

Abstract
Background
Clinical practice guidelines suggest that magnetic resonance imaging (MRI) of the brain should be performed at certain time points or intervals distant from diagnosis (interval or surveillance imaging) of cerebral glioma, to monitor or follow up the disease; it is not known, however, whether these imaging strategies lead to better outcomes among patients than triggered imaging in response to new or worsening symptoms.
Objectives
To determine the effect of different imaging strategies (in particular, pre‐specified interval or surveillance imaging, and symptomatic or triggered imaging) on health and economic outcomes for adults with glioma (grades 2 to 4) in the brain.
Search methods
The Cochrane Gynaecological, Neuro‐oncology and Orphan Cancers (CGNOC) Group Information Specialist searched the Cochrane Central Register of Controlled Trials (CENTRAL), MEDLINE and Embase up to 18 June 2019 and the NHS Economic Evaluation Database (EED) up to December 2014 (database closure).
Selection criteria
We included randomised controlled trials, non‐randomised controlled trials, and controlled before‐after studies with concurrent comparison groups comparing the effect of different imaging strategies on survival and other health outcomes in adults with cerebral glioma; and full economic evaluations (cost‐effectiveness analyses, cost‐utility analyses and cost‐benefit analyses) conducted alongside any study design, and any model‐based economic evaluations on pre‐ and post‐treatment imaging in adults with cerebral glioma.
Data collection and analysis
We used standard Cochrane review methodology with two authors independently performing study selection and data collection, and resolving disagreements through discussion. We assessed the certainty of the evidence using the GRADE approach.
Main results
We included one retrospective, single‐institution study that compared post‐operative imaging within 48 hours (early post‐operative imaging) with no early post‐operative imaging among 125 people who had surgery for glioblastoma (GBM: World Health Organization (WHO) grade 4 glioma). Most patients in the study underwent maximal surgical resection followed by combined radiotherapy and temozolomide treatment. Although patient characteristics in the study arms were comparable, the study was at high risk of bias overall.
Evidence from this study suggested little or no difference between early and no early post‐operative imaging with respect to overall survival (deaths) at one year after diagnosis of GBM (risk ratio (RR) 0.86, 95% confidence interval (CI) 0.61 to 1.21; 48% vs 55% died, respectively; very low certainty evidence) and little or no difference in overall survival (deaths) at two years after diagnosis of GBM (RR 1.06, 95% CI 0.91 to 1.25; 86% vs 81% died, respectively; very low certainty evidence). No other review outcomes were reported.
We found no evidence on the effectiveness of other imaging schedules. In addition, we identified no relevant economic evaluations assessing the efficiency of the different imaging strategies.
Authors' conclusions
The effect of different imaging strategies on survival and other health outcomes remains largely unknown. Existing imaging schedules in glioma seem to be pragmatic rather than evidence‐based. The limited evidence suggesting that early post‐operative brain imaging among GBM patients who will receive combined chemoradiation treatment may make little or no difference to survival needs to be further researched, particularly as early post‐operative imaging also serves as a quality control measure that may lead to early re‐operation if residual tumour is identified. Mathematical modelling of a large glioma patient database could help to distinguish the optimal timing of surveillance imaging for different types of glioma, with stratification of patients facilitated by assessment of individual tumour growth rates, molecular biomarkers and other prognostic factors. In addition, paediatric glioma study designs could be used to inform future research of imaging strategies among adults with glioma.
Keywords: Humans, Brain Neoplasms, Brain Neoplasms/diagnostic imaging, Brain Neoplasms/mortality, Brain Neoplasms/surgery, Controlled Before‐After Studies, Glioma, Glioma/diagnostic imaging, Glioma/mortality, Glioma/surgery, Magnetic Resonance Imaging, Magnetic Resonance Imaging/methods, Randomized Controlled Trials as Topic, Reoperation
Plain language summary
Brain scans for people with cerebral gliomas
Background
Gliomas are brain tumours arising from the supporting brain tissues. They are termed low grade (WHO 1 and 2) or high grade (WHO 3 and 4), depending on their cell activity and how aggressive the tumour is. Gliomas are diagnosed in 4 to 11 people for every 100,000 each year, more commonly in high‐income countries. Whilst low‐grade, slow‐growing gliomas may be watched before deciding the most appropriate treatment (active surveillance), most people with gliomas will eventually undergo surgery to safely remove as much of the tumour as possible. After surgery, other treatments such as radiation and chemotherapy might be suitable, depending on the tumour grade and other aspects. The standard treatment for grade 4 tumours, known as glioblastomas, is radiotherapy and an anti‐cancer medicine (temozolomide).
In people with slow‐growing tumours, magnetic resonance imaging (MRI) scans are usually performed at regular intervals to check tumour growth before treatment. After surgery to remove gliomas of any grade, regular scans check how tumours are responding to added treatment and whether the disease is coming back. Scans are mostly performed at set timings rather than happening due to changes in a patient's condition. This is known as surveillance imaging. An MRI is often done within three days of high‐grade glioma surgery to check how much tumour has been removed.
Instead of having regular scans, you could scan when someone experiences changes suggesting that the tumour has grown. This is known as symptomatic or triggered imaging. Brain scans can be expensive and regular scans when a person feels well may cause anxiety. Also, if a brain scan will not change treatment it might not be needed. We undertook this review because it is not known whether different timings of imaging have an impact on the time a person will survive after diagnosis. We also wanted to see which approach was better in identifying concerning tumour changes, and effects on quality of life, anxiety and depression. We also searched to see which approach would provide better value to the health service.
How we conducted the review and what we found
We looked for studies involving adults with gliomas that compared current practice of doing scans at specific time points with other approaches. We found only one study meeting our criteria. This was from a cancer centre in the USA, looking at glioblastoma patients (those with the most aggressive gliomas) who had been treated between 2006 and 2016. The study involved 125 people and split them into those scanned within two days of surgery (early scan) with those who were not. They showed that doing the early scan made no change to the chance of being alive at one and two years after diagnosis. This might have been because the early scans were not used to change treatments, which mainly were to receive standard radiotherapy and temozolomide, and we could not tell if the patients' surgeon(s) were different or had different approaches to care. We judged this suggestion of little change in survival time with or without early scanning to be very uncertain. The number of people included over 10 years was small, and the decision whether or not to have a scan after surgery was based on surgeon's choice. It was not clear whether the surgeon(s) involved or their approaches to care differed, nor whether a person's care might have been changed in light of early imaging. The other search did not find any studies looking at the value of different imaging approaches.
Conclusions
We still do not know whether doing scans regularly at specific times after glioma diagnosis changes how well patients do. The limited evidence, suggesting early scans after operations do not affect survival, is unreliable and more research is needed, especially as early scans may also help surgeons improve their practice, and decide whether to repeat the operation earlier than they might otherwise have chosen to do.
The best timings and reasons for scanning brain gliomas in adults are not known. Lessons might be learned from studies involving children, and by looking at large collections of clinical trials. It is also important to study the potential costs and benefits of different strategies.
Summary of findings
for the main comparison.
| Early post‐operative MRI compared with no early post‐operative MRI for glioblastoma | |||||
|
Patient or population: adults with glioblastoma Settings: tertiary care Intervention: early post‐operative MRI (within 48 hours of tumour resection) Comparison: no early post‐operative MRI | |||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | |
| Assumed risk | Corresponding risk | ||||
| No early MRI | Early MRI | ||||
| Overall survival (deaths at 1 year) | 554 per 1000 | 476 per 1000 (338 to 670) | (RR 0.86, 95% CI 0.61 to 1.21) | 125 (1) | ⊕⊕⊝⊝ very low1 |
| Overall survival (deaths at 2 years) | 804 per 1000 | 852 per 1000 (732 to 1000) | (RR 1.06, 95% CI 0.91 to 1.25) | 125 (1) | ⊕⊕⊝⊝ very low1 |
| Progression free survival | no evidence | ||||
| Anxiety | no evidence | ||||
| Quality of life | no evidence | ||||
| *The basis for the assumed risk is the risk in the control group of the included study. The corresponding risk (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | |||||
| GRADE Working Group grades of evidence High certainty: Further research is very unlikely to change our confidence in the estimate of effect. Moderate certainty: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low certainty: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low certainty: We are very uncertain about the estimate. | |||||
1Downgraded for risk of bias, as the patients in this study received the interventions based on surgeons' preferences and discretion
Background
Description of the condition
Brain and other central nervous system (CNS) tumours are less common than many other cancers, accounting for around 1.9% of new cancer diagnoses annually and approximately 2.3% or 189,382 deaths worldwide in 2012 (GLOBOCAN 2012). In terms of years of potential life lost (YPLL), in the USA malignant brain and other CNS tumours have the greatest impact of any cancer in adults, a reflection of their age of onset and the comparatively short survival time of some of the most common types (Rouse 2016). Gliomas are brain tumours that are believed to arise from progenitor glial lineage cells derived from neural stem cells (although this is still a matter of debate). They occur at an annual incidence of four to 11 people per 100,000 and are more frequent in high‐income, industrialised countries (Ohgaki 2009). Gliomas are graded 1 to 4 by the World Health Organization (WHO), according to their malignancy or malignant potential. The 2007 WHO classification system (Louis 2007), used in completed clinical studies since 2007, graded gliomas based on histological characteristics only. However in the 2016 WHO classification system, which is likely to be used in future studies, grading for the first time depends on both histological and molecular features, for example isocitrate dehydrogenase (IDH) status, chromosome 1p/19q co‐deletions, and other genetic parameters (Louis 2016). Molecular characterisation is now the default assessment where available, and supersedes the histological phenotype. Using the 2007 WHO classification, gliomas graded 1 and 2 are referred to as low‐grade gliomas: these included pilocytic astrocytomas (grade 1) which represent a distinct clinical entity with low malignant potential; and diffuse astrocytomas, oligodendrogliomas and mixed oligoastrocytomas (grade 2). High‐grade gliomas included anaplastic astrocytomas, anaplastic oligodendrogliomas (grade 3) and glioblastoma multiforme (GBM; grade 4). The 2016 classification separates pilocytic astrocytoma into "other astrocytic tumours" and it is the category of "diffuse astrocytic and oligodendroglial tumours" comprising grades 2 to 4 that are the focus of this review. As of 2016, true mixed oligoastrocytomas are now exceedingly rare, since detection of both canonical molecular signatures is required within the same tumour. Such changes are indicative of the challenges faced in interpreting previous studies in glioma in the current clinical context. While in the literature malignant transformation rates vary between approximately one in four to three in four, there is a significant risk of malignant transformation of low‐grade to high‐grade glioma and the potential for, and timing of, malignant transformation is not well understood.
Grades correspond to prognosis. Grade 1 has a good prognosis and can often be cured with surgery alone; grade 2 gliomas have a median survival of five to fifteen years; anaplastic astrocytoma (grade 3) between two and five years; whereas grade 4 has the poorest prognosis, with a median survival of one to two years in the era of safe maximal resection and temozolomide chemoradiotherapy (Louis 2007). Even in the molecular era, therefore, tumour grade is a key factor in deciding how to treat gliomas. It also plays a role in determining the timing and frequency of radiologic follow‐up. It is increasingly recognised, however, that molecular characteristics also play a role, with 1p/19q co‐deleted IDH mutant oligodendrogliomas having a more favourable prognosis than 1p/19q intact IDH mutant diffuse astrocytomas, which in turn demonstrate longer survival than IDH‐wildtype diffuse astrocytomas. It is expected that further molecular characterisation of the last group will officially enter WHO classification in a future update, identifying those with GBM‐like markers, which can be managed in a similar way to high‐grade glioma despite the traditional histological appearance of low‐grade glioma. It is currently unclear how these molecular features will dovetail with traditional 'high risk' stratification of low‐grade glioma (e.g. Pignatti 2002), such as aged older than 40 years and achieving less than gross total resection. This evolving clinical and molecular risk stratification will have implications for future guidelines and research in how gliomas are treated and imaged.
Description of the intervention
Magnetic resonance imaging (MRI) or computed tomography (CT) of the brain is key to the diagnosis of a glioma. MRI is the preferred technique as it has superior soft tissue resolution and gives a better definition of tumour extent (NCCN 2018; NCI 2017; NICE 2006). These imaging techniques can, however, be challenging to interpret when distinguishing the type of tumour and grade, or occasionally when differentiating tumours from inflammatory or ischaemic lesions (e.g. following a stroke or radiation). This is particularly the case during follow‐up of treated gliomas where the most commonly used criterion of enlarging contrast‐enhancing lesions can be non‐specific and is also seen with treatment‐related effects such as pseudoprogression or radiation necrosis. Other imaging techniques, such as magnetic resonance spectroscopy (MRS), MR perfusion imaging, single photon emission computed tomography (SPECT) and positron emission photometry (PET), can be used to provide additional information prior to surgery to improve the accuracy of histopathological examination and provide increased specificity during follow‐up, although variation in training, performance, and availability of these techniques limits widespread use (NCCN 2018; NICE 2018).
