

Abstract
Single-chain antibodies from camelids have served as powerful tools ranging from diagnostics and therapeutics to crystallization chaperones meant to study protein structure and function. In this study, we isolated a single-chain antibody from an Indian dromedary camel (ICab) immunized against a bacterial 14TM helix transporter, NorC, from Staphylococcus aureus. We identified this antibody in a yeast display screen built from mononuclear cells isolated from the immunized camel and purified the antibody from Escherichia coli after refolding it from inclusion bodies. The X-ray structure of the antibody at 2.15 Å resolution revealed a unique feature within its CDR3 loop, which harbors a Zn2+-binding site that substitutes for a loop-stabilizing disulfide bond. We performed mutagenesis to compromise the Zn2+-binding site and observed that this change severely hampered antibody stability and its ability to interact with the antigen. The lack of bound Zn2+ also made the CDR3 loop highly flexible, as observed in all-atom simulations. Using confocal imaging of NorC-expressing E. coli spheroplasts, we found that the ICab interacts with the extracellular surface of NorC. This suggests that the ICab could be a valuable tool for detecting methicillin-resistant S. aureus strains that express efflux transporters such as NorC in hospital and community settings.
Keywords: single-domain antibody (sdAb, nanobody); zinc; multidrug transporter; isothermal titration calorimetry (ITC); X-ray crystallography; drug resistance; antimicrobial efflux; camelid antibody; major facilitator superfamily (MFS); MRSA diagnosis; NorC; efflux pump
Introduction
Multidrug efflux has emerged as a major strategy employed by drug-resistant pathogens to overcome antibiotic and biocide stress that they encounter in their environments (1). Integral membrane transporters belonging to five major families, including the major facilitator superfamily (MFS),6 multidrug and toxic compound extrusion (MATE) family, small multidrug resistance (SMR) family, ATP-binding cassette (ABC) transporters, and resistance nodulation division (RND) superfamily, are involved in the process of active efflux of antibacterial compounds (2). Among the numerous efflux pumps observed in bacterial species, the major facilitator superfamily comprises the largest class of secondary active transporters (3). Efflux transporters within this superfamily are grouped into drug:H+ antiporters (DHA) containing 12 or 14 transmembrane (TM) helices referred to as DHA1 and DHA2 families, respectively (3, 4). The DHA2 family members are capable of transporting a wide array of substrates, including antibiotics, bile salts, and lipophilic cations (5, 6). A large number of DHA2 efflux transporters exist within pathogenic bacteria, although, unlike the DHA1 members, there is no representative atomic structure available for any of them. Efflux pumps have gained increased attention in recent times as a potential target for efflux pump inhibitors that could serve as adjuvants enhancing the efficacy of existing antibiotics (7, 8).
The role of DHA members acquires prominence in Gram-positive pathogens like methicillin-resistant S. aureus (MRSA), which lack RND family transporters due to the absence of an outer membrane (9). Numerous DHA2 efflux transporters are observed in MRSA, including QacA, NorB, and NorC, that are proposed to efflux antibacterial compounds (10) and quinolone antibiotics, respectively (11, 12). MRSA is known to cause infections ranging from minor skin infections like boils and abscesses to lethal infections like bacteremia, sepsis, and endocarditis (13). Also, detection of efflux pumps in the bacterial membrane would aid in ascertaining whether the superbug could efflux drugs of certain chemical classes. Any tool that either detects or blocks efflux in pathogens would serve as a diagnostic or therapeutic agent to test for the presence and treatment of superbugs, respectively. In this study, we employ NorC as a test case to generate single domain camelid antibodies from an Indian dromedary camel (Camelus dromedarius) for performing structural studies.
Single-domain camelid antibodies, also known as variable heavy chain antibodies (VHHs) or nanobodies, consist of only a heavy chain in their structure with a single domain retaining the epitope-binding complementarity-determining regions (CDRs) instead of an antibody fragment that comprises a light chain and a heavy chain (14). VHH domains were identified in dromedaries (15) and in sharks (16) and have become powerful tools due to their small size (∼15 kDa), high stability, greater permeability and ease of heterologous expression compared with conventional antibodies (∼150 kDa) (17, 18).
The paratope in VHH domains comprises three hypervariable CDRs that link the framework regions of the single-domain antibody as opposed to conventional antibody or Fabs that have six CDRs, three from the heavy and three from the light chain that constitute the antigen-binding site. The three CDRs of VHH are also longer than those of conventional antibodies. In particular, the CDR3 of VHH domains between strands F and G displays a stretched twisted turn conformation to shield the FR2 region, which is involved in the formation of the heterodimer interface between VH and VL domains in conventional antibodies (14). The CDR3 loop is typically stabilized by an additional disulfide between the CDR1 and -3 loops, particularly in camel VHH domains that have a longer CDR3 compared with that of llamas. The long CDRs in VHH domains compensate for the absence of the VL domain and its CDRs that form the large antigen-binding site in conventional antibodies. This property gives the VHH domains their characteristic prolate ellipsoid shape that allows them to target cavities deep within their target antigens (19). VHH domains are frequently observed to target active sites of enzymes (20) and specific conformations of receptors (21) and transporters (22) and interact with structured epitopes of their targets (23). Owing to their unique properties, VHH domains have become increasingly popular for applications in medicine, biotechnology, and research as therapeutics, diagnostics, and research tools (24–26). They have also been used successfully as crystallization chaperones to obtain diffraction quality crystals of protein targets that are otherwise difficult to crystallize like integral membrane proteins (27, 28). As a result, many of the transporters (22, 29, 30), channels (31, 32), and G protein–coupled receptors (33) that are considered recalcitrant targets could be successfully crystallized and their structures determined with the use of VHHs.
Isolation of VHH domains against a target antigen was primarily done through antigen immunization of llamas or camels followed by construction of phage or yeast display libraries for screening of high-affinity binders against the target antigen (21, 34, 35). More recently, naive synthetic nanobody yeast libraries have been constructed and used to isolate high-affinity binders against antigens (36). In this study, we followed the conventional route of immunizing an Indian camel against a putative multidrug efflux transporter, NorC, and built a yeast display library using Saccharomyces cerevisiae to isolate single-domain VHH antibodies that we refer to as ICabs (Indian camelid antibodies). We isolated a high-affinity ICab against NorC that displays a rather interesting Zn2+-binding site in the CDR3 region to compensate for the absence of the disulfide. We observe that this Zn2+-binding site plays a vital role in determining the stability of the antibody and its affinity to NorC. All atom simulations clearly reveal enhanced flexibility in the CDR3 region upon removal of the Zn2+ ion. The ICab also demonstrates an ability to bind NorC at the extracellular surface that could serve to develop efflux pump detection assays in pathogenic organisms.
Results and discussion
Camel immunization and preparation of yeast display library
A 4-year-old male Indian Camel (C. dromedarius) was immunized with purified WT NorC isolated through heterologous expression in Escherichia coli membranes. The transporter was extracted from crude membranes using dodecyl-β-d-maltopyranoside (DDM) followed by exchange into lauryl maltose neopentlyglycol during size-exclusion chromatography (Fig. S1). Purified NorC was reconstituted into proteoliposomes prior to immunization using GERBU-FAMA as adjuvant. Sera from the immunized animal were used to check for peak shifts of NorC-CGFP in fluorescence-detection size-exclusion chromatography (FSEC) (Fig. S2B). Repeated immunization was carried out to increase the titer against NorC (Fig. S2A). The pre-immune serum displayed weak binding against NorC-CGFP, evident in the shift of the NorC-GFP peak to a higher hydrodynamic size as compared to the control. However, the binding was enhanced by the sixth week of immunization, with the serum having an enriched antibody titer compared with the pre-immune serum (Fig. S2, C and D). After six rounds of immunization with the antigen, total mRNA from peripheral blood mononuclear cells (PBMCs) were isolated, and cDNA was prepared using RT-PCR. The VHH regions were amplified from the cDNA using standard methods (35). The amplified VHH sequences were cloned into pPNL6 vector (a kind gift from Prof. Rajan Dighe, IISc) followed by electroporation into S. cerevisiae (EBY100) cells to build the yeast display library (Fig. 1A). The VHH fragment was cloned 3′ to the aga2 gene in between hemagglutinin and c-Myc tags. Expression of VHH on yeast surface was followed using c-Myc tag at the C terminus.
Figure 1.

Screening of VHH against NorC using yeast display. A, schematic of the yeast surface display strategy used for screening of VHH (ICab) binders against NorC (T4C) labeled with fluorescein (excitation, 494 nm; emission, 521 nm). A yeast display library built using vhh genes from PBMCs of an immunized camel was screened using anti-c-Myc antibody labeled with Alexa Fluor 647 (excitation, 650 nm; emission, 668 nm). B, FACS screening of uninduced yeast cells displays no shifts when incubated with fluorescein-labeled NorC and displays no nonspecific binding to anti-c-Myc antibody. Induced cells expressing ICab3 display diagonal shifts, signifying binding of NorC to surface-displayed ICab3.
