

Abstract
Psychosocial stress—especially when chronic, excessive, or occurring early in life—has been associated with accelerated aging and increased disease risk. With rapid aging of the world population, the need to elucidate the underlying mechanisms is pressing, now more so than ever. Among molecular mechanisms linking stress and aging, the present article reviews evidence on the role of epigenetics, biochemical processes that can be set into motion by stressors and in turn influence genomic function and complex phenotypes, including aging-related outcomes. The article further provides a conceptual mechanistic framework on how stress may drive epigenetic changes at susceptible genomic sites, thereby exerting systems-level effects on the aging epigenome while also regulating the expression of molecules implicated in aging-related processes. This emerging evidence, together with work examining related biological processes, begins to shed light on the epigenetic and, more broadly, molecular underpinnings of the long-hypothesized connection between stress and aging.
Keywords: aging, DNA methylation, epigenetics, inflammation, psychosocial stress, telomere
Abstract
El estrés psicosocial -especialmente cuando es crónico, excesivo o se presenta en los inicios de la vida- se ha asociado con un envejecimiento acelerado y un mayor riesgo de enfermedad. Ya que existe un rápido envejecimiento de la población mundial, se hace necesario dilucidar cuanto antes los mecanismos subyacentes. Este artículo, considerando los mecanismos moleculares que vinculan el estrés con el envejecimiento, revisa la evidencia acerca del papel de la epigenética, los procesos bioquímicos activados por los estresores que pueden influir en la función genómica y los fenotipos complejos, incluyendo los resultados relacionados con el envejecimiento. El artículo entrega además un marco conceptual mecanicista sobre cómo el estrés puede conducir a cambios epigenéticos en sitios genómicos susceptibles, provocando así efectos a nivel de sistemas del epigenoma del envejecimiento, a la vez que regula la expresión de moléculas implicadas en procesos relacionados con el envejecimiento. Esta reciente evidencia, junto con el trabajo que examina los procesos biológicos relacionados, comienza a dar luces sobre los fundamentos epigenéticos y, más ampliamente, sobre las bases moleculares de la antigua hipotética conexión entre el estrés y el envejecimiento.
Abstract
Le stress psychosocial, surtout lorsqu’il est chronique, excessif ou si il survient précocement dans la vie, a été associé à un vieillissement accéléré et un risque augmenté de maladie. Le vieillissement de la population s’accélérant dans le monde, il devient plus que jamais urgent d’en identifier les mécanismes sous-jacents. Parmi les mécanismes moléculaires liant le stress au vieillissement, cet article analyse les preuves du rôle de l’épigénétique, ces processus biochimiques activés par des agents stresseurs qui peuvent influer sur la fonction génomique et les phénotypes complexes, dont les événements liés à l’âge. Nous proposons ensuite un cadre mécanistique conceptuel sur la façon dont le stress peut induire des modifications épigénétiques au niveau des sites génomiques sensibles : le stress a donc des effets multi-systémiques sur l’épigénome vieillissant et régule aussi l’expression des molécules impliquées dans les processus liés à l’âge. Ces nouvelles données, ajoutées aux travaux sur les mécanismes biologiques, commencent à faire la lumière sur l’épigénétique et, plus largement, sur les fondements moléculaires de la connexion depuis longtemps supposée entre stress et vieillissement.
“It was he [Jean Valjean] in fact. The clerk’s lamp illuminated his countenance. He was pale and he trembled slightly. His hair, which had still been gray at his arrival [to the court], was now entirely white; it had turned white during the hour he had sat there.”
Victor Hugo, Les Misérables
Introduction
In Hugo’s fictional work Les Misérables , an extreme stressor causes the main character, Jean Valjean, to undergo accelerated aging, depicted as rapid whitening of his hair. 1 This dramatic depiction is just one among innumerable examples—found in literary works, movies, and folklore legends—of individuals whose “biological clocks” appear to tick fast in the face of life adversity. Beyond fiction, however, the connection between psychosocial stress and rate of biological aging is also seen in everyday life and clinical practice; for example, past US presidents notoriously exhibited signs of accelerated aging during their time in office, 2 and individuals with stress-related psychiatric disorders often appear older than their stated age. 3 Furthermore, several epidemiological studies have now grounded these observations on scientific evidence, linking psychosocial stress and related psychiatric conditions, such as major depressive and posttraumatic stress disorder, with increased risk for a number of aging-related disease states. 4 - 7
Together these observations raise important questions: Can the effects of stress on the aging process be quantified with biological measures? Do such biological effects shape health and disease outcomes? And could we, one day, prevent or even reverse stress-related aging for the benefit of individuals and societies?