In addition to primary diagnosis, brain imaging is an integral part of management and follow‐up of glioma and informs treatment decisions in the period immediately after diagnosis. After surgery, MRI has been shown to more accurately identify the extent of residual tumour — an important prognostic factor — than the neurosurgeon's estimation (Albert 1994). The National Comprehensive Cancer Network (NCCN) guidelines recommends post‐operative brain MRI within 24 to 72 hours of glioma surgery for this purpose (NCCN 2018). Imaging is also used universally to assess the tumour response among people actively receiving treatment and, in clinical studies of glioma treatment, imaging before the start of adjuvant therapy is common practice, which can serve as the baseline for detecting tumour progression (e.g. Malmström 2017; Wick 2012). In a study of bevacizumab for glioblastoma, performing a follow‐up MRI from as early as four weeks after starting treatment has been reported to be an accurate predictor of both treatment response and survival (Field 2017).
Outside the immediate treatment period, people with glioma will also be offered imaging at regular intervals. Interval imaging, sometimes referred to as surveillance imaging, refers to imaging at defined future time points as requested by the clinical team, for monitoring tumour appearance. Outside periods of treatment, this is referred to as active surveillance. It is not known at that specific time point in the future whether the person affected will be less symptomatic, the same (i.e. stable), or be more symptomatic at the imaging visit. The imaging is not, therefore, based on a clinical deterioration of the person’s symptoms and signs. An alternative approach to interval imaging would be to arrange imaging when a person, carer, or clinician notes a deterioration in signs or symptoms (or both); this is called symptomatic or triggered imaging. Current practice represents a combination of these, with surveillance imaging as the default, and triggered imaging when a significant clinical change is encountered between planned imaging visits.
There are several scenarios in which interval imaging is used among people with a glioma. Before surgical biopsy or treatment among those with a suspected low‐grade glioma, interval imaging might be performed to ensure that the actual growth of the tumour is not faster than that expected from the anticipated grade. It might also be considered for lesions with radiological features of a low‐grade tumour that do not necessitate immediate treatment, for example an optic pathway glioma (NICE 2018). After treatment, interval imaging is usually performed for both low‐grade and high‐grade gliomas to check for any interval tumour growth and to determine whether treatment has been successful at slowing or halting growth. The NCCN recommends MRI follow‐up of low‐grade gliomas every three to six months for five years, then at least annually thereafter; whereas for GBM, an MRI two to six weeks after radiotherapy, then every two to four months for three years, then every six months indefinitely, is recommended (NCCN 2018). Suggested imaging intervals in National Institute for Health and Clinical Excellence (NICE) guidance are slightly longer (e.g. for GBM the suggested intervals are three to six months up to two years after treatment, then six to 12 months up to three years, then annually indefinitely (Table 2)). In general, though, for both low‐grade and high‐grade gliomas, interval duration increases over time from diagnosis and treatment.
1. National Institute for Health and Clinical Excellence (NICE) guidance on interval imaging.
| Tumour grade | Years after end of treatment | |||||
| 0 to 1 | 1 to 2 | 2 to 3 | 3 to 4 | 5 to 10 | > 10 (for the rest of life) | |
| Grade 1 | Scan at 12 months, then:
|
|||||
| Grade II 1p/19q non‐codeleted, IDH mutated | Scan at 3 months, then every 6 months | Annually | Every 1 to 2 years | Consider ongoing imaging every 1 to 2 years | ||
| Grade II 1p/19q codeleted | ||||||
| Grade III 1p/19q codeleted | ||||||
| Grade II IDH wildtype | Every 3 to 6 months |
Every 6 to 12 months |
Annually | Consider ongoing imaging every 1 to 2 years |
||
| Grade III 1p/19q non‐codeleted | ||||||
| Grade IV (glioblastoma) | ||||||
From NICE 2018 (p17) Table 3: Possible regular clinical review schedule for people with glioma depending on grade of tumour
SPECT and PET can also be helpful in the post‐treatment scenario to distinguish between tumour recurrence and treatment effects such as pseudoprogression and radiation necrosis (NCCN 2018; NCI 2017). This is an important role of imaging, since treatment‐related effects can be symptomatic, and either neutral (radiation necrosis) or favourable (pseudoprogression) when deciding on whether to maintain or change treatment.
Why it is important to do this review
To our knowledge, there are no existing systematic reviews of this topic. The relative benefits of surveillance imaging, if any, over other imaging approaches among people with glioma are currently not known. In addition, whilst clinical guidelines recommend routine MRI follow‐up or active monitoring, the optimal frequency of routine active monitoring has not been rigorously established. In existing clinical practice guidelines, it is recommended that the frequency of routine active monitoring decreases with time (NCCN 2018; NICE 2018; SEOM 2017). However, since low‐grade glioma can present at any time during a natural history that might span over a decade, and the risk and timing of recurrence is unpredictable, frequent imaging early on might be unnecessary.
Diagnostic uncertainties raised by brain imaging performed within the first three to six months due to treatment, such as true tumour progression versus pseudoprogression, might increase patient and clinician anxiety during this monitoring period, and negatively affect quality of life. In addition, potential health risks from repeat gadolinium administration has come under scrutiny recently, especially in people with longer‐term survival. Brain imaging is potentially costly, both for the health system and the individual and, while performing it routinely has not been robustly assessed from a health economic perspective, there are also limited data on the psychological impact experienced by patients and carers during treatment from repeated cycles of imaging and uncertainty while waiting on the result.
Surveillance imaging was established as a follow‐up strategy early in the development of readily available non‐invasive imaging investigation, based on expert consensus. This basis has become questionable, since this expert opinion was derived from experience gained during the 'pre‐molecular' era when recent standards of care for glioma treatment were not yet available or established. The aim of this review, therefore, is to systematically evaluate the evidence for different imaging strategies to inform clinical practice guidelines and research agendas.
Objectives
To determine the effect of different imaging strategies (in particular, pre‐specified interval or surveillance imaging, and symptomatic or triggered imaging) on health and economic outcomes for adults with glioma (grades 2 to 4) in the brain.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs), non‐randomised controlled trials (NRCTs), and controlled before‐after studies (CBAS) with concurrent comparison groups.
Full economic evaluations (cost‐effectiveness analyses, cost‐utility analyses and cost‐benefit analyses), conducted alongside any study design and any model‐based economic evaluations.
Types of participants
Adults with a histologically confirmed diagnosis of cerebral glioma or suspected low‐grade infiltrating glioma on imaging. Given the known biological differences between adult and childhood gliomas, in addition to the different logistic challenges and economic aspects of imaging adult and paediatric populations, this review addresses adults with glioma.
Types of interventions
Pre‐ or post‐treatment brain imaging, including MRI and other types of imaging, performed at regular intervals (interval or surveillance imaging) compared with pre‐ or post‐treatment brain imaging performed upon the development of new or worsening symptoms (symptomatic or triggered imaging) or other imaging schedules, such as hybrids, with a defined surveillance hiatus.
Types of outcome measures
Primary outcomes
Overall survival (as time‐to‐event or dichotomous outcomes or both)
Secondary outcomes
Progression‐free survival (as time‐to‐event or dichotomous outcomes or both)
Health‐related quality of life measured by a standardised instrument, such as the European Organisation for Research and Treatment of Cancer (EORTC) QLQ‐C30 or QLQ‐BN20 (specific for brain cancer), or the Functional Assessment of Cancer Therapy scale (FACT)‐G (general) or FACT‐Br (specific for brain cancer)
Anxiety, measured by a standardised instrument, such as the Hospital Anxiety and Depression Scale (HADS)
Depression, measured by a standardised instrument, such as HADS and the Beck Depression Inventory (BDI)
Economic data (cost, cost effectiveness, cost utility and cost benefits)
Search methods for identification of studies
Electronic searches
The Cochrane Gynaecological, Neuro‐oncology and Orphan Cancer (CGNOC) Information Specialist searched the following databases on 26 April 2018 and topped up the search for us on the 18 June 2019.
-
For intervention studies
Cochrane Central Register of Controlled Trials (CENTRAL), in the Cochrane Library;
MEDLINE Ovid (from 1946 onwards);
Embase Ovid (from 1980 onwards).
-
For economic evidence
MEDLINE Ovid (from 2015);
Embase Ovid (from 2015);
NHS Economic Evaluation Database (EED).
The EED database was searched up to the end of December 2014 (when the last records were added to that database) and MEDLINE and Embase from 1 January 2015, as NHS EED already included comprehensive searches of these databases prior to 2015. We also considered relevant grey literature, such as health technology assessments, reports and working papers, for inclusion.
Please refer to Appendix 1 for draft MEDLINE search strategies. We did not apply language restrictions to any of the searches.
Searching other resources
We searched the following for ongoing studies.
ClinicalTrials.gov (clinicaltrials.gov)
International Clinical Trials Registry Platform (ICTRP) (apps.who.int/trialsearch)
We used the 'related articles' feature of PubMed and handsearched the reference lists of included studies to identify newly published articles and additional studies of relevance. If through these searches we had identified any ongoing studies that had not been published, we planned to approach the principal investigators to ask for an update on the study status and any available relevant data.
Data collection and analysis
Selection of studies
The CGNOC Information Specialist downloaded all titles and abstracts retrieved by electronic searching for intervention studies to Endnote® and removed duplicates. Two review authors (GT and TL) independently screened the remaining records on title and abstract. For potentially eligible records, we obtained copies of the full texts and independently assessed them for eligibility. The two review authors resolved any disagreements by discussion. We used Covidence to facilitate this study selection process and documented reasons for exclusion accordingly.
In our search for economic evidence, two review authors independently screened for eligible studies using the same methods as above.
Data extraction and management
Intervention studies
Two review authors (TL and GT) independently extracted data from the included intervention study to a pre‐designed data extraction form that included the following.
Author contact details
Country
Setting
Dates of participant accrual
Funding source
Inclusion and exclusion criteria
Study design
-
Study population and baseline characteristics
number of participants enrolled;
number of participants analysed;
age of participants;
gender of participants;
type of glioma (low grade or high grade; tumour size, molecular markers);
type of glioma treatment (surgery; radiotherapy; chemotherapy).
-
Intervention details
type of intervention (pre‐specified, interval or surveillance imaging), including whether it occurred pre‐treatment or post‐treatment, the interval period, and the type of scan (e.g. MRI);
type of comparator (symptomatic imaging or other imaging schedule), including indications for imaging, information on the timing of the imaging (e.g. median time to first scan post‐treatment), and the type of scan.
'Risk of bias' assessment (see below)
Duration of follow‐up
Primary outcome(s) of the study
-
Review outcomes
For time‐to‐event outcomes (overall and progression‐free survival), we planned to extract the hazard ratio (HR) with its 95% confidence interval (CI) for time points as reported by the study authors. We would have noted the definition of progression and the procedure used to identify it. Where reported, we also planned to extract dichotomous data for these outcomes at author‐specified time points.
For dichotomous outcomes (e.g. death, anxiety, depression), we extracted the number of participants in each treatment arm that experienced the outcome of interest and the number of participants assessed.
For continuous outcomes (e.g. quality‐of‐life scores, anxiety, depression), we planned to extract the value and standard deviation of the outcome of interest and the number of participants assessed at the relevant time point in each group. We also planned to extract change‐from‐baseline score data where reported and to note the type of scale used.
We planned to extract adjusted statistics where reported.
Where possible, all data extracted were those relevant to an intention‐to‐treat analysis, in which participants were analysed in the groups to which they were assigned.
We resolved differences between review authors by discussion or by appeal to a third review author when necessary.
Studies of economic evaluation
We developed a data extraction form for economic evaluations based on the format and guidelines used to produce structured abstracts of economic evaluations for inclusion in the NHS Economic Evaluation Database (NHS EED), adapted to the specific requirements of this review. In addition to the outcomes described above, we planned to extract the following data.
Type of evaluations
Sources of effectiveness data
Cost data
Sources of cost data
Sources of outcome valuations
Analytical approach
Assessment of risk of bias in included studies
For studies of clinical effects, we assessed the risk of bias of the included study using Cochrane's tool and the criteria specified in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2017). This included assessment of:
random sequence generation;
allocation concealment;
blinding of participants and healthcare providers;
blinding of outcome assessors;
incomplete outcome data (more than 20% missing data considered high risk);
selective reporting of outcomes;
other possible sources of bias, such as lack of a power calculation, baseline differences in group characteristics.
We assessed according to whether there was a risk of bias for the following criteria.
How participants were selected or allocated to the intervention/comparison groups.
Whether the intervention participants were representative of the population of interest.
Whether the comparison participants were representative of the population of interest.
Whether differences between the intervention and comparison groups were controlled for or absent.
Any other risk of bias.
Two review authors (TL and GT) assessed risk of bias independently and resolved any differences by discussion. We summarised judgements in a 'Risk of bias' table along with the characteristics of the included study and interpreted the review results in light of the overall 'Risk of bias' assessment. For more details about assessing risk of bias, see Appendix 2.
