

Abstract
Background
Bipolar disorder is a severe and common mental disorder where patients experience recurrent symptoms of elevated or irritable mood, depression, or a combination of both. Treatment is usually with psychiatric medication, including mood stabilisers, antidepressants and antipsychotics. Valproate is an effective maintenance treatment for bipolar disorder. However, evidence assessing the efficacy of valproate in the treatment of acute mania is less robust, especially when comparing it to some of the newer antipsychotic agents. This review is an update of a previous Cochrane Review (last published 2003) on the role of valproate in acute mania.
Objectives
To assess the efficacy and tolerability of valproate for acute manic episodes in bipolar disorder compared to placebo, alternative pharmacological treatments, or a combination pharmacological treatments, as measured by the treatment of symptoms on specific rating scales for individual episodes in paediatric, adolescent and adult populations.
Search methods
We searched Ovid MEDLINE (1950‐ ), Embase (1974‐ ), PsycINFO (1967‐ ) and the Cochrane Central Register of Controlled Trials (CENTRAL) to 28 September 2018. We had also conducted an earlier search of these databases in the Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR) (all years to 6 June 2016). We also searched the World Health Organization (WHO) trials portal (ICTRP) and clinicaltrials.gov in September 2018, to identify any additional unpublished or ongoing studies.
Selection criteria
Single‐ and double‐blind, randomised controlled trials comparing valproate with placebo, alternative antimanic treatments, or a combination of pharmacological treatments. We also considered studies where valproate was used as an adjunctive treatment in combination with another agent separately from studies where it was used in monotherapy. We included male and female patients of all ages and ethnicity with bipolar disorder.
Data collection and analysis
Two review authors independently performed data extraction and methodological quality assessment. For analysis, we used the odds ratio (OR) for binary efficacy outcomes and the mean difference (MD) or standardised mean difference (SMD) for continuously distributed outcomes.
Main results
Twenty‐five trials (3252 participants) compared valproate with either placebo or alternative antimanic treatments to alleviate the symptoms of acute mania. For efficacy, our primary outcome was response rate. For tolerability, our primary outcome was the number of participants with any adverse effect. This meta‐analysis included studies focusing on children, adolescents, as well as adults with a range of severity of manic symptoms. The majority of studies focused on adult men and women (aged 18 and above), were conducted in inpatient settings and completed in the US. Five studies in this review focused on children and adolescents (aged 18 and under) so that the review covers an age range from 3 ‐ 82 years. Seven studies contained outpatient participants in some form. Nine studies included data that has been collected outside the US, namely Iran (4 studies), India (3 studies), China (1 study), or across several international countries (1 study).
In adults, high‐quality evidence found that valproate induces a slightly higher response compared to placebo (45% vs 29%, OR 2.05, 95% CI 1.32 to 3.20; 4 studies, 869 participants). Moderate‐quality evidence found there was probably little or no difference in response rates between valproate and lithium (56% vs 62%, OR 0.80, 95% CI 0.48 to 1.35; 3 studies, 356 participants). In adults, low‐quality evidence found there may be little or no difference in response rate between valproate and olanzapine (38% vs 44%, OR 0.77, 95% CI 0.48 to 1.25; 2 studies, 667 participants).
In the children and adolescent population, the evidence regarding any difference in response rates between valproate and placebo was uncertain (23% vs 22%, OR 1.11, 95% CI 0.51 to 2.38; 1 study, 151 participants, very low‐quality evidence). Low‐quality evidence found that the response rate of participants receiving valproate may be lower compared to risperidone (23% vs 66%, OR 0.16, 95% CI 0.08 to 0.29; 1 study, 197 participants). The evidence regarding any difference in response rates between valproate and lithium was uncertain (23% vs 34%, OR 0.57, 95% CI 0.31 to 1.07; 1 study, 197 participants, very low‐quality evidence).
In terms of tolerability in adults, moderate‐quality evidence found that there are probably more participants receiving valproate who experienced any adverse events compared to placebo (83% vs 75%, OR 1.63, 95% CI 1.13 to 2.36; 3 studies, 745 participants). Low‐quality evidence found there may be little or no difference in tolerability between valproate and lithium (78% vs 86%, OR 0.61, 95% CI 0.25 to 1.50; 2 studies, 164 participants). We did not obtain primary tolerability outcome data on the olanzapine comparison.
Within the children and adolescent population, the evidence regarding any difference between valproate or placebo was uncertain (67% vs 60%, OR 1.39, 95% CI 0.71 to 2.71; 1 study, 150 participants, very low‐quality evidence). We did not obtain primary tolerability outcome data on the lithium or risperidone comparisons.
Authors' conclusions
There is evidence that valproate is an efficacious treatment for acute mania in adults when compared to placebo. By contrast, there is no evidence of a difference in efficacy between valproate and placebo for children and adolescents. Valproate may be less efficacious than olanzapine in adults, and may also be inferior to risperidone as a monotherapy treatment for paediatric mania. Generally, there is uncertain evidence regarding whether valproate causes more or less side effects than the other main antimanic therapies. However, evidence suggests that valproate causes less weight gain and sedation than olanzapine.
Plain language summary
Valproate for acute mania
Headline: Valproate is an effective antimanic treatment. Valproate may be inferior to olanzapine in adults. Valproate may be inferior to risperidone in acute mania in paediatric and adolescent populations.
Who may be interested in this review? People with bipolar disorder and their healthcare providers.
Why is this review important? Bipolar disorder is a mood disorder that is a common mental health problem. Patients may experience recurrent symptoms of elevated or irritable mood, depression, or a combination of both. Treatment is usually with psychiatric medication, including mood stabilisers, antidepressants and antipsychotics. Valproate is a drug traditionally used in the treatment of mania, but its effectiveness compared to some of the newer antipsychotics is yet to be firmly established.
What questions does this review aim to answer? This review investigates the effectiveness and acceptability of valproate compared to placebo and other drugs in the treatment of acute manic episodes in bipolar disorder.
Which studies were included in the review? The authors searched medical databases to find reports of clinical trials (specifically randomised controlled trials) published up to date. We identified 25 studies that involved 3252 participants as relevant. The studies compared the effects of valproate with placebo or other conventional medications, both on its own and in combination with other treatments.
What does the evidence from the review tell us?
We found high‐quality evidence showing that valproate is more effective than placebo when used alone in adults. There is mixed evidence comparing olanzapine and valproate. Low‐quality evidence did not find a difference in response rate of olanzapine compared to valproate. However, high‐quality evidence suggests that olanzapine is better at reducing manic symptoms. This suggests olanzapine may be more effective. Moderate‐quality evidence shows no difference in response rates between lithium and valproate. There is insufficient evidence to confidently assess any difference between valproate and other antimanic drugs in adults.
In children and adolescents, we found low‐quality evidence that valproate is inferior to risperidone. The evidence is of insufficient quality to confidently assess any difference between valproate and other antimanic drugs in children and adolescents.
In terms of tolerability in adults, there is moderate‐quality evidence that valproate causes more side effects than placebo and low‐quality evidence it causes more side effects than oxcarbazepine. There is low‐quality evidence that valproate may cause fewer side effects than carbamazepine. Low‐quality evidence found no difference in the number of individuals with side effects when taking valproate compared to lithium. There is insufficient evidence to confidently assess any difference between valproate and other antimanic drugs in adults.
In children and adolescents, very‐low quality evidence found no difference in the number of individuals with side effects when taking valproate compared to placebo.
Summary of findings
Summary of findings for the main comparison. Valproate compared to placebo for acute mania in adults.
| Valproate compared to placebo for acute mania | ||||||
| Patient or population: Adults (aged 18 and over) with acute mania Setting: Mixed inpatient and outpatient Intervention: Valproate Comparison: Placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with valproate | |||||
| Response rate at 3 weeks (primary efficacy outcome) |
Study population | OR 2.05 (1.32 to 3.20) | 869 (4 RCTs) | ⊕⊕⊕⊕ HIGH | Valproate results in a higher response rate than placebo at three weeks | |
| 287 per 1000 | 452 per 1000 (347 to 563) | |||||
| Number with any adverse event after 3 weeks (primary tolerability outcome) |
Study population | OR 1.63 (1.13 to 2.36) | 745 (3 RCTs) | ⊕⊕⊕⊝ MODERATEb | Valproate may increase the number of individuals experiencing adverse effects compared to placebo, although the evidence is uncertain. | |
| 754 per 1,000 | 833 per 1,000 (777 to 878) | |||||
| Individual adverse events ‐ Sedation At 3‐8 weeks (secondary tolerability outcome) |
Study population | OR 1.76 (0.95 to 3.24) |
932 (5 RCTs) |
⊕⊕⊕⊕ HIGH | No clear evidence of difference in sedation rates compared to placebo, although a clear trend favouring placebo. | |
| 122 per 1,000 | 197 per 1,000 (117 to 310) |
|||||
| Change in symptom severity at 3 weeks (secondary efficacy outcome) |
SMD ‐ 0.23 (‐ 0.45 to 0 ) | ‐ | 907 (4 RCTs) | ⊕⊕⊕⊝ MODERATEc,d | Valproate decreases manic symptoms compared to placebo. Based on Cohen's effect sizes, a standard deviation of 0.2 represents a small difference between groups. | |
| Dropout rate ‐ All‐cause 3‐8 weeks (secondary acceptability outcome) |
Study population | OR 0.83 (0.64 to 1.07) | 1156 (6 RCTs) | ⊕⊕⊕⊝ MODERATEd | No evidence of a difference in overall dropout rates in individuals using valproate compared to placebo. | |
| 520 per 1,000 | 474 per 1,000 (410 to 537) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; RCT: randomised controlled trial; SMD: standardised mean difference. | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aEvidence downgraded by two levels for imprecision, due to single study and small study size. bEvidence downgraded by one level as > 30% of studies in comparison rated as "at serious risk of bias". Hirschfeld 2010 was at serious risk of bias as it was considered at high risk for reporting and attrition biases, and at unclear risk of bias for all other domains assessed except other biases. cSignificant heterogeneity (I2 > 50%). However this is largely driven by one poor quality study (Pope 1991), the removal of which does not materially affect results (Before: −0.23 (95% CI −0.45 to −0.00); After: −0.16 (95% CI −0.30 to −0.02)). Therefore, as this heterogeneity was explained and does not alter the main character of the results, we felt justified in not downgrading for this. dEvidence downgraded by one level as > 30% of studies in comparison rated as at "at serious risk of bias". Hirschfeld 2010 was at serious risk of bias as it was considered at high risk of bias for reporting and attrition biases, and at unclear risk of bias for all other domains assessed except other biases. Pope 1991 was at serious risk of bias, as it was at high risk for attrition bias and other biases, and at unclear risk of bias for selection and reporting biases.
Summary of findings 2. Valproate compared to placebo for acute mania in children and adolescents.
| Valproate compared to placebo for acute mania | ||||||
| Patient or population: Children and adolescents with acute mania Setting: Outpatient Intervention: Valproate Comparison: Placebo | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with placebo | Risk with valproate | |||||
| Response rate at 4 weeks (primary efficacy outcome) | Study population | OR 1.11 (0.51 to 2.38) | 151 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b,c | The evidence is very uncertain about the relative effects of valproate and placebo on response rates. | |
| 216 per 1000 | 234 per 1000 (123 to 396) | |||||
| Number with any adverse event at 4 weeks (primary tolerability outcome) | Study population | OR 1.39 (0.71 to 2.71) | 150 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b,c | The evidence is very uncertain about the relative effects of valproate and placebo on the number of individuals with adverse effects | |
| 595 per 1000 | 671 per 1000 (510 to 799) | |||||
| Individual adverse events ‐ Sedation At 4 weeks (secondary tolerability outcome) |
Study population | OR 0.40 (0.12 to 1.37) |
150 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b,c | The evidence is very uncertain about the relative effects of valproate and placebo on the number of individuals with sedation | |
| 122 per 1,000 | 52 per 1,000 (16 to 159) |
|||||
| Change in symptom severity at 4 weeks (secondary efficacy outcome) |
SMD ‐ 0.09 (‐ 0.41 to 0.24) | ‐ | 144 (1 RCTs) | ⊕⊝⊝⊝ VERY LOWa,b,c | The evidence is very uncertain about the relative effects of valproate and placebo on decreasing manic symptoms at 4 weeks. Based on Cohen's effect sizes, a standard deviation of under 0.2 represents a small difference between groups. | |
| Dropout rate ‐ All‐cause At 4‐6 weeks (secondary acceptability outcome) |
Study population | OR 1.77 (0.83 to 3.78) | 179 (2 RCTs) | ⊕⊕⊝⊝ LOWc,e | The evidence is uncertain about the relative effects of valproate and placebo on dropout rate. | |
| 160 per 1000 | 256 per 1000 (138 to 424) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; RCT: randomised controlled trial; SMD: standardised mean difference. | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aEvidence downgraded by one level as > 30% of studies in comparison rated as at "at serious risk of bias". Wagner 2009 at serious risk of bias. Wagner 2009 at unclear risk of selection, detection and other biases. bEvidence downgraded by one level for imprecision, as single study only. cEvidence downgraded by one level for imprecision, as total number of participants in both comparisons < 100. dEvidence downgraded by one level as > 30% of studies in comparison rated as at "at serious risk of bias". Kowatch 2015 at serious risk of bias. Kowatch 2015 at serious risk of detection bias and unclear risk of other biases. eEvidence downgraded by one level as > 30% of studies in comparison rated as at "at serious risk of bias". Kowatch 2015 and Wagner 2009 at serious risk of bias. Kowatch 2015 at serious risk of detection bias and at unclear risk of other biases. Wagner 2009 at unclear risk of selection, detection and other biases.
Summary of findings 3. Valproate compared to lithium for acute mania in adults.
| Valproate compared to lithium for acute mania | ||||||
| Patient or population: Adults (aged 18 and over) with acute mania Setting: Inpatient Intervention: Valproate Comparison: Lithium | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with lithium | Risk with valproate | |||||
| Response rate at 3 weeks (primary efficacy outcome) |
Study population | OR 0.80 (0.48 to 1.35) | 356 (3 RCTs) | ⊕⊕⊕⊝ MODERATEa | No evidence of difference in response rate between valproate and lithium. | |
| 615 per 1000 | 561 per 1000 (434 to 683) | |||||
| Number of participants with any adverse event at 2‐4 weeks (primary tolerability outcome) | Study population | OR 0.61 (0.25 to 1.50) | 164 (2 RCTs) | ⊕⊝⊝⊝ LOWb,c | The evidence is uncertain about the relative effects of valproate and lithium on number of individuals with side effects. | |
| 855 per 1000 | 782 per 1000 (595 to 898) | |||||
| Individual adverse events ‐ Sedation At 3 weeks (secondary tolerability outcome) |
Study population | OR 0.96 (0.35 to 2.67) | 105 (1 RCT) | ⊕⊕⊝⊝ LOWd,e | The evidence is uncertain about the relative effects of valproate and lithium on sedation. | |
| 194 per 1000 | 188 per 1000 (78 to 392) | |||||
| Change in symptom severity at 3 weeks (secondary efficacy outcome) |
SMD 0.69 (0.14 to 1.25) | ‐ | 57 (2 RCTs) | ⊕⊝⊝⊝ VERY LOWa,d,e,f | Lithium may cause a greater decrease in manic symptoms than valproate, but the evidence is very uncertain. Based on Cohen's effect sizes, a standard deviation of 0.6 represents a moderate difference between groups. | |
| Dropout rate ‐ All‐cause At 2‐4 weeks (secondary acceptability outcome) |
Study population | OR 0.83 (0.47 to 1.45) | 388 (3 RCTs) | ⊕⊕⊕⊝ MODERATEb | No evidence of difference in all‐cause dropout rate between valproate and lithium | |
| 281 per 1,000 | 245 per 1,000 (155 to 362) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; RCT: randomised controlled trial; SMD: standardised mean difference. | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aEvidence downgraded by one level as > 30% of trials at serious risk of bias, Freeman 1992 at serious risk of bias, as selection, detection, reporting and other biases are all at uncertain risk of bias. bEvidence downgraded by one level as > 30% of trials at serious risk of bias, Hirschfeld 1999 at serious risk of bias, as reporting at high risk of bias and selection and other biases at uncertain risk of bias. cEvidence downgraded by one level for imprecision, due to wide confidence interval. dEvidence downgraded by one level for imprecision due to single study. eEvidence downgraded by one level for imprecision due to small study size, fEvidence downgraded by one level for indirectness; in Freeman 1992 only able to use endpoint measures as statistics for differences not provided.