The concept of accelerated biological and epigenetic aging: links with disease risk and stress burden
Aging is the single most important risk factor for several disease states that are currently the leading causes of morbidity and mortality, including cardiovascular disease, neurodegenerative disorders, and cancer. 8 Yet individuals of the same age exhibit substantial variability in their propensity to develop aging-related disease. 9 This variability could be in part explained by individual discrepancies between chronological age, the units of time elapsed since birth, and biological age, the wear-and-tear biological events thought to accrue throughout life and confer disease risk. 10 , 11 The idea that biological aging may better reflect disease risk has spurred efforts to develop molecular measures of biological age using telomere length, 12 epigenomic patterns, 13 , 14 transcriptomic signatures, 15 or proteomic profiles. 16 Moreover, accelerated biological aging—ie, having biological age more advanced than chronological age—has been hypothesized to confer disease risk beyond that associated with chronological age itself. This hypothesis has been vigorously tested using the so-called measures of epigenetic aging, composite markers that accurately predict age by combining in a statistical regression model the DNA methylation levels of multiple genomic sites. Such studies have indeed linked accelerated epigenetic aging with a host of aging-related disease outcomes, including all-cause mortality in late life, 17 physical and cognitive impairment, 18 cancer incidence, 19 frailty in older ages, 20 dementia, 21 and others. 22 Similar associations have been observed with other measures of accelerated biological aging, such as telomere shortening. 23 Overall this work supports a model whereby variability in the rate of biological aging may underlie differences in disease risk across individuals.
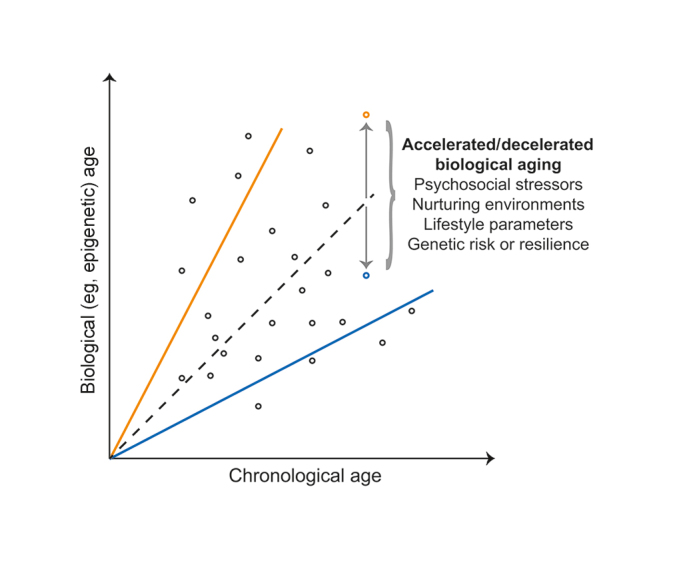
Therefore, uncovering factors that explain interindividual variance in biological aging can yield novel insights into the pathogenesis of aging-related disease. As conceptually depicted on Figure 1 , age predicted with biological measures, such as epigenetic age, generally correlates well with chronological age at the population level; yet the two can substantially differ for certain individuals, even when using the most accurate multi-tissue predictor of epigenetic age, developed by Steve Horvath. 13 Individuals can thus be conceptualized to follow different trajectories, depending on whether their biological aging is accelerated (biological age > chronological age) or decelerated (biological age < chronological age). Because aging is a complex phenotype, biological age acceleration and associated disease risk can be conferred by multiple genetic and environmental factors. 24 Environmental contribution is supported by several studies showing that lifestyle parameters and behaviors—including smoking, unhealthy diets, sedentary lifestyle, and alcohol—are major risk factors for aging-related diseases, together accounting for 70% of deaths worldwide. 25 Psychosocial stress, the focus of the present review, is another key environmental factor that not only predisposes to health- damaging behaviors but also directly affects body physiology, contributing to negative outcomes. 26 Given that stress is ubiquitous in modern fast-paced societies, gaining mechanistic insights into how it influences biological aging has important implications for prevention and treatment.
Figure 1. Conceptual graph showing the relation between chronological and biological age across individuals (depicted as circles). Although age predicted with biological measures, such as DNA methylation, generally correlates well with chronological age at the population level (dotted black line), the two can substantially differ for certain individuals, who can thus follow trajectories with either accelerated (biological age > chronological age; orange continuous line) or decelerated biological aging (biological age < chronological age; blue continuous line). Such interindividual variance in biological aging may be explained by genetic factors that confer risk or resilience, environmental factors such as psychosocial stress, and their complex interplay. For illustrative purposes, the figure highlights two individuals with the same chronological age and either accelerated (orange circle) or decelerated biological aging (blue circle).
Among metrics of accelerated biological aging that may be influenced by stress exposure, the largest body of work to date has examined telomere length. Following the seminal work by Epel and colleagues linking chronic caregiver stress with telomere shortening, 27 several studies found similar associations with other types of stress. A detailed account of these studies is beyond the scope of this article, but types and measures of stress examined to date in relation to telomere function include perceived stress, 28 intrauterine and early-life stress, 29 , 30 work-related exhaustion, 31 and major depression. 32 Although these associations have not been replicated in all populations studied, 33 , 34 meta-analyses have corroborated that higher stress burden and related phenotypes are associated with telomere attrition. 35 - 37
More recently, a number of studies have linked psychosocial stress with biological aging assessed using metrics of epigenetic age. In particular, acceleration in epigenetic aging has been observed with various types of early-life stress, including exposure to harsh parenting during childhood and adolescence, 38 perceived racial discrimination during adolescence, 39 childhood exposure to violence and threat, 40 , 41 early-life socioeconomic disadvantage, 42 - 44 and childhood abuse. 45 Although lack of association with early-life stress has also been reported, 46 , 47 a recent meta-analysis corroborated an overall positive but small relation between epigenetic age acceleration and childhood stress. 48 Likewise, accelerated epigenetic aging has been associated with stress exposure in adulthood such as deployment to combat, 47 financial hardship and socioeconomic disadvantage, 49 , 50 and cumulative lifetime stress 46 ; however, lack of relation has been also reported with adulthood socioeconomic status and lifetime trauma. 42 , 43 , 48 Lastly, some studies have found positive relations of epigenetic age acceleration with diagnosis of major depression and lifetime severity of posttraumatic stress disorder, 45 , 48 whereas others noted no relation with major depression 46 and nonsignificant or even inverse relations with posttraumatic stress diagnosis and symptoms. 47 , 48 Such discrepancies across studies could result from heterogeneity in their design and methodology, including differences in the sociodemographic characteristics of the study participants, the definition of stress exposure and related outcomes, and the time lag between stress exposure and epigenetic assessment.