Screening and isolation of ICabs against NorC
The yeast display library was constructed to screen binders against NorC using fluorescently labeled protein that was cross-linked with fluorescein maleimide after creating a substitution (cysteine) at the N terminus (T4C). FACS was performed to screen and isolate yeast cells displaying binders against NorC with high library expression and showing upward shifts in the mean fluorescence intensity when incubated with the labeled antigen (Fig. S3, A–D). About 28 of the 60 clones used for screening displayed promising FACS profiles. Sanger sequencing yielded four unique clones (ICab1 to -4) (Fig. S3). We could overexpress three of the four ICabs in E. coli Rosetta cells that yielded protein in inclusion bodies. We were successful at refolding one of the three proteins, ICab3 (Fig. 1B), using urea denaturation and refolding by gradually reducing urea concentration in a stepwise dialysis at 4 °C. Interestingly, ICab3 lacks a Cys residue in the CDR3 region, unlike the remaining ICabs identified in this study (Fig. 2F and Fig. S3E). The refolded protein was homogeneous (Fig. S6A) and pure (Fig. S6B) and gave a typical β-sheet secondary structure profile in CD suggesting proper folding (Fig. S6C). The leader sequence cleaved post-expression as the estimated mass of ICab3 correlated to protein without the pelB signal sequence in MALDI-TOF. The pure protein behaved as a monomer in solution when analyzed by FSEC and displayed shifts in the FSEC when incubated with NorC-CGFP, suggesting formation of a stable NorC ICab3 complex (Fig. S6D).
Figure 2.

Structural features of ICab3. A, overall structure of ICab3. CDR1, CDR2, and CDR3 regions are represented by red, green, and cyan, respectively; transparent spheres depict the disulfide bond between Cys26 and Cys103. B, anomalous map (magenta mesh) of ICab3 contoured at 6.0σ showing a strong density in the CDR3 of each chain. C, 2Fo − Fc map contoured at 3.0σ showing Zn2+-coordinating residues in the Zn2+-binding pocket. D, Zn2+ is bound with a tetrahedral coordination geometry, outlined with gray lines; dashes show distances between Zn2+ and coordinating atoms (Å). E, structural superposition of ICab3 (light blue) with another camelid VHH (gray; PDB code 3JBE) showing overlap of the Zn2+-binding region of ICab3 (light blue) with that of the disulfide bond of 3JBE. F, multiple-sequence alignment of ICab3 with other VHH sequences whose structures are known. β-Strands are represented by overhead arrows, whereas CDR1 to -3 are highlighted in the same color as in A. Cysteines that form disulfides have been shown with connectors (-S-S-), whereas the region in CDR3 that harbors additional cysteine is demarcated with a dashed line. Conservation scores are given in the histogram below. 0, least conservation; *, total conservation. −, unassigned region. FR1–4 represent framework regions 1–4 in the VHH.
Crystallization and structure determination of ICab3
ICab3 was crystallized by hanging-drop vapor diffusion method at 4 °C, and the structure was determined through molecular replacement using a related VHH structure (PDB code 3JBE) as a template to a resolution of 2.15 Å (Table 1).
Table 1.
Crystallographic data and refinement statistics
| Native | Zn2+ anomalous | |
|---|---|---|
| Data collection statistics | ||
| X-ray source | CuKα | ESRF ID29 |
| Temperature | 100 K | 100 K |
| Space group | P32 2 1 | P32 2 1 |
| Cell dimensions | ||
| a, b, c (Å) | 106.9, 106.9, 70.6 | 106.7, 106.7, 71.4 |
| α, β, γ (degrees) | 90, 90, 120 | 90, 90, 120 |
| Wavelength (Å) | 1.5417 | 1.2814 |
| Resolution (Å) | 53.44–2.15 (2.22–2.15) | 71.40–2.10 (2.16–2.10) |
| Total number of observations | 225,745 (17,407) | 231,995 (15,128) |
| Unique reflections | 25,502 (2171) | 27,730 (2218) |
| Multiplicity | 8.9 (8.0) | 8.4 (6.8) |
| Data completeness | 99.6 (98.7) | 99.9 (99.1) |
| I/σI | 11.1 (1.9) | 17.0 (3.0) |
| Rpim (%) | 4.5 (42.2) | 2.8 (21.7) |
| CC½ | 0.997 (0.624) | 0.998 (0.862) |
| Anomalous completeness (%) | 99.7 (98.2) | |
| Anomalous multiplicity | 4.1 (3.4) | |
| DelAnom CC½ | 0.625 (0.044) | |
| Refinement statistics | ||
| Resolution (Å) | 46.28–2.15 | |
| Unique reflections | 25,479 | |
| Rwork (%)/Rfree (%) | 19.32/23.24 | |
| Non-hydrogen atoms/molecules | ||
| Protein | 2008 | |
| Ethylene glycol | 18 | |
| Water | 121 | |
| Sodium | 1 | |
| Zinc | 2 | |
| Average B-factor (Å2) | ||
| Protein | 59.4 | |
| Ethylene glycol | 67.4 | |
| Zinc | 39.2 | |
| Sodium | 49.12 | |
| Water | 60.9 | |
| RMSD, bond lengths (Å) | 0.006 | |
| RMSD, bond angles (°) | 0.914 | |
| Ramachandran plot | ||
| Favored (%) | 98.47 | |
| Allowed (%) | 1.53 | |
| Outliers (%) | 0.00 |
| (Eq. 1) |
| (Eq. 2) |
The refined X-ray structure of ICab3 displays a canonical immunoglobulin fold of VHH with nine β-strands organized into two β-sheets (Fig. 2A). The β-sandwich comprises five and four β-strands in each of the two β-sheets. The conserved disulfide bond, a defining feature of immunoglobulin structure, is present between Cys26 and Cys103 connecting the two β-sheets. The uniqueness of any VHH is determined by its hypervariable loops or CDRs because they constitute the structural determinants forming the paratope that defines the unique binding propensities with the cognate epitope. The length and sequence variability of CDR loops determine the binding affinity and specificity. The regions between the CDRs, referred to as “framework regions” (FRs), are made up of highly conserved sequences. A unique feature of VHHs that differentiates them from conventional antibodies is the presence of long CDR loops 1 and 3. Interestingly, in the ICab3 structure, CDR1 is unusually long compared with most of the VHH structures available in the Protein Data Bank. CDR1 to -3 comprise 13 residues (Gly30–Gly42), 6 residues (Lys59–Ser64), and 19 residues (Asp106–Tyr124), respectively. A close inspection of the loop sequence reveals that the additional length of CDR1 loop is due to the repetition of the motif STY (Fig. 2F), making the loop polar and rich in aromatic residues. Tyrosines were found to have a higher representation in the CDRs of many VHHs and are known to mediate strong interactions with the antigen in many of the known VHH-antigen complexes (37). Despite having a longer length, the B-factors of atoms in the additional residues in CDR1 are comparable with the rest of the framework regions, likely due to their close interactions with symmetry-related partners. As mentioned earlier, VHHs bind to their antigen with high shape surface complementarity, and the long protruding CDR1 suggests the likelihood that it could closely interact with crevices or the vestibular region in NorC. Interestingly, CDR1 and -2 of ICab3 are not only enriched in Tyr but also have several Ser and His residues. Thus, the paratope of ICab3 has a more hydrophilic character in contrast to the paratope of other VHHs characterized so far, which have a more hydrophobic surface.
The long CDR3 loop is a hallmark feature of camel VHHs and is usually shorter in the case of llama VHHs. The long loop folds back over the FR2 region, assuming a typical stretched-twist conformation (38) covering a part of the FR2 region. In conventional antibody, FR2 of the VH domain has residues Val42, Gly49, Leu50, and Trp52 that make the surface hydrophobic, thus readily allowing VL and VH domains to dimerize and exist as a stable dimer in solution (39). The corresponding residues of FR2 of ICab3 are Phe44, Glu51, Arg52, and Gly54, allowing it to exist as a stable monomer. These amino acid substitutions are a characteristic feature of VHHs and are highly conserved (40, 41).
CDR3 reveals a Zn2+-binding site
During the refinement of the ICab3 structure, we encountered a strong electron density in the midst of the CDR3 loop and suspected it to be a metal ion. Considering that tetrahedral coordination geometry is possible for transition metal ions (42), we modeled it as a Zn2+ ion and confirmed the same by an X-ray energy scan at the synchrotron that yielded a strong anomalous signal for Zn2+ at its absorption edge (9675 eV) (Fig. 2B and Fig. S4). Asp106, His109, and His116 from CDR3 and Cys40 from CDR1 coordinate the Zn2+ ion with a tetrahedral geometry (Fig. 2, C and D). The Zn2+ ion was observed in the structure despite no extraneous addition of metal ion during refolding, purification, or crystallization. Subsequently, we added Zn2+ during the refolding process and found that the refolding efficiency improved. The presence of a metal ion in the CDR3 loop is unique and has not been reported for any of the VHH structures solved so far. Upon comparison of the ICab3 structure with other VHH structures, we observed that the Zn2+ occupies the same position that harbors a disulfide bond in many dromedarian VHHs that possess long CDR3 loops (Fig. 2, E and F) (43). This disulfide bond is formed between an additional pair of cysteines present in the CDR1 and CDR3 loop and likely restrains their flexibility (44). The additional cysteine in the CDR1 region generally occurs at a conserved position and is encoded in the germ line genes of camels, whereas the one in the CDR3 region is variable in its position (Fig. 2F) and probably is the result of recombination or hypersomatic mutation (45, 46). This conserved disulfide bond, however, is absent in llama VHHs as the loops are relatively shorter in them (41). Restraining the long CDR3 loop with an additional disulfide reduces the entropic cost of binding with the antigen, thus increasing the binding affinity (47). In the ICab3 structure, Zn2+ ion acts as a bridge between CDR1 and CDR3 loops, imparting conformational rigidity to them. Hence, it seems to play the same role as is provided by the disulfide bond in other VHHs. Also, the presence of a bound Zn2+ likely provides greater rigidity to the CDR3 loop conformation compared with the disulfide bond, as it involves coordination with four residues, three of which come from different parts of the CDR3 loop.
