

Abstract
Insulin action in adipose tissue is crucial for whole-body glucose homeostasis, with insulin resistance being a major risk factor for metabolic diseases such as type 2 diabetes. Recent studies have proposed mitochondrial oxidants as a unifying driver of adipose insulin resistance, serving as a signal of nutrient excess. However, neither the substrates for nor sites of oxidant production are known. Because insulin stimulates glucose utilization, we hypothesized that glucose oxidation would fuel respiration, in turn generating mitochondrial oxidants. This would impair insulin action, limiting further glucose uptake in a negative feedback loop of “glucose-dependent” insulin resistance. Using primary rat adipocytes and cultured 3T3-L1 adipocytes, we observed that insulin increased respiration, but notably this occurred independently of glucose supply. In contrast, glucose was required for insulin to increase mitochondrial oxidants. Despite rising to similar levels as when treated with other agents that cause insulin resistance, glucose-dependent mitochondrial oxidants failed to cause insulin resistance. Subsequent studies revealed a temporal relationship whereby mitochondrial oxidants needed to increase before the insulin stimulus to induce insulin resistance. Together, these data reveal that (a) adipocyte respiration is principally fueled from nonglucose sources; (b) there is a disconnect between respiration and oxidative stress, whereby mitochondrial oxidant levels do not rise with increased respiration unless glucose is present; and (c) mitochondrial oxidative stress must precede the insulin stimulus to cause insulin resistance, explaining why short-term, insulin-dependent glucose utilization does not promote insulin resistance. These data provide additional clues to mechanistically link nutrient excess to adipose insulin resistance.
Keywords: adipocyte, glucose, insulin, mitochondria, respiration, oxidative stress, insulin resistance
Introduction
Adipose tissue is a key nutrient sensor in mammals (1). Being highly sensitive to insulin, adipocytes respond to nutrient-replete conditions by increasing energy intake and storage as lipid. Conversely, under nutrient excess, adipocytes become insulin-resistant, leading to increased lipid utilization and reduced glucose intake. Indeed, impaired glucose uptake into adipose tissue is one of the earliest defects observed in whole-body insulin resistance, in both rodents and humans (e.g. see Refs. 2–4). Furthermore, knocking out glucose transporter 4 (GLUT4)6 specifically in the adipose tissue results in whole-body insulin resistance in mice (5). Thus, adipocyte insulin sensitivity appears critical for whole-body glucose homeostasis.
Insulin resistance in adipose tissue can be caused by numerous insults, such as chronic hyperinsulinemia, inflammation, and corticosteroids. Each of these have been examined in isolation through in vivo (mouse) and in vitro (adipocyte) models (e.g. see Refs. 6 and 7), revealing that they all produce mitochondrial oxidants (mitoROS) as a potential unifying cause of insulin resistance (reviewed in Ref. 8). Although impaired mitochondrial function has been observed in insulin resistance (9, 10), we have recently shown that acute treatment with mitochondria-targeted paraquat (mPQ) increased mitoROS and caused insulin resistance, without impacting mitochondrial respiration (11). This supports the notion that mitoROS alone, without defects in mitochondrial respiration, are sufficient to cause insulin resistance. This raised the possibility that under physiological circumstances, insulin resistance may arise without defects in mitochondrial respiration, but rather as a result of oxidation of specific substrates (e.g. glucose and fatty acids) generating mitoROS. This suggests that mitoROS might represent a mechanistic sensor for nutrient excess (12), particularly in the adipocyte.
Because insulin potently promotes glucose uptake into adipocytes, we considered the role of glucose utilization in adipose insulin resistance. We have previously observed that insulin stimulates glucose oxidation (13), glucose-fueled tricarboxylic acid cycle flux,7 and respiration (14) in adipocytes. Thus, we hypothesized that insulin stimulates respiration and subsequent mitoROS production in a glucose-dependent manner, with insulin resistance stemming from glucose oversupply. Such “glucose-dependent” insulin resistance would limit further glucose uptake as a form of negative feedback.
Using respirometry and metabolic labeling, we found that glucose was not required for insulin-stimulated respiration in cultured and primary adipocytes. In contrast, the insulin-stimulated increase in mitoROS was glucose-dependent. This presents a physiological disconnect between mitochondrial respiration and oxidative stress; although both increase in response to insulin, they have distinct substrate requirements. Furthermore, insulin-stimulated oxidant generation did not cause insulin resistance, at least acutely, despite mitoROS rising to levels observed with other agents that induce insulin resistance (11). This was because mitoROS needed to increase prior to the insulin stimulus, demonstrating that the timing of oxidative stress is crucial in the development of insulin resistance. These observations provide additional clues to mechanistically link nutrient excess to adipose insulin resistance.
Results
Glucose is dispensable for insulin-responsive respiration
In this study, we sought to understand the role of glucose metabolism in adipose insulin resistance. We hypothesized that insulin resistance was caused by mitoROS generated from increased glucose metabolism. Because mitoROS is generated by enzymes that participate in mitochondrial respiration, we reasoned that glucose would fuel insulin-stimulated respiration to generate mitoROS. Thus, in the first instance, we examined the role of glucose in adipocyte respiration.
We delineated glucose metabolism from insulin action using galactose, a “no glucose” sugar analogue previously shown to impair glycolysis (e.g. see Refs. 15–17). We confirmed that galactose ablated insulin-stimulated lactate production (Fig. 1A). However, contrary to our hypothesis, insulin increased respiration (oxygen consumption in intact cells) both in the presence and absence of glucose (Fig. 1B). In contrast, both glucose and insulin were required for maximal respiration, induced by a maximal dose of the mitochondrial uncoupler (BAM15). This suggests that insulin-dependent glucose metabolism can fuel respiration but is not necessary for respiration under insulin-stimulated conditions.
Figure 1.

Glucose is dispensable for insulin-stimulated respiration in adipocytes. A, 3T3-L1 adipocytes were treated for 1 h with or without 100 nm insulin (Ins) in Medium BS, supplemented with 25 mm glucose (Glc) or galactose (Gal). Following treatment, lactate production was quantified enzymatically as described under “Experimental procedures.” Data are presented as mean ± S.E. (error bars) from three separate experiments. B, 3T3-L1 adipocytes were incubated in Medium CX supplemented with 25 mm Glc or Gal, and JO2 was measured using the XFp Analyzer following sequential treatment with or without Ins (100 nm), oligomycin (Oligo; 10 μg/ml), BAM15 (10 μm), rotenone (Rot; 5 μm), and antimycin A (AA; 10 μm) at the time points indicated. Data are presented as mean ± S.E. from four separate experiments. C, 3T3-L1 adipocytes were preloaded with TMRM dye, and fluorescence was measured following incubation in Medium CX supplemented with 25 mm Glc or Gal, treatment with Ins (100 nm), and treatment with Oligo (10 μg/ml) and BAM15 (10 μm) as indicated. Fluorescence was normalized to the Glc/control condition (for details, see “Experimental procedures”). Data are presented as mean ± S.E. from five separate experiments. D–F, mature primary adipocytes were isolated from rats and incubated in assay buffer supplemented with 5 mm Glc or Gal. In D, adipocytes were treated without (control; Ctrl) or with Ins (20 nm) for 1 h. Following treatment, lactate content was determined enzymatically following organic extraction and normalized to the Glc/Ins condition. Data are presented as mean ± S.E. from three separate experiments. In E, respiration was measured using the Oroboros O2K following the addition of Ins (20 nm), Oligo (5 μg/ml), FCCP (concentrations as indicated), Rot (10 μm), and AA (25 μm). Respiration was normalized to the baseline respiration of the Glc/Ins condition. Data are presented as mean ± half-range from two separate experiments. F, respiration was measured as in E. Data are presented as mean ± S.E. from four separate experiments. G, schematic depicting how the contribution of glucose to respiration-derived CO2 was determined using ECAR, lactate production, and glucose oxidation data. For details, see “Experimental Procedures” and “Results.” Data are presented as mean ± S.E., derived from experiments described in the “Experimental Procedures” and “Results.” AUC, area under the curve.