Economic evaluations would be assessed for bias in two stages. The first stage would involve assessing risk of bias from the sources of the effectiveness data. We would assess economic evaluations carried out alongside clinical studies using the Cochrane 'Risk of bias' tool, as described above. If the economic evaluation was model‐based, we would use the ROBIS tool to assess bias in the effectiveness studies (Whiting 2016). The second stage involves assessing the risk of bias of the economic evidence (i.e. assessing the overall methodological quality). We would use the CHEERS and Evers checklists to do this (hsr.mumc.maastrichtuniversity.nl/consensus‐health‐economic‐criteria‐chec‐list), (Husereau 2013; Thielen 2016; Van Mastrigt 2016; Wijnen 2016).
Measures of treatment effect
We only found data for dichotomous outcomes, for which we calculated the effect size as a risk ratio (RR) with its 95% CI.
Unit of analysis issues
Two review authors (TL and GT) would have reviewed unit of analysis issues according to the criteria presented in Deeks 2017 and would have resolved any disagreements by discussion. Unit of analysis issues might have included reports where there were multiple observations for the same outcome (e.g. repeated measurements with different scales or at different time points, recurring events).
Dealing with missing data
We did not impute missing data. In the event of missing data, we planned to write to study authors to request the data and describe in the 'Characteristics of included studies' tables how any missing data were obtained.
Assessment of heterogeneity
We planned to assess heterogeneity between intervention studies in each meta‐analysis by visual inspection of forest plots; by estimation of the percentage heterogeneity between studies that cannot be ascribed to sampling variation (Higgins 2003); by a formal statistical test of the significance of the heterogeneity (Deeks 2008); and, where possible, by subgroup analyses. If there was evidence of substantial heterogeneity, we would have investigated and reported the possible reasons for it.
Assessment of reporting biases
If there were 10 or more intervention studies in meta‐analyses we planned to investigate reporting biases (such as publication bias) using funnel plots and planned to assess asymmetry visually. If asymmetry was suggested by a visual assessment, we planned to perform exploratory analyses to investigate it (Sterne 2017).
Data synthesis
For interventions studies
We planned to conduct meta‐analyses if we judged participants, comparisons and outcomes to be sufficiently similar to ensure an answer that was clinically meaningful. We planned to use the random‐effects model with inverse variance weighting for all meta‐analyses. If any studies had multiple intervention groups, we planned to divide the ‘shared’ comparison group into the number of treatment groups and comparisons between each treatment group, and treat the split comparison group as independent comparisons. We planned to perform meta‐analysis of the results assuming that we found at least two included studies that were sufficiently similar for the findings to be clinically meaningful. When a meta‐analysis was not possible due to the availability of single studies only, we planned to enter the data from single studies into Review Manager 5 (Review Manager 2014), without totals, and grade the findings as described below.
For studies of economic evaluation
We planned to summarise characteristics and results of included economic evaluations using additional tables, supplemented by a narrative summary that would compare and evaluate methods used and principal results between studies. We would also tabulate unit cost data, when available. We would report the currency and price year applicable to measures of costs in each original study, alongside measures of costs, incremental costs and incremental cost effectiveness, by study. Where details of currency and price year were available in original studies, we would convert measures of costs, incremental costs and cost effectiveness to (latest year) International Dollars value using implicit price deflators for gross domestic product (GDP) and GDP purchasing power parities (eppi.ioe.ac.uk/costconversion/default.aspx; Shemlit 2011). We would summarise details of the methodological characteristics of individual included health economics studies in the ‘Characteristics of included studies’ tables. We would conduct all elements of the economics component of this review according to current guidance on the use of economics methods in the preparation and maintenance of Cochrane Reviews (Shemlit 2011).
GRADE, 'Summary of findings' tables and results reporting
For evidence from intervention studies, based on the methods described in Chapter 11 of the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2017a), we prepared a 'Summary of findings' table to present the results of the following outcomes.
Overall survival
Progression‐free survival
Quality of life
Anxiety
For each assumed risk cited in the tables, we provided a rationale, and used the GRADE system to rank the quality of the evidence (Schünemann 2017b). Where the evidence was based on single studies, or where there was no evidence on a specific outcome, we included the outcome in the 'Summary of findings' tables and graded or explained accordingly. Two review authors would grade the evidence together. We would consider downgrading evidence of a clear effect derived from single studies with small sample sizes or few events and would resolve any differences of opinion by discussion and, if necessary, by involving a third review author. We planned to report the results of the meta‐analyses in the text based on the guidance from Cochrane Effective Practice and Organisation of Care on review results reporting and interpretation (EPOC 2015).
For the economic evaluation evidence, we planned to present the following findings in a table.
Method of economic evaluation
Costs
Outcomes
Incremental cost‐effectiveness ratio
Subgroup analysis and investigation of heterogeneity
For intervention studies, we planned to perform subgroup analysis by the glioma grade (low grade and high grade) and molecular markers, if possible. We planned to use formal tests for subgroup differences to determine whether the effect of interventions differs according to these subgroups. Depending on these findings, we would consider whether an overall summary was meaningful. We would have also considered factors such as age, gender, type of treatment, and risk of bias in interpretation of any heterogeneity. If we had identified substantial heterogeneity, we planned to investigate it in sensitivity analyses.
Sensitivity analysis
For intervention studies, we planned to perform sensitivity analysis to investigate substantial heterogeneity identified in meta‐analyses of primary outcomes and also to evaluate the effect after excluding studies at high risk of bias, to investigate how study quality affects the certainty of the findings.
Results
Description of studies
Results of the search
Intervention study searches
Electronic searches for intervention studies yielded 1891 deduplicated records. Of these, we selected 57 studies on title and abstract as potentially eligible studies. At this level of screening we included paediatric studies and certain other studies that would clearly be excluded based on study design because we expected included studies to be few and wanted to identify as many studies relevant to the research question as possible. We subsequently found no studies (including an additional abstract identified from the 2016 BNOS conference proceedings) that met our study selection criteria and 58 studies were excluded. We identified no eligible studies on www.ClinicalTrials.gov.
A subsequent top‐up search conducted on 18 June 2019 yielded 173 records and led to the retrieval of two full texts: we included Mrowczynski 2018 and excluded Zaazoue 2019. See Figure 1.
1.
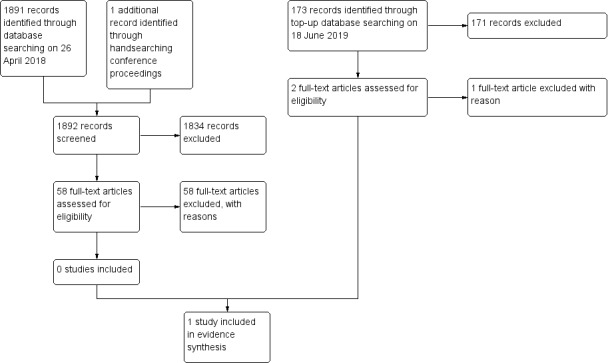
Study flow diagram.
Economic evaluation study searches
The electronic searches for studies of economic evaluation yielded a list of 58 deduplicated records; of these, we identified one study for potential eligibility. After examining the full text of Heinzel 2012 we agreed that the evaluation was not assessing imaging approaches for monitoring or follow‐up but approaches for diagnosis: we therefore excluded this study (i.e. we had not identified any economic evaluations). A top‐up search was carried out in June 2019, which identified a further four records: one of these was potentially eligible (Zaazoue 2019). We considered this study for inclusion as, although it was a paediatric study, some of the participants were over the age of 18. There was, however, no specific subgroup analysis for participants over 18 and ultimately we excluded the study. We therefore have no economic evaluations to include.
Included studies
Intervention evaluation studies
Only one study was included (Mrowczynski 2018). This was a retrospective series of 125 patients with GBM who were treated at the Pennsylvania State University Department of Neurosurgery between 2006 and 2016. The study compared early post‐operative MRI (within 48 hours of surgery) with no early post‐operative MRI. Patients were excluded if they had had a previous low‐grade glioma, and if date of death could not be accurately determined. The primary outcome in this study was overall survival, which was reported in terms of the probability of surviving one and two years, and median survival in days. Mean age was approximately 60 years overall, with slightly more men than women in the cohort. Most patients were treated with maximal safe resection and received radiotherapy (60 Gy) combined with temozolomide chemotherapy, with approximately 80% in both imaging groups completing these treatments. Just over half the patients (56%) also received adjuvant temozolomide or another treatment (e.g. bevacizumab, Optune), or both. The group characteristics were largely comparable between the intervention arms, with a slightly lower mean age and higher treatment completion rates in the early post‐operative imaging group than the no post‐operative imaging group. Authors did not report the timing of the first post‐operative (pre‐radiotherapy) MRI in the 'no early post‐operative imaging' group.
Economic evaluation studies
We identified no economic evaluations for this review.
Excluded studies
Intervention studies
We excluded the 59 studies (58 from the April 2018 search and one from the June 2019 top‐up search) for the following reasons.
Different study question (Albert 1994; Chang 2014; Chataway 1999; Danchaivijitr 2008; Farace 2013; Field 2017; Fujimura 2004; Galban 2011; Gittleman 2016; Gorlia 2013; Klingelhofer 2015; Li 2011; Merkel 2017; Milano 2010; Patil 2017; Sreenivasan 2016; Wick 2016).
-
Wrong type of study
Case report (Cochereau 2016; Galldiks 2010; Singh 2012)
Non‐comparative case series (Gui 2018; Leonard 1975; Lorimer 2017; Quinn 1971; Spitaels 2017)
Other non‐comparative study (Hamdan 2017; Mahaley 1983; Meyers 2003; Scoccianti 2010; Seiz 2011; Wang 2009; Weizman 2014)
Review (Abrigo 2018; Christie 1975; Cihoric 2017; Fouke 2015; Oberheim Bush 2016; Sanghera 2012; Shah 2011; Shiroishi 2013).
Commentary/opinion paper (Easaw 2011; Macdonald 1990; Neagu 2015; Reardon 2014; Sabattini 1980).
Qualitative study (Geer 2012)
Wrong study population (Ali 2014; de Graaf 2002; Kim 2014; Korones 2001; Perreault 2014; Poussaint 2011; Saunders 2005; Sethi 2011; Sutton 1996; Udaka 2012; Zaazoue 2019).
Several studies had more than one reason for exclusion. For two older studies (Aoyama 1982; Mueller 1981), we were unable to obtain the full text.
In addition, we excluded Heinzel 2012 from the economic search as it related to a different study question, making the total number of excluded studies 59.
Risk of bias in included studies
The only included intervention evaluation study was a non‐randomised retrospective study that we assessed as being at high risk of bias overall, mainly due to high risk of selection bias because the decision about whether or not a patient received the intervention was left to the preference and discretion of the attending clinician (Mrowczynski 2018). Thus, intervention groups might have been divided across different practitioners and have been managed differently. The study population was, however, appropriate in terms of disease evaluated (GBM) and treatment received; and available patient baseline characteristics were comparable between the groups and any differences that might have favoured better outcomes in the intervention group (e.g. lower mean age) did not do so. Molecular information, early re‐operation for residual enhancing disease, and second line and subsequent therapies were not reported. In addition, the reviewer team had concerns about attrition bias from patients who did not have their date of death recorded in the site's system, because the number of patients eligible and included in the study from a 10‐year period at the neuro‐oncology centre seemed low.
Effects of interventions
See: Table 1
Evidence from intervention evaluation studies
Comparison: Early post‐operative imaging versus no early post‐operative imaging
The evidence is derived from one included study only (Mrowczynski 2018).
Overall survival
Findings from the single study with 125 subjects suggested little or no difference between early and no early post‐operative imaging with respect to the proportion of deaths at one year after diagnosis (RR 0.86, 95% CI 0.61 to 1.21; 48% vs 55% had died, respectively; very low certainty evidence; Analysis 1.1) or two years after diagnosis (RR 1.06, 95% CI 0.91 to 1.25; 86% vs 81% had died, respectively; very low certainty evidence; Analysis 1.2).
1.1. Analysis.
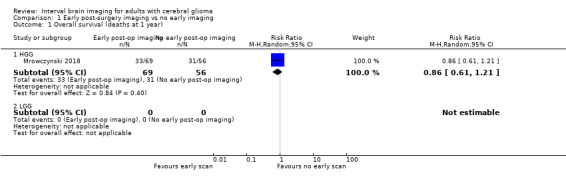
Comparison 1 Early post‐surgery imaging vs no early imaging, Outcome 1 Overall survival (deaths at 1 year).
1.2. Analysis.
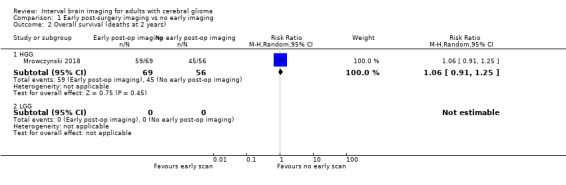
Comparison 1 Early post‐surgery imaging vs no early imaging, Outcome 2 Overall survival (deaths at 2 years).
We found no data related to other review outcomes or comparisons.
Evidence from economic evaluation studies
There was no evidence regarding the cost‐effectiveness of different imaging strategies for monitoring glioma in adults.