High-affinity ICab3-NorC interaction requires the Zn2+-binding site
The affinity of ICab3 for NorC was measured using isothermal titration calorimetry (ITC), wherein we titrated NorC with ICab3 and observed a binding affinity of 38 nm (Fig. 3A and Table 2). Such high binding affinity suggests high specificity and a strong affinity of ICab3 for NorC.
Figure 3.

Binding analysis of ICab3 and its mutants with NorC. A, ITC profile (top, differential power; bottom, binding isotherms with integrated peaks normalized to moles of injectant and offset-corrected) showing nanomolar affinity of WT ICab3 with NorC. B, the binding affinity decreases 100-fold in the case of ICab3 V115C mutant. C, ICab3 H116C has no affinity for NorC.
Table 2.
ITC data
| WT ICab3 | ICab3 V115C | ICab3 H116C | |
|---|---|---|---|
| n (sites) | 0.997 | 1.16 | NDa |
| KD (nm) | 38.2 ± 5.7 | 4060 ± 591 | ND |
| ΔH (kcal/mol) | −7.0 ± 0.07 | −10.0 ± 0.45 | ND |
| TΔS (kcal/mol) | 3.1 | −2.6 | ND |
| ΔG (kcal/mol) | −10.1 | −7.4 | ND |
| Model | One set of sites | One set of sites | ND |
a ND, not determined.
To further understand the role and importance of Zn2+ ion in structural and functional properties of ICab3, we created point mutations to abolish Zn2+ binding. We also wanted to see the effect of engineering a disulfide bond back into the CDR3 region and evaluate its effect on the WT ICab3 structure. Therefore, we mutated His116, which coordinates with Zn2+, to a Cys residue. Upon analyzing all of the reported structures of VHH, we found few structures (PDB entries 3JBE, 2X6M, 1RI8, an 1RJC) whose CDR3 was similar to that of the ICab3 with respect to the size and overall conformation but comprised of an interloop disulfide bond between the Cys residues that corresponded to Cys40 and Val115, respectively, of the ICab3 structure (Fig. 2, E and F). Given the flexibility in the position of the cysteine residue that forms a disulfide in the CDR3 of numerous VHH structures, we also substituted Val115 to a Cys residue. We purified both of these mutants by refolding with a similar protocol as was optimized for WT ICab3 but with few modifications. As we expected these to be devoid of Zn2+, we did not add any external Zn2+. We also maintained 1 mm β-mercaptoethanol in the dialysis buffers to avoid disulfide bond formation among mismatched cysteines during the refolding process.
We analyzed the formation of disulfide bonds using MS and PEG maleimide cross-linking. As expected, the ESI-MS spectrum of WT ICab3 conformed to a zinc-free species with two oxidized cysteines corresponding to a disulfide bond between Cys26 and Cys103 (Fig. S5A). On the other hand, the masses of both of the mutants, ICab3 V115C and ICab3 H116C, corresponded to zinc-free species with four oxidized cysteines, plausibly harboring two disulfide bridges. This premise was evaluated through cross-linking free cysteine(s) with PEG(5000) maleimide (Fig. S5B). Whereas SDS and heat-denatured WT ICab3 formed a PEG maleimide adduct of higher molecular weight, the same was not observed in the case of either ICab3 V115C or H116C, thereby suggesting formation of disulfide linkages between Cys40 and V115C or H116C, respectively.
The effect of both these mutations on the binding affinity with NorC was measured using FSEC and ITC. Interestingly, in FSEC, there was no change in elution volume of NorC upon incubation with ICab3 H116C, suggesting a lack of interaction between them. However, incubation of ICab3 V115C with NorC yielded a reduced change in the elution volume of NorC-ICab3 V115C complex, compared with the NorC-WT ICab3 complex, suggesting weakened interaction (Fig. S6, E and F). To further quantitate the binding affinity, we performed ITC and titrated both of the mutants against NorC. The results revealed that H116C mutation completely abolished the binding, whereas substituting Val115 with a Cys residue decreased the affinity by about 100-fold compared with the WT (Fig. 3, B and C), as initially suggested by FSEC (Fig. S6, E and F). Also, the addition of Zn2+ enhanced the affinity of WT ICab3 for NorC by 2-fold to about 19 nm, whereas it did not have any effect on affinity of mutants for NorC (Fig. S7). It can therefore be inferred that Zn2+ binding is indispensable for retaining the binding affinity of ICab3 with NorC. Mutating the Zn2+-binding residue likely distorts the CDR3 loop, resulting in the loss of affinity with NorC. It is therefore plausible that the C40-V115C and C40-H116C disulfide bonds contort the CDR3 to varying extents, resulting in a significant reduction or complete loss of the ability to interact with the transporter, respectively.
Effect of substitution of Val115 and His116 to Cys on the stability of ICab3
As mentioned earlier, having additional disulfides introduces rigidity in the CDR loops, and in one of the previous studies, it was found that camel VHHs with an additional disulfide bond had higher Tm values compared with llama VHH domains that lack a disulfide in CDR3 (48). Also, altering this disulfide with different pairs of residues lowered the thermal stability (ΔTm ∼10–12 °C) and reduced the affinity toward the antigen by ∼2–10-fold (47). In this context, it is interesting to look at the extent of thermostabilization achieved by having a Zn2+ ion in the place of a disulfide bond that conventional dromedary VHHs possess. Hence, we carried out thermal denaturation studies of WT ICab3 and its mutants by nano-differential scanning fluorimetry (nano-DSF) that measures small changes in intrinsic tryptophan fluorescence in a label-free manner as a protein molecule unfolds with increasing temperature (49). Thermal melting temperature (Tm) of WT ICab3 was found to be ∼75.0 °C, with a mild increment in Tm to ∼78 °C with the addition of Zn2+ (Fig. 4A and Table 3). However, this high stability of WT ICab3 was compromised in the presence of EDTA, resulting in a Tm value of ∼64 °C. This suggests a vital effect of Zn2+ binding on the stability of WT ICab3. We further observe a reduced stability in V115C and H116C mutants, with Tm values at 70 and 64 °C, respectively (Fig. 4, B and C). This reduction in stability remains unaffected in the presence or absence of excess Zn2+ or EDTA.
Figure 4.
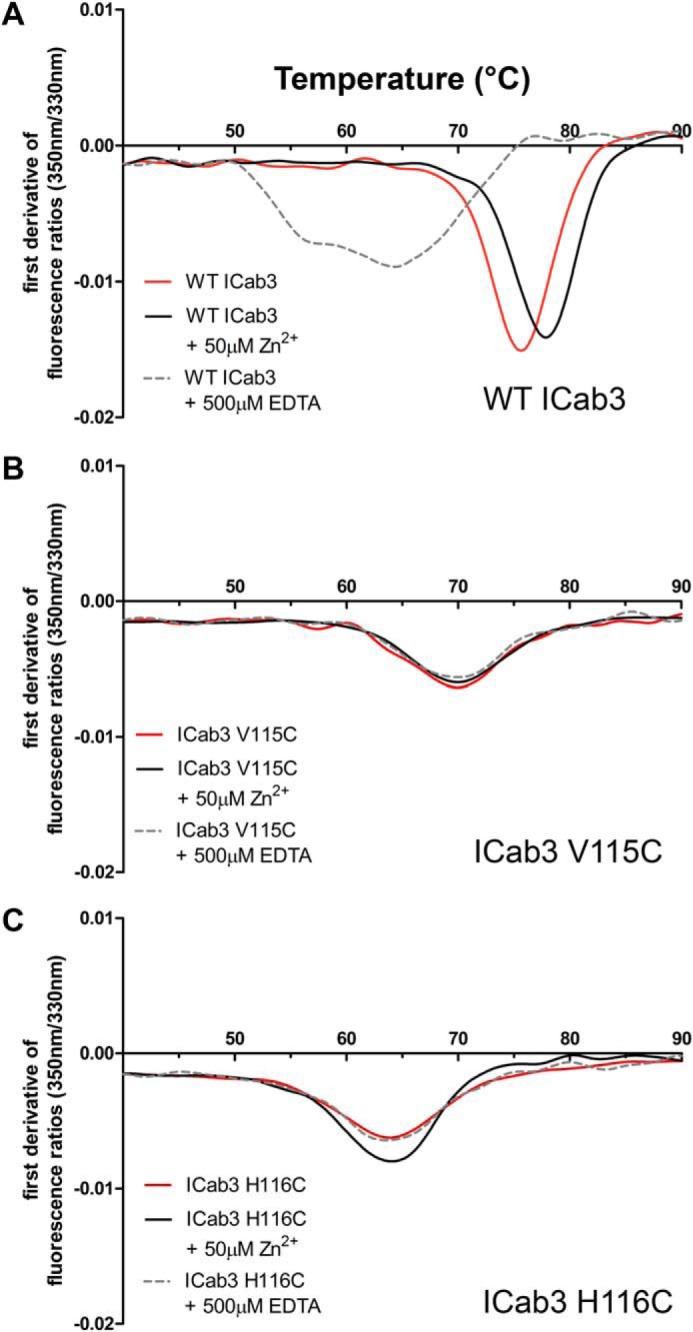
Thermal stability of ICab3 and its mutants. Nano-DSF profiles of WT ICab3 and the mutants showing highest thermal stability for WT ICab3 (A) followed by ICab3 V115C (B) and ICab3 H116C (C). The Tm of WT ICab3 resembles that of ICab3 H116C upon the addition of 0.5 mm EDTA. While, Tm of WT ICab3 increases upon the addition of Zn2+, neither its excess nor the presence of EDTA as its chelator alters the Tm of ICab3 V115C or ICab3 H116C.
