essentially the same location in the L1 pocket. There, the nitrogen of the pyridine ring makes a water-mediated hydrogen bond with R660 of a symmetry mate. The fluorine atoms of the trifluoromethyl moiety make no hydrogen bond interactions with protein residues, but they do make van der Waals interactions with H463, P464, and L712 in the L1 pocket. Despite the ability of electronegative aromatic ring substituents to influence intermolecular interactions,37,38 the trifluoromethylpyridine ring of 3 does not appear to make any interactions that would benefit from such electrostatic modulation.
Interestingly, one of the fluorine atoms of the trifluoromethyl moiety appears to make an n→π* interaction with the amide carbonyl group of the inhibitor. The F---C distance is 2.7 Å and the F---C=O angle is 86° in monomer A; the corresponding values are 2.8 Å and 118° in monomer B. While this interaction is likely to be weak, it may contribute to the orientation of the trifluoromethylpyridine capping group.
The 1.79 Å-resolution crystal structure of the HDAC6 CD2–4 complex contains one monomer in the asymmetric unit. Unique to the structure of this inhibitor is the monoterpene capping group, a pinene moiety. This is the first crystal structure of an HDAC–inhibitor complex containing a terpene derivative. The structural and stereochemical diversity of terpenoid natural products (which currently number more than 80,000)39 points toward vast chemodiversity readily incorporated in the design of new inhibitor capping groups. The hydroxamate group of 4 coordinates to Zn2+ in monodentate fashion, and the benzyl linker makes favorable offset π–π interactions with F643 and F583 (Figure 5). The secondary amino group of the linker engages in a hydrogen bond network with S531 through two intervening water molecules, and the nonpolar pinene moiety sits in the L1 pocket and makes van der Waals interactions with H463, P464, and L712.
Figure 5.

Polder omit map of the HDAC6 CD2–4 complex (PDB 6PZS; contoured at 4.0 σ). Atoms are color-coded as follows: C = light blue (HDAC6 CD2) or wheat (inhibitor), N = blue, O = red, Zn2+ = gray sphere, and solvent = small red spheres. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. Locations of the L1 and L2 pockets are indicated.
The 1.74 Å-resolution crystal structure of the HDAC6 CD2–5 complex contains 2 monomers in the asymmetric unit. Electron density corresponding to bidentate hydroxamate-Zn2+ coordination interactions is observed in both monomers, although the average Zn2+---O separation of 2.6 Å for the hydroxamate C=O group is somewhat long to be considered inner sphere coordination. The benzyl linker of 5 makes favorable offset aromatic π–π interactions with F643 and F583.
Inhibitor 5 is one of the largest inhibitors studied in complex with HDAC6 CD2 and highlights the feasibility of designing a peptide to bind to a new protein landscape. The capping group of 5 is the N-protected dipeptide Cbz-Leu-Ala (Cbz = carbobenzyloxy), and this peptide sequence maps out three different conformations as it interacts with the L1 pocket.
In monomer A, the alanine residue of the Cbz-Leu-Ala moiety adopts a single conformation, but the Cbz-Leu segment adopts two different conformations as indicated by somewhat noisy but nonetheless interpretable electron density (Figure 6a). In one conformation, the Cbz phenyl group makes a weakly polar C–H hydrogen bond with D460. In the alternate conformation, inhibitor binding is stabilized by water-mediated hydrogen bonds with H614 and the backbone NH group of F643.
Figure 6.

(a) Polder omit map of the HDAC6 CD2–5 complex (PDB 6PZU; monomer A, contoured at 3.0 σ). Atoms are color-coded as follows: C = light blue (monomer A) or wheat (inhibitor), N = blue, O = red, Zn2+ = gray sphere, and solvent = small red spheres. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. (b) Polder omit map of the HDAC6 CD2–5 complex (PDB 6PZU; monomer B, contoured at 3.0 σ). Atoms are color-coded as follows: C = light blue (monomer B) or wheat (inhibitor), N = blue, O = red, Zn2+ = gray sphere, and solvent = small red spheres. Metal coordination and hydrogen bond interactions are indicated by solid and dashed black lines, respectively. Locations of the L1 and L2 pockets are indicated.
In monomer B, the inhibitor conformation is such that the Cbz-Leu-Ala moiety extends somewhat further into the L1 pocket, although electron density for the Cbz protecting group is noisy (Figure 6b). This presumably signifies molecular disorder. Inhibitor binding is stabilized by water-mediated hydrogen bonds with H614 and the backbone NH group of F643. No other hydrogen bond interactions are observed between the inhibitor and enzyme residues. It is interesting that the Cbz-Leu-Ala capping group is reasonably ordered despite the lack of direct enzyme-inhibitor hydrogen bonds in this complex.
To further explore structure-affinity relationships for inhibitors bearing dipeptide capping groups related to that of inhibitor 5, compounds 6 and 7 were synthesized and compared to compound 5 for inhibitory activity (Figure 1). Compound 6 contained a Cbz-Gly-Gly capping group and hence lacked substitution at the amino acid Cα atoms, but was otherwise isosteric with compound 5. Notably, compound 6 was essentially equipotent to 5 with respect to HDAC6 CD2 inhibitory activity. Thus, the amino acid side chains of 5 do not contribute substantially to inhibitory potency. In compound 7, the Cbz-Leu-Ala capping group is identical to that of 5, but it is substituted at the meta position of the phenyl ring. This structural variation resulted in more than a 10-fold loss of inhibitory potency against HDAC6 CD2, thereby confirming the strict steric requirements for entry into the zinc binding group pocket with a para-substituted aromatic linker as found in compounds 5 and 6.
Structural determinants of enzyme-Inhibitor affinity.
The binding interactions observed for all five enzyme-inhibitor complexes in this study are highly informative regarding intermolecular interactions of the linker segment and the capping group of the inhibitor in the active site of HDAC6 CD2. It is notable that without exception, the aromatic ring linker of each inhibitor binds in an aromatic crevice formed by the side chains of F583 and F643. Regardless of the ring size (5- or 6-membered) or composition (phenyl, oxazole, 3-vinylpyrrole), the aromatic linker of the inhibitor makes favorable offset π–π interactions in the aromatic crevice.
The linker moiety of each inhibitor plays an important role in orienting the capping group in the outer active site cleft. As such, it is intriguing that the 3-vinylpyrrole linker of inhibitor 1 directs the capping group toward the L2 pocket, where it displaces solvent and makes van der Waals interactions with various residues lining the pocket. Inhibitor capping groups rarely bind in this region in crystal structures reported to date, and until now has only been observed for inhibitors with bifurcated, sterically bulky capping groups.33 There are some polar residues in or near this pocket that could be targeted for hydrogen bond interactions, such as S531. The side chain of this residue is at the edge of the L2 pocket and hydrogen bonds to the backbone NH group of acetyllysine substrates.27 S531 is unique to HDAC6 CD2, and inhibitors that engage in hydrogen bond interactions with this residue tend to exhibit selectivity for binding to HDAC6 CD2.29,31,33,34 The lack of such an interaction in the binding of 1, also known as the cancer chemotherapy drug Resminostat, is consistent with the broad isozyme selectivity observed for this inhibitor, i.e., it is a pan-HDAC inhibitor.40
The binding of two iodide ions along with inhibitor 1 in the HDAC6 CD2 active site results from the crystallization conditions used to prepare crystals of the enzyme-inhibitor complex. It is not clear whether iodide binding is a cause or a consequence of the binding conformation of 1. However, in the structure of the HDAC6 CD1–1 complex (PDB 6UOB), recently determined in the absence of iodide ions, the inhibitor capping group adopts an alternate conformation that places it in the L1 pocket. This could suggest that iodide binding influences the binding conformation of 1 as observed in the HDAC6 CD2 active site.