One possibility is that insulin stimulates respiration by different mechanisms in the presence and absence of glucose. For instance, glucose utilization may generate more reducing equivalents, increasing the proton gradient and respiration (increased respiration through increased supply). Alternatively, respiration would also increase in response to higher energy demand, which may manifest as a depleted proton gradient in the absence of glucose, if glucose is required for insulin-stimulated respiration. Thus, we examined mitochondrial membrane potential (MMP), a surrogate for the proton gradient (Fig. 1C). Insulin did not alter MMP in the presence of glucose (p > 0.05, two-way ANOVA comparing glucose with/without insulin over time). Removing glucose led to a reduction in MMP (∼2% difference between galactose/insulin and glucose/insulin; p < 0.01, two-way ANOVA). However, this change was mild compared with the hypo- or hyperpolarization observed with BAM15 and oligomycin, respectively (Fig. 1C). This suggests that substrate supply and energy demand are synchronized in response to insulin, without substantial energetic imbalance, even in the absence of glucose.
We next sought to translate our findings ex vivo. Because adipose tissue consists of various cell types in addition to adipocytes (1), we tested whether glucose was required for respiration in mature adipocytes isolated from rat adipose tissue. We confirmed that galactose blunted insulin-responsive lactate levels (Fig. 1D). With this tool, we found that insulin slightly increased respiration (Fig. 1E), but this did not require glucose except in the presence of the mitochondrial uncoupler, carbonyl cyanide-4-(trifluoromethoxy)phenylhydrazone (FCCP) (Fig. 1F). Lower doses of FCCP had little effect on respiration (data not shown). Overall, this demonstrates that our findings in cultured adipocytes are conserved in primary adipocytes ex vivo.
Glucose only contributes a portion of respiration-derived CO2
Given that insulin could stimulate respiration without glucose, we predicted that glucose would not be a significant contributor to respiration-derived CO2 (i.e. CO2 from pyruvate dehydrogenase (PDH) and the Krebs cycle). It is theoretically possible to calculate this from the cellular respiration rate (JO2) if glucose is the sole substrate (18). However, there are other substrates present in our experiments, such as amino acids in the media and fatty acids from intracellular lipid stores. Each substrate varies in their respiratory quotient (CO2 produced/O2 consumed), rendering this approach challenging.
To overcome this, we measured JO2 from cultured adipocytes using the XFp Analyzer (Seahorse Biosciences), which simultaneously measures the extracellular acidification rate (ECAR) within the same experiment (Fig. S1A). Importantly, ECAR is influenced by not only lactate production, but also bicarbonate formed by CO2 production (18). We utilized this to calculate the contribution of glucose to cellular respiration (Fig. 1G, detailed under “Experimental procedures”). Briefly, we used the buffering capacity of the medium (Fig. S1B) to convert the ECAR into the proton production rate. Next, we subtracted the lactate production rate from the proton production rate to obtain media acidification due to CO2, from which we inferred total CO2 production (Fig 1G). Finally, we compared this value with the incorporation of (14C-labeled) glucose into CO2, determining that glucose contributes only 19.8% of respiration-derived CO2 upon insulin stimulation. These experiments were conducted in the presence of bicarbonate, which we have previously found to influence cellular respiration (14). In the absence of bicarbonate, we found a similar contribution of glucose to respiration-derived CO2 (26.8%; data not shown).
Thus, glucose contributes a relatively minor portion of the oxidizable carbon for cellular respiration under insulin-stimulated conditions. Conversely, insulin can stimulate adipocyte respiration without glucose (Fig. 1, B and F). Collectively, this suggests that glucose is dispensable for the increase in mitochondrial respiration upon insulin exposure.
Glucose is required for insulin-dependent mitoROS production
Because respiratory complexes also participate in mitoROS production (e.g. see Refs. 19 and 20), this raised the possibility that insulin treatment may similarly increase mitoROS levels independently of insulin-stimulated glucose metabolism. To maximize glucose oxidation, we utilized dichloroacetate (DCA), which causes the dephosphorylation of PDH to promote the activity of this enzyme (21). Interestingly, although DCA rapidly decreased medium acidification (Fig. 2A), DCA treatment had little effect on global respiration (Fig. 2B), even under maximal bioenergetic demand (BAM15). This provided further evidence that global respiration is not influenced by changes in glucose flux.
Figure 2.

Glucose is required for insulin to increase mitochondrial oxidant levels. A and B, 3T3-L1 adipocytes were incubated in Medium CX supplemented with 25 mm glucose (Glc) and ECAR (A) or JO2 (B) was measured using the XFp Analyzer following sequential treatment with insulin (Ins, 100 nm), DCA (1 mm), oligomycin (Oligo; 10 μg/ml), and BAM15 (10 μm). Data are presented as mean ± S.E. (error bars) from four separate experiments. C, 3T3-L1 adipocytes were serum-starved in Medium BS with 25 mm Glc. During the last 1 h of serum starvation, cells were pretreated with DCA (1 mm) or antimycin A (AA; 50 nm). Cells were then incubated in Medium BS, supplemented with 25 mm Glc or galactose (Gal), with Ins (100 nm) and DCA (1 mm) for the indicated times. Following treatment, cells were harvested, and lysates were subjected to Western blotting with antibodies against the indicated proteins. Data are representative of four separate experiments. D, dimer; M, monomer. D, densitometry of PRDX2 and PRDX3 bands from C, expressed as a ratio of dimer to monomer bands and made relative to the treatment with Glc at t = 0 min. Data are presented as mean ± S.E. from four separate experiments. E, densitometry of the indicated proteins from C, normalized to α-tubulin and made relative to the Glc/t = 20 min condition. Data are presented as mean ± S.E. from four separate experiments. F, 3T3-L1 adipocytes were serum-starved and then incubated in Medium BS with Ins (100 nm), AA (50 nm), and/or cytochalasin B (CB; 25 μm). This treatment medium was also supplemented with the indicated concentration of Glc, with the final volume balanced with either water or Gal (for the latter, the final combined sugar concentration was 25 mm). Following 1 h of treatment, cells were harvested and lysates were subjected to Western blotting with antibodies against the indicated proteins. Data are representative of four separate experiments. G, densitometry of PRDX3 bands from F, expressed as the ratio of dimer to monomer bands and made relative to the treatment with 25 mm Glc without Ins (second lane in F). Data are presented as mean ± S.E. from four separate experiments. H, PRDX3 dimerization (G) plotted against Glc uptake (Fig. S2C) for Ins-treated cells exposed to varying medium Glc concentrations (where the final volume was balanced with water). The Hill equation was fitted based on individual data points and overlaid onto the data, which are presented as mean ± S.E. from four separate experiments.
We treated adipocytes with insulin in the presence or absence of glucose and DCA, and examined mitoROS. We initially utilized matrix-roGFP, a mitochondrially targeted ROS-sensitive probe, which did not exhibit substantial changes in signal upon treatment with either insulin or mPQ (Fig. S2, A and B). Subsequently, we assessed the dimer/monomer status of peroxiredoxin 3 (PRDX3) and PRDX2, which are robust, sensitive indicators of relative oxidant exposure in the mitochondria and cytosol, respectively (11). For instance, pretreatment with antimycin A significantly increased PRDX3 dimerization (Fig. 2, C (first two lanes) and D) but had a less pronounced effect on PRDX2 dimerization (Fig. 2, C and D; p ∼0.060 versus control, two-sample t test); this demonstrates that antimycin A more readily increases mitoROS, consistent with its known mechanism of action.
Insulin increased PRDX3 dimerization, but only when glucose was present (Fig. 2, C and D). This was not due to an impairment of insulin signaling in the absence of glucose, as indicated by similar levels of phosphorylation of Akt kinase and its substrate, AS160 (Fig. 2, C and E). The insulin- and glucose-dependent increase in PRDX3 dimerization was enhanced in the presence of DCA, which caused a sustained reduction in PDH phosphorylation throughout the time course (Fig. 2, C and E). In contrast, the change in PRDX2 dimerization was less consistent over time (Fig. 2, C and D). Overall, these data demonstrate that insulin increases mitoROS levels in a glucose-dependent manner.
Consequently, we examined the sensitivity of mitoROS to glucose uptake. As observed previously (21), glucose uptake increased with insulin and higher medium glucose concentrations (Fig. S2C). PRDX3 dimerization also increased under these conditions (Fig. 2, F and G), particularly when physiological glucose concentrations (and the Km of GLUT4 (22)) were reached. Inhibiting glucose uptake with cytochalasin B (Fig. S2C) reduced PRDX3 dimerization (Fig. 2, F and G), but did not affect respiration (Fig. S2D). Furthermore, plotting PRDX3 dimerization against glucose uptake in insulin-stimulated adipocytes revealed a sigmoidal trend (Fig. 2H). This suggests that substantial PRDX3 dimerization occurs once a threshold of glucose uptake is reached. Thus, mitoROS is sensitive to insulin-stimulated glucose uptake.