Table 3.
Thermal stability data
| ICab3 WT | ICab3 V115C | ICab3 H116C | |
|---|---|---|---|
| °C | °C | °C | |
| Native | 75.6 | 70.5 | 63.9 |
| +50 μm Zn2+ | 77.8 | 70.5 | 64.0 |
| +500 μm EDTA | 64.1 | 69.8 | 63.7 |
Zn2+ restrains the CDR3 loop to a fixed conformation
As mentioned earlier, CDR3 in camelid VHHs is generally large and is required to be restrained for better binding to the antigen. Therefore, a disulfide bond has been evolutionarily selected in the CDR3 loop of many camelid VHHs that prevents the loop from acquiring greater mobility. In the case of ICab3, the equivalent site contains a Zn2+ ion. The presence of Zn2+ restrains the flexibility of CDR3 loop, as reflected by the low B-factor values for the residues of these loops as compared with the B-factors in the framework regions. The CDR1 loop, which is longer than usual, is also highly restrained, as evidenced by the low B-factor. To further understand the conformational landscape of the CDR loops and the role and importance of the Zn2+ ion in limiting their conformational freedom, we carried out all-atom molecular dynamics simulations for 100 ns using Zn2+-bound as well as Zn2+-free ICab3. During the entire run of both the simulations, the ICab3 molecule remained very stable, as is evident from the RMSD plot of all residue Cα atoms (Fig. S8A). When simulation was carried out in the presence of Zn2+ ion, the CDR3 loop remained spatially confined and Zn2+-coordinating residues retained their positions, suggesting a strong stabilizing effect of Zn2+ on CDR3 (Fig. 5A). However, removing Zn2+ from CDR3 results in its destabilization (Fig. 5B), as can be seen in the increased root mean square fluctuation values of its residues (Fig. S8B). As a result, Zn2+-coordinating residue His116 moves away from the Cys40, and by the end of 20 ns, it flips to the outside toward the solvent (Fig. 5B). The residues that lie in the vicinity of His116 also show a marked increase in fluctuation, suggesting that Zn2+ helps to restrict the movement of the CDR3 loop, thereby increasing stability and restricting the conformational mobility (Fig. 5, C–F). As mentioned earlier, the CDR1 loop makes extensive hydrogen bonding with CDR3 loop and contains amino acids with heavy side chains; it does not fluctuate much from its mean position in the presence or absence of Zn2+ during the entire run of the simulation. Thus, the presence of Zn2+ affects conformational freedom of CDR3 loop only (Fig. S8, C and D).
Figure 5.

Molecular dynamics simulations highlighting the role of Zn2+ in providing conformational stability to CDR3. A and B, representation of structure along the trajectory at different time points in the Zn2+-bound (A) and Zn2+-free (B) state. In the presence of Zn2+, CDR3 loop conformation is restrained, whereas in the absence of Zn2+, it becomes more mobile, which allows the His residue (involved in coordination to Zn2+) to flip outside. C and D, RMSD of residues in the CDR3 region over the course of trajectory in the presence of Zn2+ (C) and in the absence of Zn2+ (D). E and F, RMS flexibility of residues in the CDR3 region in Å/ns in the presence of Zn2+ (E) and in the absence of Zn2+ (F); only side-chain atoms were taken for calculation. Conformational flexibility of residues from 115 to 121 significantly increases in the absence of Zn2+ coordination.
ICab3 interacts with the extracellular surface of NorC
ICab generation and selection criterion, employed in this study, leads to selection of ICabs that can access both extra- and intracellular surfaces of the transporter. Consequently, the selected ICab may potentially bind to either the extracellular or cytosolic face of the transporter. However, antibodies that display high-affinity interactions to the extracellular side of the transporter could serve as good candidates to develop screening and detection tools to identify efflux pumps in S. aureus strains.
We carried out confocal microscopy studies to investigate whether ICab3 binds to the extracellular or cytosolic face of NorC (Fig. 6). NorC-GFP was expressed in E. coli LMG194 cells. The outer membrane of the E. coli was digested with lysozyme-EDTA to make spheroplasts (50). WT ICab3 and ICab3 H116C were coupled to NHS-rhodamine by amine coupling, and spheroplasts expressing NorC-GFP was incubated with these fluorescently labeled ICabs and visualized under a confocal microscope. GFP fluorescence localized at the cell membrane of the spheroplasts was indicative of NorC's expression in E. coli's inner cell membrane. Spheroplasts incubated with rhodamine-labeled WT ICab3 displayed co-localization of GFP and rhodamine signals on the cell membrane, suggesting binding of WT ICab3 to the outer side of the expressed NorC. For negative control, uninduced E. coli spheroplasts were used instead, where no fluorescence was observed at the cell membrane. Also, as another negative control, we used rhodamine-labeled ICab3 H116C, as we knew a priori that it does not interact with NorC. Neither of the cases exhibited any rhodamine localization on the spheroplast surface, confirming that WT ICab3 specifically binds to NorC. DAPI was used to search for intact spheroplasts, and anti-His–anti-mouse-Alexa Fluor 647, which binds to the C-terminal His tag of NorC-GFP, was used to make sure that the spheroplasts were not ruptured or clumped with other membrane debris, as ICab3 can readily stick onto it. This ensured that the topology of NorC inferred for the experiment was consistent with the conclusion that ICab3 binds to the extracellular side of NorC.
Figure 6.

Confocal imaging showing WT ICab3 binding to the extracellular face of NorC in E. coli spheroplasts. Images were acquired after incubating E. coli spheroplasts expressing NorC-GFP with rhodamine-labeled WT ICab3 (A) and rhodamine-labeled ICab3 H116C (B). C, images acquired with uninduced E. coli spheroplasts incubated with rhodamine-labeled WT ICab3. Total number of cells counted for A = 40, and that for B and C was 15 each in four trials. Scale bar, 1 μm. DIC, differential interference contrast.
Conclusions
The structure of ICab3 displays a previously unknown facet of CDR3 having a Zn2+-binding site. Although we have clearly established the metal ion to be a zinc ion through crystallographic methods, the site could potentially bind other transition metals. The Zn2+ site, in addition to being important to retain the stability of the single domain antibody, is also vital for controlling the interactions with the antigen NorC. This novel CDR3 modification could also be used to engineer synthetic versions of VHHs to expand the repertoire of individual naive VHH libraries against clinically relevant targets. In the context of ICab3 NorC interaction, the antibody could serve as potential proof-of-principle to use VHH domains as detection tools to evaluate the presence of efflux pumps of certain types in pathogenic superbugs. Although we could not demonstrate the effect of ICab3 on the transport properties of NorC due to lack of any biochemical transport assays, VHH-based inhibition of efflux pumps could be a potential line of exploration for developing efflux pump inhibitors for use as antibiotic adjuvants in the not so distant future.
Experimental procedures
Cloning, expression, and purification of NorC to use as antigen
NorC was cloned into pET-16b vector between restriction sites NcoI and KpnI with a His8 tag at its C terminus. The BL21-C41 cells were transformed with the vector containing NorC and plated on an LB-agar plate containing 100 μg/ml ampicillin. A single colony was used to grow primary culture in LB broth (Himedia) for 10–12 h at 37 °C. Large-scale cultures (2–3 liters) were grown at 37 °C until the OD600 reached 0.4, following which protein expression was induced using β-d-1-thiogalactopyranoside at a final concentration of 0.2 mm at 20 °C. The cells were further grown for about 15 h. Cells were harvested by pelleting and resuspended in a buffer containing 20 mm Hepes, pH 7.0, 200 mm NaCl and lysed at high pressure of about 800 bars using a homogenizer (GEA Niro Soavi PandaPlus 1000 homogenizer). The cell lysate was spun at 108,000 × g for 1 h at 4 °C. The pelleted membranes were dissolved in a buffer containing 20 mm Hepes, pH 7.0, 200 mm NaCl, 1 mm DDM (Anatrace), and 1 mm phenylmethylsulfonyl fluoride (buffer A) using a rotor-stator homogenizer, and the final concentration of DDM was increased to 20 mm. The solution was kept for nutation for about 2 h at 4 °C to extract NorC into micelles, followed by ultracentrifugation at 100,000 × g for 1 h at 4 °C to remove the insoluble debris. The supernatant was incubated with nickel-nitrilotriacetic acid beads pre-equilibrated with buffer A at 4 °C for 1 h. The bead-containing solution was transferred to a Bio-Rad gravity column and washed with 50 column volumes of buffer A having 30 mm imidazole. The protein was eluted with 300 mm imidazole in buffer A and further purified by size-exclusion chromatography using Superdex S-200 increase 10/300 column (GE Healthcare). The purity of the protein was analyzed by running it on SDS-PAGE. Similarly, NorC-T4C without GFP was cross-linked with N-(5-fluoresceinyl)maleimide (1:1.2 ratio) at Cys4 and SEC-purified.
