

Summary
Trichomes are storage compartments for specialized metabolites in many plant species. In trichome, plant primary metabolism is significantly changed, providing substrates for downstream secondary metabolism. However, little is known of how plants coordinate trichome formation and primary metabolism regulation. In this report, tomato (Solanum lycopersicum) trichome regulator SlMIXTA‐like is indicated as a metabolic regulation gene by mGWAS analysis. Overexpression of SlMIXTA‐like in tomato fruit enhances trichome formation. In addition, SlMIXTA‐like can directly bind to the promoter region of gene encoding 3‐deoxy‐7‐phosphoheptulonate synthase (SlDAHPS ) to activate its expression. Induction of SlDAHPS expression enhances shikimate pathway activities and provides substrates for downstream secondary metabolism. Our data provide direct evidence that trichome regulator can directly manipulate primary metabolism, in which way plants can coordinate metabolic regulation and the formation of storage compartments for specialized metabolites. The newly identified SlMIXTA‐like can be used for future metabolic engineering.
Keywords: tomato, mGWAS, SlMIXTA‐like, primary metabolism, secondary metabolism, phenylpropanoid, 3‐Deoxy‐d‐arabinoheptulosonate 7‐phosphate synthase
Introduction
Phenylpropanoid compounds are ubiquitous in plant kingdom. They are associated with almost all important physiological processes: from the formation of plant architecture to stress response, as well as plant reproduction and symbiosis (Vogt, 2010). In addition, the health benefits of phenylpropanoids have been extensively investigated during the past 20 years. Many studies have provided evidences that phenylpropanoid compounds play positive roles in human health such as chronic disease preventing, anticancer and anti‐ageing (Butelli et al., 2008; Carmona‐Gutierrez et al., 2019; Martin and Li, 2017; Scarano et al., 2017). As a result, phenylpropanoids with health benefits are often the main targets of metabolic engineering. During the past decade, the development of tools and experiences enable us to engineer various secondary metabolites in plant systems (Patron et al., 2015). Among popular plant production platforms, tomato is one of the most desirable chassis, due to its high yield and short life cycle, as well as its capability to apply most modern biotechnologies (Li et al., 2018).
One common strategy to improve the efficiency of metabolic engineering in plant chassis is the application of transcription factors (TFs) (Century et al., 2008; Fu et al., 2018). Previous studies indicate MYB proteins are main regulators for phenylpropanoid metabolism and MYBs with activation function are found to mainly belong to subgroups 5, 6, 7 and 27 (Liu et al., 2015). Fruit‐specific expression of these TFs can significantly improve the production of valuable metabolites in tomato fruit (Bovy et al., 2002; Butelli et al., 2008; Gonzali et al., 2009; Luo et al., 2008).
It was not until recently did we begin to realize the importance of primary metabolism regulation in metabolic engineering (Morandini, 2013). Phenylpropanoids are synthesized from phenylalanine, which are the main products of shikimate pathway (Vogt, 2010). Phosphoenolpyruvate from glycolysis and erythrose‐4‐phosphate from pentose phosphate pathway are the substrates for the first step of shikimate pathway, and 3‐Deoxy‐d‐arabinoheptulosonate 7‐phosphate synthase (DAHPS) catalysed this reaction. Previously indicated overexpression of DAHPS in plants can significantly enhance the activities of shikimate pathway, leading the metabolic flux towards phenylpropanoids biosynthesis (Tzin et al., 2012, 2013). Coincidentally, overexpression of AtMYB12, a flavonol regulator in Arabidopsis thaliana, in tomato fruit was found to significantly enhance the expression of genes involved in glycolysis, pentose phosphate pathway and shikimate pathway (Fu et al., 2018; Luo et al., 2008; Zhang et al., 2015). Further investigation indicates that AtMYB12 directly activates the expression of genes encoding DAHPS and enolase (ENO). And activation of these two genes redirects the carbon flux towards aromatic amino acid biosynthesis, providing substrates for downstream secondary metabolic pathways. (Fu et al., 2018; Luo et al., 2008; Zhang et al., 2015). All these indicate that reprogramme of primary metabolism can be achieved by manipulating the activities of several key enzymes. And some specific TFs can regulate the expression of genes encoding these enzymes therefore guide the carbon flux towards certain pathways.
Based on this theory, it is possible to design the plant carbon flux once regulatory mechanism for key genes in primary metabolism been identified (Tzin et al., 2012, 2013). And there are examples that a single TF can activate those key genes (Zhang et al., 2015). In addition to pathway reconstruction, using TFs to control both primary and secondary metabolic pathways has become a new generation of methodology in plant metabolic engineering (Fu et al., 2018). As a result, the discovery and identification of new TFs are vital for successful metabolic engineering. So far, the most successful strategies are using natural mutants and through mutagenesis approaches (Adato et al., 2009; Ballester et al., 2009; Borevitz et al., 2000; Schwinn et al., 2006). Recently, multi‐omics integration analysis was used to link genetic basis and metabolic changes in tomato breeding. The genome, transcriptome and metabolome of 610 tomato germplasms revealed the molecular basis for the domestication of important agronomic traits in tomato (Zhu et al., 2018). In addition to its importance in studying tomato domestication process, this data set is a vital resource for dissecting plant metabolic regulation in high throughput.
Compared to microbial system, one unique character of plant chassis is that the production of metabolites is tissue‐specific. Trichomes are common structures for plants to store specialized metabolites, particularly chemicals involved in stress and disease tolerance (Glas et al., 2012; Huchelmann et al., 2017). In order to produce large amount of metabolites, carbon flux has been significantly changed in trichomes (Balcke et al., 2017). Therefore, the plants may have a regulatory mechanism to coordinate primary metabolism regulation and trichome formation. Previous studies indicate MIXTA‐like MYB TFs are responsible for trichome formation in many plants (Glover et al., 1998; Lashbrooke et al., 2015; Perez‐Rodriguez et al., 2005). And recent study in Artemisia annua indicates trichome and artemisinin regulator 1 (TAR1), an AP2 transcription factor, is required for both trichome development and artemisinin biosynthesis (Tan et al., 2015). However, how can plant significantly reprogramme the primary metabolism in trichome is still unknown. In this study, we performed mGWAS for a recently published tomato multi‐omics data set (Zhu et al., 2018). We identified SlMIXTA‐like (Solyc02g088190), a gene encoding a MIXTA‐like TF, which belongs to subgroup 9 MYB family, as a direct regulator of tomato primary metabolism. Our finding provides evidence that trichome regulator has direct roles in primary metabolic regulation and this can be used for future metabolic engineering.
Results
SlMIXTA‐like is the candidate gene for a major phenylpropanoid QTN
In order to screen new TFs regulating important metabolites in tomato, we checked through the tomato multi‐omics data set from our co‐authors (see Experimental procedures) (Zhu et al., 2018). We noticed a significant quantitative trait nucleotide (QTN) (P = 1.93E‐10) between the levels of an important phenylpropanoid compound—ρ‐coumaric acid (SlFM0124) and a SNP (sf0250278120) on chromosome 2 (Figure 1a, b). This SNP is located 74 Kb away from gene Solyc02g088190 (Data S1). Using haplotype analysis, we found that the two SNPs in the CDS region of Solyc02g088190 gene have significant correlation with the content of ρ‐coumaric acid (sf0250353631 and sf0250353789; P = 2.17E‐16 and 4.71E‐18, respectively) (Figure S1), Molecular phylogenetic analysis of the R2R3‐MYB transcription factors from Arabidopsis thaliana and tomato revealed that the Solyc02g088190 belongs to the subgroup 9 (Figure S2 and S3) (Liu et al., 2015). It has previously been reported as SlMIXTA‐like, a MIXTA‐like R2R3 MYB TF, which was found to be linked to conical cell development and trichome formation (Ewas et al., 2016; Lashbrooke et al., 2015). As trichomes have been shown to produce and store various metabolites, phenylpropanoids, included, we predict SlMIXTA‐like is the candidate gene underlining this locus.
Figure 1.
