

Abstract
Sepsis-associated encephalopathy (SAE) is commonly complicated by septic conditions, and is responsible for increased mortality and poor outcomes in septic patients. Uncontrolled neuroinflammation and ischemic injury are major contributors to brain dysfunction, which arises from intractable immune malfunction and the collapse of neuroendocrine immune networks, such as the cholinergic anti-inflammatory pathway, hypothalamic-pituitary-adrenal axis, and sympathetic nervous system. Dysfunction in these neuromodulatory mechanisms compromised by SAE jeopardizes systemic immune responses, including those of neutrophils, macrophages/monocytes, dendritic cells, and T lymphocytes, which ultimately results in a vicious cycle between brain injury and a progressively aberrant immune response. Deep insight into the crosstalk between SAE and peripheral immunity is of great importance in extending the knowledge of the pathogenesis and development of sepsis-induced immunosuppression, as well as in exploring its effective remedies.
Keywords: Sepsis-associated encephalopathy, Immune suppression, Vicious cycle, Therapeutic target
Background
Sepsis is one of the major threats to the survival and prognosis of patients in intensive care units (ICUs) but lacks specific and effective treatments. According to the new definition of sepsis 3.0, life-threatening organ dysfunction caused by an aberrant immune response to infection is responsible for the pathogenesis and progression of sepsis [1]. The interplay between malfunctions in the immune system and multiple organ damage is deemed the major cause of poor outcomes among sepsis cases. Maddux and colleagues reported a close association between disturbed innate immune responses and organ failure in adults with sepsis [2]. Dysfunction of multiple organs results in extensive aberration of the immune response, which may accelerate the progression of sepsis. For example, brain injury is commonly complicated by the septic state and is usually identified as the first organ exposed to an inflammatory episode [3]. Of note, sepsis-associated encephalopathy (SAE) is one of the most common etiological factors for febrile encephalopathy, especially in elderly people. Approximately 70% of patients with bacteremia develop neurological symptoms ranging from lethargy to coma, and over 80% suffer from abnormalities as measured by electroencephalogram (EEG) [3, 4]. Moreover, it has been identified that SAE is critically involved in increased mortality, extensive in-hospital cost, and prolonged hospitalization, followed by persistent cognitive impairment and limitations in physical function [5, 6]. Therefore, early recognition and prompt interference for brain injury are of great importance for the survival and prognosis of septic patients.
As we know, dysfunction of the central nervous system is responsible for the collapse of the peripheral immune system because of its central role in multiple types of neuroendocrine immune networks, including the hypothalamic-pituitary-adrenal (HPA) axis and the sympathetic and parasympathetic nervous systems. The inflammatory signals reach the different brain regions mainly through two disparate ways: the humoral and neural pathways, which involve a compromised blood–brain barrier (BBB) and activation of afferent fibers of the vagus nerve, respectively [7]. The brain further processes and manipulates the peripheral inflammatory response by initiating the neural reflex and promoting the release of neurotransmitters. For example, the cholinergic anti-inflammatory pathway (CAP), which is composed of brain cholinergic nuclei, efferent vagus nerve, and peripheral α7 nicotinic acetylcholine receptors (α7nAchRs), is reportedly beneficial for various diseases because of its anti-inflammatory capacity [8]. It has been demonstrated that activation of the CAP, either by stimulation of vagus nerve or administration of agonists for α7nAchR, significantly alleviates multiple organ damage and improves survival of septic animals [9, 10]. However, the corruption in any component of the CAP directly leads to its unresponsiveness or an aberrant response. In traumatic brain injury (TBI), for instance, the vagus nerve presents with obvious overactivity, which is responsible for the development of immune paralysis, suggesting that there is feedback for the loss of brain cholinergic nuclei [11]. The pathophysiological progression of TBI reportedly results from dysregulation of the cholinergic and inflammatory systems, while modulation of cholinergic activity shows great benefit for brain injury, and it serves as a promising remedy with neuroprotection [12, 13]. Thus, the viability and functional homeostasis of the brain cholinergic system are essential for the integrity of CAP activity. In fact, extensive apoptosis of cholinergic neurons was observed under septic exposure, which showed a close connection to an unresolved inflammatory response, as reported by Zaghloul et al., suggesting that dysfunction of the central nervous system might be an important contributor to the collapse of neuroendocrine immune networks, as well as a potential therapeutic target for sepsis-induced immune depression [14]. Even worse, brain injury might act as a vicious cycle for sepsis-induced immunosuppression because of its pivotal role in neuroendocrine immune networks.
The role of the immune response in the pathogenesis of sepsis associated encephalopathy
Multiple factors are reportedly involved in the pathogenesis of SAE (Fig. 1), including inflammatory cytokines, collapse of the BBB, ischemic processes, alterations in neurotransmitters, and mitochondrial dysfunction, while the specific mechanism has not yet been established. Two types of mechanisms have been identified with critical involvement in the development of brain injury: uncontrolled neuroinflammation and ischemic injury, which are common presentations among patients with severe sepsis [15]. Notably, the dysregulated immune response is confirmed to be a major contributor to the onset of sepsis, which highlights its pivotal role in the progression of multiple organ dysfunction syndrome (MODS), especially for the central nervous system, which is vulnerable to inflammatory insults [1, 2]. In addition, the interplay between uncontrolled inflammation and ischemic injury makes SAE a difficult issue.
Fig. 1.
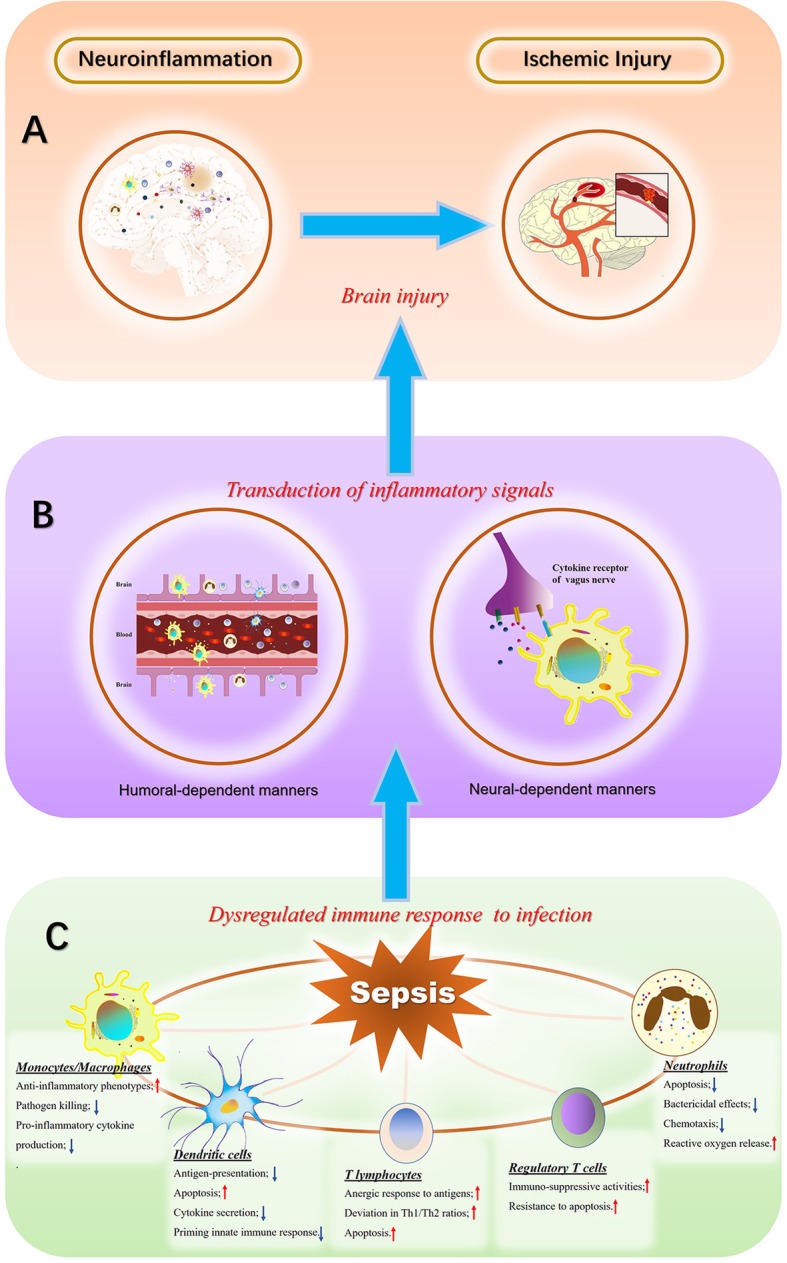
Pathogenesis of sepsis associated encephalopathy (SAE). Neuroinflammation and ischemic injury are considered as major causes of SAE (a), which arises from a dysregulated peripheral response to infection. The source of neuroinflammation includes both resident immune cells, such as microglia and astrocytes, and infiltration of peripheral inflammatory mediators and immune cells. In addition, inflammatory insults are responsible for the ischemic process. The inflammatory signals can reach different brain regions in both neural- and humoral-dependent manners after the initiation of septic challenge (b) and involve aberrant infiltration of blood–brain barrier (BBB), saturable transportation and specific areas without covering of BBB as well as neuro-inflammatory receptors. The abnormal immune response to infection is closely associated with the pathogenesis and progression of sepsis, according to sepsis 3.0 definition (c), and it is also a major contributor to irreversible brain damage
Neuroinflammatory insults
Inflammatory signals can reach different brain regions in both neural- and humoral- dependent manners after initiation of septic syndrome. For example, visceral inflammation can be detected by the vagus nerve via terminal cytokine receptors, which further stimulates brain cholinergic nuclei and acts as negative feedback for the inflammatory response by releasing acetylcholine (Ach) at the terminal efferent nerve [16]. Other mechanisms contribute to the transmission of peripheral inflammatory signals, including aberrant infiltration of the BBB, saturable transportation, and specific areas without covering of BBB [17]. In fact, the brain is the first organ that suffers from septic challenge, which might result in different impacts on the peripheral immune system in sepsis [18]. In the initial stage of sepsis, activation of the functional nucleus in the central nervous system provides an essential feedback for the peripheral inflammatory response, and it serves as a crucial interchange for multiple neuroendocrine immune networks. The CAP, as an example, has been demonstrated to have beneficial effects on various critical illnesses via inhibiting the inflammatory response [8]. However, excessive production of inflammatory cytokines, such as tumor necrosis factor (TNF)-α and high mobility group box-1 protein (HMGB1), results in deteriorative neuroinflammation and extensive loss of brain cells, as noted with ablation under specific antagonism [19, 20]. TNF-α appears to be a key mediator of SAE due to its direct correlation with BBB corruption, brain edema, neutrophil infiltration, astrocytosis, and apoptosis of brain cells, which do not occur in TNFR1-deficient mice [19]. HMGB1, commonly regarded as a later lethal mediator in sepsis, is significantly increased in different brain regions under expose to sepsis [20]. Antagonism of both blood and cerebral HMGB1 is beneficial in SAE by preventing the loss of brain cells and restoring neurocognitive function, indicating an important role for the release of inflammatory cytokines in the development of SAE [20–22].
