

Abstract
Background
Spirochetal organisms of the Treponema genus are responsible for causing Treponematoses. Pathogenic treponemes is a Gram-negative, motile, spirochete pathogen that causes syphilis in human. Treponema pallidum subsp. endemicum (TEN) causes endemic syphilis (bejel); T. pallidum subsp. pallidum (TPA) causes venereal syphilis; T. pallidum subsp. pertenue (TPE) causes yaws; and T. pallidum subsp. Ccarateum causes pinta. Out of these four high morbidity diseases, venereal syphilis is mediated by sexual contact; the other three diseases are transmitted by close personal contact. The global distribution of syphilis is alarming and there is an increasing need of proper treatment and preventive measures. Unfortunately, effective measures are limited.
Results
Here, the genome sequences of 53 T. pallidum strains isolated from different parts of the world and a diverse range of hosts were comparatively analysed using pan-genomic strategy. Phylogenomic, pan-genomic, core genomic and singleton analysis disclosed the close connection among all strains of the pathogen T. pallidum, its clonal behaviour and showed increases in the sizes of the pan-genome. Based on the genome plasticity analysis of the subsets containing the subspecies T pallidum subsp. pallidum, T. pallidum subsp. endemicum and T. pallidum subsp. pertenue, we found differences in the presence/absence of pathogenicity islands (PAIs) and genomic islands (GIs) on subsp.-based study.
Conclusions
In summary, we identified four pathogenicity islands (PAIs), eight genomic islands (GIs) in subsp. pallidum, whereas subsp. endemicum has three PAIs and seven GIs and subsp. pertenue harbours three PAIs and eight GIs. Concerning the presence of genes in PAIs and GIs, we found some genes related to lipid and amino acid biosynthesis that were only present in the subsp. of T. pallidum, compared to T. pallidum subsp. endemicum and T. pallidum subsp. pertenue.
Keywords: Pan-genome, Core genome, Singletons, Treponema pallidum, Syphilis
Background
Spirochetal organisms of the Treponema genus are responsible for causing Treponematoses. Pathogenic treponemes cause multi-stage infections like endemic syphilis, venereal syphilis, yaws and pinta. These infections have many similarities, but they can be differentiated based on epidemiological, clinical and geographical criteria [1–3]. Primarily, the pathogenic treponemes can be classified based on the clinical symptoms of the respective disease they cause. Treponema pallidum subsp. endemicum causes endemic syphilis; T. pallidum subsp. pallidum causes venereal syphilis; T. pallidum subsp. pertenue causes yaws; and T. pallidum subsp. carateum causes pinta. Out of these four high morbidity diseases, venereal syphilis is only transmitted by sexual contact; the other three diseases are transmitted by close personal contact [2].
It is estimated by the World Health Organization (WHO) that there are 12 million new cases of syphilis annually and the aggregated cases of yaws, bejel, and pinta (the endemic treponematoses) are approximately 2.5 million globally, although good surveillance data is not available. The infections caused by T. pallidum are characterized by periods of active clinical disease interrupted by episodes of asymptomatic latent infection and may cause life-long infections in untreated individuals [4, 5]. Treponema pallidum is a Gram-negative, motile, spirochete human pathogen. Syphilis is a multistage infectious disease that can be communicated between sexual partners through active lesions or from an infected woman to her fetus during pregnancy [6, 7]. Syphilis has a worldwide distribution (e.g. Africa has a high incidence), affecting every country and continent except perhaps Antarctica [8–12]. The stages of syphilis have been divided on the basis of clinical findings that lead to treatment and follow-up. Syphilis chancres may go unnoticed primarily due to their well-documented painless nature and if they are present in those parts of the body that are difficult to visualize (e.g. cervix, throat or anus/rectum) [13]. Furthermore, due to pleomorphic appearance and lack of physician familiarity with the expressions of syphilis, their lesions may be misdiagnosed. Secondary, syphilis may manifest itself through severe rashes that may go unobserved by the patient or may mimic an extensive condition [8]. T. pallidum is completely sensitive to penicillin treatment, despite the use of this antibiotic for seven decades in treating syphilis infections. Standard treatment of uncomplicated syphilis with parenteral Benzathine penicillin G is highly effective at all stages. Many antibiotics’ resistance (e.g Macrolide and Clindamycin resistance) has been reported in several countries [6]. The ongoing high rate of syphilis worldwide, despite the availability of inexpensive and effective treatment, presents the most convincing argument for the need of developing new and potent vaccine against syphilis [14]. Despite the WHO’s Initiative for the Global Elimination of Congenital Syphilis, an intensive syphilis-targeted public health control has been undertaken to reduce the incidence; however, it has not been achieved yet [14]. Specifically, the reasons for failure are multifactorial; some of the responsibility can be attributed to the difficulty in the diagnosis of syphilis and treatment, and lack of access or use of prenatal screening programs [15]. The advancement in the field of genomics and cost-effective sequencing technologies has transformed the human bacterial pathogens study and helped in the improvement of vaccine designing technologies. A new and emerging methodology to get deep insight of the genome of a species or genus is the pan-genomics approach, which was introduced by Tettelin and collaborators in 2005 working with Streptococcus agalactiae [16]. Pan-genome provides us with the complete and non-redundant collection of genes from a species or genus and is composed of three subsets (core genome, shared genome and singletons): the core genome, which is the collection of all the genes commonly shared between all the genomes used as dataset; the shared genome, which contains only the genes shared between two or more strains, which are not present in all strains of the dataset; and, the singletons, which are present only in one strain and are referred to as strain-specific genes.
The first genome of T. pallidum subsp. pallidum (strain Nichols) was sequenced in 1998. The organism has a comparatively small genome and only 55% of T. pallidum’s 1041 open reading frames are recognized to have a biological function, which indicates that it uses host biosynthesis to complete some of its metabolic needs [3]. The DNA-DNA hybridization studies showed homology between DNA of venereal syphilis spirochete and DNA of culturable treponemes (T. phagedenis and its biotypes Reiter and Kazan) was less than 5% identical, but was indistinguishable from DNA of the yaws spirochete T. pallidum [3, 17, 18]. This study led to the reclassification of the agents of endemic syphilis, venereal syphilis and yaws as T. pallidum subsp. endemicum, Treponema pallidum subsp. pallidum and T. pallidum subsp. pertenue, respectively. Genomic sequencing has recognized these subspecies as clonal, but forming distinct genetic clusters [2, 3].
In this work, we perform a pan-genome approach to better understand the differences of Treponema pallidum infections in the broad spectrum and how genome plasticity is related to the symptom patterns. For pan-genomic comparative analyses, we used 53 T. pallidum strains. We present phylo-genomic correlations between all T. pallidum strains. Furthermore, we describe the “pan-genome”, which is the complete inventory of genes found in any member of the species; the “core genome”, which is important for basic life processes; and the “singletons”, which are normally related to environmental fitness and adaptation to host. Finally, we provide insights into the specific subsets (singletons and the pan- and core genomes) of 53 genomes of T pallidum strains and correlate these subsets with the plasticity of pathogenicity islands and virulence genes.
Results
Phylogenomics study of Treponema pallidum strains
The phylogenomics relationships between T. pallidum strains were determined using Gegenees [19]. Furthermore, all genome sequences were cross-compared to generate a phylogenomic tree and to plot a heatmap. According to the generated phylogenomic tree, closely related strains appeared in the same cluster. The subspecies responsible for non-venereal syphilis is Treponema pallidum subsp. endemicum (TEN) and T. pallidum subsp. pertenue (TPE) strains appeared in closely related clusters (Fig. 1). The T. pallidum subspecies strains responsible for venereal syphilis formed different clusters. Additionally, T. pallidum strain BosniaA (subsp. endemicum) was positioned between the clusters of Treponema pallidum subsp. Pertenue and venereal syphilis (Treponema pallidum subsp. pallidum). According to the heatmap, the non-venereal isolates are 100% similar to each other and many of the venereal isolates are 100% similar to each other, but the two groups show some difference (Additional file 1: Figure S2). Moreover, the heatmap indicated the clonal-like behavior of T. pallidum subsp., compared with the isolates other than genital, anal or Neurosyphilitic samples, which showed similarities ranging from 97 to 100%.
