

Abstract
Human cytomegalovirus (HCMV), strain AD169, contains four genes (US27, US28, UL33, and UL78) that encode putative homologues of cellular G protein-coupled receptors (GCRs). GCRs transduce extracellular signals to alter intracellular processes, and there is evidence that HCMV may elicit such changes at early times following infection. The US27, US28, and UL33 genes are transcribed during infection, and the US28 gene product has been found to be a functional receptor for the β-chemokine class of immune modulators. The US27, UL33, and UL78 gene products have not been described and we have concentrated on identifying the UL33 protein because it is the most highly conserved of the GCR homologues among the human β and γ herpesviruses. We report here cloning UL33 into a recombinant baculovirus (rBV) and expressing it in insect cells; constructing a mutant HCMV with a disrupted UL33 gene; and identifying the UL33 protein in HCMV-infected cells and virus particles. Our results demonstrate that the UL33 protein (i) is expressed as a ≈36-kDa, heat-aggregatable protein in rBV-infected cells, (ii) is modified heterogeneously by asparagine-linked glycosylation and expressed as a ≥58-kDa glycoprotein that is present in the region of the cytoplasmic inclusions in HCMV-infected fibroblasts, (iii) is present in virions and two other enveloped virus particles, and (iv) is not essential for growth of HCMV in human foreskin fibroblast cultures.
INTRODUCTION
Human cytomegalovirus (HCMV) is a member of the herpesvirus family (human herpesvirus 5) and is a ubiquitous pathogen that causes many disorders in immunodeficient or immunocompromised individuals and in neonates (Alford and Britt, 1990; Bruggeman, 1993). Analysis of the nucleotide sequence of the 230-kb genome of HCMV strain AD169 (Chee et al., 1990a) revealed four open reading frames (ORFs), UL33, UL78, US27, and US28, that potentially encode homologues of cellular G protein-coupled receptors (GCRs) (Attwood and Findlay, 1994; Chee et al., 1990b), a family of signal transducing proteins found in numerous biological systems (Attwood and Findlay, 1994; Kolakowski, 1994).
GCRs characteristically have seven membrane-spanning domains and generally contain two disulfide-linked cysteines in the second and third extracytoplasmic loops (Fraser et al., 1994); multiple serines and threonines (Fraser et al., 1994) and a palmitoylated cysteine (Papac et al., 1992) in the intracellular, carboxyl tail; and asparagine-linked glycosylation on extracellular domains (Fraser et al., 1994). In addition, an N-P-X-X-Y motif in the seventh transmembrane domain (Berlose et al., 1994), a conserved D-R-Y sequence in the second intracellular loop, thought to be necessary for the receptor–G protein interaction (Oliveira et al., 1993), and other conserved residues throughout are commonly found (Fraser et al., 1994; Horuk, 1994).
GCRs are typically the first members involved in a signal transduction cascade initiated by ligand binding and receptor activation; these ligands can be biogenic amines, peptides, odorants, tastants, or other signals (Fraser et al., 1994). Ligand binding is predicted to alter the conformation of a GCR, generating an active form of the receptor, and dissociation of the ligand returns the GCR to an inactive state (Fraser et al., 1994). There are also examples of constitutively activated, mutant GCRs that are apparently fixed in an active conformation; these do not require ligand binding for activation (Shenker, 1994).
An activated GCR transmits its signal to a coupled heterotrimeric G protein, which itself becomes activated and, in turn, alters the activity of target proteins; for example, upregulating phosphatidylinositol phospholipase C (Sleight and Lieberman, 1995), modulating adenylate cyclase (Sleight and Lieberman, 1995), or gating ion channels (Sleight and Lieberman, 1995; Yatani, 1995) are common effects. These affected proteins amplify and propagate the signal by altering the levels of small intracellular second messenger molecules, such as inositol trisphosphate, diacylglycerol, arachidonic acid, cyclic AMP (cAMP), cyclic GMP, calcium, potassium, or sodium (Sleight and Lieberman, 1995). Examples of signal transmission by second messengers include (i) liberating free calcium, to coordinate with calcium-binding proteins and increase calcium-dependent processes (Dedman and Kaetzel, 1995), (ii) interacting with protein kinase C (PKC), to activate other proteins (Sleight and Lieberman, 1995), and (iii) interacting with cAMP-dependent proteins, to regulate transcription or other intracellular processes (Lalli et al., 1993; Sleight and Lieberman, 1995).
Changes in some second messengers have been noted during HCMV infection of human fibroblasts. In creases of inositol trisphosphate, diacylglycerol, arachidonic acid, cAMP, calcium, and sodium, as well as in creases of the transcription factors c-jun, c-myc, and c-fos, have all been detected early in infection (Albrecht et al., 1991), leading to the hypothesis that one or more of the virally encoded GCRs is responsible for these changes. Supporting such a relationship are data showing that the HCMV US28 ORF encodes a functional β-chemokine receptor that facilitates an increase of intracellular calcium upon stimulation (Gao and Murphy, 1994; Neote et al., 1993). The other HCMV GCRs encoded by US27 and UL33 are also most homologous to chemokine receptors (Horuk, 1994; this report), proteins in volved in chemotaxis of monocytes and granulocytes (Ahuja et al., 1994; Durum and Oppenheim, 1993). Since HCMV can be found in these cell types in vivo (Bruggeman, 1993; von Laer et al., 1995), it is possible that HCMV uses its own GCR homologues to take advantage of signal transduction pathways normally used by the host immune system. Because of the potential involvement of these GCRs in the pathology of HCMV infection, it is important to determine their function and mechanism of action.
We (Welch et al., 1991) and others (Davis-Poynter et al., 1995; Jones et al., 1995) have previously shown that the three ORFs, UL33, US27, and US28, are transcribed maximally in HCMV-infected fibroblasts at late times after infection. In this report we have used Western immunoassays and indirect immunofluorescence to identify and to begin to characterize the protein product of the HCMV strain AD169 UL33 ORF.
(Initial reports of this work were presented at the 17th International Herpesvirus Workshop in Edinburgh, Scotland, August 1–7, 1992, and at the 20th International Herpesvirus Workshop in Groningen, the Netherlands, July 30–August 3, 1995.)
MATERIALS AND METHODS
Cells and viruses
The propagation of human foreskin fibroblasts (HFF cells) and MRC5 cells and their infection with HCMV strain AD169 (ATCC VR-538) have been described before (Gibson, 1981b; Kaye et al., 1992; Weiner and Gibson, 1983). HCMV virions, noninfectious enveloped particles (NIEPs), and dense bodies (DBs) were effectively purified, separated from one another, and concentrated by banding three times in potassium tartrate gradients as documented previously (Irmiere and Gibson, 1983; Talbot and Almeida, 1977), except that the virus pellets were stored at −80° without solubilization. Spodoptera frugiperda (Sf9) cells (ATCC CRL-1711) were grown and infected with baculovirus Autographa californica nuclear polyhedrosis virus (AcNPV) as described (Gruenwald and Heitz, 1993; O’Reilly et al., 1992). The recombinant baculoviruses (rBVs) rBV-33, rBV-Tag33, and rBV-N33 were prepared using the baculovirus transfer plasmids described below and plaque purified at least twice (Gruenwald and Heitz, 1993; O’Reilly et al., 1992).
Construction of rBV transfer plasmids
The HCMV ORF UL33 (Chee et al., 1990a,b) was cloned through the polymerase chain reaction (PCR) (Scharf, 1990) by using a plasmid containing the HindIII J fragment of HCMV, strain AD169 (gift of Marc Chee, Medical Research Council), as a template and synthetic primers, with BamHI ends, which consisted of nucleotides 43246–43269 for the coding strand and 44429–44453 for the complementary strand of the AD169 genome (Chee et al., 1990a). The plasmid BJM1 was made by inserting the resulting 1.2-kb UL33 PCR product into the BamHI site of the vector pcDNA1 (Invitrogen, San Diego, CA). Subsequent dideoxy sequencing of the PCR-generated insert (Sequenase, United States Biochemical, Cleveland, OH) (Kraft et al., 1988) revealed one point mutation (base number 840 of the UL33 coding sequence, C for T), probably introduced by PCR (Smith et al., 1993), that was not repaired because it did not change the codon specificity for asparagine.
A second plasmid, pTag33, that contained the ORF for an epitope-tagged version of the UL33 protein was made. The UL33 ORF from BJM1 was first cloned into the BamHI site of the vector pET-11c (Novagen, Madison, WI), creating pET-33. BJM1 was then cut with HindIII, the 5′ overhang filled with the large fragment of DNA polymerase I (Klenow fragment, New England Biolabs, Beverly, MA), and cut with PvuI. The 5′ coding region of the UL33 ORF from pET-33 (containing the coding sequence for an 11-amino-acid addition) was cut with NdeI, treated as above with a Klenow fragment, and subsequently cut with PvuI. The blunt-to-PvuI fragment from pET-33 was subcloned in place of the excised region of the UL33 ORF in BJM1, preserving the HindIII site. The plasmid pK-Tag33 was created by inserting a complementary pair of oligonucleotides containing a Kozak-consensus ATG start codon in place of the existing ATG initiating codon in pTag33 (Kaufman, 1990): pTag33 was partially digested with NheI (Parker et al., 1977) and then cut with HindIII. The double stranded oligonucleotide, containing ends compatible with these same restriction sites and an internal BglII site 5′ to the ATG, was then cloned into the prepared pTag33 vector.
pVL-33, the transfer plasmid for rBV-33, was constructed by cloning the UL33 ORF from BJM1 into the BamHI site of the baculovirus transfer vector pVL1393 (Pharmingen, San Diego, CA). pVL-Tag33, the transfer plasmid for rBV-Tag33, was constructed by inserting the 1.3-kb BglII–EcoRI extended UL33 ORF from pK-Tag33 into the corresponding sites of the baculovirus transfer vector pVL1392 (Pharmingen). pVL-N33 is the rBV transfer plasmid for rBV-N33 and codes for a different amino-terminally extended version of the UL33 protein; it was constructed as follows. First, an amino-terminal extension of UL33 was generated by PCR; the template was the HCMV HindIII J fragment, cleaved with BfaI; the coding strand primer included a BamHI site followed by the first 21 codons predicted in the longer, spliced mRNA for UL33 (Davis-Poynter et al., 1995); the other primer for the complementary strand was an internal 20-mer previously used for sequencing. The final PCR product was cut with BamHI and DraIII and used to replace the BglII–DraIII fragment in pVL-Tag33.