Discussion
Summary of main results
This systematic review found one retrospective, comparative study (125 patients) of early post‐operative imaging compared with no early post‐operative magnetic resonance imaging for the management of GBM. Evidence suggested that there may be little or no difference with early imaging after surgery compared with no early imaging in patient survival at one and two years after diagnosis when the standard of care for GBM patients was combined radiotherapy and temozolomide after maximal surgical resection. However, we assessed the study as having a high risk of bias for various reasons mainly related to its design, and hence we assessed the evidence as very low quality/certainty.
We found no evidence on the comparative benefits and risks of other types of imaging strategies that could inform guidance on the optimal timing of surveillance imaging.
Overall completeness and applicability of evidence
There is little evidence to inform clinical practice guidelines with respect to the effectiveness of different imaging strategies to improve health outcomes among people with gliomas.
This review has identified no evidence to inform decision makers on the efficiency of different imaging strategies in the management of adults with glioma.
Quality of the evidence
The evidence on the relative effectiveness of early post‐operative imaging is of very low quality/certainty and further research is needed. Evidence on other imaging strategies is absent.
This review has identified no evidence on the cost effectiveness of different imaging strategies in the management of adults with glioma.
Potential biases in the review process
We followed Cochrane methodology for reviews of interventions and are unaware of any potential biases in the review process. We found only one comparative study of the different imaging approaches for GBM in the early post‐operative interval, and none for other types of glioma. We were unable therefore to synthesize any evidence by meta‐analysis. Although we found some studies that had interesting data on imaging time points (for example, from certain studies conducted in paediatric populations), none fulfilled the review eligibility criteria and could inform the review question directly. Whilst we considered deviating from the protocol to allow for potential inclusion of these studies, we decided rather to remain true to the protocol and discuss the strengths and limitations of findings from these 'indirect' studies in the 'Discussion' section of the review. This seemed appropriate as Cochrane Reviews intrinsically evaluate quality/certainty of evidence, and we considered that any evidence potentially derived from such indirect studies would most likely be very low certainty in the context of the review question. This is particularly the case for paediatric studies, since the disease processes and issues surrounding imaging, such as risk from anaesthesia and added parental anxieties, are sufficiently different from the context in adults.
Whilst some might question the value of a relatively 'empty' review with only one included study of limited quality, we believed that identifying knowledge gaps in clinical practice is as important as finding evidence to support a clinical practice. Without identifying the knowledge gaps, meaningful discussions and research planning on how high‐quality studies can be designed to fill the gaps are precluded.
Agreements and disagreements with other studies or reviews
The primary aim of imaging in glioma management is to identify definitive disease progression, as this leads to a change in management. As with any surveillance examination, there will be many visits in which no change of treatment is instigated. To our knowledge, the first review that looked at potential benefits of serial imaging in neuro‐oncology was Christie 1975, which introduced the concept that early transient changes do not reflect true disease progression. A more recent systematic review and evidence‐based clinical practice guideline on low‐grade gliomas in adults did not attempt to answer questions around the effectiveness of imaging (Fouke 2015). One recent review highlighted that trials of imaging are under‐represented in current research prioritisation (Cihoric 2017), and another called for rigorous clinical evaluation of advanced imaging techniques that might distinguish pseudoprogression and pseudoresponse from true progression and response (Shiroishi 2013).
In the absence of evidence on imaging strategies for low‐grade gliomas, a few excluded studies deserve mention: Kim 2014 is a paediatric study that demonstrates the type of research that could potentially be undertaken in adults to generate evidence, particularly since it considers both clinical and economic aspects of imaging options. This retrospective study of 67 children with recurrent grade 1 gliomas evaluated the timing and frequency of post‐operative imaging with respect to recurrence‐free survival and cost in the health system in the USA. Thirteen asymptomatic recurrences occurred and all were detected by imaging, with a mean time to recurrence of 32.4 months (ranging from 2.9 to 128.5 months). Comparing a standard imaging schedule (comprising an MRI every 3 months for the first year, 6 months for the second year, yearly until year 5 and then 2 to 3 yearly thereafter) with a tailored, less frequent schedule (comprising imaging at 0 months, 3 months, 1 year, 2 years, and 5 years after surgery), the authors found that the less frequent schedule (5 scans instead of 10) was sufficient. It is not known whether detection of asymptomatic recurrence improved survival outcomes in this cohort, however, given the slow‐growing nature of the tumour. The authors proposed the less intensive imaging schedule as having potential psychosocial and economic benefits.
Similarly, another study conducted in the USA analysed detection efficiency of different post‐operative surveillance imaging protocols for paediatric low‐grade gliomas (Zaazoue 2019). Based on retrospective data of 517 patients who underwent 8061 scans, they reported that an 8‐image surveillance protocol (comprising imaging at 0 months, 3 months, 6 months, 1 year, 2 years, 3 years, 5 years and 6 years) detected progression at comparable rates to the 15‐image protocol currently in practice in that setting. For patients who underwent gross total resection, this could be further reduced to six scans.
By comparison, for adults with less aggressive gliomas (excluding grade 1) the post‐operative imaging surveillance schedule currently suggested by NICE comprises eight or nine scans in the first five years after surgery. Thus retrospective reviews of adult data similar to the paediatric studies could help to clarify whether this type of schedule is effective, particularly since numbers attainable from several co‐ordinated multi‐centre and international clinical trials would be larger than those available in paediatric practice.
With respect to high‐grade gliomas another excluded study, the findings of which were reported at the 2016 British Neuro‐Oncology Society conference and available in abstract form only, compared recurrence detection rates of interval scans with those of symptomatic scans among 38 GBM patients who had completed the full standard of care chemoradiotherapy regimen (Hamdan 2017). Authors reported that symptomatic scans (n = 12) detected more instances of recurrence than interval scans (n = 145) in this selected group of patients (58.3% vs 22.7%, respectively). In addition, none of these optimally treated patients had disease progression after a fifth stable routine interval scan (presumably within the study period, which was 2004 to 2012). While comparing symptomatic and surveillance imaging, these scans were being carried out in the same cohort, not between two cohorts of differently‐managed patients, and there is no indication of whether this partitioning was done based on actual clinical presence or absence of symptoms and signs, or whether this was based on imaging being on or off schedule ‐ some surveillance imaging is still performed when patients are symptomatic or clinically worsening. There is also no indication of how progressive disease was diagnosed, and whether confirmation of progression on symptomatic imaging was back‐dated to the time of initial ‐ sometimes asymptomatic ‐ suspected progression, as happens in clinical trials using response assessment in neuro‐oncology (RANO) criteria for example. The number of patients (n = 38) and number of symptomatic scans (n = 12) are also low, and there is no way to determine the differential impact of symptomatic versus surveillance imaging on the summary data included in the abstract. Surveillance imaging detected a higher absolute number of progression events than symptomatic imaging (33 versus 7, which of note does not sum to the quoted number of 38 participants). There are no results presented which address whether the asymptomatic progression was associated with different overall outcomes. However, mathematical modelling using a larger database, as suggested by these authors, would appear a reasonable approach to aid in determining the optimal timing of interval scans for low‐ and high‐grade gliomas. Such modelling could use prognostic algorithms to stratify patients into surveillance imaging pathways, and tailoring these pathways to risk factors should optimise resource use (Perreault 2014).
Several studies have evaluated the predictive value of various clinical (patient age, gender, performance status, extent of resection, radiotherapy and chemotherapy) and tumour characteristics (location, volume, histopathology), as well as imaging and molecular biomarkers (Gorlia 2013; Lorimer 2017; Pignatti 2002; Rees 2009). Assessing individual tumour growth rates through serial imaging in the first year after diagnosis could also be helpful by distinguishing those low‐grade gliomas that have a higher risk of progression from those that are in a slow growth phase (Gui 2018; Weizman 2014). A prognostic algorithm that incorporates some or all these factors might, therefore, reliably predict tumour behaviour and facilitate stratification of patients into different surveillance imaging pathways. It could also be important to consider patient preferences as a component of any potential algorithm. Whilst there is currently little known about the potential adverse psychological effects of surveillance imaging, Kim 2014 and others have pointed out that less frequent imaging might reduce its psychological burden, and hence might be preferred by patients, although the obverse may also be the case for some patients and carers who find regular imaging reassuring. Preferences could be assessed in a future survey or study.
In most developed country settings, people receiving treatment for glioma are encouraged to participate in clinical trials (NCCN 2018), which often employ imaging to detect progressive disease at more frequent intervals than suggested in clinical guidance. Thus, it may be challenging to conduct a randomised trial solely to answer the question of the optimal timing of imaging in glioma, particularly if a comparison of generic schedules may be contrary to a more individualised approach.
Finally, in addition to 'when' to image, 'how' to image needs research. We may not be realising any benefit from early/pre‐symptomatic diagnosis of progression because we lack the appropriate stratification tools and treatments. If we had improved techniques that could detect meaningful changes in tumour behaviour before any symptoms present, it might lead to improved decision‐making with regards to continuing or changing treatment and subsequently better patient outcomes. One such example is 2‐hydroxyglutarate (2HG) magnetic resonance spectroscopy, which can provide a non‐invasive biomarker of IDH mutation status and could be used to inform follow‐up approaches (Leather 2017). Biopsies of blood or cerebrospinal fluid may also be used to direct such decisions as this technology improves (Shankar 2017). In the future, these could complement (or even partly replace) surveillance imaging and should be borne in mind in the design of future studies.
Economic evidence
There is a paucity of evidence regarding the efficiency and cost effectiveness of different imaging strategies. We found no studies that examined the cost effectiveness of different imaging strategies for monitoring and follow‐up of adults with glioma but there was one excluded study that built a decision model assessing different imaging strategies for diagnosing glioma (Heinzel 2012). This study used a decision tree model to assess whether the cost effectiveness of the combined use of 18F‐fluoroethyl‐L‐tyrosine (FET) PET and MRI may be superior to MRI alone. The study concludes that the combined strategy had an 18.5% increase in the likelihood of correct diagnosis. If the time horizon of this model could be extended for the entire course of the disease, the costs and benefits of these two imaging strategies could be explored. Another excluded study examined post‐operative interval imaging in a paediatric population (Zaazoue 2019). The study concluded that fewer post‐operative surveillance imaging intervals were necessary to manage low‐grade glioma in a paediatric population compared to standard practice. As discussed above the post‐operative imaging surveillance schedule for adults suggests at least eight scans. If these results were generalisable to an adult population this could result in a more efficient use of imaging resources in the health care system. Future studies modelling the disease progression with different imaging strategies and intervals would aid decision makers in understanding the optimum way to manage adults with glioma from the perspective of both the patient and the health care system.
Authors' conclusions
Implications for practice.
Current approaches to imaging gliomas are based on plausibility and pragmatism through expert consensus rather than a body of high‐level research evidence. It remains unknown whether identification of progression through surveillance imaging instead of symptomatic imaging improves glioma survival or other health outcomes. Limited evidence suggesting that early post‐operative brain imaging among GBM patients who receive combined chemoradiation treatment could make little or no difference to survival needs robust assessment through further research, particularly as early post‐operative imaging also serves as a quality control measure that may lead to early re‐operation if residual tumour is identified.
We expect that as further evidence on prognostic indicators comes to light, it might be possible to stratify people with glioma to different imaging schedules based on assessment of tumour behaviour and other prognostic factors particular to each individual. A clear example of this would be the recognition of molecular high‐risk, histologically low‐grade glioma, which would be managed as per GBM. In the absence of good‐quality evidence on the effectiveness and optimal frequency of surveillance imaging, a multi‐disciplinary team approach and shared decision‐making is essential in the context of existing broad consensus recommendations.
Implications for research.
There are efforts across neuro‐oncology to standardise or harmonise both the type and timing of imaging although, as described above, this is not based on any high‐quality evidence. Researching management of gliomas is challenging because it is an uncommon disease covering a variety of age groups and prognostic types, for some of whom the optimal treatment strategy remains uncertain. There is also the added challenge of a shifting diagnostic and prognostic landscape with recent additions of molecular characterisation which is expected to expand in the near future. Collaborative, multicentre studies are now considered essential to sufficiently power relevant clinical questions. There is also the philosophical challenge of investigating higher versus lower frequency imaging; particularly in the oncology sphere, where in general there is an evolution in management aimed at replacing or adding to regimens rather than removing from them. There are relevant precedents, however, including alterations to surveillance imaging practice, such as regular chest radiographs and bone scintigraphy in breast cancer having demonstrated a lack of value (Lam 2017), and the paediatric studies discussed above, which recommended a significant reduction in the follow‐up imaging schedule. It should be possible to prospectively determine a research paradigm into imaging schedules in glioma in the era of shared decision‐making.
It would also be of potential value to retrospectively evaluate individual patient data from trials that have been conducted in the post‐Stupp regimen RANO era for GBM, given the natural jitter in timing of follow‐up (i.e. not every patient is imaged precisely at 3‐monthly intervals), missed or delayed imaging visits for a variety of reasons, and different imaging frequencies in trials (e.g. 2‐ versus 3‐monthly) that likely have used the Stupp regimen in their control arms. These data could be analysed to determine if there are any impacts on outcome of different imaging schedules. Challenges to this would include the exclusion of any trial participants if being imaged off schedule represents a protocol violation and avoiding selection bias of those eligible to enter trials.