Fig. 1.
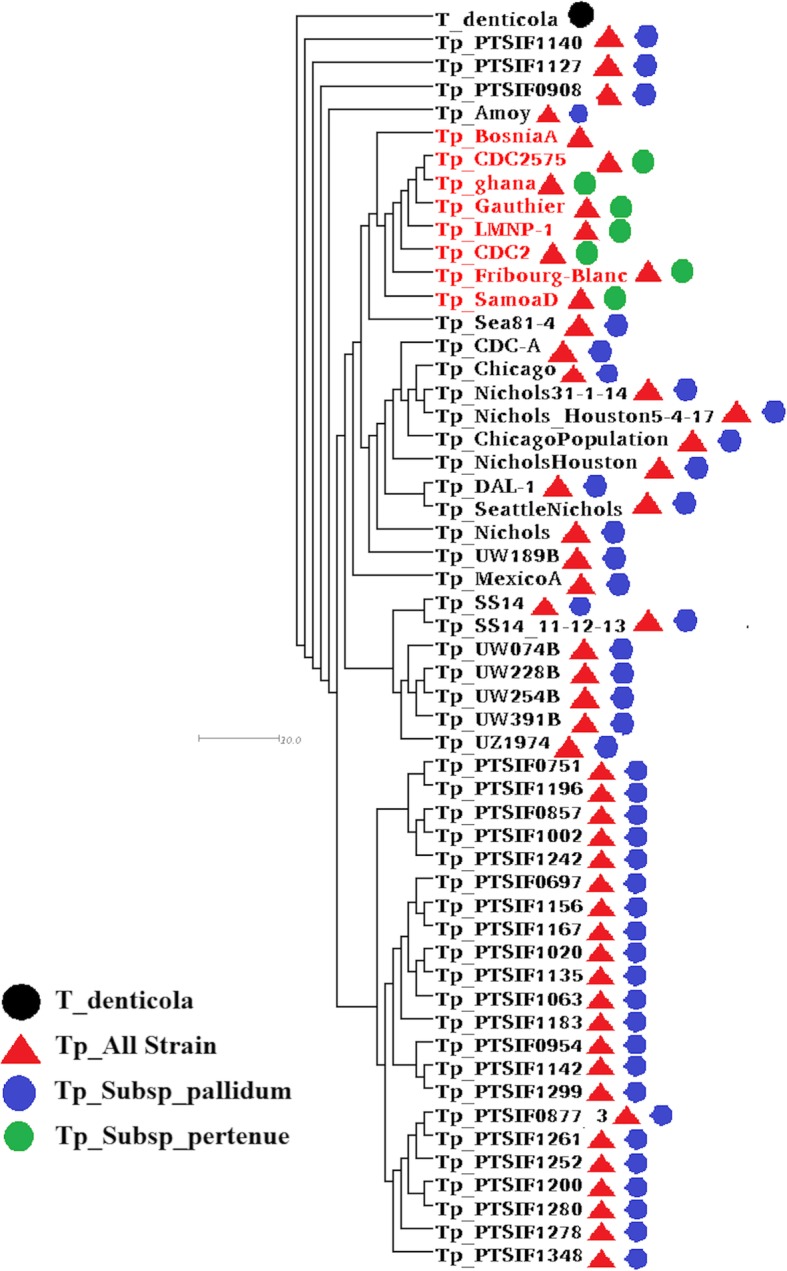
Phylogenomic tree analysis of 53 Strains of Treponema pallidum. The generated distance matrix data from Gegenees was used to generate a phylogenomic tree with SplitsTree (version 4.14.5) using neighbour joining method to create a dendogram. The strains name in the clade represented in red and black showed the Non-venereal and venereal strains of Treponema pallidum, respectively. Non-venereal Treponema pallidum strains are present in same clade. The shapes (circle and triangle) next to the name of the strain indicate the subset of strains used for Pangenome analysis according to the color of the legend respectively
The Pan-genome, Core genome and singletons of Treponema pallidum
The main goal of the pan-genome is the comparison of different strains of the same species or even genus at the genomic level. The resulting pan-genome of Pan All (Fig. 2A1-A3), Pan Subsp_pallidum (Fig. 3B1-B3), and Pan_subsp_pertenue (Fig. 4C1-C3), of T. pallidum contains a total of 2112, 982, and 1049 genes respectively. The formula (α =1-γ) inferred that the pan-genome of T. pallidum is increasing with an α of 0.9435. The extrapolation was also separately calculated for all divided subsets for the analysis in this work. The α value for each subset Pan Subsp_pallidum and Pan_subsp_pertenue, were 0.916 and 0.999329 respectively. The α values for all datasets used in this work are less than 1 which indicates that all have an open pan-genome. However, although the pan-genome is still open, it increases at a very low rate [20, 21].
Fig. 2.

Pan-genome, core genome and singletons of T. pallidum. A1/A2/A3, respectively, showing the pan-genome, core genome and singletons development using 53 strains of T. pallidum
Fig. 3.

Pan-genome, core genome and singletons of T. pallidum Subsp_pallidum. B1/B2/B3, respectively, showing the pan-genome, core genome and singletons development using 45 strains belonging to subspecies pallidum
Fig. 4.

Pan-genome, core genome and singletons of T. pallidum Pan_subsp_pertenue. C1/C2/C3, respectively, showing the pan-genome, core genome and singletons development using 7 strains belonging to subspecies pertenue
The core genome and singletons of the complete dataset and all the subsets of T. pallidum were calculated by the least-squares fit of the exponential regression decay to the mean values, as represented by the formula n = k * exp[―x/τ] + tg(θ), where n is the expected subset of genes for a given number of genomes, x is the number of genomes, exp is Euler’s number, and the other terms are constants defined to fit the specific curve. The resulting core genome of the complete dataset (Pan All), the subsets Pan Subsp_pallidum and Pan Subsp_pertenue, have the following tg(θ) values, respectively: ~ 318, ~ 627, and ~ 1038. Concerning the Singletons of the complete dataset (Pan All) and the subsets Pan Subsp_pallidum, and Pan Subsp_pertenue, have the following tg(θ) values, respectively: ~ 1, ~ 0.1, and ~ 0.025. According to the least-squares fit of the exponential regression decay, the tg(θ) represents the point where the curve stabilizes, which may be translated to the number of genes in the core genome after stabilization and the number of singletons that will be added to the pan-genome for each newly sequenced genome. Considering this rule, the core genome of the subset Subsp_pertenue have higher number of core genes (1038-number of core genes) after stabilization, whereas, the complete dataset haS the smallest number of core genes (318-number of core genes). For the Singletons, the tg(θ) value for all the dataset indicates only one gene will be added, whereas, the subsets from Pan Subsp_pallidum and Pan Subsp_pertenue will have 1 and 0.025 newly added genes respectively.
The core genes of the complete dataset, the subsets Pan Subsp_pallidum and Pan Subsp_pertenue, of T. pallidum were classified by COG (Cluster of Orthologous Genes) functional category. According to the chart in Fig. 5a-c, the core genome of all the strains had many genes related to the “Metabolism” and “Information storage and processing” categories. Moreover, the majority of the core genome of all the strains were classified as “poorly characterized” (Additional file 1: Table S2A-C).
Fig. 5.