NorC reconstitution into proteoliposomes
Purified NorC was reconstituted into E. coli polar lipids (Avanti Polar Lipids) at a weight ratio of 1:20 protein/lipid. 70 mg of E. coli polar lipids were dried in a continuous stream of N2 and subsequently dried under vacuum. The lipid layer was hydrated in 20 mm Hepes, pH 7.0, 200 mm NaCl buffer. Vesicles formation was induced through repeated rounds of freeze-thaw cycles after which proteoliposomes were made by incubating the liposome suspension with 3.5 mg of purified NorC and 8 mm DDM for 30 min at 4 °C. Excess detergent was removed through overnight incubation with polystyrene Bio-Beads (Bio-Rad). After detergent removal, the proteoliposomes were pelleted through ultracentrifugation (190,000 × g, 2 h) and evaluated for incorporation of NorC using FSEC.
Camel immunization and generation of immune library against NorC
The immunization was performed according to the guidelines at the National Research Center on Camels (NRCC), Bikaner, Rajasthan, India and protocols detailed for llama immunization (35). Each immunization consisted of a 600-μl mixture of reconstituted proteoliposomes (total antigen concentration was 0.3 mg/dose) with an equal amount of GERBU-FAMA adjuvant. NorC in proteoliposomes was mixed with an equal volume of GERBU-FAMA adjuvant and injected subcutaneously on either side of the neck base of the camel near the bow lymph node. A similar preparation was used for weekly booster doses for five continuous weeks. Blood samples (10 ml) were collected just before the start of the experiment, and on day 21, without anticoagulant, serum was harvested and stored at −80 °C until further analysis. On day 38, 100 ml of blood was collected using heparin as anticoagulant for isolation of PBMCs (35). The procedure for camel immunization was approved by the Institute Animal Ethics Committee, ICAR-NRCC, Bikaner. The pre-immune serum and sixth week serum were collected and mixed with antigen NorC-CGFP in a ratio of 1:5 and analyzed for shifts using FSEC.
Total RNA was isolated using the TRIzol reagent and used to generate cDNA using a Superscript II reverse transcriptase kit (Invitrogen). Subsequently, two-step nested PCR was used to amplify the final products of VHH fragments. In the first step PCR, two fragments of about 1000 and 700 bp in length were obtained. The 700-bp fragment was gel-purified and used for VHH domain amplification with VHH-specific primers, which contained homologous sequences to the yeast display vector (pPNL6) added at the 5′ ends. The yeast surface display vector and vhh fragments were digested with NheI and SalI, and the digested product was gel-purified and precipitated. About 1 μg of vector DNA was mixed with 3 μg of vhh insert and electroporated into yeast cells in 3-ms pulses at 1.5 kV. The electroporated cells were recovered in YPD medium for 2 h, centrifuged, and transferred to SDCAA medium, and the library size was estimated on SDCAA-agar.
Selection of NorC-specific VHH
The yeast display library was subjected to FACS-based cell sorting using a BD FACSAriaTM III. About 108 yeast cells were incubated with 5 μm NorC-T4C-FM antigens for binding along with anti-Myc antibody conjugated to Alexa Fluor 647 for expression, and the double positives were isolated through FACS. The isolated yeast display sublibrary was enriched by two subsequent passages, and the enrichment was analyzed by FACS (BD AccuriTM C6).
VHH domain–displaying positive clones were grown in 5 ml of SDCAA medium overnight. The cells were centrifuged at 3000 × g for 5 min and washed with autoclaved water. Thereafter, the cells were washed with 1 ml of incubation buffer (25 mm Tris, pH 7.5, 0.9 m sorbitol, 1 mm EDTA, 14 mm β-mercaptoethanol). The cell pellet was resuspended in 0.4 ml of incubation buffer and 20 μl of 100 units of lyticase was added and incubated at 37 °C overnight. The suspension was centrifuged at 1800 × g for 5 min, and the supernatant was discarded. The spheroplasts were resuspended from the pellet in suspension buffer provided in the plasmid purification kit (Qiagen), and the plasmid was extracted as specified by the instructions. VHH displaying vectors were transformed into TOP10 E. coli cells through the heat shock method. The transformed cells were plated on LB-ampicillin agar plates and incubated overnight at 37 °C. Each colony was transferred into separate LB-ampicillin broth medium and grown at 37°C overnight. The plasmids isolated from all of the clones were checked for the presence of vhh insert. All of the plasmids showing positive vhh amplification were submitted for DNA sequencing to further confirm the clones and variability.
Cloning, protein expression, and purification of ICab
The vhh was subcloned in pET-22b vector between restriction sites NcoI and XhoI in frame with pelB leader sequence to the 5′ end of the gene. E. coli Rosetta cells were transformed with vhh-containing vector and plated on LB-agar medium containing 100 μg/ml ampicillin and 34 μg/ml chloramphenicol. A single colony of the transformed cell was picked and inoculated in LB broth and grown at 37 °C for 10–12 h. Large-scale culture was grown at 37 °C until the OD600 reached 0.6, when the protein expression was induced by adding β-d-1-thiogalactopyranoside to a final concentration of 0.5 mm, and cells were further grown at 37 °C for 5–6 h. Cells were spun down and resuspended in a lysis buffer containing 20 mm Hepes, pH 7.0, 200 mm NaCl, and 1 mm phenylmethylsulfonyl fluoride and sonicated. The cell lysate was spun for 1 h at 17,000 × g at 4 °C. The supernatant was discarded, and the pellet, containing the protein of interest, was washed by dissolving in the lysis buffer, followed by centrifugation at 17,000 × g for 30 min. The pellet was dissolved in a denaturation buffer containing 20 mm Hepes, pH 7.0, 200 mm NaCl, and 6 m urea and kept for nutation for 6 h. The suspension was centrifuged at 17,000 × g for 15 min to remove any undissolved part or debris, and the supernatant was transferred in a dialysis bag with a size cutoff of 3 kDa and dialyzed against a buffer containing 20 mm Hepes, pH 7.0, 200 mm NaCl, 1 mm ZnCl2. The urea concentration was decreased in a stepwise manner to 3, 1.5, and finally to 0 m while keeping the rest of the buffer unchanged. Each dialysis step was carried out for 10–12 h at 4 °C. After the final round of dialysis, the protein solution was centrifuged at 17,000 × g for 1 h at 4 °C. The supernatant was concentrated and further purified by size-exclusion chromatography using Superdex S-75 10/300 GL column (GE Healthcare) in a buffer containing 20 mm Hepes, pH 7.0, and 200 mm NaCl. The purified protein was run on SDS-PAGE to check for purity and integrity and subjected to MALDI-TOF to check for molecular mass.
Site-directed mutagenesis of ICab3
The site-directed mutagenesis of ICab3 was carried out by the single-primer extension method. A single primer was designed for each mutant by changing the codon to introduce the desired mutation at the specific site. The primer was used to amplify the entire plasmid carrying the vhh gene by PCR. The amplicon was treated with Dpn1 at 37 °C for 2–3 h to degrade the WT plasmid and transformed into TOP10 E. coli cells. The transformed cells were plated on LB-ampicillin agar plates and incubated overnight at 37 °C. Individual colonies were used for plasmid isolation. The positive clones were confirmed by DNA sequencing. E. coli Rosetta cells were transformed with the mutant plasmid. The cells were plated on an LB-agar plate containing 100 μg/ml ampicillin and 34 μg/ml chloramphenicol and grown at 37 °C for 10–12 h. The mutant proteins were expressed and purified in the same manner as the WT ICab3 except with certain modifications. β-Mercaptoethanol was kept at a concentration of 1 mm in all of the buffers except the final dialysis buffer and the SEC buffer. Also, ZnCl2 was not added at any of the steps.
Isothermal titration calorimetry
The binding affinity of ICab3 and its mutants with NorC was determined by ITC using Microcal PEAQ-ITC (Malvern Panalytical) at 25 °C. The purified WT ICab3 and its mutants were dialyzed against a buffer containing 20 mm Hepes, pH 7.0, 200 mm NaCl, and 4 mm decyl-β-d-maltopyranoside detergent. The NorC protein was purified in the same buffer. All of the titrations consisted of 18 injections, where the first injection was set up at 0.4 μl and all the subsequent injections were set up at 2 μl with a time duration of 4 s for each injection. The time between two consecutive injections was kept at 150 s, and the sample in the cell was stirred at 750 rpm during the entire run. 319 μm WT ICab3 was titrated against 300 μl of 30 μm NorC. For mutants, 350 and 317 μm ICab3-V115C and ICab3-H116C, respectively, were titrated against 300 μl of 25 μm NorC. For blank, WT ICab3 and mutants were titrated against buffer, and heat of mixing and dilution were subtracted from the titration data against NorC. All of the data were fit using Microcal PEAQ-ITC analysis software with one set of sites model.
Thermal stability measurements
Thermal stability measurements were carried out using nano-DSF using a Prometheus NT.48 instrument (NanoTemper Technologies). This measures the change in intrinsic tryptophan fluorescence as the protein molecule unfolds with increasing temperature. Fluorescence at 330 and 350 nm is recorded, and a ratio of F350/330 is plotted as a function of temperature. About 0.5 mg/ml protein solution in a buffer containing 20 mm Hepes, pH 7.0, and 200 mm NaCl was put into nano-DSF NT.48 capillaries (nanoTemper) and placed on the instrument capillary holder. A temperature scan was carried out between 25 and 95 °C with a scan rate of 1 °C/min. All measurements were carried out in duplicates. The first derivative of the ratio of F350/330 was plotted against temperature, where the critical point gives the Tm.