Cloning and plasmid construction and purification was done by standard techniques with strains MC1061/P3 (Invitrogen) and XL1-Blue (Stratagene, La Jolla, CA) of Escherichia coli (Sambrook et al., 1989; Scharf, 1990). The composition and orientation of each construct was verified by restriction analyses and dideoxy sequencing (Kraft et al., 1988) when necessary.
Construction of UL33-disrupted HCMV
A mutant of HCMV that contains a disrupted UL33 ORF and is called HCMV Δ33 (Δ 33) was made as follows. A fragment of the AD169 genome (nucleotides 38517–46434) containing the UL33 coding region was cloned in pUC13 to give pHBHJ. This plasmid was digested with AsuII which cleaves it once, 23 bp 3′ of the ATG of the UL33 ORF. A linker, constructed by annealing the oligonucleotides 5′-CGAAAGATCTAGGCCTTT-3′ and 5′-CGAAAGGCCTAGAT-3′ was ligated into AsuII-cleaved and phosphatased pHBHJ to give pAsuII. The BglII site present in the linker is unique in pAsuII, and a 3.8-kb BamHI fragment containing E. coli lacZ sequences down stream of the CMV major immediate-early promoter derived from the plasmid pMV1 (Browne et al., 1992) was ligated into BglII-cut and phosphatased pAsuII to give pUL33ΔgalE. This plasmid contains the lacZ gene in the opposite transcription polarity to that of UL33. pUL33ΔgalE (2.5 μg) was linearized with BamHI and cotransfected with 20 μg of AD169-infected cell DNA into subconfluent monolayers of MRC5 cells by a modified calcium phosphate precipitation method (Chen and Okayama, 1987). Resulting recombinant viruses were identified by their blue-plaque phenotype, isolated, and plaque-purified as previously described for the UL18 and UL16 HCMV insertion mutants (Browne et al., 1992; Kaye et al., 1992). The correct insertion of lacZ sequences within the UL33 gene and the absence of contaminating wild-type virus were established by Southern hybridization (Sambrook et al., 1989). HCMV plaque assays were done as described elsewhere (Browne et al., 1992).
Preparation of UL33 antiserum and F(ab)2 fragments
Approximately 2 mg of a synthetic peptide corresponding to the predicted 17 carboxy-terminal amino acids of the protein encoded by UL33 (UL33 peptide, NH2-TKI-PHRLSQSHHNLSGV-CO2H) was coupled to 2 mg of key hole limpet hemocyanin (KLH; Pierce, Rockford, IL) via an EDC (1-ethyl-3-(dimethylaminopropyl) carbodiimide) bridge, according to the manufacturer (Imject kit; Pierce); this immunogen was named KLH-UL33. Also, 4 mg of UL33 peptide was crosslinked to itself with 0.1% glutaraldehyde, essentially as described (Collawn and Paterson, 1995), to create UL33-UL33. One female New Zealand white rabbit (Hazleton Research Products, Inc., Denver, PA) was immunized with a primary injection of 100 μg of KLH-UL33 in Freund’s complete adjuvant, followed by two similar boosts in Freund’s incomplete adjuvant (IFA) (Harlow and Lane, 1988). Subsequent boosts consisted of 40 μg of UL33-UL33 in IFA. Serum was collected from the immunized animal periodically after at least six UL33-UL33 booster injections and stored at −80° until use; this antiserum was called rabbit anti-33
F(ab)2 fragments were prepared as follows. The IgG fraction of the rabbit anti-33 antiserum was isolated by binding to a Fast-Flow Protein G Sepharose column (Pharmacia, Piscataway, NJ) in 20 mM sodium phosphate buffer (pH 7.3), washing with 1 M NaCl in 20 mM sodium phosphate buffer (pH 7.3), and eluting the IgG with 50 mM glycine–HCl (pH 3.0) in 5-ml fractions into tubes containing 500 μl of 1 M Tris–HCl (pH 7.5) (Harlow and Lane, 1988). Fractions containing IgG were identified by sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS–PAGE) and staining with Coomassie brilliant blue (CBB), and the amount of protein present in each was measured by the BCA protein assay (Pierce). IgG-containing fractions were pooled, dialyzed overnight against 200 mM sodium acetate (pH 4.0), and treated with 100 μg pepsin (Boehringer-Mannheim, Indianapolis, IN)/mg IgG for 4 hr at 37° (Harlow and Lane, 1988; Hildreth and Hyman, 1989). The preparation was neutralized with 0.05 vol of 1 M Tris–HCl (pH 7.5) and dialyzed overnight against calcium- and magnesium-free PBS (CMFPBS). Fc fragments and incompletely cleaved IgG mole cules were removed from the F(ab)2 preparation by their binding to protein A–Sepharose beads (Sigma, St. Louis, MO) in CMF-PBS. The remaining, unbound F(ab)2 fragments, called rabbit F(ab)2 Anti-33, were concentrated (Centricon 30; Amicon, Beverly, MA) and determined to be free of Fc fragments and IgG by SDS–PAGE and CBB staining.
SDS–PAGE, peptide comparisons, and Western immunoassays
SDS–PAGE was done essentially as described previously (Gibson, 1981b). Exceptions were using 10 mM dithiothreitol (DTT) instead of 10% β-mercaptoethanol in sample solubilizing buffer (Parker et al., 1993) and heating samples in a 42° water bath (instead of 100°) for 3 min (Parker et al., 1993), unless stated otherwise in the text. Protein separations were in 12% polyacrylamide gels, and peptide-separating, second-dimension gels were 18% polyacrylamide, crosslinked with diallyltartardiamide (DATD; Bio-Rad, Melville, NY) (Anker, 1970; Gibson, 1981b). Peptide comparisons were done by two-dimensional SDS–PAGE followed by Western immunoassay, as previously described (Schenk et al., 1991). Proteins were cleaved at tryptophan residues, after excising the lanes containing samples, by treatment in situ with N-chlorosuccinimide (NCS; Aldrich, Milwaukee, WI) (Lischwe and Ochs, 1982; Schenk et al., 1991). Immobilon-P membranes were stained for the presence of protein with 0.1% CBB in 50% acetic acid and 10% methanol, as indicated.
Western immunoassays were done essentially as described by Towbin et al. (1979), using rabbit anti-33 (1:40), rabbit anti-minor capsid protein (anti-miCP, 1:50) (Gibson et al., 1996), or mouse monoclonal anti-lower matrix protein (Anti-LM, 1:100; DuPont No. 9220), prepared in 5% BSA, 0.9% NaCl, 0.02% NaN3, and 10 mM Tris–HCl (pH 7.4) (Western blocking buffer), and detected with 125I-labeled protein A (Amersham, Arlington Heights, IL) (Welch et al., 1993). Specific electrotransfer conditions were: Immobilon-P membrane (Millipore, Bedford, MA); 20% methanol, 50 mM Tris buffer; and transfer time calculated from the equation: transfer time equals gel length × width × 2.5 mA per 0.5 hr (Welch et al., 1993). The mouse monoclonal antibody, anti-Tag (recognizes the T7-Tag epitope on the amino terminus of the UL33 protein coded in the construct pET-33; Novagen), was diluted 1:10,000 in Western blocking buffer and detected with rabbit anti-mouse IgG (10 μg/ml, Jackson ImmunoResearch, West Grove, PA), followed by 125I-labeled protein A. Fluorograms were prepared from the processed Immobilon-P sheets by using XAR-5 film (Eastman-Kodak, Rochester, NY) and a calcium-tungstate intensifying screen at −80° (Laskey and Mills, 1977).
Indirect immunofluorescence
Each chamber of an Erie SuperCell 8 chamber slide (VWR No. 48393–406) was seeded with ≈6.25 × 104 HFF cells or Sf9 cells. HFF cells were infected the next day with wild-type or Δ33 HCMV or left uninfected; Sf9 cells were infected with rBVs 20 min after seeding. Virus stocks were clarified by centrifugation at ≈8000 g for 1 min in a microfuge to pellet cellular debris before inoculating cultures. Immunofluorescence assays were done 8 days after infection with HCMV or 3 days after infection with rBVs.
Cells were fixed with 3% paraformaldehyde (Polysciences, Inc., Warrington, PA) in CMF-PBS and permeabilized with 0.5% Triton X-100 in TNgly (10 mM Tris–HCl (pH 7.4), 0.9% NaCl, and 10 mM glycine) (Machamer and Rose, 1988; Rose and Bergmann, 1982). All subsequent incubations were 35 min long at 25° and were followed by three washes with TNgly (Harlow and Lane, 1988). Slides were incubated with one of the following primary reagents, diluted in Western blocking buffer: nonspecific rabbit serum (1:40), nonspecific rabbit F(ab)2 (50 μg/ml), nonspecific rabbit Fc (25 μg/ml), rabbit anti-33 antiserum (1:40), rabbit F(ab)2 anti-33 (50 μg/ml), or mouse anti-Tag (1:100). Peptide competition experiments were done by adding a 1000-fold molar excess of either UL33 peptide or C1 peptide (Schenk et al., 1991) to diluted rabbit F(ab)2 anti-33, rocking the solution for 30 min at 25°, and subjecting the solution to centrifugation (≈8000 g, 25°, 1 min) to pellet insoluble material, all prior to use in the assay (Jemmerson and Hutchinson, 1990). The primary reagents were detected as specified, by incubation with goat IgG anti-rabbit (GAR)-lissamine rhodamine sulfonyl chloride (LRSC) (1:125), goat IgG anti-mouse (GAM)-fluorescein isothiocyanate (FITC) (1:50), goat F(ab)2 anti-rabbit F(ab)2 (GFARF)-LRSC (1:125), or goat F(ab)2 anti-rabbit Fc (GFARFc)-FITC (1:50). Each secondary reagent reacted species- or antibody-fragment-specifically, and neither fluorochrome exhibited significant cross-excitation. All nonspecific sera, nonspecific IgG subfragments, and secondary reagent conjugates were purchased from Jackson ImmunoResearch. Coverslips were attached to the processed slides with a drop of Mowiol mounting medium, containing Mowiol 4–88 (Calbiochem, San Diego, CA No. 475904) and the antifade agent DABCO (Sigma), prepared as described (Harlow and Lane, 1988), and cells were examined and photographed with an Olympus BH-2 microscope, equipped with a 40X SPlan objective lens, using Kodak Ektachrome 400 ASA color slide film
Preparation of cells and membranes and treatment with peptide N-glycosidase F
HCMV-infected, HCMV Δ33-infected, or noninfected HFF cells were recovered from 32-oz glass bottles (≈5 × 107 cells/bottle) (Gibson, 1981b) 8 days after infection, resuspended in CMF-PBS (1 ml/bottle), and frozen at −80°. rBV-infected cells were prepared similarly (≈6 × 106 cells/1 ml of CMF-PBS) and frozen at −80°. Cellular membranes were prepared from fresh or0frozen cells (Sung et al., 1991) that had been disrupted by forcing through a 27-gauge needle (Parker et al., 1993).