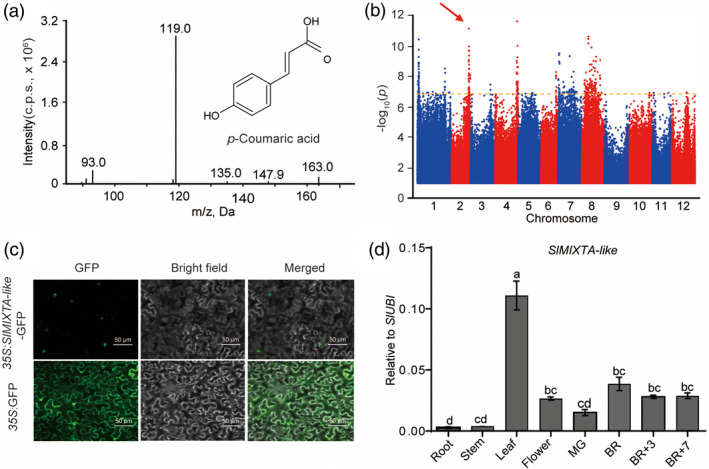
SlMIXTA‐like is the candidate gene for a major phenylpropanoid QTN. (a) Structure and MS/MS profile of compound SlFM0124, ρ‐coumaric acid. (b) Manhattan plot for the metabolite of SlFM0124, ρ‐coumaric acid. A significant QTN was shown on chromosome 2. (c) SlMIXTA‐like is a nuclei localized protein. Full‐length SlMIXTA‐like cDNA was fused with GFP. Agroinfiltrated Nicotiana benthamiana leaves were analysed at 3dpi. (d) RT‐qPCR data indicate tissue‐specific expression pattern of SlMIXTA‐like in MicroTom. Error bars show SEM (n = 3). Different letters indicate significantly different values at P < 0.05 (one‐way ANOVA, Tukey's post hoc test).
As previously described, SlMIXTA‐like is a nucleus‐localized protein (Figure 1c) (Dubos et al., 2010; Ewas et al., 2016; Lashbrooke et al., 2015). To better investigate the role of SlMIXTA‐like, we analysed SlMIXTA‐like expression pattern in different tissues using RT‐qPCR. The transcript level of SlMIXTA‐like reaches its highest in leaves while remains a relative high level in flower and different fruit stages (Figure 1d). Similar result has also been shown from TomExpress website (Figure S4) (Zouine et al., 2017).
In order to verify SlMIXTA‐like's function, we first overexpressed SlMIXTA‐like under CaMV 35S promoter. Compared to MicroTom, the SlMIXTA‐like expression levels are significantly increased in the seedlings of transgenic lines (Figure 2a). As previously reported, enhanced trichome formation was observed in the stem of transgenic lines (Figure 2c and Figure S5). To analyse effects of SlMIXTA‐like overexpression in tomato fruit, we then checked the expression level of SlMIXTA‐like in transgenic fruit. However, due to the limitation of 35S promoter, the expression of SlMIXTA‐like was very low in transgenic fruit and no significant phenotypic changes were observed (Figure 2b, d).
Figure 2.
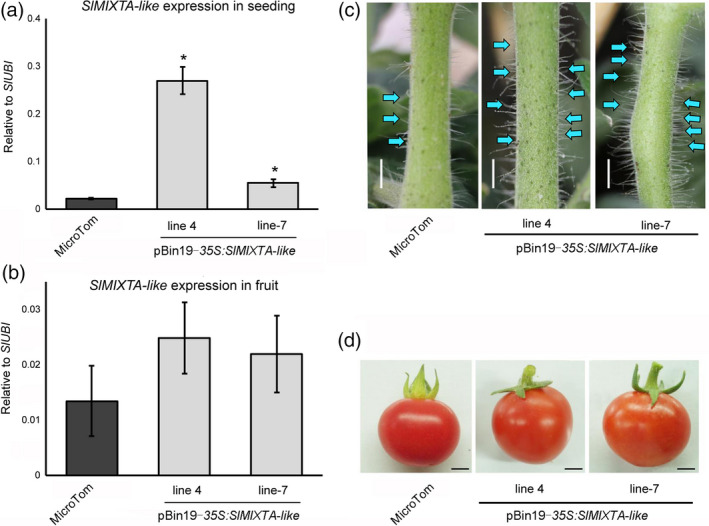
Constitutive expression of SlMIXTA‐like in tomato plant enhances trichome formation in seedlings. (a) Expression levels of SlMIXTA‐like in MicroTom and 35S:SlMIXTA‐like seedlings. (b) Enhanced trichome formation on the stem of 35S:SlMIXTA‐like seedlings. Scale bars show 4 mm. (c) SlMIXTA‐like expression levels in the fruit of MicroTom and 35S:SlMIXTA‐like plants. (d) Phenotype of ripe fruit of MicroTom and 35S:SlMIXTA‐like. Scale bars show 4 mm. *P < 0.05.
Fruit‐specific expression of SlMIXTA‐like significantly changes fruit phenotype
To better characterize the potential function of SlMIXTA‐like in tomato fruit, we then overexpressed SlMIXTA‐like under fruit‐specific E8 promoter (see Experimental procedures). A total of 14 T0 lines were obtained and preliminary screening indicated 13 of them had significantly higher SlMIXTA‐like expression in ripe fruit compared to MicroTom (Figure S6). High expression level was found in the fruit of lines A and B, and these lines were chosen to grow to T1 generation for further investigation (Figure S6). Compared to MicroTom plants, both E8:SlMIXTA‐like‐A and E8:SlMIXTA‐like‐B have no significant difference by mature green (MG) stage. However, at MG, as the induction of E8 promoter (Deikman, 1996; Fischer, 1988), SlMIXTA‐like begins to express and both transgenic lines begin to accumulate trichomes (Figure 3a and Figure S7). Further investigation using scanning electron microscopy (SEM) confirmed that the E8:SlMIXTA‐like fruit produces higher density of trichome on fruit surface (Figure 3b). Compared to 35S:SlMIXTA‐like plant, the fruit of E8:SlMIXTA‐like transgenic lines has significantly higher expression of SlMIXTA‐like (Figure 2b and Figure S6). Thus, the trichome density on the surface of latter is significantly increased (Figure 3). This matches previous conclusion that SlMIXTA‐like is associated with trichome formation in tomato (Ewas et al., 2016; Lashbrooke et al., 2015).
Figure 3.

Fruit‐specific expression of SlMIXTA‐like enhances trichome formation on tomato fruit. (a) Phenotypes of transgenic and MicroTom tomato fruit at different stages. MG, mature green; BR, breaker; BR+7, 7 days post breaker. Scale bars show 4 mm. (b) Scanning electron microscope observation of trichome structures on the surface of T1 generation transgenic and MicroTom tomato fruit. Samples were analysed at BR+7. Scale bars show 500 μm.
In addition to trichomes, we also noticed significant changes in the colour of E8:SlMIXTA‐like ripe fruit. Compared to MicroTom and 35S:SlMIXTA‐like fruit, E8:SlMIXTA‐like fruit shows orange colour, indicating changes in the contents of metabolites due to the overexpression of SlMIXTA‐like in fruit (Figures 2d and 3a). All of the results above indicate that the overexpression of SlMIXTA‐like in tomato fruit not only induces trichome formation on fruit surface, but also alters metabolic patterns in fruit.