Glucose-dependent mitoROS does not cause insulin resistance
These data are consistent with the notion that glucose-dependent mitoROS is an endogenous signal to cause insulin resistance under conditions of excess nutrient supply (Fig. 3A). If true, we would predict that sustained insulin exposure would increase glucose oxidation, mitoROS, and lead to acute insulin resistance. Furthermore, this would be exacerbated in the presence of DCA.
Figure 3.

Glucose-dependent mitochondrial oxidants do not cause insulin resistance. A, schematic depicting how insulin-dependent glucose oxidation could lead to oxidant-induced insulin resistance. Glucose is taken up (1), and a majority is diverted to fates such as lipid or lactate production (2) (21).7 A small portion of glucose is oxidized (3), and although this does not contribute substantially to respiration, it increases mitochondrial ROS levels (4), eventually leading to insulin resistance (5). B–D, 3T3-L1 adipocytes were treated in Medium B with mPQ (20 μm), DCA (1 mm), and insulin (Ins, 100 nm) at the times indicated in B. Cells were also treated with auranofin (AF; 5 μm) at t = 30 min (concurrently with mPQ treatment) in C. Cells in C and D were treated in parallel. Following treatment, cells were either harvested for protein and lysates subjected to Western blotting with antibodies against the indicated proteins (C) or assayed for 2-deoxyglucose (2DOG) uptake under cold conditions (D). In C, densitometric analysis was performed as described in the legend to Fig. 2D. Data are presented as mean ± S.E. (error bars) from four separate experiments. *, p < 0.05; ns, p > 0.05 by two-sample t test; D, dimer; M, monomer. E, 3T3-L1 adipocytes constitutively expressing HA-tagged GLUT4 were incubated in Medium B and stimulated with Ins (100 nm) for the indicated times, after which GLUT4 translocation to the PM was measured. Data are presented as mean ± S.E. from three separate experiments.
Thus, we exposed adipocytes to insulin, with or without DCA (Fig. 3B). We included mPQ as a positive control for specifically increasing mitochondrial oxidant levels and causing insulin resistance (Fig. S2E) (11) and the thioreductase inhibitor, auranofin, as a control for PRDX3 dimerization. MitoROS levels rose with prolonged insulin treatment (Fig. 3C), and co-treatment with DCA and insulin increased mitoROS to a greater extent than observed with mPQ treatment (Fig. 3C).
Because insulin-responsive GLUT4 trafficking is selectively impaired in adipocyte insulin resistance (23), we assessed insulin action by cell-surface glucose transporter activity (24, 25). Contrary to our model (Fig. 3A), we did not observe any detectable inhibition of insulin action by DCA treatment, with only mPQ causing insulin resistance within this acute time frame (Fig. 3D). This concurred with the presence of glucose and/or DCA having no consistent effect on insulin-stimulated kinase signaling (Fig. 2, C and E). Furthermore, in the presence of glucose, insulin rapidly stimulated GLUT4 translocation to the plasma membrane (PM), and the proportion of cell-surface GLUT4 did not decrease within 1 h (Fig. 3E), which was a sufficient time frame for glucose-dependent mitoROS levels to rise (Fig. 2D). Thus, glucose-dependent mitoROS does not appear to cause acute insulin resistance.
MitoROS needs to increase before the insulin stimulus to impair insulin action
Although insulin-dependent glucose metabolism increases mitoROS levels comparable to mPQ treatment (Fig. 3C), mPQ was added prior to the insulin treatment (as in Ref. 11). This would have allowed mitoROS to increase prior to the insulin stimulus, unlike glucose-dependent mitoROS (Fig. 2C). Thus, mitoROS may need to increase prior to insulin stimulation to cause insulin resistance. To test this, we treated adipocytes with mPQ before or after insulin stimulation. Strikingly, mPQ pretreatment impaired insulin-stimulated GLUT4 translocation (Fig. 4A), but this was not observed when cells were treated with mPQ after insulin stimulation (Fig. 4B). However, in each case, mPQ increased mitoROS levels, assessed by PRDX3 dimerization (Fig. 4, A and B). Thus, our data suggest that mitoROS only leads to insulin resistance if mitoROS increases prior to insulin stimulation (Fig. 4C).
Figure 4.
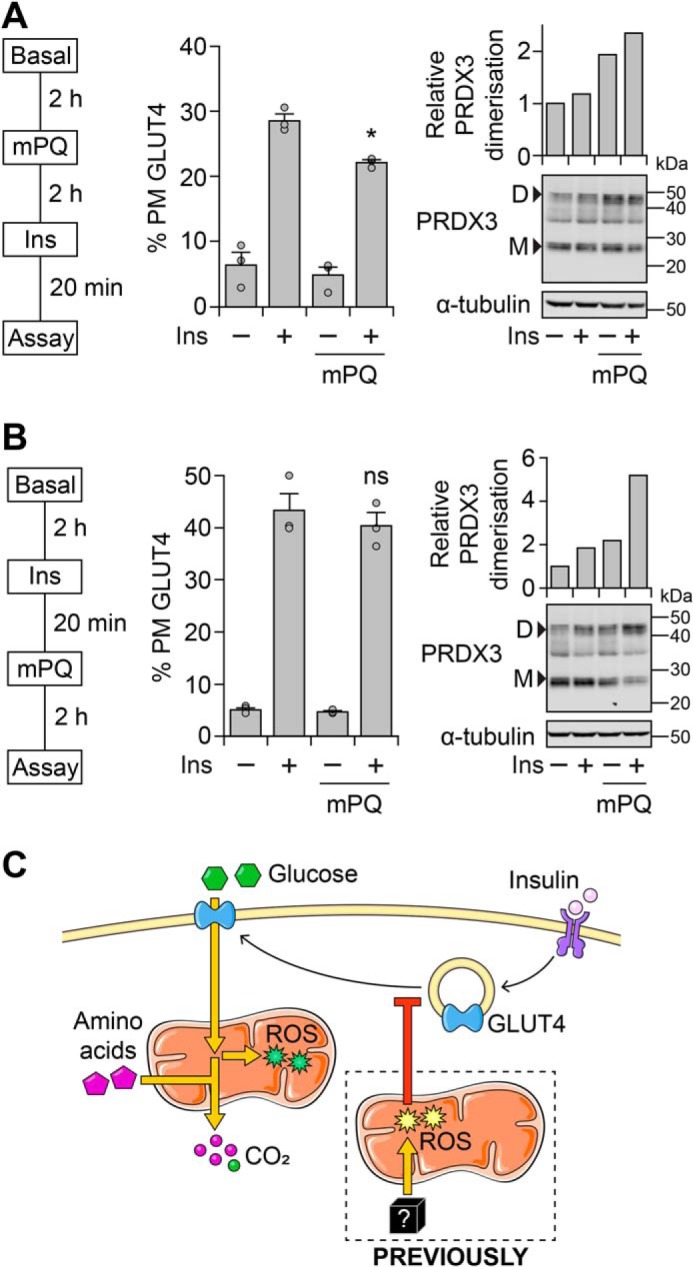
Mitochondrial oxidants need to increase prior to the insulin stimulus to cause insulin resistance. A and B, 3T3-L1 adipocytes constitutively expressing HA-tagged GLUT4 were incubated in Medium B and treated with mPQ (20 μm) and insulin (Ins, 100 nm) at the indicated time points (left). Once a drug was added, it remained in the medium for the remainder of the treatment period. Following treatment, cells were assayed for GLUT4 translocation to the PM (middle) or harvested, and lysates were subjected to Western blotting with antibodies against the indicated proteins (right). Densitometric analysis (right) was performed as described in the legend to Fig. 2D. Data in the middle panel are presented as mean ± S.E. (error bars) from three separate experiments. Data in the right panel are representative of two separate experiments. D, dimer; M, monomer. *, p < 0.05; ns, p > 0.05, compared with Ins-treated cells, by two-sample t test. C, schematic depicting how glucose metabolism demonstrates a disconnect between insulin-stimulated mitochondrial respiration and oxidant production in adipocytes. For details, see “Results” and “Discussion.”
Discussion
In this study, we examined the relationship between nutrient excess and insulin resistance in adipocytes. We tested the concept of “glucose-dependent” insulin resistance, whereby insulin-stimulated glucose metabolism would fuel respiration, generate mitoROS, and feed back to reduce glucose uptake. Although insulin stimulated respiration in the absence of glucose (Fig. 1), glucose was required for insulin to increase mitoROS (Fig. 2). However, glucose-dependent mitoROS did not acutely cause insulin resistance (Fig. 3) because mitoROS needed to increase before the insulin stimulus for insulin resistance to occur (Fig. 4). This may explain why insulin-stimulated glucose utilization does not acutely cause insulin resistance in adipocytes (Fig. 4C).