Graphical representation of COG (Cluster of Orthologous Genes) functional categories of identified core genes. a, b, c showing the core genes belonging to the Information storage and processing, cellular processing and signaling, Metabolism and poorly characterized functional categories for complete dataset, the subsets Pan Subsp_pallidum and Pan Subsp_pertenue of T. pallidum, respectively
Detection of PAIs in the Treponema pallidum genome
The presence of pathogenicity islands (PAIs) is generally related to evolution in a different genomic environment [22]. However, it may only be the effect of relaxation of purifying selection genes involved in increasing the range of environmental responses. Interspecies genome plasticity may result from several events, of which horizontal gene transfer is particularly important because it can cause the acquisition of blocks of genes (genomic islands, or GIs), producing evolution by quantum leaps [23]. These genes are often flanked by transposases (insertion elements), have altered G + C content and skew, suggesting their acquisition through Horizontal Gene Transfer (HGT), intermediated by phages or recombination [22]. PAIs are important in this context because they represent a class of GIs that carry virulence genes, i.e., factors that enable or enhance the parasitic growth of an organism inside a host [24]. The genome plasticity of all 53 T. pallidum strains was determined by using GIPSy (Genomic Island Prediction Software) on subspecies-based study. The software BRIG (BLAST Ring Image Generator) [25] was used for the circular genome comparison visualization. Some of the other strains from the representing cluster of the dendogram were also used for the circular genome visualization. We found differences in the presence/absence of pathogenicity islands (PAIs) and genomic islands (GIs) on subspecies-based study: four Pathogenicity Islands (PAIs) eight genomic islands (GIs) in subsp. pallidum (Fig. 6); three PAIs and seven GIs in subsp. endemicum (Fig. 7); and, three PAIs and eight GIs in subsp. pertenue (Fig. 8).
Fig. 6.

Circular genomic representation of islands (PAIs and GIs) in the genomes of T. pallidum subsp. pallidum strain Nichols as a reference. All genomes were aligned using strain Tp_Nichols as a reference. From the inner to outer circle the strains were represented in different colours. The outer most dotted circle represents the genome of T. denticola. The figure represents the coding sequences (CDS); GIs (Genomics islands) and PAIs (Pathogenicity Island)
Fig. 7.

Circular genomic representation of islands (PAIs and GIs) in the genomes of T. pallidum subsp. pertenue strain SamoaD as a reference. All genomes were aligned using strain Tp_SamoaD as a reference. From the inner to outer circle the strains were represented in different colours. The outer most dotted circle represents the genome of T. denticola. The figure represents the coding sequences (CDS); GIs (Genomics islands) and PAIs (Pathogenicity Island)
Fig. 8.

Circular genomic representation of islands (PAIs and GIs) in the genomes of T. pallidum subsp. endemicum strain BosniaA as a reference. All genomes were aligned using strain Tp_BosniaA as a reference. From the inner to outer circle the strains were represented in different colours. The outer most dotted circle represents the genome of T. denticola. The figure represents the coding sequences (CDS); GIs (Genomics islands) and PAIs (Pathogenicity Island)
Variations in pathogenicity and Genomic Island in subspecies group
Regarding the presence of genes in PAIs and GIs, we compared the genes in all the subsp. of T. pallidum to each other. When compared to each other, we found high similarity of the genes in all the subsp. of T. pallidum. The genomic region related to PAIs 2 and PAIs 3 of subsp. pertenue and endemicum (Non- venereal subsp.) were similar to the PAIs 1 and PAIs 4 of subsp. Pallidum. When we compared the genes related to PAIs 2 of subsp. pertenue and endemicum, there were differences of three genes found that were only present in subsp. pertenue. Out of those three genes, two were hypothetical proteins and one was RNA polymerase sigma factor. Furthermore, the genes clusters related to the PAIs 3 of subs. Pertenue and endemicum were similar to PAIs 4 of subsp. Pallidum. Interestingly, we found the genomic region related to PAIs 1 of subsp. pertenue and endemicum (Non- venereal subsp.) were not present in any of the GIs or PAIs of subsp. pallidum. The list of genes related to PAI 1 of subsp. pertenue and endemicum is mentioned in Table 1.
Table 1.
The list of genes related to PAI 1 of subs. Pertenue and endemicum
| Protein ID | PAIs Coordinates | Protein Name | Functions (MF: Molecular Function, BP: Biological Process) |
|---|---|---|---|
| WP_012460510.1 | 22,054–24,177 | VWFA domain-ontaining protein | – |
| WP_010881470.1 | 25,115–2538 | Hypothetical protein | |
| WP_014342234.1 | 25,459–27,504 | Hypothetical protein | – |
| WP_010881472.1 | 27,565–28,896 | Sodium-dependent tryptophan transporter |
MF: Neurotransmitter:sodium symporter activity BP |
| WP_010881473.1 | 29,076–29,786 | Potassium transporter Trk |
MF: Cation transmembrane transporter activity BP: Potassium ion transport |
| WP_010881474.1 | 29,865–32,936 | M16C subfamily peptidase |
MF: Catalytic activity, Metal ion binding BP: Proteolysis |
| WP_010881475.1 | 32,965–33,987 | Flagellar motor switch protein FliG |
MF: Motor activity BP: Bacterial-type flagellum-dependent cell motility, Chemotaxis |
| WP_014342235.1 | 34,060–35,433 | Putative hemolysin HlyC |
MF: Flavin adenine dinucleotide binding BP |
| WP_010881477.1 | 35,447–36,808 | Putative hemolysin HlyC |
MF: Flavin adenine dinucleotide binding BP |
| WP_039487502.1 | 36,916–38,193 | UDP-N-acetylglucosamine 1-carboxyvinyltransferase | – |
| WP_010881479.1 | 38,312–39,946 | 60 kDa chaperonin |
MF: ATP binding Source, Unfolded protein binding BP: Protein refolding |
| WP_014342237.1 | 40,010–40,288 | Hypothetical protein | |
| WP_010881481.1 | 40,285–41,085 | Ribosomal RNA small subunit methyltransferase E |
MF: Methyltransferase activity BP: rRNA processing |
| WP_014342238.1 | 41,082–41,750 | Hypothetical protein | |
| WP_039486943.1 | 41,794–42,924 | Zinc (Zn2+) ABC superfamily ATP binding cassettetransporter, binding protein |
MF: Metal ion binding BP: Cell adhesion, Metal ion transport |
| WP_010881484.1 | 42,942–43,658 | Zinc (Zn2+) ABC superfamily ATP binding cassettetransporter, ABC protein |
MF: ATPase activity, ATP binding BP |
| WP_010881485.1 | 43,658–44,458 | Zinc (Zn2+) ABC superfamily ATP binding cassettetransporter, membrane protein |
MF: ATPase-coupled transmembrane transporter activity BP: |
| WP_010881486.1 | 44,567–45,562 | Lactate dehydrogenase |
MF: D-lactate dehydrogenase activity, NAD binding BP |
| WP_014342240.1 | 45,659–46,711 | Putative regulatory protein PfoR |
MF: Protein-N (PI)-phosphohistidine-sugar phosphotransferase activity BP: Phosphoenolpyruvate-dependent sugar phosphotransferase system |
| WP_014342241.1 | 46,739–46,918 | Hypothetical protein | |
| WP_039486948.1 | 46,945–49,392 | Putative methyl-accepting chemotaxis protein |
MF: Transmembrane signaling receptor activity BP: Chemotaxis, Signal transduction |
| WP_010881491.1 | 49,513–50,445 | Peptidoglycan-binding protein LysM | – |
On the other hand, we found that the genes present in PAIs 2 of subsp. pallidum were not present in any of the GIs or PAIs of subsp. pertenue and endemicum (Non- venereal subsp.). This may reflect the fact that the genomic signature of those regions has already adapted in subsp. pallidum to cause different modes of transmission. The list of genes related to PAI 2 of subsp. pallidum is mentioned in Table 2 excluding the hypothetical genes.
Table 2.