PEG-maleimide cross-linking
The WT ICab3 and the mutant proteins (∼30 μm) in 50 mm Hepes, pH 7.0, were mixed with PEG(5000) maleimide (Sigma) in native and denatured state in a molar ratio of 1:20 (protein/PEG(5000) maleimide) and incubated at room temperature for 3 h. The denatured sample was prepared by mixing the proteins with 0.5% SDS and incubating at 90 °C for 10 min. Samples also contained 0.5 mm EDTA to chelate any free Zn2+ ion. All of the samples were then cooled to room temperature, and PEG(5000) maleimide was added. The reaction was quenched by adding 3 mm DTT. All of the samples were run on SDS-PAGE to check for labeling-induced gel shifts.
Mass spectrometry
The mass of the intact protein was determined by LC-ESI MS. The samples were analyzed on an ESI-MS, Maxis Impact (Bruker Daltonics, Bremen, Germany), which was coupled with an HPLC 1260 Infinity (Agilent Technologies). The protein samples were passed through C8 Zorbax 300SB, 3.1 × 100-mm RP column. The mobile phase consisted of water and acetonitrile with 0.1% formic acid. The linear gradient of 10–90% acetonitrile in 25 min was run with a flow rate of 0.2 ml/min. For ESI, nebulizer pressure was kept at 26 p.s.i., dry gas flow at 9 liters/min, dry temperature at 250 °C, and capillary voltage at 3500 V. Data were analyzed using the Bruker software Data Analysis 4.1.
Crystallization, data collection, and structure determination
The ICab3 was crystallized by the hanging-drop vapor diffusion method at 4 °C. 0.75 μl of protein (5 mg/ml) was mixed with 0.50 μl of the crystallization condition containing 0.1 m Tris-Cl, pH 8.25, 3.5 m sodium formate, and 5% ethylene glycol and placed on a siliconized coverslip. The crystals appeared within a week. The crystals were flash-frozen in liquid N2, and X-ray diffraction data were collected at the home source at the CuKα X-ray radiation of 1.5417 Å wavelength, generated by a Rigaku FR-E+ Superbright X-ray generator operating at 45 kV and 55 mA and focused with an osmic mirror system. A total of 180 frames were collected at 100 K using a Rigaku R-axis IV detector, with a 1° oscillation range and 1° frame width. The crystal-to-detector distance was kept at 180 mm. The diffraction data were processed using iMosflm (51) and scaled using AIMLESS (52) of the CCP4i2 software suite (53). The data collection and processing statistics are given in Table 1. To check for the presence of metal ion, an X-ray energy scan was carried out at the ID-29 beamline at the ESRF synchrotron (Grenoble, France). The structure was determined by molecular replacement using the program PHASER (54) of the CCP4i2 suite. A known VHH structure (PDB code 3JBE) was used as a model to search for two molecules in the asymmetric unit, resulting in structure solution in space group P3221. The model obtained from molecular replacement was manually built using COOT (55) and refined by PHENIX Refine (56) iteratively until the refinement converged to R and Rfree values of 19 and 24%, respectively. The refinement statistics are given in Table 1. The final structure was validated using MOLPROBITY (57).
Confocal microscopy
E. coli LMG194 cells, harboring pBAD-norc-gfp, were grown in 30–40 ml of LB broth. When the OD600 of the culture reached 0.4, the culture was split into two equal parts; one part was induced with 0.05% (w/v) l-arabinose, whereas the other part was kept uninduced. Both of the cultures were grown further for 4–5 h at 25 °C, after which they were pelleted down, suspended in 4 ml of 1× PBS containing 0.01 mg/ml DAPI, and incubated in the dark for 20 min. Excess DAPI was washed off of cultures by resuspending cells in 1× PBS.
To make spheroplasts, the cells were pelleted and suspended in 800–850 μl of 0.5 mg/ml lysozyme, 10 mm EDTA (pH 8.0), 800 mm sucrose, and 1000 units of DNase. The cell suspensions were incubated at 37 °C with mild shaking for 15–20 min. To the cell suspension, a 100 μl solution of 10 mm Tris-HCl, pH 8.0, 700 mm sucrose, and 20 mm MgCl2 was added to stabilize them. All of the cultures were immediately processed for microscopy.
Induced spheroplasts were split into two equal parts, and the test sample was prepared by adding 1 μm NHS-rhodamine–coupled ICab3 anti-His antibody and anti-mouse antibody Alexa Fluor 647 (Thermo Fisher Scientific) to one of the suspensions and incubated in the dark for 10 min. The spheroplast suspension was then spun at 800 × g for 10 min and was resuspended in 0.7 m sucrose, 10 mm Tris-HCl, and 2 mm MgCl2. The spheroplasts were washed again and then resuspended by gentle pipetting for 4–5 min. Each of the samples was mixed with Fluoroshield (Sigma-Aldrich) before visualization under a confocal microscope (Olympus FV3000). For the control, the other induced fraction was treated similarly, but replacing WT ICab3-NHS rhodamine with ICab3-H116C-NHS rhodamine. As another control, spheroplasts made using uninduced culture were processed with the same set of fluorophore-tagged proteins as used in the case of the samples.
Molecular dynamics
An all-atom molecular dynamics simulation was carried out using the GROMACS 2018.4.2 package (58) with CHARMM36m force field (59). For simulation in the Zn2+-bound state, a refined structure of the ICab3 was used as an initial model, and nomenclature for Zn and Cys in the pdb file was changed accordingly. The Zn2+ ion was deleted from the pdb file before running the simulation in the apo state without the bound Zn2+ ion. The protein was solvated using a TIP4P water model in a cubic box. A minimum distance of 15 Å was kept between any of the protein atom and box wall. NaCl was added at a concentration of 150 mm to the system to mimic the physiological condition while keeping the system electrically neutral. Long-range electrostatic interactions were calculated with the particle mesh Ewald method (60), and bonds were constrained using the LINCS algorithm (61). Energy minimization was carried out by the steepest-descent method. The system was equilibrated under the NVT ensemble for 1 ns, followed by the NPT ensemble for 2 ns, keeping the protein atoms restrained by applying a force constant of 1000 kJ mol−1 nm−2. To perform temperature equilibration, a modified Berendsen thermostat (62) was used with a coupling time constant of 0.1 ps and a reference temperature of 300 K. Pressure equilibration was done using the Parrinello–Rahman method (63) with a coupling time of 2 ps and a reference pressure of 1 bar. Leap-frog integrator was used for equilibration and production MD with a time step of 2 fs. Each of the two simulations was run for 100 ns and then analyzed for structural changes using PyMOL (Schrodinger). The coordinates and energies were written every 10 ps.
Author contributions
S. K., R. R., and A. P. resources; S. K., M. I., and A. A. data curation; S. K. and A. A. software; S. K., M. I., and A. A. formal analysis; S. K., M. I., A. A., and A. P. validation; S. K. and A. A. visualization; S. K., M. I., A. A., and R. R. methodology; S. K. and A. P. writing-original draft; M. I. and A. A. investigation; M. I., A. A., and R. R. writing-review and editing; A. P. conceptualization; A. P. supervision; A. P. funding acquisition; A. P. project administration.
Supplementary Material
Acknowledgments
We acknowledge members of the Penmatsa laboratory, particularly Ashutosh Gulati and Rahul Nair, for optimization of NorC purification and help with camel immunization. The kind gift of the pPNL6 vector by Prof. Rajan Dighe (MRDG, IISc) is acknowledged. The Director of NRCC is gratefully acknowledged for support and access to facilities for camel immunization. Work in this paper was performed under the aegis of an MoU between the Indian Institute of Science and National Research Centre on Camel. Camel immunization was done through the NRCC animal ethics committee (NRCC/PME/6(141)/2000-Tech (vol II)), and the protocol was approved through the Committee for the Purpose of Control and Supervision of Experiments on Animals (CPCSEA), India. The confocal microscopy protocol was optimized in the Bioimaging Facility, Division of Biological Sciences, IISc, whereas the confocal imaging experiments were done in the Department of Biochemistry, Division of Biological Sciences, IISc. We thank Prof. Raghavan Varadarajan for access to the FACS facility and Sunita Prakash for help with MS at the Proteomics Facility, Molecular Biophysics Unit, IISc. Prof. B. Gopal is acknowledged for access to PEAQ ITC. The X-ray diffraction facility for macromolecular crystallography at the Indian Institute of Science is supported by Department of Science and Technology–Science and Engineering Research Board (DST-SERB) Grant IR/SO/LU/0003/2010-PHASE-II. We acknowledge the ESRF access program of RCB (Grant BT/INF/22/SP22660/2017) of the Department of Biotechnology, India.
This work was supported by Wellcome Trust/DBT India Alliance Intermediate Fellowship IA/1/15/2/502063 and Department of Biotechnology, India, Grant BT/PR31976/MED/29/1421/2019 (to A. P.). This work was also supported by DBT-IISc partnership program phase I and phase II. The authors declare that they have no conflicts of interest with the contents of this article.
This article contains Figs. S1–S8.
The atomic coordinates and structure factors (code 6KSN) have been deposited in the Protein Data Bank (http://wwpdb.org/).
- MFS
- major facilitator superfamily
- MATE
- multidrug and toxic compound extrusion
- SMR
- small multidrug resistance
- ABC
- ATP-binding cassette
- RND
- resistance nodulation division
- DHA
- drug:H+ antiporter(s)
- TM
- transmembrane
- MRSA
- methicillin-resistant S. aureus
- VHH
- variable heavy chain antibody
- CDR
- complementarity-determining region
- DDM
- dodecyl-β-d-maltopyranoside
- SEC
- size-exclusion chromatography
- FSEC
- fluorescence-detection size-exclusion chromatography
- PBMC
- peripheral blood mononuclear cell
- FR
- framework region
- ITC
- isothermal titration calorimetry
- nano-DSF
- nano-differential scanning fluorimetry
- DAPI
- 4′,6-diamidino-2-phenylindole
- OD
- optical density
- LB
- Luria broth
- ESI
- electrospray ionization
- RMSD
- root mean square deviation.