For peptide N-glycosidase F (PNGase F) treatment, membrane preparations (20 μg of protein) were first incubated with 1/5 vol of freshly prepared 60 mM DTT, 12% SDS, and 300 mM Tris–HCl (pH 7.0) (i.e., 6× solubilizing buffer without glycerol or bromophenol blue) at 42° for 5 min. Pelleted virus particles were solubilized in 60 μl of fresh 2× solubilizing buffer, diluted 1:1 with water, and warmed as above. Solubilized membranes and virus particles were combined with 1/5 vol of 6× PNGase “B” buffer, consisting of 90 mM EDTA, 20% Nonidet P-40 (NP-40), 24 mM o-phenanthriline, 6 mM PMSF, and 450 mM glycine–HCl (pH 9.3), and incubated with or without 200 mU PNGase F (Boehringer-Mannheim No. 1365–169) overnight at 37° (Haltiwanger and Hart, 1993; Sung et al., 1991; Tarentino and Plummer, 1994). Treated samples were then prepared for SDS–PAGE and Western immunoassay by adding 1/3 vol of 4× solubilizing buffer.
RESULTS
Expression of the UL33 protein from recombinant baculoviruses
We began this work by first attempting to clone and identify the UL33 protein in a high-expression system. We used the recombinant baculovirus system (O’Reilly et al., 1992) because of its ability to carry out many post-translational protein modifications and because it has been used successfully to express other cloned GCRs (Mouillac et al., 1992; Parker et al., 1993; Zastawny et al., 1994). We used the rabbit anti-33 anti-peptide antiserum to detect the protein by Western immunoassay and immunofluorescence. We made and compared two rBVs, one expressing the UL33 ORF (rBV-33, producing recombinant UL33 protein, pUL33) and one expressing pUL33 with an 11-amino-acid epitope (T7 gene 10 sequence, detectable with a commercial mouse monoclonal antibody, anti-Tag) (Lutz-Freyermuth et al., 1990; Tsai et al., Keene, 1992) fused to its amino end (rBV-Tag33, producing pTag33). Construction and use of these reagents is described under Materials and Methods.
Sf9 cells were infected with the recombinant baculoviruses rBV-33 or rBV-Tag33, harvested in 2× SDS–PAGE sample buffer 3 days later, and analyzed by SDS–PAGE and Western immunoassays (Fig. 1). Two sets of samples were analyzed in parallel; one was probed with rabbit anti-33 (Fig. 1, lanes 1–6) and the other with mouse anti-Tag (Fig. 1, lanes 7–12). rBV-33 expressed ≈36-kDa protein (Fig. 1, lane 2), reactive with the anti-33 antiserum. rBV-Tag33 also expressed a protein that was immunoreactive with the anti-33 antiserum, but its size was slightly larger (i.e., ≈39 kDa, circle in Fig. 1, lane 3) due to the 11-amino-acid epitope added to its amino end. When heated in a boiling water bath, as usual prior to SDS–PAGE (Fig. 1, lanes 4–6 and 10–12), both pUL33 and pTag33 formed high-molecular-weight aggregates that barely entered the resolving gel (e.g., Fig. 1, lanes 5 and 6), a phenomenon observed with other GCRs and highly hydrophobic proteins analyzed by SDS–PAGE (Semenza et al., 1990). Only pTag33, whether monomeric (circle, Fig. 1, lane 9) or heat-aggregated (Fig. 1, lane 12), reacted with the anti-Tag antibody, confirming its identity as the recombinant pTag33. The higher-molecular-weight immunoreactive material (e.g., see arrow, Fig. 1) is most likely multimers of pUL33 and pTag33 that did not dissociate during SDS–PAGE (Sung et al., 1991). No similar protein was detected in wild-type BV-infected Sf9 cells (BV; lanes 1, 4, 7, 10). The presence of this protein only in rBV-33-infected cells, its reaction with the anti-33 antiserum, and its size difference and reaction with the anti-Tag antibody when fused with the 11-amino-acid epitope establish its identity as pUL33.
FIG. 1.

Expression of recombinant pUL33. Western immunoassay shows proteins produced by Sf9 cells that were infected with wild-type baculovirus (BV; lanes 1, 4, 7, and 10), rBV-33 (lanes 2, 5, 8, and 11), or rBV-Tag33 (lanes 3, 6, 9, and 12) and harvested 3 days after infection. Whole cell lysates were prepared in solubilizing buffer and not heated (lanes 1–3, 7–9) or heated in a boiling water bath (lanes 4–6, 10–12) prior to SDS–PAGE. Western immunoassays were done with the anti-33 antiserum or anti-Tag antibody, as indicated. Circles are shown next to the Tag33 protein, detected by both anti-33 and anti-Tag. Arrow indicates GCR multimers. Molecular weight (in kDa) was estimated from molecular weight markers included during SDS–PAGE (Mark 12; Novex, San Diego, CA) that were visualized by staining that portion of the Immobilon-P with Coomassie brilliant blue (CBB) immediately following electrotransfer.
pUL33 expression from a recombinant baculovirus
To investigate the localization of the recombinant UL33 protein, the previously characterized anti-33 antiserum and anti-Tag antibody (Fig. 1) were used in immunofluorescence assays of rBV-infected insect cells. Sf9 cells were infected with either wild-type baculovirus (BV), rBV-33, or rBV-Tag33 and 3 days later were subjected to immunofluorescence assays with the anti-33 antiserum or anti-Tag antibody (Fig. 2). rBV-33-infected cells showed a pattern of generalized extranuclear fluorescence (Fig. 2A) that was absent from BV-infected cells assayed with the rabbit anti-33 antiserum (Fig. 2B). rBV-Tag33-infected cells show the same pattern of fluorescence with both anti-33 and anti-Tag antibodies (Figs. 2C and 2D, respectively). The anti-Tag antibody also did not react with BV-infected cells (Fig. 2E). The unequivocal identification of pTag33 by its unique epitope, and its coincident distribution, when visualized with the anti-33 or anti-Tag antibodies, establish both the specificity of the anti-33 antiserum for pUL33 and the nonnuclear localization of pUL33.
FIG. 2.

Recombinant pUL33 expression in rBV-infected Sf9 cells shown by indirect immunofluorescence. Bound primary antibodies, anti-33 or anti-Tag, were detected by indirect immunofluorescence after incubation with either GAR-LRSC (for rabbit anti-33) or GAM-FITC (for mouse anti-Tag) as secondary reagents. See text for abbreviations. (A) rBV-33-infected cells assayed with rabbit anti-33, as primary, and GAR-LRSC, as secondary, antibodies. (B) BV-infected cells assayed with rabbit anti-33, as primary, and GAR-LRSC, as secondary, antibodies. (C and D) rBV-Tag33-infected cells assayed with both rabbit anti-33 and mouse anti-Tag, as primary reagents and with both GAR-LRSC and GAM-FITC, as secondary reagents. (E) rBV-infected cells assayed with anti-Tag antibody, as primary reagent, and GAM-FITC, as secondary antibody. LRSC fluorescence is shown in A–C, FITC fluorescence in D and E.
FIG. 3. GCR33 localizes to the region of the HCMV-infected cell cytoplasmic inclusion. HFFs were infected with wild-type (strain AD169) HCMV (WT; A, D–K, M, and N), HCMV Δ33 (Δ33; B and L), or not infected (Mock; C) and processed for indirect immunofluorescence. Primary reagents used were rabbit F(ab)2 anti-33 (A–C, E, and F), nonspecific rabbit F(ab)2 (D), nonspecific rabbit serum (G and H), nonspecific rabbit Fc (L), a mixture of nonspecific rabbit Fc and rabbit F(ab)2 anti-33 (J and K), C1 peptide-competed rabbit F(ab)2 anti-33 (M), or UL33 peptide-competed rabbit F(ab)2 anti-33 (N). Secondary reagents used were GFARF-LRSC (A–D, M, and N), GFARFc-FITC (L), or a mix of both (E–K). LRSC fluorescence is shown in A–E, G, K, M, and N, FITC fluorescence in F, H, J, and L.
GCR33 localizes to cytoplasmic inclusion structures in HCMV-infected cells
Having verified the specificity of the anti-33 antiserum and determined some properties of pUL33 expressed in rBVs, we next attempted to identify the UL33 protein in HCMV-infected fibroblasts. The UL33 protein expressed in HCMV-infected cells was called GCR33, to distinguish it from the recombinant form of the protein, pUL33. Immunofluorescence was done first as a way of verifying that GCR33 is expressed in detectable amounts during infection. Because IgG nonspecifically binds to cytoplasmic inclusion (CI) structures in HCMV-infected cells via the Fc portion of the antibody molecule (Furukawa et al., 1975; Keller et al., 1976), immunofluorescence was done using F(ab)2 fragments (prepared from the anti-33 antiserum as described under Materials and Methods) and F(ab)2-fluorochrome conjugates of the secondary antibodies.