Summary of findings 4. Valproate compared to lithium for acute mania in children and adolescents.
| Valproate compared to lithium for acute mania | ||||||
| Patient or population: Children and adolescents with acute mania Setting: Outpatient Intervention: Valproate Comparison: Lithium | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with lithium | Risk with valproate | |||||
| Response rate at 8 weeks (primary efficacy outcome) | Study population | OR 0.57 (0.31 to 1.07) | 197 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | The evidence is very uncertain about the relative effects of valproate and lithium on response rates. | |
| 344 per 1000 | 230 per 1000 (140 to 360) | |||||
| Number of participants with any adverse event (primary tolerability outcome) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Individual adverse events ‐ Sedation At 8 weeks (secondary tolerability outcome) |
Study population | OR 1.59 (0.84 to 3.00) | 190 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | The evidence is very uncertain about the relative effects of valproate and lithium on sedation. | |
| 244 per 1000 | 340 per 1000 (214 to 493) | |||||
| Change in symptom severity of mania rating scale at 8 weeks (secondary efficacy outcome) |
The mean in symptom severity for lithium at 8 weeks was 26.2. | MD 1.40 (‐ 2.03 to 4.83) | ‐ | 190 (1 RCT) |
⊕⊝⊝⊝ VERY LOWa,b | The evidence is very uncertain about the relative effects of valproate and lithium on manic symptoms. |
| Dropout rate ‐ All‐cause At 8 weeks (secondary acceptability outcome) |
Study population | OR 0.74 (0.39 to 1.37) | 197 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | The evidence is very uncertain about the relative effects of valproate and lithium on dropout rates. | |
| 312 per 1,000 | 251 per 1,000 (150 to 383) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; RCT: randomised controlled trial; MD: mean difference. | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aEvidence downgraded by two levels as > 30% of trials at very serious risk of bias. Geller 2012 at very serious risk of bias, as performance, attrition and reporting biases were all assessed as high risk of bias. bEvidence downgraded by one level for imprecision due to single study.
Summary of findings 5. Valproate compared to olanzapine for acute mania in adults.
| Valproate compared to olanzapine for acute mania | ||||||
| Patient or population: Adults (aged 18 and over) with acute mania Setting: Mixed inpatient and outpatient Intervention: Valproate Comparison: Olanzapine | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with olanzapine | Risk with valproate | |||||
| Response rate at endpoint at 3 weeks (primary efficacy outcome) | Study population | OR 0.77 (0.48 to 1.25) | 667 (2 RCTs) | ⊕⊕⊝⊝ LOWa,b | The evidence is uncertain about the relative effects of valproate and olanzapine on response rates. | |
| 441 per 1000 | 378 per 1000 (275 to 497) | |||||
| Number of participants with any adverse event (primary tolerability outcome) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Adverse events ‐ Sedation At 3‐12 weeks (secondary tolerability outcome) |
Study population | OR 0.50 (0.28 to 0.91) | 536 (2 RCTs) | ⊕⊕⊕⊕ HIGH | Evidence of decreased rate of sedation with valproate compared to olanzapine. | |
| 143 per 1000 | 77 per 1000 (45 to 132) | |||||
| Change in symptom severity at 3 weeks (secondary efficacy outcome) | SMD 0.25 (0.11 to 0.39) |
‐ | 826 (4 RCTs) | ⊕⊕⊕⊕ HIGH | Evidence of larger decrease in manic symptoms with olanzapine compared to valproate at 3 weeks. Based on Cohen's effect sizes, a standard deviation of 0.2 represents a small difference between groups. | |
| Dropout rate ‐ All‐cause At 3‐12 weeks (secondary acceptability outcome) |
Study population | OR 1.04 (0.71 to 1.52) | 616 (3 RCTs) | ⊕⊕⊕⊕ HIGH | No evidence of difference between valproate and olanzapine. | |
| 304 per 1000 | 313 per 1000 (237 to 400) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; RCT: randomised controlled trial; SMD: standardised mean difference. | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aDowngraded by one level for significant heterogeneity (I2 > 50%). This heterogeneity may be partially explained by different methodologies of the studies within Tohen 2002 and Tohen 2008, but we felt that as these are the only two studies in this comparison and this only partially explains the heterogeneity it was still warranted to downgrade the quality of this rating. bEvidence downgraded by one level for risk of bias, as primary outcome reported by half or fewer of the studies found. cEvidence downgraded by one level for imprecision due to single study and less then 100 participants.
Summary of findings 6. Valproate compared to risperidone for acute mania in children and adolescents.
| Valproate compared to risperidone for acute mania | ||||||
| Patient or population: Children and adolescents with acute mania Setting: Outpatient Intervention: Valproate Comparison: Risperidone | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with risperidone | Risk with valproate | |||||
| Response rate at 8 weeks (primary efficacy outcome) | 656 per 1000 | 234 per 1000 (132 to 356) |
OR 0.16 (0.08 to 0.29) |
197 (1 RCT) | ⊕⊕⊝⊝ LOWa,b,c | Risperidone may increase response rate compared to valproate at 8 weeks, but the evidence is uncertain. |
| Number of participants with any adverse event (primary tolerability outcome) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Adverse events ‐ Sedation At 8 weeks (secondary tolerability outcome) |
Study population | OR 0.50 (0.28 to 0.91) | 189 (1 RCT) | ⊕⊝⊝⊝ VERY LOWb,c | Risperidone may cause a higher sedation rate compared to valproate but the evidence is very uncertain. | |
| 484 per 1000 | 328 per 1000 (214 to 463) | |||||
| Change in symptom severity at 5 ‐ 12 weeks (secondary efficacy outcome) | SMD 1.01 (0.74 to 1.29 ) |
‐ | 228 (2 RCTs) |
⊕⊕⊝⊝ LOWb | Risperidone may decrease manic symptoms compared to valproate after 5 ‐ 12 weeks, but the evidence is uncertain. Based on Cohen's effect sizes, a standard deviation of 1.0 represents a large difference between groups. | |
| Dropout rate ‐ All‐cause (secondary acceptability outcome) At 5 ‐ 12 weeks |
Study population | OR 1.96 (1.00 to 3.82) | 236 (2 RCTs) | ⊕⊕⊝⊝ LOWb | Risperidone may decrease dropout rates compared to valproate, but the evidence is uncertain. | |
| 144 per 1000 | 248 per 1000 (144 to 391) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; RCT: randomised controlled trial; SMD: standardised mean difference. | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aEvidence upgraded by one level for large effect, OR < 0.5.
bEvidence downgraded by two levels as > 30% of studies at very high risk of bias. Geller 2012 at very serious risk of bias, as performance, attrition and reporting biases were all assessed as high risk of bias. Kowatch 2015 was at serious risk of bias, as detection bias was at high risk of bias and other biases were at unclear risk of bias.
cEvidence downgraded by one level for imprecision due to single study.
Background
Description of the condition
Bipolar disorder (also known as bipolar affective disorder, or manic depressive disorder) describes a group of mood disorders of which the key identifying feature is repeated, erratic shifts between elevated mood (mania) and depression (Philips 2013). More than 1% of the world’s population is affected by bipolar disorder “irrespective of nationality, ethnic origin, or socioeconomic status” (Findling 2018; Grande 2016). Bipolar disorder largely impairs the ability to carry out normal daily activities (Alonso 2011) and is among the leading causes of years lived in disability worldwide (GBD 2013 Risk Factors Collaborators 2015). The disorder is not only associated with an increased risk in suicide and suicide attempts (Merikangas 2011) but also shortens life expectancy as a result of prevalent comorbid medical disorders (Carr 2018; Laursen 2011).
In the DSM‐5, a 'manic episode' is defined as a distinct abnormally and persistently elevated, expansive, or irritable mood, which lasts at least one week (or shorter if hospitalisation is necessary), with three (four if only irritability is present) or more of the following symptoms present to a significant degree: inflated self‐esteem or grandiosity, decreased need for sleep, more talkative than usual, flight of ideas or a subjective experience that thoughts are racing, distractibility by unimportant and irrelevant external stimuli, increase in goal‐directed activity or psychomotor agitation, and an excessive involvement in pleasurable activities that have a high potential for painful or negative consequences (APA 2013). Unlike DSM‐IV, DSM‐5 allows a diagnosis of bipolar disorder I in people with major depression whose mania emerges during treatment (e.g. during medication or electroconvulsive therapy (ECT)) and persists at a fully syndromal level beyond the physiological effect of the treatment (APA 2000; APA 2013). Hypomania may occur as part of a bipolar II disorder or as a transitional state towards full‐blown mania. By definition, the symptoms of hypomania differ only quantitatively from those of mania: A hypomanic episode is diagnosed when one presents with an elevated, expansive mood, and three (four, if only irritability is present) additional manic symptoms are present for a minimum of four days (Yatham 2002).
Bipolar I disorder is diagnosed in individuals who experience major depressive episodes in combination with full manic episodes (abnormally elevated mood or irritability and related symptoms with severe functional impairments or psychotic symptoms for seven days or more). Bipolar II disorder is characterised by hypomanic episodes (abnormally elevated mood or irritability and related symptoms with decreased or increased function for four days or more) which alternate with depressive episodes (APA 2013; WHO 1992). In a less common pattern, called rapid cycling type, the person switches back and forth between depressive and manic or mixed episodes (with at least four episodes a year) with little or no 'normal' functioning (Leibenluft 2000). Although manic episodes are often thought of as the 'mirror image' to depression in which people enjoy their heightened mood and act self‐confidently, the detrimental consequences of manic episodes must not be underestimated. In fact, self‐reported data from bipolar people support the conceptualisation that mania and hypomania are syndromes characterised by a reduced rather than an increased sense of well‐being and quality of life (Vojta 2001). These findings are not surprising, in light of the strong link between manic episodes and impulsive behaviour and substance abuse (Swann 2007), and the perceived negative experiences regarding psychosocial functioning (Judd 2005). These behaviours can potentially lead to devastating economic, occupational, and interpersonal problems, as often at the stage of treatment engagement the manic syndrome is generally already escalated (Kendall 2014).
Description of the intervention
Valproate has been used in the treatment of acute mania since 1966 (Lambert 1966) and is mentioned as an effective treatment in many official guidelines (CANMAT 2018; Goodwin 2016; Malhi 2015; NICE 2014). Whilst a plethora of mechanisms that are at work have been reported in the literature (see below), the exact mechanism of action underlying the effect of valproate on any specific clinical effects has not yet been fully understood (Chateauvieux 2010; Marson 2015). The multifaceted workings underlying the effect of valproate may not only explain the drug's effect on a large spectrum of neurological and mood disorders but also account for the diverse adverse events associated with this medication (see below).
Valproate is a term that is used to describe valproic acid as well as its derivatives sodium valproate and semi‐sodium valproate (Taylor 2009). It is a fatty acid containing eight carbon atoms and is referred to as dipropylpentanoic acid (FDA 2015). Originally used as an anticonvulsant for epilepsy, valproate was trialled as an alternative to lithium in the treatment of bipolar disorder during the 1980s and was approved by the Food and Drug Administration for the treatment of acute mania in 1995 (Emrich 1980; Leo 1999). It became part of the group of mood stabilisers, which also include other anticonvulsants, antipsychotics and lithium (Geddes 2013). Mood stabilisers are used in both maintenance and acute stabilisation of bipolar disorder (Geddes 2013).
An increasingly recognised important side effect of valproate is its teratogenic effects. Recent meta‐analyses have estimated that around 11% of children exposed to valproate in the womb have malformations such as neural tube defects and cleft palate at birth, compared with a 2% to 3% risk for children in the general population (Meador 2008). This represents a substantial increase (Meador 2008). Furthermore, there is evidence of a three‐fold increase in the incidence of pervasive developmental disorders such as autism spectrum disorders (Christensen 2013), as well as developmental issues such as delayed walking and talking, memory problems, difficulty with speech and language, and lower intellectual ability (Bromley 2014; Cummings 2011). However recent evidence suggests that rates of prescribing in women of reproductive age remain high. One study looking specifically at valproate use in acute mania through the Arzneimittelsicherheit in der Psychiatry database (International Drug Safety Program in Psychiatry: AMSP) suggests that 36.8% of women aged under 40 received valproate which, while significantly less than in men (47.8%), is still high (Kleimann 2016) and an Irish study showed very low rates of folic acid and contraceptive co‐prescription with valproate (Murphy 2016). The concern about the effects of valproate triggered a review of the medication by the European Medication Agency (EMA) that recommended avoiding the use of valproate in pregnancy for bipolar and that valproate should not be used in the absence of a pregnancy prevention programme (. Mania is an especially troublesome condition with regards to teratogenicity, as the condition both greatly impairs judgement and increases the likelihood of risky sexually promiscuous behaviour. This suggests that it is extremely important to fully inform women of the risks of valproate before prescription and that valproate should not be used in women of child bearing age unless necessary, as reflected in recently updated NICE guidelines (NICE 2018a). Indeed new 2018 MHRA guidance contra‐indicates valproate use in women of child‐bearing age unless a pregnancy prevention programme is in place (MHRA 2018).
Prescribing trends reflect the decreasing popularity of valproate as a first‐line treatment. Recent data from a US study indicates that in longer‐term treatment for mania a bipolar specialist clinic prescription for valproate fell from 24.2% (2000 to 2005) to 14.9% (2006 to 2011) (Hooshmand 2014). This contrasts with the previous trends of increasing valproate prescription. A further study examined prescriptions specifically for acute mania, examining the AMSP database, which monitors over 430,000 patients at 114 hospitals in Germany, Austria, Switzerland, Belgium and Hungary. This study similarly found a decrease in valproate prescriptions, albeit a minor one, from the start of their study in 2005 (41.3%) to 2011 (38.5%) (Kleimann 2016). This was mirrored by about 80% of manic patients receiving SGAs in 2011/12 (Kleimann 2016). This may be in part due to concerns about teratogenicity.
How the intervention might work
Valproate is absorbed in the gastrointestinal tract and enters the bloodstream where 80% to 90% is protein‐bound, depending on plasma concentration (FDA 2015). A small proportion of valproate in plasma (10%) will cross the blood‐brain barrier where it acts on neuronal cells (FDA 2015). It is generally accepted that valproate reduces neuronal excitability, by increasing synaptic concentrations of the inhibitory neurotransmitters as well as blocking voltage‐gated ion channels (Rosenberg 2007). Eventually, it undergoes glucuronidation or oxidation in the liver and is finally excreted through the kidneys. Valproate is usually administered orally and for mania is started at a dose of 250 mg given three times a day (Taylor 2009), whereas for epilepsy it is started at 600 mg daily (Joint Formulary Committee 2015). The dose is then adjusted depending on tolerability and patient response, with the normal treatment dose lying between 1000 and 2000 mg a day (Joint Formulary Committee 2015).
Specifically, valproate increases brain concentrations of the inhibitory neurotransmitter gamma‐aminobutyric acid (GABA), probably through increased GABA synthesis, decreased GABA turnover, and inhibition of GABA degradation (Chateauvieux 2010; Marson 2015; Perruca 2002). One potential beneficial effect of the increase of GABA might be that it quickly leads to a slightly sedative state and lessens anxiety, which may help moderate mania (Freeman 2002; Li 2002). Furthermore, valproate blocks voltage‐sensitive sodium ion channels in several pathways in the brain, suppressing high‐frequency firing of neurons, and possibly indirect effects on non‐GABA‐ergic neurotransmission (Johannessen 2000; Rho 1999). Valproate is also thought to reduce the release of the excitatory aminoacid β‐hydroxybutyric acid, inhibit N‐Methyl‐D‐aspartate (NMDA receptor‐mediated excitatory transmissions), blocks of calcium channels, potentiation of calcium‐activated potassium currents, modulation of serotonergic and dopaminergic neurotransmission; and inhibition of histone deacetylases (Chateauvieux 2010; Marson 2015; Perruca 2002). In addition, it has been shown that valproate may attenuate the activity of an enzyme (active protein kinase), which shows an increased cell surface in people with bipolar disorder (Hahn 2005; Wang 1996; Wang 1999; Wang 2001). The hypothesis that valproate, a fatty acid, might alter the brain’s lipid metabolism has also been suggested (Bazinet 2006). In addition to short‐term biochemical effects, there also seems to be consistent, strong evidence that valproate works through exerting long‐term effects at the genomic level (Bosetti 2005; Tang 2004).