Mitochondrial respiration is governed by nutrient supply and energy demand (12), permitting the mitochondrion to serve as a cellular sensor for energetic imbalance. Several observations suggest that for adipocytes exposed to insulin, respiration is driven by increased energy demand rather than substrate (glucose) supply. First, insulin stimulated respiration in both the absence or presence of glucose (Fig. 1) and upon inhibition of glucose uptake (Fig. S2D), and DCA treatment had no impact on respiration (Fig. 3). Second, we previously showed that insulin stimulates anabolism before glucose uptake is maximal (24), corresponding with a transient decrease in the global energy charge in adipocytes. Here, we found that insulin treatment did not alter MMP (Fig. 1), suggesting that supply and demand are synchronized and hence respiration likely increases in response to the energetic demands of the cell. Third, although insulin stimulated glucose transport by severalfold (24), glucose only contributed a quarter of respiration-derived CO2 upon insulin stimulation (Fig. 1 and Fig. S1). Finally, glucose only impacted respiration under maximal respiratory demand, whereby both insulin and glucose were required to increase further respiration in the presence of an uncoupler (Fig. 1). Indeed, treatment with a mitochondrial uncoupler increased glucose oxidation substantially higher than insulin alone (21). Collectively, this suggests that insulin stimulates energy demand in adipocytes, but this increased demand is not enough to require substrate supply from exogenous glucose.
The minor contribution of glucose to respiration-derived CO2 (Fig. 1G) concurs with our previous tracer metabolomics analysis of insulin-stimulated adipocytes (24); for instance, with insulin treatment, the unlabeled isotopologue of acetyl-CoA was dominant, suggesting that other carbon sources contributed substantially to the Krebs cycle. Given that insulin inhibits glycogen breakdown and lipolysis, this leaves amino acids as a potential respiratory substrate. Indeed, previous stable-isotope tracer studies have demonstrated that in adipocytes, the Krebs cycle can be fueled by branched-chain amino acids and glutamine (26, 27). This provides a likely explanation for how insulin stimulated respiration in the absence of glucose (Fig. 4C), by accessing carbon from amino acids for the Krebs cycle. Overall, these findings suggest that insulin plays a much larger role in directing cellular metabolism beyond stimulating glucose uptake, coordinating the oxidation of other substrates to fuel the energy demands of anabolism.
In contrast, glucose was specifically required for the insulin-stimulated increase in mitoROS, which was ablated by the absence of glucose or inhibition of glucose uptake, and accentuated by DCA treatment (Fig. 2). Given that insulin suppresses lipid breakdown by anti-lipolysis, this together implies that the remaining substrates (e.g. amino acids in the DMEM buffer) do not contribute to insulin-stimulated oxidant production. Furthermore, PRDX3 dimerization only increased when a threshold of glucose uptake was reached (Fig. 2H), suggesting that mitoROS generated below this threshold can be adequately buffered by the adipocyte. Upon acute insulin stimulation, we speculate that sites of glucose-dependent mitoROS production would either be PDH or mitochondrial glycerol 3-phosphate dehydrogenase (19, 28), which represent more “glucose-specific” sites of entry into mitochondrial respiration.
Collectively, these data demonstrate a disconnect between mitochondrial respiration and oxidant production (Fig. 4C). Although both respiration and mitoROS generation occur in the mitochondria, they differ in their substrate requirements, with only mitoROS levels increasing in the presence of glucose. Specifically, respiration responds to increases in energy demand, whereas mitoROS levels respond to increases in glucose supply. We believe that this disconnect between respiration and mitoROS enables adipocytes to distinguish between energetic demand and nutrient supply, fine-tuning its metabolism and insulin sensitivity to different nutritional and hormonal cues.
Although mitoROS are implicated in insulin resistance (8), glucose-dependent mitoROS did not acutely lead to insulin resistance (Fig. 3). In contrast, treatment with mPQ in a similarly short time frame was sufficient to induce insulin resistance (8). We subsequently found that mitoROS needed to increase prior to the insulin stimulus to induce insulin resistance (Fig. 4). Although we cannot exclude the possibility that glucose oxidation and mPQ treatment generate different oxidant species and/or oxidants that arise from different cellular locations, our findings suggest that there is a temporal requirement for mitoROS to cause insulin resistance. Specifically, as mitoROS outweighs endogenous scavenging systems, the mitoROS likely reacts with a macromolecule required in the initial stages of insulin-stimulated glucose transport. Furthermore, because oxidative stress does not require long-term adaptive changes such as transcriptional modification to cause insulin resistance in adipocytes (29), it is likely to cause rapid changes in kinase signaling and/or GLUT4 trafficking. However, mitoROS did not affect Akt signaling (Fig. 3), concurring with previous studies (11). Thus, it is tempting to speculate that mitoROS specifically blocks the insulin-dependent mobilization of GLUT4 to the cell surface (e.g. exocytosis of GLUT4-containing vesicles), rather than steps involved in the maintenance of enhanced PM GLUT4 during an insulin stimulus, such as the recycling of PM GLUT4 via endosomes.
Teleologically, this temporal requirement for mitoROS suggests that the adipocyte needs to sense excess nutrient supply prior to the insulin stimulus to trigger insulin resistance. This is consistent with a model whereby consumption of a meal will not impair insulin action, whereas oxidative stress upon chronic overnutrition would lead to a persistent decrease in insulin action. This raises the question of what physiological substrate(s) is oxidized to cause oxidant-induced insulin resistance (Fig. 4C). One strong candidate is fatty acids, because fatty acid oxidation has been demonstrated to impair insulin-stimulated glucose uptake in myotubes, and in muscle tissue in rodents and human subjects (e.g. see Refs. 6, 30, and 31). Also, insulin suppresses lipolysis; conversely, fatty acid oxidation would be elevated when insulin levels are low, providing a source of mitoROS prior to an insulin stimulus. This may explain why both long-term fasting (sustained lipolysis) and Western diets (high fat) both cause insulin resistance, in isolated rat adipocytes (32–35) and at the whole-body level (reviewed in Ref. (8). Ultimately, this would mechanistically link nutrient excess with insulin resistance.
Experimental procedures
Reagents
The following chemicals were used in this study: insulin (Sigma–Aldrich #I5500), dichloroacetate (sodium salt, Sigma–Aldrich #347795), oligomycin (Sigma–Aldrich #O4876), rotenone (Sigma–Aldrich #R8875), antimycin A (Sigma–Aldrich #A8674), FCCP (Sigma-Aldrich #C2920), and cytochalasin B (Sigma–Aldrich #C6762). In addition, BAM15 (36) was a gift from Dr. Kyle Hoehn (University of New South Wales, Sydney, Australia), and mPQ (37) was a gift from Dr. Mike Murphy (University of Cambridge, Cambridge, UK). Matrix-roGFP was a gift from Paul Schumacker (Addgene plasmid #49437) (46).
Cell culture
3T3-L1 fibroblasts were cultured as described previously (38), using Medium A, consisting of bicarbonate-buffered DMEM (Life Technologies, Inc., #11960), supplemented with 10% (v/v) fetal bovine serum (Life Technologies, #16000044) and 2 mm GlutaMAX (Life Technologies, #35050061). 3T3-L1 fibroblasts were differentiated into adipocytes as described previously (24, 38). Adipocytes were used between days 9 and 12 after the initiation of differentiation. At least 90% of the cells were differentiated prior to experiments. These cells were routinely tested for mycoplasma infection.
Unless otherwise specified, prior to insulin stimulation treatments, cells were serum-starved for at least 2 h. This involved washing cells three times with PBS and incubating them in basal medium. By default, the basal medium was Medium B, which consisted of bicarbonate-buffered DMEM (Life Technologies #11960), supplemented with 0.2% (w/v) BSA (Bovostar) and 2 mm GlutaMAX.
For experiments within the CO2 incubator that involved treatment with different sugars, cells were washed twice after serum starvation: once with PBS and then with BSF-DMEM, which consisted of substrate-free DMEM (Sigma–Aldrich #D5030), supplemented with 44 mm NaHCO3 and adjusted to pH 7.4 with CO2 (dry ice). Cells were then incubated in Medium BS, which consisted of BSF-DMEM, supplemented with 0.2% (w/v) BSA, 1 mm GlutaMAX, 1 mm glutamine, and sugar (glucose/galactose) as specified in the figure legends. The supplementation of glutamine in addition to GlutaMAX served to provide an immediate source of glutamine substrate for experiments involving early time points.