Results of the experiment are summarized in Fig. 3. The rabbit F(ab)2 anti-33 fragments showed a pattern of fluorescence that localized to the region of the CI in wild-type HCMV-infected fibroblasts (Fig. 3A). Fluorescence in cells infected with the UL33 mutant virus, Δ33 (Fig. 3B), or in noninfected cells (Fig. 3C) was essentially at the level of background. This infected-cell-specific fluorescence was not due to nonspecific binding of F(ab)2 fragments (Fig. 3D); nor was it due to binding of residual Fc in the rabbit F(ab)2 anti-33 preparation, as evidenced by detection with an anti-F(ab)2 reagent (Fig. 3E) but not with an anti-Fc reagent (Fig. 3F). The typical pattern of CI fluorescence by nonspecific IgG binding in wild-type-infected cells is shown for comparison (Figs. 3G and 3H). The juxtanuclear location and generally spherical shape of the nonspecific, Fc-binding CIs (Fig. 3J) appeared to be the same as the pattern of specific binding by the rabbit F(ab)2 anti-33 fragments, shown in the same field (Fig. 3K). Nonspecific Fc binding is also shown in Δ33-infected cells (Fig. 3L), demonstrating that although there is no GCR33 in these cells, Δ33 still produces an Fc-binding component in the CIs during infection. This last control also shows that if Fc were present and the cause of reactivity in Figs. 3A and 3E, it would have been detected with the anti-Fc-FITC reagent in Fig. 3F.
The specificity of the interaction seen in Fig. 3A was also established by peptide competition experiments. An unrelated 21-amino-acid peptide, C1 (Schenk et al., 1991), was unable to compete rabbit anti-33 F(ab)2 binding at a 1,000-fold molar excess (Fig. 3M), but the 17-amino-acid UL33 peptide at the same concentration completely blocked the interaction (Fig. 3N). Together, these results show that the rabbit F(ab)2 anti-33 preparation specifically reacts with GCR33 in immunofluorescence experiments, that GCR33 localizes to the CI region of HCMV-infected cells, and that GCR33 is not responsible for the Fc binding that defines the region of the infected-cell CIs.
GCR33 is ≈43 kDa and exhibits heterogeneous asparagine-linked glycosylation (N-glycosylation) in HCMV-infected HFFs
We next used Western immunoassays to identify the protein by size. Initial experiments with whole cell lysates gave high backgrounds of nonspecific immunoreactivity, so we used membrane preparations as an alternate and potentially enriched source of material (see Materials and Methods). Membrane preparations (20 μg protein) from cells infected with wild-type HCMV or with Δ33, or from noninfected cells, were subjected to SDS–PAGE followed by Western immunoassay using anti-33 (Fig. 4, “Membranes”). Immunoreactive material (see asterisked bar, Fig. 4, lane 2), was detected in the preparation from WT-infected cells, but not in those from Δ33-infected (Fig. 4, lane 3) or noninfected (Fig. 4, lane 1) cells. The broad size range of the immunoreactive material suggested that it was heterogeneously glycosylated or aggregated, or both. To test for the presence of asparagine-linked oligosaccharides, an amount of sample equal to that used in lanes 1–3 was treated with PNGase F (Tarentino and Plummer, 1994). This treatment yielded a strong new band in the WT HCMV-infected cell preparation (Fig. 4, lane 5) that was absent from both the Δ33-infected and noninfected cell preparations (Fig. 4, lanes 6 and 4, respectively). The ≈43-kDa size of this band approximates the computer-predicted molecular weight of the protein encoded by UL33. The generally increased level of immunoreactivity observed in the PNGase F-treated HCMV-infected cell preparation (Fig. 4, lane 5) likely results from better electrotransfer of the 43-kDa deglycosylated protein from the gel, compared to the higher-molecular-weight glycosylated form (Fig. 4, lane 2) (Gibson, 1981a), or to comparatively better retention of the deglycosylated form on the Immobilon-P membrane (Gibson, 1981a), or both. We attribute the small amount of large, electrophoretically heterogeneous material that remained after PNGase F treatment (Fig. 4, lane 5; also see Fig. 7, lane 9) to incompletely deglycosylated GCR33 or the tendency of the protein to aggregate, or both.
FIG. 4.

HCMV-infected HFF cells express glycosylated GCR33. HFF cells were infected with WT HCMV (WT-Inf.; lanes 2, 5, and 8), HCMV Δ33 (Δ33-Inf.; lanes 3, 6, and 9), or not infected (Mock; lanes 1, 4, and 7). Total cell membranes (Membranes) or whole cells (Cells) were isolated 8 days after infection and incubated with or without PNGase F (+/− PNGase). Membrane protein (20 μg) or whole cell protein (15 μg) was subjected to SDS–PAGE and analyzed by Western immunoassay; the Membranes blot was probed with rabbit anti-33, and the cells blot was probed with an antiserum to the basic phosphoprotein (BPP, protein product of HCMV UL32 ORF) (Greis et al., 1994). The asterisked bar represents a molecular weight range of ≈58 to ≈100 kDa; molecular weight (in kDa) was estimated from molecular weight markers included during SDS–PAGE that were visualized by staining that portion of the Immobilon-P with CBB immediately following electrotransfer.
FIG. 7.

Size comparison of different forms of the protein encoded by UL33. Sf9 cells infected with WT baculovirus (BV; lane 1), rBV-33 (lane 2), or rBV-N33 (lanes 3, 4, and 5) were harvested 3 days after infection; membranes from noninfected (Mock; lanes 6 and 7) or WT HCMV-infected (WT-Inf.; lanes 8 and 9) HFFs were prepared and treated or not treated with PNGase F (+/− PNGase). Lane 4 (rBV-N33 1:1) is a twofold dilution, and lane 3 (rBV-N33 1:3) a fourfold dilution, of the material in lane 5. All samples were then analyzed by SDS–PAGE and Western immunoassay with the anti-33 antiserum. Circles indicate the three forms of the protein encoded by UL33. The asterisked bar indicates a broad molecular weight range of glycosylated GCR33 and aggregates of GCR33 that are visible near the top of the gel; molecular weights (in kDa) were estimated from molecular weight markers included during SDS–PAGE that were visualized by staining that portion of the Immobilon-P with CBB immediately following electrotransfer.
Whole cell lysates (Fig. 4, “Cells”) were also prepared (15 μg protein per lane) and subjected to SDS–PAGE in the same gel. Western immunoassays were done on these lysates to establish that the inability to detect a UL33 protein in Δ33-infected cells was not due to a generally lower amount of protein in those preparations, compared to WT-infected cells. An anti-peptide antiserum to the HCMV basic phosphoprotein (BPP, pp150, 149 kDa, product of UL32 ORF) (Greis et al., 1994) was used and showed BPP in both Δ33- and WT-infected HFFs (Fig. 4, lanes 8 and 9, respectively), indicating that (i) the Δ33 infections had progressed to approximately the same extent and (ii) insertion of lacZ into the UL33 ORF was not deleterious to expression of the adjacent UL32 ORF. We have not further investigated the possible over-expression of BPP in Δ33-infected cells (i.e., see Fig. 4; compare lane 8 with lane 9). These data indicate that GCR33 is a heterogeneously N-glycosylated protein in HCMV-infected cells.
HCMV-infected cell-specific GCR33 shows peptide similarities to rBV-expressed pUL33
Peptide comparisons were used to establish that the anti-33-reactive, infected-cell-specific protein (Fig. 4, lanes 2 and 5) is GCR33. Membranes from HCMV-infected HFF cells (treated or not treated with PNGase F) or rBV-33-infected insect cells were subjected to SDS–PAGE, followed by in situ protein cleavage with NCS, separation of the resulting peptides in a second-dimension gel, and finally Western immunoassay with anti-33 to identify related peptides, all as explained under Methods and Materials. All three preparations gave rise to five comigrating, immunoreactive fragments (Fig. 5, numbered 1–5). Peptides 4 and 5, although not well demonstrated in this figure (Fig. 5, circles, lanes 1 and 2), were readily detected in longer exposures of this immunoassay. Correspondence of the peptide patterns for HCMV-infected-cell GCR33 (with or without PNGase F treatment) and rBV-33-infected-cell pUL33 provides direct evidence that the anti-33 reactive HCMV-infected-cell protein is the product of the UL33 ORF.
FIG. 5.
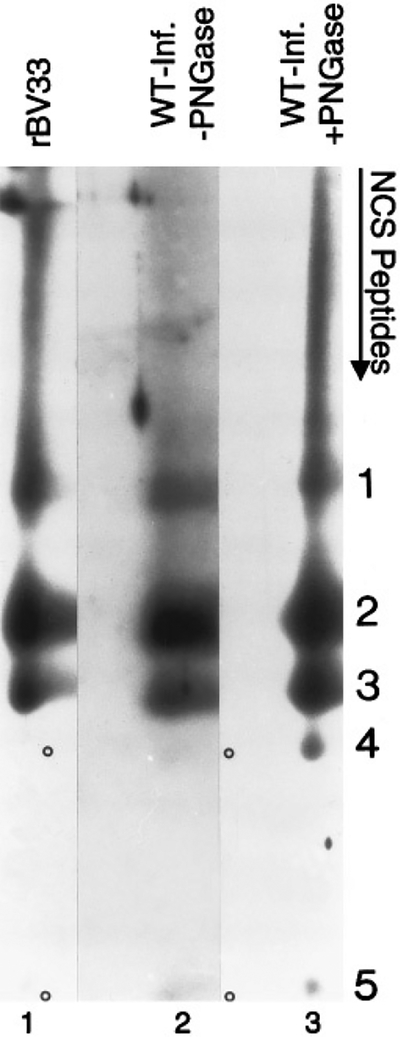
Peptide comparisons show similarities between rBV (pUL33)- and HCMV-infected cell (GCR33) UL33 proteins. Samples of rBV-33-infected Sf9 cell proteins (lane 1) and membrane preparations from WT HCMV-infected HFF cells without (lane 2) or with (lane 3) PNGase F treatment were subjected to first-dimension SDS–PAGE in a single 12% polyacrylamide gel. The region of each sample spanning the size range of GCR33 and pUL33 was excised from the gel, treated with NCS to cleave the proteins in situ, and the resulting peptides were compared by SDS–PAGE in an 18% DATD-crosslinked polyacrylamide gel by electrophoresis perpendicular to the original direction of migration (i.e., top to bottom), all as described under Materials and Methods. Peptides in the second-dimension gel were detected by Western immunoassay with the anti-33 antiserum (to the carboxyl end of UL33 protein). Immunoreactive peptides are labeled 1 to 5. Circles indicate peptides 4 and 5 in lanes 1 and 2.