Ectopic expression of SlMIXTA‐like in tomato fruit alters primary metabolism
To better investigate the potential roles of SlMIXTA‐like in metabolic regulation, we performed both transcriptome and metabolic profiling for both E8:SlMIXTA‐like‐A and E8:SlMIXTA‐like‐B fruit (Data S2). Compared to MicroTom, there are 1696 differently expressed genes (DEGs) (fold change ≥ 1.5, FDR < 0.05) in the fruit of E8:SlMIXTA‐like line A and 2670 DEGs in line B. In total, 948 differently expressed genes are shared for both lines (Figure S8a). Among the shared DEGs, we found many genes involved in glycolysis, and pentose phosphate pathway and shikimate pathway are up‐regulated in both E8:SlMIXTA‐like fruits (Figure 4, Figure S8b and Table S2). We further checked the changes in gene expression levels by RT‐qPCR and confirmed genes involved in glycolysis (SlSUS1, SlHK, SlGPI, SlPFP, SlPFK1, SlTPI, SlENO), pentose phosphate pathway (SlPGLS, SlPGD1 and SlRpe) and shikimate pathway (SlDAHPS, SlDHQS, SlDHQD, SlSHD, SlSK, SlEPSPS, SlCS, SlCM) are significantly induced in the fruit of both E8:SlMIXTA‐like lines (Figure 5a–c). In addition to that, downstream general phenylpropanoid pathway genes (PAL5A, PAL5B and PAL5C) are also up‐regulated (Figure 5d). To exclude the influence of trichomes’ presence to the expression of primary metabolic genes, we removed the trichomes of E8:SlMIXTA‐like‐B fruit and measured gene expression pattern in the pericarp (Figure S9a). Still, we can see the induction of key primary metabolic genes is directly associated with SlMIXTA‐like overexpression (Figure S9b). All these results indicate overexpression of SlMIXTA‐like in tomato fruit reprogrammes primary metabolism, by inducing the expression of genes involved in glycolysis, pentose phosphate pathway and shikimate pathway.
Figure 4.
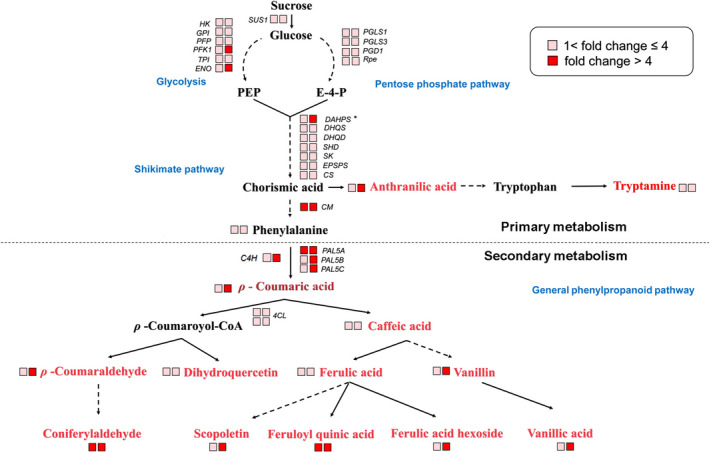
Overexpression of SlMIXTA‐like in tomato fruit significantly changes the dynamic of primary and secondary metabolism. Schematic representation of metabolic changes in tomato fruits expressing SlMIXTA‐like. Based on transcriptome and metabolomics data, genes and metabolites which are significantly increased in the transgenic fruits (line A and line B) are coloured. Data are represented as fold change compared to MicroTom. Detailed data are shown in Tables S2 and S3.
Figure 5.
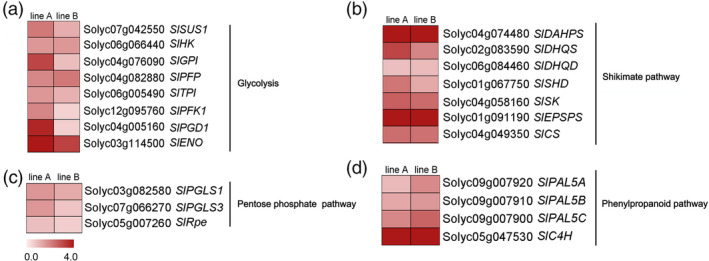
RT‐qPCR confirmation of differently expressed genes. Genes involved in glycolysis (a), pentose phosphate pathway (b), shikimate pathway (c) and phenylpropanoid pathway (d) are analysed. Data are represented in fold changes compared to MicroTom in log2 scale for both lines A and B. Gene IDs and abbreviations are explained in Table S4.
In order to confirm how fruit‐specific expression of SlMIXTA‐like alters primary metabolism, we performed widely targeted metabolomics analysis to MicroTom and E8:SlMIXTA‐like tomato fruit at ripe stage (7 days after breaker). Compared to MicroTom fruit, there are 208 metabolites significantly changed in E8:SlMIXTA‐like‐A, while for E8:SlMIXTA‐like‐B the number is 237. In total, 109 metabolites are significantly changed in both E8:SlMIXTA‐like lines (Figure S10a and Data S3). In addition to previously reported compounds in lipid biosynthesis (Lashbrooke et al., 2015), there is also significant increase in aromatic amino acid derivatives and general phenylpropanoids (Figure 4a, Figure S10b–c and Table S3). We further confirmed these using UPLC and found phenylpropanoid compounds such as ρ‐coumaric acid, chlorogenic acid and ferulic acid are significantly enriched in E8:SlMIXTA‐like, fruit while rutin, a major flavonol, is not changed (Figure S11a–d). Noticeably, ρ‐coumaric acid is the compound we used to perform mGWAS analysis (Figure 1a, b) and these data further attest SlMIXTA‐like is the gene responsible for the regulation of ρ‐coumaric acid content in tomato.
As fruit of both E8:SlMIXTA‐like lines A and B showed orange colour (Figure 3), we also checked the expression of carotenoid biosynthetic genes and the contents of major carotenoids in both MicroTom and transgenic fruit. Compared to MicroTom, we saw significant induction of carotenoid biosynthesis genes in E8:SlMIXTA‐like fruit (Figure S12a). Metabolic analysis also indicates there are increased contents of lycopene and β‐carotene (Figure S12b). This matches previous observation on overexpression of SlMX1 in Ailsa Craig tomato (Ewas et al., 2016). All these data above indicate instead of the inhibition of carotenoid biosynthesis, and the orange colour of E8:SlMIXTA‐like fruit is mainly due to the accumulation of phenylpropanoid compounds.
Taken together, all these data indicate ectopic expression of SlMIXTA‐like in tomato fruit significantly changes transcriptional level of primary metabolic genes, resulting in the change in contents of related metabolites: the dynamics of primary metabolism has been significantly changed by overexpression of SlMIXTA‐like in tomato fruit.
SlMIXTA‐like directly interacts with SlDAHPS to change the dynamics of primary metabolism
Previous study indicates AtMYB12, a TF from Arabidopsis thaliana, can directly binds to the promoter region of genes encoding enolase (SlENO) and 3‐deoxy‐d‐arabinoheptulosonate 7‐phosphate synthase (SlDAHPS). As a result, genes involved in glycolysis, pentose phosphate pathway and shikimate pathway are up‐regulated, redirecting carbon flux towards aromatic amino acid biosynthesis (Zhang et al., 2015). As similar changes were seen in E8:SlMIXTA‐like fruit, we predict SlMIXTA‐like uses a similar mechanism to control primary metabolism.
We first performed yeast one‐hybrid (Y1H) assay to test whether SlMIXTA‐like can directly bind to the promoter region of SlENO and SlDAHPS. Compared to AtMYB12, SlMIXTA‐like showed similar binding ability to proSlDAHPS (Figure 6a). For proSlENO, although direct binding activity was found for AtMYB12, no direct binding activity was shown for SlMIXTA‐like (Figure S13a). We further confirmed this by dual‐luciferase report system. SlMIXTA‐like can directly activate SlDAHPS promoter in Nicotiana benthamiana protoplast, whereas mutation of the MYB recognition element (MRE) in SlDAHPS promoter significantly reduces the activity (Figure 6b). For proSlENO, however, no significant induction by SlMIXTA‐like was observed (Figure S13b, c). To further confirm SlMIXTA‐like can directly bind to the promoter of SlDAHPS, we generated transgenic plants with FLAG tagged SlMIXTA‐like driven by E8 promoter. The E8:FLAG‐SlMIXTA‐like fruit showed similar trichome enrichment, and chromatin immunoprecipitation (ChIP)‐qPCR shows significant enrichment of FLAG‐SlMIXTA‐like binding around the MRE of proSlDAHPS (Figure S14). All these data together indicate SlMIXTA‐like directly binds to the MRE in the promoter of SlDAHPS to induce its expression.
Figure 6.