References
- 1. Walsh C. (2000) Molecular mechanisms that confer antibacterial drug resistance. Nature 406, 775–781 10.1038/35021219 [DOI] [PubMed] [Google Scholar]
- 2. Webber M. A., and Piddock L. J. (2003) The importance of efflux pumps in bacterial antibiotic resistance. J. Antimicrob. Chemother. 51, 9–11 10.1093/jac/dkg050 [DOI] [PubMed] [Google Scholar]
- 3. Reddy V. S., Shlykov M. A., Castillo R., Sun E. I., and Saier M. H. Jr. (2012) The major facilitator superfamily (MFS) revisited. FEBS J. 279, 2022–2035 10.1111/j.1742-4658.2012.08588.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Saier M. H. Jr., Reddy V. S., Tamang D. G., and Västermark A. (2014) The transporter classification database. Nucleic Acids Res. 42, D251–D258 10.1093/nar/gkt1097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Paulsen I. T., Brown M. H., and Skurray R. A. (1996) Proton-dependent multidrug efflux systems. Microbiol. Rev. 60, 575–608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Mallonee D. H., and Hylemon P. B. (1996) Sequencing and expression of a gene encoding a bile acid transporter from Eubacterium sp. strain VPI 12708. J. Bacteriol. 178, 7053–7058 10.1128/jb.178.24.7053-7058.1996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Wright G. D. (2016) Antibiotic adjuvants: rescuing antibiotics from resistance (Trends in Microbiology 24, 862–871; October 17, 2016). Trends Microbiol. 24, 928 10.1016/j.tim.2016.07.008 [DOI] [PubMed] [Google Scholar]
- 8. Douafer H., Andrieu V., Phanstiel O. 4th, and Brunel J. M. (2019) Antibiotic adjuvants: make antibiotics great again! J. Med. Chem. 62, 8665–8681 10.1021/acs.jmedchem.8b01781 [DOI] [PubMed] [Google Scholar]
- 9. Piddock L. J. (2006) Multidrug-resistance efflux pumps—not just for resistance. Nat. Rev. Microbiol. 4, 629–636 10.1038/nrmicro1464 [DOI] [PubMed] [Google Scholar]
- 10. Paulsen I. T., Brown M. H., Littlejohn T. G., Mitchell B. A., and Skurray R. A. (1996) Multidrug resistance proteins QacA and QacB from Staphylococcus aureus: membrane topology and identification of residues involved in substrate specificity. Proc. Natl. Acad. Sci. U.S.A. 93, 3630–3635 10.1073/pnas.93.8.3630 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Costa S. S., Viveiros M., Amaral L., and Couto I. (2013) Multidrug efflux pumps in Staphylococcus aureus: an update. Open Microbiol. J. 7, 59–71 10.2174/1874285801307010059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Truong-Bolduc Q. C., Strahilevitz J., and Hooper D. C. (2006) NorC, a new efflux pump regulated by MgrA of Staphylococcus aureus. Antimicrob. Agents Chemother. 50, 1104–1107 10.1128/AAC.50.3.1104-1107.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Tong S. Y., Davis J. S., Eichenberger E., Holland T. L., and Fowler V. G. Jr. (2015) Staphylococcus aureus infections: epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 28, 603–661 10.1128/CMR.00134-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Muyldermans S. (2013) Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem. 82, 775–797 10.1146/annurev-biochem-063011-092449 [DOI] [PubMed] [Google Scholar]
- 15. Hamers-Casterman C., Atarhouch T., Muyldermans S., Robinson G., Hamers C., Songa E. B., Bendahman N., and Hamers R. (1993) Naturally occurring antibodies devoid of light chains. Nature 363, 446–448 10.1038/363446a0 [DOI] [PubMed] [Google Scholar]
- 16. Greenberg A. S., Avila D., Hughes M., Hughes A., McKinney E. C., and Flajnik M. F. (1995) A new antigen receptor gene family that undergoes rearrangement and extensive somatic diversification in sharks. Nature 374, 168–173 10.1038/374168a0 [DOI] [PubMed] [Google Scholar]
- 17. Beghein E., and Gettemans J. (2017) Nanobody technology: a versatile toolkit for microscopic imaging, protein-protein interaction Analysis, and protein function exploration. Front. Immunol. 8, 771 10.3389/fimmu.2017.00771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Baral T. N., MacKenzie R., and Arbabi Ghahroudi M. (2013) Single-domain antibodies and their utility. Curr. Protoc. Immunol. 103, Unit 2.17 10.1002/0471142735.im0217s103 [DOI] [PubMed] [Google Scholar]
- 19. De Genst E., Silence K., Decanniere K., Conrath K., Loris R., Kinne J., Muyldermans S., and Wyns L. (2006) Molecular basis for the preferential cleft recognition by dromedary heavy-chain antibodies. Proc. Natl. Acad. Sci. U.S.A. 103, 4586–4591 10.1073/pnas.0505379103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Kromann-Hansen T., Oldenburg E., Yung K. W., Ghassabeh G. H., Muyldermans S., Declerck P. J., Huang M., Andreasen P. A., and Ngo J. C. (2016) A camelid-derived antibody fragment targeting the active site of a serine protease balances between inhibitor and substrate behavior. J. Biol. Chem. 291, 15156–15168 10.1074/jbc.M116.732503 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Rasmussen S. G., Choi H. J., Fung J. J., Pardon E., Casarosa P., Chae P. S., Devree B. T., Rosenbaum D. M., Thian F. S., Kobilka T. S., Schnapp A., Konetzki I., Sunahara R. K., Gellman S. H., Pautsch A., Steyaert J., Weis W. I., and Kobilka B. K. (2011) Structure of a nanobody-stabilized active state of the β(2) adrenoceptor. Nature 469, 175–180 10.1038/nature09648 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Jiang X., Smirnova I., Kasho V., Wu J., Hirata K., Ke M., Pardon E., Steyaert J., Yan N., and Kaback H. R. (2016) Crystal structure of a LacY-nanobody complex in a periplasmic-open conformation. Proc. Natl. Acad. Sci. U.S.A. 113, 12420–12425 10.1073/pnas.1615414113 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Mitchell L. S., and Colwell L. J. (2018) Comparative analysis of nanobody sequence and structure data. Proteins 86, 697–706 10.1002/prot.25497 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Wesolowski J., Alzogaray V., Reyelt J., Unger M., Juarez K., Urrutia M., Cauerhff A., Danquah W., Rissiek B., Scheuplein F., Schwarz N., Adriouch S., Boyer O., Seman M., Licea A., et al. (2009) Single domain antibodies: promising experimental and therapeutic tools in infection and immunity. Med. Microbiol. Immunol. 198, 157–174 10.1007/s00430-009-0116-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Harmsen M. M., and De Haard H. J. (2007) Properties, production, and applications of camelid single-domain antibody fragments. Appl. Microbiol. Biotechnol. 77, 13–22 10.1007/s00253-007-1142-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Hassanzadeh-Ghassabeh G., Devoogdt N., De Pauw P., Vincke C., and Muyldermans S. (2013) Nanobodies and their potential applications. Nanomedicine (Lond.) 8, 1013–1026 10.2217/nnm.13.86 [DOI] [PubMed] [Google Scholar]
- 27. Desmyter A., Spinelli S., Roussel A., and Cambillau C. (2015) Camelid nanobodies: killing two birds with one stone. Curr. Opin. Struct. Biol. 32, 1–8 10.1016/j.sbi.2015.01.001 [DOI] [PubMed] [Google Scholar]
- 28. Mujić-Delić A., de Wit R. H., Verkaar F., and Smit M. J. (2014) GPCR-targeting nanobodies: attractive research tools, diagnostics, and therapeutics. Trends Pharmacol. Sci. 35, 247–255 10.1016/j.tips.2014.03.003 [DOI] [PubMed] [Google Scholar]
- 29. Ehrnstorfer I. A., Geertsma E. R., Pardon E., Steyaert J., and Dutzler R. (2014) Crystal structure of a SLC11 (NRAMP) transporter reveals the basis for transition-metal ion transport. Nat. Struct. Mol. Biol. 21, 990–996 10.1038/nsmb.2904 [DOI] [PubMed] [Google Scholar]
- 30. Kumar H., Finer-Moore J. S., Jiang X., Smirnova I., Kasho V., Pardon E., Steyaert J., Kaback H. R., and Stroud R. M. (2018) Crystal structure of a ligand-bound LacY-nanobody complex. Proc. Natl. Acad. Sci. U.S.A. 115, 8769–8774 10.1073/pnas.1801774115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Löw C., Yau Y. H., Pardon E., Jegerschöld C., Wåhlin L., Quistgaard E. M., Moberg P., Geifman-Shochat S., Steyaert J., and Nordlund P. (2013) Nanobody mediated crystallization of an archeal mechanosensitive channel. PLoS One 8, e77984 10.1371/journal.pone.0077984 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Hassaine G., Deluz C., Grasso L., Wyss R., Tol M. B., Hovius R., Graff A., Stahlberg H., Tomizaki T., Desmyter A., Moreau C., Li X. D., Poitevin F., Vogel H., and Nury H. (2014) X-ray structure of the mouse serotonin 5-HT3 receptor. Nature 512, 276–281 10.1038/nature13552 [DOI] [PubMed] [Google Scholar]