Why it is important to do this review
Increasing evidence from network meta‐analyses suggests that valproate monotherapy is inferior to antipsychotic drugs such as olanzapine, risperidone and quetiapine both for efficacy and for acceptability (Cipriani 2011; Yildiz 2015). Yet valproate (either alone or in combination with other drugs) is used in clinical practice for acute mania, especially in severe and treatment‐resistant cases (NICE 2014). Moreover, valproate monotherapy is still frequently listed in treatment guidelines as a first‐line option for acute mania (CANMAT 2018; Goodwin 2016). Historically, it has been assumed that valproate shows greater efficacy than lithium in people who suffer from bipolar disorder with mixed features (Swann 1997). Support for valproate is offered by more recent studies, including a subgroup analysis by Bowden 2006 which showed a decrease in manic symptoms in people with a mixed episode compared with placebo, and results by Del Grande 2014 which suggest that valproate added to lithium treatment is superior to lithium monotherapy in those suffering from acute mania and mixed features. Although other studies indicate that lithium and valproate show comparable efficacy and tolerability for mixed episodes (Bowden 2010), valproate is still considered one of the preferred treatments for mixed mania (Fountoulakis 2012). It is important for practitioners to have access to up‐to‐date, comprehensive, synthesised evidence that can inform their treatment decisions (Williams 2018).
In terms of tolerability, many potentially significant adverse drug reactions with valproate have been reported in the medical literature. Side effects relating to the digestive system commonly occur. These range from relatively benign effects, such as nausea, vomiting, changes in bowel habits or dyspepsia (Bowden 2006; Nanau 2013) to pancreatitis (Gerstner 2007). In comparison to lithium, for instance, which has a narrow therapeutic range lying close to toxic levels and requires frequent monitoring of drug levels in the blood, thereby potentially reducing acceptability for patients, valproate is preferred in some cases due to reduced monitoring requirements (Taylor 2009). However, amongst the most frequently used mood stabilisers and benzodiazepines, valproate is associated with the greatest risk of potential liver toxicity (hepatotoxicity), which is rare but potentially life‐threatening (Telles‐Correia 2017). Moreover, valproate can adversely affect the central nervous system and lead to tremors, dizziness, sedation and confusion, and has been shown to be teratogenic in pregnancy (Morrow 2006). Given the high risk of adverse events associated with valproate, it is vital for practitioners to be fully informed about its effectiveness in order to make an accurate cost‐benefit judgement.
Despite the publication of a recent network meta‐analysis (Yildiz 2015), the knowledge base of valproate treatment in acute mania is still incomplete. The network meta‐analysis by Yildiz 2015 included 11 studies, but notably the authors did not include one study that was included in a previous network meta‐analysis (Cipriani 2011). Neither meta‐analysis has included studies involving children or adolescents (DelBello 2006; Geller 2012; Wagner 2009), even though they are an important patient population in clinical practice (Doherty 2018). Finally, new studies on the effects of valproate in mania continue to be published (e.g. Ahmad 2016; Xu 2015), so that a comprehensive update of the previous Cochrane Review is needed, to be followed at a later late by a formal Cochrane network meta‐analysis if deemed appropriate and necessary.
Objectives
To assess the efficacy and tolerability of valproate for acute manic episodes in bipolar disorder compared to placebo, to alternative pharmacological treatments and to combination pharmacological treatments, as measured by the treatment of symptoms on specific rating scales for individual episodes in paediatric, adolescent and adult populations.
Methods
Criteria for considering studies for this review
Types of studies
Randomised trials comparing valproate in the treatment of acute mania (including mixed‐mood episodes) with alternative antimanic drug treatments or placebo in bipolar disorder. Cross‐over studies were eligible for inclusion, although we planned to include data only from the first phase of randomisation. We also planned to include cluster‐randomised controlled trials, with assessment of their potential for unit‐of‐analysis errors (Higgins 2011). We excluded all quasi‐randomised studies, such as those allocating participants by using alternate days of the week, but included effectiveness trials (Tansella 2006). We considered both published and unpublished trials.
Types of participants
Participant characteristics Male and female participants of all ages and of any ethnicity.
Diagnosis
We included studies when participants had a primary diagnosis of bipolar disorder corresponding to the Feighner criteria (Feighner 1972), Research Diagnostic Criteria (Spitzer 1978), DSM‐III (APA 1980), DSM‐III‐R (APA 1987), DSM‐IV (APA 1994), DSM‐IV‐TR (APA 2000) DSM‐5 (APA 2013), or ICD‐10 (WHO 1992). We excluded studies using ICD‐9, as it has only disease names and no diagnostic criteria. We included people with the following subtypes of bipolar disorder:
Manic episodes, with or without psychotic symptoms, approximating to the respective codes mentioned in the above guidelines
Mixed episodes, with or without psychotic symptoms, approximating to the respective codes mentioned in the above guidelines
Treatment‐resistant mania (defined as an unsatisfactory response to at least two trials of two different medications (Poon 2012), with or without psychotic symptoms
Rapid cycling disorder, with or without psychotic symptoms
We excluded cyclothymia, as well as studies that defined mania as scoring above a certain cut‐off on a screening questionnaire.
Comorbidities
We did not consider a concurrent secondary diagnosis of another psychiatric disorder an exclusion criterion. However, we excluded studies in which all participants had a concurrent primary diagnosis of a DSM‐IV Axis I and II disorder. We also excluded studies in which participants had a serious concomitant medical illness or active postpartum depression. This is because the presence of active postpartum depression would alter the management of any active concomitant mania (Furukawa 2016b; NICE 2018b).
Setting
We included studies from all settings, including inpatients and outpatients.
Types of interventions
Experimental Intervention
Valproate in the treatment of acute manic or mixed episodes in the context of bipolar disorder.
We defined 'acute treatment' as treatment instituted specifically to alleviate symptoms of an existing acute episode. We would not analyse the second phase of a discontinuation trial, in which participants received open‐label valproate prior to blind randomisation, and trials that only concerned the maintenance phase. When trials combined acute treatment and maintenance phases, we analysed the data from the acute phase whilst disregarding data points from the maintenance phase.
Comparator intervention
Placebo (either as monotherapy or as adjunctive treatment)
Alternative antimanic treatments (either as monotherapy or as adjunctive treatment)
Combination of pharmacological treatments
We also considered studies where valproate was used as an adjunctive treatment in combination with another agent separately from studies where it was used in monotherapy. Lastly, we included trials that allow rescue medications (e.g. hypnotics) as long as these medications were equally distributed among the randomised arms.
Types of outcome measures
We included studies that meet the above inclusion criteria, regardless of whether they reported on the following outcomes.
Primary outcomes
1. Efficacy (dichotomous): We defined response to treatment as a 50% or greater reduction in mean score on the Young Mania Rating Scale (YMRS) ‐ or any other equivalent standardised rating scale ‐ from baseline. 2. Tolerability (dichotomous): We assessed tolerability using the number of participants experiencing adverse events of any nature. In order to avoid missing any relatively rare or unexpected but important side effects, in the data extraction phase, we collected information on all side‐effects data reported in the studies and discussed ways to summarise them post hoc. We extracted descriptive data for adverse effect profiles from all available studies. In cases where reporting was inconsistent, we combined terms describing similar side effects (Caddy 2015; McCloud 2015). For example, we combined 'dry mouth’, 'reduced salivation’, and 'thirst’ into 'dry mouth’.
Secondary outcomes
1. Remission (dichotomous): We defined remission as a score of 12 or less on the YMRS (or equivalent on other validated mania rating scales).
2. Efficacy (continuous): We assessed the efficacy of valproate by assessing the change in symptom severity, using mean endpoint scores or mean change scores on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline to the time point in question. We allowed a looser form of intention‐to‐treat (ITT) analysis, whereby all the participants with at least one post‐baseline measurement were represented by their last observations carried forward (LOCF), but in any pooled analysis we examined the impact of the LOCF in a sensitivity analysis.
3. Acceptability (dichotomous): We assessed the acceptability of valproate using dichotomous information on:
Overall number of participants who dropped out during the trial as a proportion of the total number of randomised participants.
Number of participants who dropped out due to lack of efficacy during the trial as a proportion of the total number of randomised participants.
Number of participants who dropped out due to side effects during the trial as a proportion of the total number of randomised participants.
4. Global functioning: We assessed global functioning using the Clinical Global Impressions‐Improvement scale (CGI‐I) or Clinical Global Impression ‐ Bipolar Disorder‐ Improvement (CGI‐BP‐I), considered at time points closest to three weeks. We considered the proportion of participants who improved at endpoint based on the final CGI‐I score of 1 to 2.
5. Tolerability (dichotomous): We assessed tolerability by collecting data on individual side effects to help compare side effect profiles of different comparisons.
Hierarchy of outcome measures
If data on more than one measure of efficacy of treatment were provided for a trial, we extracted data according to the following hierarchy:
YMRS
Other outcome measure of efficacy of treatment with manic symptom rating scales.
Timing of outcome assessment
Outcomes will be measured at the following time points:
at four days (if not available, any duration less than 1 week)
at 1 week (if not available, any duration between 1 and 2 weeks);
at 3 weeks (if not available, any duration between more than 2 weeks and up to 4 weeks)
at 8 weeks (if not available, any duration between more than 5 weeks and up to 12 weeks)
Search methods for identification of studies
Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR)
The Cochrane Common Mental Disorders Group (CCMD) retains two clinical trials registers at its editorial base (current to June 2016); a references register and a studies‐based register. The CCMDCTR‐References Register contains over 40,000 reports of randomised controlled trials (RCTs) in depression, anxiety and neurosis. Approximately half of these references have been tagged to individual, coded trials. The coded trials are held in the CCMDCTR‐Studies Register and records are linked between the two registers through the use of unique Study ID tags. Coding of trials is based on the EU‐Psi coding manual, using a controlled vocabulary. (Please contact the CCMD Information Specialists for further details). We collated reports of trials for inclusion in the Group's registers from routine (weekly), generic searches of MEDLINE (1950 ‐), Embase (1974 ‐) and PsycINFO (1967 ‐), quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL) and review‐specific searches of additional databases. We also sourced reports of trials from international trial registers through the World Health Organization's trials portal (the International Clinical Trials Registry Platform (ICTRP)), pharmaceutical companies, conference proceedings and other (non‐Cochrane) systematic reviews and meta‐analyses.
Details of CCMD's generic search strategies (used to identify RCTs) can be found on the Group's website, with an example of the core MEDLINE search displayed in Appendix 1. The register was up‐to‐date only to June 2016.
Electronic searches
1. We searched the CCMDCTR‐Studies Register on 6 June 2016, using the following controlled search terms: Condition = (bipolar or mani* or schizoaffective) AND Intervention = (divalproex or valpro*)
2. We searched the CCMDCTR‐References Register on 6 June 2016, using a more sensitive set of free‐text terms to identify additional untagged/uncoded reports of RCTs: Free‐text = (valpro* or divalpro*) and (bipolar or mania or manic or hypomani* or psychos* or psychotic or postpsycho* or post‐psycho* or "rapid cycling" or schizoaffective)
3. In September 2018, CCMD's Information Specialist ran an update search on the following bibliographic databases (as the CCMDCTR was out of date at the time). The search strategies are reported in Appendix 2.
Ovid MEDLINE (2016 to 28 September 2018)
Ovid Embase (2016 to 2018 week 39).
Ovid PsycINFO (2016 to September week 4, 2018)
Cochrane Central Register of Controlled Trials (CENTRAL) (via the Cochrane Register of Studies (CRSO)) (all years to 28 September 2018)
4. We also searched international trial registries (28 September 2018) through the World Health Organization's trials portal (ICTRP) and ClinicalTrials.gov to identify unpublished or ongoing studies.
We applied no restrictions by date, language or publication status to the searches.
Searching other resources
Grey literature
We conducted complementary searches on the websites of the following drug regulatory authorities for additional unpublished data: the US Food and Drug Administration, the Medicines and Healthcare products Regulatory Agency in the UK, the European Medicines Agency in the EU, the Pharmaceuticals and Medical Devices Agency in Japan, and the Therapeutic Goods Administration in Australia.
Reference lists
We checked the reference lists of all included studies and relevant systematic reviews to identify additional studies missed from the original electronic searches (for example, unpublished or in‐press citations).
Data collection and analysis
Selection of studies
Two review authors (JJ and RRZ) independently screened for inclusion titles and abstracts of all the references retrieved by the search strategy. We subsequently retrieved full‐text study reports/publications, which the two review authors (JJ and RRZ) also independently screened for inclusion. At this stage, we recorded the reasons for excluding the ineligible studies, resolving any disagreement through discussion or, if required, by consulting a third person of the review team (AC). We identified and removed duplicate records, and collated multiple reports that related to the same study so that each study, rather than each report, is the unit of interest in the review. We recorded the selection process in sufficient detail to complete a PRISMA flow diagram (Moher 2009) and the Characteristics of excluded studies table.
Data extraction and management
We used a data collection form to extract study characteristics and outcome data that was piloted on at least one study in the review. Two review authors (JJ and RRZ) extracted study characteristics and outcome data from each included study independently and compared the results. We resolved any disagreement through discussion with a third member of the team (AC). We contacted study authors if necessary, to acquire supplemental information. We extracted the following study characteristics:
Eligibility: confirm eligibility for the review, and reason for exclusion.
Methods: study design,the total duration of the study, study setting, withdrawals, date of study, sequence generation, allocation sequence concealment, blinding, and other concerns about bias.
Participants: total number, age range, gender, diagnostic criteria, country.
Interventions: intervention, comparison, concomitant medication, and excluded medications.
Outcomes: primary and secondary outcomes specified and collected, and time points reported.
Notes: funding for trial and notable conflicts of interest of trial authors.
We noted in the Characteristics of included studies table if outcome data were not reported in a useable way, resolving disagreements by consensus or by involving a third person (AC). One review author (JJ) transferred data into the Review Manager 5 file (RevMan 5). We double‐checked that data were entered correctly by comparing the data presented in the review with the study reports. A second review author (RRZ) spot‐checked study characteristics for accuracy against the trial report.
Assessment of risk of bias in included studies
Two review authors (JJ and RRZ) independently assessed the risks of bias for each study using the criteria outlined in the Cochrane Handbook for Systematic Reviews of Interventions (Cochrane Handbook; Higgins 2017). We resolved any disagreements by discussion or by involving another review author (AC). We assessed the risks of bias of the included studies according to the following domains.
Random sequence generation
Allocation concealment
Blinding of participants and personnel
Blinding of outcome assessment
Incomplete outcome data
Selective outcome reporting
Other bias
We judged each potential source of bias as high, low, or unclear, and provided a supporting quotation from the study report together with a justification for our judgement in the 'Risk of bias' tables. We summarise the 'Risk of bias' judgements across different studies for each of the domains listed. Where information on the risk of bias was informed by unpublished data or correspondence with an author, we have noted this in the 'Risk of bias' table.
As suggested in Higgins 2017, the authors have estimated the risk of bias associated with the blinding of the outcome assessment depending on the impact on the respective outcome. For instance, knowledge of the assignment of the intervention may lead to high bias when manic symptoms are assessed using a questionnaire, whilst other outcomes such as 'suicide' would not be affected. For the biases concerned with incomplete outcome reporting, we judged the risk of bias as low if the dropout rate was lower than 30% in all study arms. We also rated the risk of bias as low when the dropout rate was higher than 30% in one or more study arms, but the difference in dropout between study arms was not two‐fold or more. We estimated the risk of bias to be high for incomplete outcome reporting when the overall dropout rate was above 75%, or when the dropout rate of one study arm was twice as high (or higher) than in any of the other study arms. We assessed the bias associated with selective outcome reporting by comparing the measures mentioned in the published protocol with the measures provided in the published study report. In cases where no protocol was available, we rated this bias as 'unclear'.