HCMV enveloped virus particles contain glycosylated GCR33
Because GCRs are membrane proteins, enveloped virus particles were examined for the presence of GCR33 by SDS–PAGE and Western immunoassay. HCMV noninfectious enveloped particles (NIEP or N), virions (VIR or V), and dense bodies (DB or D) were prepared from WT HCMV-infected HFFs (WT) or from Δ33-infected (Δ33) HFFs. A small portion of the pellet from each particle type was either treated or not treated with peptide N-glycosidase F (+/− PNGase) and then tested by SDS–PAGE and Western immunoassay with the anti-33 antiserum (Fig. 6, anti-33). Although two background bands are present in all samples, detracting from the data, the anti-33 antiserum specifically reacted with material in all three WT HCMV enveloped particles (indicated by circles, Fig. 6, lanes 5, 9, and 13) but in none of the corresponding Δ33 particles (Fig. 6, lanes 7, 11, and 15). The diffuse pattern of anti-33 reactivity observed in the WT HCMV particles (Fig. 6, circles, lanes 5, 9, and 13) was consistent with that seen in membrane preparations from WT-infected cells (Fig. 4, lane 2; Fig. 6, lane 3). Also as observed with the membrane preparations (Fig. 6, lane 4), treatment of the WT particles with PNGase F (Fig. 6, lanes 6, 10, and 14) decreased the heterogeneity and size of the immunoreactive material to approximate the computer-predicted 43-kDa molecular weight of the protein product of UL33.
FIG. 6.

HCMV enveloped particles contain glycosylated GCR33. Cell membranes were isolated from noninfected (Mock; lanes 1 and 2) or WT HCMV-infected (WT; lanes 3 and 4) HFF cells. WT HCMV (WT; lanes 5, 6, 9, 10, 13, 14, 17, 19, and 21) or HCMV Δ33 (Δ33; lanes 7, 8, 11, 12, 15, 16, 18, 20, and 22) enveloped extracellular virus particles were used in these experiments; NIEPs (lanes 5–8, 17, and 18), virions (lanes 9–12, 19, and 20), or dense bodies (lanes 13–16, 21, and 22) were purified from infected cells, treated or not treated with PNGase F (+/− PNGase), and subjected to SDS–PAGE, followed by Western immunoassay with rabbit anti-33. Circles (lanes 5, 9, and 13) indicate a broad, immunoreactive band of material in WT NIEPs, virions, and dense bodies. Another set of samples, not treated with PNGase F (lanes 17–22), was heated in a boiling water bath prior to SDS–PAGE in the same gel and then subjected to Western immunoassay with a mixture of anti-miCP (to minor capsid protein) and anti-LM (to abundant lower matrix tegument protein). The positions of the minor capsid protein (miCP) and lower matrix protein (LM) are indicated; molecular weight (in kDa) was estimated from molecular weight markers included during SDS–PAGE that were visualized by staining that portion of the Immobilon-P with CBB immediately following electrotransfer.
As part of this experiment, a similar portion of each of the samples described above was solubilized more completely by heating in a boiling water bath prior to SDS–PAGE and was subjected to electrophoresis in the same gel. A Western immunoassay was done on these samples with a mixture of a rabbit anti-peptide antiserum, called anti-miCP, against a capsid component, called the “minor capsid protein” (miCP, pUL85), and a mouse monoclonal antibody, called anti-LM, against an abundant tegument phosphoprotein called the “lower matrix protein” (LM or pp65, pUL83) (Fig. 6, anti-miCP/anti-LM). The reactivity of these antibodies with Δ33 particle preparations (Fig. 6, lanes 18, 20, and 22) and WT particle preparations (Fig. 6, lanes 17, 19, and 21) demonstrated that the presence of the miCP and LM proteins was qualitatively the same in both sets of particle preparations. Therefore, the absence of anti-33 reactivity with Δ33 particles seen in the anti-33 immunoassay (Fig. 6, lanes 5–16) was not due to a significantly reduced amount of Δ33 virus particles. The ratio of LM to miCP differed, however, between the WT and Δ33 particles, suggesting either that the Δ33 NIEP and virion preparations were partially contaminated with dense bodies or that the absence of GCR33 in some way altered the protein composition of these Δ33 particles. The anti-miCP/anti-LM panel also demonstrates that the dense body preparations (Fig. 6, lanes 21 and 22) contained comparatively large amounts of LM but no miCP, as expected for these particles whose protein composition is approximately 90% LM and which contain no capsid structure or capsid protein constituents (Gibson, 1993; Irmiere and Gibson, 1983). These experiments show that GCR33 is present in all three types of WT HCMV enveloped particles.
GCR33 from HCMV-infected cells and virus particles is larger than proteins expressed from UL33 or “spliced UL33”
Recent work showed that one of the mRNAs generated from UL33 during HCMV infection is a spliced transcript (Davis-Poynter et al., 1995). This spliced mRNA would code for a variant of GCR33 extending 22 amino acids beyond the amino terminus of the protein encoded by the originally defined ORF (Chee et al., 1990b). Because the recombinant pUL33, which lacks this amino extension, migrates with a smaller apparent molecular weight than the deglycosylated GCR33 from WT HCMV-infected cells during SDS–PAGE, the possibility was tested that this discrepancy was due to the presence of an amino-terminal extension on the protein made in WT HCMV-infected cells. An rBV (rBV-N33) was engineered that encodes the 22-amino-acid-extended splice variant of pUL33, called pUL33-XTND, for this purpose.
The proteins produced by wild-type baculovirus (BV), rBV-33, rBV-N33, and wild-type HCMV were then compared by SDS–PAGE and Western immunoassay using the anti-33 antiserum (Fig. 7). Membrane preparations from WT HCMV-infected (WT-Inf.) or noninfected (Mock) cells were treated or not treated with PNGase F (+/− PNGase). As expected, pUL33-XTND (produced by rBV-N33, Fig. 7, lanes 3, 4, and 5, circle) had a larger apparent molecular weight (i.e., ≈39 kDa, versus ≈36 kDa) than the original pUL33 (Fig. 7, lane 2, circle). The glycosylated and deglycosylated GCR33 (Fig. 7, lane 8, and Fig. 7, circle, lane 9, respectively) migrated as in Fig. 4, with apparent molecular weights of ≥58 and ≈43 kDa, respectively. No similar immunoreactive material was de tected in WT baculovirus-infected cells (Fig. 7, BV, lane 1) or noninfected HFFs (Fig. 7, lanes 6 and 7). Thus, the amino extension of pUL33-XTND does not fully account for the discrepancy between the observed sizes of pUL33 and deglycosylated GCR33.
UL33 ORF is not essential for HCMV growth in cell culture
To determine whether UL33 is required for HCMV replication in cell culture, we disrupted the UL33 gene by insertional mutagenesis (with the lacZ gene) and introduced the mutant gene into the HCMV genome by homologous recombination, as described under Materials and Methods. A viable mutant, called Δ33, was selected on the basis of its blue-plaque phenotype. The time course of replication for Δ33 and WT HCMV were compared under single-cycle growth conditions (Fig. 8). The results showed that Δ33 yields approximately the same titer of progeny virus by 4 to 6 days after infection, but Δ33 titers were about 10-fold lower than those of WT HCMV at the four earlier time points (i.e., 16, 24, 48, and 72 hr after infection).
FIG. 8.

The UL33 ORF is dispensable for growth in cell culture. Monolayers of MRC5 cells were infected with WT HCMV or Δ33 viruses at a multiplicity of infection of 5 and washed three times with medium containing 10% FCS after adsorption. Supernatant virus was collected at 16, 24, 48, 72, 96, and 144 hr after infection and was assayed by plaquing on a fresh monolayer of MRC5 cells (Browne et al., 1992; Kaye et al., 1992). Duplicate assays were performed from duplicate infections for each time point. Shown here is a one-step growth curve; standard errors were not greater than 6% between the two sets of measurements that were averaged.
DISCUSSION
The primary findings in this report are that the HCMV GCR, GCR33 (i) is expressed from recombinant baculovirus in insect cells as a ≈36-kDa, heat-aggregatable protein (pUL33) that is localized to nonnuclear regions of the cell, (ii) is present in HCMV-infected HFFs as a heterogeneously N-glycosylated protein that localizes to the same region as the Fc-binding cytoplasmic inclusion, (iii) is also present in three types of HCMV enveloped virus particles, and (iv) is not essential for replication of HCMV in cell culture.
We have shown that pUL33 can be expressed from rBVs in insect cells. Both the wild-type and T7-epitope-tagged forms (Kolodziej and Young, 1991) were excluded from the nucleus, but a more precise localization was not achieved due to the small cytoplasmic space in these spherical Sf9 cells. rBV-encoded and in vitro-translated pUL33 (data not shown) comigrated during SDS–PAGE, with an estimated size of 36 kDa (smaller than the computer-predicted 43 kDa). The electrophoretic mobility of the rBV product was not affected by treatment with PNGase F (data not shown), indicating that it is not appreciably glycosylated in insect cells. This finding was surprising given that GCRs are typically N-glycosylated (Fraser et al., 1994) and that insect cells are competent for the initial steps of this post-translational modification (O’Reilly et al., 1992); the lack of glycosylation may relate to interference with cellular N-glycosylation observed during the late stages of baculovirus infection (Jarvis and Summers, 1989). Like other GCRs and highly hydrophobic proteins (Semenza et al., 1990), pUL33 is heat sensitive and aggregated when heated above 60°; aggregation was to the extent that essentially none of the protein was detected in the resolving gel when the sample was heated with solubilizing buffer in a boiling water bath.