Advanced imaging techniques to identify true progression (and not pseudoprogression) and true response (not pseudoresponse) also require rigorous evaluation in the relevant clinical contexts.
Other research questions for high‐grade gliomas are as follows.
Based on clinicoradiological or molecular markers (or both), can high‐grade gliomas be stratified into different interval imaging schedules to rationalise the frequency of imaging visits?
What is the impact of interval imaging frequency on time to progression during first line treatment?
How does interval imaging affect the entry to second line trials?
Does the frequency of interval imaging influence overall survival?
What are the cost effectiveness and diagnostic performance of additional imaging (e.g. MRS/perfusion/permeability MRI/PET) to determine response to treatment when progression is suspected in order to minimise repeated standard MRI during the current period of diagnostic uncertainty?
Other research questions for low‐grade gliomas are as follows.
Based on clinicoradiological or molecular markers (or both), can low‐grade gliomas be stratified into different interval imaging schedules to rationalise the frequency of imaging visits (e.g. IDH mutation status from 2HG spectroscopy)?
Does interval imaging lead to earlier detection of progression and malignant transformation?
Does interval imaging influence overall survival?
What are the cost effectiveness and diagnostic performance of additional imaging (e.g. MRS/perfusion/permeability MRI/PET) to stratify patients into different prognostic groups requiring different imaging schedules?
To improve decision‐making and promote good practice and external validity, protocols of future studies on imaging should adhere to prevailing guidance (e.g. CONSORT, QUADAS and STARD) to ensure the quality of the study, and should include economic and psychosocial outcomes whenever possible, in this era of shared decision‐making.
Acknowledgements
We thank Robin Grant (Co‐ordinating Editor) and Gail Quinn and Clare Jess (Managing Editors) from Cochrane Gynaecological, Neuro‐oncology and Orphan Cancers, for their advice and support in the preparation of this review. We would also like to thank the Information Specialist, Jo Platt, for designing the search strategy; and the library staff at the Royal United Hospital for sourcing many of the articles cited.
This project was supported by the National Institute for Health Research (NIHR), via Cochrane Programme Grant funding ‒ 16/144 to Cochrane Gynaecological, Neuro‐oncology and Orphan Cancers. The views and opinions expressed herein are those of the review authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, National Health Service, or the Department of Health.
The authors and Cochrane Gynaecological, Neuro‐oncology and Orphan Cancers Team are grateful to the following peer reviewers for their time and comments: Jill Abrigo, Helen Bulbeck and Fiona McKevitt.
Appendices
Appendix 1. MEDLINE search strategy
MEDLINE strategy
1. exp Glioma/ 2. (glioma* or astrocytoma* or ependymoma* ganglioglioma* or gliosarcoma* or oligodendroglioma* or glioblastoma* or oligoastrocytoma* or GBM*).ti,ab. 3. 1 or 2 4. neuroimaging/ 5. exp Magnetic Resonance Imaging/ 6. (MRI or MRi or magnetic resonance imag*).ti,ab. 7. (brain* adj5 (imag* or scan*)).ti,ab. 8. Tomography, X‐Ray Computed/ 9. ((CT or CAT or compute* tomograph*) adj5 (scan* or imag*)).ti,ab. 10. exp Positron‐Emission Tomography/ 11. (positron‐emission tomography* or PET).ti,ab. 12. exp Magnetic Resonance Spectroscopy/ 13. (magnetic resonance spectroscop* or MR*).ti,ab. 14. contrast‐enhanced computerised tomography or CT.ti,ab. 15. Perfusion Imaging/ 16. exp Tomography, Emission‐Computed, Single‐Photon/ 17. SPEC or SPECT.ti,ab. 18. 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 or 13 or 14 or 15 or 16 or 17 19. 3 and 18 20. ((inter* or routin* or regular* or frequen* or sequential or continuous* or serial* or recur*or longitudinal* or repeat*) adj5 (scan* or imag*)).ti,ab. 21. 19 and 20
Key
mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept word, rare disease supplementary concept word, unique identifier ab=abstract sh=subject heading ti=title pt=publication type
Draft MEDLINE strategy with economic filter
1. exp Glioma/ 2. (glioma* or astrocytoma* or ependymoma* ganglioglioma* or gliosarcoma* or oligodendroglioma* or glioblastoma* or oligoastrocytoma* or GBM*).ti,ab. 3. 1 or 2 4. neuroimaging/ 5. exp Magnetic Resonance Imaging/ 6. (MRI or MRi or magnetic resonance imag*).ti,ab. 7. (brain* adj5 (imag* or scan*)).ti,ab. 8. Tomography, X‐Ray Computed/ 9. ((CT or CAT or compute* tomograph*) adj5 (scan* or imag*)).ti,ab. 10. exp Positron‐Emission Tomography/ 11. (positron‐emission tomography* or PET).ti,ab. 12. exp Magnetic Resonance Spectroscopy/ 13. (magnetic resonance spectroscop* or MR*).ti,ab. 14. contrast‐enhanced computerised tomography.mp. or CT.ti,ab. [mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept word, rare disease supplementary concept word, unique identifier, synonyms] 15. Perfusion Imaging/ 16. exp Tomography, Emission‐Computed, Single‐Photon/ 17. SPEC.mp. or SPECT.ti,ab. [mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept word, rare disease supplementary concept word, unique identifier, synonyms] 18. 4 or 5 or 6 or 7 or 8 or 9 or 10 or 11 or 12 or 13 or 14 or 15 or 16 or 17 19. 3 and 18 20. ((inter* or routin* or regular* or frequen* or sequential or continuous* or serial* or recur*or longitudinal* or repeat*) adj5 (scan* or imag*)).ti,ab. 21. 19 and 20 22. Economics/ 23. exp "costs and cost analysis"/ 24. Economics, Dental/ 25. exp economics, hospital/ 26. Economics, Medical/ 27. Economics, Nursing/ 28. Economics, Pharmaceutical/ 29. (economic$ or cost or costs or costly or costing or price or prices or pricing or pharmacoeconomic$).ti,ab. 30. (expenditure$ not energy).ti,ab. 31. value for money.ti,ab. 32. budget$.ti,ab. 33. 22 or 23 or 24 or 25 or 26 or 27 or 28 or 29 or 30 or 31 or 32 34. ((energy or oxygen) adj cost).ti,ab. 35. (metabolic adj cost).ti,ab. 36. ((energy or oxygen) adj expenditure).ti,ab. 37. 34 or 35 or 36 38. 33 not 37 39. letter.pt. 40. editorial.pt. 41. historical article.pt. 42. 39 or 40 or 41 43. 38 not 42 44. 21 and 43
Key
mp=title, abstract, original title, name of substance word, subject heading word, keyword heading word, protocol supplementary concept word, rare disease supplementary concept word, unique identifier ab=abstract sh=subject heading ti=title pt=publication type
Appendix 2. Assessment of risk of bias
For randomised controlled trials
We will assess the risk of bias according to the following criteria.
(1) Random sequence generation
Low risk of bias e.g. participants assigned to treatments on basis of a computer‐generated random sequence or a table of random numbers
High risk of bias e.g. participants assigned to treatments on basis of date of birth, clinic ID‐number or surname, or no attempt to randomise participants
Unclear risk of bias e.g. not reported, information not available
(2) Allocation concealment
Low risk of bias e.g. where the allocation sequence could not be foretold
High risk of bias e.g. allocation sequence could be foretold by participants, investigators or treatment providers
Unclear risk of bias e.g. not reported
(3) Blinding of participants and personnel
Low risk of bias if participants and personnel were adequately blinded
High risk of bias if participants and/or personnel were not blinded to the intervention that the participant received
Unclear risk of bias if this was not reported or unclear
(4) Blinding of outcome assessors
Low risk of bias if outcome assessors were adequately blinded to the intervention that the participant received
High risk of bias if outcome assessors were not blinded to the intervention that the participant received
Unclear risk of bias if this was not reported or unclear
(5) Incomplete outcome data
We will record the proportion of participants whose outcomes were not reported at the end of the study. We will code a satisfactory level of loss to follow‐up for each outcome as:
low risk of bias, if fewer than 20% of participants were lost to follow‐up and reasons for loss to follow‐up were similar in both treatment arms
High risk of bias, if more than 20% of participants were lost to follow‐up or reasons for loss to follow‐up differed between treatment arms
Unclear risk of bias if loss to follow‐up was not reported
(6) Selective reporting of outcomes
Low risk of bias e.g. review reports all outcomes specified in the protocol
High risk of bias e.g. It is suspected that outcomes have been selectively reported
Unclear risk of bias e.g. It is unclear whether outcomes had been selectively reported
(7) Other bias
Low risk of bias, i.e.no other source of bias suspected and the study appears to be methodologically sound
High risk of bias: we suspect that the study was prone to an additional bias
Unclear risk of bias: we are uncertain whether an additional bias may have been present
For non‐randomised controlled trials and controlled before‐after studies
We will assess the risk of bias in accordance with four criteria concerning sample selection comparability of treatment groups:
(1) Relevant details of criteria for assignment of patients to treatments
Low risk of bias, e.g. yes, details provided
High risk of bias, e.g. no details provided
Unclear risk of bias, e.g. details unclear
(2) Representative group of people who received the experimental intervention
Low risk of bias, if representative of participants with low and/or high grade gliomas who receive interval brain imaging to assess their condition
High risk of bias, if groups of participants were selected (non‐consecutive)
Unclear, if selection of the group was not described
(3) Representative group of people who received the comparison intervention
Low risk of bias, if drawn from the same population as the experimental group
High risk of bias, if drawn from a different source
Unclear risk of bias, if selection of group not described
(4) Baseline differences between groups controlled for, in particular with reference to age, gender, grade of glioma and glioma treatment
Low risk of bias, if at least three of these characteristics were reported
High risk of bias, if the groups differed in these baseline characteristics and differences were not controlled for
Unclear risk of bias, if fewer than three of these characteristics were reported even if there were no other differences between the groups, and other characteristics were controlled for
Data and analyses
Comparison 1. Early post‐surgery imaging vs no early imaging.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Overall survival (deaths at 1 year) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 HGG | 1 | 125 | Risk Ratio (M‐H, Random, 95% CI) | 0.86 [0.61, 1.21] |
| 1.2 LGG | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2 Overall survival (deaths at 2 years) | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 HGG | 1 | 125 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.91, 1.25] |
| 2.2 LGG | 0 | 0 | Risk Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Mrowczynski 2018.