Neuroinflammation is critically involved in the pathogenesis of SAE, as uncontrolled inflammation is a primary indicator of septic conditions. Neuroinflammation is responsible for dysfunction and massive apoptosis of brain cells, including microglial cells, neurons, and endothelial cells [23]. Both peripheral inflammation and local inflammation are induced by activation of resident brain immune cells, such as microglial cells, and astrocytes, and reportedly accounts for the induction of neuroinflammatory response and worse outcomes due to septic complications [24–26]. Over activation of microglia, for instance, is involved in the progression of brain dysfunction by deteriorating the BBB and enhancing the release of reactive oxygen species (ROS) [24, 27]. In contrast, inhibition of microglia is beneficial for abating brain oxidative damage and inflammation in sepsis, along with improvements in long-term cognitive function [24]. Astrocytes also play pivotal roles in driving inflammatory brain injury due to critical involvement in brain-immune interfaces [28]. It is reported to orchestrate the effects of immune cells in central nervous system by acting as a surveillant yet integrate center for inflammatory signals [26]. Under septic exposure, however, astrocytes present with aberrant responses that promote intractable neuroinflammation and cognitive impairment [26, 29]. The activation of astrocytes is detected in brain tissues as early as 4 h following sepsis, which attains the peak at 24 h [28]. Activated astrocytes are capable of releasing multiple kinds of inflammatory mediators, such as TNF-α, interleukin (IL)-1β, IL-6, and IL-18, and further facilitating the development of neuroinflammation, which are interpreted by enhancing expressions of p21 and nuclear factor (NF)-κB [29]. In addition, the structure of astrocytes reveals extensive changes under LPS challenge, as evidenced by structural remodeling and loss of end feet, which are responsible for the collapse of BBB [30]. Abnormal performance and loss of endothelial cells also show great potential in augmenting deterioration of the brain after the onset of sepsis through a direct link to the collapse of BBB, increased infiltration of inflammatory cells, aberrant migration of microglia, and excessive formation of ROS and nitric oxide (NO) [31]. Injury of endothelial cells as a result of neuroinflammation leads to derangement of cerebral perfusion, which renders the ischemic processes of SAE an intractable problem [32].
Ischemic processes
Reduction in brain perfusion plays an important role in the development of SAE, and it can directly result in the abnormal cellular metabolism and oxidative stress [32]. Cerebral blood flow (CBF) is found to be significantly lower in patients with sepsis compared to that of normal controls, which shows a tight association with brain metabolism impairment [33, 34]. Similarly, cerebrovascular autoregulation presents with marked impairment and acts as one of the major triggers of SAE [35]. However, the precise mechanism of inadequate cerebral perfusion and abnormal autoregulation have not been elucidated. Multiple factors are involved in the pathogenesis of disturbed cerebral perfusion and microcirculation, such as abnormal vasoconstriction, vasopressors, decreased mean arterial pressure, and inflammatory insults [36]. The effects of other factors, including coagulation and platelets, are also noteworthy but remain controversial in the development of ischemia during SAE. A prothrombogenic phenotype is noticed at 4 h after operation by cecal ligation and puncture, accompanied by outward signs of brain dysfunction [37]. Indeed, disseminated intravascular coagulation is reportedly responsible for extensive cerebral ischemia and poor outcomes in fatal septic shock [38]. The count and mass of platelets are critically involved in maintaining the integrity of cerebral microcirculation. However, further researches did not reveal a significant difference between profound behavioral deficits and platelet recruitment in cerebral venules [37]. These conclusions were further validated by postmortem analysis of brain tissues from patients who died from septic shock. Sharshar and colleagues observed that all patients with septic shock presented with cerebral ischemia, which ranked the first in neuropathologic features, followed by hemorrhages and hypercoagulability syndrome [39]. Nevertheless, the latter two pathologies revealed no close relationship to the incidence of cerebral clotting disturbance [39]. A study by Feng et al. showed that the platelet counts were not significantly different between the SAE and non-SAE groups, or between the survival and non-survival groups in the setting of sepsis [40]. The interplay between neuroinflammation and ischemic injury is a great threat to the central nervous system following sepsis. Uncontrolled neuroinflammation jeopardizes the cerebrovascular system by inducing damage to vascular endothelial cells and imbalances in neurotransmitters, resulting in thrombogenesis and abnormal vasoconstriction, thereby further exacerbating the ischemic brain [41]. In return, the ischemic process worsens local inflammation by either increasing the infiltration of inflammatory cells or inducing dysfunction of resident immune cells that is difficult to resolve, indicating that the crosstalk between neuroinflammation and ischemic insult is a potential accelerator for the development of SAE and forms a vicious cycle [42]. Therefore, homeostasis of the immune response is of great significance in maintaining the functional integrity of the central nervous system after septic challenge.
The impact of brain injury on the response of various immune cells
The dysregulated immune response is considered as a major cause for the development of sepsis and is a critical contributor in the pathogenesis of SAE. The influence of brain dysfunction on the systemic immune response is also a noteworthy issue for the progression of sepsis, as it plays an integral role in the neuroimmune networks (Table 1). In this regard, disruption of the HPA axis is commonly complicated by sepsis, accounts for disruption of vascular sensitivity and increases the likelihood of death [59]. The HPA axis plays an essential role in resolving systemic inflammation and immune responses by releasing glucocorticoid, while insufficient activation is considered a pivotal predictor for poor outcomes of critically ill patients [60, 61]. Therefore, reconstruction of the HPA axis may act as a promising therapeutic strategy for improving the survival and prognosis of septic patients [62]. Deep insight into the potential mechanism of the impact of brain injury on the responses of multiple types of immune cells is essential for understanding the pathophysiology of sepsis-induced immunosuppression.
Table 1.
The function and activity of immune cells exposed to brain injury
| Types of immune cells | Changes in function and activity | References |
|---|---|---|
| Monocytes/macrophages | Decreasing the number of monocytes; polarization of M2 phenotype; increasing production of IL-10; reducing phagocytic capability | [43], [44], [45] |
| Neutrophils | Impairment in ROS production; reduction of phagocytosis; shortening life-span as a result of spontaneous apoptosis | [43, 46–49] |
| Dendritic cells | Decreasing number of circulating DCs; anergic response to TLR3 and TLR4 stimulations; incapable of priming effective cellular immune response; disturbed infiltration and aberrant phenotypic differentiation | [50–53] |
| T lymphocytes | Reducing proportion of peripheral T cells; disturbing response to antigens; polarization of anti-inflammatory phenotypes; inducing imbalance between Tregs and proinflammatory phenotypes of T cells | [54–58] |
Monocytes and macrophages
The functional status and differential phases of monocytes or macrophages reveal the responsive capacity of the innate immune system, which serves as the main repository for multiple types of inflammatory mediators. Several studies have documented that monocytes and macrophages are either hyporesponsive or hyperactive during sepsis and have dysregulated cytokine production, resulting in compromised survival of septic animals [63–65]. Inflammatory monocytes and macrophages constitute a large proportion of the initial infiltrates in the central nervous system after septic exposure [66]. The activation of both infiltrated monocytes or macrophages and resident microglial cells is closely associated with excessive neuroinflammation [46, 67]. Thus, targeting the infiltration of inflammatory monocytes and inhibiting the activation of microglial cells might resolve the neuroinflammatory response and improve cognitive performance in sepsis, suggesting the potential therapeutic significance of monocytes or macrophages in SAE [46, 66].
The function and polarization of monocytes are compromised by different types of brain dysfunction. For instance, nosocomial infection is commonly complicated and regarded as a major cause of mortality secondary to severe TBI, which brings about outward signs of systemic immune depression [43, 68]. The number of monocytes was markedly decreased at 24 h post TBI, resulting in immune suppression accompanied by dominant differentiation of M2 phenotype cells [44]. Moreover, expression of intracellular IL-10 was significantly upregulated in monocytes resulting from TBI, suggesting the prominent capacity of brain dysfunction in eliciting anti-inflammatory responses [45]. Other conditions that cause a spectrum of brain dysfunction, such as neuroinflammation, brain edema, cognitive impairment, and locomotor dysregulation, involve aberrant responses of monocytes and macrophages. In addition, the phagocytic capability of alveolar macrophages was shown to be markedly reduced and augmented systemic inflammation and pulmonary damage [69]. Collectively, SAE might act as a second hit for compromised viability and the dysfunction of monocytes and macrophages in sepsis.
However, the specific mechanism underlying the unresponsiveness and apoptosis of monocytes and macrophages in acute TBI remains elusive. In physiological conditions, the central nervous system manipulates systemic inflammatory signals and immunomodulation mainly through the following three pathways: HPA axis, CAP, and sympathetic nervous system. In brain injury, however, these mechanisms act as feedback for the loss or dysfunction of central nuclei and elicit outward signs of hyperactivation, which are responsible for the abnormal response of the immune system. It has been documented that the activity of the HPA axis is enhanced after brain injury, and it relates to marked immunosuppression by increasing the production of corticosteroid [70]. A similar tendency was also observed with the CAP and the sympathetic nervous system, both of which exhibit hyperactivity during brain injury and result in immune dissonance of monocytes and macrophages. For example, the vagus nerve is overactive in brain injury, which accounts for the disturbed chemotaxis and decline in production of proinflammatory cytokines by monocytes and macrophages [11, 71]. Other factors, such as the failure in locomotor activity due to unconsciousness status, sedation, and inappropriate administration of glucocorticoids, are potential contributors to the compromised viability and function of monocytes and macrophages during SAE and should be addressed promptly.
Neutrophils
Neutrophils are an active participant in the innate immune response and are mobilized early after infection. Reprograming of neutrophils is regarded as one of the major features of sepsis-induced immune dysfunction, as evidenced by abnormal accumulation and inefficient chemotaxis and bactericidal effects in sepsis [72]. Restoring the viability and functional homeostasis of neutrophils has beneficial effects on the survival and prognosis of septic animals [73]. Neutrophils display increased infiltration in the central nervous system and are associated with inflammatory insults at the early stage of sepsis, but they are rarely found in the normal brain [66]. Neutrophils accumulating in the central nervous system jeopardize brain cells by releasing inflammatory cytokines, increasing the activity of myeloperoxidase, and promoting oxidative damage [74]. Taken together, the balance between infiltration and withdrawal of neutrophils is of great importance for the development of SAE.
Peripheral neutrophils suffer from disorder and dysregulated apoptosis after exposure to different brain injuries. For example, in the early stage of TBI, peripheral neutrophils have remarkable infiltration in the central nervous system, which is deemed a protective behavior by eliminating damaged neurons and improving tissue repair [75]. Nevertheless, persistent accumulation of neutrophils was reported to be related to cell death, brain edema, and tissue loss, which was abated by neutrophil depletion [47]. In addition, neutrophils are implicated in early TBI and are characterized by increased ROS generation partly due to the abnormal regulatory feedback of the brain injury [48, 68]. Therefore, the vicious cycle between overactivation of neutrophils and dysfunction of the central nervous system might result in difficulties in developing efficient treatments for SAE. Persistent exposure to brain disorders, however, brings about noteworthy suppression of the function and activity of peripheral neutrophils. ROS production in neutrophils was significantly impaired in the days following brain injury, accompanied by a marked reduction in phagocytosis [49, 68]. Even though no obvious loss of neutrophil counts was found at any time point post brain injury, the lifespan of neutrophils was demonstrably shorter than those from uninjured patients as a result of spontaneous apoptosis [44, 48]. It has been demonstrated that activation of the complement system, such as C5a, is responsible for disturbed neutrophil phagocytosis that is implicated in brain injury [76], but the mechanisms of the impaired ROS production and spontaneous apoptosis remain unknown.