Additional work has linked stress with other measures of biological aging, such as oxidative stress, mitochondrial dysfunction, and N-glycosylation. Oxidative stress, a process that can impact aging through cumulative damage in macromolecules and cells, has been shown to increase with stress exposure and in stress-related psychiatric disorders. 51 Mitochondrial dysfunction, determined with measures such as mitochondrial DNA levels and enzymatic activity, has been associated with both stress exposure and depressive symptoms. 52 , 53 N-glycosylation, a marker that accumulates with advancing age, was found to increase in trauma-exposed individuals and persons with post-traumatic stress disorder. 54
Taken together and notwithstanding discordances across studies, this body of work links certain types of stress—most notably early-life, cumulative, chronic, and excessive stress—and related psychiatric disorders with accelerated aging determined using diverse molecular measures. This conclusion is important because it elucidates—at a deeper, molecular level—the previously known epidemiological connection between stress burden and risk for aging- related disease. 4 - 7 It further supports the notion that future research endeavors should aim to determine the extent to which psychosocial and other environmental stressors as well supportive and nurturing environments contribute to interindividual variance in biological aging ( Figure 1 ). While the multilayered associations reported above suggest a complex interplay among many molecular processes, the following section will discuss in more depth the role of epigenetics as a mechanistic link between stress and aging.
Epigenetic mechanisms linking psychosocial stress and aging
Epigenetics—a composite Greek word derived from the prefix “epi,” meaning “upon,” and “genetics”—encompasses an ever-growing repertoire of biochemical processes that together regulate gene function without changing the genetic code itself. These processes include covalent DNA modifications, such as methylation of cytosine-guanine dinucleotides (CpG), posttranslational histone modifications, regulation of transcription by noncoding RNAs, and higher-order changes in chromatin conformation. 55 These processes are known to not only direct cellular differentiation but to also respond to environmental triggers, including psychosocial stressors at various life stages, 56 and to regulate genomic function, thereby acting as a molecular interface between environments and genomes. Epigenetic changes can thus be conceptualized as molecular switches or, better still, as rheostats that are set into motion by environmental stressors and can in turn fine-tune gene activity and cell function ( Figure 2 ). While these “genomic rheostats” can respond dynamically to stressors, 57 stress-related and other epigenetic signatures can also persist in time, 56 having potentially lasting consequences on cell function and complex phenotypes. Epigenetic regulation has been implicated in many such phenotypes and is thought to represent a hallmark of the aging process. 58 Although the various players of the epigenetic machinery act in concert, the rest of this section will largely focus on CpG methylation, which with the advent of microarray technology has become the most studied epigenetic modification in humans.
Psychosocial stressors trigger a set of behavioral, neural, hormonal, and molecular processes. Among such processes, previous work supports a key role for glucocorticoids, hormones secreted in the peripheral circulation upon exposure to stress. 59 The genomic effects of glucocorticoids are largely driven by the glucocorticoid receptor (GR), a ligand-dependent transcription factor found in essentially every body tissue. These genomic effects can be mediated either by direct binding of the GR to glucocorticoid response elements (GRE)—thousands of conserved DNA sequences scattered throughout the genome—or by interactions with other transcription factors via GRE-dependent or -independent mechanisms. 56 Notably, both stress and glucocorticoids can induce not only acute changes in gene transcription but also lasting epigenetic modifications, most notably changes in DNA methylation. These lasting effects are thought to represent a form of epigenetic memory that influences subsequent genomic function and stress-related outcomes. 56 , 60 , 61
The role of glucocorticoid signaling as a link between stress and epigenetic aging has been supported by two studies utilizing the Horvath measure of epigenetic age. In the first study, cumulative lifetime stress was positively associated with accelerated epigenetic aging in a highly traumatized cohort, and the CpGs comprising the Horvath measure were found to colocalize with GREs more often than expected by chance, indicating their susceptibility to stress and glucocorticoid exposure. 46 Accordingly, treatment with a synthetic glucocorticoid induced DNA methylation changes at several epigenetic age CpGs as well as dynamic transcriptional changes in the majority of their neighboring genes. Corroborating these observations, another study found that greater total daily cortisol, the primary glucocorticoid in humans, is associated with accelerated epigenetic aging in a cohort of adolescent females. 62 Notably, the glucocorticoid-regulated genes neighboring the epigenetic age CpGs were also associated with aging-related diseases, including coronary artery disease, arteriosclerosis, and leukemias. 46 Together these findings support a model whereby stress and the associated increased glucocorticoid burden may accelerate epigenetic aging and contribute to disease risk ( Figure 2 ).