In the HDAC6 CD2–1 complex, each iodide ion is stabilized by a single hydrogen bond and an array of van der Waals interactions with the steric bulk of surrounding protein residues and inhibitor atoms (Figure 2). It is interesting to speculate that iodide ions could play some role in facilitating the binding of inhibitors with suitable steric bulk in their linker segments and capping groups. If so, this might enhance HDAC6 CD2 inhibition in iodide-rich tissues such as the thyroid. The pan-HDAC inhibitor Vorinostat does not exhibit a therapeutic effect in patients diagnosed with radioiodine-refractory thyroid cancer,41 but then again Vorinostat contains a slender hydrocarbon linker segment and a smaller capping group in comparison with inhibitor 1.
From the array of HDAC6 CD2–inhibitor binding conformations observed to date, it appears that the L1 pocket is preferentially occupied by the capping groups of phenylhydroxamate inhibitors, unless the binding of other species, e.g., iodide ions, leads to the binding of the capping group in the L2 pocket. Even so, inhibitor capping groups can make favorable van der Waals interactions in either pocket. Interactions in both pockets are simultaneously exploited by the phenylhydroxamate inhibitor RTS-V5, which bears a bifurcated capping group such that one substituent binds in the L2 pocket and the other substituent binds in the L1 pocket (Figure 1b).33 Since the three-dimensional contours of the L1 and L2 pockets at the mouth of the active site can vary from one HDAC isozyme to another, the incorporation of bifurcated capping groups in phenylhydroxamate inhibitors may serve as a useful molecular strategy for the improvement of isozyme selectivity as well as affinity.
In addition to alternative capping group binding conformations in the HDAC6 CD2 active site, it is interesting to consider the utility of halogen atoms as incorporated into various inhibitor designs, such as 2 and 3. For instance, the para-bromophenyl capping group of 2 contributes to selectivity for HDAC6 CD2 inhibition over other HDAC isozymes, and the substitution of the para-bromine atom with a chlorine or a fluorine atom enhances selectivity for HDAC6 CD2 over HDAC8; substitution with a hydrogen atom decreases selectivity in isosteres of 2.42 However, the crystal structure of the HDAC6 CD2–2 complex (Figure 3) reveals that the para-bromo group is oriented out toward solvent, making no specific interactions in the outer active site cleft. This could suggest that the effect of the para-substituted halogen atom on the capping group of 2 and its derivatives arises from electrostatic inductive effects, perhaps enhanced by the near planarity of the π systems of the para-bromophenyl ring and the oxazole ring. The more electron-withdrawing this linker-capping group combination becomes by substitution of increasingly electronegative halogen atoms for the para-bromo group, the more the anionic Zn2+-bound hydroxamate moiety will be stabilized.
Consider, too, the trifluoromethyl group of inhibitor 3. Here, the fluorine atoms make van der Waals interactions with residues in the L1 pocket, and these interactions presumably contribute to potent inhibitory activity. Additionally, the trifluoromethyl group serves as a steric protrusion that locks the trifluoropyridyl ring in a conformation that is 125° on average out of the plane of the adjacent amide linker (Figure 4). Consequently, the π systems of the amide group and the pyridyl ring are not in resonance with each other, so the ability of the trifluoromethyl group to inductively stabilize the Zn2+-bound hydroxamate moiety will be minimal. However, the trifluoromethyl group is perfectly pre-oriented for making van der Waals interactions in the L1 pocket.
The pinene capping group of inhibitor 4 binds in the L1 pocket, which appears to be the preferred binding conformation for a phenylhydroxamate inhibitor bearing a single capping group. The pinene capping group is a unique feature of this inhibitor and to our knowledge represents the first use of a terpene as an inhibitor capping group. Although the inhibitory potency of 4 is very good (although relatively modest with IC50 = 149 nM), the impressive chemodiversity represented by tens of thousands of terpene natural products suggests that there is vast untapped potential for molecular diversification of HDAC inhibitor capping groups using terpene derivatives for the improvement of inhibitor affinity and selectivity.39
The peptide-based capping group of inhibitor 5 exhibits some variability in its conformation as it binds in the L1 pocket, but it is notable that the Cbz-Leu-Ala moiety is reasonably ordered even though the dipeptide did not naturally evolve to bind to this protein surface. In a study of para-substituted phenylsulfonamide inhibitors bound in the active site of carbonic anhydrase II, the use of flexible glycine residues resulted in mainly disordered binding of the para-substituted Gly-Gly-Gly-Bz moiety (Bz = benzyl).43 Thus, it is feasible for a de novo designed peptide to achieve binding when anchored to a protein surface of choice, but the peptide is more likely to bind in ordered fashion if its glycine content is limited. It is interesting that despite lacking amino acid side chains in the Cbz-Gly-Gly capping group, compound 6 is essentially equipotent to inhibitor 5 (Figure 1). This is consistent with the crystal structure of the HDAC6 CD2–5 complex, which shows that the amino acid side chains of the Cbz-Leu-Ala capping group do not make substantial interactions in the active site. The 20-fold loss of inhibitory potency against HDAC6 CD2 measured for compound 7 indicates that the steric requirements for optimal binding in the HDAC6 CD2 active site favor a para-substituted over a meta-substituted phenyl linker group.
Conclusions
The current study demonstrates how a variety of aromatic and heteroaromatic linkers direct inhibitor capping group interactions toward different regions of the HDAC6 CD2 active site spanning the L1 and the L2 pockets. Ideal aromatic linkers for binding to HDAC6 CD2 are para-substituted phenylhydroxamates, i.e., inhibitors in which the capping group extends out away from the hydroxamate group in linear fashion. Inhibitors in which the capping group extends out at an angle, such as that found in the oxazole inhibitor 2 or the meta-substituted phenylhydroxamate inhibitor 7, appear to be less well-suited for capturing affinity interactions at the mouth of the active site in the L1 or L2 pockets.
Phenylhydroxamate inhibitors generally bind with their capping groups oriented into the L1 pocket, but occasional examples exist where capping groups bind in the L2 pocket, e.g., as observed for inhibitor 1 in the presence of iodide ions. Both the L1 and L2 pockets can be simultaneously occupied by the substituents of bifurcated capping groups to enhance affinity and selectivity, as exemplified by the binding of RTS-V5 (Figure 1b).33 Intermolecular interactions of inhibitor capping groups provide a means of structural diversification that can be tailored to a specific HDAC isozyme, since the three-dimensional contour of the mouth of the active site tends to vary from one isozyme to another. Structural studies of HDAC6 CD2-inhibitor complexes continue to reveal fundamental structural features underlying enzyme-inhibitor affinity and selectivity, and thus promise to inform current efforts in medicinal chemistry to optimize the properties of HDAC inhibitors for use in biology and medicine.
Experimental Section
Reagents.
All buffers and chemicals were purchased from Fisher, Millipore Sigma, or Hampton Research and used without further purification. The purity of each final tested compound is as follows: compound 1, as purchased from APExBIO, purity = 98%; compound 2, as previously reported,42 purity > 95%; compounds 3–7, as determined using LC-MS, purity > 95% (Supporting Information, Figure S1).
N-(4-(hydroxycarbamoyl)benzyl)-2-(trifluoromethyl)nicotinamide (3).
To a stirred solution of 11 (50 mg; 0.12 mmol) in 4 mL of methanol at 0 oC was added dropwise 0.5 mL of 4 N HCl in dioxane. The reaction mixture was slowly warmed to room temperature and stirred for an additional 4 h. Completion of the reaction was confirmed by LC-MS. The solvent was then evaporated and dried under high vacuum to afford compound 3 as a light-yellow solid (32 mg; 0.094 mmol, 80%). Product was confirmed by 1H NMR and MS.