More importantly, we have identified the UL33 GCR protein, GCR33, in HCMV-infected fibroblasts. Unlike recombinant pUL33, GCR33 in HCMV-infected cells was larger and more heterogeneous in size than the computer-predicted 43 kDa, due to asparagine-linked glycosylation. Treatment with PNGase F reduced the size to ≈43 kDa, close to the predicted size, but larger than the ≈36-kDa size of rBV pUL33. This mobility difference could be due to several causes. First, infected-cell GCR33 may bear additional modifications (Fraser et al., 1994) that are absent from the rBV product and possibly produced by infected-cell-specific enzymes. If such differences are present, they are not likely to be at the carboxyl end of GCR33 because peptide comparisons revealed no mobility differences among the five carboxy-coterminal peptides detected (Fig. 5). Second, the recombinant protein may have an increased electrophoretic mobility due to misfolding (e.g., intramolecular hydrophobic interactions of transmembrane domains) or incomplete denaturation. A third explanation is that the in fected-cell protein is actually larger. This possibility is attractive in view of the recent finding that the UL33 mRNA exists in an additional spliced form (Davis-Poynter et al., 1995) that would give rise to a GCR33 that is 22 amino acids longer at its amino end than rBV pUL33. To test this last explanation, we cloned and expressed the ORF that would code for this splice variant from an rBV and found that, although larger than pUL33, as expected, it still migrated somewhat faster than deglycosylated GCR33 during SDS-PAGE (Fig. 7). Thus, the protein detected in HCMV-infected HFFs differs in some way from the proteins expressed from both the spliced and nonspliced recombinant forms of UL33 tested here.
Glycosylated GCR33 is also present in virions and two types of enveloped virus particles called noninfectious enveloped particles and dense bodies. Because dense bodies are composed primarily of lower matrix protein surrounded by the viral envelope (Gibson, 1993; Irmiere and Gibson, 1983), and GCRs are characteristically membrane proteins, this finding is consistent with GCR33 being a virus envelope glycoprotein. The presence of GCR33 in the viral envelope would allow for it being carried to cells with the virus and being deposited in the cellular plasma membrane with the rest of the viral envelope during entry. If introduced into the cell membrane by this mechanism, a functionally competent HCMV GCR could hypothetically induce the signaling changes observed in fibroblasts immediately following HCMV infection (Albrecht et al., 1991), as suggested previously (Welch et al., 1991).
As shown in Fig. 3, GCR33 localizes to the region of the cytoplasmic inclusions (CIs). This result is intriguing, considering that GCRs normally appear at the cell surface (Zastawny et al., 1994). Several explanations could account for the predominant CI localization of GCR33. First, its distribution may not be accurately reflected by immunofluorescence. For example, plasma membrane-localized GCR33 may be partially extracted or denatured during fixation and permeabilization; it may not be well presented for the antibodies to react; or it may be as abundant as it is in CIs, but less regionally concentrated, and hence less readily detected. A second explanation is that GCR33 has an intracellular function, perhaps in addition to its surface role. HSV-1 glycoprotein gK offers a possible parallel in that it appears to localize predominantly intracellularly by immunofluorescence and yet is involved in syncytia formation, suggesting a surface location (Hutchinson et al., 1995). Third, if GCR33 is a functional receptor, it may be internally sequestered as a means of downregulating its activity (Fraser et al., 1994; Saffitz and Liggett, 1992; Zastawny et al., 1994); such a sequestration could hypothetically involve translocation of GCR33 from the cell surface to the CIs. A fourth explanation, based on the presence of GCR33 in enveloped virus particles, is that the protein may be targeted to this region to be incorporated into maturing virions.
A ligand for GCR33 has not yet been identified, but the protein products from both its nonspliced and especially its spliced mRNA show homology with known β-chemokine receptors (see FASTA scores, Table 1) (Kolakowski, 1994). The longer amino terminus of the protein encoded by the spliced mRNA, GCR33-XTND (Davis-Poynter et al., 1995), would contain four more asparagine-linked glycosylation sites than GCR33, one more cysteine, and would have a slightly acidic charge, all characteristics of the amino termini of chemokine receptors (Ahuja and Murphy, 1993; Horuk, 1994; Murphy, 1994a,b). Also, sequences in the second transmembrane domain and third transmembrane domain extending into the second intracellular loop are 55% homologous (18% identical) to amino acid stretches found in those regions of chemokine receptors (Charo et al., 1994). Thus, either the spliced or nonspliced mRNA from UL33 may encode a chemokine receptor (Table 1).
TABLE 1.
FASTA Comparison of GCR33, Other Viral GCRs, and Representative Chemokine Receptors
| Class | Species | ORF/Receptora | Closest relative (class)b | Scorec |
|---|---|---|---|---|
| β-Herpes | HCMV | GCR33 | RANTES (β) | 313 |
| β-Herpes | HCMV | GCR33-XTND | RANTES (β) | 313 |
| β-Herpes | HCMV | US27 | RANTES (β) | 420 |
| β-Herpes | HCMV | US28 | RANTES (β) | 562 |
| β-Herpes | HCMV | UL78 | f-MLP (N/A) | 157 |
| β-Herpes | HHV-6 | U12 | RANTES (β) | 245 |
| β-Herpes | HHV-6 | U51 | μ-opioid (N/A) | 182 |
| β-Herpes | HHV-7 | ORF 17 | RANTES (β) | 224 |
| β-Herpes | HHV-7 | ORF 57 | IL-8RB (α) | 185 |
| (γ-Herpes) | (Human) | EBI-1 | IL-8RB (α) | 681 |
| (γ-Herpes) | (Human) | EBI-2 | RANTES (β) | 533 |
| γ-Herpes | EHV-2 | E1 | RANTES (β) | 1087 |
| γ-Herpes | EHV-2 | 74 | RANTES (β) | 312 |
| γ-Herpes | HVS | ECRF3 | IL-8RB (α) | 428 |
| Poxvirus | Swinepox | K2R | RANTES (β) | 573 |
| Poxvirus | Capripox | Q2/3L | RANTES (β) | 705 |
The ORF or the receptor is given by locus number or common receptor name (Attwood and Findlay, 1994; Birkenbach et al., 1993; Chee et al., 1990a; Davis-Poynter et al., 1995; Gompels et al., 1995; Horuk, 1994; Massung et al., 1993; Murphy, 1994a; Nicholas et al., 1992; Telford et al., 1995). The EBI-1 and −2 ORFs are human genes induced by EBV, HHV-6, and HHV-7 infection.
The most closely related human GCR of known function is listed. The chemokine receptor class for each relative is also shown in parentheses. HCMV UL78 and HHV-6 U51 are marked N/A because the closest relatives are not chemokine receptors.
Optimized FASTA score of closest relative. Scores above 100 are considered significant.
Other viral GCRs also show similarities to chemokine receptors. Both HCMV US28 and herpesvirus saimiri ECRF3 (Nicholas et al., 1992) encode functional chemokine receptors (Ahuja and Murphy, 1993; Gao and Murphy, 1994; Kuhn et al., 1995; Neote et al., 1993; Vieira et al., 1995). Human herpesviruses 6 and 7 (HHV-6 and −7), other members of the human β-herpesvirus group like HCMV, encode GCR homologues related to chemokine receptors (Gompels et al., 1995) (Table 1), and the rodent β herpesviruses murine CMV and rat CMV encode positionally conserved homologues of UL33 (Beisser et al., 1995; Davis-Poynter et al., 1995). In addition, homologues of chemokine receptors are encoded by the Epstein–Barr virus-induced cellular GCR mRNAs (Birkenbach et al., 1993), one of which is also induced by HHV-6 and −7 (Hasegawa et al., 1994) (Table 1), by the genome of the recently described human γ herpesvirus, Kaposi’s sarcoma herpes virus (HHV-8) (Chang et al., 1994), and by the genome of equine herpesvirus 2 (Telford et al., 1995) (Table 1). The conservation of these ORFs among the β and γ herpesviruses implies that these GCR homologues may have important functions during infection. Recently, swinepox and capripox viruses have also been shown to encode chemokine receptor homologues (Cao et al., 1995; Massung et al., 1993) (Table 1), suggesting that acquisition of genes encoding such proteins that are capable of utilizing molecules normally employed by the immune system may give these large DNA viruses a survival advantage.
Given that the HCMV US27, US28, and UL33, either individually or in combination, are all dispensable for virus replication in cell culture (Jones et al., 1995), it will be interesting and important to determine their biological function and clinical relevance.
ACKNOWLEDGMENTS
We thank Jenny Borchelt and Rebecca Magno for their excellent technical assistance and James Hildreth for help in preparing the rabbit F(ab)2 anti-33 fragments. We also thank Phil Murphy, Frank Kolakowski, Terri Attwood, and John Nicholas for helpful discussions on GCR homology. John Nicholas also provided the protein sequences of the HHV-7 GCR homologues. B.J.M. was a student in the Biochemistry, Cellular, and Molecular Biology Training Program and was supported, in part, by Grant GM07445 from USPHS; H.B. is a postdoctoral fellow in the laboratory of Tony Minson. This work was supported in part by USPHS Grants AI13718 and AI22711 to W.G. and by Wellcome Trust UK grants to Tony Minson, in whose lab the Δ33 mutant was made.