Dendritic cells
Dendritic cells (DCs) are essential in maintaining homeostasis of the immune response, serving as a bridge connecting innate immunity to the adaptive immune system. Functional integrity and activity of DCs are closely related to the survival and prognosis of septic patients, while dysfunction of DCs has been identified as one of the major contributors to sepsis-induced immunosuppression and accounts for increased mortality and poor outcomes [77, 78]. DCs show evident signs of activation and maturation in the initial phase of sepsis, and this effectively primes the response of the adaptive immune system via upregulating the expression of surface molecules, including CD80, CD86, CD40, and major histocompatibility complex (MHC)-II, as well as enhancing the release of IL-12 [79]. Under persistent exposure to septic challenge, however, both the phenotypes and IL-12 secretion of DCs suffer from significant alterations, thereby resulting in disturbed antimicrobial defense [78]. The decreased number of DCs is one of the main causes of suppressed priming of the adaptive immune system, which was initially noted at 12 h post-sepsis [80, 81]. Currently, no direct evidence on the function and viability of DCs has been provided in the development of SAE, but the disorder of DCs is related to multiple organ dysfunction and poor outcomes of septic animals [82]. In addition, the abnormal response of DCs was found to contribute to the initiation and development of neuroinflammation in autoimmune encephalomyelitis, and it was also regarded as a potential therapeutic target [83, 84]. Therefore, it is reasonable to infer that dysfunction of DCs also accounts for the development of SAE due to its critical involvement in neuroinflammation.
It has been documented that the function and activity of DCs are affected by different types of brain dysfunction. In patients with severe head injury, for example, the host response to common antigens showed significant suppression, along with a marked increase in opportunistic infections [85]. The number of circulating DCs was significantly lower in patients suffering from aneurysmal subarachnoid hemorrhage compared to that of healthy controls [50]. These DCs exhibited a remarkable anergic response to Toll-like receptor (TLR)3 and TLR4 stimulation, as evidenced by diminished production of TNF-α and IL-2, suggesting that aneurysmal subarachnoid hemorrhage-induced brain dysfunction compromised the function and viability of DCs [50]. Yilmaz and colleagues found that circulating DC precursors underwent transient decrease after acute stroke, which was correlated with the severity of brain injury [51]. Furthermore, disturbed infiltration and aberrant phenotypic differentiation of DCs were also characterized under brain dysfunction [52, 53]. However, the precise mechanisms of the dysfunction of DCs and its significance in host immunosuppression remain to be clarified. Nonetheless, the inability to mount an effective cellular immune response is complicated in severe TBI, which is partly due to the impaired priming activity of DCs.
T cells
T lymphocytes are classically representatives of the adaptive immune system, which are important for the prognosis of septic complications, and their impaired activity and aberrant differentiation are noteworthy in sepsis [86]. This indicates that the number of T cells is a prerequisite for the efficient response of the adaptive immune system. Extensive apoptosis of T cells was observed at 24 h following septic challenge, which was associated with poor outcomes and further identified as a rational and optimal therapeutic target for sepsis-induced immunosuppression, as successful remedy was achieved using anti-apoptosis drugs [87, 88]. Likewise, imbalanced differentiation of helper T (Th) cells is critically involved in the development of immune paralysis resulting from sepsis [89, 90]. Based on the types of cytokines specifically secreted by cells, Th cells are classified into three subgroups, Th1, Th2, and Th17, and proinflammatory mediators are produced by Th1 and Th17 cells, while anti-inflammatory cytokines are produced by Th2 cells. In the late stage of sepsis, Th cells are predominantly polarized to Th2 and show outward signs of anti-inflammatory status, while differentiation of Th1 cells occurs primarily during the initial phase of sepsis [91]. Failure to maintain homeostasis between Th1 and Th2 cells might be a major indicator of immune system dysfunction under septic conditions [89]. Depletion of peripheral T lymphocytes is independently associated with the development of SAE, as reported by Lu and colleagues [92]. Similarly, the types of T cells and their polarization status accounts for sepsis-induced brain injury and should be taken into consideration for effective treatments [93]. However, the specific role and significance of impaired viability and the dysfunction of T lymphocytes in the pathogenesis of SAE remain indistinct, and an imbalanced inflammatory response as a result of deviated Th1/Th2 ratios leads to brain dysfunction by augmenting local inflammation [54].
The recruitment of T cells occurs during the early stages of TBI, which controls damage and tissue repair through the release of anti-inflammatory cytokines [55]. However, the proportion and function of peripheral T cells are significantly suppressed during persistent severe head injury, followed by a marked impairment in cell-mediated immunity [56]. Previous studies showed that a reduction in peripheral T lymphocytes was observed at 24 h and remained low for 4 days post TBI, which resulted in an inadequate number of cells to exert an effective immune response [94]. Moreover, increased levels of serum catecholamines are related to a decreased percentage of peripheral T cells partly through suppressing egress from the lymph nodes due to enhanced stimulation of the β2-adrenergic receptor [95, 96]. Splenic CD4+ T cells play a crucial role in maintaining the integrity of the CAP, which is rendered incapable of potent anti-inflammatory effects either through splenectomy or depletion of CD4+ T cells [57]. Thus, a shortage of peripheral T lymphocytes due to TBI may be a vicious cycle for suppressed immune function, as it promotes feedback for the CAP by enhancing the activity of vagus nerve [11]. Peripheral blood T lymphocytes have disturbed responses to antigens and polarization of anti-inflammatory phenotypes, thereby exacerbating the undermined stability of the immune system and its ability to eliminate invading pathogens [56, 97]. Mitigation of brain injury either through downregulating neuroinflammation or reducing apoptosis of brain cells is an effective strategy for reversing the dysfunction of peripheral T cells [98].
The enhanced activity of the HPA axis and the production of glucocorticoids are theoretically responsible for the abnormal function and polarization of peripheral T cells that have anti-inflammatory capacities, which might act as feedback for the loss of effective brain nuclei [7, 99]. Regulatory T cells (Tregs), one of the major subtypes of T lymphocytes with potent anti-inflammatory activity, are extensively involved in sepsis-induced immunosuppression due to delayed apoptosis and enhanced activity [58]. Although the role of Tregs in the pathogenesis and progression of SAE has not been established, Tregs do show benefits for TBI patients via alleviating local inflammation and promoting tissue repair [100]. Nevertheless, evidence on changes in the immune function of Tregs in brain injury has not yet been provided, and deep insight into the pathophysiology of immunosuppression due to dysfunction of the central nervous system is important.
Regulatory mechanisms
The aberrant responses of multiple neuroendocrine immune networks are reportedly responsible for the disorders in host immunity that are secondary to brain injury. The central nervous system plays an essential role in maintaining the functional integrity of the immune system, as the command center of various neuromodulatory pathways and has close connections to peripheral organs. Thus, clarifying the specific mechanism with regard to both neurologically dependent and independent pathways is of great importance for timely recognition and prompt interference in the vicious cycle between SAE and intractable immunosuppression.
Cholinergic anti-inflammatory pathway
CAP represents an important branch of the parasympathetic nervous system and serves as an effective therapeutic target for various inflammatory diseases. It mainly constitutes the following three parts: brain for signal integration, processing and transition, efferent vagus nerve for signal transmission, and cellular α7nAchR for initiating intracellular machinery [8]. Peripheral inflammatory signals are captured by receptors of immune-transmitter in afferent vagus nerve, such as receptors of IL-1β and prostaglandins, and then reach the nucleus tractus solitarius of the central nervous system [101, 102]. The dorsal motor nucleus is activated after interconnecting with nucleus tractus solitarius, and further drives the excitation of efferent vagus nerve for Ach release [102, 103]. The anti-inflammatory effects of CAP are achieved after activation of α7nAchR, which disrupts activation of NF-κB and inflammasomes by promoting phosphorylation of the Janus kinase 2 (JAK2)-signal transducer and activator of transcription 3 (STAT3) pathway, inhibiting the TLR4-myeloid differentiation factor 88 (MyD88)-interleukin-1 receptor-associated kinase (IRAK) cascade and the release of mitochondrial DNA [104–107]. It has been reported that activation of α7nAchR is capable of blocking activation and entry of NF-κB via interfering phosphorylation of inhibitor κB (IκB) [108, 109]. Furthermore, microRNA124 is identified involving in the anti-inflammatory mechanism of CAP after increased expression by α7nAchR activation, which is evidenced by inhibiting production of TNF-α and IL-6 [110]. Hamano and colleagues found that stimulation of α7nAchR suppressed immune responses of human monocytes by downregulating the expressions of CD14, TLR4, intercellular adhesion molecule 1, and CD40 [111]. Abnormal response of the CAP jeopardizes the peripheral immune system, and contributes to intractable immunosuppression after brain injury. For example, the vagus nerve is overactivated during the course of TBI, and it is a major cause of immune paralysis [11]. Dysfunction of the CAP is also commonly complicated in severe septic condition, as evidenced by a massive loss of brain cholinergic neurons and vagal tone together with disturbed responses of Ach-α7nAchR machinery, which fails at inhibiting inflammation and protecting organs [14, 112, 113]. A close relationship has been noted between poor outcomes and indicators of CAP malfunction, such as a decrease in heart rate variability, an increase in cholinesterase activity, and downregulation of α7nAchR mRNA expression in peripheral immune cells [113–115]. Manipulation of CAP activity through stimulating brain cholinergic nuclei or vagus nerve, inhibiting cholinesterase activity and administering agonists of α7nAchR, is beneficial for organ function and survival of septic animals by reconstruction or simulation of CAP effects [9, 10, 116, 117]. In addition, the CAP is recognized as a neuroprotective mechanism that is capable of reducing both systemic and cerebral inflammation, which further attenuates brain damage after activation [118, 119]. Intriguingly, the vagus nerve is constitutively activated in septic survivors and appears to be associated with immune impairment and increased vulnerability to infections [120]. Therefore, the bidirectional changes in CAP activity deserve timely recognition and interference to eliminate potential hazards of the immune response. In addition, the response of the CAP can be achieved by affecting the sympathetic nervous system which to some extent acts as a feedback for the circuit of CAP [121, 122].
Sympathetic nervous system
Activation of sympathetic nervous system (SNS) represents the “fight or flight” response to external threats. It is essential for inflammatory control and modulation of the immune response by releasing norepinephrine (NE) at terminal nerves, which is a pivotal part in the inflammatory reflex. The SNS shows critical involvement in the development and response of immune cells. For example, failure of intact SNS signaling impairs the steady-state circadian rhythmicity of hematopoietic stem cells and disturbs their mobilization [123]. Basic β-adrenoreceptor (β-AR) activity is programmed to retain lymphocytes within the lymph nodes, while activation of β-AR results in a rapid decline in lymphocytes in peripheral blood and lymphatic fluid by blocking efficient emigration [96, 124]. Further studies provided evidence that activation of β-AR in T cells promotes interactions between β-AR and C-C motif chemokine receptor (CCR)7 and CCR4 [96]. Hyperactivation of SNS has also been identified as a common characteristic of brain insults, such as TBI, cerebral ischemia and stroke, and constitutes a great threat to the immune response [125–127]. It has been demonstrated that overactivation of SNS induces extensive immunosuppression by driving a negative immunomodulatory phenotype. Di Battista and colleagues reported that exaggerated SNS activation was responsible for dysregulated production of peripheral inflammatory mediators during the course of TBI, suggesting a potential target for orchestrating the inflammatory response [125]. Activation of β2-AR by NE triggers increased levels of cyclic AMP (cAMP) and activation of protein kinase A (PKA), which results in downregulation of proinflammatory cytokines by suppressing translocation of NF-κB [128]. Moreover, enhanced SNS activity is a major cause of the decreased ratio and anti-inflammatory phenotype polarization of blood T cells through upregulating expression of cellular programmed cell death 1 (PD1) and promoting emigration and activation of Tregs in stroke [126, 129]. Furthermore, these effects are established by increasing prostaglandin E2 (PGE2) levels and disrupting the stromal cell-derived factor-1 (SDF-1) axis after enhancing β-AR activation [126]. Other signaling pathways, such as the fibroblast growth factor (FGF) 21-extracellular signal-regulated kinase (ERK) 1/2-CCL11 axis, are critically involved in SNS-driven type-2 immunity [130]. In sepsis, distinct changes in SNS activity are a failure of an efficient neuroendocrine immune network and a potential remedy for the immune response [131]. However, the regulatory mechanism underlying SNS activity in sepsis-induced immunosuppression remains largely unknown, even though its immunomodulatory impact has been extensively studied. Furthermore, no evidence has been noted implicating SNS in the pathophysiology of SAE. Given its potent immune-modulatory effect, it might be assumed that dysregulated response of SNS do jeopardize brain function by disturbing peripheral immune system during septic exposure.