For metabolic assays performed outside of the CO2 incubator, cells were washed twice after serum starvation: once with PBS and then with HSF-DMEM, which consisted of substrate-free DMEM (Sigma–Aldrich #D5030) supplemented with 30 mm Na-HEPES (pH 7.4) and adjusted to pH 7.4 with NaOH. Cells were then incubated in Medium C, which consisted of HSF-DMEM, supplemented with 0.2% (w/v) BSA, 1 mm NaHCO3 (added fresh), 1 mm glutamine, 1 mm GlutaMAX, and sugars (glucose/galactose) as specified in the figure legends or below. Alternatively, cells were assayed in Medium CX, which consisted of Medium C without BSA, as specified in the figure legends.
Quantification of lactate content
Lactate content was assayed enzymatically using the hydrazine sink method, as described previously (21, 39).
Quantification of glucose uptake
Glucose uptake was determined by measuring the disappearance of medium glucose, as described previously (21, 24). This differs from the measurement of glucose transport, which is described below.
Respirometry using the XFp Analyzer (Seahorse BioSciences)
On day 7–8 of differentiation, 3T3-L1 adipocytes were seeded into XFp plates as described previously (14). Cells were prepared for the assay as described previously (14), with minor modifications. Specifically, cells were washed 4 times with PBS before incubation in Medium C, supplemented with 25 mm glucose and without NaHCO3, for at least 1.5 h in a non-CO2 incubator at 37 °C. Cells were then washed once with PBS, once with HSF-DMEM, and incubated in Medium CX (150 μl/well), supplemented with 25 mm sugar (glucose/galactose). Cells were then assayed in the XFp Analyzer; following a 12-min equilibration period, respiration and extracellular acidification rates were measured with mix/wait/read cycles of 3/0/2 min. Following stabilization of baseline rates, compounds were injected sequentially as indicated in the figure legends. Flux rates were normalized to cellular DNA content, the latter being quantified as described below.
Measurement of mitochondrial membrane potential
Mitochondrial membrane potential was assessed using the “quenching method” with tetramethylrhodamine methyl ester (TMRM; Sigma-Aldrich, #T5428) dye (40). 3T3-L1 adipocytes were seeded into black 96-well plates (Corning), precoated with Matrigel (Corning), in a similar manner as seeding into XFp plates (described above). On the day of the experiment, cells were washed four times with PBS (150 μl/wash) before incubation in Medium C, supplemented with 25 mm glucose and without NaHCO3, for at least 1.5 h in a non-CO2 incubator at 37 °C. In the last 30 min of this serum starvation period, cells were labeled with 500 nm TMRM (or DMSO control (“TMRM-free”)).
Cells were then washed twice with PBS and once with HSF-DMEM and then incubated in 90 μl of Medium CX (without glucose/galactose) per well. The plate was incubated at 37 °C in the FLUOstar Omega microplate reader (BMG Labtech). Fluorescence was measured every 90 s using the top optic at 544-nm excitation and 590-nm emission, with 20 flashes/well. This preread period was conducted for 15 min to ensure that a stable baseline measurement was achieved for each well. Drugs were added in the following sequence in 10 μl volumes, followed by fluorescence measurement (with final drug concentrations and treatment times indicated in the figure): glucose or galactose; insulin or naive medium control; oligomycin, BAM15, or DMSO control. At the end of the experiment, aliquots of media were taken from the wells treated with DMSO (i.e. glucose/galactose, insulin/control) and assessed for lactate content (described above) to check that insulin and sugar responses were similar to Fig. 1A (data not shown).
Fluorescence measurements were first adjusted by the fluorescence of identically treated cell-free and TMRM-free controls, to account for background fluorescence. Next, measurements were normalized to the last measurement of the preread period, to account for well-to-well differences in TMRM labeling. Finally, these values were normalized to the glucose-control-DMSO condition, to account for changes in signal independent of drug treatments. This normalized measurement was denoted “relative TMRM signal.”
Measurement of glucose oxidation and pentose phosphate pathway (PPP) flux
Following serum starvation in Medium B, cells were incubated with Medium C supplemented with 10 mm glucose and either 0.5 μCi/ml [1-14C]glucose, 1 μCi/ml [6-14C]glucose, or 1 μCi/ml [U-14C]glucose tracer. Following the addition of drug treatments, glucose oxidation was assessed as described previously (13, 21). Briefly, a gas trap was installed in each well, plates were sealed, and following treatment, cells were quenched by acidification. Radioactivity of the gas trapping solution was measured by liquid scintillation counting and adjusted to cell-free controls.
Total glucose oxidation was derived from the use of [U-14C]glucose tracer, and PPP flux was derived from the use of [1-14C]glucose and [6-14C]glucose tracers (41, 42). Calculating PPP flux also required the rate of glucose uptake and protein content, which were derived from cells treated in parallel (in the absence of radiolabel or gas trap installed). Glucose uptake was determined as described above, and protein content was determined as described below after washing cells three times with ice-cold PBS and lysis in PBS containing 1% (v/v) Triton X-100.
Assessment of insulin action by glucose transporter activity
Glucose transporter activity was measured as described previously (24). Briefly, following treatment, cells were washed with ice-cold PBS, incubated in cold Krebs–Ringer phosphate buffer (24) without any drugs, and assessed for 2-deoxyglucose uptake on ice. These cold conditions ensured that the rate of uptake was solely dependent on the membrane trafficking and movement of glucose transporters to the cell surface, which had occurred during the treatment period (but not the 2-deoxyglucose uptake period).
Assessment of insulin action by GLUT4 translocation
GLUT4 translocation in 3T3-L1 adipocytes was measured as described previously (11). Briefly, 3T3-L1 fibroblasts were retrovirally infected with a construct expressing HA-tagged GLUT4, differentiated, and seeded into black 96-well plates. Following treatment, the quantity of cell-surface HA was assessed by immunofluorescence staining and normalized to total HA content (11).
Normalization for cell culture experiments
Flux rates were normalized to cellular protein or DNA content, using cell lysate derived from the respective assay or from cells treated in parallel. Unless otherwise specified, flux rates were normalized to protein content, determined using the Pierce bicinchoninic acid assay kit (Thermo Fisher Scientific) according to the manufacturer's instructions. If flux rates were normalized to DNA, the DNA quantification was performed following cell lysis by repeated freeze-thaw, using Hoechst staining as described previously (21).
Western blotting
Following treatment, cells were harvested for protein, and lysates were subjected to Western blotting as described previously (11). Antibodies detecting pS473-Akt (clone 587F11, catalogue #4051), pT308-Akt (catalogue #9275), Akt (clone 11E7, catalogue #4685), and pT642-AS160 (catalogue #4288) were obtained from Cell Signaling Technology. The antibodies detecting PDH-E1α (clone H-131, catalogue #sc292543) and 14-3-3 (clone K-19, catalogue #sc-629) were obtained from Santa Cruz Biotechnology. The antibody detecting pS293-PDH-E1α (catalogue #ABS204) was obtained from Millipore. The antibody detecting PRDX3 (catalogue #LF-PA0030) was obtained from Life Technologies. The antibody detecting PRDX2 (clone EPR5154, catalogue #ab109367) was obtained from Abcam. Densitometric analysis was performed using either Fiji software (43) or LI-COR Image Studio.
Assessment of mitochondrial oxidative stress by microscopy
3T3-L1 adipocytes were electroporated and imaged as described previously (44). On day 7 of differentiation, 3T3-L1 adipocytes were tryspinized, pelleted at 150 × g for 5 min, and washed three times with PBS before resuspension in electroporation solution (20 mm HEPES, 135 mm KCl, 2 mm MgCl2, 0.5% (w/v) Ficoll 400, 1% (v/v) DMSO, 2 mm ATP, and 5 mm GSH, pH 7.6) and 5 μg of plasmid DNA (matrix-roGFP). Cells were then electroporated at 200 mV for 20 ms and plated onto Matrigel-coated 35-mm Ibidi glass-bottom m-Dishes.
Imaging experiments were performed 24 h post-electroporation. Cells were serum-starved in Medium B and imaged in Fluorobrite DMEM (Life Technologies), supplemented with 0.2% (w/v) BSA and 2 mm GlutaMAX, under 10% CO2. Imaging was performed on a Nikon Ti inverted microscope equipped with a Spectral Applied Research Diskovery spinning disk module and an Okolab cage incubator maintained at 37 °C. Drugs were added using a custom-made perfusion system. Image analysis was performed in Fiji (43) using a scripted analysis pipeline.