- 33. Manglik A., Kobilka B. K., and Steyaert J. (2017) Nanobodies to study G protein-coupled receptor structure and function. Annu. Rev. Pharmacol. Toxicol. 57, 19–37 10.1146/annurev-pharmtox-010716-104710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Uchański T., Zögg T., Yin J., Yuan D., Wohlkönig A., Fischer B., Rosenbaum D. M., Kobilka B. K., Pardon E., and Steyaert J. (2019) An improved yeast surface display platform for the screening of nanobody immune libraries. Sci. Rep. 9, 382 10.1038/s41598-018-37212-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Pardon E., Laeremans T., Triest S., Rasmussen S. G., Wohlkönig A., Ruf A., Muyldermans S., Hol W. G., Kobilka B. K., and Steyaert J. (2014) A general protocol for the generation of Nanobodies for structural biology. Nat. Protoc. 9, 674–693 10.1038/nprot.2014.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. McMahon C., Baier A. S., Pascolutti R., Wegrecki M., Zheng S., Ong J. X., Erlandson S. C., Hilger D., Rasmussen S. G. F., Ring A. M., Manglik A., and Kruse A. C. (2018) Yeast surface display platform for rapid discovery of conformationally selective nanobodies. Nat. Struct. Mol. Biol. 25, 289–296 10.1038/s41594-018-0028-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Zavrtanik U., Lukan J., Loris R., Lah J., and Hadži S. (2018) Structural basis of epitope recognition by heavy-chain camelid antibodies. J. Mol. Biol. 430, 4369–4386 10.1016/j.jmb.2018.09.002 [DOI] [PubMed] [Google Scholar]
- 38. Sircar A., Sanni K. A., Shi J., and Gray J. J. (2011) Analysis and modeling of the variable region of camelid single-domain antibodies. J. Immunol. 186, 6357–6367 10.4049/jimmunol.1100116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chothia C., Novotný J., Bruccoleri R., and Karplus M. (1985) Domain association in immunoglobulin molecules. The packing of variable domains. J. Mol. Biol. 186, 651–663 10.1016/0022-2836(85)90137-8 [DOI] [PubMed] [Google Scholar]
- 40. Muyldermans S., Atarhouch T., Saldanha J., Barbosa J. A., and Hamers R. (1994) Sequence and structure of VH domain from naturally occurring camel heavy chain immunoglobulins lacking light chains. Protein Eng. 7, 1129–1135 10.1093/protein/7.9.1129 [DOI] [PubMed] [Google Scholar]
- 41. Vu K. B., Ghahroudi M. A., Wyns L., and Muyldermans S. (1997) Comparison of llama VH sequences from conventional and heavy chain antibodies. Mol. Immunol. 34, 1121–1131 10.1016/S0161-5890(97)00146-6 [DOI] [PubMed] [Google Scholar]
- 42. Barber-Zucker S., Shaanan B., and Zarivach R. (2017) Transition metal binding selectivity in proteins and its correlation with the phylogenomic classification of the cation diffusion facilitator protein family. Sci. Rep. 7, 16381 10.1038/s41598-017-16777-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Conrath K. E., Wernery U., Muyldermans S., and Nguyen V. K. (2003) Emergence and evolution of functional heavy-chain antibodies in Camelidae. Dev. Comp. Immunol. 27, 87–103 10.1016/S0145-305X(02)00071-X [DOI] [PubMed] [Google Scholar]
- 44. De Genst E., Handelberg F., Van Meirhaeghe A., Vynck S., Loris R., Wyns L., and Muyldermans S. (2004) Chemical basis for the affinity maturation of a camel single domain antibody. J. Biol. Chem. 279, 53593–53601 10.1074/jbc.M407843200 [DOI] [PubMed] [Google Scholar]
- 45. Nguyen V. K., Muyldermans S., and Hamers R. (1998) The specific variable domain of camel heavy-chain antibodies is encoded in the germline. J. Mol. Biol. 275, 413–418 10.1006/jmbi.1997.1477 [DOI] [PubMed] [Google Scholar]
- 46. Nguyen V. K., Hamers R., Wyns L., and Muyldermans S. (2000) Camel heavy-chain antibodies: diverse germline V(H)H and specific mechanisms enlarge the antigen-binding repertoire. EMBO J. 19, 921–930 10.1093/emboj/19.5.921 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Govaert J., Pellis M., Deschacht N., Vincke C., Conrath K., Muyldermans S., and Saerens D. (2012) Dual beneficial effect of interloop disulfide bond for single domain antibody fragments. J. Biol. Chem. 287, 1970–1979 10.1074/jbc.M111.242818 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Kunz P., Flock T., Soler N., Zaiss M., Vincke C., Sterckx Y., Kastelic D., Muyldermans S., and Hoheisel J. D. (2017) Exploiting sequence and stability information for directing nanobody stability engineering. Biochim. Biophys. Acta Gen. Subj. 1861, 2196–2205 10.1016/j.bbagen.2017.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Niesen F. H., Berglund H., and Vedadi M. (2007) The use of differential scanning fluorimetry to detect ligand interactions that promote protein stability. Nat. Protoc. 2, 2212–2221 10.1038/nprot.2007.321 [DOI] [PubMed] [Google Scholar]
- 50. Futai M. (1974) Orientation of membrane vesicles from Escherichia coli prepared by different procedures. J. Membr. Biol. 15, 15–28 10.1007/BF01870079 [DOI] [PubMed] [Google Scholar]
- 51. Powell H. R., Battye T. G. G., Kontogiannis L., Johnson O., and Leslie A. G. W. (2017) Integrating macromolecular X-ray diffraction data with the graphical user interface iMosflm. Nat. Protoc. 12, 1310–1325 10.1038/nprot.2017.037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Evans P. (2006) Scaling and assessment of data quality. Acta Crystallogr. D Biol. Crystallogr. 62, 72–82 10.1107/S0907444905036693 [DOI] [PubMed] [Google Scholar]
- 53. Winn M. D., Ballard C. C., Cowtan K. D., Dodson E. J., Emsley P., Evans P. R., Keegan R. M., Krissinel E. B., Leslie A. G., McCoy A., McNicholas S. J., Murshudov G. N., Pannu N. S., Potterton E. A., Powell H. R., et al. (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr. D Biol. Crystallogr. 67, 235–242 10.1107/S0907444910045749 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. McCoy A. J., Grosse-Kunstleve R. W., Adams P. D., Winn M. D., Storoni L. C., and Read R. J. (2007) Phaser crystallographic software. J. Appl. Crystallogr. 40, 658–674 10.1107/S0021889807021206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Emsley P., and Cowtan K. (2004) Coot: model-building tools for molecular graphics. Acta Crystallogr. D Biol. Crystallogr. 60, 2126–2132 10.1107/S0907444904019158 [DOI] [PubMed] [Google Scholar]
- 56. Adams P. D., Afonine P. V., Bunkóczi G., Chen V. B., Davis I. W., Echols N., Headd J. J., Hung L. W., Kapral G. J., Grosse-Kunstleve R. W., McCoy A. J., Moriarty N. W., Oeffner R., Read R. J., Richardson D. C., et al. (2010) PHENIX: a comprehensive Python-based system for macromolecular structure solution. Acta Crystallogr. D Biol. Crystallogr. 66, 213–221 10.1107/S0907444909052925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Williams C. J., Headd J. J., Moriarty N. W., Prisant M. G., Videau L. L., Deis L. N., Verma V., Keedy D. A., Hintze B. J., Chen V. B., Jain S., Lewis S. M., Arendall W. B. 3rd, Snoeyink J., Adams P. D., et al. (2018) MolProbity: More and better reference data for improved all-atom structure validation. Protein Sci. 27, 293–315 10.1002/pro.3330 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Pronk S., Páll S., Schulz R., Larsson P., Bjelkmar P., Apostolov R., Shirts M. R., Smith J. C., Kasson P. M., van der Spoel D., Hess B., and Lindahl E. (2013) GROMACS 4.5: a high-throughput and highly parallel open source molecular simulation toolkit. Bioinformatics 29, 845–854 10.1093/bioinformatics/btt055 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Huang J., Rauscher S., Nawrocki G., Ran T., Feig M., de Groot B. L., Grubmüller H., and MacKerell A. D. Jr. (2017) CHARMM36m: an improved force field for folded and intrinsically disordered proteins. Nat. Methods 14, 71–73 10.1038/nmeth.4067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Darden T., York D., and Pedersen L. (1993) Particle mesh Ewald—an N.Log(N) method for Ewald sums in large systems. J. Chem. Phys. 98, 10089–10092 10.1063/1.464397 [DOI] [Google Scholar]
- 61. Hess B., Bekker H., Berendsen H. J. C., Fraaije J. G. E. M. (1997) LINCS: a linear constraint solver for molecular simulations. J. Comput. Chem. 18, 1463–1472 [DOI] [Google Scholar]
- 62. Berendsen H. J. C., Postma J. P. M., Vangunsteren W. F., Dinola A., and Haak J. R. (1984) Molecular-dynamics with coupling to an external bath. J. Chem. Phys. 81, 3684–3690 10.1063/1.448118 [DOI] [Google Scholar]
- 63. Parrinello M., and Rahman A. (1981) Polymorphic transitions in single-crystals—a new molecular-dynamics method. J. Appl. Phys. 52, 7182–7190 10.1063/1.328693 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.