Hypothalamic-pituitary-adrenal axis
HPA axis belongs to the neurological branch of stress system that is programmed for maintaining functional homeostasis. The hypothalamus and brain stem constitute a central component of the HPA axis and induce a cascade of endocrine hormones, including corticotropin-releasing hormone (CRH), adrenocorticotropic hormone (ACTH), and glucocorticoid [132]. The level of cortisone is a basic indicator of HPA activity and the key executor of immunomodulation. It was reported that the HPA axis is initiated early after the onset of neuroinflammation and returns to baseline levels by 24 h, which serves as a negative mechanism for the inflammatory response [133]. However, the corticosteroids are present and have ongoing release, and dominant immunosuppressive effects occur with exposure to prolonged inflammation, suggesting a pivotal role of the HPA axis in inflammatory balance [134]. The anti-inflammatory and negatively immunomodulatory effects of the HPA axis have been extensively recognized and applied in multiple diseases. Deficiency in the HPA axis, as a result of impaired production of the adrenal hormone, contributes to the deterioration of septic conditions [135]. The adrenal insufficiency is commonly complicated in critically ill patients, which presents with decreased response to exogenous ACTH stimulation from 10% to 20%, but attains 60% in patients with septic shock [60]. Activation of HPA axis is capable of restricting the inflammatory response by driving a dominant anti-inflammatory phenotype, interfering with proinflammatory intracellular signaling, and promoting negative immunomodulation [132]. For instance, stimulation of HPA system significantly downregulates NF-κB activation and further inhibits the production of proinflammatory cytokines, such as TNF-α, IL-6 and IL-1, but enhances the expression of IL-10, followed by Th2 polarization [70, 136].
Glucocorticoids reportedly enable the anti-inflammatory effects of the HPA axis by either directly forming complex with transcription factors, e.g., NF-κB, to interfere its binding with proinflammatory genes, or promoting expression of anti-inflammatory proteins, such as annexin A1, mitogen-activated protein kinase phosphatase-1 (MKP-1), and glucocorticoid-induced leucine zipper protein (GILZ) [137]. Reduction of mitochondrial ROS is also involved in the anti-inflammatory effects of the HPA axis, which is associated with upregulated expression of uncoupling protein-2 (UCP2) [138]. Over-production of corticosteroids inhibits the activity of T cells by inducing apoptosis in a Fas/FasL-dependent manner [139]. It has been documented that the HPA axis disturbs rolling and adhesion of leukocytes by reducing l-selectin activity [140]. The distribution and status of glucocorticoid receptors (GRs) also constitute important factors for the function and fitness of immune cells. Activation of GRs in DCs, for example, represses DC maturation by controlling c-jun amino-terminal kinase (JNK) activation after TLR7 and TLR8 stimulation [141]. Aberrant expression of GRs is responsible for disturbed cellular response to corticosteroids [142]. The corticosteroid system shows dysregulated performance in severe septic conditions, and it is responsible for intractable immunosuppression and poor outcomes associated with the immune response [143]. Though multiple factors are identified interfering with efficient immunomodulation of the HPA axis, the specific mechanism for dysfunction of the HPA axis under severe sepsis exposure has not been established. Indeed, abnormal response of the immune system is a great threat to the functional integrity of the HPA axis, from collapsed cerebral nuclei to dysfunction of peripheral adrenal gland during septic course [135]. It has been documented that increased production of proinflammatory cytokines, such as TNF-α and IL-1β, contributes to remarkable suppression in the production of pituitary hormones and response of corticotrope cells [144]. In addition, increased infiltration of immune cells in the adrenal gland, especially neutrophils, causes unresponsiveness of adrenal cells by inducing hemorrhages and cell death [135]. Therefore, the interplay between abnormal immune response and dysfunction of the HPA axis is a vicious cycle for collapsed modulation of the HPA axis and intractable immunosuppression secondary to sepsis. However, no direct evidence has been provided for the relationship between a malfunctioning HPA axis and SAE-associated immunosuppression. The HPA axis is noted with significant dysfunction in critically ill patients with TBI and accounts for dysregulated inflammatory responses [145]. In addition, the involvement of HPA axis in the pathophysiology of SAE should be elucidated with regard to its essential participation in modulating peripheral immune response, while it remains speculative as a result of lacking direct evidence.
SAE is a vicious cycle for immunosuppression and a future perspective
The interplay between host immune depression and development of SAE is responsible for the deteriorating outcome of sepsis due to uncontrolled neuroinflammation and disorders of systemic immunity (Fig. 2). The infiltration of immune cells at the initial stage of sepsis is a protective mechanism for the central nervous system by eliminating damaged brain cells, maintaining homeostasis of local inflammatory response, and promoting tissue repair [23, 25, 100, 146]. Neuroendocrine immune networks are extensively activated to limit excessive inflammation and maintain balance of the host immune response (Fig. 3). For instance, stimulation of HPA axis is capable of restricting production of proinflammatory mediators and downregulating function of immune cells with proinflammatory phenotypes [137]. Persistent exposure to sepsis, however, drives irreversible brain injury by manipulating uncontrolled neuroinflammation and disturbing brain perfusion, which in turn acts as a vicious cycle for immunosuppression, as brain is the common center for multiple types of neuroendocrine immune networks [15, 18]. Sepsis-induced brain dysfunction reportedly comes with abnormal responses of these neuromodulatory mechanisms, as evidenced by either significant suppression or unexpected hyperactivity as a result of robust feedback [11, 14, 147]. For example, the HPA axis has markedly suppressed activation under prolonged exposure to severe sepsis, followed by strikingly low levels of plasma corticosterone [147]. In addition, the CAP is remarkably unresponsive due to the loss of brain cholinergic nuclei during sepsis [14]. The failure of efficient neuromodulation can lead to uncontrolled inflammation and irreversible damage to the central nervous system. Reconstruction of either HPA axis or CAP through administration of corticosteroids or stimulation of vagus efferent nerve, respectively, show great benefits for septic animals by ameliorating multiple organ damage and improving the survival [62, 120]. In fact, deteriorative neuroinflammation is the center of the vicious cycle of SAE and immunosuppression because it arises from the aberrant response of immune system and acts as a key contributor to brain dysfunction. Our previous study revealed that inhibition of cerebral HMGB1, a pivotal etiology for the late peak of inflammatory response and late mortality of septic cases, showed potent protective effects against brain injury and suppressive responses of splenic T cells [20, 148]. Taken together, these findings might provide a novel strategy not only for interfering with sepsis-induced brain injury but also a potential target for reversing intractable immunosuppression via blocking this unexpected vicious cycle.
Fig. 2.

SAE acts as a vicious cycle of sepsis-induced immunosuppression. In the physiological condition, the brain is essential for the homeostasis of systemic immune response due to its central role in multiple types of neuroendocrine immune networks, including hypothalamic-pituitary-adrenal (HPA) axis, sympathetic and parasympathetic nervous system, which are capable of inhibiting excessive inflammation and enabling efficient immunomodulation. Appropriate infiltration of immune cells into the central nervous system is beneficial for functional integrity and viability of neurons. However, sepsis-induced immunosuppression contributes to intractable neuroinflammation which results in massive loss of effective nuclei and serious cognitive impairment. Furthermore, persistent brain injury impairs peripheral immune response due to abnormal response of these neuromodulatory mechanisms which present with either significant suppression or unexpected hyperactivity
Fig. 3.

Intracellular signaling pathways of CAP, SNS, and HPA axis for anti-inflammatory responses. Acetylcholine (Ach) can be released from the terminal efferent vagus nerve and further interacts with α7 nicotinic acetylcholine receptor (α7nAChR) on immune cells. Activation of α7nAChR triggers multiple intracellular signaling pathways, including promoting phosphorylation of Janus kinase 2 (JAK2)-signal transducer and activator of transcription 3 (STAT3) pathway, inhibiting Toll-like receptor (TLR)4-myeloid differentiation factor 88 (MyD88)-interleukin-1 receptor-associated kinase (IRAK) cascade as well as declining release of mitochondrial DNA, which contribute to suppression of proinflammatory phenotypes by disturbing activation of nuclear factor-κB (NF-κB) and inflammasomes. The interaction between glucocorticoids and glucocorticoid receptors (GRs) is capable of inhibiting production of proinflammatory cytokines by downregulating NF-κB activity and disturbing c-jun amino-terminal kinase (JNK) cascade after Toll-like receptor (TLR)7 and TLR8 stimulation. Activation of β2-adrenergic receptor with norepinephrine promotes the expression of cyclic AMP (cAMP) and activation of protein kinase A (PKA), which results in decreased production of proinflammatory cytokines by suppressing translocation of NF-κB. It also increases expression of fibroblast growth factor 21 (FGF21) which further suppresses NF-κB activation by promoting activation of extracellular signal regulated kinase (ERK) 1/2-STAT3 cascades in an autocrine manner
Nonetheless, specific mechanisms of both SAE and its impacts on peripheral immune response have not yet been established, even though dysfunction of the brain has been identified, which impairs immune homeostasis and worsens outcomes in septic cases. Moreover, the specific point at which the initiation of SAE becomes a vicious cycle of immunosuppression must be clarified for timely recognition and prompt treatment. Therefore, the interplay between the pathogenesis of SAE and the abnormal response of immune cells is noteworthy and can be used for constructing a predictive algorithm after confluence analysis, given the participation of multiple immune effectors. In addition, exploration of a set would be beneficial for preventing such a vicious cycle.
Conclusions
SAE is commonly yet severely complicated by sepsis but is prone to be neglected in clinical practice. Brain damage plays a critical role in the survival and prognosis of septic patients, which should be recognized as not only a compromised organ but also an essential participant in impaired immunomodulation secondary to sepsis because brain is the command center for multiple types of neuroendocrine immune networks, such as CAP, HPA axis, and sympathetic nervous system. The impaired activity of these neuromodulatory mechanisms reportedly contributes to decreased counts and abnormal responses of peripheral immune cells, including neutrophils, macrophages/monocytes, DCs, and T lymphocytes, which might drive brain injury into a vicious cycle of sepsis-induced immunosuppression as a result of uncontrolled neuroinflammation in the wake of progressively aberrant immunity. Therefore, it may be an efficient strategy for the sepsis-induced immunosuppressive state to block the vicious cycle between SAE and peripheral immune dissonance.
Acknowledgments
Not applicable.