NMR: 1H NMR (400 MHz, MeOD-D4): δ 8.75 (d, J = 4.5 Hz, 1H), 8.00 (d, J = 7.8 Hz, 1H), 7.81 – 7.65 (m, 3H), 7.48 (d, J = 8.1 Hz, 2H), 4.60 (s, 2H).
Mass: m/z calc’d for C15H13F3N3O3 [M+H]+; 340.1; found 340.1.
4-(((6,6-dimethylbicyclo[3.1.1]heptan-2-yl)amino)methyl)-N-hydroxybenzamide (4).
To a solution of 12 (73 mg; 0.24 mmol, 1 eq) in 2 mL 1:1 THF/MeOH was added 0.50 mL hydroxylamine (50% aq, 0.25 g; 3.6 mmol, 15 eq) and aqueous NaOH (5.0 M, 0.10 mL; 0.50 mmol, 2 eq). The reaction mixture was stirred for 3 h at room temperature. The product was purified by preparative reversed-phase HPLC with a gradient of 0.025% TFA in water and methanol. Upon drying in vacuo product 4 (29 mg; 0.096 mmol, 40%) was obtained as a white solid. Product was confirmed by 1H NMR and MS.
1H NMR (DMSO-d6): δ 11.3 (s, 1H), 9.1 (s, 1H), 9.0 (s, 2H), 7.78 (d, 2H), 7.54 (d, 2H), 4.2 (m, 2H), 3.34 (s, 2H), 2.91 (s, 2H), 2.58–2.23 (m, 7H), 2.03–1.71 (m, 5H), 1.73–1.38 (m, 4H), 1.59–1.37 (m, 1H), 1.23–1.07 (m, 3H), 0.99–0.72 (m, 4H).
Mass: m/z calc’d for C18H26N2O2 [M+H]+, 303.2; found 303.2.
Benzyl ((S)-1-(((S)-1-((4-(hydroxycarbamoyl)benzyl)amino)-1-oxopropan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (5).
To a stirred solution of 15 (80 mg; 0.12 mmol) in 4 mL of methanol at 0 oC was added 0.5 mL of 4 N HCl in dioxane dropwise slowly. The reaction mixture was warmed to room temperature and stirred for an additional 4 h. Completion of the reaction was confirmed by LC-MS. The solvent was evaporated to yield the crude product, which was purified by preparative reverse phase HPLC to afford 5 (30 mg; 0.061 mmol, 51%). Product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, DMSO): δ 11.59 (s, 1H), 11.17 (s, 1H), 8.40 (t, J = 6.2 Hz, 1H), 8.03 (d, J = 7.3 Hz, 1H), 7.70 (t, J = 7.7 Hz, 2H), 7.44 (d, J = 8.3 Hz, 1H), 7.41 – 7.17 (m, 7H), 5.13 – 4.88 (m,2H), 4.42 – 4.22 (m, 2H), 4.14 – 3.94 (m, 1H), 3.83 – 3.63 (m, 1H), 3.63 – 3.34 (m, 1H), 1.80 – 1.34 (m, 1H), 1.31 – 1.15 (m, 4H), 0.89 – 0.70 (m, 6H).
Mass: m/z calc’d for C25H33N4O6 [M+H]+; 285.2; found 285.3.
Benzyl (2-((2-((4-(hydroxycarbamoyl)benzyl)amino)-2-oxoethyl)amino)-2-oxoethyl)-carbamate (6).
To a stirred solution of 18 (120 mg; 0.24 mmol) in 3 mL of methanol at 0 oC was added 0.5 mL of 4 N HCl in dioxane dropwise. The reaction mixture was slowly warmed to room temperature and stirred for 4 h. Completion of the reaction was confirmed by LC-MS. Solvent was evaporated to yield crude product, which was purified by reverse phase preparative HPLC to afford 6 (40 mg, 0.096, 40%). Product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, MeOD): δ 7.71 (t, J = 9.0 Hz, 2H), 7.38 (d, J = 8.1 Hz, 2H), 7.34 – 7.25 (m, 5H), 5.03 (s, 2H), 4.44 (s, 2H), 3.92 (s, 2H), 3.81 (s, 2H).
Mass: m/z calc’d for C20H22N4NaO6 [M+Na]+; 437.1; found 437.2.
Benzyl ((S)-1-(((S)-1-((3-(hydroxycarbamoyl)benzyl)amino)-1-oxopropan-2-yl)amino)-4-methyl-1-oxopentan-2-yl)carbamate (7).
To a stirred solution of 21 (150 mg; 0.26 mmol) in 4 mL of methanol at 0 oC was added 0.5 mL of 4 N HCl in dioxane dropwise. The reaction mixture was slowly warmed to room temperature and stirred for 4 h. Completion of the reaction was confirmed by LC-MS. Solvent was evaporated to yield crude product, which was purified by reverse phase preparative HPLC to afford 7 (45 mg; 0.092 mmol, 35%). Product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, DMSO): δ 11.18 (s, 1H), 8.40 (t, J = 5.9 Hz, 1H), 8.03 (d, J = 7.2 Hz, 1H), 7.66 (s, 1H), 7.60 (d, J = 4.0 Hz, 1H), 7.44 – 7.27 (m, 7H), 5.08 – 4.94 (m, 2H), 4.30 (dd, J = 12.7, 6.3 Hz, 3H), 4.05 (dd, J = 15.1, 8.1 Hz, 1H), 1.70 – 1.54 (m, 1H), 1.44 (dd, J = 18.0, 10.4 Hz, 2H), 1.25 (d, J = 6.9 Hz, 3H), 0.84 (dd, J = 12.5, 6.4 Hz, 6H).
Mass: m/z calc’d for C25H33N4O6 [M+H]+; 485.2; found 485.3.
4-((((9H-Fluoren-9-yl)methoxy)carbonylamino)methyl)benzoic acid (8).
To a stirred solution of 4-(aminomethyl) benzoic acid (2 g; 13.23 mmol) in 10 mL (1:1 dioxane-H2O) at 0 oC were added Na2CO3 (4.19 g; 39.7 mmol) and Fmoc-Cl (3.59 g; 13.23 mmol) simultaneously. The reaction mixture was slowly brought to room temperature and stirred for 12 h. Completion of the reaction was confirmed by liquid chromatography-mass spectrometry (LC-MS). The reaction mixture was then cooled to 0 oC in an ice bath and acidified with 1 N HCl to pH 5. The precipitated solid was filtered and washed with H2O and hexane, and then dried under high vacuum to afford 8 (4.4 g; 11.78 mmol, 90%) as a white solid. Product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, DMSO): δ 12.87 (s, 1H), 8.03 – 7.78 (m, 3H), 7.69 (t, J = 10.3 Hz, 2H), 7.49 – 7.37 (m, 2H), 7.33 (t, J = 8.0 Hz, 3H), 4.38 (d, J = 6.8 Hz, 2H), 4.24 (m, 3H).
Mass: m/z calc’d for C23H18NO4 [M-H]+; 372.1; found 372.3.
(9H-Fluoren-9-yl) methyl 4-(tetrahydro-2H-pyran-2-yloxycarbamoyl)-benzylcarbamate (9).
To a stirred solution of 8 (1.0 g; 2.55 mmol) and O-(tetrahydro-2H-pyran-2-yl)hydroxylamine (328 mg; 2.8 mmol) in 20 mL of dry CH2Cl2 at 0 oC were added diisopropyl ethyl amine (0.58 mL; 3.31 mmol) and T3P 50% solution in CH2Cl2 by weight (2.10 g; 3.31 mmol) dropwise simultaneously. The reaction mixture was allowed to stir for 30 min at 0 oC. Completion of the reaction was confirmed by LC-MS. The reaction mixture was quenched with cold water and the product was extracted with CH2Cl2 and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure and the crude product was purified by flash column chromatography to afford 9 (1.02 g; 2.16 mmol, 85%). Product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, DMSO): δ 11.59 (s, 1H), 7.99 – 7.84 (m, 3H), 7.71 (dd, J = 7.4, 5.6 Hz, 4H), 7.42 (t, J = 7.4 Hz, 2H), 7.30 (dt, J = 20.7, 10.0 Hz, 3H), 4.99 (s, 1H), 4.35 (dd, J = 10.7, 6.0 Hz, 2H), 4.23 (t, J = 7.0 Hz, 3H), 4.06 (s, 1H), 3.51 (t, J = 12.3 Hz, 1H), 1.67 (d, J = 42.9 Hz, 3H), 1.55 (s, 3H).