Quantification of the contribution of glucose to respiration-derived CO2
Our approach (Fig. 1G) was based on the principle that ECAR is influenced by both lactate production and bicarbonate derived from CO2 release (18). This required several sets of flux data (ECAR, lactate production, and glucose oxidation), each derived from separate batches of cells. These flux parameters, when measured in insulin-stimulated 3T3-L1 cells (in Medium C), typically varied by <20% between batches of cells (data not shown).
To quantify the contribution of glucose to respiration-derived CO2, the following steps were performed. (i) The total ECAR (mpH/h) was derived from flux data (e.g. Fig. S1A). Because ECAR initially rises before reaching a plateau with insulin treatment, total ECAR was determined by calculating the area under the curve using the trapezoidal rule. Because maximal ECAR remains constant for at least 1 h after insulin stimulation (data not shown), ECAR flux was extrapolated to 1 h using the last three time points at the maximal rate if complete data were not available. (ii) The buffering capacity (BF) of the medium (Fig. S1B) was determined by incubating naive medium in the Seahorse XFp and measuring pH changes following repeated injections of 1 m NaOH or HCl. Close to the pKa, the buffering curve is “pseudo-linear” (18), and thus the gradient can be used to determine buffering capacity (μm H+/mpH; i.e. change in [H+] per change in pH). (iii) The proton production rate (PPR; changes in medium [H+] in an XFp well) was determined from the ECAR (changes in medium pH), using the medium buffering capacity, measurement chamber volume, and Kvol adjustment factor (45)(Eq. 1),
| (Eq. 1) |
(iv) Next, we obtained lactate production data (21), which involved 3T3-L1 adipocytes treated with 100 nm insulin for 1 h in Medium C, supplemented with 25 mm glucose. The cells were treated under slightly different conditions (e.g. plate format, presence of BSA in assay medium), but these parameters did not impact insulin-stimulated lactate production (Fig. S1C). The lactate production data were scaled down to the cell number in a well of an XFp plate. (v) Bicarbonate production was determined from the PPR and lactate production data. Because each lactate molecule is effluxed with 1 proton and the conversion of CO2 into NaHCO3 generates 1 proton, these outputs are stoichiometrically 1:1 with PPR. Thus, we calculated bicarbonate production as the difference between PPR and lactate production (Eq. 2).
| (Eq. 2) |
We assumed that other sources of proton efflux were negligible (18). (vi) Total CO2 production was determined from bicarbonate production by accounting for the solubility of bicarbonate in water (18). (vii) The contribution of glucose to CO2 production was calculated using measurements of the incorporation of 14C-radiolabeled glucose into CO2 (21). Briefly, 3T3-L1 adipocytes were treated with 100 nm insulin for 1 h in Medium C, supplemented with 25 mm glucose and [U-14C]glucose tracer. This value was scaled down to the cell number in a well of an XFp plate and multiplied by 6 (1 glucose generates 6 CO2) to obtain the production rate of glucose-derived CO2. (viii) Because CO2 can be generated by the PPP in addition to respiration, we compared the incorporation of glucose into CO2 versus PPP flux (Fig. S1D). PPP flux was stimulated by insulin and abolished with an inhibitor of 6-phosphogluconate dehydrogenase, 6-aminonicotinamide (Fig. S1D). Nevertheless, PPP-derived CO2 was negligible compared with the total incorporation of glucose into CO2 (performed in parallel; Fig. S1D), considering that PPP generates 1 CO2 per glucose molecule compared with 6 CO2 via respiration. Thus, it was assumed that glucose-derived CO2 was solely derived from respiration. (ix) Glucose-derived CO2 production was compared with total CO2 production, yielding the contribution of glucose to respiration-derived CO2.
Metabolic assays in primary adipocytes
Isolation and assay buffers
Buffers included Krebs–Henseleit buffer (KHB: 120 mm NaCl, 4.7 mm KCl, 1.18 mm KH2PO4, 1.17 mm MgSO4, 1.8 mm CaCl2, 30 mm Na-HEPES, 1 mm NaHCO3 (added fresh), 1% (w/v) fatty acid–free BSA (Sigma), pH adjusted to 7.4 with NaOH), KHB-Glc buffer (KHB, supplemented with 10 mm glucose), and assay buffer (HSF-DMEM, supplemented with 1 mm NaHCO3 (added fresh), 1 mm glutamine, 1 mm GlutaMAX, and 1% (w/v) fatty acid–free BSA, with pH adjusted to 7.4 with NaOH). All buffers were maintained at 37 °C throughout the experiment.
Isolation of mature primary adipocytes
Male Sprague–Dawley rats were housed at 23 °C in individually ventilated cages on a 12-h light/dark cycle with ad libitum access to standard rodent chow and water. Rat experiments were approved by the University of Sydney Animal Ethics Committee (project #2018/1418). At 7–8 weeks of age (200–300 g), rats were sacrificed with CO2. Mature primary adipocytes were isolated as described previously (21). Briefly, epidydimal fat pads were isolated and collagenase-digested in KHB-Glc buffer. The isolated adipocytes were washed twice with KHB and then twice with assay buffer, to remove collagenase, nonadipocyte cells, and glucose from the digestion buffer. After the final wash, the infranatant was removed, and aliquots of adipocyte suspension were used in the assays below.
Respirometry of primary adipocytes using the O2K oxygraph (Oroboros)
High-resolution oxygen consumption measurements were conducted using the O2K oxygraph (series H). 200–400 μl of primary adipocyte suspension were loaded into each O2K chamber, made up to a final volume of 2.1 ml with assay buffer supplemented with 5 mm (final concentration) glucose or galactose. Experiments were conducted at 37 °C with stirring at 750 rpm and data recording at 2-s intervals. Cells were allowed to equilibrate prior to the sequential addition of pharmacological agents as indicated in the figure legends. Respiratory flux data were analyzed using DatLab 7.3.0.3 (Oroboros).
Following the experiment, aliquots (3 × 500 μl) of cell suspension were taken from each chamber and immediately frozen. DNA was isolated under high-salt conditions with organic extraction, similar in principle to what has been described previously (21). After the addition of 0.2 volume of 5 m NaCl and mixing, an equal volume (600 μl) of CHCl3 was added. Samples were mixed thoroughly and centrifuged for 20 min at 16,000 × g and 4 °C. The supernatant was assayed for DNA content using SyBr stain (Life Technologies), according to the manufacturer's instructions. Fluorescence measurements were compared with a standard curve of salmon sperm DNA, spiked with naive assay buffer extracted alongside adipocyte samples. Furthermore, extraction efficiency was determined by spiking one aliquot of adipocyte suspension from each O2K chamber (of three) with salmon sperm DNA prior to extraction. Respiration fluxes were normalized to DNA content and then to the baseline respiration of the chamber receiving the glucose + insulin condition.
Lactate measurement in primary adipocytes
Adipocytes were assessed for lactate production as described previously (21), with slight modifications. Briefly, 200 μl of adipocyte suspension was dispensed into wells of 24-well polystyrene, tissue culture microplates (Corning). To each well, 50 μl of prewarmed assay buffer, containing glucose/galactose, PBS, and/or insulin, was added. As a result, each well contained a final volume of 250 μl, with 5 mm sugar (glucose or galactose) and either PBS control or 20 nm insulin. Plates were then immediately sealed with TopSeal-A PLUS (PerkinElmer Life Sciences) and gently rocked for 1 h with a rocking platform in a 37 °C incubator.
Following incubation, cells were immediately quenched by the addition of 400 μl of MeOH, followed by 400 μl of water. Lysates were transferred to tubes, 800 μl of CHCl3 was added, and samples were vortexed briefly prior to centrifugation for 20 min at 16,000 × g and 4 °C. The supernatant was lyophilized using an EZ-2 centrifugal evaporator (GeneVac), resuspended in water, and assayed for lactate content enzymatically as described above and previously (21).
Author contributions
J. R. K., D. J. F., and D. E. J. conceptualization; J. R. K. formal analysis; J. R. K., G. J. C., D. J. F., and D. E. J. funding acquisition; J. R. K., S. D. E., A. D.-V., K. C. C., J. G. B., and D. J. F. investigation; J. R. K. and K. H. F.-W. methodology; J. R. K., D. J. F., and D. E. J. writing-original draft; J. R. K., S. D. E., A. D.-V., K. C. C., K. H. F.-W., G. J. C., D. J. F., and D. E. J. writing-review and editing; G. J. C., D. J. F., and D. E. J. supervision.