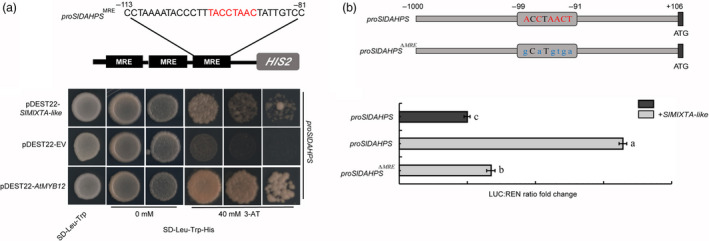
SlMIXTA‐like directly binds to the promoter of SlDAHPS to induce its expression. (a) Yeast one‐hybrid indicates SlMIXTA‐like directly interacts with the MRE in SlDAHPS promoter. The SlDAHPS promoter sequence from −113 to −81 containing the MRE motif was repeated three times and fused to the HIS2 reporter gene in Y1H assay. AtMYB12 was used a positive control. (b) SlMIXTA‐like directly binds to MRE in proSlDAHPS to induce its expression. The SlDAHPS promoter sequence (−1000 to + 106) containing the MRE motif (from −99 to −91) was inserted into the reporting system. A mutated version (∆ MRE) was designed as a negative control. Error bars show SEM (n = 3). Different letters indicate significantly different values at P < 0.05 (one‐way ANOVA, Tukey's post hoc test).
3‐Deoxy‐d‐arabinoheptulosonate 7‐phosphate synthase is reported as rate‐limiting enzyme for shikimate pathway. And overexpression of DAHPS alone is enough to induce the expression of genes involved in shikimate pathway, resulting enhanced production of aromatic amino acids and downstream secondary metabolites (Tzin et al., 2012, 2013). SlMIXTA‐like can directly bind to the MRE in the promoter region of SlDAHPS and induce its expression. The induction of SlDAHPS significantly changes the dynamics of primary metabolism, redirecting carbon flux towards the synthesis of aromatic amino acid, providing substrate for downstream secondary metabolism (Zhang et al., 2015) (Figure 7).
Figure 7.
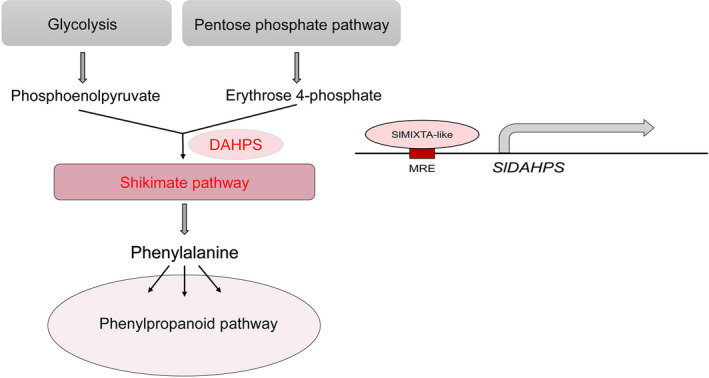
Proposed model for the molecular mechanism of SlMIXTA‐like mediated primary metabolic regulation in tomato. SlMIXTA‐like directly binds to proSlDAHPS to induce its expression. Enhanced DAHPS activities direct carbon flux towards shikimate pathway and downstream phenylpropanoid pathway.
In addition to regulating trichome formation, SlMIXTA‐like manipulates the dynamics of primary metabolism by directly inducing the transcriptional level of SlDAHPS. Through one TF, tomato can reprogramme primary metabolism for the production of specialized metabolites and form trichomes as storage compartments.
Discussion
One of the biggest challenges for plant metabolic engineering is to identify transcription factors regulating key steps. Important metabolic pathways often have multiple levels of regulation to maintain its stability. Compared to structural genes, mutations in regulatory genes might not have significant phenotype due to the redundancy. Although there are examples of using natural mutants or mutagenesis approaches to identify TFs (Adato et al., 2009; Ballester et al., 2009; Borevitz et al., 2000; Schwinn et al., 2006), new high‐throughput methodologies are needed. The development of broad profiling technologies such as genome sequencing, transcriptome analysis and metabolomics profiling provides a perfect solution to this situation (Fang et al., 2019). Population genetic combined with omics can help us to detect minor content changes and link them to nucleic acid sequence difference at base pair level. As this forward genetics strategy is unbiased and accurate, it can provide more confident candidates for further study (Luo, 2015).
In plants, specialized metabolites are not synthesized in all cells. This is largely due to the variation of activities of TFs in different tissues. As a result, the barriers of cell types can be broken to produce specialized metabolites if right regulators can be identified (Fu et al., 2018). Recent studies reveal that glandular trichomes (GTs) of tomato contain large amount of secondary metabolites (Balcke et al., 2017; Glas et al., 2012; Huchelmann et al., 2017). In order to manage such significant change in secondary metabolism, the primary metabolism in trichome has been significantly altered (Balcke et al., 2017). As there are already reports about TFs regulating trichome formation (Glover et al., 1998; Lashbrooke et al., 2015; Perez‐Rodriguez et al., 2005), as well as TFs regulating primary metabolism (Zhang et al., 2015), we are wondering if any of them can directly regulate both trichome formation and primary metabolism. SlMIXTA‐like was reported as regulator for conical cell development and trichome formation (Ewas et al., 2016; Lashbrooke et al., 2015). In this study, our mGWAS analysis indicated that it is also a regulator for plant metabolism. We found SlMIXTA‐like can directly bind to the MRE in the promoter region of SlDAHPS to activate its expression. Overexpression of SlDAHPS can enhance the activities of shikimate pathway to reprogramme primary metabolism, redirecting carbon flux towards the production of aromatic amino acids and their derivatives, which are the major compounds found to be stored in trichomes. By using the same TF to control trichome formation and primary metabolic regulation, plants can coordinate the production of specialized compounds and the initiation of storage compartments. As a result, they can provide accurate regulation to the production and storage of toxic specialized metabolites without impairing normal growth.
Once a TF being identified, changing its expression pattern can break the production barriers between different tissues (Fu et al., 2018). However, constitutive expression of TFs in plants is shown to be less effective than tissue‐specific expression (Luo et al., 2008). This is likely due to the dramatic changes in primary and secondary metabolism attenuate normal plant growth (Zhang et al., 2015). When SlMIXTA‐like is driven by 35S CaMV promoter, it maintains its regulatory function to trichome formation. However, its function on primary metabolism is attenuated by the plants and losses the significance (Ewas et al., 2016; Lashbrooke et al., 2015). Tomato fruit is a perfect chassis for metabolic engineering. This is largely due to the high content of primary metabolites (sugars, amino acids, etc.,). In addition, fruit is the reproductive organ of tomato, which is produced at the late stage of tomato growth cycle. Therefore, changing its primary metabolism has little effects on vegetative growth. When SlMIXTA‐like is expressed under fruit‐specific E8 promoter, its regulatory function to primary metabolism is maximally released (Figures 2 and 3).
Previously, MYB12 was found to directly bind to the promoter region of key primary metabolic genes (ENO and DAHPS). Overexpression of AtMYB12 in tomato fruit can guide the carbon flux towards the production of aromatic amino acids (Phe, Tyr, Trp). So, it can be used as a general tool for phenylpropanoid engineering. However, as MYB12 can also directly bind to the promoter of flavonol biosynthetic genes (CHS, F3H, FLS) (Hartmann et al., 2005; Zhang et al., 2015), AtMYB12 overexpression tomato produces mainly flavonols (Luo et al., 2008; Zhang et al., 2015). In order to produce other phenylpropanoids, flavonol biosynthesis needs to be blocked by using natural mutants or other strategies (Zhang et al., 2015). SlMIXTA‐like, however, only controls the expression of shikimate pathway genes (DAHPS) (Figure 6). So, its induction of phenylpropanoid biosynthesis has no preference, making it a better general tool for metabolic engineering.
Experimental procedure
Genome‐wide association analysis
A total of 2 037 679 SNPs (MAF > 5% and Missing rate < 10%) for 351 accessions (Table S1) were used to perform the genome‐wide association analysis. Efficient Mixed‐Model Association eXpedited (EMMAX) was used to conduct all associations (Kang et al., 2010). GWAS analysis was conducted as described previously (Zhu et al., 2018). The genome‐wide significance thresholds of all the traits were set with a uniform threshold (P = 1/n, n is the effective number of independent SNPs). The unified threshold (P = 2.87E10‐7) was used to filter the SNPs for all the metabolites. LD (linkage disequilibrium) analyses were performed based on all the SNPs (MAF > 0.05) using Haploview software. To reduce the redundancy of mGWAS signals, the lead SNP within a 1 Mb window for each metabolite was extracted as one signal.