Mass: m/z calc’d for C28H28N2NaO5 [M+Na]+; 495.2; found 495.3
4-(Aminomethyl)-N-(tetrahydro-2H-pyran-2-yloxy)benzamide (10).
To a stirred solution of 9 (700 mg; 1.48 mmol) in 20 mL of MeOH at room temperature was added 10 mL of piperidine all at once and the reaction mixture was stirred for 1 h. Completion of the reaction was confirmed by LC-MS. The volatiles were evaporated under reduced pressure to yield the crude product, which was purified by flash column chromatography to afford the desired amine 10 (343 mg; 1.37 mmol, 92%) as a white solid. Product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, CDCl3): δ 7.73 (d, J = 8.2 Hz, 2H), 7.38 (d, J = 8.1 Hz, 2H), 5.08 (d, J = 2.9 Hz, 1H), 4.07 – 3.95 (m, 1H), 3.93 (s, 2H), 3.71 – 3.62 (m, 1H), 1.99 – 1.76 (m, 3H), 1.76 – 1.52 (m, 3H).
Mass: m/z calc’d for C13H18N2NaO3 [M+Na]+; 273.1; found 273.2.
N-(4-(tetrahydro-2H-pyran-2-yloxycarbamoyl)benzyl)-2-(trifluoromethyl)-nicotinamide (11).
To a stirred solution of 2-(trifluoromethyl)nicotinic acid (50 mg; 0.2 mmol) and 10 (38 mg; 0.2 mmol) in 5 mL of dry CH2Cl2 at 0 oC was added diisopropyl ethyl amine (103 mg; 0.8 mmol) and T3P 50% solution in CH2Cl2 by weight (82 mg; 0.26 mmol) simultaneously dropwise. The reaction mixture was stirred for 1 h at 0 oC. Completion of the reaction was confirmed by LC-MS. The reaction mixture was quenched with cold water and the product was extracted with CH2Cl2 and then dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure to yield the crude product, which was purified by flash column chromatography to afford compound 11 (94 mg; 0.22 mmol, 85%). Product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, CDCl3): δ 8.90 (s, 1H), 8.75 (d, J = 4.6 Hz, 1H), 7.91 (d, J = 7.8 Hz, 1H), 7.70 (d, J = 7.7 Hz, 2H), 7.55 (dd, J = 7.8, 4.8 Hz, 1H), 7.39 (d, J = 7.8 Hz, 2H), 6.42 (s, 1H), 5.06 (s, 1H), 4.66 (d, J = 5.8 Hz, 2H), 4.05 – 3.91 (m, 1H), 3.69 – 3.59 (m, 1H), 1.86 (dd, J = 18.5, 11.2 Hz, 3H), 1.74 – 1.52 (m, 3H).
Mass: m/z calc’d for C20H20F3N3NaO4 [M+Na]+; 446.1; found 446.2.
Methyl 4-(((6,6-dimethylbicyclo[3.1.1]heptan-2-yl)amino)methyl)benzoate (12).
A solution of methyl 4-formylbenzoate (0.306 mg; 1.86 mmol, mole 1.1 eq.) and (–)-cis-myrtanylamine (247 mg; 1.62 mmol, 1 eq.) in 5 mL methanol was stirred for 1.5 h at room temperature. Then, 175 mg sodium borohydride was added portion-wise and the reaction mixture was stirred until no starting material remained as determined by thin layer chromatography (approximately 4 h). The mixture was concentrated in vacuo and partitioned between ethyl acetate and water. The aqueous layer was extracted two more times with ethyl acetate and the combined organic phases were dried over sodium sulfate. Solvent was removed under reduced pressure and product 12 was purified by column chromatography, affording 463 mg (1.54 mmol, 94%) of a yellowish oil. Product was confirmed by 1H NMR and MS.
1H NMR (chloroform-d): δ 8.0 (d, 2H), 7.6 (d, 2H), 4.17–3.77(m, 3H), 3.98 (s, 2H), 2.77–1.65 (m, 11H), 1.65–0.43 (m, 11H).
Mass: m/z calc’d for C19H27NO2 [M+H]+, 302.2; found 302.7.
(S)-tert-butyl 2-((S)-2-(benzyloxycarbonylamino)-4-methylpentanamido)propanoate (13).
To a stirred solution of (S)-2-(benzyloxycarbonylamino)-4-methylpentanoic acid (1.46 g; 5.5 mmol) and (S)-tert-butyl 2-aminopropanoate hydrochloride (1.0 g; 5.5 mmol) in 20 mL dry CH2Cl2 at 0 oC were added diisopropyl ethyl amine (3.8 mL, 2.2 mmol) and T3P 50% solution in CH2Cl2 by weight (5.45 g; 7.15 mmol) dropwise simultaneously. The reaction mixture was stirred for an additional 1 h at 0 oC, and completion of the reaction was confirmed by LC-MS. The reaction mixture was quenched with cold water and the product was extracted with CH2Cl2 and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to provide the crude reaction product, which was purified by flash column chromatography to afford 13 (1.94 mg; 4.94 mmol, 90%) as a white solid. Product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, CDCl3): δ 7.43 – 7.28 (m, 5H), 6.46 (d, J = 6.6 Hz, 1H), 5.30 (s, 1H), 5.19 (t, J = 13.3 Hz, 1H), 5.11 (s, 2H), 4.42 (p, J = 7.1 Hz, 1H), 4.18 (t, J = 14.8 Hz, 1H), 1.76 – 1.54 (m, 3H), 1.46 (s, 9H), 1.38 – 1.25 (m, 3H), 0.96 (t, J = 15.3 Hz, 6H).
Mass: m/z calc’d for C21H32N2NaO5 [M+Na]+; 415.2; found 415.2.
(S)-2-((S)-2-(benzyloxycarbonylamino)-4-methylpentanamido)propanoic acid (14).
To a stirred solution of 13 (1.8 g; 4.59 mmol) in 30 mL CH2Cl2 at 0 oC was added 10 mL of TFA. The reaction mixture was then warmed to room temperature and stirred for 2 h. Completion of the reaction was confirmed by LC-MS. Volatiles were evaporated under reduced pressure to yield crude product which was co-distilled with 20 mL of toluene to afford 14 (1.5 g; 4.45 mmol, 98%) as a viscous liquid. Product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, CDCl3): δ 7.41 – 7.28 (m, 5H), 7.11 (s, 3H), 5.70 (d, J = 8.3 Hz, 1H), 5.19 – 5.01 (m, 2H), 4.62 – 4.46 (m, 1H), 4.32 (d, J = 5.9 Hz, 1H), 1.76 – 1.48 (m, 3H), 1.48 – 1.31 (m, 3H), 0.99 – 0.82 (m, 6H).
Mass: m/z calc’d for C17H24N2NaO5 [M+Na]+; 359.2; found 359.3.
Benzyl (2S)-4-methyl-1-oxo-1-((2S)-1-oxo-1-(4-(tetrahydro-2H-pyran-2-yloxycarbamoyl)benzylamino)propan-2-ylamino)pentan-2-yl carbamate (15).