Supplementary Material
Acknowledgments
We thank the Charles Perkins Centre (CPC) Research Support team, particularly Dr. Ian Garthwaite, Dr. Natalia Magarinos, Harry Simpson, Dr. Melissa Gardiner, and Dr. Macarena Rodriguez, for technical support.
The authors declare that they have no conflicts of interest with the contents of this article. The contents of the published material are solely the responsibility of the authors and do not reflect the views of the National Health and Medical Research Council (NHMRC).
This article contains Figs. S1 and S2.
L. E. Quek, J. R. Krycer, S. Ohno, K. Yugi, D. J. Fazakerley, R. Scalzo, S. D. Elkington, Z. Dai, A. Hirayama, S. Ikeda, F. Shoji, K. Suzuki, J. W. Locasale, T. Soga, D. E. James, S. Kuroda, submitted for publication.
- GLUT4
- glucose transporter 4
- mitoROS
- mitochondrial reactive oxygen species
- ROS
- reactive oxygen species
- mPQ
- mitochondria-targeted paraquat
- MMP
- mitochondrial membrane potential
- ANOVA
- analysis of variance
- FCCP
- carbonyl cyanide p-trifluoromethoxyphenylhydrazone
- PDH
- pyruvate dehydrogenase
- ECAR
- extracellular acidification rate
- DCA
- dichloroacetate
- PRDX2 and PRDX3
- peroxiredoxin 2 and 3, respectively
- DMEM
- Dulbecco's modified Eagle's medium
- PM
- plasma membrane
- TMRM
- tetramethylrhodamine methyl ester
- PPP
- pentose phosphate pathway
- mpH
- milli-pH units
- PPR
- proton production rate
- KHB
- Krebs–Henseleit buffer
- Ins
- insulin
- Glc
- glucose
- Gal
- galactose
- Oligo
- oligomycin
- Rot
- rotenone
- AA
- antimycin A
- JO2
- respiration rate.
References
- 1. Rosen E. D., and Spiegelman B. M. (2014) What we talk about when we talk about fat. Cell 156, 20–44 10.1016/j.cell.2013.12.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Garvey W. T., and Kolterman O. G. (1988) Correlation of in vivo and in vitro actions of insulin in obesity and noninsulin-dependent diabetes mellitus: role of the glucose transport system. Diabetes Metab. Rev. 4, 543–569 10.1002/dmr.5610040602 [DOI] [PubMed] [Google Scholar]
- 3. Kraegen E. W., Clark P. W., Jenkins A. B., Daley E. A., Chisholm D. J., and Storlien L. H. (1991) Development of muscle insulin resistance after liver insulin resistance in high-fat-fed rats. Diabetes 40, 1397–1403 10.2337/diab.40.11.1397 [DOI] [PubMed] [Google Scholar]
- 4. Turner N., Kowalski G. M., Leslie S. J., Risis S., Yang C., Lee-Young R. S., Babb J. R., Meikle P. J., Lancaster G. I., Henstridge D. C., White P. J., Kraegen E. W., Marette A., Cooney G. J., Febbraio M. A., and Bruce C. R. (2013) Distinct patterns of tissue-specific lipid accumulation during the induction of insulin resistance in mice by high-fat feeding. Diabetologia 56, 1638–1648 10.1007/s00125-013-2913-1 [DOI] [PubMed] [Google Scholar]
- 5. Abel E. D., Peroni O., Kim J. K., Kim Y. B., Boss O., Hadro E., Minnemann T., Shulman G. I., and Kahn B. B. (2001) Adipose-selective targeting of the GLUT4 gene impairs insulin action in muscle and liver. Nature 409, 729–733 10.1038/35055575 [DOI] [PubMed] [Google Scholar]
- 6. Hoehn K. L., Salmon A. B., Hohnen-Behrens C., Turner N., Hoy A. J., Maghzal G. J., Stocker R., Van Remmen H., Kraegen E. W., Cooney G. J., Richardson A. R., and James D. E. (2009) Insulin resistance is a cellular antioxidant defense mechanism. Proc. Natl. Acad. Sci. U.S.A. 106, 17787–17792 10.1073/pnas.0902380106 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Fazakerley D. J., Chaudhuri R., Yang P., Maghzal G. J., Thomas K. C., Krycer J. R., Humphrey S. J., Parker B. L., Fisher-Wellman K. H., Meoli C. C., Hoffman N. J., Diskin C., Burchfield J. G., Cowley M. J., Kaplan W., et al. (2018) Mitochondrial CoQ deficiency is a common driver of mitochondrial oxidants and insulin resistance. Elife 7, e32111 10.7554/eLife.32111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fazakerley D. J., Krycer J. R., Kearney A. L., Hocking S. L., and James D. E. (2019) Muscle and adipose tissue insulin resistance: malady without mechanism? J. Lipid Res. 60, 1720–1732 10.1194/jlr.R087510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Montgomery M. K., and Turner N. (2015) Mitochondrial dysfunction and insulin resistance: an update. Endocr. Connect. 4, R1–R15 10.1530/EC-14-0092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. De Pauw A., Tejerina S., Raes M., Keijer J., and Arnould T. (2009) Mitochondrial (dys)function in adipocyte (de)differentiation and systemic metabolic alterations. Am. J. Pathol. 175, 927–939 10.2353/ajpath.2009.081155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Fazakerley D. J., Minard A. Y., Krycer J. R., Thomas K. C., Stöckli J., Harney D. J., Burchfield J. G., Maghzal G. J., Caldwell S. T., Hartley R. C., Stocker R., Murphy M. P., and James D. E. (2018) Mitochondrial oxidative stress causes insulin resistance without disrupting oxidative phosphorylation. J. Biol. Chem. 293, 7315–7328 10.1074/jbc.RA117.001254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Fisher-Wellman K. H., and Neufer P. D. (2012) Linking mitochondrial bioenergetics to insulin resistance via redox biology. Trends Endocrinol. Metab. 23, 142–153 10.1016/j.tem.2011.12.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Krycer J. R., Diskin C., Nelson M. E., Zeng X. Y., Fazakerley D. J., and James D. E. (2018) A gas trapping method for high-throughput metabolic experiments. BioTechniques 64, 27–29 10.2144/000114629 [DOI] [PubMed] [Google Scholar]
- 14. Krycer J. R., Fisher-Wellman K. H., Fazakerley D. J., Muoio D. M., and James D. E. (2017) Bicarbonate alters cellular responses in respiration assays. Biochem. Biophys. Res. Commun. 489, 399–403 10.1016/j.bbrc.2017.05.151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Eagle H., Barban S., Levy M., and Schulze H. O. (1958) The utilization of carbohydrates by human cell cultures. J. Biol. Chem. 233, 551–558 [PubMed] [Google Scholar]
- 16. Reitzer L. J., Wice B. M., and Kennell D. (1979) Evidence that glutamine, not sugar, is the major energy source for cultured HeLa cells. J. Biol. Chem. 254, 2669–2676 [PubMed] [Google Scholar]
- 17. Chang C. H., Curtis J. D., Maggi L. B. Jr., Faubert B., Villarino A. V., O'Sullivan D., Huang S. C., van der Windt G. J., Blagih J., Qiu J., Weber J. D., Pearce E. J., Jones R. G., and Pearce E. L. (2013) Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell 153, 1239–1251 10.1016/j.cell.2013.05.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Mookerjee S. A., Gerencser A. A., Nicholls D. G., and Brand M. D. (2017) Quantifying intracellular rates of glycolytic and oxidative ATP production and consumption using extracellular flux measurements. J. Biol. Chem. 292, 7189–7207 10.1074/jbc.M116.774471 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Brand M. D. (2010) The sites and topology of mitochondrial superoxide production. Exp. Gerontol. 45, 466–472 10.1016/j.exger.2010.01.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Quinlan C. L., Perevoshchikova I. V., Hey-Mogensen M., Orr A. L., and Brand M. D. (2013) Sites of reactive oxygen species generation by mitochondria oxidizing different substrates. Redox Biol. 1, 304–312 10.1016/j.redox.2013.04.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Krycer J. R., Quek L. E., Francis D., Fazakerley D. J., Elkington S. D., Diaz-Vegas A., Cooke K. C., Weiss F. C., Duan X., Kurdyukov S., Zhou P. X., Tambar U. K., Hirayama A., Ikeda S., Kamei Y., et al. (2019) Lactate production is a prioritized feature of adipocyte metabolism. J. Biol. Chem. 294, 83–98 10.1074/jbc.RA119.011178 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Palfreyman R. W., Clark A. E., Denton R. M., Holman G. D., and Kozka I. J. (1992) Kinetic resolution of the separate GLUT1 and GLUT4 glucose transport activities in 3T3-L1 cells. Biochem. J. 284, 275–282 10.1042/bj2840275 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Tan S. X., Fisher-Wellman K. H., Fazakerley D. J., Ng Y., Pant H., Li J., Meoli C. C., Coster A. C., Stöckli J., and James D. E. (2015) Selective insulin resistance in adipocytes. J. Biol. Chem. 290, 11337–11348 10.1074/jbc.M114.623686 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Krycer J. R., Yugi K., Hirayama A., Fazakerley D. J., Quek L. E., Scalzo R., Ohno S., Hodson M. P., Ikeda S., Shoji F., Suzuki K., Domanova W., Parker B. L., Nelson M. E., Humphrey S. J., et al. (2017) Dynamic metabolomics reveals that insulin primes the adipocyte for glucose metabolism. Cell Rep. 21, 3536–3547 10.1016/j.celrep.2017.11.085 [DOI] [PubMed] [Google Scholar]