Abbreviations
- Ach
Acetylcholine
- ACTH
Adrenocorticotropic hormone
- BBB
Blood–brain barrier
- cAMP
Cyclic AMP
- CAP
Cholinergic anti-inflammatory pathway
- CBF
Cerebral blood flow
- CCR
C-C motif chemokine receptor
- CRH
Corticotropin-releasing hormone
- CXCR
C-X-C motif chemokine receptor
- DC
Dendritic cell
- EEG
Electroencephalogram
- ERK
Extracellular signal regulated kinase
- FGF21
Fibroblast growth factor 21
- GR
Glucocorticoid receptor
- HMGB1
High mobility group box-1 protein
- HPA
Hypothalamic-pituitary-adrenal
- ICU
Intensive care units
- IL
Interleukin
- IRAK
Interleukin-1 receptor-associated kinase
- JAK2
Janus kinase 2
- JNK
c-Jun amino-terminal kinase
- MODS
Multiple organ dysfunction syndrome
- MyD88
Myeloid differentiation factor 88
- NE
Norepinephrine
- NF-κB
Nuclear factor-κB
- NO
Nitric oxide
- PD1
Programmed cell death 1
- PGE2
Prostaglandin E2
- PKA
Protein kinase A
- ROS
Reactive oxygen species
- SAE
Sepsis associated encephalopathy
- SDF-1
Stromal cell-derived factor-1
- SNS
Sympathetic nervous system
- STAT3
Signal transducer and activator of transcription 3
- TBI
Traumatic brain injury
- Th
Helper T cell
- TLR
Toll-like receptor
- TNF
Tumor necrosis factor
- Treg
Regulatory T cell
- UCP2
Uncoupling protein-2
- α7nAchR
α7 Nicotinic acetylcholine receptor
- β-AR
β-Adrenoreceptor
Authors’ contributions
YMY and CR conceived the idea of this review. CR and RQY performed literature searching and co-wrote this paper. HZ and YWF conducted language editing and re-checking literature. YMY checked and edited the content and format of this manuscript before submission. All authors read and approved the final manuscript.
Funding
The National Natural Science Foundation of China (Nos. 81730057, 81842025, 81801935), the National Key Research and Development Program of China (No. 2017YFC1103302), the Military Medical Innovation Program of Chinese PLA (No. 18CXZ026), and Shenzhen San-ming Project (No. SZSM20162011).
Availability of data and materials
Not applicable.
Ethics approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
Footnotes
Publisher’s Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Chao Ren and Ren-qi Yao contributed equally to this work.
References
- 1.Rhodes A, Evans LE, Alhazzani W, Levy MM, Antonelli M, Ferrer R, et al. Surviving sepsis campaign: International Guidelines for Management of Sepsis and Septic Shock: 2016. Intensive Care Med. 2017;43:304–377. doi: 10.1007/s00134-017-4683-6. [DOI] [PubMed] [Google Scholar]
- 2.Maddux AB, Hiller TD, Overdier KH, Pyle LL, Douglas IS. Innate immune function and organ failure recovery in adults with sepsis. J Intensive Care Med. 2019;34:486–494. doi: 10.1177/0885066617701903. [DOI] [PubMed] [Google Scholar]
- 3.Peidaee E, Sheybani F, Naderi H, Khosravi N, Jabbari Nooghabi M. The etiological spectrum of febrile encephalopathy in adult patients: a cross-sectional study from a developing country. Emerg Med Int. 2018;2018:3587014. doi: 10.1155/2018/3587014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Schuler A, Wulf DA, Lu Y, Iwashyna TJ, Escobar GJ, Shah NH, et al. The Impact of acute organ dysfunction on long-term survival in sepsis. Crit Care Med. 2018;46:843–849. doi: 10.1097/CCM.0000000000003023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sonneville R, de Montmollin E, Poujade J, Garrouste-Orgeas M, Souweine B, Darmon M, et al. Potentially modifiable factors contributing to sepsis-associated encephalopathy. Intensive Care Med. 2017;43:1075–1084. doi: 10.1007/s00134-017-4807-z. [DOI] [PubMed] [Google Scholar]
- 6.Iwashyna TJ, Ely EW, Smith DM, Langa KM. Long-term cognitive impairment and functional disability among survivors of severe sepsis. JAMA. 2010;304:1787–1794. doi: 10.1001/jama.2010.1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dantzer R, Konsman JP, Bluthe RM, Kelley KW. Neural and humoral pathways of communication from the immune system to the brain: parallel or convergent? Auton Neurosci. 2000;85:60–65. doi: 10.1016/S1566-0702(00)00220-4. [DOI] [PubMed] [Google Scholar]
- 8.Rosas-Ballina M, Tracey KJ. Cholinergic control of inflammation. J Intern Med. 2009;265:663–679. doi: 10.1111/j.1365-2796.2009.02098.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Huston JM, Gallowitsch-Puerta M, Ochani M, Ochani K, Yuan R, Rosas-Ballina M, et al. Transcutaneous vagus nerve stimulation reduces serum high mobility group box 1 levels and improves survival in murine sepsis. Crit Care Med. 2007;35:2762–2768. doi: 10.1097/01.CCM.0000288102.15975.BA. [DOI] [PubMed] [Google Scholar]
- 10.Tsoyi K, Jang HJ, Kim JW, Chang HK, Lee YS, Pae HO, et al. Stimulation of alpha7 nicotinic acetylcholine receptor by nicotine attenuates inflammatory response in macrophages and improves survival in experimental model of sepsis through heme oxygenase-1 induction. Antioxidants redox signaling. 2011;14:2057–2070. doi: 10.1089/ars.2010.3555. [DOI] [PubMed] [Google Scholar]
- 11.Kox M, Pompe JC, Pickkers P, Hoedemaekers CW, van Vugt AB, van der Hoeven JG. Increased vagal tone accounts for the observed immune paralysis in patients with traumatic brain injury. Neurology. 2008;70:480–485. doi: 10.1212/01.wnl.0000279479.69502.3e. [DOI] [PubMed] [Google Scholar]
- 12.Valiyaveettil M, Alamneh YA, Miller SA, Hammamieh R, Arun P, Wang Y, et al. Modulation of cholinergic pathways and inflammatory mediators in blast-induced traumatic brain injury. Chemico-biological interactions. 2013;203:371–375. doi: 10.1016/j.cbi.2012.10.022. [DOI] [PubMed] [Google Scholar]
- 13.Shin SS, Dixon CE. Alterations in cholinergic pathways and therapeutic strategies targeting cholinergic system after traumatic brain injury. J Neurotrauma. 2015;32:1429–1440. doi: 10.1089/neu.2014.3445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zaghloul N, Addorisio ME, Silverman HA, Patel HL, Valdes-Ferrer SI, Ayasolla KR, et al. Forebrain cholinergic dysfunction and systemic and brain inflammation in murine sepsis survivors. Front Immunol. 2017;8:1673. doi: 10.3389/fimmu.2017.01673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Adam N, Kandelman S, Mantz J, Chretien F, Sharshar T. Sepsis-induced brain dysfunction. Expert Rev Anti Infect Ther. 2013;11:211–221. doi: 10.1586/eri.12.159. [DOI] [PubMed] [Google Scholar]
- 16.Tracey KJ. Reflex control of immunity. Nat Rev Immunol. 2009;9:418–428. doi: 10.1038/nri2566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Licinio J, Mastronardi C, Wong ML. Pharmacogenomics of neuroimmune interactions in human psychiatric disorders. Exp Physiol. 2007;92:807–811. doi: 10.1113/expphysiol.2007.038471. [DOI] [PubMed] [Google Scholar]
- 18.Gofton TE, Young GB. Sepsis-associated encephalopathy. Nat Rev Neurol. 2012;8:557–566. doi: 10.1038/nrneurol.2012.183. [DOI] [PubMed] [Google Scholar]
- 19.Alexander JJ, Jacob A, Cunningham P, Hensley L, Quigg RJ. TNF is a key mediator of septic encephalopathy acting through its receptor, TNF receptor-1. Neurochem Int. 2008;52:447–456. doi: 10.1016/j.neuint.2007.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ren C, Tong YL, Li JC, Dong N, Hao JW, Zhang QH, et al. Early antagonism of cerebral high mobility group box-1 protein is benefit for sepsis induced brain injury. Oncotarget. 2017;8:92578–92588. doi: 10.18632/oncotarget.21502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Chavan SS, Huerta PT, Robbiati S, Valdes-Ferrer SI, Ochani M, Dancho M, et al. HMGB1 mediates cognitive impairment in sepsis survivors. Mol Med. 2012;18:930–937. doi: 10.2119/molmed.2012.00195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhang QH, Sheng ZY, Yao YM. Septic encephalopathy: when cytokines interact with acetylcholine in the brain. Mil Med Res. 2014;1:20. doi: 10.1186/2054-9369-1-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dal-Pizzol F, Tomasi CD, Ritter C. Septic encephalopathy: does inflammation drive the brain crazy? Braz J Psychiatry. 2014;36:251–258. doi: 10.1590/1516-4446-2013-1233. [DOI] [PubMed] [Google Scholar]
- 24.Michels M, Vieira AS, Vuolo F, Zapelini HG, Mendonca B, Mina F, et al. The role of microglia activation in the development of sepsis-induced long-term cognitive impairment. Brain Behav Immun. 2015;43:54–59. doi: 10.1016/j.bbi.2014.07.002. [DOI] [PubMed] [Google Scholar]
- 25.Comim CM, Vilela MC, Constantino LS, Petronilho F, Vuolo F, Lacerda-Queiroz N, et al. Traffic of leukocytes and cytokine up-regulation in the central nervous system in sepsis. Intensive Care Med. 2011;37:711–718. doi: 10.1007/s00134-011-2151-2. [DOI] [PubMed] [Google Scholar]
- 26.Shulyatnikova T, Verkhratsky A. Astroglia in sepsis associated encephalopathy. Neurochem Res. 2019. [DOI] [PMC free article] [PubMed]
- 27.Michels M, Danielski LG, Dal-Pizzol F, Petronilho F. Neuroinflammation: microglial activation during sepsis. Curr Neurovasc Res. 2014;11:262–270. doi: 10.2174/1567202611666140520122744. [DOI] [PubMed] [Google Scholar]
- 28.Hasegawa-Ishii S, Inaba M, Umegaki H, Unno K, Wakabayashi K, Shimada A. Endotoxemia-induced cytokine-mediated responses of hippocampal astrocytes transmitted by cells of the brain-immune interface. Sci Rep. 2016;6:25457. doi: 10.1038/srep25457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bellaver B, Dos Santos JP, Leffa DT, Bobermin LD, Roppa PHA, da Silva Torres IL, et al. Systemic inflammation as a driver of brain injury: the astrocyte as an emerging player. Mol Neurobiol. 2018;55:2685–2695. doi: 10.1007/s12035-017-0526-2. [DOI] [PubMed] [Google Scholar]
- 30.Cardoso FL, Herz J, Fernandes A, Rocha J, Sepodes B, Brito MA, et al. Systemic inflammation in early neonatal mice induces transient and lasting neurodegenerative effects. J Neuroinflammation. 2015;12:82. doi: 10.1186/s12974-015-0299-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Wang H, Hong LJ, Huang JY, Jiang Q, Tao RR, Tan C, et al. P2RX7 sensitizes Mac-1/ICAM-1-dependent leukocyte-endothelial adhesion and promotes neurovascular injury during septic encephalopathy. Cell Res. 2015;25:674–690. doi: 10.1038/cr.2015.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Taccone FS, Scolletta S, Franchi F, Donadello K, Oddo M. Brain perfusion in sepsis. Curr Vasc Pharmacol. 2013;11:170–186. doi: 10.2174/1570161111311020007. [DOI] [PubMed] [Google Scholar]