The list of genes related to PAI 2 of subsp. pallidum
| Protein ID | PAIs Coordinates | Protein Name | Functions (MF: Molecular Function, BP: Biological Process) |
|---|---|---|---|
| WP_010882272.1 | 895,372–895,749 | holo-ACP synthase |
MF: holo-[acyl-carrier-protein] synthase activity, magnesium ion binding. BP: Fatty acid biosynthetic process |
| WP_010882273.1 | 895,746–896,630 | membrane protein | |
| WP_010882275.1 | 897,479–899,248 | arginine--tRNA ligase |
MF: arginine-tRNA ligase activity, ATP binding BP: arginyl-tRNA aminoacylation |
| WP_010882284.1 | 910,864–913,113 | MFS transporter | |
| WP_010882286.1 | 914,944–915,711 | methionine aminopeptidase | Aminopeptidase activity, metal ion binding, metalloexopeptidase activity |
| WP_010882287.1 | 915,835–916,659 | Heat shock protein, putative | |
| WP_010882288.1 | 916,752–917,804 | glyceraldehyde-3-phosphate dehydrogenase |
MF: glyceraldehyde-3-phosphate dehydrogenase (NAD+) phosphorylating activity. BP: glucose metabolic process, glycolytic process. |
| WP_010882292.1 | 919,085–919,453 | 50S ribosomal protein L20 |
MF: rRNA binding, structural constituent of ribosome. BP: translation. |
| WP_010882293.1 | 919,486–919,686 | 50S ribosomal protein L35 |
MF: structural constituent of ribosome, BP: translation. |
| WP_010882295.1 | 920,441–922,615 | DUF4954 domain-containing protein | |
| WP_010882296.1 | 922,644–923,642 | prolipoprotein diacylglyceryl transferase |
MF: transferase activity, transferring glycosyl groups. BP: lipoprotein biosynthetic process. |
Moreover, we also compared GIs of all subspecies; as a result, we found that the genes of some GIs which are present in the GI2 and GI4 in pallidum subspecies and are not reported in any of GIs of the subspecies endemicum and pertenue (Table 3). Most of the genes present in GI2 and GI4 of pallidum subspecies are hypothetical genes but some genes are chemotaxis protein (CheA) that are associated with the transmission of sensory signals from the chemoreceptors to the flagellar motors [26]. The mechanisms by which T. pallidum sense and respond to nutrient gradients help in pathogenic processes such as crossing the endothelial barrier to reach the bloodstream.
Table 3.
The list of genes related to GI 2 and GI 4 of subsp. pallidum. The table shows the list of proteins excluding the hypothetical proteins
| Protein ID | GI 2 and GI 4 Coordinates | Protein Name | Functions (MF: Molecular Function, BP: Biological Process) |
|---|---|---|---|
| WP_010881790.1 | 365,067–365,627 | cytidylate kinase |
MF: ATP binding, cytidylate kinase activity BP: pyrimidine nucleotide metabolic process. |
| WP_010881791.1 | 365,621–366,454 | adenine glycosylase |
MF: catalytic activity. DNA binding. BP: base-excision repair. |
| WP_010881792.1 | 366,459–369,881 | transcription-repair coupling factor |
MF: ATP binding, damaged DNA binding, hydrolase activity. BP: regulation of transcription, DNA-template, transcription-coupled nucleotide-excision repair. |
| WP_010881793.1 | 370,034–371,125 | phospho-N-acetylmuramoyl-pentapeptide- transferase |
MF: phospho-N-acetylmuramoyl-pentapeptide-transferase activity, UDP-N-acetylmuramoyl-L-alanyl-D-glutamyl-meso-2, transferase activity. BP:cell cycle, cell divisio, cell wall organization. |
| WP_010881797.1 | 373,895–374,425 | FKBP-type peptidyl-prolyl cis-trans isomerase |
MF: Metal ion binding, peptidyl-prolyl sis-trans isomerase activity. BP: protein refolding |
| WP_010881798.1 | 374,639–375,925 | gamma-glutamyl-phosphate reductase |
MF: glutamate-5semialdehyde dehydrogenase activity. BP: L-proline biosynthetic process. |
| WP_010881799.1 | 375,922–376,812 | glutamate 5-kinase |
MF: ATP-binding BP: L-proline biosynthetic process. |
| WP_010881801.1 | 377,198–377,707 | ribonuclease H | MF: Metal-ion binding, nucleic acid binding |
| WP_010881802.1 | 377,970–378,596 | thymidylate kinase |
MF: ATP binding, thymidylate kinase activity. BP: dTDP biosynthetic process |
| WP_010881806.1 | 377,970–378,596 | glycosyl hydrolase |
MF: catalytic activity. BP: carbohydrate metabolic process. |
| WP_014342391.1 | 382,926–383,729 | lysophosphatidic acid acyltransferase |
MF: transferase activity. BP: metabolic process |
| WP_014505476.1 | 384,584–387,016 | chemotaxis protein CheA |
MF: ATP bindinf, phosphorelay sensor kinase activity BP: chemotaxis. |
| WP_010881907.1 | 488,333–488,911 | SMC-Scp complex subunit ScpB | BP: cell division, chromosome separation |
| WP_014342436.1 | 488,928–489,725 | RsuA family pseudouridine synthase | MF: pseudouridine synthase activity, RNA binding. |
| WP_010881910.1 | 490,476–490,835 | transcriptional regulator | |
| WP_010881913.1 | 492,278–493,030 | tRNA (guanine-N(7)-)-methyltransferase | MF: tRNA (guanine-N7-)-methyltransferase activity. |
Discussion
The subspecies T. pallidum subsp. endemicum (TEN) and T. pallidum subsp. pertenue (TPE), are reasons for the diseases bejel and yaws, respectively. In the last few years, T. pallidum subsp. pallidum (TPA), has been reported as a reemerging pathogen [1, 15]. These three subsp. of Treponema pallidum are so close to each other that they cannot be differentiated serologically, their morphology is indistinguishable and are antigenically cross-reactive [27, 28]. Mostly, the disease phenotype caused by these pathogens can only be distinguished clinically and geographically. The distribution of venereal syphilis is global, non-venereal yaws usually effect kids in hot and/or humid regions of Africa and Asia, endemic syphilis be in dry places like Sahelian Africa and Saudi Arabia [27, 29]. The nature of T. pallidum is highly invasive. It circulates through bloodstream and lymphatics and overruns a wide-ranging of tissues and organs. As demonstrated by the widespread clinical manifestations related to syphilis infections, Treponema pallidum subsp. pallidum crosses placental, endothelial and blood-brain barriers early in infection, the incidence of congenital syphilis and invasion of central nervous system has been observed in almost 40% of early syphilis patients. Though, the understanding of the mechanisms responsible for the widespread distribution capability of T. pallidum is still very limited [30, 31].
The transmission of yaws is characterized by direct contact on skin and primary cutaneous lesion. It is facilitated by damaged skin surface. Scratching or rubbing these damaged parts of the body can facilitate the lesions spread across the body [28, 29]. Contrarily, endemic syphilis is an acute infection. Primary lesions of endemic syphilis can be seen in the children of ages between 2 and 15 years in dry and arid climates. While the mode of transmission is not known, it is believed that it may occur through mucosal and skin contact, even via shared eating utensils or drinking vessels [28, 29].
The defined relationships among the bacteria are still argued. The expansion of next-generation sequencing (NGS) in last few decades influences the fields of treatment and prevention, especially about bacterial diseases [32]. The ability of genomics data of T. pallidum gives us better understanding of the biology involving its interaction with its hosts. A comprehensive in silico pan-genome study was carried out for 53 sequenced genomes of T. pallidum, which indicates that the pan-genome of T. pallidum is still open; however, it is increasing at a very low rate as represented by the α of 0.9435 for the Pan All and the α of 0.916 and 0.999329 for Pan Subsp_pallidum and Pan_subsp_pertenue, respectively. Moreover, the α of 0.999329 indicates that the Pan Subsp pertenue is almost closed, which is corroborated by the tg(θ) of ~ 0.025.