Beyond such composite effects captured by measures of epigenetic aging, it is also relevant to interrogate selected gene loci that may be both susceptible to stress and implicated in aging-related disease. One such locus, the stress- responsive FKBP5 gene, was examined by a recent study combining large-scale measurements in human blood with mechanistic experiments in cells. The study found convergent evidence that stress—measured with childhood trauma and major depression questionnaires in humans and modeled with glucocorticoid exposure in cells—may synergize with aging to decrease DNA methylation at selected CpGs located near the FKBP5 transcription start site. 63 This observation suggests that—similar to the notion that interindividual differences in stress exposure may in part explain variance in epigenetic aging ( Figure 1 )—stress and glucocorticoids can shape the epigenetic state of selected sites as life advances. Notably, the aging/stress-related FKBP5 methylation signature enhanced the gene’s expression in immune cells, an effect that in turn activated the master immune regulator NF-κB and promoted chemotaxis and inflammation 63 ; thus, stress and aging can synergize to epigenetically deregulate effector molecules and downstream biological processes that are implicated in disease pathogenesis. In line with this hypothesis, the aging/stress-related FKBP5 methylation signature was present in individuals with a history of myocardial infarction, 63 a disease state linked to inflammation. 64
Figure 2. Simplified scheme that highlights epigenetic mechanisms linking psychosocial stress and aging. Stress exposure culminates in peripheral secretion of cortisol, the primary glucocorticoid in humans. The genomic effects of cortisol are largely driven by the glucocorticoid receptor (GR), a ligand-dependent transcription factor able to induce epigenetic modifications, such as DNA methylation changes, with potentially lasting impact on genomic function. These epigenetic modifications can thus be conceptualized as switches or rheostats that are set into motion by stress and can in turn fine-tune activity of susceptible genes. Stress-induced epigenetic changes can deregulate the expression of effector molecules, eg, FKBP5, and influence downstream biological pathways implicated in aging-related processes. Beyond such effects at selected gene loci, stress can also induce composite (systems level) effects on the aging epigenome, as captured, for example, by measures of epigenetic aging. Importantly, psychosocial stress and epigenetic mechanisms also influence other processes implicated in aging-related phenotypes, including inflammation, telomere function, oxidative stress, and mitochondrial function. Many of these relations are reciprocal and subject to regulation by feedback and feedforward loops. For simplicity, cortisol is depicted as a yellow hexagon and CpG methylation as an orange circle on a stick.

Similar to these effects on inflammation, stress-induced epigenetic changes at selected sites could deregulate other downstream processes implicated in aging-related disease. Considering again the example of FKBP5, this versatile protein has been shown to influence not only NF-κB signaling and inflammation but also a host of other aging-related processes and disease phenotypes, 65 including Akt signaling and cancer, 66 tau degradation and neurodegeneration, 67 autophagy, 68 and telomere biology. 69 Versatile effectors like FKBP5 could thus be viewed as “hub molecules” that may integrate pleiotropic effects of stress on aging-related processes ( Figure 2 ). However, examining how stress-driven epigenetic changes regulates transcription of such effector molecules paints only part of the picture, given the complexity of regulatory loops and pathway crosstalk. For instance, higher NF-κB activity—which as discussed above can result from FKBP5 upregulation—can in turn trigger FKBP5 transcription through an NF-κB response element that is flanked and moderated by the age/stress-related FKBP5 methylation sites 63 ; this suggests a positive feedback loop of FKBP5-NF-κB signaling that may be prone to epigenetic deregulation by stress and aging. As another example, FKBP5 can decrease activity of DNA methyltransferase 1 (DNMT1), 70 the mammalian enzyme that maintains DNA methylation, thereby reducing global and site-specific DNA methylation levels; thus, the epigenetic programming of FKBP5 by stress and aging could have secondary effects on DNMT1 activity and in turn influence the epigenetic landscape in other parts of the genome.
While the evidence presented above supports a role for epigenetics as a link between stress and aging, this role should be put in broader context with other implicated processes—most notably telomere function, inflammation, and oxidative stress—as well as their complex interplay and reciprocal relations ( Figure 2 ). Epigenetic processes can regulate telomere function, 71 but they are also influenced by telomere regulators such as telomerase, the enzyme responsible for maintaining telomere length. 72 Accelerated epigenetic aging and telomere shortening are associated with inflammation and oxidative stress, 27 , 73 both processes that are not only influenced by psychosocial stressors but can themselves affect brain physiology and stress responses. 51 , 74 These biological processes plausibly synergize and exert effects that may accumulate and eventually reach a critical threshold as life advances. In line with this “wear and tear” notion, cumulative life stress was found to have a greater effect on epigenetic aging in older as compared with younger individuals, 46 though longitudinal studies to further support this association are lacking. Furthermore, it is currently unclear whether stress exposure simply influences the rate of aging-related epigenetic and other molecular changes or, vice versa, whether the aging process determines vulnerability or resilience of certain epigenetic sites and molecular pathways to stress. Elucidating the multifaceted mechanisms linking stress and aging will likely require orchestrated efforts that combine large-scale longitudinal studies in humans with experiments in cellular and animal model systems. Such efforts may ultimately yield deeper insights into the molecular events that shape stress-related disease along the human lifespan.