Measures of treatment effect
Dichotomous data
We analysed dichotomous data as an odds ratio (OR) with a 95% confidence interval (CI). We decided to use odds ratios since we are comparing alternative treatments directly and ORs better show the incremental benefit in comparing option A to option B. A further benefit of odds ratios is that they enable meta‐regression, which is impossible when working with risk ratios. While this does not apply directly to our review, it does apply to previous network meta‐analyses (NMAs) done in this field. Keeping our values in odds ratios enables our review to be compared to and evaluated easily against previous large‐scale NMAs, allowing understanding of similarities and differences in results. We acknowledge that odds ratios can be misinterpreted as relative risks; we counsel readers not to interpret an odds ratio as the risk ratio of an outcome, but instead to read it as the ratio of the odds of a result happening with one intervention compared to another. Please note that when an outcome is rare the odds and risk ratios are similar; the more common an outcome, the more an odds ratio will overstate the effect of the treatment on the outcome measure if interpreted as a risk ratio.
Continuous data
We analysed continuous data as a mean difference (MD) or a standardised mean difference (SMD) with a 95% CI. We use the MD to compare continuous data where every study uses the same rating scale, whereas standardised mean differences allow comparison of multiple studies using different mania scales by standardising changes across scales. We present data as a scale with a consistent direction of effect. We undertook meta‐analyses only where this was meaningful, i.e. if the treatments, participants, and underlying clinical question were similar enough for pooling to make sense. We would narratively report skewed data as medians and interquartile ranges. Where a single trial reported multiple trial arms, we have included only the relevant arms.
Unit of analysis issues
Cluster‐randomised trials
In cluster‐randomised trials, groups rather than individuals are randomised to different interventions. Cluster‐randomised trials would need to account for intra‐class correlation. To adjust for cluster effects, we would use the generic inverse variance technique, provided that cluster‐randomised trials have been appropriately analysed, taking into account an intra‐class correlation coefficient (ICC). If the necessary summary statistics were not reported, we would contact the authors; otherwise the data cannot be re‐analysed. We did not have any cluster‐randomised trials in this meta‐analysis.
Cross‐over trials
In cross‐over trials, each participant is allocated to a sequence of interventions and each participant acts as his or her own control. We used only data from the first phase of cross‐over trials because the effect of treatment in the first period can affect the outcome in the second period. We did not have any cross‐over trials in this review.
Studies with multiple treatment groups
Where a study involved more than two treatment arms, we included all relevant treatment arms in comparisons. If data were binary, we combined them into one group, if appropriate (Higgins 2011). If data were continuous, we combined data following the formula in section 7.7.3.8 of the Cochrane Handbook (Higgins 2011). Seven studies had multiple treatment arms. The studies with multiple treatment arms were; Bowden 1994 (valproate versus lithium versus placebo), Geller 2012 (valproate versus lithium versus risperidone), Kowatch 2015 (valproate versus risperidone versus placebo), Hirschfeld 1999 (valproate loading versus valproate non‐loading versus lithium), Ahmad 2016 (valproate versus endoxifen (4 mg) versus endoxifen 8 mg), Tohen 2008 (valproate versus olanzapine versus placebo), Xu 2015 (valproate versus olanzapine versus combined valproate and olanzapine). We aggregated data in two of these seven studies. In Ahmad 2016 we aggregated the two endoxifen dose arms into one comparison group, and in Hirschfeld 1999 we aggregated the loading and non‐loading valproate groups into one arm.
Dealing with missing data
Dichotomous data
Where the dichotomous outcomes were not reported, we contacted trial authors and asked them to supply the data. We calculated responders to treatment and remitters on a strict ITT basis: we included dropouts in this analysis and used the number of participants randomised as the denominator. Where participants were excluded from a trial before the endpoint, we assumed that they experienced a negative outcome by the end of the trial (e.g. failure to respond to treatment).
Continuous data
When there were missing data and the method of LOCF was used to perform an ITT analysis, we used the LOCF data. We contacted the original study authors for missing data. When only the standard error (SE) or t‐statistics or P values were reported, we calculated standard deviations (SDs) according to Altman 1996. Where SDs were not reported, we contacted trial authors and asked them to supply the data. In the absence of data from the authors, we borrowed SDs from other studies in the review (Furukawa 2006). TheCochrane Handbook (16.1.3.1 Imputing standard deviations) (citing Furukawa 2006) recommends imputing standard deviations, given that only a small proportion of studies fail to provide them. We have followed the suggested approach of using the most conservative standard deviation within similar studies.
Assessment of heterogeneity
We assessed heterogeneity between studies using the I2 statistic (Higgins 2003), and by visual inspection of the forest plot. Following recommendations in the Cochrane Handbook, we interpreted I2 values as follows: 0% to 40% might not be important; 30% to 60% may represent moderate heterogeneity; 50% to 90%: may represent substantial heterogeneity; and 75% to 100% considerable heterogeneity. If we identified significant heterogeneity, we investigated the potential sources.
Assessment of reporting biases
We entered data from included studies into a funnel plot (trial effect against trial variance) to investigate small‐study effects, only when at least 10 studies are included in the meta‐analysis (Sterne 2000). Where we produced a funnel plot, we interpreted results cautiously, with a visual inspection of the funnel plots (Higgins 2017). If we identified evidence of small‐study effects, we investigated possible reasons for funnel plot asymmetry, including publication bias (Egger 1997).
Data synthesis
At the protocol stage, we decided to present any skewed data and non‐quantitative data descriptively. We considered statistically significant a P value of less than 0.05 and a 95% CI that does not cross the line of no effect. In forest plots with two or more studies, we used a random‐effects model for both dichotomous and continuous variables. We adopted the random‐effects model under these circumstances because it has the highest generalisability for empirical examination of summary effect measures in meta‐analyses (Furukawa 2002; Wandel 2016). However, as recommended by the Cochrane Handbook (10.4.4) (Sterne 2017), when there are concerns about the influence of small‐study effects on the results of a meta‐analysis with between‐study heterogeneity, we examined the robustness by comparing the fixed‐effect model and the random‐effects model. We report any material differences between the models.
Subgroup analysis and investigation of heterogeneity
We did not plan any subgroup analysis, as the only subgroup comparison would have been children/adolescents versus adult and we have decided to analyse these groups separately due to the large apparent difference in these groups' response to medication.
During review author comments, we raised the issue of a subgroup analysis between those with rapid cycling mania and those without rapid cycling mania. We agree that it is conceivable that there are differences between these population groups and thus a subgroup analysis would be justified. Unfortunately, none of the studies included in the review separate out those with rapid‐cycling mania versus those without. Similarly, with mixed states, only one study explicitly excluded those with mixed states, which leaves the feasibility of any subgroup analysis untenable. In future updates of the review, the possibility of exploring any differences between these subgroups should be revisited.
Sensitivity analysis
We ran the following sensitivity analyses for primary outcomes:
Excluding studies that recruited participants with treatment‐resistant mania;
Excluding trials with unclear allocation concealment or unclear double‐blinding;
Excluding studies with valproate as add‐on treatment;
Excluding trials for which the SD had to be borrowed from other trials (Furukawa 2006);
Assessing for differential efficacy in participants with and without psychotic features by excluding all studies with participants with psychotic features.
Our routine comparisons of random‐effects and fixed‐effect models, as well as our secondary outcomes of remission rates and continuous severity measures, may be considered additional forms of sensitivity analyses. For the results of these analyses, see the sub‐section 'Sensitivity analyses' in the Effects of interventions section of the review.
'Summary of findings' table
We have produced a 'Summary of findings’ table for each comparison, including the following five outcomes.
Response.
Number of participants with any adverse event.
Individual adverse events ‐ sedation
Severity of manic symptoms at end of trial.
Total dropouts
In the ’Summary of findings’ tables, we have used GRADE proGDT software (GRADEpro GDT 2015) and the principles of the GRADE approach (Atkins 2004), which assess the quality of a body of evidence‐based on the extent to which we can be confident that the obtained effect estimate reflects the true underlying effect. The quality of a body of evidence is judged on the basis of the included studies’ risks of bias, the directness of the evidence, unexplained heterogeneity, imprecision, and the risk of publication bias. For details on the precise criteria used in this study please see below:
We assessed the quality of the data in the following way, as given in the flow sheet provided by the GRADE Pro software that generates 'Summary of findings' tables. We chose our measures for downgrading on each measurement.
Data begin by default as high‐quality, and are downgraded quality levels for the following issues:
Risk of Bias:
Each study was individually rated as follows:
If the study had one or more criteria at high risk of bias OR the study had four or more criteria at either high or unclear risk of bias, we rated the study as a whole at serious risk of bias
If the study had three or more criteria at high risk of bias OR the study had seven criteria at either high or unclear risk of bias we rated the study as a whole at very serious risk of bias
We rated outcome measures as a whole at serious risk if:
More than 30% of studies had outcomes at serious risk of bias
The primary outcome was reported by half or fewer studies
Outcomes were at very serious risk:
More than 30% of studies had outcome at very serious risk of bias
Two of the serious risk criteria were met
Directness of the evidence:
Studies were considered at risk of indirectness if any outcome could not be directly calculated from the data available and instead a proxy measure was used. For example; if the difference between start and end mania scores were not reported and endpoint mania rating scores had to be used as a proxy for change in mania rating scores.
Heterogeneity:
Considered at serious risk if I2 > 50% and heterogeneity was unexplained. Considered at very serious risk if I2 > 70%.
Imprecision: Considered at serious risk if one of the following, or at very serious risk if two of the following.
Total number of participants in both comparisons were fewer than 100
Single study only
95% CI covered a four‐fold increase/decrease in odds and no effect
Publication bias:
Our data were not large enough to allow assessment of publication bias – the Grade tool suggests downgrading only if there was a strong suspicion of publication bias.
Large effect size:
Following GRADE instructions, we upgraded quality by one rank if the Risk Ratio (RR) > 2 or < 0.5
Results
Description of studies
See Characteristics of included studies
Results of the search
We identified 683 references from the CCMDCTR and five references from other sources (one from searching previous meta‐analysis reference lists and four from reference lists from remaining articles searched). After duplicates were removed, 629 references remained to screen. Following a review of the abstracts, we excluded 489 references. We assessed 139 full‐text articles for eligibility, excluding 108 records and categorising seven as awaiting classification and one as ongoing. The final number of included studies at this stage was 23.
An update search in September 2018 identified 325 references from bibliographic database searches, and one reference from searching review reference lists. We assessed 29 full‐text articles for eligibility, excluding 26 records and categorising one as ongoing. The final number of additional included studies was two, including one of the previous studies identified as ongoing. Please see PRISMA flow diagram Figure 1 for details of all the searches. The total number of studies finally included in the qualitative analysis and in the meta‐analysis was 25. In any cases of missing data or information, we contacted the study authors (For a detailed list of these contacts please see Appendix 3).
1.
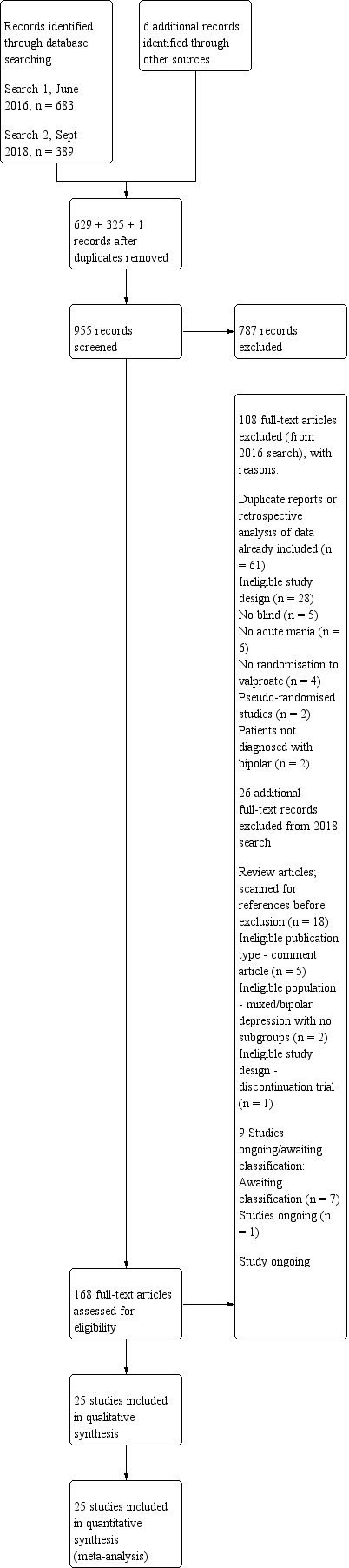
study flow diagram.
Included studies
We include 25 studies in this updated systematic review.
Study design All included studies were randomised controlled trials.
Eighteen trials were two‐armed, with valproate versus placebo in five studies (Bowden 2006; Hirschfeld 2010; McElroy 2010; Pope 1991Wagner 2009). The remaining 13 two‐armed studies compared valproate against other psychoactive medication, including carbamazepine (Vasudev 2000), haloperidol (McElroy 1996), lithium (Freeman 1992; Shafti 2008; Young 2017), olanzapine (Tohen 2002; Zajecka 2002), oxcarbazepine (Kakkar 2009), quetiapine (DelBello 2006; Feifel 2011) and topiramate (Hebrani 2009). Two out of the 13 two‐armed studies investigated the effects of valproate as adjunctive therapy. In Mahmoudi‐Gharaei 2012, valproate was compared to topiramate, each being administered as an adjunctive medication to lithium and to risperidone. In Moosavi 2014, valproate was administered as an adjunctive medication to risperidone and compared to risperidone monotherapy.
Seven studies were three‐armed. Hirschfeld 1999 compared valproate loading with valproate non‐loading and lithium; we merged the two valproate arms for the purposes of our analyses. Ahmad 2016 compared valproate with two different doses of endoxifen (4 mg versus 8 mg), which we merged for our purposes. Bowden 1994 compared valproate, lithium, and placebo. Geller 2012 compared valproate, lithium, and risperidone. Kowatch 2015 looked at valproate, risperidone, and placebo. Tohen 2008 investigated valproate, olanzapine, and placebo. Xu 2015 compared treatments of valproate, olanzapine, and combined valproate and olanzapine treatment, constituting the only three‐armed study which allowed for an analysis of the effects of valproate added on to olanzapine.
Sample size We included 3252 randomised participants, with a mean study sample size of 130 participants. Overall, Freeman 1992 included the lowest number of participants (14 randomised to valproate and 13 to placebo), while Bowden 2006 and Tohen 2008 tested the largest number of participants per study arm. The former tested 377 (192 participants randomised to valproate extended‐release and 185 participants randomised to placebo), whilst the latter included 521 participants (215 randomised to olanzapine, 201 randomised to valproate, 105 randomised to placebo).
Participants All studies included male and female participants, except for Shafti 2008, where all participants were female only. One study focused on older people, including only those aged 60 or older (Young 2017). In 10 studies, the age range for participants was 18 to 65 years (Ahmad 2016; Bowden 1994Bowden 2006; Feifel 2011; Hirschfeld 2010; McElroy 1996; Pope 1991; Tohen 2008; Vasudev 2000; Zajecka 2002). Six additional studies also focused on adults (Hirschfeld 1999; Kakkar 2009; McElroy 2010; Moosavi 2014; Tohen 2002Xu 2015), but deviated slightly from the above‐mentioned age range. Hirschfeld 1999 included participants between 18 and 60, Kakkar 2009 included participants aged between 18 and 50, Moosavi 2014 included participants aged between 20 and 60, Tohen 2002 included participants aged between 18 and 75, and Xu 2015 included participants aged between 19 and 60. McElroy 2010 only states that participants above the age of 18 are included in the study, but does not specify an upper age limit.