Plant material and growth conditions
Solanum lycopersium cv MicroTom seeds were purchased from PanAmerican Seedtm. Plants were grown in glasshouse under 16‐h light, 24°C and 60% humidity.
Subcellular localization
Full‐length SlMIXTA‐like cDNA was amplified from pBin19‐E8:SlMIXTA‐like using primer pair SalI‐SlMIXTA‐like‐F and SlMIXTA‐like‐KpnI‐R and fused with GFP in the pSuper1300 (Llave et al., 2000;; Liu et al., 2017; Ni et al., 1995); the pSuper1300‐SlMIXTA‐like and pSuper1300 were individually transient injected into leaves of Nicotiana benthamiana by Agrobacterium tumefaciens strain GV3101 as described previously (Llave et al., 2000). Microscope inspection was done at 3 dpi (day past inoculation), and the peak wavelength of GFP was 488 nm.
Plant transformation vectors’ construction and tomato transformation
To make pBin19‐E8‐GW plasmid, first, proE8 was amplified from plasmid pSLJ.E8.1500 (Butelli et al., 2008) and inserted into pJIT60 to make pJIT60‐proE8. Gateway cassette was then inserted to make pJIT60‐E8‐GW. Finally, E8‐GW fragment was inserted into pBin19 to make pBin19‐E8‐GW. Full‐length CDS of SlMIXTA‐like (Solyc02g088190) was inserted into the destination vector through Gateway Cloning to make pBin19‐E8:SlMIXTA‐like. To make pBin19‐35S:SlMIXTA‐like plasmid, the full‐length CDS of SlMIXTA‐like was inserted into destination vector pBin19‐35S‐GW through Gateway Cloning. Tomato stable transformation was done by Agrobacterium tumefaciens strain EHA105 as previously described (McCormick et al., 1986).
RNA extraction and RT‐qPCR
Both MicroTom and transgenic fruit were harvested at BR + 7. Fruit pericarp was ground into fine powder using liquid nitrogen. Total RNA samples were isolated from each sample using RNAiso Plus (Takara Bio, Kusatsu, Japan, AHF1820A) following the manufacturer's instruction. cDNA (complementary DNA) was synthesized from 1 μg of total RNA using a PrimeScripttm RT reagent Kit with gDNA Eraser (Takara Bio, Kusatsu, Japan, AK4201). cDNA products were diluted into 2.5 ng/μL and used as templates for the qPCR. RT‐qPCR was performed using the Bio‐Rad CFX384. Each reaction (10 μL) consisted of 5 μL of iTaqtm Universal SYBR Green Supermix (Bio‐Rad, Hercules, USA, #172‐5124), 1 μL each of forward and reverse primers and 3 μL of cDNA. Thirty‐nine cycles of amplification (pre‐incubation at 95°C for 2 min followed by each cycles consisting of 5 s at 95°C, 10 s at 60°C and added melting curve analysis during the 65–95°C). The results were calculated using Bio‐Rad CFX Manager software. With SlUBI as an internal control, the relative expression of each genes was calculated by the ∆∆Ct method (Schefe et al., 2006). The primer pairs for RT‐qPCR were designed using Primer3Plus (http://www.primer3plus.com) and blasted at NCBI database to ensure primer specific (see Table S4).
Scanning electron microscopy (SEM) assay
For SEM, 2 mm3 of flesh tomato peel was collected at BR + 7 stage for MicroTom and transgenic tomato and fixed with 2% glutaraldehyde for 12 h. Samples were dried using critical point drying method. Scanning electron microscopy was performed using SU3500 at 15 kV (Panikashvili et al., 2009).
Transcriptome and metabolic profiling
Both MicroTom and transgenic fruit were harvested at BR + 7. Fruit pericarp was ground into fine powder using liquid nitrogen. RNA isolation was performed using an RNeasy Plant Mini Kit (Qiagen, Stockach, Germany). The libraries were produced and sequenced by Illumina HiSeq2500/x. Raw sequences were filtered to remove the adaptor sequence, low‐quality reads (reads containing sequencing Ns > 10%) and short reads (Q < 10 nt), and the resulting sets of the high‐quality clean reads were used for transcriptome analysis. All clean reads were mapped to the reference genome using the TopHat (vision 2.1.1) (Kim et al., 2013) algorithm and conserved the mapped clean reads for the subsequent analysis. The mapped clean reads were calculated and then normalized to reads FPKM by Cuffquant and Cuffnorm software (vision 2.2.1). Differential expression analysis of two conditions/groups was performed using the DESeq2 R (vision 1.10.1) package, and DESeq2 (Anders and Huber, 2010) provides statistical routines for determining differential expression in digital gene expression data using a model based on the negative binomial distribution (Jiang and Wong, 2009) through indices of fold change (log2 ratio) and P‐value (false discovery rate, FDR). The resulting P‐values were adjusted using the Benjamini and Hochberg's approach for controlling the false discovery rate. Genes with an adjusted P‐value <0.01 and a log2 ratio >1.5 or <0.5 found by DESeq2 were assigned as differentially expressed. Clean data were deposited into the Genome Sequence Archive in Big Data Center, Beijing Institute of Genomics, Chinese Academy of Science, under accession number CRA001388 that are publicly accessible at http://bigd.big.ac.cn/gsa (Wang et al., 2017; Xu et al., 2018).
Metabolite profiling was carried out using a widely targeted metabolome method by Wuhan Metware Biotechnology Co., Ltd. (Wuhan, China) (http://www.metware.cn/). A liquid chromatography–electrospray ionization–tandem mass spectrometry (LC‐ESI‐MS/MS) system was used for the relative quantification of metabolites in dried tomato fruit samples (Zhu et al., 2018). The dried tomato fruit samples were crushed using a mixer mill (MM 400, Retsch) with a zirconia bead for 1.5 min at 30 Hz, and 100 mg dried powder was weighted and extracted overnight at 4°C with 1.0 mL pure methanol (or 70% aqueous methanol) containing 0.1 mg l−1 lidocaine (internal standard) for lipid‐solubility metabolites (or water‐solubility metabolites). Quantification of metabolites was carried out using a scheduled multiple reaction monitoring method (Chen et al., 2013).
Compound extraction and UPLC measurement
Tomatoes were harvested at 7 days after breaker (BR + 7). Fruit pericarp was freeze‐dried and ground into fine powder. Extraction was performed as previously described (Zhang et al., 2015). Briefly, 100 mg of fruit powder was extracted with 5 mL 70% (v/v) MeOH for 12 h at 4°C, under agitation. After centrifugation at 3000 g at 4°C for 15 min, the supernatant was taken. The pellets were re‐extracted 5 mL 70% (v/v) MeOH at 4°C for another 2 h. After centrifugation, the supernatant was combined and further diluted five times with 70% MeOH. The combined solution was centrifugation at 18 000 g at 4°C for 15 min, and 10 μL supernatant was injected for before UPLC analysis.
For phenylpropanoid analysis, the samples were run on a Dionex Ultimate 3000 Series UPLC (Thermo Scientific, MA, USA). Separation was on a 100 × 2.1 mm 1.9 μm Hypersil Gold C18 column (Thermo Scientific, MA, USA) using following gradient of 0.1% formic acid in ultrapure water as mobile phase A and 100% acetonitrile as mobile phase B, run at 0.5 mL/min at 40°C: 0–0.5 min, 20% B; 0.5–5.5 min, 2%–25% B; 5.5–7 min, 25%–95% B; 7–7.5 min, 95% B; 7.5–7.6 min, 95%–2% B; and 7.6–12 min, 2% B. Detection was performed at 330 nm for chlorogenic acid, ρ‐coumaric acid and ferulic acid, 350 nm for rutin. All samples were performed in biological triplicate. Compounds were quantified using standards purchased from Sigma‐Aldrich (https://www.sigmaaldrich.com/).