Concluding remarks
Gaining insights into the epigenetic and, more broadly, the molecular links between stress and aging can have major implications for developing novel prevention and treatment strategies. The field of epigenetics has generated much excitement because, unlike genetic mutations, epigenetic modifications are often acquired and, thus, potentially preventable and reversible. Accordingly, work in both rodents and humans suggests that prevention and reversal of stress-induced epigenetic modifications could be pursued by both behavioral modifications and medication management. Human studies show that supportive family environments and behavioral interventions targeting harsh parenting can ameliorate the accelerated epigenetic aging associated with early life stress. 38 , 39 Rodent studies indicate that drugs targeting the epigenetic and transcriptional machinery can rescue DNA methylation changes associated with early life stress. 75 , 76 Although the clinical significance of specifically modulating stress- induced epigenetic changes awaits to be tested, medications targeting the epigenetic machinery have been shown to improve aging-related disease trajectories, such as in certain types of cancer. 77 Moreover, when manipulating the epigenome is not feasible or not desirable, interventions can instead target effector molecules that get aberrantly expressed as a result of stress-induced epigenetic changes. For example, treating cells with selective FKBP5 antagonists was shown to prevent the effect of glucocorticoid exposure and FKBP5 upregulation on NF-κB signaling. 63 While the in vivo significance of this finding remains to be seen, it suggests FKBP5-NF-κB signaling as a tractable treatment candidate. Employing similar approaches in future studies may point to new avenues for prevention and treatment.
The need to elucidate the molecular underpinnings of stress and aging is pressing, now more so than ever. As populations age worldwide, aging-related diseases are expected to pose unprecedented challenges to individuals and societies. 78 At the same time, surveys indicate that reported stress levels increase year by year and with successive generations. 79 To face these challenges, it is critical to intervene early in the course of disease pathogenesis, ideally well before disease states set in. Molecular insights can guide the development of such interventions and their targeting to vulnerable stress-exposed individuals. Concurrently, social policies should strive to ameliorate or, when possible, prevent excessive stress in the first place.
Acknowledgments
The author declares no conflicts of interest
REFERENCES
- 1.Hugo V. Les Miserables. San Diego, CA: Canterbury Classics; 2012. [Google Scholar]
- 2.Tani M, Cranley E. Before-and-after photos show how dramatically presidents aged in office. [Accessed November 2019];Business Insider [Internet] 2019 Available from: https://www.businessinsider.fr/us/presidents-aging-before-and-after-photos-2017-6. [Google Scholar]
- 3.Akiskal HS, Fatemi SH, Clayton PJ. The Medical Basis of Psychiatry. Totowa, NJ, USA: Humana Press; 2008. The mental status examination. [Google Scholar]
- 4.Felitti VJ, Anda RF, Nordenberg D, et al Relationship of childhood abuse and household dysfunction to many of the leading causes of death in adults. The Adverse Childhood Experiences (ACE) Study. Am J Prev Med. 1998;14(4):245–258. doi: 10.1016/s0749-3797(98)00017-8. [DOI] [PubMed] [Google Scholar]
- 5.Vaccarino V, Goldberg J, Rooks C, et al Post- traumatic stress disorder and incidence of coronary heart disease: a twin study. J Am Coll Cardiol. 2013;62(11):970–978. doi: 10.1016/j.jacc.2013.04.085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Jones DJ, Bromberger JT, Sutton-Tyrrell K, Matthews KA. Lifetime history of depression and carotid atherosclerosis in middle-aged women. Arch Gen Psychiatry. 2003;60(2):153–160. doi: 10.1001/archpsyc.60.2.153. [DOI] [PubMed] [Google Scholar]
- 7.Peavy GM, Salmon DP, Jacobson MW, et al Effects of chronic stress on memory decline in cognitively normal and mildly impaired older adults. Am J Psychiatry. 2009;166(12):1384–1391. doi: 10.1176/appi.ajp.2009.09040461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niccoli T, Partridge L. Ageing as a risk factor for disease. Curr Biol. 2012;22(17):R741–752. doi: 10.1016/j.cub.2012.07.024. [DOI] [PubMed] [Google Scholar]
- 9.Blackmore HL, Ozanne SE. Programming of cardiovascular disease across the life-course. J Mol Cell Cardiol. 2015;83:122–130. doi: 10.1016/j.yjmcc.2014.12.006. [DOI] [PubMed] [Google Scholar]
- 10.Ries W. Motivations for determining biological aging. Z Gerontol. 1990;23(3):160–162. [PubMed] [Google Scholar]
- 11.Squier TC. Oxidative stress and protein aggregation during biological aging. Exp Gerontol. 2001;36(9):1539–1550. doi: 10.1016/s0531-5565(01)00139-5. [DOI] [PubMed] [Google Scholar]
- 12.Herrmann M, Pusceddu I, Marz W, Herrmann W. Telomere biology and age-related diseases. Clin Chem Lab Med. 2018;56(8):1210–1222. doi: 10.1515/cclm-2017-0870. [DOI] [PubMed] [Google Scholar]