We also included studies in children or adolescents. In Kowatch 2015, children were between 3 and 7 years of age, and Geller 2012 included children and adolescents between the ages of 6 and 15 years. Two studies (Mahmoudi‐Gharaei 2012Wagner 2009) focused solely on adolescents, including participants between 11 and 18 years and 10 and 17 years respectively. Both DelBello 2006 and Hebrani 2009 focused on participants between 12 and 18 years of age. We were unable to identify the age ranges for two studies (Freeman 1992Shafti 2008).
Sixteen studies were conducted only within the USA with most participants identifying as white (Bowden 1994; Bowden 2006; DelBello 2006; Feifel 2011; Freeman 1992; Geller 2012; Hirschfeld 1999; Hirschfeld 2010; Kowatch 2015; McElroy 1996; McElroy 2010; Pope 1991, Tohen 2002; Wagner 2009; Young 2017; Zajecka 2002). Ahmad 2016 conducted the trial at various hospitals in India with a racial makeup of 100% Asian. The studies by Kakkar 2009 and Vasudev 2000 were also conducted in India. Xu 2015 investigated the effects of valproate in a population of Chinese patients. The adolescents in Hebrani 2009 were enrolled from the adolescent ward of Mashhad University in Iran. Similarly, the lead authors of three further trials are associated with institutions in Iran (Mahmoudi‐Gharaei 2012; Moosavi 2014Shafti 2008). Tohen 2008 recruited participants from private practices, hospital clinics, and university clinics from a range of countries, including Lithuania, Puerto Rico, Russia, and the United States.
In 17 studies, participants met the criteria for bipolar disorder, type I (according to DSM‐IV or DSM‐IV‐TR), experiencing a manic episode (Ahmad 2016; Bowden 2006; DelBello 2006; Feifel 2011; Geller 2012; Hebrani 2009; Hirschfeld 1999; Hirschfeld 2010; Kakkar 2009; Mahmoudi‐Gharaei 2012; Moosavi 2014; Shafti 2008; Tohen 2002; Wagner 2009; Xu 2015; Young 2017; Zajecka 2002), whilst two studies (Kowatch 2015; McElroy 2010) included participants with bipolar disorder type I or type II (according to DSM‐IV or DSM‐IV‐TR). Participants in Freeman 1992, McElroy 1996, and Pope 1991 had been diagnosed based on the DSM‐III‐R and the authors did not specify the type of bipolar disorder for inclusion. Kakkar 2009 and Tohen 2008 did not specify the type of bipolar disorder for inclusion. Participants in Bowden 1994 were included when they met Research Diagnostic Criteria for manic disorder, based on the structured interview and rating scale of the Schedule for Affective Disorders and Schizophrenia (SADS).
A range of studies based inclusion in the study on a score on the Young Mania Rating Scale (YMRS) of ≥ 20 (Ahmad 2016; DelBello 2006; Hebrani 2009; Kakkar 2009; Kowatch 2015; Tohen 2002; Tohen 2008; Vasudev 2000; Wagner 2009), ≥ 18 (Young 2017), ≥ 17 (Feifel 2011; Xu 2015), ≥ 14 (Hirschfeld 1999), or ≥ 10 and < 21 (McElroy 2010). Other studies required a score of ≥ 14 (Bowden 1994), ≥ 18 (Bowden 2006) or ≥ 25 (Hirschfeld 2010Zajecka 2002) on the Mania Rating Scale (MRS). The remaining studies did not comment on this feature (Freeman 1992; Geller 2012; Mahmoudi‐Gharaei 2012; McElroy 1996; Moosavi 2014; Pope 1991; Shafti 2008).
Twenty studies included participants experiencing mixed episodes (Ahmad 2016; Bowden 2006; DelBello 2006; Feifel 2011; Freeman 1992; Geller 2012; Hebrani 2009; Hirschfeld 1999; Hirschfeld 2010; Kowatch 2015; McElroy 2010; Mahmoudi‐Gharaei 2012; McElroy 1996; Tohen 2002; Tohen 2008; Vasudev 2000; Wagner 2009; Xu 2015; Young 2017; Zajecka 2002). Kakkar 2009 explicitly excluded people with mixed states, and Bowden 1994, Moosavi 2014, Pope 1991, and Shafti 2008 did not specify whether or not they included or excluded patients with mixed‐episodes.
Ten studies included participants with and without psychotic features (Ahmad 2016; Bowden 2006; Feifel 2011; Geller 2012; Kowatch 2015; Mahmoudi‐Gharaei 2012; Tohen 2002; Wagner 2009; Young 2017; Zajecka 2002). McElroy 1996 only included participants with psychotic features, whereas McElroy 2010, Moosavi 2014 and Tohen 2008 excluded those with psychotic features. Eleven studies did not specifically comment on the presence of psychosis in their participant group (Bowden 1994; DelBello 2006; Freeman 1992; Hebrani 2009; Hirschfeld 1999; Hirschfeld 2010; Kakkar 2009; Pope 1991;Shafti 2008Vasudev 2000; Xu 2015;)
Most studies were conducted in an inpatient setting (Ahmad 2016; Bowden 1994; Bowden 2006; DelBello 2006; Feifel 2011; Freeman 1992; Hebrani 2009; Hirschfeld 1999; Hirschfeld 2010; Kakkar 2009; Mahmoudi‐Gharaei 2012; McElroy 1996; Moosavi 2014; Pope 1991; Shafti 2008; Tohen 2002; Xu 2015). Four studies were conducted in an outpatient setting (Geller 2012; Kowatch 2015; McElroy 2010; Wagner 2009). Participants in Tohen 2008 and Young 2017 were a mix of inpatients and outpatients. Participants in Vasudev 2000 were attending an outpatient clinic but were hospitalised for the purposes of the study. Participants in Zajecka 2002 were inpatients for a period of up to 21 days, after which they were followed up as outpatients.
Interventions/comparisons Seven studies used valproate extended‐release tablets (Ahmad 2016; Bowden 2006; Feifel 2011; Hirschfeld 2010; Kowatch 2015; McElroy 2010; Wagner 2009). Three studies used valproate as adjunctive treatment (Mahmoudi‐Gharaei 2012; Moosavi 2014; Xu 2015). In one study, valproate versus topiramate was added to lithium and risperidone (Mahmoudi‐Gharaei 2012). The second study administered valproate as an adjunctive to risperidone versus risperidone monotherapy (Moosavi 2014). Thirdly, Xu 2015 included one treatment group which received olanzapine and valproate combined, whilst the other two groups received a monotherapy of valproate or olanzapine, respectively.
Thirteen studies did not allow participants to take any psychotropic medications (Ahmad 2016; Bowden 2006; Feifel 2011; Geller 2012; Hebrani 2009; Kakkar 2009; Kowatch 2015; McElroy 1996; McElroy 2010; Pope 1991; Vasudev 2000; Wagner 2009, Young 2017) and one study did not allow any other neuroleptic drugs (Bowden 1994). By contrast, Shafti 2008 and Mahmoudi‐Gharaei 2012 allowed antipsychotic medication (haloperidol) as an adjunctive medication. The remaining studies did not explicitly mention whether or not additional psychotropic medication was permitted during the trial. In addition, most studies did allow the use of adjunctive lorazepam (Ahmad 2016; Bowden 1994; Bowden 2006; DelBello 2006; Feifel 2011; Freeman 1992; Hebrani 2009; Hirschfeld 1999; Hirschfeld 2010; Kakkar 2009; Pope 1991; Shafti 2008; Tohen 2002; Tohen 2008; Wagner 2009; Zajecka 2002). Ahmad 2016 and Vasudev 2000 allowed the use of diazepam; Bowden 1994, Freeman 1992, Hirschfeld 2010 and Zajecka 2002 allowed chloral hydrate; McElroy 1996, Tohen 2002, and Zajecka 2002 allowed benztropine; Wagner 2009 and Zajecka 2002 allowed zolpidem tartrate, and Mahmoudi‐Gharaei 2012 allowed the use of biperiden in the event of extrapyramidal side effects.
Geller 2012 allowed the maintenance of methylphenidate, amphetamine preparations (total daily dose equivalent to < 60 mg methylphenidate) and allergy/asthma medications were allowed, to mimic usual clinical practice. In Moosavi 2014, clonazepam and trihexyphenidyl started in divided dose in both groups. All participants in both groups also received prophylactic anticholinergic drugs (trihexyphenidyl) and benzodiazepine (clonazepam) daily. In Tohen 2008, anticholinergics and ongoing thyroid supplementation therapy were permitted. Wagner 2009 allowed the ongoing treatment of attention‐deficit/hyperactivity disorder (ADHD) with stimulant medications (with the exception of pemoline). Lastly, Xu 2015 did not allow any adjunctive medication during the course of the trial.
Outcomes Freeman 1992, Kowatch 2015, Mahmoudi‐Gharaei 2012, Moosavi 2014, and Pope 1991 did not specify between primary efficacy and secondary efficacy outcomes in their published reports.
Primary outcomes Fourteen studies used scores on the YMRS as their primary outcome measure. Ahmad 2016, DelBello 2006, Feifel 2011, Hebrani 2009, Hirschfeld 1999, Kakkar 2009, McElroy 1996, McElroy 2010, Tohen 2002, Tohen 2008, Vasudev 2000, Wagner 2009, Xu 2015, and Young 2017 measured the change in YMRS scores from baseline to endpoint. Ahmad 2016, Kakkar 2009, Tohen 2002, Vasudev 2000, and Wagner 2009 also focused on clinical response as defined by a 50% or greater decrease in YMRS score from baseline. By contrast, Bowden 1994, Bowden 2006, Hirschfeld 2010 and Zajecka 2002 defined their primary efficacy measure as the change in scores on the MRS. Geller 2012 used the Clinical Global Impression for Bipolar Illness ‐ Improvement Mania (CGI‐BP‐IM) as a primary outcome measure on which ratings of 1 or 2 (very much or much improved, respectively) counted for response. Lastly, Shafti 2008 assessed the primary efficacy measure using the changes in the Manic State Rating Scale (MSRS), measuring the frequency and intensity of manic episodes, as well as changes on the Clinical Global Impression Scale ‐ Severity of Illness (CGI‐S).
Thirteen studies did not report the primary tolerability outcome of the number of participants with any side effect from medication (DelBello 2006; Feifel 2011; Freeman 1992; Geller 2012; Hebrani 2009; Kowatch 2015; Mahmoudi‐Gharaei 2012; McElroy 1996; Pope 1991; Shafti 2008; Tohen 2002; Tohen 2008; Xu 2015).
Secondary outcomes Bowden 2006, Hirschfeld 2010 and Zajecka 2002 used the Manic Syndrome Scale (MSS) as a secondary outcome.
Four studies used the change in the Montgomery–Asberg Depression Rating Scale (MADRS) total score (Ahmad 2016; Feifel 2011; Tohen 2008; Young 2017). The Hamilton Depression Rating Scale (HDRS) was used by Tohen 2002 and Zajecka 2002, whilst McElroy 2010 used the Inventory of depressive symptoms (IDS). In addition, McElroy 2010 also used the Hamilton Anxiety Rating Scale (HARS).
Four studies included the CGI‐S score (Ahmad 2016; Feifel 2011; Wagner 2009; Zajecka 2002) as a secondary outcome. Three studies used the Global Assessment Scale (GAS) (Bowden 1994; Bowden 2006; Wagner 2009) and McElroy 2010 used the Global Assessment of Functioning (GAF) scale. One study (Hebrani 2009) used the Children Global Assessment Scale (CGAS). The Clinical Global Impression ‐ Bipolar Illness (CGI‐BP) scale was used by Tohen 2008 and Xu 2015, whereas the CGI‐I was used by Ahmad 2016, Feifel 2011 and Wagner 2009. DelBello 2006 used four additional scales, including the Clinical Global Impression‐Bipolar Disorder Version Improvement scores, the Clinical Global Impression‐Bipolar Disorder Version mania scores (CGI‐BP‐I mania), the Change from baseline to endpoint in the Positive and Negative Syndrome Scale, Positive Subscale (PANSS‐P) and the Childhood Depression Rating Scale‐Revised version (CDRS).
The Brief Psychiatric Rating Scale (BPRS) was used in two studies (Hirschfeld 2010; Zajecka 2002). In addition, Ahmad 2016 used the Columbia–Suicide Severity Rating Scale (C‐SSRS) score, Bowden 1994 used the Affective Disorder Rating Scale (ADRS), and Bowden 2006 used the Changes on the Behaviour Ideation Scale (BIS) and Depressive Syndrome Scale (DSS).
Feifel 2011 used the Extra Pyramidal Symptoms Rating Scale (ESRS) and Behavioural Activity Rating Scale (BARS), and Geller 2012 used the K‐SADS Mania Rating Scale (KMRS). Hirschfeld 2010 used the Brief Agitation Rating Scale as well as the Overt Aggression Scale. Three additional secondary outcomes were assessed by McElroy 1996, including changes in the global scores of the Scale for Assessment of Positive Symptoms (SAPS) and SAPS subscale scores, total dose of adjunctive lorazepam received per participant, and length of hospital stay. Wagner 2009 included the Children’s Depression Rating Scale‐Revised (CDRS‐R), ADHD Rating Scale IV, and the Caregiver Strain Questionnaire (CGSQ). Lastly, Zajecka 2002 used the Quality of Life Enjoyment and Satisfaction Questionnaire (Q‐LES‐Q), and both Hirschfeld 2010 and Zajecka 2002 used the Behavioural and Ideation Score.
Individual adverse events were reported in all studies, except Freeman 1992, Mahmoudi‐Gharaei 2012, McElroy 2010, Shafti 2008, and Young 2017. Study withdrawals were reported in every study, except for Kakkar 2009.
Timing of outcome assessment All studies were conducted over a relatively short period. McElroy 1996 presented the shortest trial, at only six days. Most trials lasted 21 days (Ahmad 2016; Bowden 1994; Bowden 2006; Feifel 2011; Freeman 1992; Hirschfeld 2010; Pope 1991; Shafti 2008; Tohen 2002; Tohen 2008), followed by 28 days (DelBello 2006; Vasudev 2000; Wagner 2009; Xu 2015).The duration of Hirschfeld 1999 was 10 days. The studies Kowatch 2015 and Mahmoudi‐Gharaei 2012 were conducted over six weeks. The study Moosavi 2014 was conducted over seven weeks. The duration of the studies Geller 2012, Hebrani 2009, and McElroy 2010 were eight weeks. Young 2017 was conducted over a total of nine weeks, but the authors chose to use the results after week three because participants with an inadequate response received open adjunctive risperidone at this point. The studies with the longest intervention period were Kakkar 2009 and Zajecka 2002, each lasting 12 weeks.
Excluded studies
See Characteristics of excluded studies
In the first search of the 126 full articles screened, we excluded 108 studies for several reasons, such as study design, reporting results already included in the review, ineligible publication types and no randomisation to valproate. In our second search of 29 full articles screened, we excluded one study for being a discontinuation trial. We have reported all of the studies excluded for methodological flaws (n = 20) in the Characteristics of excluded studies section. The studies excluded for being duplicates, ineligible publication type or post hoc analyses of data are not listed here, as the characteristics of these studies do not add anything to the review. No studies were excluded due to participants having active postpartum depression.
Three excluded studies are deserving of special explanation. Pavuluri and colleagues conducted two RCTs of adolescent bipolar disorder, comparing the effectiveness of valproate to risperidone. The two studies were Pavuluri 2010 (n = 66) and West 2011 (n = 24). We initially selected them for inclusion in the final list of studies to be analysed and extracted data. However, on clarification with the author of the two studies, it emerged that the method of randomisation used was pseudo‐random (participants were randomised consecutively as they came). This meant that these studies fulfilled the exclusion criteria as detailed in our protocol.
Walkup 2015 was an extension of the TEAM study (Geller 2012) that used a novel study design to interrogate the effectiveness of treatments in partial and non‐responders. They combined recruited participants who met their criteria with participants who had either completely or partially failed to respond to treatment in Geller 2012. This was done in a way that merged these two data sets. As we had already included Geller 2012 in our meta‐analysis, it would have biased the sample to include participants who had failed in one study in subsequent analyses. We, therefore, excluded Walkup 2015 from our analysis.