Yeast one‐hybrid assay
DNA fragment consisting of three copies of the SlDAHPS (3‐deoxy‐7‐phosphoheptulonate synthase, Solyc04g074480) and SlENO (Enolase, Solyc03g114500) promoter sequence containing the R2R3‐MYB core binding domain (−99 to −91 of SlDAHPS and −67 to −59 of SlENO) were chemically synthesized and then cloned into the pHis2‐Leu‐GW through Gateway Cloning (Zhang et al., 2015). The pHis2‐Leu‐SlDAHPS and pHis2‐Leu‐SlENO vectors were transformed into the yeast strain Y187 cells with the LiCl‐PEG method, respectively (Shim et al., 2013). The positive clones were selected on SD/‐Leu medium. Full‐length CDS of SlMIXTA‐like was introduced into pDEST22 vector to make the yeast expression vector pDEST22‐SlMIXTA‐like and transformed into yeast strains containing pHis2‐Leu‐SlDAHPS or pHis2‐Leu‐SlENO. The transformants were further grown on SD/‐Leu/‐Trp medium. Positive yeast clones were picked and grown in liquid culture and diluted into different concentrations (OD600 = 100, 10−2, 10−4). Eight microlitres of suspension was spotted on the SD/‐Leu/‐Trp/‐His medium, with or without 3‐AT (0 or 30 mm). For comparison, full‐length cDNA of AtMYB12 was used to replace SlMIXTA‐like as positive control (Zhang et al., 2015).
Transient dual‐luciferase assays
The plasmid construction of dual‐luciferase assay was done by Golden Braid 2.0 cloning strategy (Sarrion‐Perdigones et al., 2013). Firstly, the 1 kb region promoter of SlDAHPS (−1000 to +106) and SlENO (−1021 to +150) was cloned and inserted into pUPD2 plasmid. The promoters were then inserted into GB_3‐α1 vector with LUC gene and Tnos to make proSlDAHPS:LUC:Tnos or proSlENO:LUC:Tnos. In the meantime, the SlMIXTA‐like gene under the control of a 35S promoter was inserted into GB_3‐α2 vector to make pro35S:SlMIXTA‐like:T35S. The two α plasmids were combined by inserted into the Ω2 plasmid through new round of Golden Braid reaction. In the same construct, we combined the pGB3‐α1_ pro35S:REN:Tnos and pGB3‐α2_ pro35S:P19:T35S and converted to the Ω1 plasmid. Finally, the Ω1 and Ω2 plasmids were combined to make the α‐level plasmid (pro35S:SlMIXTA‐like:T35S/proSlENO:LUC:Tnos/pro35S:REN:Tnos/pro35S:P19:Tnos and pro35S:SlMIXTA‐like:T35S/proSlDAHPS:LUC:Tnos/pro35S:REN:Tnos/pro35S:P19:Tnos). The Ω‐level plasmid without the pro35S:SlMIXTA‐like:T35S part was used as the control (Sarrion‐Perdigones et al., 2013). The final binary vectors were directly transformed into Agrobacterium tumefaciens strain GV3101. The Agrobacterium cultures were grown to an OD600 of 0.8–1.0 and infiltrated into tobacco (Nicotiana benthamiana) leaves. Infiltrated leaves were harvested at 3dpi, and today protein was isolated by PBS solution. The transient expression was assayed using the Dual‐Luciferase Reporter Assay System (Promega, Madison, USA) (Llave et al., 2000). The ratio of LUC/REN was measured by a Synergy ™ H1 hybrid multimode microplate reader (BioTek) with according to the manufacturer's instructions.
Chromatin immunoprecipitation (ChIP)
The N‐terminal of SlMIXTA‐like was fused with the FLAG‐tag (DYKDDDK) to generate FLAG‐SlMIXTA‐like vector and constructed pBin19‐E8:FLAG‐SlMIXTA‐like through Gateway Cloning. ChIP analysis was performed as previously described (Zhang et al., 2015).
ChIP‐qPCR was performed on three independent replicates with appropriate primers. The ACTIN gene from tomato was used as the internal control for the ChIP‐qPCR experiments. Data were represented as the ratio of target genes/ACTIN in ChIPed DNA to target genes/ACTIN in input DNA.
Statistics
Unpaired, two‐tailed Student's t‐tests were used for comparison of individual lines with their relevant controls; P < 0.05 were considered significant. To compare measurements of multiple experiment designs with each other, we performed univariate ANOVA followed by the post hoc Tukey test of multiple pairwise comparisons to determine group differences; P < 0.05 were recognized as significant.
Conflicts of interests
All authors declare no competing interests.
Author contributions
YZ, SCW and JL designed the experiments. SY and MS performed the experiments, with help of YW, LZ, RF, YL and HG. SY, MS, SCW and YZ analysed the data and wrote the manuscript.
Supporting information
Figure S1 The effect of different alleles on the content of ρ‐coumaric acid (SlFM0124).
Figure S2 Phylogenetic tree of SlMIXTA‐like protein and R2R3‐MYB protein family from Arabidopsis thaliana.
Figure S3 Molecular characterization of SlMIXTA‐like.
Figure S4 SlMIXTA‐like expression pattern in tomato.
Figure S5 Overexpression of SlMIXTA‐like in tomato seedlings enhances trichome formation on stems.
Figure S6 Screening of SlMIXTA‐like expression in T0 E8:SlMIXTA‐like tomato fruits.
Figure S7 The expression of SlMIXTA‐like at different fruit development stages for both MicroTom and E8:SlMIXTA‐like‐B fruit.
Figure S8 The analysis of RNA‐seq data in E8:SlMIXTA‐like line A and B.
Figure S9 SlMIXTA‐like changes the expression of primary metabolic genes in the pericarp of E8:SlMIXTA‐like fruit.
Figure S10 The analysis of metabolism data in E8:SlMIXTA‐like line A and B.
Figure S11 The contents of rutin (a), ρ‐coumaric acid (b), chlorogenic acid (c) and ferulic acid (d) in E8:SlMIXTA‐like‐B and MicroTom.
Figure S12 Expression of carotenoid pathway genes (a) and carotenoid contents (b) in E8:SlMIXTA‐like‐B and MicroTom fruit.
Figure S13 SlMIXTA‐like can't directly bind to the promoter of SlENO.
Figure S14 ChIP‐qPCR indicates SlMIXTA‐like directly binds to the promoter of SlDAHPS in vivo.
Table S1 The list of collected 351 tomato varieties.
Table S2 The list of E8:SlMIXTA‐like‐A and B common DEGs in RNA‐seq.
Table S3 The list of SlMIXTA‐like‐A and B common DEMs in metabolic profiling.
Table S4 The list of oligonucleotids used in this study.
Data S1 The list of total 53 SNPs significantly associated with ρ‐coumaric acid in this study.
Data S2 All sample RNA‐seq fpkm.
Data S3 All sample metabolomics data.
Acknowledgements
This work is supported by a grant from National Natural Science Foundation of China (31701255). YZ was also supported by the National One Thousand Young Talents programme from China and the Fundamental Research Funds for the Central Universities (2019SCUD013, YJ201640 and 2017SCU04A11). SCW was supported by the Hainan Provincial Natural Science Foundation of China (319QN155) and Hainan University Startup Fund (KYQD(ZR)1916).
Contributor Information
Shouchuang Wang, Email: shouchuang.wang@hainanu.edu.cn.
Yang Zhang, Email: yang.zhang@scu.edu.cn.