REFERENCES
- Ahuja SK, Gao J-L, and Murphy PM (1994). Chemokine receptors and molecular mimicry. Immunol. Today 15, 281–287. [DOI] [PubMed] [Google Scholar]
- Ahuja SK, and Murphy PM (1993). Molecular piracy of mammalian interleukin-8 receptor type B by herpesvirus saimiri. J. Biol. Chem 268, 20691–20694. [PubMed] [Google Scholar]
- Albrecht T, Boldogh I, Fons MP, Deng CZ, AbuBakar S, and Millinoff D (1991). Role of immediate early cellular responses in the initiation of cytomegalovirus infection In “Progress in Cytomegalovirus Research” (Landini MP, Ed.), pp. 239–257. Excerpta Medica, Amsterdam. [Google Scholar]
- Alford CA, and Britt WJ (1990). Cytomegalovirus In “Virology” (Fields BN and Knipe DM, Eds.), 2nd ed., pp. 1981–2010. Raven Press, New York. [Google Scholar]
- Anker HS (1970). A solubilizable acrylamide gel for electrophoresis. FEBS Lett. 7, 293. [DOI] [PubMed] [Google Scholar]
- Attwood TK, and Findlay JBC (1994). Fingerprinting G-protein-coupled receptors. Protein Eng. 7, 195–203. [DOI] [PubMed] [Google Scholar]
- Beisser PS, Vink C, and Bruggeman CA (1995). Characterization of a rat cytomegalovirus gene encoding a G protein-coupled receptor 20th International Herpesvirus Workshop, Groningen, The Netherlands, Abstract 75. [Google Scholar]
- Berlose J-P, Convert O, Brunissen A, Chassaing G, and Lavielle S (1994). Three-dimensional structure of the highly conserved seventh transmembrane domain of G-protein-coupled receptors. Eur. J. Biochem 225, 827–843. [DOI] [PubMed] [Google Scholar]
- Birkenbach M, Josefsen K, Yalamanchili R, Lenoir G, and Kieff E (1993). Epstein-Barr virus-induced genes: First lymphocyte-specific G protein-coupled peptide receptors. J. Virol 67, 2209–2220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Browne H, Churcher M, and Minson T (1992). Construction and characterisation of a human cytomegalovirus with the UL18 (class I homologue) gene deleted. J. Virol 66, 6784–6787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruggeman CA (1993). Cytomegalovirus and latency: An overview. Virchows Arch. B Cell Pathol. Incl. Mol. Pathol 64, 325–333. [DOI] [PubMed] [Google Scholar]
- Cao JX, Gershon PD, and Black DN (1995). Sequence analysis of HindIII Q2 fragment of capripoxvirus reveals a putative gene encoding a G-protein-coupled chemokine receptor homologue. Virology 209, 207–212. [DOI] [PubMed] [Google Scholar]
- Chang Y, Cesarman E, Pessin MS, Lee F, Culpepper J, Knowles DM, and Moore PS (1994). Identification of herpesvirus-like DNA sequences in AIDS-associated Kaposi’s sarcoma. Science 266, 1865–1869. [DOI] [PubMed] [Google Scholar]
- Charo IF, Myers SJ, Herman A, Franci C, Connolly AJ, and Coughlin SR (1994). Molecular cloning and functional expression of two monocyte chemoattractant protein 1 receptors reveals alternative splicing of the carboxy-terminal tails. Proc. Natl. Acad. Sci. USA 91, 2752–2756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chee MS, Bankier AT, Beck S, Bohni R, Brown CM, Cerny R, Horsnell T, Hutchison CA, Kouzarides T, Martignetti JA, Preddie E, Satchwell SC, Tomlinson P, Weston KM, and Barrell BG (1990a). Analysis of the protein-coding content of the sequence of human cytomegalovirus strain AD169. Curr. Top. Microbiol. Immu nol 154, 125–169. [DOI] [PubMed] [Google Scholar]
- Chee MS, Satchwell SC, Preddie E, Weston KM, and Barrell BG (1990b). Human cytomegalovirus encodes three G protein-coupled receptor homologues. Nature (London) 344, 774–777. [DOI] [PubMed] [Google Scholar]
- Chen C, and Okayama H (1987). High-efficiency transformation of mammalian cells by plasmid DNA. Mol. Cell. Biol 7, 2745–2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collawn JF, and Paterson Y (1995). Chemical coupling of synthetic peptide to carrier protein using glutaraldehyde In “Current Protocols in Molecular Biology” (Ausubel FM, Brent R, Kingston RE, Moore DD, Seidman JG, Smith JA, and Struhl K, Eds.), pp. 11.15.2–11.15.4. Wiley, New York. [Google Scholar]
- Davis-Poynter NJ, Rawlinson WD, Barrell BG, Shellam GR, and Farrell HE (1995). Identification and characterisation of a G-protein coupled receptor homologue encoded by murine cytomegalovirus 20th International Herpesvirus Workshop, Groningen, The Nether lands, Abstract 88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedman JR, and Kaetzel MA (1995). Calcium as an intracellular second messenger: Mediation by calcium binding proteins In “Cell Physiology Source Book” (Sperelakis N, Ed.), pp. 128–136. Academic Press, San Diego. [Google Scholar]
- Durum SK, and Oppenheim JJ (1993). Proinflammatory cytokines and immunity In “Fundamental Immunology” (Paul WE, Ed.), 3rd ed., pp. 801–826. Raven Press, New York. [Google Scholar]
- Fraser CM, Lee NH, Pellegrino SM, and Kerlavage AR (1994). Molecular properties of G-protein-coupled receptors. Prog. Nucleic Acid Res. Mol. Biol 49, 113–156. [DOI] [PubMed] [Google Scholar]
- Furukawa T, Hornberger E, Sakuma S, and Plotkin SA (1975). Demonstration of immunoglobulin G receptors induced by human cytomegalovirus. J. Clin. Microbiol 2, 332–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J-L, and Murphy PM (1994). Human cytomegalovirus open reading frame US28 encodes a functional β chemokine receptor. J. Biol. Chem 269, 28539–28542. [PubMed] [Google Scholar]
- Gibson W (1981a). Protease-facilitated transfer of high-molecular-weight proteins during electrotransfer to nitrocellulose. Anal. Bio chem 118, 1–3. [DOI] [PubMed] [Google Scholar]
- Gibson W (1981b). Structural and nonstructural proteins of strain Colburn cytomegalovirus. Virology 111, 516–537. [DOI] [PubMed] [Google Scholar]
- Gibson W (1993). Molecular biology of human cytomegalovirus In “Molecular Aspects of Human Cytomegalovirus Diseases” (Becker Y, Darai G, and Huang E-S, Eds.), pp. 303–329. Springer-Verlag, Berlin. [Google Scholar]
- Gibson W, Baxter MK, and Clopper KS (1996). Cytomegalovirus “missing” capsid protein identified as heat-aggregatable product of HCMV UL46. J. Virol, in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gompels UA, Nicholas J, Lawrence G, Jones M, Thomson BJ, Martin MED, Efstathiou S, Craxton M, and Macaulay HA (1995). The DNA sequence of human herpesvirus-6: Structure, coding content, and genome evolution. Virology 209, 29–51. [DOI] [PubMed] [Google Scholar]
- Greis KD, Gibson W, and Hart GW (1994). Site-specific glycosylation of the human cytomegalovirus tegument basic phosphoprotein (UL32) at serine 921 and serine 952. J. Virol 68, 8339–8349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruenwald S, and Heitz J (1993). “Baculovirus Expression Vector System: Procedures and Methods Manual,” 2nd ed. Pharmingen, San Diego, CA. [Google Scholar]
- Haltiwanger RS, and Hart GW (1993). Glycosyltransferases as tools in cell biological studies. Methods Mol. Biol 14, 175–187. [DOI] [PubMed] [Google Scholar]
- Harlow E, and Lane D (1988). “Antibodies: A Laboratory Manual.” Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Hasegawa H, Utsunomiya Y, Yasukawa M, Yanagisawa K, and Fujita S (1994). Induction of G protein-coupled peptide receptor EBI 1 by human herpesvirus 6 and 7 infection in CD4+ T cells. J. Virol 68, 5326–5329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hildreth JEK, and Hyman JA (1989). Production and characterization of monoclonal and anti-CD18 anti-idiotype antibodies. Mol. Immunol 26, 1155–1167. [DOI] [PubMed] [Google Scholar]
- Horuk R (1994). Molecular properties of the chemokine receptor family. Trends Pharmacol. Sci 15, 159–165. [DOI] [PubMed] [Google Scholar]
- Hutchinson L, Roop-Beauchamp C, and Johnson DC (1995). Herpes simplex virus glycoprotein K is known to influence fusion of infected cells, yet is not on the cell surface. J. Virol 69, 4556–4563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irmiere A, and Gibson W (1983). Isolation and characterization of a noninfectious virion-like particle released from cells infected with human strains of cytomegalovirus. Virology 130, 118–133. [DOI] [PubMed] [Google Scholar]
- Jarvis DJ, and Summers MD (1989). Glycosylation and secretion of human tissue plasminogen activator in recombinant baculovirus-infected insect cells. Mol. Cell. Biol 9, 214–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jemmerson R, and Hutchinson RM (1990). Fine manipulation of antibody affinity for synthetic epitopes by altering peptide structure: Anti body binding to looped peptides. Eur. J. Immunol 20, 579–585. [DOI] [PubMed] [Google Scholar]
- Jones TR, Stenberg RM, and Sun L (1995). Recombinant HCMV mutants with deletion of the three G protein-coupled receptor genes The Fifth International Cytomegalovirus Conference, Stockholm, Abstract P066. [Google Scholar]
- Kaufman RJ (1990). Vectors used for expression in mammalian cells. Methods Enzymol. 185, 487–511. [DOI] [PubMed] [Google Scholar]
- Kaye J, Browne H, Stoffel M, and Minson T (1992). The UL16 gene of human cytomegalovirus encodes a glycoprotein that is dispensable for growth in vitro. J. Virol 66, 6609–6615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keller R, Pietchel R, Goldman JN, and Goldman M (1976). An IgGFc receptor induced in cytomegalovirus-infected human fibroblasts. J. Immunol 116, 772–777. [PubMed] [Google Scholar]