- 13.Horvath S. DNA methylation age of human tissues and cell types. Genome Biol. 2013;14(10):R115. doi: 10.1186/gb-2013-14-10-r115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hannum G, Guinney J, Zhao L, et al Genome-wide methylation profiles reveal quantitative views of human aging rates. Mol Cell. 2013;49(2):359–367. doi: 10.1016/j.molcel.2012.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Fleischer JG, Schulte R, Tsai HH, et al Predicting age from the transcriptome of human dermal fibroblasts. Genome Biol. 2018;19(1):221. doi: 10.1186/s13059-018-1599-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanaka T, Biancotto A, Moaddel R, et al Plasma proteomic signature of age in healthy humans. Aging Cell. 2018;17(5):e12799. doi: 10.1111/acel.12799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chen BH, Marioni RE, Colicino E, et al DNA methylation-based measures of biological age: meta-analysis predicting time to death. Aging (Albany NY) 2016;8(9):1844–1865. doi: 10.18632/aging.101020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Marioni RE, Shah S, McRae AF, et al The epigenetic clock is correlated with physical and cognitive fitness in the Lothian Birth Cohort 1936. Int J Epidemiol. 2015;44(4):1388–1396. doi: 10.1093/ije/dyu277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Levine ME, Hosgood HD, Chen B, Absher D, Assimes T, Horvath S. DNA methylation age of blood predicts future onset of lung cancer in the women’s health initiative. Aging (Albany NY) 2015;7(9):690–700. doi: 10.18632/aging.100809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Breitling LP, Saum KU, Perna L, Schottker B, Holleczek B, Brenner H. Frailty is associated with the epigenetic clock but not with telomere length in a German cohort. Clin Epigenetics. 2016;8:21. doi: 10.1186/s13148-016-0186-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Levine ME, Lu AT, Bennett DA, Horvath S. Epigenetic age of the pre-frontal cortex is associated with neuritic plaques, amyloid load, and Alzheimer’s disease related cognitive functioning. Aging (Albany NY) 2015;7(12):1198–1211. doi: 10.18632/aging.100864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Horvath S, Raj K. DNA methylation-based biomarkers and the epigenetic clock theory of ageing. Nat Rev Genet. 2018;19(6):371–384. doi: 10.1038/s41576-018-0004-3. [DOI] [PubMed] [Google Scholar]
- 23.Zhu X, Han W, Xue W, et al The association between telomere length and cancer risk in population studies. Sci Rep. 2016;6:22243. doi: 10.1038/srep22243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Jylhava J, Hjelmborg J, Soerensen M, et al Longitudinal changes in the genetic and environmental influences on the epigenetic clocks across old age: Evidence from two twin cohorts. EBioMedicine. 2019;40:710–716. doi: 10.1016/j.ebiom.2019.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Noncommunicable Diseases Progress Monitor. Geneva, Switzerland: World Health Organization World Health Organization; 2017. [Google Scholar]
- 26.Danese A, Pariante CM, Caspi A, Taylor A, Poulton R. Childhood maltreatment predicts adult inflammation in a life-course study. Proc Natl Acad Sci U S A. 2007;104(4):1319–1324. doi: 10.1073/pnas.0610362104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Epel ES, Blackburn EH, Lin J, et al Accelerated telomere shortening in response to life stress. Proc Natl Acad Sci U S A. 2004;101(49):17312–17315. doi: 10.1073/pnas.0407162101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parks CG, Miller DB, McCanlies EC, et al Telomere length, current perceived stress, and urinary stress hormones in women. Cancer Epidemiol Biomarkers Prev. 2009;18(2):551–560. doi: 10.1158/1055-9965.EPI-08-0614. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Entringer S, Epel ES, Kumsta R, et al Stress exposure in intrauterine life is associated with shorter telomere length in young adulthood. Proc Natl Acad Sci U S A. 2011;108(33):E513–518. doi: 10.1073/pnas.1107759108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kananen L, Surakka I, Pirkola S, et al Childhood adversities are associated with shorter telomere length at adult age both in individuals with an anxiety disorder and controls. PLoS One. 2010;5(5):e10826. doi: 10.1371/journal.pone.0010826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Ahola K, Siren I, Kivimaki M, et al Work-related exhaustion and telomere length: a population-based study. PLoS One. 2012;7(7):e40186. doi: 10.1371/journal.pone.0040186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wolkowitz OM, Mellon SH, Epel ES, et al Leukocyte telomere length in major depression: correlations with chronicity, inflammation and oxidative stress--preliminary findings. PLoS One. 2011;6(3):e17837. doi: 10.1371/journal.pone.0017837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jodczyk S, Fergusson DM, Horwood LJ, Pearson JF, Kennedy MA. No association between mean telomere length and life stress observed in a 30 year birth cohort. PLoS One. 2014;9(5):e97102. doi: 10.1371/journal.pone.0097102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shalev I, Moffitt TE, Braithwaite AW, et al Internalizing disorders and leukocyte telomere erosion: a prospective study of depression, generalized anxiety disorder and post-traumatic stress disorder. Mol Psychiatry. 2014;19(11):1163–1170. doi: 10.1038/mp.2013.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ridout KK, Ridout SJ, Price LH, Sen S, Tyrka AR. Depression and telomere length: A meta-analysis. J Affect Disord. 2016;191:237–247. doi: 10.1016/j.jad.2015.11.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Li X, Wang J, Zhou J, Huang P, Li J. The association between post-traumatic stress disorder and shorter telomere length: A systematic review and meta-analysis. J Affect Disord. 2017;218:322–326. doi: 10.1016/j.jad.2017.03.048. [DOI] [PubMed] [Google Scholar]