Studies awaiting classification:
See Characteristics of studies awaiting classification.
We identified seven studies that are awaiting classification (see above). For all of these studies, we were unable to obtain the full texts, despite having a clinical trials database link. Four of these studies have originally been published in Spanish, Chinese, and Russian (two studies), respectively. We will continue to contact the authors in order to locate (and translate) these articles for the next update of this review.
Ongoing studies:
See Characteristics of ongoing studies.
One study was still ongoing (NCT01893229) examining the efficacy of valproate compared to several other antimanic agents (Lithium, Oxcarbazepine, Quetiapine, Olanzapine, Ziprasidone)
Risk of bias in included studies
See Characteristics of included studies and Figure 2; Figure 3
2.
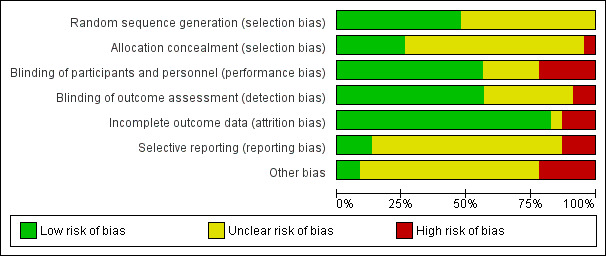
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.
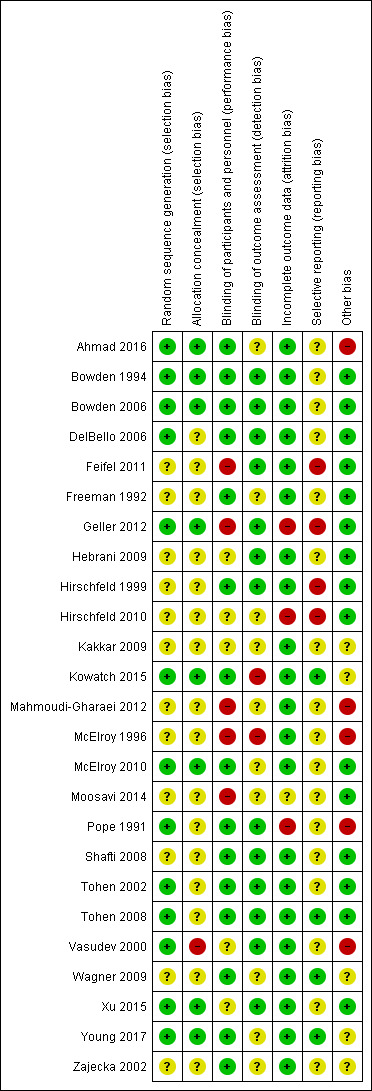
Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
The evaluation of risks of bias of included studies varied greatly, including a number of severe study violations and a plethora of studies where the risks of bias were unclear as a result of limited reported information. Two studies were at low risk for almost all domains, except reporting bias and other bias (Bowden 1994; Bowden 2006). DelBello 2006, Tohen 2002, Tohen 2008, and Xu 2015 did not provide sufficient information on selection bias, detection bias, reporting bias, and other biases respectively. We were largely unable to properly assess the risk of bias for Kakkar 2009, as the study report lacked most information necessary to make judgements on biases. In addition, Hirschfeld 2010, Mahmoudi‐Gharaei 2012; McElroy 1996, and Moosavi 2014 were characterised by a similar lack of important details on study characteristics but were also at high risk for different individual biases. For instance, Hirschfeld 2010 is characterised by dropout rates higher than 75%, resulting in a high risk of attrition bias.
Allocation
We were unable to judge the risk of bias for random sequence generation for most studies because they did not provide sufficient information (Feifel 2011; Freeman 1992; Hebrani 2009; Hirschfeld 1999; Hirschfeld 2010; Kakkar 2009; Mahmoudi‐Gharaei 2012; McElroy 1996Moosavi 2014; Shafti 2008; Wagner 2009; Zajecka 2002). We rated the remaining studies at low risk of bias because they used randomisation schedules generated by a computer (Ahmad 2016; Bowden 1994; Bowden 2006; DelBello 2006; Geller 2012; Kowatch 2015; McElroy 2010; Pope 1991; Tohen 2002; Tohen 2008; Vasudev 2000; Xu 2015; Young 2017), for instance according to a table of random numbers (Vasudev 2000; Xu 2015) or through permutated block randomisation (Young 2017). For one study (Bowden 2006) we judged the random sequence generation as low risk since previous trials published by the same authors (Bowden 1994) had adequate methods in place to generate random sequences.
In addition, most studies at low risk for random sequence generation were also considered low‐risk in allocation concealment, except for five studies. Specifically, four studies (DelBello 2006; Pope 1991; Tohen 2002; Tohen 2008) did not provide any information on allocation concealment and we were unable to judge the risk of bias. We estimated the risk of bias as high for one further study (Vasudev 2000) because medications were assigned to participants consecutively using an open random‐number table. This means that the next randomisation allocation is potentially predictable when deciding on a participant's eligibility for the trial.
Blinding
The risk of both performance bias (blinding of participants and personnel) and detection bias (blinding of outcome assessment) was low for a number of studies, including Bowden 1994, Bowden 2006, DelBello 2006, Hirschfeld 1999, Pope 1991, Shafti 2008, Tohen 2002, and Tohen 2008. Six studies (Ahmad 2016; Freeman 1992; McElroy 2010; Wagner 2009; Young 2017; Zajecka 2002) were at low risk for performance bias but did not provide sufficient information to draw conclusions about potential detection bias. Vasudev 2000 and Xu 2015 showed the reverse pattern, indicating low risk of detection bias but insufficient information to judge for performance bias. The risk of performance bias is high in Feifel 2011, because participants were not blind. Similarly, the risk of performance bias is high in Geller 2012 as participants, family members, and treating clinicians were aware of treatment assignment. We also considered Mahmoudi‐Gharaei 2012 and Moosavi 2014 to have high risks of performance bias because they describe their studies as 'single blind', which means that either participants or personnel were not blind. Lastly, McElroy 1996 shows high risk of performance bias because participants were not blind to treatment procedure, and high risk of detection bias as the higher rates of extrapyramidal side effects amongst participants in the haloperidol group are likely to have compromised the raters' blindness.
Incomplete outcome data
We decided to adopt the following approach to decide whether a study was at low or high risk of attrition bias:
If the dropout rate for all groups was less than 30%, the study was at low risk of attrition bias
If the dropout rate for the study as a whole was greater than 75%, the study was at high risk of attrition bias
If 1 and 2 did not apply, we considered a study to be at a high risk of attrition bias if one of the treatment groups had a dropout rate more than double that of another group, as this would indicate a significant difference in dropout rates between groups.
We rated most studies at low risk for attrition bias, except for Moosavi 2014, where the risk is unclear. In addition, Geller 2012 had a high risk of attrition bias because dropout rates were twice as high in the lithium group compared to the risperidone group. Lastly, the combined dropout rates in Hirschfeld 2010 and Pope 1991 were higher than 75%, which led us to rate these studies at high risk of bias.
Our review examines acute mania, a volatile condition which results in higher rates of dropout than might be expected for other conditions. (Yildiz 2011) We chose the level of 75% with these high dropout rates in mind. It is also important to note that the figure of 75% represents the limit for automatically classifying a study as high risk, rather than a permissive number below which a study is at low risk of attrition bias. At dropout rates of less than 75%, we checked the study for unbalanced dropout rates between groups, which would bias the samples. This acts as a double‐check of any risk of attrition bias.
Selective reporting
For most studies, the risk of reporting bias was unclear because we were unable to find a pre‐published protocol, and were therefore unable to assess the risk. Kowatch 2015, Tohen 2008, Wagner 2009 and Young 2017 were at low risk of reporting bias, as their pre‐published protocols outlined all reported measures. We considered Feifel 2011, Hirschfeld 2010 and Hirschfeld 1999 to be at high risk of bias: Feifel 2011 and Hirschfeld 2010 reported mean change in YMRS score from baseline to endpoint without standard deviations, and all other data were represented in graphical form only; Hirschfeld 1999 did not report means and standard deviations for all outcomes, which meant we were unable to conduct any analyses on the study. We estimated the standard deviations of Hirschfeld 2010, as discussed in our protocol. To do this, we followed the suggested tactic of using the most conservative standard deviation within similar studies. Similarly, Geller 2012 only reported adverse events for each group if they occurred above a certain frequency level. This meant some adverse events were reported for one of the trial groups but not for the others. This made meta‐analysis on large sections of the data impossible.
Other potential sources of bias
The majority of studies were free of other biases (Bowden 1994; Bowden 2006; DelBello 2006; Feifel 2011; Freeman 1992; Geller 2012; Hebrani 2009; Hirschfeld 1999; Hirschfeld 2010; McElroy 2010; Moosavi 2014; Shafti 2008; Tohen 2002; Tohen 2008; Xu 2015). Five studies (Ahmad 2016, Kakkar 2009; Kowatch 2015; Wagner 2009; Zajecka 2002) mentioned that participants received rescue medication (e.g. lorazepam) but did not elaborate on whether or not the medication was balanced between groups, not allowing us to draw conclusions on potential biases. We considered five studies as being at high risk for various other biases. First, the study Ahmad 2016 deployed a two‐stage design in which a higher endoxifen dose was trialled if a lower dose failed to show a 50% or greater improvement for YMRS scores. This is a biased approach and this trial should have been run in a parallel design. Additionally, baseline characteristics of groups are not reported, nor were any information on whether the concomitant medication was balanced between the two groups provided. Second, study groups in Mahmoudi‐Gharaei 2012 had significantly different baseline characteristics in some areas (i.e. more hospitalisations in one study arm). Third, McElroy 1996, Pope 1991 and Vasudev 2000 described unbalanced rescue medication between study arms. However, it is important to note that in Pope 1991 it is the placebo group that used more rescue medication and showed less recovery, indicating that this bias does not seem to be causing the result.
Effects of interventions
See: Table 1; Table 2; Table 3; Table 4; Table 5; Table 6
Because of the broad nature of the present meta‐analyses, we generated a large number of comparisons. However, the usefulness of the data for each comparison varied. Many comparisons for adults only contain one study and several studies did not report the specified primary outcomes.
'Summary of findings' tables
Valproate versus placebo ‐ for adults see Table 1; for children and adolescents see Table 2
Valproate versus carbamazepine ‐ for adults see Table 7
1. Valproate compared to carbamazepine for acute mania in adults.
| Valproate compared to carbamazepine for acute mania | ||||||
| Patient or population: Adults (aged 18 and over) with acute mania Setting: Inpatient Intervention: Valproate Comparison: Carbamazepine | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with carbamazepine | Risk with valproate | |||||
| Response rate at 4 weeks (primary efficacy outcome) | Study population | OR 2.41 (0.52 to 11.10) | 30 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | The evidence is very uncertain about the relative effects of valproate and carbamazepine on response rates. | |
| 533 per 1000 | 734 per 1000 (373 to 927) | |||||
| Number with any adverse event at 4 weeks (primary tolerability outcome) | Study population | OR 0.13 (0.02 to 0.82) | 30 (1 RCT) | ⊕⊕⊝⊝ LOWa,b,c | Valproate may cause fewer people to have side effects than carbamazapine, but the evidence is uncertain. | |
| 533 per 1000 | 129 per 1000 (22 to 484) | |||||
| Individual adverse events ‐ Lethargy At 4 weeks (secondary tolerability outcome) |
Study population | OR 0.14 (0.01 to 1.42) |
30 (1 RCT) | ⊕⊕⊝⊝ LOWa,b,c | Valproate may cause fewer people to have less lethargy than carbamazapine, but the evidence is uncertain. | |
| 333 per 1000 | 65 per 1000 (5‐415) |
|||||
| Change in symptom severity at 4 weeks (secondary efficacy outcome) | The mean change in symptom severity for carbamazepine at 4 weeks was ‐ 20.8. | MD ‐ 12.00 (‐ 21.82 to ‐ 2.18 ) | ‐ | 30 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | Valproate may cause a greater reduction in manic symptoms than carbamazapine, but the evidence is very uncertain. |
| Dropout rate ‐ All‐cause At 4 weeks (secondary acceptability outcome) |
Study population | OR 1.00 (0.17 to 5.98) | 30 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b,d | The evidence is very uncertain about the relative effects of valproate and carbamazapine on dropout rates. | |
| 200 per 1000 | 200 per 1000 (41 to 599) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; RCT: randomised controlled trial | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aEvidence downgraded by one level as > 30% of trials were at serious risk of bias. Vasudev 2000 at serious risk of bias, as at high risk of selection and other bias and at unclear risk of bias on performance and reporting biases. bEvidence downgraded by two levels for imprecision, due to single study and small study size. cEvidence upgraded by one level as large effect, RR < 0.5. dEvidence downgraded by one level for imprecision due to wide confidence interval ‐ Includes both no effect and OR of 4/0.25.
Valproate versus endoxifen ‐ for adults see Table 8
2. Valproate compared to endoxifen for acute mania in adults.
| Valproate compared to endoxifen for acute mania | ||||||
| Patient or population: Adults (aged 18 and over) with acute mania Setting: Inpatient Intervention: Valproate Comparison: Endoxifen | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with endoxifen | Risk with valproate | |||||
| Response rate at 3 weeks (primary efficacy outcome) | Study population | OR 2.19 (0.83 to 5.78) | 84 (1 RCT) | ⊕⊝⊝⊝ VERY LOW a,b,c | The evidence is very uncertain about the relative effects of valproate and endoxifen on response rates. | |
| 545 per 1000 | 724 per 1000 (499 to 874) | |||||
| Number of participants with any adverse event at 3 weeks (primary tolerability outcome) | Study population | OR 1.88 (0.73 to 4.86) | 84 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b,c | The evidence is very uncertain about the relative effects of valproate and endoxifen on causing adverse effects. | |
| 273 per 1000 | 413 per 1000 (215 to 646) | |||||
| Individual adverse events ‐ Nausea At 3 weeks |
Study population | OR 5.52 (1 to 30.5) |
84 (1 RCT) | ⊕⊕⊝⊝ LOWa,b,c,d | Valproate may cause more people to have less nausea than endoxifen, but the evidence is uncertain. | |
| 36 per 1000 | 172 per 1000 (36 to 535) |
|||||
| Change in symptom severity (secondary efficacy outcome) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Dropout rate – all‐cause (secondary acceptability outcome) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; RCT: randomised controlled trial | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aEvidence downgraded by two levels for imprecision, due to single study and small study size. bEvidence downgraded by one level as > 30% of trials at serious risk of bias. Ahmad 2016 at serious risk of bias, at high risk of other bias and at unclear risk of detection and reporting biases. cEvidence downgraded by one level for imprecision due to wide confidence interval ‐ Includes both no effect and OR of 4/0.25.
dEvidence upgraded by two levels as large effect, RR > 4.