| Methods | Design: retrospective series from a single institution Country: USA Accrual dates: 2006 to 2016 Trial reg: N/A Funding: N/R |
|
| Participants | No. analysed: 125 Inclusion/exclusion criteria: all patients with GBM at the Pennsylvania State University Department of Neurosurgery between 2006 and 2016 were included unless they had had a previous low‐grade glioma, and if date of death could not be accurately determined Age: mean 59.98 (range 5 to 82) Gender: 53% male, 47% female Glioma type: GBM Glioma grade: 4 Treatment: most patients were treated with maximal safe resection and received radiotherapy (60 Gy) combined with temozolomide chemotherapy, with approximately 80% in both imaging groups completing these treatments. Just over half the patients (56%) also received adjuvant temozolomide or other treatment (bevacizumab, Optune) |
|
| Interventions | Arm 1: post‐operative imaging within 48 hours of resection based on surgeon preference and discretion (n = 69) Arm 2: no post‐operative imaging (n = 56) |
|
| Outcomes | Overall survival at 1 year and 2 years; median survival | |
| Notes | It is not clear in the report when patients in Arm 2 had their first post‐operative MRI, i.e. for radiation planning. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Selection bias | High risk | "The decision of post‐operative imaging was largely based on surgeon preference and discretion." |
| Intervention participants representative | Low risk | These were patients with GBM, most of whom received chemoradiation after maximal resection. |
| Comparison participants representative | Low risk | These were patients with GBM, most of whom received chemoradiation after maximal resection. |
| Baseline differences absent or controlled for | Unclear risk | Baseline patient characteristics are comparable and any differences that might favour better outcomes in the intervention group (e.g. lower mean age and slightly less treatment completion) did not do so. However, there was no information on molecular characteristics. |
| Other bias | Unclear risk | Because the number of patients eligible and included in the study from a 10‐year period at the neuro‐oncology centre seemed low, the reviewer team had concerns about possible attrition bias arising from patients who did not have their date of death recorded in the site's system. |
N/A: not applicable
N/R: not reported
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Abrigo 2018 | Wrong type of study ‒ a diagnostic test accuracy review of magnetic resonance perfusion. |
| Albert 1994 | Wrong study question ‒ a non‐comparative observational study on early post‐operative imaging. |
| Ali 2014 | Wrong study population ‒ a study conducted in children with suspected LGG. |
| Aoyama 1982 | Unable to obtain full text. |
| Chang 2014 | Wrong study question ‒ this observational study aimed to evaluate the survival difference between early‐ versus late‐diagnosed progressive disease. |
| Chataway 1999 | Wrong study question ‒ a retrospective analysis of an MRC trial that compared the predictive value of two different clinically‐assessed scores (not imaging techniques/intervals) for determining progression. |
| Christie 1975 | Wrong type of study ‒ an older review of brain imaging. |
| Cihoric 2017 | Wrong type of study ‒ a review of registered trials in GBM management (see Agreements and disagreements with other studies or reviews). |
| Cochereau 2016 | Wrong type of study ‒ a case report of one case reflecting that progression can occur without clinical manifestation. |
| Danchaivijitr 2008 | Wrong study question ‒ a prospective cohort study looking at correlation between cerebral blood volume changes and malignant transformation. |
| de Graaf 2002 | Wrong study population ‒ a non‐comparative study in children on the predictive role of clinical assessment to inform symptomatic imaging. |
| Easaw 2011 | Wrong type of study ‒ this is a guideline consensus paper that concluded that the optimal timing of MRI after chemoradiation has not been established. |
| Farace 2013 | Wrong study question ‒ this is a non‐comparative observational study looking at whether another imaging time point is needed before starting adjuvant treatment in GBM. |
| Field 2017 | Wrong study question ‒ a substudy of an RCT looking at the prognostic association of early progression on MRI among recurrent GBM patients receiving bevacizumab vs bevacizumab plus carboplatin. |
| Fouke 2015 | Wrong type of study ‒ a review of the role of imaging in the management of glioma. |
| Fujimura 2004 | Wrong study question ‒ a retrospective study of pre‐operative MRI imaging vs no pre‐operative MRI imaging in the elderly. Control group is historical and only a single pre‐operative time point is measured (i.e. it is not about interval imaging or symptomatic imaging). |
| Galban 2011 | Wrong study question ‒ a study that looked at a new MRI technique to predict outcomes and facilitate earlier second‐line treatment decisions. |
| Galldiks 2010 | Wrong type of study ‒ a study of two GBM patients who underwent complex PET imaging. |
| Geer 2012 | Wrong type of study ‒ qualitative study about whether perfusion MRI increased confidence in diagnosis and management plan. |
| Gittleman 2016 | Wrong study question ‒ a study to design a nomogram to predict survival in GBM. |
| Gorlia 2013 | Wrong study question ‒ a study on prognostic indicators for survival in grade 3 gliomas. |
| Gui 2018 | Wrong type of study ‒ a retrospective case series on the growth pattern/dynamics of slow‐growing tumours. |
| Hamdan 2017 | Wrong type of study ‒ a retrospective study reporting the proportion of symptomatic (12 scans) and interval MRIs (145 scans) that detected disease progression in a group of 38 fully treated glioblastoma patients. |
| Heinzel 2012 | Wrong intervention ‒ a cost‐effectiveness study of a decision tree model of imaging for diagnosis (not monitoring and follow‐up) of glioma |
| Kim 2014 | Wrong study population ‒ a retrospective cohort study on the frequency and timing of post‐operative imaging. |
| Klingelhofer 2015 | Wrong study question ‒ prognostic study of MRI versus histology to predict survival. |
| Korones 2001 | Wrong study population ‒ retrospective study among children with low‐ or high‐grade gliomas to quantify the proportion of asymptomatic progression. |
| Leonard 1975 | Wrong type of study ‒ one of the earliest papers to show asymptomatic biological progression of brain tumours. |
| Li 2011 | Wrong study question ‒ prognostic study that examines serial MRI for stratifying patients. There are potential imaging implications from the findings. |
| Lorimer 2017 | Wrong type of study ‒ retrospective case series of elderly people with GBM. |
| Macdonald 1990 | Wrong type of study ‒ a report on formalised clinical radiological response criteria to identify tumour response to treatment. |
| Mahaley 1983 | Wrong type of study ‒ a substudy of a clinical trial of levisamole in which all participants received serial CT scans. Authors concluded that, for first 6 months, clinical findings were most useful for response assessment, and that (CT) imaging could be started from 6 months. Cost of CT influenced conclusions. |
| Merkel 2017 | Wrong type of study ‒ a prognostic study that found that 60% of GBM patients are early progressors and the timing of imaging pre‐chemoradiotherapy should needs formal research. |
| Meyers 2003 | Wrong type of study ‒ a retrospective study of 56 patients with recurrent brain tumours that were recruited to clinical trials and who had scans at regular intervals — usually monthly — and cognitive function testing. Cognitive deterioration preceded radiographic evidence of tumour progression by 6 weeks on average, suggesting that clinical surveillance using cognitive domain scoring might be of value in addition to other follow‐up measures to identify progression. |
| Milano 2010 | Wrong study question ‒ a study of the patterns of recurrence of GBM, and not about imaging. |
| Mueller 1981 | Unable to obtain this paper. |
| Neagu 2015 | Wrong type of study ‒ an opinion paper on management of GBM, with discussion around advanced imaging at follow‐up but nothing on the frequency of follow‐up (see Agreements and disagreements with other studies or reviews). |
| Oberheim Bush 2016 | Wrong type of study ‒ a review of treatment strategies for LGG. |
| Patil 2017 | Wrong study question ‒ an RCT of video follow‐up versus clinical follow‐up of people with glioma. |
| Perreault 2014 | Wrong study population ‒ a study of surveillance imaging in children with CNS tumours, which discusses the role of tailoring spinal MRI according to the risk of spinal relapse to improve the allocation of resources. |
| Poussaint 2011 | Wrong study population ‒ a retrospective study of data from 2 clinical trials conducted among children with brainstem glioma that employed frequent MRI surveillance (post‐op and every 8 weeks in year 1, 3‐monthly thereafter, and at the end of treatment or disease progression); however, no evidence for the number of images obtained and their timeframe was given. |
| Quinn 1971 | Wrong type of study ‒ an older case series of 14 patients that introduces the concept of serial imaging in GBM management. |
| Reardon 2014 | Wrong type of study ‒ this is a consensus report that does not address frequency of imaging; rather, it is more concerned with increasing the accuracy of imaging to identify progression. Authors suggest introducing more complex imaging (at the very least T1w subtraction maps) but not reducing imaging frequency. |
| Sabattini 1980 | Wrong type of study ‒ this is an older opinion paper that proposes a protocol for serial CT scans (before and after surgery, before chemo‐radiotherapy and after each treatment cycle) based on the authors' experience. |
| Sanghera 2012 | Wrong type of study ‒ a review on distinguishing pseudoprogression from true progression in patients with glioblastoma |
| Saunders 2005 | Wrong study population ‒ a retrospective study on surveillance imaging strategies conducted among children with cerebellar astrocytoma, which discusses the frequency and timing of surveillance imaging in this context. |
| Scoccianti 2010 | Wrong type of study ‒ a retrospective pattern of care study about changes in GBM care over time in Italy. |
| Seiz 2011 | Wrong type of study ‒ a survey of diagnosis and treatment practices related to LGG in Germany, which revealed an approximate 50:50 split over 'wait and see' and early intervention approaches. |
| Sethi 2011 | Wrong study population ‒ a retrospective non‐comparative study of 16 paediatric patients with diffuse intrinsic pontine glioma who underwent serial 4‐monthly neuraxial MRIs. Authors concluded that it led to a high detection rate of leptomeningeal dissemination (and earlier intervention) and suggested that such serial scanning should therefore be considered in future clinical trials. |
| Shah 2011 | Wrong type of study ‒ a systematic review of the management of incidental suspected LGG. Authors conclude that "the asymptomatic patient may be monitored safely with serial MR imaging and occasionally PET scanning before treatment is initiated." The 'optimal treatment paradigm' proposed includes serial MRI every 4 months. |
| Shiroishi 2013 | Wrong type of study ‒ a review on post‐treatment assessment of central nervous system tumours that highlights issues such as pseudoprogression and pseudoresponse and the need for rigorous clinical evaluation of promising advanced imaging techniques such as perfusion MR, diffusion MR and permeability MR imaging, MR spectroscopy and PET in this context. |
| Singh 2012 | Wrong type of study ‒ a case report of a glioma patient with pseudoprogression and clinical deterioration on treatment. |
| Spitaels 2017 | Wrong type of study ‒ a case series describing the management of 35 people with recurrent diffuse LGG. |
| Sreenivasan 2016 | Wrong study question ‒ a retrospective archive‐based study that evaluated glioma volume measurement techniques. |
| Sutton 1996 | Wrong study population ‒ a retrospective study of the records of 93 children with cerebellar astrocytomas from 1975 to 1993, which suggested that routine surveillance imaging might not be necessary in completely resected tumours because recurrence is small; however, it is probably of value in incompletely resected tumours. |
| Udaka 2012 | Wrong study population ‒ a restrospective case series of 102 children with LGG, which suggests that routine surveillance imaging up to 5 years is beneficial, even for completely resected tumours. Authors suggest the possibility of shortening the imaging protocol, which may represent some form of rationalisation, but not reducing the number of visits (i.e. just reducing the time taken for each). The median number of scans per year in this study was 3.4. |
| Wang 2009 | Wrong type of study ‒ a mathematical modelling paper based on 32 patients with GBM aimed at stratifying GBM by behaviour. |
| Weizman 2014 | Wrong type of study ‒ this study of 10 patients with LGGs evaluates different techniques for tracking the progress of slow‐growing tumours. |
| Wick 2016 | Wrong study question ‒ substudy of an RCT of bevacizumab added to chemoradiation looking at pseudoprogression rates and tumour progression patterns. |
| Zaazoue 2019 | Wrong study population ‒ paediatric population |
Contributions of authors
Gerard Thompson and Theresa Lawrie wrote the first draft of the review. Ashleigh Kernohan wrote the economic content. All authors advised on and approved the final version.
Sources of support
Internal sources
No sources of support supplied
External sources
NIHR 16/144 Cochrane Programme Grant Scheme, UK, Other.