The genome plasticity analysis reveals the differences in the presence and absence of some genome regions when compared at the subspecies level. Pathogenicity islands carry the genes related to the virulence, which are essential and characterize a class of Genomics Island [33]. The comparative analysis of PAIs and GIs showed the absence of genes at the subspecies level. We found gene clusters, that are related to amino acid and lipid biosynthesis, belonging to PAIs 2 of T. pallidum subsp. pallidum have not been identified in any PAIs or GIs of T. pallidum subsp. endemicum and T. pallidum subsp. pertenue. It might be possible that these genes help bacteria to execute different modes of infection at subsp. level of T. pallidum. Acyl carrier protein (ACP) synthase (AcpS) catalyzes the transfer of the 4′-phosphopantetheine moiety from coenzyme A (CoA) onto a serine residue of apo-ACP, to convert apo-ACP to the functional holo-ACP. During the biosynthesis of fatty acids and phospholipids, the holo form of bacterial ACP plays a vital role in mediating the transfer of acyl fatty acid. AcpS is therefore an attractive target for therapeutic interpolation. It has been reported that, AcpS enzymes from Mycoplasma pneumoniae and S. pneumoniae may play a crucial role in the acylation of fatty acids derived from human tissues for their lipid biosynthesis, suggesting that AcpS is a more striking antimicrobial target for discovery of novel antibiotics than bacterial fatty acid biosynthetic enzymes [34, 35].
Moreover, the presence of chemotaxis protein (CheA) in different GIs of T. pallidum subsp. pallidum might be responsible for different molecular modes of infection as T. pallidum genome contains two operons for the Che response regulators [31, 36]. The bacterial transcription-repair coupling factor (TRCF) is a large, multi-domain, SF2 ATPase that is generally conserved. It forms the dual of nucleotide excision repair with transcription by dislodging inactive RNA polymerase molecules stalled at template DNA lesions, and by increasing the rate at which the Uvr(A) BC exonuclease acts at these sites [37].
Pathogens are frequently using antigenic variation mechanisms to elude the adaptive immune response that ultimately results in persistent infection [38]. It might be because of the variation in expression of different Tpr proteins in the syphilis spirochete, Treponema pallidum subsp. pallidum, that have important implications in its ability to elude host immune detection [39]. A 12-membered protein family Treponema pallidum repeat (tpr) has been identified in T. pallidum subsp. Pallidum, which may be concerned in the pathogenesis of T. pallidum [38, 40]. On the basis of amino acid homology, these 12 Tprs are further divided into three subfamilies: subfamily I (TprC, D, F, I), subfamily II (TprE, G, J), and subfamily III (TprA, B, H, K, L) [40] [41].
Despite the host’s efforts to eliminate the infection, mechanisms of T. pallidum’s persistence include residence within intracellular or immune-privileged positions to hide from the immune effectors. T. pallidum‘s has the ability to cape its surface with host serum proteins or mucopolysaccharides to dodge immune response and immunosuppression of the host triggered by syphilis infection [38]. Freeze-fracture electron microscopy of T. pallidum has revealed lack of integral membrane proteins in the outer membrane (OM) of T. pallidum, conceivably accounting for the reasonably poor antigenicity of this spirochete’s surface [38, 42, 43].
However, as T. pallidum could be phagocytized in the presence of opsonic antibody, antibody targets must be present on the surface of the bacterium. Furthermore, the treponemes harvested from the tissues of later stage infections after the elimination of majority of treponemes are resistant to opsonophagocytosis. It raised the likelihood of antigenic variation occurring in T. pallidum, but no exact variable antigen was identified [38, 44]. Following the identification and investigation of TprK, provides the first candidate antigen of T. pallidum that might function in fudging the immune response. TprK vary among and within T. pallidum strains, with diversity of sequence localized in seven distinct regions (V1-V7) bordered by conserved domains [38, 43, 45, 46]. During experimental infection, these V regions are the main targets of the host humoral immune response [38]. Antigenic variation of the TprK antigen has been acknowledged to explain the persistence of T. pallidum in the host.
Recent work of Dan Liu et al. [47] has recognized an improved number of variants within these seven V regions of the tprK gene in the samples of secondary syphilis. A 3-bp changing pattern was observed in the sequences within each V region of the protein. However, same pattern of change was observed in variable sequences within the V regions of tprK in the secondary syphilis. Notably, the amino acid sequences IASDGGAIKH and IASEDGSAGNLKH in V1 are not only present in high proportion in inter-strain comparison but also were found at a quite high frequency in the populations. The alignment of all amino acid sequences revealed some really stable pattern within each V region of the primary and secondary syphilis samples, particularly the amino acid sequences IASDGGAIKH and IASEDGSAGNLKH in V1 region. The highly stable peptides found in V1 region are likely promising vaccine components. The highly heterogenetic regions (e.g., V6) could help to understand the role of tprK in fudging immune response. However, in our analysis, we found that some of tpr genes (tprC, tprD,tprF,tprI, trpJ) were present in some of PAIs or GIs T. pallidum subsp. endemicum (TEN) and T. pallidum subsp. pertenue (TPE). While, the GIs and PAIs related to T. pallidum subsp. pallidum we only identified some tpr domain proteins. It has been reported by Maděrankova et al. 2019, tpr genes responsible for the adaptive evolution of the pathogen [48].
Conclusions
Apart from establishing phylogenetic relationships among treponemal species and subspecies, the addition of comparative genomics was also required to illuminate the lower degree of virulence associated with T. pallidum subsp. pertenue than with T. pallidum subsp. pallidum. Unlike syphilis, it is said that yaws cannot be transmitted vertically or affect the central nervous system. It is rather limited to skin, bones, joints and soft tissues. In the 1980s, a very limited genetic diversity between these pathogens was established when hybridization experiments were carried out with DNA isolated from yaws and syphilis strains [29]. Our work also showed that genomes of syphilis, yaws, and Bejel treponemes share 97–100% overall similarity, as well as the identical organization. This evidence proposes that small genetic changes in key genes among these organisms could be responsible for the reported differences in disease pathogenesis. Considering the genes in PAIs and GIs, we identified some absence of pathogenicity islands in all subspecies. Genes which are present in pallidum subspecies pathogenicity islands (PAIs) or genomic islands (GIs) are absent in the subspecies endemicum and pertenue. The findings of this analysis are very important, as it can help in the understanding of molecular basis of infections from T. pallidum subsps. Furthermore, the core genes represent the most desirable source for the selection of conserved genes; therefore, characterization of such poorly studied proteins helps in understanding the cellular metabolism, mode of infection and regulation of gene expression of Treponema pallidum. Hence, this study can help to better understand the molecular modes of bacterial infection and are significance for vaccine development for syphilis.
Methods
Genome sequences
The genome sequences of 53 T. pallidum strains were retrieved from the NCBI (National Centre for Biotechnology Information) database (https://www.ncbi.nlm.nih.gov/genome/genomes/741?) (Accessed June 2018): 46 genomes of T. pallidum subsp. pallidum were isolated from different parts of human body, rabbits and baboons (USA, China & Portugal). Six genomes from Africa and Australia/ Oceania continents (strain SamoaD, CDC2, Gauthier, CDC2575, Ghana051 and LMNP-1) from subsp. pertenue were isolated from humans, baboons and rabbits (Additional file 1: Table S1). One genome of Treponema pallidum subsp. endemicum (strain BosniaA) was isolated in Europe from human tongue and tonsils. The genome of Treponema denticola strain ATCC 35405 was used as non-pathogenic bacteria in this work. The general information about all T. pallidum strains and the Complete workflow applied in this work are given in Additional file 1: Table S1 and Figure S1, respectively.