- 37.Hanssen LM, Schutte NS, Malouff JM, Epel ES. The relationship between childhood psychosocial stressor level and telomere length: a meta-analysis. Health Psychol Res. 2017;5(1):6378. doi: 10.4081/hpr.2017.6378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brody GH, Yu T, Chen E, Beach SR, Miller GE. Family-centered prevention ameliorates the longitudinal association between risky family processes and epigenetic aging. J Child Psychol Psychiatry. 2016;57(5):566–574. doi: 10.1111/jcpp.12495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Brody GH, Miller GE, Yu T, Beach SR, Chen E. Supportive family environments ameliorate the link between racial discrimination and epigenetic aging: a replication across two longitudinal cohorts. Psychol Sci. 2016;27(4):530–541. doi: 10.1177/0956797615626703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sumner JA, Colich NL, Uddin M, Armstrong D, McLaughlin KA. Early Experiences of Threat, but Not Deprivation, Are Associated With Accelerated Biological Aging in Children and Adolescents. Biol Psychiatry. 2019;85(3):268–278. doi: 10.1016/j.biopsych.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Jovanovic T, Vance LA, Cross D, et al Exposure to violence accelerates epigenetic aging in children. Sci Rep. 2017;7(1):8962. doi: 10.1038/s41598-017-09235-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Austin MK, Chen E, Ross KM, et al Early-life socioeconomic disadvantage, not current, predicts accelerated epigenetic aging of monocytes. Psychoneuroendocrinology. 2018;97:131–134. doi: 10.1016/j.psyneuen.2018.07.007. [DOI] [PubMed] [Google Scholar]
- 43.Hughes A, Smart M, Gorrie-Stone T, et al Socioeconomic position and DNA methylation age acceleration across the life course. Am J Epidemiol. 2018;187(11):2346–2354. doi: 10.1093/aje/kwy155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chen E, Miller GE, Yu T, Brody GH. The Great Recession and health risks in African American youth. Brain Behav Immun. 2016;53:234–241. doi: 10.1016/j.bbi.2015.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Han LKM, Aghajani M, Clark SL, et al Epigenetic aging in major depressive disorder. Am J Psychiatry. 2018;175(8):774–782. doi: 10.1176/appi.ajp.2018.17060595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Zannas AS, Arloth J, Carrillo-Roa T, et al Lifetime stress accelerates epigenetic aging in an urban, African American cohort: relevance of glucocorticoid signaling. Genome Biol. 2015;16(1):266. doi: 10.1186/s13059-015-0828-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Boks MP, van Mierlo HC, Rutten BP, et al Longitudinal changes of telomere length and epigenetic age related to traumatic stress and post-traumatic stress disorder. Psychoneuroendocrinology. 2015;51:506–512. doi: 10.1016/j.psyneuen.2014.07.011. [DOI] [PubMed] [Google Scholar]
- 48.Wolf EJ, Maniates H, Nugent N, et al Traumatic stress and accelerated DNA methylation age: A meta-analysis. Psychoneuroendocrinology. 2018;92:123–134. doi: 10.1016/j.psyneuen.2017.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Simons RL, Lei MK, Beach SR, et al Economic hardship and biological weathering: The epigenetics of aging in a U.S. sample of black women. Soc Sci Med. 2016;150:192–200. doi: 10.1016/j.socscimed.2015.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fiorito G, Polidoro S, Dugue PA, et al Social adversity and epigenetic aging: a multi-cohort study on socioeconomic differences in peripheral blood DNA methylation. Sci Rep. 2017;7(1):16266. doi: 10.1038/s41598-017-16391-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Miller MW, Sadeh N. Traumatic stress, oxidative stress and post-traumatic stress disorder: neurodegeneration and the accelerated-aging hypothesis. Mol Psychiatry. 2014;19(11):1156–1162. doi: 10.1038/mp.2014.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Trumpff C, Marsland AL, Basualto-Alarcon C, et al Acute psychological stress increases serum circulating cell-free mitochondrial DNA. Psychoneuroendocrinology. 2019;106:268–276. doi: 10.1016/j.psyneuen.2019.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Picard M, Prather AA, Puterman E, et al A mitochondrial health index sensitive to mood and caregiving stress. Biol Psychiatry. 2018;84(1):9–17. doi: 10.1016/j.biopsych.2018.01.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moreno-Villanueva M, Morath J, Vanhooren V, et al N-glycosylation profiling of plasma provides evidence for accelerated physiological aging in post-traumatic stress disorder. Transl Psychiatry. 2013;3:e320. doi: 10.1038/tp.2013.93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Telese F, Gamliel A, Skowronska-Krawczyk D, Garcia-Bassets I, Rosenfeld MG. “Seq-ing” insights into the epigenetics of neuronal gene regulation. Neuron. 2013;77(4):606–623. doi: 10.1016/j.neuron.2013.01.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zannas AS, Chrousos GP. Epigenetic programming by stress and glucocorticoids along the human lifespan. Mol Psychiatry. 2017;22(5):640–646. doi: 10.1038/mp.2017.35. [DOI] [PubMed] [Google Scholar]