Valproate versus haloperidol ‐ for adults see Table 9
3. Valproate compared to haloperidol for acute mania in adults.
| Valproate compared to haloperidol for acute mania | ||||||
| Patient or population: Adults (aged 18 and over) with acute mania Setting: Inpatient Intervention: Valproate Comparison: Haloperidol | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with haloperidol | Risk with valproate | |||||
| Response rate at 1 week (primary efficacy outcome) |
Study population | OR 1.82 (0.46 to 7.18) | 36 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b,c | The evidence is very uncertain about the relative effects of valproate and haloperidol on response rates. | |
| 333 per 1000 | 476 per 1000 (187 to 782) | |||||
| Number of participants with any adverse event (primary tolerability outcome) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Individual adverse events ‐ Extra‐pyramidal side effects At 1 week |
Study population | OR 0.02 (0.00 to 0.40) | 36 (1 RCT) | ⊕⊕⊝⊝ LOWa,b,d | Valproate may cause fewer individuals to have extra‐pyramidal side effects compared to haloperidol, but the evidence is uncertain. | |
| 533 per 1000 | 22 per 1000 (0 to 314) | |||||
| Individual adverse events ‐ Sedation At 1 week |
Study population | OR 0.14 (0.01 to 1.39) | 36 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b,c | The evidence is very uncertain about the relative effects of valproate and haloperidol on sedation. | |
| 267 per 1000 | 48 per 1000 (4 to 336) | |||||
| Change in symptom severity at 1 week (secondary efficacy outcome) |
The mean in symptom severity for haloperidol at 1 week was 24.3. | MD ‐ 3.60 (‐ 11.48 to 4.28) | ‐ | 36 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | The evidence is very uncertain about the relative effects of valproate and haloperidol on reducing manic symptoms. |
| Dropout rate – all‐cause (secondary acceptability outcome) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; RCT: randomised controlled trial | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aEvidence downgraded by two levels as > 30% of studies at very serious risk of bias. McElroy 1996 was at very serious risk of bias as it was at high risk of performance, detection and other biases, while the rest of the assessed biases were at uncertain risk of bias. bEvidence downgraded by two levels for imprecision, due to single study and small study size. cEvidence downgraded by one level for imprecision, due to wide confidence interval ‐ Includes both no effect and OR of 4/0.25. dEvidence upgraded by two levels as very large effect, RR < 0.25
Valproate versus. lithium ‐ for adults see Table 3; for children and adolescents see Table 4
Valproate versus olanzapine ‐ for adults see Table 5
Valproate add‐on versusolanzapine alone ‐ for adults see Table 10
4. Valproate + olanzapine compared to olanzapine alone for acute mania in adults.
| Valproate + olanzapine compared to olanzapine alone for acute mania | ||||||
| Patient or population: Adults (aged 18 and over) with acute mania Setting: Inpatient Intervention: Valproate and olanzapine Comparison: Olanzapine alone | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with olanzapine alone | Risk with valproate + olanzapine | |||||
| Response rate (primary efficacy outcome) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Number of participants with any adverse event (primary tolerability outcome) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Individual adverse events ‐ Somnolence At 3 weeks |
Study population | OR 0.79 (0.30 to 2.06) | 80 (1 RCT) | ⊕⊕⊝⊝ LOWa | The evidence is uncertain about the relative effects of valproate with olanzapine compared to olanzapine alone on somnolence. | |
| 325 per 1,000 | 276 per 1,000 (126 to 498) | |||||
| Individual adverse events ‐ Weight gain At 3 weeks |
Study population | OR 1.31 (0.47 to 3.61) | 80 (1 RCT) | ⊕⊕⊝⊝ LOWa | The evidence is uncertain about the relative effects of valproate with olanzapine compared to olanzapine alone on weight gain. | |
| 725 per 1000 | 775 per 1000 (553 to 905) | |||||
| Change in symptom severity at 3 weeks (secondary efficacy outcome) |
The mean change in symptom severity for olanzapine alone at 3 weeks was ‐ 20.74. | MD ‐ 2.76 (‐ 9.17 to 3.65) | ‐ | 76 (1 RCT) | ⊕⊕⊝⊝ LOWa,b | The evidence is uncertain about the relative effects of valproate with olanzapine compared to olanzapine alone on decreasing manic symptoms. |
| Dropout ‐ All‐cause At 3 weeks (secondary acceptability outcome) |
Study population | OR 2.05 (0.18 to 23.59) | 80 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | The evidence is very uncertain about the relative effects of valproate with olanzapine compared to olanzapine alone on dropout rates. | |
| 25 per 1000 | 50 per 1000 (5 to 377) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; RCT: randomised controlled trial | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aEvidence downgraded by two levels for imprecision, due to single study and small study size. bEvidence downgraded by one level for imprecision, due to wide confidence interval; OR includes both 4 and 0.25
Valproate versus oxcarbazepine ‐ for adults see Table 11
5. Valproate compared to oxcarbazepine for acute mania in adults.
| Valproate compared to oxcarbazepine for acute mania | ||||||
| Patient or population: Adults (aged 18 and over) with acute mania Setting: Inpatient Intervention: Valproate Comparison: Oxcarbazepine | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with oxcarbazepine | Risk with valproate | |||||
| Response rate (primary efficacy outcome) | ‐ | ‐ | ‐ | ‐ | ‐ | Not measured. |
| Number of participants with any adverse event at 3 weeks (primary tolerability outcome) | Study population | OR 4.67 (1.57 to 13.87) | 60 (1 RCT) | ⊕⊕⊝⊝ LOWa,b,d | Oxcarbazepine may cause fewer side effects than valproate, but the evidence is uncertain. | |
| 300 per 1000 | 667 per 1000 (402 to 856) | |||||
| Individual adverse events ‐ Nausea At 3 weeks |
Study population | OR 1.80 (0.39 to 8.32) |
60 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | The evidence is very uncertain about the relative effects of valproate compared to oxcarbazepine on sedation. | |
| 100 per 1000 | 167 per 1000 (42 to 480) |
|||||
| Change in symptom severity at 3 weeks (secondary efficacy outcome) | The mean in symptom severity for oxcarbazepine at 3 weeks was 23.9. | MD 0.73 (‐ 2.17 to 3.63 ) | ‐ | 60 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | The evidence is very uncertain about the relative effects of valproate and oxcarbazepine on reducing manic symptoms at 3 weeks. |
| Dropout ‐ All‐cause (secondary acceptability outcome) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; RCT: randomised controlled trial | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aEvidence downgraded by one level as > 30% of trials at serious risk of bias. Kakkar 2009 was at serious risk of bias, as selection, detection, performance, reporting and other biases were all at unclear risk of bias. bEvidence downgraded by two levels for imprecision, due to single study and small study size. cEvidence downgraded by one level for imprecision, due to wide confidence interval ‐ Includes both no effect and OR of 4/0.25. dEvidence upgraded by one level due to large effect ‐ RR > 2.
Valproate versus quetiapine ‐ for adults see Table 12; for children and adolescents see Table 13
6. Valproate compared to quetiapine for acute mania in adults.
| Valproate compared to quetiapine for acute mania | ||||||
| Patient or population: Adults (aged 18 and over) with acute mania Setting: Inpatient Intervention: Valproate Comparison: Quetiapine | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with quetiapine | Risk with valproate | |||||
| Response rate (primary efficacy outcome) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Number of participants with any adverse event (primary tolerability outcome) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Change in symptom severity score (secondary efficacy outcome) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Dropout rate ‐ All‐cause At 3 weeks (secondary acceptability outcome) |
Study population | OR 1.73 (0.31 to 9.57) | 30 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b,c | The evidence is very uncertain about the relative effects of valproate and quetiapine on dropout rate. | |
| 188 per 1000 | 285 per 1000 (67 to 688) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; RCT: randomised controlled trial | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aEvidence downgraded by one level for > 30% of studies at serious risk of bias. Feifel 2011 was at serious risk of bias, as performance and reporting were both at high risk of bias and selection and other biases were at unclear risk of bias. bEvidence downgraded by two levels for imprecision, due to single study and small study size. cEvidence downgraded by one level for imprecision, due to wide confidence interval ‐ Includes both no effect and OR of 4/0.25.
7. Valproate compared to quetiapine for acute mania in children and adolescents.
| Valproate compared to quetiapine for acute mania | ||||||
| Patient or population: Children and adolescents with acute mania Setting: Inpatient Intervention: Valproate Comparison: Quetiapine | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with quetiapine | Risk with valproate | |||||
| Response rate (primary efficacy outcome) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Number of participants with any adverse event (primary tolerability outcome) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Individual adverse events ‐ Increased appetite At 4 weeks |
Study population | OR 1.57 (0.24 to 10.30) | 50 (1 RCT) | ⊕⊕⊝⊝ LOWa,c | The evidence is uncertain about the relative effects of valproate and quetiapine on increasing appetite. | |
| 80 per 1000 | 120 per 1000 (20 to 472) | |||||
| Individual adverse events ‐ Sedation or lethargy At 4 weeks |
Study population | OR 0.38 (0.12 to 1.18) | 50 (1 RCT) | ⊕⊕⊝⊝ LOWa,c | The evidence is uncertain about the relative effects of valproate and quetiapine on sedation. | |
| 600 per 1000 | 363 per 1000 (153 to 639) | |||||
| Change in symptom severity at 4 weeks (secondary efficacy outcome) |
The mean change in symptom severity for quetiapine at 4 weeks was ‐23. | MD 4.00 (‐ 2.10 to 10.10) | ‐ | 50 (1 RCT) | ⊕⊕⊝⊝ LOWa | The evidence is uncertain about the relative effects of valproate and quetiapine on decreasing manic symptoms. |
| Dropout rate ‐ All‐cause at 4 weeks (secondary acceptability outcome) | Study population | OR 1.00 (0.27 to 3.66) | 50 (1 RCT) | ⊕⊕⊝⊝ LOWa | The evidence is uncertain about the relative effects of valproate and quetiapine on dropout rates. | |
| 240 per 1000 | 240 per 1000 (79 to 536) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; RCT: randomised controlled trial | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aEvidence downgraded by two levels for imprecision, due to single study and small study size. bEvidence upgraded by one level for large effect, RR < 0.5. cEvidence downgraded by one level for imprecision, due to wide confidence interval; OR includes both 4 and 0.25.
Valproate versus risperidone ‐ for children and adolescents see Table 6
Valproate add‐on versus risperidone alone ‐ for adults see Table 14
8. Valproate + risperidone compared to risperidone alone for acute mania in adults.
| Valproate + risperidone compared to risperidone alone for acute mania | ||||||
| Patient or population: Adults (aged 18 and over) with acute mania Setting: Inpatient Intervention: Valproate and risperidone Comparison: Risperidone alone | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with risperidone alone | Risk with valproate + risperidone | |||||
| Response rate (primary efficacy outcome) |
‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Number of participants with any adverse event at 7 weeks (primary tolerability outcome) | Study population | OR 0.93 (0.27 to 3.16) | 48 (1 RCT) | ⊕⊝⊝⊝ VERY LOWa,b | The evidence is very uncertain about the relative effects of risperidone and valproate compared to risperidone alone on total number with any adverse event. | |
| 320 per 1000 | 304 per 1000 (113 to 598) | |||||
| Change in symptom severity (secondary efficacy outcome) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Dropout rate (secondary acceptability outcome) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; RCT: randomised controlled trial | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aEvidence downgraded by one level as > 30% of studies at serious risk of bias. Moosavi 2014 at serious risk of bias, as performance bias at high risk of bias and all other biases assessed were at unclear risk of bias except other biases. bEvidence downgraded by two levels for imprecision, due to single study and small study size. cEvidence downgraded by one level for imprecision, due to wide confidence interval; OR includes both 4 and 0.25.
Valproate versus topiramate ‐ for adults see Table 15
9. Valproate compared to topiramate for acute mania in children and adolescents.
| Valproate compared to Topiramate for acute mania | ||||||
| Patient or population: Children and adolescents with acute mania Setting: Inpatient Intervention: Valproate Comparison: Topiramate | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Risk with topiramate | Risk with valproate | |||||
| Response rates (primary efficacy outcome) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Number of participants with any adverse event (primary tolerability outcome) | ‐ | ‐ | ‐ | ‐ | ‐ | Not reported |
| Individual adverse events ‐ Drowsiness At 12 weeks |
Study population | OR 0.34 (0.15 to 0.79) |
142 (1 RCT) |
⊕⊝⊝⊝ VERY LOWb,d | The evidence is very uncertain about the relative effects of valproate and topiramate on drowsiness | |
| 324 per 1000 | 140 per 1000 (67 to 275) |
|||||
| Change in symptom severity at endpoint at 5 ‐ 12 weeks (secondary efficacy measures) |
SMD ‐ 0.41 (‐ 1.16 to 0.35 higher) |
‐ | 149 (2 RCTs) |
⊕⊝⊝⊝ VERY LOWa,b,c | The evidence is very uncertain about the relative effects of valproate and topiramate on reducing manic symptoms. Based on Cohen's effect sizes, a standard deviation of 0.4 represents a small to moderate difference between groups. | |
| Dropout rates ‐ All cause At 6 weeks (secondary acceptability outcome) |
Study population | OR 0.31 (0.01 to 8.28) | 30 (1 RCT) | ⊕⊝⊝⊝ VERY LOWb,d,e | The evidence is very uncertain about the relative effects of valproate and topiramate on dropout rates. | |
| 67 per 1000 | 22 per 1000 (1 to 372) | |||||
| *The risk in the intervention group (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio; RCT: randomised controlled trial | ||||||
| GRADE Working Group grades of evidence High quality: We are very confident that the true effect lies close to that of the estimate of the effect Moderate quality: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low quality: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low quality: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
aEvidence downgraded by two levels for very significant heterogeneity (I2 > 75%). bEvidence downgraded by one level as > 30% of studies at high risk of bias (Hebrani 2009; Mahmoudi‐Gharaei 2012). cEvidence downgraded by one level for imprecision, due to small total study sizes. dEvidence downgraded by two levels for imprecision, due to single study and small study size. eEvidence downgraded by one level for imprecision, due to wide confidence interval; OR includes both 4 and 0.25.
Comparison 1: Valproate versus placebo
Eight studies contributed to this comparison (Bowden 1994; Bowden 2006; Hirschfeld 2010; Kowatch 2015; McElroy 2010; Pope 1991; Tohen 2008; Wagner 2009), with two of them aiming to investigate the effects of valproate in children and adolescents (Kowatch 2015; Wagner 2009).
Primary outcomes
1.1. Efficacy (dichotomous): number of participants experiencing a 50% or greater reduction in mean score on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: adults
There was high‐quality evidence for an effect of valproate on the response rate at three weeks, although the magnitude of this effect is uncertain: odds ratio (OR) 2.05, 95% confidence interval (CI) 1.32 to 3.20; P = 0.001, I2 = 46%; 4 studies, 869 participants. In addition, low‐quality evidence did not find strong evidence for a difference in the response rate between the two groups at eight weeks (OR 1.50, 95% CI 0.54 to 4.15; P = 0.44; 1 study, 62 participants). See Analysis 1.1.
1.1. Analysis.
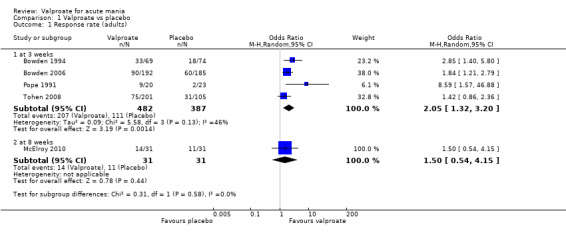
Comparison 1 Valproate vs placebo, Outcome 1 Response rate (adults).
1.2. Efficacy (dichotomous): number of participants experiencing a 50% or greater reduction in mean score on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: children and adolescents
Very low‐quality evidence found that there was not strong evidence for a difference in the response rate between the two groups at four weeks: OR 1.11, 95% CI 0.51 to 2.38; P = 0.80; 1 study, 151 participants. See Analysis 1.2.
1.2. Analysis.
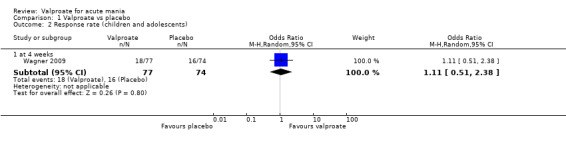
Comparison 1 Valproate vs placebo, Outcome 2 Response rate (children and adolescents).
1.3. Tolerability (dichotomous): number with any adverse event
1.3.1. Adults
There was moderate‐quality evidence for an effect of valproate on experiencing any adverse events, although the magnitude of this effect is uncertain: OR 1.63, 95% CI 1.13 to 2.36; P = 0.009, I2 = 0%; 3 studies, 745 participants. See Analysis 1.3.1.
1.3. Analysis.
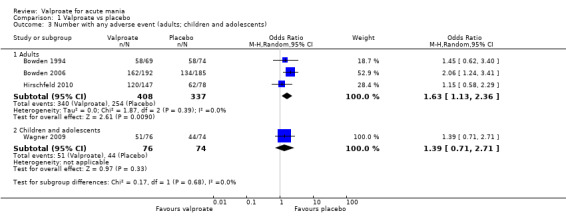
Comparison 1 Valproate vs placebo, Outcome 3 Number with any adverse event (adults; children and adolescents).