Phylogenomic analysis of all Treponema pallidum strains
For phylogenomic analysis of all Treponema pallidum strains, Gegenees (version 2.1) [19] was used. The Gegenees software was used to perform an all-versus-all similarity search. It divides the genomes into small sequences and determines the minimum content shared by all the genomes. Subsequently, the obtained minimum shared contents were subtracted from all the genomes resulting in the variable contents, which were eventually compared with all the other strains for the calculation of the percentages of similarity. Finally, these percentages were plotted in a heatmap chart with a spectrum ranging from low similarity (red) to high similarity (green). The Gegenees data was exported as a distance matrix file in nexus format (.nex) and, further, the generated distance matrix was used as an input file in SplitsTree software (version 4.14.5) [49] using neighbour joining method to create a dendogram [50, 51].
Prediction of Pan-genome, Core-genome and singleton
We divided 53 strains of T. pallidum in 3 subsets for pan-genome calculation. We performed Pan All (with all 53 strains of T. pallidum), Pan Subsp_pallidum and Pan_subsp_pertenue (based on subspecies). For the identification of core genome (commonly shared by all strains), shared genome (genes present in two or more than two strains but not shared by all strains) and singletons (strain specific genes), we used OrthoFinder [52]. Briefly, OrthoFinder uses the .faa amino acid sequence file for each genome to perform all-vs-all BLASTp for the Orthologous analysis. It uses MCL (Markov Clustering algorithm) program to determine the Orthologous genes [53]. The cut-off value of 1e− 10 was used for Pan-genome, Core-genome and singletons identification for all the subsets. Furthermore, in-house scripts were used to estimate the fixed parameters for Heap’s Law (pan-genome analyses) [20, 51] and least-squares fit of the exponential regression decay (core-genome and singletons). The extrapolations of the pan-genomes from the complete dataset and all subsets were calculated based on Heap’s Law [20, 51], which was used to calculate whether the pan-genome was open or closed. Heap’s Law is an empirical law represented by the formula n = k*Nγ; it describes the number of distinct words in a document (or set of documents) as a function of the document length. In a genetic context, n is the expected number of genes for a given number of genomes, N determines the number of genomes, and the k and γ (α =1-γ) are free parameters that are determined empirically. According to Heap’s Law, when α > 1 (γ < 0), the pan-genome is considered to be closed, and there will be no significant increase in the number of genes with the addition of a new genome. On the other hand, when α < 1 (0 < γ < 1), the pan-genome is open and there will be a significant increase in the number of genes for each newly added genome.
Genomic and Pathogenicity Islands prediction
This section describes the analyses that were performed for the prediction of genomic and pathogenicity Islands following three datasets based on the subspecies: A) using T. pallidum subsp. pallidum strain Nichols as a reference; B) using T. pallidum subsp. pertenue strain SamoaD as a reference; and C) using T. pallidum subsp. endemicum strain BosniaA as a reference. The islands predictions for three datasets were determined by using GIPSy (Genomic Island Prediction Software) [33]. GIPSy is a multi-step approach that predicts Genomic islands (GIs) and Pathogenicity islands (PAIs). PAIs and GIs predictions are based on commonly shared features such as genomic signature deviation (anomalous G + C content and codon usage deviation), presence of transposase genes; metabolism, virulence, antibiotic resistance, or symbiosis-related genes; flanking tRNA genes; and absence in other organisms of the same genus or closely related species [33]. T. denticola strain ATCC 35405 was used as a non-pathogenic species from the same Treponema genus for GIs and PAIs prediction [54]. The sizes of the islands were compared with all the other strains via ACT (Artemis Comparison Tool) software [55]. PAIs regions were plotted using the software BRIG [25]. Following the curation of the PAIs, the genes of all the islands in each strain were assessed for their presence/absence in all the other strains.
Supplementary information
Additional file 1: Table S1. General information about 53 Treponema pallidum Strains used in this work. List of all Treponema pallidum strains (with features) retrieved from the NCBI (National Center for Biotechnology Information) database. Table S2A. The COG functional categories with detailed description of Core genes: The table showing the number of core genes of the complete dataset were classified by COG (Cluster of Orthologous Genes) functional category. Table S2B. The COG functional categories with detailed description of Core genes: The table showing the number of core genes of the Pan Subsp_pallidum dataset were classified by COG (Cluster of Orthologous Genes) functional category. Table S2C. The COG functional categories with detailed description of Core genes: The table showing the number of core genes of the Pan Subsp_pertenue dataset were classified by COG (Cluster of Orthologous Genes) functional category. Figure S1. The Complete workflow applied in this work. The figure represent the methodology and software were used in this analysis. Figure S2. The heatmap analysis of 53 Strains of Treponema pallidum.The figure represents the comparison between the variable content of all strains. The percentages were plotted in the heatmap with a spectrum ranging from red (low similarity) to green (high similarity). The names of the strains on the left side of the figure (vertically) are organized in the same order in the top part of the figure (horizontally). Once Gegenees uses the similarities in the variable contents, the outgroup normally presents a very small percentage of similarity to the other strains.
Acknowledgments
We acknowledge the collaboration and assistance of all team members and the Brazilian funding agencies CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brasil), and FAPEMIG (Fundação de Amparo à Pesquisa de Minas Gerais). Arun Kumar Jaiswal was supported by the CAPES (Coordenação de Aperfeiçoamento de Pessoal de Nível Superior, Brasil) fellowship for doctoral studies. Syed Babar Jamal acknowledges the “TWAS-CNPq Postgraduate Fellowship Programme” for granting a fellowship for doctoral studies.
Abbreviations
- CDS
Coding sequences
- COG
Cluster of Orthologous Genes
- GIs
Genomic islands
- PAIs
Pathogenicity islands
- TEN
Treponema pallidum subsp. endemicum
- TPA
Treponema pallidum subsp. pallidum
- TPE
Treponema pallidum subsp. pertenue
- WHO
World Health Organization
Authors’ contributions
AKJ, ST, SBJ, LCO,LGA, SCS conceived, designed the protocol, collected and analysed initial data, wrote the paper: ST, SCS, VA, CJFO coordinated and led the entire project: AKJ, ST, SBJ, SCS, PG, VA, CJFO Cross-checked all data, analysis, wrote the paper: All authors read and approved the manuscript.
Funding
No funding supported this research.
Availability of data and materials
All data generated and analysed during this study are included in this published article and its supplementary information files.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Arun Kumar Jaiswal, Email: arunjaiswal1411@gmail.com.
Sandeep Tiwari, Email: sandip_sbtbi@yahoo.com.
Syed Babar Jamal, Email: syedbabar.jamal@gmail.com.
Letícia de Castro Oliveira, Email: letcastrol@gmail.com.
Leandro Gomes Alves, Email: leandrogomesalv@gmail.com.
Vasco Azevedo, Email: vascoariston@gmail.com.
Preetam Ghosh, Email: preetam.ghosh@gmail.com.
Carlo Jose Freira Oliveira, Email: oliveiracjf@gmail.com.
Siomar C. Soares, Email: siomars@gmail.com
Supplementary information
Supplementary information accompanies this paper at 10.1186/s12864-019-6430-6.