To a stirred solution of 14 (67 mg; 0.2 mmol) and 10 (50 mg; 2.0 mmol) in 3 mL of dry CH2Cl2 at 0 oC were added diisopropyl ethyl amine (0.14 mL; 0.8 mmol) and T3P 50% solution in CH2Cl2 by weight (165 mg; 2.6 mmol) simultaneously dropwise. The reaction mixture was stirred for an additional 1 h at 0 oC. Completion of the reaction was confirmed by LC-MS. The reaction mixture was quenched with cold water. The product was extracted with CH2Cl2 and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure to yield the crude product, which was purified by flash column chromatography to afford 15 (91 mg; 0.160 mmol, 80%) as confirmed by 1H NMR and MS.
1H NMR (400 MHz, DMSO): δ 11.59 (s, 1H), 8.41 (t, J = 5.7 Hz, 1H), 8.03 (d, J = 7.3 Hz, 1H), 7.71 (d, J = 8.2 Hz, 2H), 7.44 (d, J = 8.2 Hz, 1H), 7.39 – 7.23 (m, 7H), 5.13 – 4.88 (m, 3H), 4.39 – 4.19 (m, 3H), 4.05 (dd, J = 14.1, 8.8 Hz, 2H), 3.69 – 3.45 (m, 2H), 1.68 (d, J = 30.2 Hz, 3H), 1.45 (dd, J = 11.1, 6.5 Hz, 2H), 1.24 (d, J = 6.9 Hz, 6H), 0.85 (t, J = 6.4 Hz, 6H).
Mass: m/z calc’d for C30H40N4NaO7 [M+Na]+; 591.3; found 591.5.
Methyl 2-(2-(benzyloxycarbonylamino)acetamido)acetate (16).
To a stirred solution of 2-(benzyloxycarbonylamino)acetic acid (1.0 g; 4.78 mmol) and methyl 2-aminoacetate hydrochloride (600 mg; 4.78 mmol) in 20 mL dry CH2Cl2 at 0 oC were added diisopropyl ethyl amine (3.43 mL; 19.12 mmol) and T3P 50% solution in CH2Cl2 by weight (3.95 g; 6.21 mmol) dropwise simultaneously. The reaction mixture was stirred for an additional 1 h at 0 oC, and completion of the reaction was confirmed LC-MS. The reaction mixture was quenched with cold water, and the product was extracted with CH2Cl2 and dried over anhydrous Na2SO4. The solvent was removed under reduced pressure to provide the crude reaction product, which was purified by flash column chromatography to afford 16 (1.18 g; 4.21 mmol, 88%) as a white solid. Product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, CDCl3): δ 7.45 – 7.29 (m, 5H), 6.61 (s, 1H), 5.51 (s, 1H), 5.13 (s, 2H), 4.05 (d, J = 5.2 Hz, 2H), 3.92 (d, J = 5.6 Hz, 2H).
Mass: m/z calc’d for C13H16N2NaO5 [M+Na]+; 303.1; found 303.1.
2-(2-(Benzyloxycarbonylamino)acetamido)acetic acid (17).
To a stirred solution of 16 (1.18 g; 4.21 mmol) in 30 mL of MeOH and H2O (5:1) at room temperature was added LiOH•H2O (707 mg; 16.84 mmol). The reaction mixture was stirred for 6 h. Completion of the reaction was confirmed by thin layer chromatography. Volatiles were evaporated by reduced pressure, and the crude product was acidified with 1 N HCl at 0 oC to obtain a white precipitate which was filtered and dried under high vacuum to afford 17 (890 mg; 3.34 mmol, 80%). Product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, DMSO): δ 12.56 (s, 1H), 8.15 (t, J = 5.7 Hz, 1H), 7.48 (t, J = 6.1 Hz, 1H), 7.41 – 7.25 (m, 5H), 5.03 (s, 2H), 3.76 (d, J = 5.8 Hz, 2H), 3.65 (d, J = 6.2 Hz, 2H).
Mass: m/z calc’d for C12H14N2NaO5 [M+Na]+; 289.1; found 289.1.
Benzyl 2-oxo-2-(2-oxo-2-(4-(tetrahydro-2H-pyran-2-yloxycarbamoyl)benzylamino) ethylamino)ethylcarbamate (18).
To a stirred solution of 17 (70 mg; 0.26 mmol) and 10 (65 mg; 0.26 mmol) in 5 mL dry CH2Cl2 at 0 oC were added diisopropyl ethyl amine (0.19 mL; 1.05 mmol) and T3P 50% solution in CH2Cl2 by weight (217 mg; 0.34 mmol) dropwise. The reaction mixture was stirred for 1 h at 0 oC. Completion of the reaction was confirmed by LC-MS. The reaction was quenched with cold water, and the product was extracted with CH2Cl2 and dried over anhydrous Na2SO4. Solvent was evaporated under reduced pressure to provide crude product, which was purified by flash column chromatography to afford 18 (120 mg; 0.24 mmol, 91%). The product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, DMSO): δ 11.62 (s, 1H), 8.41 (t, J = 5.5 Hz, 1H), 8.23 (d, J = 5.6 Hz, 1H), 7.72 (d, J = 8.1 Hz, 2H), 7.53 (t, J = 5.5 Hz, 1H), 7.34 (d, J = 9.6 Hz, 7H), 5.13 – 4.87 (m, 3H), 4.33 (d, J = 5.8 Hz, 2H), 3.99 (d, J = 52.6 Hz, 1H), 3.76 (d, J = 5.7 Hz, 2H), 3.68 (d, J = 5.9 Hz, 2H), 3.51 (d, J = 11.1 Hz, 1H), 1.72 (m, 3H), 1.54 (m, 3H).
Mass: m/z calc’d for C25H30N4NaO7 [M+Na]+; 521.2; found 521.2.
Methyl 3-((5S,8S)-5-isobutyl-8-methyl-3,6,9-trioxo-1-phenyl-2-oxa-4,7,10-triazaundecan-11-yl)benzoate (19).
To a stirred solution of 14 (500 mg; 1.5 mmol) and methyl 3-(aminomethyl)benzoate (245 mg; 1.5 mmol) in 10 mL dry CH2Cl2 at 0 oC were added diisopropyl ethyl amine (1.05 mL; 5.95 mmol) and T3P 50% solution in CH2Cl2 by weight (1.225 g, 1.95 mmol) simultaneously dropwise. The reaction mixture was stirred for 30 min at 0 oC. Completion of the reaction was confirmed by LC-MS. The reaction was quenched with cold water, and the product was extracted with CH2Cl2 and then dried over anhydrous Na2SO4. Solvent was evaporated under reduced pressure to provide crude product, which was purified by flash column chromatography to afford 19 (610 mg; 1.26 mmol, 85%). Product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, CDCl3): δ 7.91 (dd, J = 10.4, 9.2 Hz, 2H), 7.53 – 7.42 (m, 1H), 7.42 – 7.28 (m, 6H), 6.88 (s, 1H), 6.56 (d, J = 7.4 Hz, 1H), 5.18 (d, J = 6.4 Hz, 1H), 5.02 (q, J = 12.2 Hz, 2H), 4.57 – 4.35 (m, 3H), 4.10 (d, J = 29.3 Hz, 1H), 3.88 (s, 3H), 1.71 – 1.55 (m, 4H), 1.52 – 1.43 (m, 1H), 1.39 (d, J = 6.8 Hz, 3H), 0.90 (d, J = 6.1 Hz, 6H).
Mass: m/z calc’d for C26H34N3O6 [M+H]+; 484.2; found 484.3.
3-((5S,8S)-5-Isobutyl-8-methyl-3,6,9-trioxo-1-phenyl-2-oxa-4,7,10-triazaundecan-11-yl)benzoic acid (20).