- 33.Bowton DL, Bertels NH, Prough DS, Stump DA. Cerebral blood flow is reduced in patients with sepsis syndrome. Crit Care Med. 1989;17:399–403. doi: 10.1097/00003246-198905000-00004. [DOI] [PubMed] [Google Scholar]
- 34.Taccone FS, Su F, De Deyne C, Abdellhai A, Pierrakos C, He X, et al. Sepsis is associated with altered cerebral microcirculation and tissue hypoxia in experimental peritonitis. Crit Care Med. 2014;42:e114–e122. doi: 10.1097/CCM.0b013e3182a641b8. [DOI] [PubMed] [Google Scholar]
- 35.Schramm P, Klein KU, Falkenberg L, Berres M, Closhen D, Werhahn KJ, et al. Impaired cerebrovascular autoregulation in patients with severe sepsis and sepsis-associated delirium. Crit Care. 2012;16:R181. doi: 10.1186/cc11665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burkhart CS, Siegemund M, Steiner LA. Cerebral perfusion in sepsis. Crit Care. 2010;14:215. doi: 10.1186/cc8856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vachharajani V, Russell JM, Scott KL, Conrad S, Stokes KY, Tallam L, et al. Obesity exacerbates sepsis-induced inflammation and microvascular dysfunction in mouse brain. Microcirculation. 2005;12:183–194. doi: 10.1080/10739680590904982. [DOI] [PubMed] [Google Scholar]
- 38.Mazeraud A, Pascal Q, Verdonk F, Heming N, Chretien F, Sharshar T. Neuroanatomy and physiology of brain dysfunction in sepsis. Clin Chest Med. 2016;37:333–345. doi: 10.1016/j.ccm.2016.01.013. [DOI] [PubMed] [Google Scholar]
- 39.Sharshar T, Annane D, de la Grandmaison GL, Brouland JP, Hopkinson NS, Francoise G. The neuropathology of septic shock. Brain Pathol. 2004;14:21–33. doi: 10.1111/j.1750-3639.2004.tb00494.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Feng Q, Ai YH, Gong H, Wu L, Ai ML, Deng SY, et al. Characterization of sepsis and sepsis-associated encephalopathy. J Intensive Care Med. 2017;34:938–945. doi: 10.1177/0885066617719750. [DOI] [PubMed] [Google Scholar]
- 41.Pfister D, Siegemund M, Dell-Kuster S, Smielewski P, Ruegg S, Strebel SP, et al. Cerebral perfusion in sepsis-associated delirium. Crit Care. 2008;12:R63. doi: 10.1186/cc6891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maekawa T, Fujii Y, Sadamitsu D, Yokota K, Soejima Y, Ishikawa T, et al. Cerebral circulation and metabolism in patients with septic encephalopathy. Am J Emerg Med. 1991;9:139–143. doi: 10.1016/0735-6757(91)90175-J. [DOI] [PubMed] [Google Scholar]
- 43.Vermeij JD, Aslami H, Fluiter K, Roelofs JJ, van den Bergh WM, Juffermans NP, et al. Traumatic brain injury in rats induces lung injury and systemic immune suppression. J Neurotrauma. 2013;30:2073–2079. doi: 10.1089/neu.2013.3060. [DOI] [PubMed] [Google Scholar]
- 44.Schwulst SJ, Trahanas DM, Saber R, Perlman H. Traumatic brain injury-induced alterations in peripheral immunity. J Trauma Acute Care Surg. 2013;75:780–788. doi: 10.1097/TA.0b013e318299616a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Shimonkevitz R, Bar-Or D, Harris L, Dole K, McLaughlin L, Yukl R. Transient monocyte release of interleukin-10 in response to traumatic brain injury. Shock. 1999;12:10–16. doi: 10.1097/00024382-199907000-00002. [DOI] [PubMed] [Google Scholar]
- 46.Tian M, Qingzhen L, Zhiyang Y, Chunlong C, Jiao D, Zhang L, et al. Attractylone attenuates sepsis-associated encephalopathy and cognitive dysfunction by inhibiting microglial activation and neuroinflammation. J Cell Biochem. 2019;120:7101–7108. doi: 10.1002/jcb.27983. [DOI] [PubMed] [Google Scholar]
- 47.Kenne E, Erlandsson A, Lindbom L, Hillered L, Clausen F. Neutrophil depletion reduces edema formation and tissue loss following traumatic brain injury in mice. J Neuroinflammation. 2012;9:17. doi: 10.1186/1742-2094-9-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Junger WG, Rhind SG, Rizoli SB, Cuschieri J, Baker AJ, Shek PN, et al. Prehospital hypertonic saline resuscitation attenuates the activation and promotes apoptosis of neutrophils in patients with severe traumatic brain injury. Shock. 2013;40:366–374. doi: 10.1097/SHK.0000000000000038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Liao Y, Liu P, Guo F, Zhang ZY, Zhang Z. Oxidative burst of circulating neutrophils following traumatic brain injury in human. PloS One. 2013;8:e68963. doi: 10.1371/journal.pone.0068963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Roquilly A, Braudeau C, Cinotti R, Dumonte E, Motreul R, Josien R, et al. Impaired blood dendritic cell numbers and functions after aneurysmal subarachnoid hemorrhage. PloS One. 2013;8:e71639. doi: 10.1371/journal.pone.0071639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Yilmaz A, Fuchs T, Dietel B, Altendorf R, Cicha I, Stumpf C, et al. Transient decrease in circulating dendritic cell precursors after acute stroke: potential recruitment into the brain. Clin Sci. 2009;118:147–157. doi: 10.1042/CS20090154. [DOI] [PubMed] [Google Scholar]
- 52.Posel C, Uri A, Schulz I, Boltze J, Weise G, Wagner DC. Flow cytometric characterization of brain dendritic cell subsets after murine stroke. Exp Transl Stroke Med. 2014;6:11. doi: 10.1186/2040-7378-6-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Israelsson C, Kylberg A, Bengtsson H, Hillered L, Ebendal T. Interacting chemokine signals regulate dendritic cells in acute brain injury. PloS One. 2014;9:e104754. doi: 10.1371/journal.pone.0104754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Tan M, Zhu JC, Du J, Zhang LM, Yin HH. Effects of probiotics on serum levels of Th1/Th2 cytokine and clinical outcomes in severe traumatic brain-injured patients: a prospective randomized pilot study. Crit Care. 2011;15:R290. doi: 10.1186/cc10579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Gadani SP, Cronk JC, Norris GT, Kipnis J. IL-4 in the brain: a cytokine to remember. J Immunol. 2012;189:4213–4219. doi: 10.4049/jimmunol.1202246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Quattrocchi KB, Frank EH, Miller CH, Dull ST, Howard RR, Wagner FC., Jr Severe head injury: effect upon cellular immune function. Neurol Res. 1991;13:13–20. doi: 10.1080/01616412.1991.11739959. [DOI] [PubMed] [Google Scholar]
- 57.Pena G, Cai B, Ramos L, Vida G, Deitch EA, Ulloa L. Cholinergic regulatory lymphocytes re-establish neuromodulation of innate immune responses in sepsis. J Immunol. 2011;187:718–725. doi: 10.4049/jimmunol.1100013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nascimento DC, Melo PH, Pineros AR, Ferreira RG, Colon DF, Donate PB, et al. IL-33 contributes to sepsis-induced long-term immunosuppression by expanding the regulatory T cell population. Nat Commun. 2017;8:14919. doi: 10.1038/ncomms14919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Annane D, Sebille V, Troche G, Raphael JC, Gajdos P, Bellissant E. A 3-level prognostic classification in septic shock based on cortisol levels and cortisol response to corticotropin. JAMA. 2000;283:1038–1045. doi: 10.1001/jama.283.8.1038. [DOI] [PubMed] [Google Scholar]
- 60.Annane D, Maxime V, Ibrahim F, Alvarez JC, Abe E, Boudou P. Diagnosis of adrenal insufficiency in severe sepsis and septic shock. American journal of respiratory and critical care medicine. 2006;174:1319–1326. doi: 10.1164/rccm.200509-1369OC. [DOI] [PubMed] [Google Scholar]
- 61.Bornstein SR. Predisposing factors for adrenal insufficiency. N Engl J Med. 2009;360:2328–2339. doi: 10.1056/NEJMra0804635. [DOI] [PubMed] [Google Scholar]
- 62.Annane D, Bellissant E, Bollaert PE, Briegel J, Keh D, Kupfer Y. Corticosteroids for treating sepsis. Cochrane Database Syst Rev. 2015;2015:CD002243. doi: 10.1002/14651858.CD002243.pub3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sfeir T, Saha DC, Astiz M, Rackow EC. Role of interleukin-10 in monocyte hyporesponsiveness associated with septic shock. Crit Care Med. 2001;29:129–133. doi: 10.1097/00003246-200101000-00026. [DOI] [PubMed] [Google Scholar]
- 64.O'Riordain MG, Collins KH, Pilz M, Saporoschetz IB, Mannick JA, Rodrick ML. Modulation of macrophage hyperactivity improves survival in a burn-sepsis model. Arch Surg. 1992;127:152–157. doi: 10.1001/archsurg.1992.01420020034005. [DOI] [PubMed] [Google Scholar]
- 65.Munoz C, Carlet J, Fitting C, Misset B, Bleriot JP, Cavaillon JM. Dysregulation of in vitro cytokine production by monocytes during sepsis. J Clin Invest. 1991;88:1747–1754. doi: 10.1172/JCI115493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Andonegui G, Zelinski EL, Schubert CL, Knight D, Craig LA, Winston BW, et al. Targeting inflammatory monocytes in sepsis-associated encephalopathy and long-term cognitive impairment. JCI Insight. 2018;3:99364. doi: 10.1172/jci.insight.99364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.McDermott AJ, Falkowski NR, McDonald RA, Frank CR, Pandit CR, Young VB, et al. Role of interferon-gamma and inflammatory monocytes in driving colonic inflammation during acute Clostridium difficile infection in mice. Immunology. 2017;150:468–477. doi: 10.1111/imm.12700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hazeldine J, Lord JM, Belli A. Traumatic brain injury and peripheral immune suppression: primer and prospectus. Front Neurol. 2015;6:235. doi: 10.3389/fneur.2015.00235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Samary CS, Ramos AB, Maia LA, Rocha NN, Santos CL, Magalhaes RF, et al. Focal ischemic stroke leads to lung injury and reduces alveolar macrophage phagocytic capability in rats. Crit Care. 2018;22:249. doi: 10.1186/s13054-018-2164-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen AL, Sun X, Wang W, Liu JF, Zeng X, Qiu JF, et al. Activation of the hypothalamic-pituitary-adrenal (HPA) axis contributes to the immunosuppression of mice infected with Angiostrongylus cantonensis. J Neuroinflammation. 2016;13:266. doi: 10.1186/s12974-016-0743-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Hall S, Kumaria A, Belli A. The role of vagus nerve overactivity in the increased incidence of pneumonia following traumatic brain injury. Br J Neurosurg. 2014;28:181–186. doi: 10.3109/02688697.2013.835373. [DOI] [PubMed] [Google Scholar]
- 72.Kovach MA, Standiford TJ. The function of neutrophils in sepsis. Curr Opin Infect Dis. 2012;25:321–327. doi: 10.1097/QCO.0b013e3283528c9b. [DOI] [PubMed] [Google Scholar]