Declarations of interest
Gerard Thompson: none declared Theresa Lawrie: none declared Ashleigh Kernohan: none declared Michael Jenkinson: none declared
Authors 1 & 2 contributed equally to this work
Authors 1 & 2 contributed equally to this work
New
References
References to studies included in this review
Mrowczynski 2018 {published data only}
- Mrowczynski OD, Zammar S, Bourcier AJ, Langan ST, Liao J, Specht CS, et al. Utility of early post‐operative magnetic resonance imaging after glioblastoma resection: implications on patient survival. World Neurosurgery 2018;120:e1171‐4. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Abrigo 2018 {published data only}
- Abrigo JM, Fountain DM, Provenzale JM, Law EK, Kwong JS, Hart MG, et al. Magnetic resonance perfusion for differentiating low‐grade from high‐grade gliomas at first presentation. Cochrane Database of Systematic Reviews 2018, Issue 1. [DOI: 10.1002/14651858.CD011551.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Albert 1994 {published data only}
- Albert FK, Forsting M, Sartor K, Adams HP, Kunze S. Early postoperative magnetic resonance imaging after resection of malignant glioma: objective evaluation of residual tumor and its influence on regrowth and prognosis. Neurosurgery 1994;34(1):45‐60. [DOI] [PubMed] [Google Scholar]
Ali 2014 {published data only}
- Ali ZS, Lang SS, Sutton LN. Conservative management of presumed low‐grade gliomas in the asymptomatic pediatric population. World Neurosurgery 2014;81(2):368‐73. [DOI] [PubMed] [Google Scholar]
Aoyama 1982 {published data only}
- Aoyama I. Usefulness of serial CT scans for evaluation of histology and prognosis in gliomas. Nippon geka hokan ‐ Archiv für japanische Chirurgie 1982;51(4):566‐82. [PubMed] [Google Scholar]
Chang 2014 {published data only}
- Chang JH, Kim CY, Choi BS, Kim YJ, Kim JS, Kim IA. Pseudoprogression and pseudoresponse in the management of high‐grade glioma: optimal decision timing according to the response assessment of the Neuro‐Oncology Working Group. Journal of Korean Neurosurgical Society 2014;55(1):5‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chataway 1999 {published data only}
- Chataway J, Stenning S, Bleehen N, Grant R. Use of different outcome measures in randomised studies of malignant glioma can significantly alter the interpretation of time to progression: reanalysis of the MRC BR2 study. Journal of Neuro‐oncology 1999;43(1):87‐92. [DOI] [PubMed] [Google Scholar]
Christie 1975 {published data only}
- Christie JH, Go RT, Schapiro RL. The diagnostic value of serial brain scanning. CRC Critical Reviews in Clinical Radiology and Nuclear Medicine 1975;7(2):161‐85. [PubMed] [Google Scholar]
Cihoric 2017 {published data only}
- Cihoric N, Tsikkinis A, Minniti G, Lagerwaard FJ, Herrlinger U, Mathier E, et al. Current status and perspectives of interventional clinical trials for glioblastoma – analysis of ClinicalTrials.gov. 2017 Radiation Oncology;12(1):1. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cochereau 2016 {published data only}
- Cochereau J, Herbet G, Rigau V, Duffau H. Acute progression of untreated incidental WHO Grade II glioma to glioblastoma in an asymptomatic patient. Journal of Neurosurgery 2016;124(1):141‐5. [DOI] [PubMed] [Google Scholar]
Danchaivijitr 2008 {published data only}
- Danchaivijitr N, Waldman AD, Tozer DJ, Benton CE, Brasil Caseiras G, Tofts PS, et al. Low‐Grade Gliomas: Do changes in rCBV measurements at longitudinal perfusion‐weighted MR imaging predict malignant transformation?. Radiology 2008;247(1):170‐8. [DOI] [PubMed] [Google Scholar]
de Graaf 2002 {published data only}
- Graaf N, Hew JM, Fock JM, Kamps WA, Graaf SS. Predictive value of clinical evaluation in the follow‐up of children with a brain tumor. Medical and Pediatric Oncology 2002;38(4):254‐7. [DOI] [PubMed] [Google Scholar]
Easaw 2011 {published data only}
- Easaw JC, Mason WP, Perry J, Laperriere N, Eisenstat DD, Maestro R, et al. Canadian recommendations for the treatment of recurrent or progressive glioblastoma multiforme. Current Oncology 2011;18(3):e126‐36. [DOI] [PMC free article] [PubMed] [Google Scholar]
Farace 2013 {published data only}
- Farace P, Amelio D, Ricciardi GK, Zoccatelli G, Magon S, Pizzini F, et al. Early MRI changes in glioblastoma in the period between surgery and adjuvant therapy. Journal of Neuro‐oncology 2013;111(2):177‐85. [DOI] [PubMed] [Google Scholar]
Field 2017 {published data only}
- Field KM, Phal PM, Fitt G, Goh C, Nowak AK, Rosenthal MA, et al. The role of early magnetic resonance imaging in predicting survival on Bevacizumab for recurrent glioblastoma: results from a prospective clinical trial (CABARET). Cancer 2017;123(18):3576‐82. [DOI] [PubMed] [Google Scholar]
Fouke 2015 {published data only}
- Fouke SJ, Benzinger T, Gibson D, Ryken TC, Kalkanis SN, Olson JJ. The role of imaging in the management of adults with diffuse low grade glioma: a systematic review and evidence‐based clinical practice guideline. Journal of Neuro‐oncology 2015;125(3):457‐79. [DOI] [PubMed] [Google Scholar]
Fujimura 2004 {published data only}
- Fujimura M, Kumabe T, Tominaga T, Jokura H, Shirane R, Yoshimoto T. Routine clinical adoption of magnetic resonance imaging was associated with better outcome after surgery in elderly patients with a malignant astrocytic tumour: a retrospective review. Acta Neurochirurgica 2004;146(3):251‐5. [DOI] [PubMed] [Google Scholar]
Galban 2011 {published data only}
- Galban CJ, Chenevert TL, Meyer CR, Tsien C, Lawrence TS, Hamstra DA, et al. Prospective analysis of parametric response map‐derived MRI biomarkers: identification of early and aistinct glioma response patterns not predicted by standard radiographic assessment. Clinical Cancer Research 2011;17(14):4751‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Galldiks 2010 {published data only}
- Galldiks N, Kracht LW, Burghaus L, Ullrich RT, Backes H, Brunn A, et al. Patient‐tailored, imaging‐guided, long‐term temozolomide chemotherapy in patients with glioblastoma. Molecular Imaging 2010;9(1):40‐6. [PubMed] [Google Scholar]
Geer 2012 {published data only}
- Geer CP, Simonds J, Anvery A, Chen MY, Burdette JH, Zapadka ME, et al. Does MR perfusion imaging impact management decisions for patients with brain tumors? A prospective study. American Journal of Neuroradiology 2012;33(3):556‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gittleman 2016 {published data only}
- Gittleman HR, Lim D, Kattan M, Chakravarti A, Gilbert MR, Lassman AB, et al. An independently validated nomogram for individualized estimation of survival among patients with newly diagnosed glioblastoma: NRG oncology/RTOG 0525 and 0825. International Journal of Radiation Oncology 2016; Vol. 96, issue 2 Supplement 1:S92. [DOI] [PMC free article] [PubMed]
Gorlia 2013 {published data only}
- Gorlia T, Delattre JY, Brandes AA, Kros JM, Taphoorn MJ, Kouwenhoven MC, et al. New clinical, pathological and molecular prognostic models and calculators in patients with locally diagnosed anaplastic oligodendroglioma or oligoastrocytoma. European Journal of Cancer 2013;49(16):3477‐85. [DOI] [PubMed] [Google Scholar]
Gui 2018 {published data only}
- Gui C, Kosteniuk SE, Lau JC, Megyesi JF. Tumor growth dynamics in serially‐imaged low‐grade glioma patients. Journal of Neuro‐oncology 2018;139:167‐75. [DOI] [PubMed] [Google Scholar]
Hamdan 2017 {published data only}
- Hamdan A, Kane P. Is routine interval surveillance imaging after completion of primary treatment effective at detecting recurrence in GBM patients?. Neuro‐oncology. BNOS Conference proceedings 2016. Jan 2017; Vol. Abstract pp73:i20.
Heinzel 2012 {published data only}
- Heinzel A, Stock S, Langen KJ, Müller D. Cost‐effectiveness analysis of FET PET guided target selection for the diagnosis of gliomas. European Journal of Nuclear Medicine and Molecular Imaging 2012;39(7):1089‐96. [DOI] [PubMed] [Google Scholar]
Kim 2014 {published data only}
- Kim AH, Thompson EA, Governale LS, Santa C, Cahill K, Kieran MW, et al. Recurrence after gross total resection of low‐grade pediatric brain tumors: The frequency and timing of postoperative imaging. Journal of Neurosurgery. Pediatrics 2014;14(4):356‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Klingelhofer 2015 {published data only}
- Klingelhofer L, Mucha D, Geiger K, Koch R, Kummer R. Prognostic value of conventional magnetic resonance imaging for adult patients with brain tumors. Clinical Neuroradiology 2015;25(3):281‐9. [DOI] [PubMed] [Google Scholar]
Korones 2001 {published data only}
- Korones DN, Butterfield R, Meyers SP, Constine LS. The role of surveillance magnetic resonance imaging (MRI) scanning in detecting recurrent brain tumors in asymptomatic children. Journal of Neuro‐oncology 2001;53(1):33‐8. [DOI] [PubMed] [Google Scholar]
Leonard 1975 {published data only}
- Leonard JR, Witherspoon LR, Mahaley MS Jr, Goodrich JK. Value of sequential postoperative brain scans in patients with anaplastic gliomas. Journal of Neurosurgery 1975;42(5):551‐6. [DOI] [PubMed] [Google Scholar]
Li 2011 {published data only}
- Li Y, Lupo JM, Polley MY, Crane JC, Bian W, Cha S, et al. Serial analysis of imaging parameters in patients with newly diagnosed glioblastoma multiforme. Neuro‐oncology 2011;13(5):546‐57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lorimer 2017 {published data only}
- Lorimer CF, Hanna C, Saran F, Chalmers A, Brock J. Challenges to treating older glioblastoma patients: the influence of clinical and tumour characteristics on survival outcomes. Clinical Oncology 2017;29(11):739‐47. [DOI] [PubMed] [Google Scholar]
Macdonald 1990 {published data only}
- Macdonald DR, Cascino TL, Schold SC Jr, Cairncross JG. Response criteria for phase II studies of supratentorial malignant glioma. Journal of Clinical Oncology 1990;8(7):1277‐80. [DOI] [PubMed] [Google Scholar]
Mahaley 1983 {published data only}
- Mahaley MS Jr, Mitchell WG, Whaley R, Dudka LF, Symons MJ. The relative roles of neurological examination, functional abilities, and computed tomography in the definition of treatment failure in patients with anaplastic gliomas. Surgical Neurology 1983;20(4):297‐300. [DOI] [PubMed] [Google Scholar]
Merkel 2017 {published data only}
- Merkel A, Soeldner D, Wendl C, Urkan D, Kuramatsu JB, Seliger C, et al. Early postoperative tumor progression predicts clinical outcome in glioblastoma ‐ implication for clinical trials. Journal of Neuro‐oncology 2017;132(2):249‐54. [DOI] [PMC free article] [PubMed] [Google Scholar]
Meyers 2003 {published data only}
- Meyers CA, Hess KR. Multifaceted end points in brain tumor clinical trials: cognitive deterioration precedes MRI progression. Neuro‐oncology 2003;5(2):89‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Milano 2010 {published data only}
- Milano MT, Okunieff P, Donatello RS, Mohile NA, Sul J, Walter KA, et al. Patterns and timing of recurrence after Temozolomide‐based chemoradiation for glioblastoma. International Journal of Radiation Oncology, Biology, Physics 2010;78(4):1147‐55. [DOI] [PubMed] [Google Scholar]
Mueller 1981 {published data only}
- Mueller H, Kaernbach A. Serial CT scans in the treatment of brain gliomas with CCNU and radiation ‐ with or without operation. Neurochirurgia 1981;24(Suppl):416. [Google Scholar]
Neagu 2015 {published data only}
- Neagu MR, Huang RY, Reardon DA, Wen PY. How treatment monitoring is influencing treatment decisions in glioblastomas. Current Treatment Options in Neurology 2015;17:1‐15. [DOI] [PubMed] [Google Scholar]
Oberheim Bush 2016 {published data only}
- Oberheim Bush NA, Chang S. Treatment strategies for low‐grade glioma in adults. Journal of Oncology Practice 2016;12(12):1235‐41. [DOI] [PubMed] [Google Scholar]
Patil 2017 {published data only}
- Patil VM, Pande N, Chandrasekharan A, Tonse R, Chandrakanth MV, Krishnatry R, et al. SHADOW Study: Comparison of conventional clinical follow‐up with clinician led video follow‐up in newly diagnosed patients with intermediate and high grade glioma receiving adjuvant Temozolomide therapy. Journal of Clinical Oncology 2017;35(15 suppl):2024. [Google Scholar]
Perreault 2014 {published data only}
- Perreault S, Lober RM, Carret AS, Zhang G, Hershon L, Decarie JC, et al. Surveillance imaging in children with malignant CNS tumors: low yield of spine MRI. Journal of Neuro‐oncology 2014;116:617‐23. [DOI] [PubMed] [Google Scholar]
Poussaint 2011 {published data only}
- Poussaint TY, Kocak M, Vajapeyam S, Packer RI, Robertson RL, Geyer R, et al. MRI as a central component of clinical trials analysis in brainstem glioma: a report from the Pediatric Brain Tumor Consortium (PBTC). Neuro‐oncology 2011;13(4):417‐27. [DOI] [PMC free article] [PubMed] [Google Scholar]
Quinn 1971 {published data only}
- Quinn JL, 3rd. Serial brain scans in glioblastoma multiforme. Radiology 1971;101(2):367‐70. [DOI] [PubMed] [Google Scholar]
Reardon 2014 {published data only}
- Reardon DA, Ballman KV, Buckner JC, Chang SM, Ellingson BM. Impact of imaging measurements on response assessment in glioblastoma clinical trials. Neuro‐oncology 2014;16 Suppl 7:vii24‐35. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sabattini 1980 {published data only}
- Sabattini L, Piazza GC, Finizio FS. Programmed serial CT scans in the surgical and radio‐chemotherapeutic management of cerebral gliomas. Journal of Neurosurgical Sciences 1980;24(1):33‐6. [PubMed] [Google Scholar]
Sanghera 2012 {published data only}
- Sanghera P, Rampling R, Haylock B, Jefferies S, McBain C, Rees JH, et al. The concepts, diagnosis and management of early imaging changes after therapy for glioblastomas. Clinical Oncology 2012;24(3):216–27. [DOI] [PubMed] [Google Scholar]
Saunders 2005 {published data only}
- Saunders DE, Phipps KP, Wade AM, Hayward RD. Surveillance imaging strategies following surgery and/or radiotherapy for childhood cerebellar low‐grade astrocytoma. Journal of Neurosurgery 2005;102 Pediatrics(SUPPL 2):172‐8. [DOI] [PubMed] [Google Scholar]
Scoccianti 2010 {published data only}
- Scoccianti S, Magrini SM, Ricardi U, Detti B, Buglione M, Sotti G, et al. Patterns of care and survival in a retrospective analysis of 1059 patients with glioblastoma multiforme treated between 2002 and 2007: A multicenter study by the central nervous system study group of Airo (Italian Association of Radiation Oncology). Neurosurgery 2010;67(2):446‐58. [DOI] [PubMed] [Google Scholar]
Seiz 2011 {published data only}
- Seiz M, Freyschlag CF, Schenkel S, Weiss C, Thome C, Schmieder K, et al. Management of patients with low‐grade gliomas ‐ A survey among german neurosurgical departments. Central European Neurosurgery 2011;72(4):186‐91. [DOI] [PubMed] [Google Scholar]
Sethi 2011 {published data only}
- Sethi R, Allen J, Donahue B, Karajannis M, Gardner S, Wisoff J, et al. Prospective neuraxis MRI surveillance reveals a high risk of leptomeningeal dissemination in diffuse intrinsic pontine glioma. Journal of Neuro‐oncology 2011;102(1):121‐7. [DOI] [PubMed] [Google Scholar]
Shah 2011 {published data only}
- Shah AH, Madhavan K, Heros D, Raper DM, Iorgulescu JB, Lally BE, et al. The management of incidental low‐grade gliomas using magnetic resonance imaging: systematic review and optimal treatment paradigm. Neurosurgical Focus 2011;31(6):E12. [DOI] [PubMed] [Google Scholar]
Shiroishi 2013 {published data only}
- Shiroishi MS, Booker MT, Agarwal M, Jain N, Naghi I, Lerner A, et al. Post treatment evaluation of central nervous system gliomas. Magnetic Resonance Imaging Clinics of North America 2013;21(2):241‐68. [DOI] [PubMed] [Google Scholar]
Singh 2012 {published data only}
- Singh AD, Easaw JC. Does neurologic deterioration help to differentiate between pseudoprogression and true disease progression in newly diagnosed glioblastoma multiforme?. Current Oncology 2012;19(4):e295‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Spitaels 2017 {published data only}
- Spitaels J, Devriendt D, Sadeghi N, Luce S, Witte O, Goldman S, et al. Management of supratentorial recurrent low‐grade glioma: A multidisciplinary experience in 35 adult patients. Oncology Letters 2017;14(3):2789‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sreenivasan 2016 {published data only}
- Sreenivasan S, Madhugiri V, Sasidharan G, Kumar RVR. Measuring glioma volumes: A comparison of linear measurement based formulae with the manual image segmentation technique. Journal of Cancer Research and Therapeutics 2016;12(1):161‐8. [DOI] [PubMed] [Google Scholar]
Sutton 1996 {published data only}
- Sutton LN, Cnaan A, Klatt L, Zhao H, Zimmerman R, Needle M, et al. Postoperative surveillance imaging in children with cerebellar astrocytomas. Journal of Neurosurgery 1996;84(5):721‐25. [DOI] [PubMed] [Google Scholar]
Udaka 2012 {published data only}
- Udaka YT, Yeh‐Nayre LA, Amene CS, VandenBerg SR, Levy ML, Crawford JR. Recurrent pediatric central nervous system low‐grade gliomas: The role of surveillance neuroimaging in asymptomatic children. Journal of Neurosurgery. Pediatrics 2012;11(2):119‐26. [DOI] [PubMed] [Google Scholar]
Wang 2009 {published data only}
- Wang CH, Rockhill JK, Mrugala M, Peacock DL, Lai A, Jusenius K, et al. Prognostic significance of growth kinetics in newly diagnosed glioblastomas revealed by combining serial imaging with a novel biomathematical model. American Association for Cancer Research 2009;69(23):9133‐40. [DOI] [PMC free article] [PubMed] [Google Scholar]
Weizman 2014 {published data only}
- Weizman L, Sira LB, Joskowicz L, Rubin DL, Yeom KW, Constantini S, et al. Semiautomatic segmentation and follow‐up of multicomponent low‐grade tumors in longitudinal brain MRI studies. Medical Physics 2014;41(5):052303‐14. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wick 2016 {published data only}
- Wick W, Gorlia T, Bady P, Platten M, Bent MJ, Taphoorn MJB, et al. Phase II study of radiotherapy and temsirolimus versus radiochemotherapy with temozolomide in patients with newly diagnosed glioblastoma without MGMT promoter hypermethylation (EORTC 26082). Clinical Cancer Research 2016;22(19):4797‐806. [DOI] [PubMed] [Google Scholar]
Zaazoue 2019 {published data only}
Additional references
Covidence [Computer program]
- Veritas Health Innovation. Covidence. Melbourne, Australia: Veritas Health Innovation.