To a stirred solution of 19 (450 mg; 0.93 mmol) in 10 mL of (THF:MeOH:H2O) (5:5:1) at 0 oC was added LiOH•H2O (117 mg; 2.79 mmol). The reaction mixture was warmed to room temperature and stirred for 6 h. Completion of the reaction was confirmed by LC-MS. Volatiles were evaporated under reduced pressure to provide crude product, which was neutralized with 1 N HCl, washed with water, and dried under vacuum to afford 20 (380 mg; 0.809 mmol, 87%). Product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, DMSO): δ 12.91 (s, 1H), 8.44 (t, J = 5.8 Hz, 1H), 8.01 (d, J = 7.4 Hz, 1H), 7.88 – 7.75 (m, 2H), 7.53 – 7.39 (m, 3H), 7.39 – 7.25 (m, 5H), 5.10 – 4.93 (m, 2H), 4.42 – 4.20 (m, 3H), 4.04 (dd, J = 14.7, 8.4 Hz, 1H), 1.66 – 1.54 (m, 1H), 1.49 – 1.35 (m, 2H), 1.32 – 1.19 (m, 3H), 0.83 (dd, J = 19.0, 13.3 Hz, 6H).
Mass: m/z calc’d for C25H32N3O6 [M+H]+; 470.2; found 470.3.
Benzyl (2S)-4-methyl-1-oxo-1-((2S)-1-oxo-1-(3-(tetrahydro-2H-pyran-2-yloxycarbamoyl)-benzylamino)propan-2-ylamino)pentan-2-yl carbamate (21).
To a stirred solution of 20 (300 mg; 0.63 mmol) and O-(tetrahydro-2H-pyran-2-yl)hydroxylamine (74 mg; 0.63 mmol) in 20 mL dry CH2Cl2 at 0 oC were added diisopropyl ethyl amine (0.44 ml; 2.56 mmol) and T3P 50% solution in CH2Cl2 by weight (528 mg; 0.83 mmol) dropwise. The reaction mixture was stirred for 1 h at 0 oC. Completion of the reaction was confirmed by LC-MS. The reaction was quenched with cold water, and the product extracted with CH2Cl2 and dried over anhydrous Na2SO4. The solvent was evaporated under reduced pressure to provide crude product, which was purified by flash column chromatography to afford 21 (298 mg; 0.524 mmol, 82%). Product was confirmed by 1H NMR and MS.
1H NMR (400 MHz, DMSO): δ 11.62 (s, 1H), 8.41 (s, 1H), 8.06 (t, J = 6.3 Hz, 1H), 7.65 (d, J = 14.2 Hz, 2H), 7.50 – 7.15 (m, 7H), 5.01 (t, J = 12.3 Hz, 3H), 4.30 (dd, J = 17.6, 6.1 Hz, 3H), 4.06 (d, J = 6.2 Hz, 2H), 3.51 (d, J = 11.3 Hz, 1H), 1.71 (m, 3H), 1.61 – 1.35 (m, 3H), 1.19 (dd, J = 48.0, 6.7 Hz, 3H), 0.84 (dd, J = 11.7, 6.0 Hz, 6H).
Mass: m/z calc’d for C30H40N4NaO7 [M+Na]+; 591.3; found 591.3.
Inhibitory activity.
The inhibitory potencies (IC50) of compounds 1 (Resminostat) and 2 against HDAC6 were previously reported by others.40,42 The inhibitory potencies of compounds 3 and 5 against HDAC6 were determined at Reaction Biology Corporation (Malvern, PA), where each compound was tested in a 10-point dose-response curve and compared with inhibition by Trichostatin A as a positive control (IC50 = 5 nM). The inhibitory potency of compound 4 against HDAC6 was determined at Nanosyn.
Crystallization and X-ray diffraction data collection.
The expression and purification of histone deacetylase 6 CD2 from Danio rerio (referred to as HDAC6 CD2) was achieved by modifying the originally reported27 preparation as recently described.44 For cocrystallization of HDAC6 CD2 complexes with inhibitors 1 and 3–5 by the sitting drop method at 4° C using a Mosquito crystallization robot (TTP Labtech), a 100-nL drop of protein solution [10 mg/mL HDAC6, 50 mM 4-(2-hydroxyethyl)-1–1piperazineethanesulfonic acid (HEPES) (pH 7.5), 100 mM KCl, 5% glycerol (v/v), 1 mM tris(2-carboxyethyl)phosphine (TCEP), and 2 mM inhibitor] was added to a 100-nL drop of precipitant solution and equilibrated against 70 μL of precipitant solution in the well reservoir. For cocrystallization of HDAC6 CD2 complexes with inhibitor 2, 500-nL drop sizes were used for both the protein and precipitant solution. Prior to flash-cooling, all crystals were soaked in their respective precipitation solutions augmented with 20% ethylene glycol to serve as a cryoprotectant.
For cocrystallization of the HDAC6 CD2–1 complex, the precipitant solution was 0.2 M potassium iodide and 20% w/v PEG 3350. X-ray diffraction data were collected on Northeastern Collaborative Access Team (NE-CAT) beamline 24-ID-C at the Advanced Photon Source (APS).
For cocrystallization of the HDAC6–2 complex, the precipitant solution was 0.2 M sodium citrate and 20% w/v PEG 3350. X-ray diffraction data were collected on beamline 9–2 at the Stanford Synchrotron Radiation Lightsource (SSRL).
For cocrystallization of the HDAC6–3 complex, the precipitant solution was 0.2 M succinic acid (pH 7.0) and 20% w/v PEG 3350. X-ray diffraction data were collected on NE-CAT beamline 24-ID-E at APS.
For cocrystallization of the HDAC6–4 complex, the precipitant solution was 0.2 M LiNO3 and 20% w/v PEG 3350. X-ray diffraction data were collected from crystals on beamline 9–2 at SSRL.
For cocrystallization of the HDAC6 CD2–5 complex, the precipitant solution was 0.2 M sodium acetate trihydrate (pH 7.0) and 20% w/v PEG 3350. X-ray diffraction data were collected on beamline 4.2.2 at the Advanced Light Source (ALS).
Data reduction, phasing, and structure refinement.
Programs implemented in the CCP4 program suite45 were used for data reduction: data indexing was achieved with iMosflm46 and Aimless47 was utilized for data scaling. Molecular replacement was used to phase the initial electron density map of each enzyme-inhibitor complex using routines implemented in Phaser.48 The atomic coordinates of unliganded HDAC6 CD2 (PDB 5EEM)27 were used as a search probe for rotation and translation function calculations. The interactive graphics program Coot49 was used to build and manipulate atomic models of each enzyme-inhibitor complex, and Phenix50 was used for crystallographic refinement. Inhibitors were added to each model in the final stages of refinement. MolProbity51 was used to validate the final structures prior to deposition in the Protein Data Bank. All data reduction and refinement statistics are recorded in Table S1.
Supplementary Material
Acknowledgements
We would like to gratefully acknowledge Reaction Biology Corporation, Malvern, PA, for providing inhibitory activity data for some of the HDAC inhibitors described herein. Additionally, we thank synchrotron beamline staff for assistance, especially Narayanasami Sukumar and Kay Perry (APS), Jay Nix (ALS), and Jennifer Wierman (SSRL). Specifically, we thank the Northeastern Collaborative Access Team (NE-CAT) which is funded by the National Institute of General Medical Sciences (NIGMS) from the NIH (P30 GM124165). The Pilatus 6M detector on beamline 24-ID-C is funded by a NIH-ORIP HEI grant (S10 RR029205). This research used resources of the Advanced Photon Source (APS), a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. We also thank the Stanford Synchrotron Radiation Lightsource (SSRL), SLAC National Accelerator Laboratory, which is supported by the DOE Office of Science, Office of Basic Energy Sciences, under Contract No. DE-AC02-76SF00515. The SSRL Structural Molecular Biology Program is supported by the DOE Office of Biological and Environmental Research, and by NIGMS (including P41GM103393). Finally, we thank the Advanced Light Source (ALS; University of California, Berkeley) which is a DOE Office of Science User Facility under contract DE-AC02-05CH11231.