- 73.Shen XF, Cao K, Jiang JP, Guan WX, Du JF. Neutrophil dysregulation during sepsis: an overview and update. J Cell Mol Med. 2017;21:1687–1697. doi: 10.1111/jcmm.13112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Zarbato GF, de Souza Goldim MP, Giustina AD, Danielski LG, Mathias K, Florentino D, et al. Dimethyl Fumarate Limits Neuroinflammation and Oxidative Stress and Improves Cognitive Impairment After Polymicrobial Sepsis. Neurotox Res. 2018;34:418–430. doi: 10.1007/s12640-018-9900-8. [DOI] [PubMed] [Google Scholar]
- 75.Roth TL, Nayak D, Atanasijevic T, Koretsky AP, Latour LL, McGavern DB. Transcranial amelioration of inflammation and cell death after brain injury. Nature. 2014;505:223–228. doi: 10.1038/nature12808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sewell DL, Nacewicz B, Liu F, Macvilay S, Erdei A, Lambris JD, et al. Complement C3 and C5 play critical roles in traumatic brain cryoinjury: blocking effects on neutrophil extravasation by C5a receptor antagonist. J Neuroimmunol. 2004;155:55–63. doi: 10.1016/j.jneuroim.2004.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kumar V. Dendritic cells in sepsis: Potential immunoregulatory cells with therapeutic potential. Mol Immunol. 2018;101:615–626. doi: 10.1016/j.molimm.2018.07.007. [DOI] [PubMed] [Google Scholar]
- 78.Poehlmann H, Schefold JC, Zuckermann-Becker H, Volk HD, Meisel C. Phenotype changes and impaired function of dendritic cell subsets in patients with sepsis: a prospective observational analysis. Crit Care. 2009;13:R119. doi: 10.1186/cc7969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Fan X, Liu Z, Jin H, Yan J, Liang HP. Alterations of dendritic cells in sepsis: featured role in immunoparalysis. Biomed Res Int. 2015;2015:903720. doi: 10.1155/2015/903720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Strother RK, Danahy DB, Kotov DI, Kucaba TA, Zacharias ZR, Griffith TS, et al. Polymicrobial sepsis diminishes dendritic cell numbers and function directly contributing to impaired primary CD8 T cell responses in vivo. J Immunol. 2016;197:4301–4311. doi: 10.4049/jimmunol.1601463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Tinsley KW, Grayson MH, Swanson PE, Drewry AM, Chang KC, Karl IE, et al. Sepsis induces apoptosis and profound depletion of splenic interdigitating and follicular dendritic cells. J Immunol. 2003;171:909–914. doi: 10.4049/jimmunol.171.2.909. [DOI] [PubMed] [Google Scholar]
- 82.Liu Q, Lu JY, Wang XH, Qu BJ, Li SR, Kang JR. Changes in the PD-1 and PD-L1 expressions of splenic dendritic cells in multiple-organ dysfunction syndrome mice and their significance. Genet Mol Res. 2014;13:7666–7672. doi: 10.4238/2014.September.26.4. [DOI] [PubMed] [Google Scholar]
- 83.Luessi F, Zipp F, Witsch E. Dendritic cells as therapeutic targets in neuroinflammation. Cell Mol Life Sci. 2016;73:2425–2450. doi: 10.1007/s00018-016-2170-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sie C, Korn T. Dendritic cells in central nervous system autoimmunity. Semin Immunopathol. 2017;39:99–111. doi: 10.1007/s00281-016-0608-7. [DOI] [PubMed] [Google Scholar]
- 85.Miller CH, Quattrocchi KB, Frank EH, Issel BW, Wagner FC., Jr Humoral and cellular immunity following severe head injury: review and current investigations. Neurol Res. 1991;13:117–124. doi: 10.1080/01616412.1991.11739977. [DOI] [PubMed] [Google Scholar]
- 86.Jensen IJ, Sjaastad FV, Griffith TS, Badovinac VP. Sepsis-induced T cell immunoparalysis: the Ins and outs of impaired t cell immunity. J Immunol. 2018;200:1543–1553. doi: 10.4049/jimmunol.1701618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Niu R, Gao H, Zhou Y, Zhang J. Ouabain attenuates sepsis-induced immunosuppression in mice by activation and anti-apoptosis of T cells. Med Sci Monit. 2018;24:2720–2727. doi: 10.12659/MSM.906889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Oami T, Watanabe E, Hatano M, Sunahara S, Fujimura L, Sakamoto A, et al. Suppression of T cell autophagy results in decreased viability and function of T cells through accelerated apoptosis in a murine sepsis model. Crit Care Med. 2017;45:e77–e85. doi: 10.1097/CCM.0000000000002016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Yoon SJ, Kim SJ, Lee SM. Overexpression of HO-1 contributes to sepsis-induced immunosuppression by modulating the Th1/Th2 balance and regulatory T-cell function. J Infect Dis. 2017;215:1608–1618. doi: 10.1093/infdis/jix142. [DOI] [PubMed] [Google Scholar]
- 90.Wu HP, Chung K, Lin CY, Jiang BY, Chuang DY, Liu YC. Associations of T helper 1, 2, 17 and regulatory T lymphocytes with mortality in severe sepsis. Inflamm Res. 2013;62:751–763. doi: 10.1007/s00011-013-0630-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Cabrera-Perez J, Condotta SA, Badovinac VP, Griffith TS. Impact of sepsis on CD4 T cell immunity. J Leukoc Biol. 2014;96:767–777. doi: 10.1189/jlb.5MR0114-067R. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lu CX, Qiu T, Tong HS, Liu ZF, Su L, Cheng B. Peripheral T-lymphocyte and natural killer cell population imbalance is associated with septic encephalopathy in patients with severe sepsis. Exp Ther Med. 2016;11:1077–1084. doi: 10.3892/etm.2016.3000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang X, Rocha-Ferreira E, Li T, Vontell R, Jabin D, Hua S, et al. gammadeltaT cells but not alphabetaT cells contribute to sepsis-induced white matter injury and motor abnormalities in mice. J Neuroinflammation. 2017;14:255. doi: 10.1186/s12974-017-1029-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Mrakovcic-Sutic I, Tokmadzic VS, Laskarin G, Mahmutefendic H, Lucin P, Zupan Z, et al. Early changes in frequency of peripheral blood lymphocyte subpopulations in severe traumatic brain-injured patients. Scand J Immunol. 2010;72:57–65. doi: 10.1111/j.1365-3083.2010.02407.x. [DOI] [PubMed] [Google Scholar]
- 95.Hamill RW, Woolf PD, McDonald JV, Lee LA, Kelly M. Catecholamines predict outcome in traumatic brain injury. Ann Neurol. 1987;21:438–443. doi: 10.1002/ana.410210504. [DOI] [PubMed] [Google Scholar]
- 96.Nakai A, Hayano Y, Furuta F, Noda M, Suzuki K. Control of lymphocyte egress from lymph nodes through beta2-adrenergic receptors. J Exp Med. 2014;211:2583–2598. doi: 10.1084/jem.20141132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Shein SL, Shellington DK, Exo JL, Jackson TC, Wisniewski SR, Jackson EK, et al. Hemorrhagic shock shifts the serum cytokine profile from pro- to anti-inflammatory after experimental traumatic brain injury in mice. J Neurotrauma. 2014;31:1386–1395. doi: 10.1089/neu.2013.2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Zhang QH, Chen Q, Kang JR, Liu C, Dong N, Zhu XM, et al. Treatment with gelsolin reduces brain inflammation and apoptotic signaling in mice following thermal injury. J Neuroinflammation. 2011;8:118. doi: 10.1186/1742-2094-8-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Steer JH, Kroeger KM, Abraham LJ, Joyce DA. Glucocorticoids suppress tumor necrosis factor-alpha expression by human monocytic THP-1 cells by suppressing transactivation through adjacent NF-kappa B and c-Jun-activating transcription factor-2 binding sites in the promoter. J Biol Chem. 2000;275:18432–18440. doi: 10.1074/jbc.M906304199. [DOI] [PubMed] [Google Scholar]
- 100.Yu Y, Cao F, Ran Q, Sun X. Regulatory T cells exhibit neuroprotective effect in a mouse model of traumatic brain injury. Mol Med Rep. 2016;14:5556–5566. doi: 10.3892/mmr.2016.5954. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 101.Ek M, Kurosawa M, Lundeberg T, Ericsson A. Activation of vagal afferents after intravenous injection of interleukin-1beta: role of endogenous prostaglandins. J Neurosci. 1998;18:9471–9479. doi: 10.1523/JNEUROSCI.18-22-09471.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Bonham AC, Hasser EM. Area postrema and aortic or vagal afferents converge to excite cells in nucleus tractus solitarius. Am J Physiol. 1993;264:H1674–H1685. doi: 10.1152/ajpheart.1993.264.5.H1674. [DOI] [PubMed] [Google Scholar]
- 103.Wang DW, Yin YM, Yao YM. Vagal modulation of the inflammatory response in sepsis. Int Rev Immunol. 2016;35:415–433. doi: 10.3109/08830185.2015.1127369. [DOI] [PubMed] [Google Scholar]
- 104.Ren C, Tong YL, Li JC, Lu ZQ, Yao YM. The Protective Effect of Alpha 7 Nicotinic acetylcholine receptor activation on critical illness and its mechanism. Int J Biol Sci. 2017;13:46–56. doi: 10.7150/ijbs.16404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Kox M, van Velzen JF, Pompe JC, Hoedemaekers CW, van der Hoeven JG, Pickkers P. GTS-21 inhibits pro-inflammatory cytokine release independent of the Toll-like receptor stimulated via a transcriptional mechanism involving JAK2 activation. Biochem Pharmacol. 2009;78:863–872. doi: 10.1016/j.bcp.2009.06.096. [DOI] [PubMed] [Google Scholar]
- 106.Li Q, Zhou XD, Kolosov VP, Perelman JM. Nicotine reduces TNF-alpha expression through a alpha7 nAChR/MyD88/NF-kB pathway in HBE16 airway epithelial cells. Cell Physiol Biochem. 2011;27:605–612. doi: 10.1159/000329982. [DOI] [PubMed] [Google Scholar]
- 107.Lu B, Kwan K, Levine YA, Olofsson PS, Yang H, Li J, et al. Alpha7 nicotinic acetylcholine receptor signaling inhibits inflammasome activation by preventing mitochondrial DNA release. Mol Med. 2014;20:350–358. doi: 10.2119/molmed.2013.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Saeed RW, Varma S, Peng-Nemeroff T, Sherry B, Balakhaneh D, Huston J, et al. Cholinergic stimulation blocks endothelial cell activation and leukocyte recruitment during inflammation. J Exp Med. 2005;201:1113–1123. doi: 10.1084/jem.20040463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Wang H, Liao H, Ochani M, Justiniani M, Lin X, Yang L, et al. Cholinergic agonists inhibit HMGB1 release and improve survival in experimental sepsis. Nat Med. 2004;10:1216–1221. doi: 10.1038/nm1124. [DOI] [PubMed] [Google Scholar]
- 110.Ulloa L. The cholinergic anti-inflammatory pathway meets microRNA. Cell Res. 2013;23:1249–1250. doi: 10.1038/cr.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Hamano R, Takahashi HK, Iwagaki H, Yoshino T, Nishibori M, Tanaka N. Stimulation of alpha7 nicotinic acetylcholine receptor inhibits CD14 and the toll-like receptor 4 expression in human monocytes. Shock. 2006;26:358–364. doi: 10.1097/01.shk.0000228168.86845.60. [DOI] [PubMed] [Google Scholar]
- 112.Schulte A, Lichtenstern C, Henrich M, Weigand MA, Uhle F. Loss of vagal tone aggravates systemic inflammation and cardiac impairment in endotoxemic rats. J Surg Res. 2014;188:480–488. doi: 10.1016/j.jss.2014.01.022. [DOI] [PubMed] [Google Scholar]