References
- Adato, A. , Mandel, T. , Mintz‐Oron, S. , Venger, I. , Levy, D. , Yativ, M. , Dominguez, E. et al. (2009) Fruit‐surface flavonoid accumulation in tomato is controlled by a SlMYB12‐regulated transcriptional network. PLoS Genet. 5, e1000777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders, S. and Huber, W. (2010) Differential expression analysis for sequence count data. Genome Biol. 11, R106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balcke, G.U. , Bennewitz, S. , Bergau, N. , Athmer, B. , Henning, A. , Majovsky, P. , Jimenez‐Gomez, J.M. et al. (2017) Multi‐omics of tomato glandular trichomes reveals distinct features of central carbon metabolism supporting high productivity of specialized metabolites. Plant Cell, 29, 960–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ballester, A.R. , Molthoff, J. , de Vos, R. , Hekkert, B.L. , Orzaez, D. , Fernandez‐Moreno, J.P. , Tripodi, P. et al. (2009) Biochemical and molecular analysis of pink tomatoes: deregulated expression of the gene encoding transcription factor SlMYB12 leads to pink tomato fruit color. Plant Physiol. 152, 71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borevitz, J.O. , Xia, Y. , Blount, J. , Dixon, R.A. and Lamb, C. (2000) Activation tagging identifies a conserved MYB regulator of phenylpropanoid biosynthesis. Plant Cell, 12, 2383–2394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bovy, A. , de Vos, R. , Kemper, M. , Schijlen, E. , Almenar Pertejo, M. , Muir, S. , Collins, G. et al. (2002) High‐flavonol tomatoes resulting from the heterologous expression of the maize transcription factor genes LC and C1. Plant Cell, 14, 2509–2526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butelli, E. , Titta, L. , Giorgio, M. , Mock, H.P. , Matros, A. , Peterek, S. , Schijlen, E.G. et al. (2008) Enrichment of tomato fruit with health‐promoting anthocyanins by expression of select transcription factors. Nat. Biotechnol. 26, 1301–1308. [DOI] [PubMed] [Google Scholar]
- Carmona‐Gutierrez, D. , Zimmermann, A. , Kainz, K. , Pietrocola, F. , Chen, G. , Maglioni, S. , Schiavi, A. et al. (2019) The flavonoid 4,4’‐dimethoxychalcone promotes autophagy‐dependent longevity across species. Nat. Commun. 10, 651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Century, K. , Reuber, T.L. and Ratcliffe, O.J. (2008) Regulating the regulators: the future prospects for transcription‐factor‐based agricultural biotechnology products. Plant Physiol. 147, 20–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, W. , Gong, L. , Guo, Z. , Wang, W. , Zhang, H. , Liu, X. , Yu, S. et al. (2013) A novel integrated method for large‐scale detection, identification, and quantification of widely targeted metabolites: application in the study of rice metabolomics. Mol. Plant, 6, 1769–1780. [DOI] [PubMed] [Google Scholar]
- Deikman, M.L.K.A.J. (1996) The tomato E8 gene lnfluences ethylene biosynthesis in fruit but not in flowers. Plant Physiol., 112, 537–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubos, C. , Stracke, R. , Grotewold, E. , Weisshaar, B. , Martin, C. and Lepiniec, L. (2010) MYB transcription factors in Arabidopsis. Trends Plant Sci. 15, 573–581. [DOI] [PubMed] [Google Scholar]
- Ewas, M. , Gao, Y. , Wang, S. , Liu, X. , Zhang, H. , Nishawy, E.M.E. , Ali, F. et al. (2016) Manipulation of SlMX1 for enhanced carotenoids accumulation and drought resistance in tomato. Sci. Bulletin, 61, 1413–1418. [Google Scholar]
- Fang, C. , Fernie, A.R. and Luo, J. (2019) Exploring the diversity of plant metabolism. Trends Plant Sci. 24, 83–98. [DOI] [PubMed] [Google Scholar]
- Fischer, J.E.L.A.R.L. (1988) Diverse mechanisms for the regulation of ethylene‐inducible gene expression. Mol. Gen. Genet. 212, 71–75. [DOI] [PubMed] [Google Scholar]
- Fu, R. , Martin, C. and Zhang, Y.J.M.P. (2018) Next‐generation plant metabolic engineering, inspired by an ancient Chinese irrigation system. Mol Plant, 11, 47–57. [DOI] [PubMed] [Google Scholar]
- Glas, J.J. , Schimmel, B.C.J. , Alba, J.M. , Escobar‐Bravo, R. , Schuurink, R.C. and Kant, M.R. (2012) Plant glandular trichomes as targets for breeding or engineering of resistance to herbivores. Int. J. Mol. Sci. 13, 17077–17103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glover, B.J. , Perez-Rodriguez, M. and Martin, C. (1998) Development of several epidermal cell types can be specified by the same MYB‐related plant transcription factor. Development, 127, 3497–3508. [DOI] [PubMed] [Google Scholar]
- Gonzali, S. , Mazzucato, A. and Perata, P. (2009) Purple as a tomato: towards high anthocyanin tomatoes. Trends Plant Sci. 14, 237–241. [DOI] [PubMed] [Google Scholar]
- Hartmann, U. , Sagasser, M. , Mehrtens, F. , Stracke, R. and Weisshaar, B. (2005) Differential combinatorial interactions of cis‐acting elements recognized by R2R3‐MYB, bZIP, and bHLH factors control light‐responsive and tissue‐specific activation of phenylpropanoid biosynthesis genes. Plant Mol. Biol. 57, 155–171. [DOI] [PubMed] [Google Scholar]
- Huchelmann, A. , Boutry, M. and Hachez, C. (2017) Plant glandular trichomes: natural cell factories of high biotechnological interest. Plant Physiol. 175, 6–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang, H. and Wong, W.H. (2009) Statistical inferences for isoform expression in RNA‐Seq. Bioinformatics, 25, 1026–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang, H.M. , Sul, J.H. , Service, S.K. , Zaitlen, N.A. , Kong, S.Y. , Freimer, N.B. , Sabatti, C. et al. (2010) Variance component model to account for sample structure in genome‐wide association studies. Nat. Genet. 42, 348–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, D. , Pertea, G. , Trapnell, C. , Pimentel, H. , Kelley, R. and Salzberg, S.L.J.G.B. (2013) TopHat2: accurate alignment of transcriptomes in the presence of insertions, deletions and gene fusions. Genome Biol. 14, R36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lashbrooke, J. , Adato, A. , Lotan, O. , Alkan, N. , Tsimbalist, T. , Rechav, K. , Fernandez‐Moreno, J.P. et al. (2015) The tomato MIXTA‐Like transcription factor coordinates fruit epidermis conical cell development and cuticular lipid biosynthesis and assembly. Plant Physiol. 169, 2553–2571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, Y. , Wang, H. , Zhang, Y. and Martin, C. (2018) Can the world's favorite fruit, tomato, provide an effective biosynthetic chassis for high‐value metabolites? Plant Cell Rep. 37, 1443–1450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, J. , Osbourn, A. and Ma, P. (2015) MYB transcription factors as regulators of phenylpropanoid metabolism in plants. Mol Plant, 8, 689–708. [DOI] [PubMed] [Google Scholar]
- Liu, Z. , Jia, Y. , Ding, Y. , Shi, Y. , Li, Z. , Guo, Y. , Gong, Z. et al. (2017) Plasma membrane CRPK1‐mediated phosphorylation of 14‐3‐3 proteins induces their nuclear import to fine‐tune CBF signaling during cold response. Mol. Cell, 66, 117–128 e115. [DOI] [PubMed] [Google Scholar]
- Llave, C. , Kasschau, K.D. and Carrington, J.C. (2000) Virus‐encoded suppressor of posttranscriptional gene silencing targets a maintenance step in the silencing pathway. Plant Biol. 97, 13401–13406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo, J. (2015) Metabolite‐based genome‐wide association studies in plants. Curr. Opin. Plant Biol. 24, 31–38. [DOI] [PubMed] [Google Scholar]