Funding Sources
This research was supported by US National Institutes of Health (NIH) grant GM49758 to D.W.C., and J.M.S. is partially supported by the NIH through grant P30 CA010815. N.J.P. received financial support from the NIH through Chemistry-Biology Interface Training Grant T32 GM071339. M.J. thanks the Deutsche Forschungsgemeinschaft for support (DFG, Ju295/13–1).
J.D.O., N.J.P., P.A.N.R., Y.-C. X, J.R., M.J., and D.W.C. declare no competing financial interests. J.M.H. is a co-founder of Eikonizo Therapeutics, Inc., which has licensed intellectual property related to HDAC inhibitors developed in his laboratory. J.M.S. was a paid consultant for Reaction Biology Corporation, and is President of Alliance Discovery, Inc.
Abbreviations
- Bz
benzyl
- Cbz
carbobenzyloxy
- CD1
catalytic domain 1
- CD2
catalytic domain 2
- HDAC
histone deacetylase
- HDAC6
histone deacetylase 6
- HEPES
4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid
- HPLC
high performance liquid chromatography
- LC-MS
liquid chromatography-mass spectrometry
- MeOH
methanol
- MS
mass spectrometry
- NMR
nuclear magnetic resonance spectroscopy
- PDB
Protein Data Bank
- T3P
propylphosphonic anhydride solution
- TCEP
tris-(2-carboxyethyl)phosphine
- TFA
trifluoroacetic acid
- THF
tetrahydrofuran
Footnotes
Supporting Information
The Supporting Information is available free of charge on the ACS Publications website at DOI: 10.1021/acs.jmedchem.9b01540
Table S1, X-ray crystallographic data collection and refinement statistics
Figure S1, mass spectra and chromatographic traces establishing the purity of newly synthesized compounds 3–7.
Molecular formula strings (CSV)
Accession codes
The atomic coordinates and crystallographic structure factors of HDAC6 complexes with inhibitors 1, 2, 3, 4, and 5 have been deposited in the Protein Data Bank (www.rcsb.org) with accession codes 6PZR, 6Q0Z, 6PZO, 6PZS, and 6PZU, respectively. Authors will release the atomic coordinates and experimental data upon article publication.
References
- (1).Allfrey VG; Faulkner R; Mirsky AE Acetylation and methylation of histones and their possible role in the regulation of RNA synthesis. Proc. Natl. Acad. Sci. U. S. A 1964, 51, 786–794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Kouzarides T Acetylation: a regulatory modification to rival phosphorylation? EMBO J. 2000, 19, 1176–1179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Sterner DE; Berger SL Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev 2000, 64, 435–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Gantt SL; Gattis SG; Fierke CA Catalytic activity and inhibition of human histone deacetylase 8 is dependent on the identity of the active site metal ion. Biochemistry 2006, 45, 6170–6178. [DOI] [PubMed] [Google Scholar]
- (5).López JE; Sullivan ED; Fierke CA Metal-dependent deacetylases: cancer and epigenetic regulators. ACS Chem. Biol 2016, 11, 706–716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Hornbeck PV; Zhang B; Murray B; Kornhauser JM; Latham V; Skrzypek E PhosphoSitePlus, 2014: mutations, PTMs and recalibrations. Nucleic Acids Res. 2015, 43, D512–D520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Gregoretti IV; Lee YM; Goodson HV Molecular evolution of the histone deacetylase family: functional implication of phylogenetic analysis. J. Mol. Biol 2004, 338, 17–31. [DOI] [PubMed] [Google Scholar]
- (8).Hernick M; Fierke CA Zinc hydrolases: the mechanisms of zinc-dependent deacetylases. Arch. Biochem. Biophys 2005, 433, 71–84. [DOI] [PubMed] [Google Scholar]
- (9).Lombardi PM; Cole KE; Dowling DP; Christianson DW Structure, mechanism, and inhibition of histone deacetylases and related metalloenzymes. Curr. Opin. Struct. Biol 2011, 21, 735–743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Porter NJ; Christianson DW Structure, mechanism, and inhibition of the zinc-dependent histone deacetylases. Curr. Opin. Struct. Biol 2019, 59, 9–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Haberland M; Montgomery RL; Olsen EN The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat. Rev. Genet 2009, 10, 32–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Grozinger CM; Hassig CA; Schreiber SL Three proteins define a class of human histone deacetylases related to yeast Hda1p. Proc. Natl. Acad. Sci. U. S. A 1999, 86, 4868–4873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Verdel A; Khochbin S Identification of a new family of higher eukaryotic histone deacetylases. Coordinate expression of differentiation-dependent chromatin modifiers. J. Biol. Chem 1999, 274, 2440–2445. [DOI] [PubMed] [Google Scholar]
- (14).Zhang Y; Gilquin B; Khochbin S; Matthias P Two catalytic domains are required for protein deacetylation. J. Biochem 2006, 281, 201–204. [DOI] [PubMed] [Google Scholar]
- (15).Zou H; Wu Y; Navre M; Sang BC Characterization of the two catalytic domains in histone deacetylase 6. Biochem. Biophys. Res. Commun 2006, 341, 45–50. [DOI] [PubMed] [Google Scholar]
- (16).Hubbert C; Guardiola A; Shao R; Kawaguchi Y; Ito A; Nixon A; Yoshida M; Wang XF; Yao TP HDAC6 is a microtubule-associated deacetylase. Nature 2002, 23, 455–458. [DOI] [PubMed] [Google Scholar]
- (17).Haggarty SJ; Koeller KM; Wong JC; Grozinger CM; Schreiber SL Domain-selective small-molecule inhibitor of histone deacetylase 6 (HDAC6)-mediated tubulin deacetylation. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 4389–4394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Jordan MA; Wilson L Microtubules as a target for anticancer drugs. Nat. Rev. Cancer 2004, 4, 253–265. [DOI] [PubMed] [Google Scholar]
- (19).Dallavalle S; Pisano C; Zunino F Development and therapeutic impact of HDAC6-selective inhibitors. Biochem. Pharmacol 2012, 84, 756–765. [DOI] [PubMed] [Google Scholar]
- (20).Santo L; Hideshima T; Kung AL; Tseng J,C; Tamang D; Yang M; Jarpe M; van Duzer JH; Mazitschek R; Ogier WC; Cirstea D; Rodig S; Eda H; Scullen T; Canavese M; Bradner J; Anderson KC; Jones SS; Raje N. Preclinical activity, pharmacodynamics, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Jochems J; Boulden J; Lee BG; Blendy JA; Jarpe M; Mazitschek R; van Duzer JH; Jones S; Berton O Antidepressant-like properties of novel HDAC6-selective inhibitors with improved brain bioavailability. Neuropsychopharmacology 2014, 39, 389–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wang Z; Leng Y; Wang J; Liao HM; Bergman J; Leeds P; Kozikowski A; Chuang DM Tubastatin A, an HDAC6 inhibitor, alleviates stroke-induced brain infarction and functional deficits: potential roles of alpha-tubulin acetylation and FGF-21 up-regulation. Sci. Rep 2016, 6, 19626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Pinho BR; Reis SD; Guedes-Dias P; Leitao-Rocha A; Quintas C; Valentao P; Andrade PB; Santos MM; Oliveira JM Pharmacological modulation of HDAC1 and HDAC6 in vivo in a zebrafish model: Therapeutic implications for Parkinson’s disease. Pharmacol. Res 2016, 103, 328–339. [DOI] [PubMed] [Google Scholar]