- 113.Cedillo JL, Arnalich F, Martin-Sanchez C, Quesada A, Rios JJ, Maldifassi MC, et al. Usefulness of alpha7 nicotinic receptor messenger RNA levels in peripheral blood mononuclear cells as a marker for cholinergic antiinflammatory pathway activity in septic patients: results of a pilot study. J Infect Dis. 2015;211:146–155. doi: 10.1093/infdis/jiu425. [DOI] [PubMed] [Google Scholar]
- 114.Papaioannou V, Pnevmatikos I. Heart rate variability: A potential tool for monitoring immunomodulatory effects of parenteral fish oil feeding in patients with sepsis. Nutr Metab Insights. 2019;12:1178638819847486. doi: 10.1177/1178638819847486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zhang QH, Li AM, He SL, Yao XD, Zhu J, Zhang ZW, et al. Serum total cholinesterase activity on admission is associated with disease severity and outcome in patients with traumatic brain injury. PloS One. 2015;10:e0129082. doi: 10.1371/journal.pone.0129082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhai Q, Lai D, Cui P, Zhou R, Chen Q, Hou J, et al. Selective activation of basal forebrain cholinergic neurons attenuates polymicrobial sepsis-induced inflammation via the cholinergic anti-inflammatory pathway. Crit Care Med. 2017;45:e1075–e1e82. doi: 10.1097/CCM.0000000000002646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Hofer S, Eisenbach C, Lukic IK, Schneider L, Bode K, Brueckmann M, et al. Pharmacologic cholinesterase inhibition improves survival in experimental sepsis. Crit Care Med. 2008;36:404–408. doi: 10.1097/01.CCM.0B013E31816208B3. [DOI] [PubMed] [Google Scholar]
- 118.Huang W, Zhu S, Liu X, Huang L, Han Y, Han Q, et al. Cholinergic anti-inflammatory pathway involves in the neuroprotective effect of huperzine A on sepsis-associated encephalopathy. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2016;28:450–454. [PubMed] [Google Scholar]
- 119.Li N, Li Z, Xiang H, Wang X, Zhang X, Li J. Protective effects of vagus nerve stimulation on rats with sepsis-associated encephalopathy. Zhonghua Wei Zhong Bing Ji Jiu Yi Xue. 2015;27:509–513. [PubMed] [Google Scholar]
- 120.Rana M, Fei-Bloom Y, Son M, La Bella A, Ochani M, Levine YA, et al. Constitutive vagus nerve activation modulates immune suppression in sepsis survivors. Front Immunol. 2018;9:2032. doi: 10.3389/fimmu.2018.02032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Rosas-Ballina M, Olofsson PS, Ochani M, Valdes-Ferrer SI, Levine YA, Reardon C, et al. Acetylcholine-synthesizing T cells relay neural signals in a vagus nerve circuit. Science. 2011;334:98–101. doi: 10.1126/science.1209985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Murray K, Godinez DR, Brust-Mascher I, Miller EN, Gareau MG, Reardon C. Neuroanatomy of the spleen: mapping the relationship between sympathetic neurons and lymphocytes. PloS One. 2017;12:e0182416. doi: 10.1371/journal.pone.0182416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mendez-Ferrer S, Battista M, Frenette PS. Cooperation of beta(2)- and beta(3)-adrenergic receptors in hematopoietic progenitor cell mobilization. Ann N Y Acad Sci. 2010;1192:139–144. doi: 10.1111/j.1749-6632.2010.05390.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kerage D, Sloan EK, Mattarollo SR, McCombe PA. Interaction of neurotransmitters and neurochemicals with lymphocytes. J Neuroimmunol. 2019;332:99–111. doi: 10.1016/j.jneuroim.2019.04.006. [DOI] [PubMed] [Google Scholar]
- 125.Di Battista AP, Rhind SG, Hutchison MG, Hassan S, Shiu MY, Inaba K, et al. Inflammatory cytokine and chemokine profiles are associated with patient outcome and the hyperadrenergic state following acute brain injury. J Neuroinflammation. 2016;13:40. doi: 10.1186/s12974-016-0500-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Wang J, Yu L, Jiang C, Fu X, Liu X, Wang M, et al. Cerebral ischemia increases bone marrow CD4+CD25+FoxP3+ regulatory T cells in mice via signals from sympathetic nervous system. Brain Behav Immun. 2015;43:172–183. doi: 10.1016/j.bbi.2014.07.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Prass K, Meisel C, Hoflich C, Braun J, Halle E, Wolf T, et al. Stroke-induced immunodeficiency promotes spontaneous bacterial infections and is mediated by sympathetic activation reversal by poststroke T helper cell type 1-like immunostimulation. J Exp Med. 2003;198:725–736. doi: 10.1084/jem.20021098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Sternberg EM. Neural regulation of innate immunity: a coordinated nonspecific host response to pathogens. Nat Rev Immunol. 2006;6:318–328. doi: 10.1038/nri1810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Yang Y, Ye Y, Chen C, Kong C, Su X, Zhang X, et al. Acute traumatic brain injury induces CD4+ and CD8+ T cell functional impairment by upregulating the expression of PD-1 via the activated sympathetic nervous system. Neuroimmunomodulation. 2019;26:43–57. doi: 10.1159/000495465. [DOI] [PubMed] [Google Scholar]
- 130.Huang Z, Zhong L, Lee JTH, Zhang J, Wu D, Geng L, et al. The FGF21-CCL11 axis mediates beiging of white adipose tissues by coupling sympathetic nervous system to type 2 immunity. Cell Metabol. 2017;26:493–508. doi: 10.1016/j.cmet.2017.08.003. [DOI] [PubMed] [Google Scholar]
- 131.Ramchandra R, Wan L, Hood SG, Frithiof R, Bellomo R, May CN. Septic shock induces distinct changes in sympathetic nerve activity to the heart and kidney in conscious sheep. Am J Physiol Regul Integr Comp Physiol. 2009;297:R1247–R1253. doi: 10.1152/ajpregu.00437.2009. [DOI] [PubMed] [Google Scholar]
- 132.Chrousos GP. The hypothalamic-pituitary-adrenal axis and immune-mediated inflammation. N Engl J Med. 1995;332:1351–1362. doi: 10.1056/NEJM199505183322008. [DOI] [PubMed] [Google Scholar]
- 133.Brown R, Li Z, Vriend CY, Nirula R, Janz L, Falk J, et al. Suppression of splenic macrophage interleukin-1 secretion following intracerebroventricular injection of interleukin-1 beta: evidence for pituitary-adrenal and sympathetic control. Cell Immunol. 1991;132:84–93. doi: 10.1016/0008-8749(91)90008-Y. [DOI] [PubMed] [Google Scholar]
- 134.Juif PE, Anton F, Hanesch U. Pain behavior and spinal cell activation due to carrageenan-induced inflammation in two inbred rat strains with differential hypothalamic-pituitary-adrenal axis reactivity. Physiol Behav. 2012;105:901–908. doi: 10.1016/j.physbeh.2011.11.008. [DOI] [PubMed] [Google Scholar]
- 135.Kanczkowski W, Sue M, Zacharowski K, Reincke M, Bornstein SR. The role of adrenal gland microenvironment in the HPA axis function and dysfunction during sepsis. Mol Cell Endocrinol. 2015;408:241–248. doi: 10.1016/j.mce.2014.12.019. [DOI] [PubMed] [Google Scholar]
- 136.Griffin AC, Zhao W, Wegmann KW, Hickey WF. The T-cell repertoire contains cells reactive with hormones of the hypothalamic-pituitary-adrenal axis: recognition of synthetic peptide fragments of corticotropin-releasing hormone (CRH) and pro-opiomelanocortin (POMC) in the Lewis rat. Brain Behav Immun. 1994;8:313–326. doi: 10.1006/brbi.1994.1029. [DOI] [PubMed] [Google Scholar]
- 137.Kasahara E, Inoue M. Cross-talk between HPA-axis-increased glucocorticoids and mitochondrial stress determines immune responses and clinical manifestations of patients with sepsis. Redox Rep. 2015;20:1–10. doi: 10.1179/1351000214Y.0000000107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Kasahara E, Sekiyama A, Hori M, Kuratsune D, Fujisawa N, Chida D, et al. Stress-induced glucocorticoid release upregulates uncoupling protein-2 expression and enhances resistance to endotoxin-induced lethality. Neuroimmunomodulation. 2015;22:279–292. doi: 10.1159/000368802. [DOI] [PubMed] [Google Scholar]
- 139.Memon SA, Moreno MB, Petrak D, Zacharchuk CM. Bcl-2 blocks glucocorticoid- but not Fas- or activation-induced apoptosis in a T cell hybridoma. J Immunol. 1995;155:4644–4652. [PubMed] [Google Scholar]
- 140.Davenpeck KL, Zagorski J, Schleimer RP, Bochner BS. Lipopolysaccharide-induced leukocyte rolling and adhesion in the rat mesenteric microcirculation: regulation by glucocorticoids and role of cytokines. J Immunol. 1998;161:6861–6870. [PubMed] [Google Scholar]
- 141.Larange A, Antonios D, Pallardy M, Kerdine-Romer S. Glucocorticoids inhibit dendritic cell maturation induced by Toll-like receptor 7 and Toll-like receptor 8. J Leukoc Biol. 2012;91:105–117. doi: 10.1189/jlb.1110615. [DOI] [PubMed] [Google Scholar]
- 142.Oh KS, Patel H, Gottschalk RA, Lee WS, Baek S, Fraser IDC, et al. Anti-inflammatory chromatinscape suggests alternative mechanisms of glucocorticoid receptor action. Immunity. 2017;47:298–309. doi: 10.1016/j.immuni.2017.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Annane D. Adrenal insufficiency in sepsis. Curr Pharmaceutical Design. 2008;14:1882–1886. doi: 10.2174/138161208784980626. [DOI] [PubMed] [Google Scholar]
- 144.Gaillard RC, Turnill D, Sappino P, Muller AF. Tumor necrosis factor alpha inhibits the hormonal response of the pituitary gland to hypothalamic releasing factors. Endocrinology. 1990;127:101–106. doi: 10.1210/endo-127-1-101. [DOI] [PubMed] [Google Scholar]
- 145.Dimopoulou I, Tsagarakis S, Kouyialis AT, Roussou P, Assithianakis G, Christoforaki M, et al. Hypothalamic-pituitary-adrenal axis dysfunction in critically ill patients with traumatic brain injury: incidence, pathophysiology, and relationship to vasopressor dependence and peripheral interleukin-6 levels. Crit Care Med. 2004;32:404–408. doi: 10.1097/01.CCM.0000108885.37811.CA. [DOI] [PubMed] [Google Scholar]
- 146.Shimada A, Hasegawa-Ishii S. Histological architecture underlying brain-immune cell-cell interactions and the cerebral response to systemic inflammation. Front Immunol. 2017;8:17. doi: 10.3389/fimmu.2017.00017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Flierl MA, Rittirsch D, Weckbach S, Huber-Lang M, Ipaktchi K, Ward PA, et al. Disturbances of the hypothalamic-pituitary-adrenal axis and plasma electrolytes during experimental sepsis. Ann Intensive Care. 2011;1:53. doi: 10.1186/2110-5820-1-53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Ren C, Li XH, Wu Y, Dong N, Tong YL, Yao YM. Inhibition of cerebral high-mobility group box 1 protein attenuates multiple organ damage and improves T cell-mediated immunity in septic rats. Mediators Inflamm. 2019;2019:6197084. doi: 10.1155/2019/6197084. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Not applicable.