References
- 1.Peeling RW, Hook EW., 3rd The pathogenesis of syphilis: the great mimicker, revisited. J Pathol. 2006;208(2):224–232. doi: 10.1002/path.1903. [DOI] [PubMed] [Google Scholar]
- 2.Mitja O, Smajs D, Bassat Q. Advances in the diagnosis of endemic treponematoses: yaws, bejel, and pinta. PLoS Negl Trop Dis. 2013;7(10):e2283. doi: 10.1371/journal.pntd.0002283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Radolf JD, Deka RK, Anand A, Smajs D, Norgard MV, Yang XF. Treponema pallidum, the syphilis spirochete: making a living as a stealth pathogen. Nat Rev Microbiol. 2016;14(12):744–759. doi: 10.1038/nrmicro.2016.141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Centurion-Lara A, Molini BJ, Godornes C, Sun E, Hevner K, Van Voorhis WC, Lukehart SA. Molecular differentiation of Treponema pallidum subspecies. J Clin Microbiol. 2006;44(9):3377–3380. doi: 10.1128/JCM.00784-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nyatsanza F, Tipple C. Syphilis: presentations in general medicine. Clin Med (Lond) 2016;16(2):184–188. doi: 10.7861/clinmedicine.16-2-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Stamm LV. Global challenge of antibiotic-resistant Treponema pallidum. Antimicrob Agents Chemother. 2009;54(2):583–589. doi: 10.1128/AAC.01095-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stamm LV. Global challenge of antibiotic-resistant Treponema pallidum. Antimicrob Agents Chemother. 2010;54(2):583–589. doi: 10.1128/AAC.01095-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Celum CL. Sexually transmitted infections and HIV: epidemiology and interventions. Top HIV Med. 2010;18(4):138–142. [PubMed] [Google Scholar]
- 9.Dhawan J, Gupta S, Kumar B. Sexually transmitted diseases in children in India. Indian J Dermatol Venereol Leprol. 2010;76(5):489–493. doi: 10.4103/0378-6323.69056. [DOI] [PubMed] [Google Scholar]
- 10.Kumar Jaiswal Arun, Tiwari Sandeep, Jamal Syed, Barh Debmalya, Azevedo Vasco, Soares Siomar. An In Silico Identification of Common Putative Vaccine Candidates against Treponema pallidum: A Reverse Vaccinology and Subtractive Genomics Based Approach. International Journal of Molecular Sciences. 2017;18(2):402. doi: 10.3390/ijms18020402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Tucker JD, Cohen MS. China's syphilis epidemic: epidemiology, proximate determinants of spread, and control responses. Curr Opin Infect Dis. 2011;24(1):50–55. doi: 10.1097/QCO.0b013e32834204bf. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Uuskula A, Puur A, Toompere K, DeHovitz J. Trends in the epidemiology of bacterial sexually transmitted infections in eastern Europe, 1995-2005. Sex Transm Infect. 2010;86(1):6–14. doi: 10.1136/sti.2009.037044. [DOI] [PubMed] [Google Scholar]
- 13.Brown DL, Frank JE. Diagnosis and management of syphilis. Am Fam Physician. 2003;68(2):283–290. [PubMed] [Google Scholar]
- 14.Cameron CE, Lukehart SA. Current status of syphilis vaccine development: need, challenges, prospects. Vaccine. 2014;32(14):1602–1609. doi: 10.1016/j.vaccine.2013.09.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peeling RW, Mabey D, Kamb ML, Chen XS, Radolf JD, Benzaken AS. Syphilis. Nat Rev Dis Primers. 2017;3:17073. doi: 10.1038/nrdp.2017.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tettelin H, Masignani V, Cieslewicz MJ, Donati C, Medini D, Ward NL, Angiuoli SV, Crabtree J, Jones AL, Durkin AS, et al. Genome analysis of multiple pathogenic isolates of Streptococcus agalactiae: implications for the microbial "pan-genome". Proc Natl Acad Sci U S A. 2005;102(39):13950–13955. doi: 10.1073/pnas.0506758102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Miao R, Fieldsteel AH. Genetics of Treponema: relationship between Treponema pallidum and five cultivable treponemes. J Bacteriol. 1978;133(1):101–107. doi: 10.1128/JB.133.1.101-107.1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Miao RM, Fieldsteel AH. Genetic relationship between Treponema pallidum and Treponema pertenue, two noncultivable human pathogens. J Bacteriol. 1980;141(1):427–429. doi: 10.1128/JB.141.1.427-429.1980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Agren J, Sundstrom A, Hafstrom T, Segerman B. Gegenees: fragmented alignment of multiple genomes for determining phylogenomic distances and genetic signatures unique for specified target groups. PLoS One. 2012;7(6):e39107. doi: 10.1371/journal.pone.0039107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Guimaraes LC, Florczak-Wyspianska J, de Jesus LB, Viana MV, Silva A, Ramos RT, Soares Sde C, Soares Sde C. Inside the Pan-genome - methods and software overview. Curr Genomics. 2015;16(4):245–252. doi: 10.2174/1389202916666150423002311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tettelin H, Riley D, Cattuto C, Medini D. Comparative genomics: the bacterial pan-genome. Curr Opin Microbiol. 2008;11(5):472–477. doi: 10.1016/j.mib.2008.09.006. [DOI] [PubMed] [Google Scholar]
- 22.Mira A, Martin-Cuadrado AB, D'Auria G, Rodriguez-Valera F. The bacterial pan-genome:a new paradigm in microbiology. Int Microbiol. 2010;13(2):45–57. doi: 10.2436/20.1501.01.110. [DOI] [PubMed] [Google Scholar]
- 23.Schmidt H, Hensel M. Pathogenicity islands in bacterial pathogenesis. Clin Microbiol Rev. 2004;17(1):14–56. doi: 10.1128/CMR.17.1.14-56.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Karaolis DK, Johnson JA, Bailey CC, Boedeker EC, Kaper JB, Reeves PR. A Vibrio cholerae pathogenicity island associated with epidemic and pandemic strains. Proc Natl Acad Sci U S A. 1998;95(6):3134–3139. doi: 10.1073/pnas.95.6.3134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alikhan NF, Petty NK, Ben Zakour NL, Beatson SA. BLAST ring image generator (BRIG): simple prokaryote genome comparisons. BMC Genomics. 2011;12:402. doi: 10.1186/1471-2164-12-402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Greene SR, Stamm LV, Hardham JM, Young NR, Frye JG. Identification, sequences, and expression of Treponema pallidum chemotaxis genes. DNA Seq. 1997;7(5):267–284. doi: 10.3109/10425179709034046. [DOI] [PubMed] [Google Scholar]
- 27.Knauf S, Liu H, Harper KN. Treponemal infection in nonhuman primates as possible reservoir for human yaws. Emerg Infect Dis. 2013;19(12):2058–2060. doi: 10.3201/eid1912.130863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Gogarten JF, Düx A, Schuenemann VJ, Nowak K, Boesch C, Wittig RM, Krause J, Calvignac-Spencer S, Leendertz FH. Tools for opening new chapters in the book of Treponema pallidum evolutionary history. Clin Microbiol Infect. 2016;22(11):916–921. doi: 10.1016/j.cmi.2016.07.027. [DOI] [PubMed] [Google Scholar]
- 29.Giacani L, Lukehart SA. The endemic Treponematoses. Clin Microbiol Rev. 2014;27(1):89–115. doi: 10.1128/CMR.00070-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lithgow KV, Hof R, Wetherell C, Phillips D, Houston S, Cameron CE. A defined syphilis vaccine candidate inhibits dissemination of Treponema pallidum subspecies pallidum. Nat Commun. 2017;8(1):1–10. [DOI] [PMC free article] [PubMed]
- 31.LaFond RE, Lukehart SA. Biological basis for syphilis. Clin Microbiol Rev. 2006;19(1):29–49. doi: 10.1128/CMR.19.1.29-49.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Arora N, Schuenemann VJ, Jager G, Peltzer A, Seitz A, Herbig A, Strouhal M, Grillova L, Sanchez-Buso L, Kuhnert D, et al. Origin of modern syphilis and emergence of a pandemic Treponema pallidum cluster. Nat Microbiol. 2016;2:16245. doi: 10.1038/nmicrobiol.2016.245. [DOI] [PubMed] [Google Scholar]
- 33.Soares SC, Geyik H, Ramos RT, de Sa PH, Barbosa EG, Baumbach J, Figueiredo HC, Miyoshi A, Tauch A, Silva A, et al. GIPSy: genomic island prediction software. J Biotechnol. 2016;232:2–11. doi: 10.1016/j.jbiotec.2015.09.008. [DOI] [PubMed] [Google Scholar]
- 34.Heath RJ, Rock CO. Fatty acid biosynthesis as a target for novel antibacterials. Curr Opin Investig Drugs. 2004;5(2):146–153. [PMC free article] [PubMed] [Google Scholar]