- 25. Tan S. X., Ng Y., and James D. E. (2010) Akt inhibitors reduce glucose uptake independently of their effects on Akt. Biochem. J. 432, 191–197 10.1042/BJ20100750 [DOI] [PubMed] [Google Scholar]
- 26. Green C. R., Wallace M., Divakaruni A. S., Phillips S. A., Murphy A. N., Ciaraldi T. P., and Metallo C. M. (2016) Branched-chain amino acid catabolism fuels adipocyte differentiation and lipogenesis. Nat. Chem. Biol. 12, 15–21 10.1038/nchembio.1961 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Liu L., Shah S., Fan J., Park J. O., Wellen K. E., and Rabinowitz J. D. (2016) Malic enzyme tracers reveal hypoxia-induced switch in adipocyte NADPH pathway usage. Nat. Chem. Biol. 12, 345–352 10.1038/nchembio.2047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Wong H. S., Dighe P. A., Mezera V., Monternier P. A., and Brand M. D. (2017) Production of superoxide and hydrogen peroxide from specific mitochondrial sites under different bioenergetic conditions. J. Biol. Chem. 292, 16804–16809 10.1074/jbc.R117.789271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Chaudhuri R., Krycer J. R., Fazakerley D. J., Fisher-Wellman K. H., Su Z., Hoehn K. L., Yang J. Y. H., Kuncic Z., Vafaee F., and James D. E. (2018) The transcriptional response to oxidative stress is part of, but not sufficient for, insulin resistance in adipocytes. Sci. Rep. 8, 1774 10.1038/s41598-018-20104-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dresner A., Laurent D., Marcucci M., Griffin M. E., Dufour S., Cline G. W., Slezak L. A., Andersen D. K., Hundal R. S., Rothman D. L., Petersen K. F., and Shulman G. I. (1999) Effects of free fatty acids on glucose transport and IRS-1-associated phosphatidylinositol 3-kinase activity. J. Clin. Invest. 103, 253–259 10.1172/JCI5001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Hoy A. J., Brandon A. E., Turner N., Watt M. J., Bruce C. R., Cooney G. J., and Kraegen E. W. (2009) Lipid and insulin infusion-induced skeletal muscle insulin resistance is likely due to metabolic feedback and not changes in IRS-1, Akt, or AS160 phosphorylation. Am. J. Physiol. Endocrinol. Metab. 297, E67–E75 10.1152/ajpendo.90945.2008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Thacker S. V., Nickel M., and DiGirolamo M. (1987) Effects of food restriction on lactate production from glucose by rat adipocytes. Am. J. Physiol. 253, E336–E342 10.1152/ajpendo.1987.253.4.E336 [DOI] [PubMed] [Google Scholar]
- 33. Newby F. D., Sykes M. N., and DiGirolamo M. (1988) Regional differences in adipocyte lactate production from glucose. Am. J. Physiol. 255, E716–E722 10.1152/ajpendo.1988.255.5.E716 [DOI] [PubMed] [Google Scholar]
- 34. Newby F. D., Wilson L. K., Thacker S. V., and DiGirolamo M. (1990) Adipocyte lactate production remains elevated during refeeding after fasting. Am. J. Physiol. 259, E865–E871 10.1152/ajpendo.1990.259.6.E865 [DOI] [PubMed] [Google Scholar]
- 35. Bernstein R. S., Zimmerman K. S., and Carney A. L. (1981) Absence of impaired glucose utilization in adipocytes from rats fed a carbohydrate-free, high protein diet. J. Nutr. 111, 237–243 10.1093/jn/111.2.237 [DOI] [PubMed] [Google Scholar]
- 36. Kenwood B. M., Weaver J. L., Bajwa A., Poon I. K., Byrne F. L., Murrow B. A., Calderone J. A., Huang L., Divakaruni A. S., Tomsig J. L., Okabe K., Lo R. H., Cameron Coleman G., Columbus L., Yan Z., et al. (2014) Identification of a novel mitochondrial uncoupler that does not depolarize the plasma membrane. Mol. Metab. 3, 114–123 10.1016/j.molmet.2013.11.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Robb E. L., Gawel J. M., Aksentijević D., Cochemé H. M., Stewart T. S., Shchepinova M. M., Qiang H., Prime T. A., Bright T. P., James A. M., Shattock M. J., Senn H. M., Hartley R. C., and Murphy M. P. (2015) Selective superoxide generation within mitochondria by the targeted redox cycler MitoParaquat. Free Radic. Biol. Med. 89, 883–894 10.1016/j.freeradbiomed.2015.08.021 [DOI] [PubMed] [Google Scholar]
- 38. Fazakerley D. J., Naghiloo S., Chaudhuri R., Koumanov F., Burchfield J. G., Thomas K. C., Krycer J. R., Prior M. J., Parker B. L., Murrow B. A., Stöckli J., Meoli C. C., Holman G. D., and James D. E. (2015) Proteomic analysis of GLUT4 storage vesicles reveals tumor suppressor candidate 5 (TUSC5) as a novel regulator of insulin action in adipocytes. J. Biol. Chem. 290, 23528–23542 10.1074/jbc.M115.657361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Prabhu A. V., Krycer J. R., and Brown A. J. (2013) Overexpression of a key regulator of lipid homeostasis, Scap, promotes respiration in prostate cancer cells. FEBS Lett. 587, 983–988 10.1016/j.febslet.2013.02.040 [DOI] [PubMed] [Google Scholar]
- 40. Perry S. W., Norman J. P., Barbieri J., Brown E. B., and Gelbard H. A. (2011) Mitochondrial membrane potential probes and the proton gradient: a practical usage guide. BioTechniques 50, 98–115 10.2144/000113610 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Katz J., and Wood H. G. (1963) The use of C14O2 yields from glucose-1- and -6-C14 for the evaluation of the pathways of glucose metabolism. J. Biol. Chem. 238, 517–523 [PubMed] [Google Scholar]
- 42. Ashcroft S. J., Weerasinghe L. C., Bassett J. M., and Randle P. J. (1972) The pentose cycle and insulin release in mouse pancreatic islets. Biochem. J. 126, 525–532 10.1042/bj1260525 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J. Y., White D. J., Hartenstein V., Eliceiri K., Tomancak P., and Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods 9, 676–682 10.1038/nmeth.2019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Norris D. M., Yang P., Krycer J. R., Fazakerley D. J., James D. E., and Burchfield J. G. (2017) An improved Akt reporter reveals intra- and inter-cellular heterogeneity and oscillations in signal transduction. J. Cell Sci. 130, 2757–2766 10.1242/jcs.205369 [DOI] [PubMed] [Google Scholar]
- 45. Romero N., Swain P., Neilson A., and Dranka B. P. (2017) Improving Quantification of Cellular Glycolytic Rate Using Agilent Seahorse XF Technology. Agilent Technologies Application Note, Agilent Technologies, Santa Clara, CA [Google Scholar]
- 46. Waypa GB, Marks JD, Guzy R, Mungai PT, Schriewer J, Dokic D, Schumacker P. T. (2009) Hypoxia triggers subcellular compartmental redox signaling in vascular smooth muscle cells. Circ Res. 106, 526–535 10.1161/CIRCRESAHA.109.206334 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.