- Luo, J. , Butelli, E. , Hill, L. , Parr, A. , Niggeweg, R. , Bailey, P. , Weisshaar, B. et al. (2008) AtMYB12 regulates caffeoyl quinic acid and flavonol synthesis in tomato: expression in fruit results in very high levels of both types of polyphenol. Plant J. 56, 316–326. [DOI] [PubMed] [Google Scholar]
- Martin, C. and Li, J. (2017) Medicine is not health care, food is health care: plant metabolic engineering, diet and human health. New Phytol. 216, 699–719. [DOI] [PubMed] [Google Scholar]
- McCormick, S. , Niedermeyer, J. , Fry, J. , Barnason, A. , Horsch, R. and Fraley, R. (1986) Leaf disc transformation of cultivated tomato (L. esculentum) using Agrobacterium tumefaciens . Plant Cell Rep. 5, 81–84. [DOI] [PubMed] [Google Scholar]
- Morandini, P. (2013) Control limits for accumulation of plant metabolites: brute force is no substitute for understanding. Plant Biotechnol. J. 11, 253–267. [DOI] [PubMed] [Google Scholar]
- Ni, M. , Cui, D. , Einstein, J. , Narasimhulu, S. , Vergara, C.E. and Gelvin, S.B.J.T.P.J. (1995) Strength and tissue specificity of chimeric promoters derived from the octopine and mannopine synthase genes. Plant J. 7, 661–676. [Google Scholar]
- Panikashvili, D. , Shi, J.X. , Schreiber, L. and Aharoni, A. (2009) The Arabidopsis DCR encoding a soluble BAHD acyltransferase is required for cutin polyester formation and seed hydration properties. Plant Physiol. 151, 1773–1789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patron, N.J. , Orzaez, D. , Marillonnet, S. , Warzecha, H. , Matthewman, C. , Youles, M. , Raitskin, O. et al. (2015) Standards for plant synthetic biology: a common syntax for exchange of DNA parts. New Phytol. 208, 13–19. [DOI] [PubMed] [Google Scholar]
- Perez‐Rodriguez, M. , Jaffe, F.W. , Butelli, E. , Glover, B.J. and Martin, C. (2005) Development of three different cell types is associated with the activity of a specific MYB transcription factor in the ventral petal of Antirrhinum majus flowers. Development, 132, 359–370. [DOI] [PubMed] [Google Scholar]
- Sarrion‐Perdigones, A. , Vazquez‐Vilar, M. , Palaci, J. , Castelijns, B. , Forment, J. , Ziarsolo, P. , Blanca, J. et al. (2013) GoldenBraid 2.0: a comprehensive DNA assembly framework for plant synthetic biology. Plant Physiol. 162, 1618–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scarano, A. , Butelli, E. , De Santis, S. , Cavalcanti, E. , Hill, L. , De Angelis, M. , Giovinazzo, G. et al. (2017) Combined dietary anthocyanins, flavonols, and stilbenoids alleviate inflammatory bowel disease symptoms in mice. Front Nutr. 4, 75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schefe, J.H. , Lehmann, K.E. , Buschmann, I.R. , Unger, T. and Funke‐Kaiser, H. (2006) Quantitative real‐time RT‐PCR data analysis: current concepts and the novel “gene expression's CT difference” formula. J. Mol. Med. (Berl) 84, 901–910. [DOI] [PubMed] [Google Scholar]
- Schwinn, K. , Venail, J. , Shang, Y. , Mackay, S. , Alm, V. , Butelli, E. , Oyama, R. et al. (2006) A small family of MYB‐regulatory genes controls floral pigmentation intensity and patterning in the genus Antirrhinum. Plant Cell, 18, 831–851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim, J.S. , Jung, C. , Lee, S. , Min, K. , Lee, Y.W. , Choi, Y. , Lee, J.S. et al. (2013) AtMYB44 regulates WRKY70 expression and modulates antagonistic interaction between salicylic acid and jasmonic acid signaling. Plant J. 73, 483–495. [DOI] [PubMed] [Google Scholar]
- Tan, H. , Xiao, L. , Gao, S. , Li, Q. , Chen, J. , Xiao, Y. , Ji, Q. et al. (2015) TRICHOME AND ARTEMISININ REGULATOR 1 is required for trichome development and artemisinin biosynthesis in Artemisia annua . Mol. Plant 8, 1396–1411. [DOI] [PubMed] [Google Scholar]
- Tzin, V. , Malitsky, S. , Ben Zvi, M.M. , Bedair, M. , Sumner, L. , Aharoni, A. and Galili, G. (2012) Expression of a bacterial feedback‐insensitive 3‐deoxy‐D‐arabino‐heptulosonate 7‐phosphate synthase of the shikimate pathway in Arabidopsis elucidates potential metabolic bottlenecks between primary and secondary metabolism. New Phytol. 194, 430–439. [DOI] [PubMed] [Google Scholar]
- Tzin, V. , Rogachev, I. , Meir, S. , Moyal Ben Zvi, M. , Masci, T. , Vainstein, A. , Aharoni, A. et al. (2013) Tomato fruits expressing a bacterial feedback‐insensitive 3‐deoxy‐D‐arabino‐heptulosonate 7‐phosphate synthase of the shikimate pathway possess enhanced levels of multiple specialized metabolites and upgraded aroma. J. Exp. Bot. 64, 4441–4452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vogt, T. (2010) Phenylpropanoid biosynthesis. Mol. Plant 3, 2–20. [DOI] [PubMed] [Google Scholar]
- Wang, Y. , Song, F. , Zhu, J. , Zhang, S. , Yang, Y. , Chen, T. , Tang, B. et al. (2017) GSA: genome sequence archive. Genom. Proteom. Bioinf. 15, 14–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, X.J. , Hao, L.L. , Zhu, J.W. , Zhou, Q. , Song, F.H. , Chen, T.T. , Zhang, S.S. et al. (2018) Database resources of the BIG data center in 2018. Nucleic Acids Res. 46, D14–D20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y. , Butelli, E. , Alseekh, S. , Tohge, T. , Rallapalli, G. , Luo, J. , Kawar, P.G. et al. (2015) Multi‐level engineering facilitates the production of phenylpropanoid compounds in tomato. Nat. Commun. 6, 8635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, G. , Wang, S. , Huang, Z. , Zhang, S. , Liao, Q. , Zhang, C. , Lin, T. et al. (2018) Rewiring of the fruit metabolome in tomato breeding. Cell, 172, 249–261 e212. [DOI] [PubMed] [Google Scholar]
- Zouine, M. , Maza, E. , Djari, A. , Lauvernier, M. , Frasse, P. , Smouni, A. , Pirrello, J. et al. (2017) TomExpress, a unified tomato RNA‐Seq platform for visualization of expression data, clustering and correlation networks. Plant J. 92, 727–735. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 The effect of different alleles on the content of ρ‐coumaric acid (SlFM0124).
Figure S2 Phylogenetic tree of SlMIXTA‐like protein and R2R3‐MYB protein family from Arabidopsis thaliana.
Figure S3 Molecular characterization of SlMIXTA‐like.
Figure S4 SlMIXTA‐like expression pattern in tomato.
Figure S5 Overexpression of SlMIXTA‐like in tomato seedlings enhances trichome formation on stems.
Figure S6 Screening of SlMIXTA‐like expression in T0 E8:SlMIXTA‐like tomato fruits.
Figure S7 The expression of SlMIXTA‐like at different fruit development stages for both MicroTom and E8:SlMIXTA‐like‐B fruit.
Figure S8 The analysis of RNA‐seq data in E8:SlMIXTA‐like line A and B.
Figure S9 SlMIXTA‐like changes the expression of primary metabolic genes in the pericarp of E8:SlMIXTA‐like fruit.
Figure S10 The analysis of metabolism data in E8:SlMIXTA‐like line A and B.
Figure S11 The contents of rutin (a), ρ‐coumaric acid (b), chlorogenic acid (c) and ferulic acid (d) in E8:SlMIXTA‐like‐B and MicroTom.
Figure S12 Expression of carotenoid pathway genes (a) and carotenoid contents (b) in E8:SlMIXTA‐like‐B and MicroTom fruit.
Figure S13 SlMIXTA‐like can't directly bind to the promoter of SlENO.
Figure S14 ChIP‐qPCR indicates SlMIXTA‐like directly binds to the promoter of SlDAHPS in vivo.
Table S1 The list of collected 351 tomato varieties.
Table S2 The list of E8:SlMIXTA‐like‐A and B common DEGs in RNA‐seq.
Table S3 The list of SlMIXTA‐like‐A and B common DEMs in metabolic profiling.
Table S4 The list of oligonucleotids used in this study.
Data S1 The list of total 53 SNPs significantly associated with ρ‐coumaric acid in this study.
Data S2 All sample RNA‐seq fpkm.
Data S3 All sample metabolomics data.