Deeks 2008
- Deeks JJ, Altman DG, Bradburn MJ. Statistical methods for examining heterogeneity and combining results from several studies in meta‐analysis. Egger M, Davey Smith G, Altman DG (eds). Systematic reviews in health care: meta‐analysis in context. onlinelibrary.wiley.com/doi/abs/10.1002/9780470693926.ch15 2008. [DOI: 10.1002/9780470693926.ch15] [DOI]
Deeks 2017
- Deeks JJ, Higgins JP, Altman DG (editors) on behalf of the Cochrane Statistical Methods Group. Chapter 9: Analysing data and undertaking meta‐analyses. In: Higgins JPT, Churchill R, Chandler J, Cumpston MS (editors), Cochrane Handbook for Systematic Reviews of Interventions version 5.2.0 (updated June 2017), Cochrane, 2017. Available from www.training.cochrane.org/handbook.
EPOC 2015
- Effective Practice, Organisation of Care (EPOC). EPOC Resources for review authors. epoc.cochrane.org/epoc‐specific‐resources‐review‐authors. Oslo: Norwegian Knowledge Centre for the Health Services;, (accessed 6 July 2016).
GLOBOCAN 2012
- International Agency for Research on Cancer. GLOBOCAN 2012: Estimated cancer incidence, mortality and prevalence worldwide in 2012. globocan.iarc.fr/Pages/fact_sheets_cancer.aspx (accessed 14 July 2016).
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2017
- Higgins JP, Altman DG, Sterne JA (editors). Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Churchill R, Chandler J, Cumpston MS (editors), Cochrane Handbook for Systematic Reviews of Interventions version 5.2.0 (updated June 2017), Cochrane, 2017. Available from www.training.cochrane.org/handbook.
Husereau 2013
- Husereau D, Drummond M, Petrou S, Carswell C, Moher D, Greenberg D, et al. Consolidated health economic evaluation reporting standards (CHEERS) Explanation and elaboration: a report of the ISPOR health economic evaluations publication guidelines good reporting practices task force. Value in Health 2013;16:231‐50. [DOI] [PubMed] [Google Scholar]
Lam 2017
- Lam DL, Houssami M, Lee JM. Imaging surveillance after primary breast cancer treatment. AJR. American Journal of Roentgenology 2017;208(3):676‐86. [DOI] [PMC free article] [PubMed] [Google Scholar]
Leather 2017
- Leather T, Jenkinson MD, Das K, Poptani H. Magnetic Resonance Spectroscopy for detection of 2‐hydroxyglutarate as a biomarker for IDH mutation in gliomas. Metabolites 2017;7(2):E29. [DOI] [PMC free article] [PubMed] [Google Scholar]
Louis 2007
- Louis DN, Ohgaki H, Wiestler OD, Cavenee WK, Burger PC, Jouvet A, et al. The 2007 WHO classification of tumours of the central nervous system. Acta Neuropathologica 2007;114(2):97‐109. [DOI] [PMC free article] [PubMed] [Google Scholar]
Louis 2016
- Louis DN, Perry A, Reifenberger G, Deimling A, Figarella‐Branger D, Cavenee WK, et al. The 2016 World Health Organization classification of tumors of the central nervous system: a summary. Acta Neuropathologica 2016;131(6):803‐20. [DOI] [PubMed] [Google Scholar]
Malmström 2017
- Malmström A, Poulsen HS, Grønberg BH, Stragliotto G, Hansen S, Asklund T, et al. Postoperative neoadjuvant temozolomide before radiotherapy versus standard radiotherapy in patients 60 years or younger with anaplastic astrocytoma or glioblastoma: a randomized trial. Acta Oncologica 2017;56(12):1776‐85. [DOI] [PubMed] [Google Scholar]
NCCN 2018
- National Comprehensive Cancer Network. NCCN Guidelines version 1. 2018. Central Nervous System Cancers. www.nccn.org/store/login/login.aspx?ReturnURL=https://www.nccn.org/professionals/physician_gls/pdf/cns.pdf (accessed 12 April 2018).
NCI 2017
- National Cancer Institute. Adult central nervous system tumors treatment (PDQ). www.cancer.gov/types/brain/hp/adult‐brain‐treatment‐pdq (accessed 20 December 2017).
NICE 2006
- National Institute for Health and Clinical Excellence. Improving outcomes for people with brain and other CNS tumours. www.nice.org.uk/guidance/csg10/resources/improving‐outcomes‐for‐people‐with‐brain‐and‐other‐central‐nervous‐system‐tumours‐update‐pdf‐27841361437 (accessed 29 November 2017).
NICE 2018
- National Institute for Health and Clinical Excellence. Brain tumours (primary) and brain metastases in adults. www.nice.org.uk/guidance/ng99. NICE, (accessed 08 August 2018).
Ohgaki 2009
- Ohgaki H. Epidemiology of brain tumours. In: Verma M editor(s). Methods of Molecular Biology, Cancer Epidemiology. Vol. 472, Totowa, NJ: Humana Press, 2009:323‐42. [DOI] [PubMed] [Google Scholar]
Pignatti 2002
- Pignatti F, Bent M, Curran D, Debruyne C, Sylvester R, Therasse P, et al. Prognostic factors for survival in adult patients with cerebral low‐grade glioma. Journal of Clinical Oncology 2002;20(8):2076‐84. [DOI] [PubMed] [Google Scholar]
Rees 2009
- Rees J, Watt H, Jäger HR, Benton C, Tozer D, Tofts P, et al. Volumes and growth rates of untreated adult low‐grade gliomas indicate risk of early malignant transformation. European Journal of Radiology 2009;72(1):54‐64. [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Rouse 2016
- Rouse C, Gittleman H, Ostrom QT, Kruchko C, Barnholtz‐Sloan JS. Years of potential life lost for brain and CNS tumors relative to other cancers in adults in the United States, 2010. Neuro‐oncology January 2016;18(1):70‐7. [DOI: ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2017a
- Schünemann HJ, Oxman AD, Higgins JP, Vist GE, Glasziou P, Akl E, et al. on behalf of the Cochrane GRADEing Methods Group and the Cochrane Statistical Methods Group. Chapter 11: Completing ‘Summary of findings’ tables and grading the confidence in or quality of the evidence. In: Higgins JPT, Churchill R, Chandler J, Cumpston MS (editors), Cochrane Handbook for Systematic Reviews of Interventions version 5.2.0 (updated June 2017). Cochrane, 2017. Available from www.training.cochrane.org/handbook.
Schünemann 2017b
- Schünemann HJ, Oxman AD, Vist GE, Higgins JP, Deeks JJ, Glasziou P, et al. on behalf of the Cochrane Applicability and Recommendations Methods Group. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Churchill R, Chandler J, Cumpston MS (editors), Cochrane handbook for systematic reviews of interventions version 5.2.0 (updated June 2017). Cochrane, 2017. Available from www.training.cochrane.org/handbook.
SEOM 2017
- Sepulveda‐Sanchez JM, Munoz Langa J, Arraez MA, Fuster J, Hernandez Lain A, Reynes G, et al. SEONM clinical guideline of diagnosis and management of low‐grade glioma. Clinical & Translational Oncology 2017;epub:ahead of print. [DOI] [PMC free article] [PubMed] [Google Scholar]
Shankar 2017
- Shankar GM, Balaj L, Stott SL, Nahed B, Carter BS. Liquid biopsy for brain tumors. Expert Review of Molecular Diagnostics 2017;17(10):943‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
Shemlit 2011
- Shemilt I, Mugford M, Vale L, Craig D, on behalf of the Campbell and Cochrane Economics Methods Group. Searching NHS EED and HEED to inform development of economic commentary for Cochrane Intervention Reviews. methods.cochrane.org/economics/workshops (accessed 29 November 2017).
Sterne 2017
- Sterne JA, Egger M, Moher D, Boutron I (editors). Chapter 10: Addressing reporting biases. In: Higgins JP, Churchill R, Chandler J, Cumpston MS (editors), Cochrane handbook for systematic reviews of interventions version 5.2.0 (updated June 2017), Cochrane, 2017. Available from www.training.cochrane.org/handbook.
Thielen 2016
- Thielen FW, Mastrigt GA, Burgers LT, Bramer WM, Majoie HJ, Evers SM, et al. How to prepare a systematic review of economic evaluations for clinical practice guidelines: database selection and search strategy development (part 2/3). Expert Review of Pharmacoeconomics & Outcomes Research 2016;16(6):705‐21. [DOI] [PubMed] [Google Scholar]
Van Mastrigt 2016
- Mastrigt GA, Hiligsmann M, Arts JJ, Broos PH, Kleijnen J, Evers SM, et al. How to prepare a systematic review of economic evaluations for informing evidence‐based healthcare decisions: a five‐step approach (part 1/3). Expert Review of Pharmacoeconomics & Outcomes Research 2016; Vol. 16, issue 6:689‐704. [DOI] [PubMed]
Whiting 2016
- Whiting P, Savović J, Higgins JP, Caldwell DM, Reeves BC, Shea B, et al. ROBIS: a new tool to assess risk of bias in systematic reviews was developed. Journal of Clinical Epidemiology 2016;69:225‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wick 2012
- Wick W, Platten M, Meisner C, Felsberg J, Tabatabai G, Simon M, et al. Temozolomide chemotherapy alone versus radiotherapy alone for malignant astrocytoma in the elderly: the NOA‐08 randomised, phase 3 trial. Lancet Oncology 2012;13(7):707‐15. [DOI] [PubMed] [Google Scholar]
Wijnen 2016
- Wijnen BF, Mastrigt GA, Redekop WK, Majoie HJ, Kinderen RJ, Evers SM. How to prepare a systematic review of economic evaluations for informing evidence‐based healthcare decisions: data extraction, risk of bias, and transferability (part 3/3). Expert review of pharmacoeconomics & outcomes research 2016;16(6):723‐32. [DOI] [PubMed] [Google Scholar]