- Kolakowski LF (1994). GCRDb: A G-protein-coupled receptor data base. Receptors Channels 2, 1–7. [PubMed] [Google Scholar]
- Kolodziej PA, and Young RA (1991). Epitope tagging and protein surveillance. Methods Enzymol. 194, 508–519. [DOI] [PubMed] [Google Scholar]
- Kraft R, Tardiff J, Krauter KS, and Leinwand LA (1988). Using mini-prep plasmid DNA for sequencing double stranded templates with Sequenase. BioTechniques 6, 544–546. [PubMed] [Google Scholar]
- Kuhn DE, Beall CJ, and Kolattukudy PE (1995). The cytomegalovirus US28 protein binds multiple CC chemokines with high affinity. Biochem. Biophys. Res. Commun 211, 325–330. [DOI] [PubMed] [Google Scholar]
- Lalli E, Lee JS, Masquilier D, Schlotter F, Foulkes NS, Molina CA, and Sassone-Corsi P (1993). Nuclear response to cyclic AMP: Central role of transcription factor CREM (cyclic-AMP-responsive-element modulator). Biochem. Soc. Trans 21, 912–917. [DOI] [PubMed] [Google Scholar]
- Laskey RA, and Mills AD (1977). Enhanced autoradiographic detection of 32P and 125I using intensifying screens and hypersensitized film. FEBS Lett. 82, 314–316. [DOI] [PubMed] [Google Scholar]
- Lischwe MA, and Ochs D (1982). A new method for partial peptide mapping using N-chlorosuccinimide/urea and peptide silver staining in sodium dodecyl sulfate–polyacrylamide gels. Anal. Biochem 127, 453–457. [DOI] [PubMed] [Google Scholar]
- Lutz-Freyermuth C, Query CC, and Keene JD (1990). Quantitative determination that one of two potential RNA-binding domains of the A protein component of the U1 small nuclear ribonucleoprotein complex binds with high affinity to stem-loop II of U1 RNA. Proc. Natl. Acad. Sci. USA 87, 6393–6397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machamer CE, and Rose JK (1988). Influence of new glycosylation sites on expression of the vesicular stomatitis virus G protein at the plasma membrane. J. Biol. Chem 263, 5948–5954. [PubMed] [Google Scholar]
- Massung RF, Jayarama V, and Moyer RW (1993). DNA sequence analysis of conserved and unique regions of swinepox virus: Identification of genetic elements supporting phenotypic observations including a novel G protein-coupled receptor homologue. Virology 197, 511–528. [DOI] [PubMed] [Google Scholar]
- Mouillac B, Caron M, Bonin H, Dennis M, and Bouvier M (1992). Agonist-modulated palmitoylation of β2-adrenergic receptor in Sf9 cells. J. Biol. Chem 267, 21733–21737. [PubMed] [Google Scholar]
- Murphy PM (1994a). The molecular biology of leukocyte chemoattractant receptors. Annu. Rev. Immunol 12, 593–633. [DOI] [PubMed] [Google Scholar]
- Murphy PM (1994b). Molecular piracy of chemokine receptors by herpesviruses. Infect. Agents Dis 3, 137–154. [PubMed] [Google Scholar]
- Neote K, DiGregorio D, Mak JY, Horuk R, and Schall TJ (1993). Molecular cloning, functional expression, and signaling characteris tics of a C-C chemokine receptor. Cell 72, 415–425. [DOI] [PubMed] [Google Scholar]
- Nicholas J, Cameron KR, and Honess RW (1992). Herpesvirus saimiri encodes homologues of G protein-coupled receptors and cyclins. Nature (London) 355, 362–365. [DOI] [PubMed] [Google Scholar]
- O’Reilly DR, Miller LK, and Luckow V (1992). “Baculovirus Expression Vectors: A Laboratory Manual.” Freeman, New York. [Google Scholar]
- Oliveira L, Paiva ACM, and Vriend G (1993). A common motif in G-protein-coupled seven transmembrane helix receptors. J. Comput. Aided Mol. Des 7, 649–658. [Google Scholar]
- Papac DI, Thornburg KR, Büllesbach EE, Crouch RK, and Knapp DR (1992). Palmitylation of a G-protein coupled receptor. J. Biol. Chem 267, 16889–16894. [PubMed] [Google Scholar]
- Parker EM, Kameyama K, Higashijima T, and Ross EM (1993). Reconstitutively active G protein-coupled receptors purified from baculovirus-infected insect cells. J. Biol. Chem 266, 519–527. [PubMed] [Google Scholar]
- Parker RC, Watson RM, and Vinogroad J (1977). Mapping of closed circular DNAs by cleavage with restriction endonucleases and calibration by agarose gel electrophoresis. Proc. Natl. Acad. Sci. USA 74, 851–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rose JK, and Bergmann JE (1982). Expression from cloned cDNA of cell-surface secreted forms of the glycoprotein of vesicular stomatitis virus in eucaryotic cells. Cell 30, 753–762. [DOI] [PubMed] [Google Scholar]
- Saffitz JE, and Liggett SB (1992). Subcellular distribution of β2-adrenergic receptors delineated with quantitative ultrastructural autoradiography of radioligand binding sites. Circ. Res 70, 1320–1325. [DOI] [PubMed] [Google Scholar]
- Sambrook J, Fritsch EF, and Maniatis T (1989). “Molecular Cloning: A Laboratory manual,” 2nd ed. Cold Spring Harbor Laboratory Press, Cold Spring Harbor, NY. [Google Scholar]
- Scharf SJ (1990). Cloning with PCR In “PCR Protocols: A Guide to Methods and Applications” (Innis MA and Gelfand DH, Eds.), pp. 84–91. Academic Press, San Diego. [Google Scholar]
- Schenk P, Woods AS, and Gibson W (1991). The 45-kilodalton protein of cytomegalovirus (Colburn) B-capsids is an amino-terminal extended form of the assembly protein. J. Virol 65, 1525–1529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenza JC, Hardwick KG, Dean N, and Pelham HRB (1990). ERD2, a yeast gene required for the receptor-mediated retrieval of luminal ER proteins from the secretory pathway. Cell 61, 1349–1357. [DOI] [PubMed] [Google Scholar]
- Shenker A (1994). Mutations in G protein-coupled signaling pathways as a cause of human disease In “Challenges of Modern Medicine: GTPase Controlled Molecular Machines” (Corda D, Hamm H, and Luini A, Eds.), Vol. 6, pp. 109–120. Ares-Serono Symposia, Rome. [Google Scholar]
- Sleight RG, and Lieberman MA (1995). Signal transduction In “Cell Physiology Source Book” (Sperelakis N, Ed.), pp. 117–127. Academic Press, San Diego. [Google Scholar]
- Smith KD, Valenzuela A, Vigna JL, AAlbers K, and Lutz CT (1993). Unwanted mutations in PCR mutagenesis: Avoiding the predictable. PCR Methods Appl. 2, 253–257. [DOI] [PubMed] [Google Scholar]
- Sung C-H, Schneider BG, Agarwal N, Papermaster DS, and Na thans J (1991). Functional heterogeneity of mutant rhodopsins re sponsible for autosomal dominant retinitis pigmentosa. Proc. Natl. Acad. Sci. USA 88, 8840–8844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talbot P, and Almeida JD (1977). Human cytomegalovirus: Purifica tion of enveloped particles and dense bodies. J. Gen. Virol 36, 345–349. [DOI] [PubMed] [Google Scholar]
- Tarentino AL, and Plummer TH Jr. (1994). Enzymatic deglycosylation of asparagine-linked glycans: Purification, properties, and specificity of oligosaccharide-cleaving enzymes from Flavobacterium men ingosepticum. Methods Enzymol. 230, 44–57. [DOI] [PubMed] [Google Scholar]
- Telford EAR, Watson MS, Aird HC, Perry J, and Davison AJ (1995). The DNA sequence of equine herpesvirus 2. J. Mol. Biol 249, 520–528. [DOI] [PubMed] [Google Scholar]
- Towbin H, Staehelin T, and Gordon J (1979). Electrophoretic transfer of proteins from polyacrylamide gels to nitrocellulose sheets: Procedure and some applications. Proc. Natl. Acad. Sci. USA 76, 4350–4354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsai DE, Kenan DJ, and Keene JD (1992). In vitro selection of an RNA epitope immunologically cross-reactive with a peptide. Proc. Natl. Acad. Sci. USA 89, 8864–8868. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vieira J, Schall T, Geballe A, and Corey L (1995). Disruption of the HCMV US28 ORF: Expression of the US28 gene from the viral genome shows the properties of a GCR for C-C chemokines 20th International Herpesvirus Workshop, Groningen, The Netherlands, Abstract 289. [Google Scholar]
- von Laer D, Serr A, Meyer-König U, Kirste G, Hufert FT, and Haller O (1995). Human cytomegalovirus immediate early and late transcripts are expressed in all major leukocyte populations in vivo. J. Infect. Dis 172, 365–370. [DOI] [PubMed] [Google Scholar]
- Weiner D, and Gibson W (1983). Phosphorylation, maturational processing, and relatedness of strain Colburn matrix proteins. Virology 129, 155–169. [DOI] [PubMed] [Google Scholar]
- Welch AR, McGregor LM, and Gibson W (1991). Cytomegalovirus homologs of cellular G protein-coupled receptor genes are transcribed. J. Virol 65, 3915–3918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Welch AR, McNally LM, Hall MRT, and Gibson W (1993). Her pesvirus proteinase: Site-directed mutagenesis used to study maturational, release, and inactivation cleavage sites of precursor and to identify a possible catalytic site serine and histidine. J. Virol 67, 7360–7372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yatani A (1995). Ion channels that are directly regulated by G proteins In “Cell Physiology Source Book” (Sperelakis N, Ed.), pp. 378–385. Academic Press, San Diego. [Google Scholar]
- Zastawny RL, Ng GYK, Trogadis JE, George SR, Stevens JK, and O’Dowd BF (1994). Expression of G protein-coupled receptors in baculovirus/Sf9 cells: Imaging receptor distribution by confocal fluorescence microscopy In “Three-Dimensional Confocal Micros copy: Volume Investigation of Biological Specimens” (Stevens JK, Mills LR, and Trogadis JE, Eds.), pp. 233–252. Academic Press, San Diego, CA. [Google Scholar]