- 57.Unternaehrer E, Luers P, Mill J, et al Dynamic changes in DNA methylation of stress-associated genes (OXTR, BDNF ) after acute psychosocial stress. Transl Psychiatry. 2012;2:e150. doi: 10.1038/tp.2012.77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Lopez-Otin C, Blasco MA, Partridge L, Serrano M, Kroemer G. The hallmarks of aging. Cell. 2013;153(6):1194–1217. doi: 10.1016/j.cell.2013.05.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chrousos GP, Gold PW. The concepts of stress and stress system disorders. Overview of physical and behavioral homeostasis. JAMA. 1992;267(9):1244–1252. [PubMed] [Google Scholar]
- 60.Thomassin H, Flavin M, Espinas ML, Grange T. Glucocorticoid-induced DNA demethylation and gene memory during development. Embo J. 2001;20(8):1974–1983. doi: 10.1093/emboj/20.8.1974. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Provençal N, Arloth J, Cattaneo A, et al Glucocorticoid exposure during hippocampal neurogenesis primes future stress response by inducing changes in DNA methylation. Proc Natl Acad Sci U S A. 2019 doi: 10.1073/pnas.1820842116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Davis EG, Humphreys KL, McEwen LM, et al Accelerated DNA methylation age in adolescent girls: associations with elevated diurnal cortisol and reduced hippocampal volume. Transl Psychiatry. 2017;7(8):e1223. doi: 10.1038/tp.2017.188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Zannas AS, Jia M, Hafner K, et al Epigenetic upregulation of FKBP5 by aging and stress contributes to NF-kappaB-driven inflammation and cardiovascular risk. Proc Natl Acad Sci U S A. 2019;116(23):11370–11379. doi: 10.1073/pnas.1816847116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Libby P, Tabas I, Fredman G, Fisher EA. Inflammation and its resolution as determinants of acute coronary syndromes. Circ Res. 2014;114(12):1867–1879. doi: 10.1161/CIRCRESAHA.114.302699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Zannas AS, Wiechmann T, Gassen NC, Binder EB. Gene-stress-epigenetic regulation of FKBP5: clinical and translational implications. Neuropsychopharmacology. 2016;41(1):261–274. doi: 10.1038/npp.2015.235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pei H, Li L, Fridley BL, et al FKBP51 affects cancer cell response to chemotherapy by negatively regulating Akt. Cancer Cell. 2009;16(3):259–266. doi: 10.1016/j.ccr.2009.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Blair LJ, Nordhues BA, Hill SE, et al Accelerated neurodegeneration through chaperone-mediated oligomerization of tau. J Clin Invest. 2013;123(10):4158–4169. doi: 10.1172/JCI69003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gassen NC, Hartmann J, Zschocke J, et al Association of FKBP51 with priming of autophagy pathways and mediation of antidepressant treatment response: evidence in cells, mice, and humans. PLoS Med. 2014;11(11):e1001755. doi: 10.1371/journal.pmed.1001755. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Lagadari M, Zgajnar NR, Gallo LI, Galigniana MD. Hsp90-binding immunophilin FKBP51 forms complexes with hTERT enhancing telomerase activity. Mol Oncol. 2016;10(7):1086–1098. doi: 10.1016/j.molonc.2016.05.002. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 70.Gassen NC, Fries GR, Zannas AS, et al Chaperoning epigenetics: FKBP51 decreases the activity of DNMT1 and mediates epigenetic effects of the antidepressant paroxetine. Sci Signal. 2015;8(404):ra119. doi: 10.1126/scisignal.aac7695. [DOI] [PubMed] [Google Scholar]
- 71.Ravlic S, Skrobot Vidacek N, Nanic L, et al Mechanisms of fetal epigenetics that determine telomere dynamics and health span in adulthood. Mech Ageing Dev. 2018;174:55–62. doi: 10.1016/j.mad.2017.08.014. [DOI] [PubMed] [Google Scholar]
- 72.Lu AT, Xue L, Salfati EL, et al GWAS of epigenetic aging rates in blood reveals a critical role for TERT. Nat Commun. 2018;9(1):387. doi: 10.1038/s41467-017-02697-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Irvin MR, Aslibekyan S, Do A, et al Metabolic and inflammatory biomarkers are associated with epigenetic aging acceleration estimates in the GOLDN study. Clin Epigenetics. 2018;10:56. doi: 10.1186/s13148-018-0481-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jones KA, Thomsen C. The role of the innate immune system in psychiatric disorders. Mol Cell Neurosci. 2013;53:52–62. doi: 10.1016/j.mcn.2012.10.002. [DOI] [PubMed] [Google Scholar]
- 75.Roth TL, Lubin FD, Funk AJ, Sweatt JD. Lasting epigenetic influence of early-life adversity on the BDNF gene. Biol Psychiatry. 2009;65(9):760–769. doi: 10.1016/j.biopsych.2008.11.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Niwa M, Jaaro-Peled H, Tankou S, et al Adolescent stress-induced epigenetic control of dopaminergic neurons via glucocorticoids. Science. 2013;339(6117):335–339. doi: 10.1126/science.1226931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Dawson MA. The cancer epigenome: Concepts, challenges, and therapeutic opportunities. Science. 2017;355(6330):1147–1152. doi: 10.1126/science.aam7304. [DOI] [PubMed] [Google Scholar]
- 78.The State of Aging and Health in America 2013. Atlanta, GA: CDC; 2013. Centers for Disease Control and Prevention. [Google Scholar]
- 79. 2015 Stress in America. [Accessed September 2019];American Psychological Association [Internet] 2015 Available from: https://www.apa.org/news/press/releases/stress/2015/snapshot.