- 35.McAllister KA, Peery RB, Zhao G. Acyl carrier protein synthases from gram-negative, gram-positive, and atypical bacterial species: biochemical and structural properties and physiological implications. J Bacteriol. 2006;188(13):4737–4748. doi: 10.1128/JB.01917-05. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fraser CM, Norris SJ, Weinstock GM, White O, Sutton GG, Dodson R, Gwinn M, Hickey EK, Clayton R, Ketchum KA, et al. Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science. 1998;281(5375):375–388. doi: 10.1126/science.281.5375.375. [DOI] [PubMed] [Google Scholar]
- 37.Deaconescu AM, Savery N, Darst SA. The bacterial transcription repair coupling factor. Curr Opin Struct Biol. 2007;17(1):96–102. doi: 10.1016/j.sbi.2007.01.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Giacani L, Molini BJ, Kim EY, Godornes BC, Leader BT, Tantalo LC, Centurion-Lara A, Lukehart SA. Antigenic variation in Treponema pallidum: TprK sequence diversity accumulates in response to immune pressure during experimental syphilis. J Immunol. 2010;184(7):3822–3829. doi: 10.4049/jimmunol.0902788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Leader BT, Hevner K, Molini BJ, Barrett LK, Van Voorhis WC, Lukehart SA. Antibody responses elicited against the Treponema pallidum repeat proteins differ during infection with different isolates of Treponema pallidum subsp. pallidum. Infect Immun. 2003;71(10):6054–6057. doi: 10.1128/IAI.71.10.6054-6057.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sun ES, Molini BJ, Barrett LK, Centurion-Lara A, Lukehart SA, Van Voorhis WC. Subfamily I Treponema pallidum repeat protein family: sequence variation and immunity. Microbes Infect. 2004;6(8):725–737. doi: 10.1016/j.micinf.2004.04.001. [DOI] [PubMed] [Google Scholar]
- 41.Centurion-Lara A, Castro C, Barrett L, Cameron C, Mostowfi M, Van Voorhis WC, Lukehart SA. Treponema pallidumMajor sheath protein homologue Tpr K is a target of opsonic antibody and the protective immune response. J Exp Med. 1999;189(4):647–656. doi: 10.1084/jem.189.4.647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Walker EM, Borenstein LA, Blanco DR, Miller JN, Lovett MA. Analysis of outer membrane ultrastructure of pathogenic Treponema and Borrelia species by freeze-fracture electron microscopy. J Bacteriol. 1991;173(17):5585–5588. doi: 10.1128/JB.173.17.5585-5588.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pinto M, Borges V, Antelo M, Pinheiro M, Nunes A, Azevedo J, Borrego MJ, Mendonca J, Carpinteiro D, Vieira L, et al. Genome-scale analysis of the non-cultivable Treponema pallidum reveals extensive within-patient genetic variation. Nat Microbiol. 2016;2:16190. doi: 10.1038/nmicrobiol.2016.190. [DOI] [PubMed] [Google Scholar]
- 44.Lukehart SAL, Shaffer JM, Zander SAB. A subpopulation of Treponema pallidum is resistant to phagocytosis: possible mechanism of persistence. J Infect Dis. 1992;166(6):1449–1453. doi: 10.1093/infdis/166.6.1449. [DOI] [PubMed] [Google Scholar]
- 45.Centurion-Lara A, Godornes C, Castro C, Van Voorhis WC, Lukehart SA. The tprK gene is heterogeneous among Treponema pallidum strains and has multiple alleles. Infect Immun. 2000;68(2):824–831. doi: 10.1128/IAI.68.2.824-831.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.LaFond RE, Centurion-Lara A, Godornes C, Rompalo AM, Van Voorhis WC, Lukehart SA. Sequence diversity of Treponema pallidum subsp. pallidum tprK in human syphilis lesions and rabbit-propagated isolates. J Bacteriol. 2003;185(21):6262–6268. doi: 10.1128/JB.185.21.6262-6268.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu Dan, Tong Man-Li, Lin Yong, Liu Li-Li, Lin Li-Rong, Yang Tian-Ci. Insights into the genetic variation profile of tprK in Treponema pallidum during the development of natural human syphilis infection. PLOS Neglected Tropical Diseases. 2019;13(7):e0007621. doi: 10.1371/journal.pntd.0007621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Maderankova D, Mikalova L, Strouhal M, Vadjak S, Kuklova I, Pospisilova P, Krbkova L, Koscova P, Provaznik I, Smajs D. Identification of positively selected genes in human pathogenic treponemes: syphilis-, yaws-, and bejel-causing strains differ in sets of genes showing adaptive evolution. PLoS Negl Trop Dis. 2019;13(6):e0007463. doi: 10.1371/journal.pntd.0007463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kloepper TH, Huson DH. Drawing explicit phylogenetic networks and their integration into SplitsTree. BMC Evol Biol. 2008;8:22. doi: 10.1186/1471-2148-8-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23(2):254–267. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 51.Soares SC, Silva A, Trost E, Blom J, Ramos R, Carneiro A, Ali A, Santos AR, Pinto AC, Diniz C, et al. The pan-genome of the animal pathogen Corynebacterium pseudotuberculosis reveals differences in genome plasticity between the biovar ovis and equi strains. PLoS One. 2013;8(1):e53818. doi: 10.1371/journal.pone.0053818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Emms DM, Kelly S. OrthoFinder: solving fundamental biases in whole genome comparisons dramatically improves orthogroup inference accuracy. Genome Biol. 2015;16:157. doi: 10.1186/s13059-015-0721-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Enright AJ, Van Dongen S, Ouzounis CA. An efficient algorithm for large-scale detection of protein families. Nucleic Acids Res. 2002;30(7):1575–1584. doi: 10.1093/nar/30.7.1575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Seshadri R, Myers GS, Tettelin H, Eisen JA, Heidelberg JF, Dodson RJ, Davidsen TM, DeBoy RT, Fouts DE, Haft DH, et al. Comparison of the genome of the oral pathogen Treponema denticola with other spirochete genomes. Proc Natl Acad Sci U S A. 2004;101(15):5646–5651. doi: 10.1073/pnas.0307639101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Carver TJ, Rutherford KM, Berriman M, Rajandream MA, Barrell BG, Parkhill J. ACT: the Artemis comparison tool. Bioinformatics. 2005;21(16):3422–3423. doi: 10.1093/bioinformatics/bti553. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Additional file 1: Table S1. General information about 53 Treponema pallidum Strains used in this work. List of all Treponema pallidum strains (with features) retrieved from the NCBI (National Center for Biotechnology Information) database. Table S2A. The COG functional categories with detailed description of Core genes: The table showing the number of core genes of the complete dataset were classified by COG (Cluster of Orthologous Genes) functional category. Table S2B. The COG functional categories with detailed description of Core genes: The table showing the number of core genes of the Pan Subsp_pallidum dataset were classified by COG (Cluster of Orthologous Genes) functional category. Table S2C. The COG functional categories with detailed description of Core genes: The table showing the number of core genes of the Pan Subsp_pertenue dataset were classified by COG (Cluster of Orthologous Genes) functional category. Figure S1. The Complete workflow applied in this work. The figure represent the methodology and software were used in this analysis. Figure S2. The heatmap analysis of 53 Strains of Treponema pallidum.The figure represents the comparison between the variable content of all strains. The percentages were plotted in the heatmap with a spectrum ranging from red (low similarity) to green (high similarity). The names of the strains on the left side of the figure (vertically) are organized in the same order in the top part of the figure (horizontally). Once Gegenees uses the similarities in the variable contents, the outgroup normally presents a very small percentage of similarity to the other strains.
Data Availability Statement
All data generated and analysed during this study are included in this published article and its supplementary information files.