- (24).Krukowski K; Ma J; Golonzhka O; Laumet GO; Gutti T; van Duzer JH; Mazitschek R; Jarpe MB; Heijnen CJ; Kavelaars A HDAC6 inhibition effectively reverses chemotherapy-induced peripheral neuropathy. Pain 2017, 158, 1126–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Benoy V; Vanden Berghe P; Jarpe M; van Damme P; Robberecht W; van Den Bosch L Development of improved HDAC6 inhibitors as pharmacological therapy for axonal Charcot-Marie-Tooth Disease. Neurotherapeutics 2017, 14, 417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Guo W; Naujock M; Fumagalli L; Vandoorne T; Baasten P; Boon R; Ordovas L; Patel A; Welters M; Vanwelden T; Geens N; Tricot T; Benoy V; Steyaert J; Lefebvre-Omar C; Boesmans W; Jarpe M; Sterneckert J; Wegner F; Petri S; Bohl D; Vanden Berghe P; Robberecht W; van Damme P; Verfaillie C; van Den Bosch L HDAC6 inhibition reverses axonal transport defects in motor neurons derived from FUS-ALS patients. Nat. Commun 2017, 8, 861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Hai Y; Christianson DW Histone deacetylase 6 structure and molecular basis of catalysis and inhibition. Nat. Chem. Biol 2016, 12, 741–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Miyake Y; Keusch JJ; Wang L; Saito M; Hess D; Wang X; Melancon BJ; Helquist P; Gut H; Matthias P Structural insights into HDAC6 tubulin deacetylation and its selective inhibition. Nat. Chem. Biol 2016, 12, 748–754. [DOI] [PubMed] [Google Scholar]
- (29).Porter NJ; Mahendran A; Breslow R; Christianson DW Unusual zinc binding mode of HDAC6-selective hydroxamate inhibitors. Proc. Natl. Acad. Sci. U. S. A 2017, 114, 13459–13464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Porter NJ; Wagner FF; Christianson DW Entropy as a driver of selectivity for inhibitor binding to histone deacetylase 6. Biochemistry 2018, 57, 3916–3924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Porter NJ; Osko JD; Diedrich D; Kurz T; Hooker JM; Hansen FK; Christianson DW Histone deacetylase 6-selective inhibitors and the influence of capping groups on hydroxamate-zinc denticity. J. Med. Chem 2018, 61, 8054–8060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Porter NJ; Shen S; Barinka C; Kozikowski AP; Christianson DW Molecular basis for the selective inhibition of histone deacetylase 6 by a mercaptoacetamide inhibitor. ACS Med. Chem. Lett 2018, 9, 1301–1305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Bhatia S; Krieger V; Groll M; Osko JD; Reβing N; Ahlert H; Borkhardt A; Kurz T; Christianson DW; Hauer J; Hansen FK Discovery of the first-in-class dual histone deacetylase-proteasome inhibitor. J. Med. Chem 2018, 61, 10299–10309. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Mackwitz MKW; Hamacher A; Osko JD; Held J; Schöler A; Christianson DW; Kassack MU; Hansen FK Multicomponent synthesis and binding mode of imidazo[1,2-α]pyridine-capped selective HDAC6 inhibitors. Org. Lett 2018, 20, 3255–3258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Vögerl K; Ong N; Senger J; Herp D; Schmidtkunz K; Marek M; Müller M; Bartel K; Shaik TB; Porter NJ; Robaa D; Christianson DW; Romier C; Sippl W; Jung M; Bracher F Synthesis and biological investigation of phenothiazine-based benzhydroxamic acids as selective histone deacetylase 6 inhibitors. J. Med. Chem 2019, 62, 1138–1166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Abendroth J; Gardberg AS; Robinson JI; Christensen JS; Staker BL; Myler PJ; Stewart LJ; Edwards TE SAD phasing using iodide ions in a high-throughput structural genomics environment. J. Struct. Funct. Genomics 2011, 12, 83–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Kim C-Y; Chang JS; Doyon JB; Baird TT; Fierke CA; Jain A; Christianson DW Contribution of fluorine to protein-ligand affinity in the binding of fluoroaromatic inhibitors to carbonic anhydrase II. J. Am. Chem. Soc 2000, 122, 12125–12134. [Google Scholar]
- (38).Kim C-Y, Chandra PP, Jain A, Christianson DW Fluoroaromatic-fluoroaromatic interactions between inhibitors bound in the crystal lattice of human carbonic anhydrase II. J. Am. Chem. Soc 2001, 123, 9620–9627. [DOI] [PubMed] [Google Scholar]
- (39).Christianson DW Structural and chemical biology of terpenoid cyclases. Chem. Rev 2017, 117, 11570–11648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Mandl-Weber S; Meinel FG; Jankowsky R; Oduncu F; Schmidmaier R; Baumann P The novel inhibitor of histone deacetylase Resminostat (RAS2410) inhibits proliferation and induces apoptosis in multiple myeloma (MM) cells. Br. J. Haematol 2010, 149, 518–528. [DOI] [PubMed] [Google Scholar]
- (41).Woyach JA; Kloos RT; Ringel MD; Arbogast D; Collamore M; Zwiebel JA; Grever M; Villalona-Calero M; Shah MH Lack of therapeutic effect of the histone deacetylase inhibitor Vorinostat in patients with metastatic radioiodine-refractory thyroid carcinoma. J. Clin. Endocrinol. Metab 2009, 94, 164–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Senger J; Melesina J; Marek M; Romier C; Oehme I; Witt O; Sippl W; Jung M Synthesis and biological investigation of oxazole hydroxamates as highly selective histone deacetylase 6 (HDAC6) inhibitors. J. Med. Chem 2016, 59, 1545–1555. [DOI] [PubMed] [Google Scholar]
- (43).Cappalonga Bunn AM; Alexander RS; Christianson DW Mapping protein-peptide affinity: binding of peptidylsulfonamide inhibitors to human carbonic anhydrase II. J. Am. Chem. Soc 1994, 116, 5063–5068. [Google Scholar]
- (44).Osko JD; Christianson DW Methods for the expression, purification, and crystallization of histone deacetylase 6-inhibitor complexes. Methods Enzymol. 2019, 626, 447–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Winn MD; Ballard CC; Cowtan KD; Dodson EJ; Emsley P; Evans PR; Keegan RM; Krissinel EB; Leslie AGW; McCoy A; McNicholas SJ; Murshudov GN; Pannu NS; Potterton EA; Powell HR; Read RJ; Vagin A; Wilson KS Overview of the CCP4 suite and current developments. Acta Cryst. 2011, D67, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (46).Battye TGG; Kontogiannis L; Johnson O; Powell HR; Leslie AGW iMOSFLM: a new graphical interface for diffraction image processing with MOSFLM. Acta Cryst. 2011, D67, 271–281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (47).Evans PR; Murshudov GN How good are my data and what is the resolution? Acta Cryst. 2013, D69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (48).McCoy AJ; Grosse-Kunstleve RW; Adams PD; Winn MD; Storoni LC; Read RJ Phaser crystallographic software. J. Appl. Crystallogr 2007, 40, 658–674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Emsley P; Lohkamp B; Scott WG; Cowtan K Features and development of Coot. Acta Cryst. 2010, D66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Adams PD; Afonine PV; Bunkóczi G; Chen VB; Davis IW; Echols N; Headd JJ; Hung L; Kapral GJ; Grosse-Kunstleve RW; McCoy AJ; Moriarty NW; Oeffner R; Read RJ; Richardson DC; Richardson JS; Terwilliger TC; Zwart PH PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Cryst. 2010, D66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (51).Chen VB; Arendall WB III; Headd JJ; Keedy DA; Immormino RM; Kapral GJ; Murray LW; Richardson JS; Richardson DC MolProbity: all-atom structure validation for macromolecular crystallography. Acta Cryst. 2010, D66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.