1.3.2. Children and adolescents
Very low‐quality evidence found that there was not strong evidence for a difference in experiencing any adverse events between the two groups: OR 1.39, 95% CI 0.71 to 2.71; P = 0.33; 1 study, 150 participants. See Analysis 1.3.2.
Secondary outcomes
1.4. Tolerability (dichotomous): individual adverse events: adults
There was evidence that more participants in the valproate group experienced adverse events, such as abdominal pain (OR 2.62, 95% CI 1.18 to 5.82; P = 0.02, I2 = 0%; 2 studies, 439 participants; Analysis 1.4.1), dizziness (OR 2.76, 95% CI 1.57 to 4.85; P < 0.001, I2 = 0%; 2 studies, 520 participants; Analysis 1.4.13), dyspepsia (OR 2.17, 95% CI 1.10 to 4.28; P = 0.02, I2 = 35%; 3 studies, 664 participants; Analysis 1.4.17), nausea (OR 2.00, 95% CI 1.38 to 2.90; P < 0.001, I2 = 0%; 5 studies, 931 participants; Analysis 1.4.25), upper respiratory chest infection (OR 3.24, 95% CI 1.30 to 8.09; P = 0.01, I2 = 20%; 2 studies, 439 participants; Analysis 1.4.35) and vomiting (OR 3.18, 95% CI 1.77 to 5.70; P < 0.001, I2 = 0%; 4 studies, 625 participants; Analysis 1.4.36), although the magnitude of these effects is uncertain. See Analysis 1.4 and Table 16 for a complete overview of all adverse events.
1.4. Analysis.
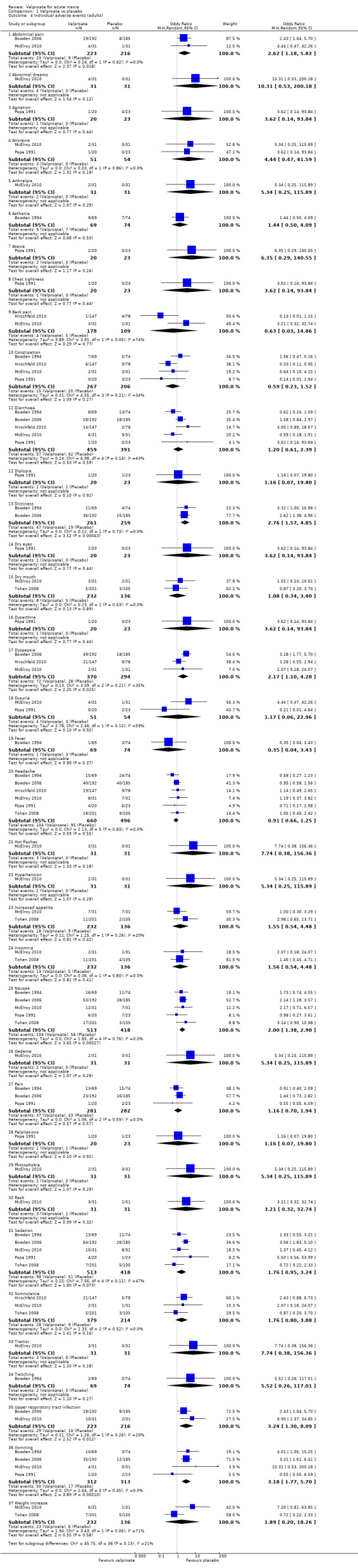
Comparison 1 Valproate vs placebo, Outcome 4 Individual adverse events (adults).
10. Adverse events ‐ valproate vs placebo (adults).
| Adverse effect | Odds ratio; 95% CI; heterogeneity; N of studies, N of participants | P value |
| Abdominal pain | OR 2.62, 95% CI 1.18 to 5.82; I2 = 0%; 2 studies, 439 participants | P = 0.02 |
| Abnormal dreams | OR 10.31, 95% CI 0.53 to 200.18; 1 study, 62 participants | P = 0.12 |
| Agitation | OR 3.62, 95% CI 0.14 to 93.84; 1 study, 43 participants | P = 0.44 |
| Anorexia | OR 4.44, 95% CI 0.47 to 41.59; I2 = 0%; 2 studies, 105 participants | P = 0.19 |
| Arthralgia | OR 5.34, 95% CI 0.25 to 115.89; 1 study, 62 participants | P = 0.29 |
| Asthenia | OR 1.44, 95% CI 0.50 to 4.09; 1 study, 143 participants | P = 0.50 |
| Ataxia | OR 6.35, 95% CI 0.29 to 140.55; 1 study, 43 participants | P = 0.24 |
| Chest tightness | OR 3.62, 95% CI 0.14 to 93.84; 1 study, 43 participants | P = 0.44 |
| Back pain | OR 0.63, 95% CI 0.03 to 14.86; I2 = 74%; 2 studies, 287 participants | P = 0.77 |
| Constipation | OR 0.59, 95% CI 0.23 to 1.52; I2 = 34%; 4 studies, 473 participants | P = 0.27 |
| Diarrhoea | OR 1.20, 95% CI 0.61 to 2.39; I2 = 43%; 5 studies, 850 participants | P = 0.59 |
| Diplopia | OR 1.16, 95% CI 0.07 to 19.80; 1 study, 43 participants | P = 0.92 |
| Dizziness | OR 2.76, 95% CI 1.57 to 4.85; I2 = 0%; 2 studies, 520 participants | P < 0.001 |
| Dry eyes | OR 3.62, 95% CI 0.14 to 93.84; 1 study, 43 participants | P = 0.44 |
| Dry mouth | OR 1.08, 95% CI 0.34 to 3.40; I2 = 0%; 2 studies, 368 participants | P = 0.89 |
| Dysarthria | OR 3.62, 95% CI 0.14 to 93.84; 1 study, 43 participants | P = 0.44 |
| Dyspepsia | OR 2.17, 95% CI 1.10 to 4.28; I2 = 35%; 3 studies, 664 participants | P = 0.02 |
| Dysuria | OR 1.17, 95% CI 0.06 to 22.96; I2 = 59%; 2 studies, 105 participants | P = 0.92 |
| Fever | OR 0.35, 95% CI 0.04 to 3.43; 1 study, 143 participants | P = 0.37 |
| Headache | OR 0.91, 95% CI 0.66 to 1.25; I2 = 0%; 6 studies, 1156 participants | P = 0.55 |
| Hot flashes | OR 7.74, 95% CI 0.38 to 156.36; 1 study, 62 participants | P = 0.18 |
| Hypertension | OR 5.34, 95% CI 0.25 to 115.89; 1 study, 62 participants | P = 0.29 |
| Increased appetite | OR 1.55, 95% CI 0.54 to 4.48; I2 = 20%; 2 studies, 368 participants | P = 0.42 |
| Insomnia | OR 1.56, 95% CI 0.54 to 4.48; I2 = 0%; 2 studies, 368 participants | P = 0.41 |
| Nausea | OR 2.00, 95% CI 1.38 to 2.90; I2 = 0%; 5 studies, 931 participants | P < 0.001 |
| Oedema | OR 5.34, 95% CI 0.25 to 115.89; 1 study, 62 participants | P = 0.29 |
| Pain | OR 1.16, 95% CI 0.70 to 1.94; I2 = 0%, 3 studies, 563 participants | P = 0.57 |
| Palpitations | OR 1.16, 95% CI 0.07 to 19.80; 1 study, 43 participants | P = 0.92 |
| Photophobia | OR 5.34, 95% CI 0.25 to 115.89; 1 study, 62 participants | P = 0.29 |
| Rash | OR 3.21, 95% CI 0.32 to 32.74; 1 study, 62 participants | P = 0.32 |
| Sedation | OR 1.76, 95% CI 0.95 to 3.24; I2 = 47%; 5 studies, 931 participants | P = 0.07 |
| Somnolence | OR 1.76, 95% CI 0.80 to 3.88; I2 = 0%; 3 studies, 593 participants | P = 0.16 |
| Tremor | OR 7.74, 95% CI 0.38 to 156.36; 1 study, 62 participants | P = 0.18 |
| Twitching | OR 5.52, 95% CI 0.26 to 117.01; 1 study, 143 participants | P = 0.27 |
| Upper respiratory tract infection | OR 3.24, 95% CI 1.30 to 8.09; I2 = 20%; 2 studies, 439 participants | P = 0.01 |
| Vomiting | OR 3.18, 95% CI 1.77 to 5.70; I2 = 0%; 4 studies, 625 participants | P < 0.001 |
| Weight increase | OR 1.89, 95% CI 0.20 to 18.26; I2 = 71%; 2 studies, 368 participants | P = 0.58 |
1.5. Tolerability (dichotomous): individual adverse events: children and adolescents
There was evidence that fewer participants in the valproate group experienced difficulties concentrating compared to participants in the placebo group: OR 0.01, 95% CI 0.00 to 0.15; P = 0.002, I2 = N/A; 1 study, 28 participants; Analysis 1.5.4. See Analysis 1.5 and Table 17 for a complete overview of all adverse events.
1.5. Analysis.
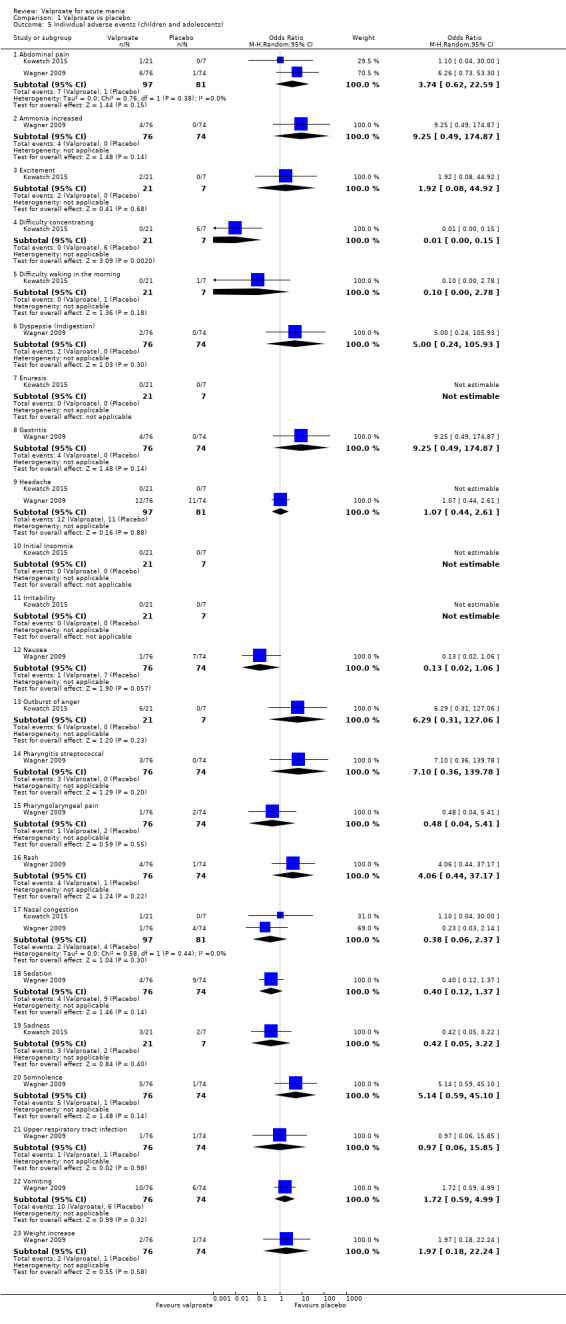
Comparison 1 Valproate vs placebo, Outcome 5 Individual adverse events (children and adolescents).
11. Adverse events ‐ valproate vs placebo (children and adolescents).
| Adverse effect | Odds ratio; 95% CI; heterogeneity; N of studies, N of participants | P value |
| Abdominal pain | OR 3.74, 95% CI 0.62 to 22.59; I2 = 0%; 2 studies, 178 participants | P = 0.15 |
| Ammonia increased | OR 9.25, 95% CI 0.49 to 174.87; 1 study, 150 participants | P = 0.14 |
| Excitement | OR 1.92, 95% CI 0.08 to 44.92; 1 study, 28 participants | P = 0.68 |
| Difficulty concentrating | OR 0.01, 95% CI 0.00 to 0.15; 1 study, 28 participants | P = 0.002 |
| Difficulty waking in the morning | OR 0.10, 95% CI 0.00 to 2.78; 1 study, 28 participants | P = 0.18 |
| Dyspepsia (Indigestion) | OR 5.00, 95% CI 0.24 to 105.93; 1 study, 150 participants | P = 0.30 |
| Enuresis | Not estimable | N/A |
| Gastritis | OR 9.25, 95% CI 0.49 to 174.87; 1 study, 150 participants | P = 0.14 |
| Headache | OR 1.07, 95% CI 0.44 to 2.61; I2 = N/A; 2 studies, 178 participants | P = 0.88 |
| Initial insomnia | Not estimable | N/A |
| Irritability | Not estimable | N/A |
| Nausea | OR 0.13, 95% CI 0.02 to 1.06; 1 study, 150 participants | P = 0.06 |
| Outburst of anger | OR 6.29, 95% CI 0.31 to 127.06; 1 study, 28 participants | P = 0.23 |
| Pharyngitis streptococcal | OR 7.10, 95% CI 0.36 to 139.78; 1 study, 150 participants | P = 0.20 |
| Pharyngolaryngeal pain | OR 0.48, 95% CI 0.04 to 5.41; 1 study, 150 participants | P = 0.55 |
| Rash | OR 4.06, 95% CI 0.44 to 37.17; 1 study, 150 participants | P = 0.22 |
| Nasal congestion | OR 0.38, 95% CI 0.06 to 2.37; I2 = 0%; 2 studies, 178 participants | P = 0.30 |
| Sedation | OR 0.40, 95% CI 0.12 to 1.37; 1 study, 150 participants | P = 0.14 |
| Sadness | OR 0.42, 95% CI 0.05 to 3.22; 1 study, 28 participants | P = 0.40 |
| Somnolence | OR 5.14, 95% CI 0.59 to 45.10; 1 study, 150 participants | P = 0.14 |
| Upper respiratory tract infection | OR 0.97, 95% CI 0.06 to 15.85; 1 study, 150 participants | P = 0.98 |
| Vomiting | OR 1.72, 95% CI 0.59 to 4.99; 1 study, 150 participants | P = 0.32 |
| Weight increase | OR 1.97, 95% CI 0.18 to 22.24; 1 study, 150 participants | P = 0.58 |
1.6. Remission (dichotomous): a score of 12 or less on the YMRS (or equivalent on other validated mania rating scales): adults
There was evidence for an effect of valproate on remission at three weeks, although the magnitude of this effect is uncertain: OR 1.61, 95% CI 1.17 to 2.22; P = 0.004, I2 = 0%; 2 studies, 683 participants. See Analysis 1.6.
1.6. Analysis.
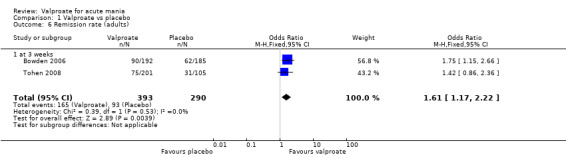
Comparison 1 Valproate vs placebo, Outcome 6 Remission rate (adults).
1.7. Remission (dichotomous): a score of 12 or less on the YMRS (or equivalent on other validated mania rating scales): children and adolescents
There was not strong evidence for a difference in remission rate between the two groups at four weeks: OR 0.87, 95% CI 0.37 to 2.04; P = 0.74; 1 study, 151 participants. See Analysis 1.7.
1.7. Analysis.
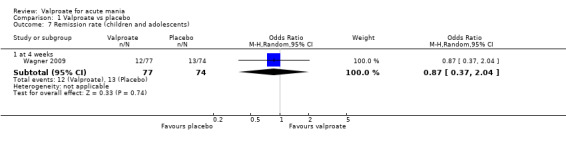
Comparison 1 Valproate vs placebo, Outcome 7 Remission rate (children and adolescents).
1.8. Efficacy (continuous): we assessed the efficacy of valproate by assessing the change in symptom severity, using mean endpoint scores or mean change scores on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: adults
There was moderate‐quality evidence for an effect of valproate on the reduction in mania rating scores at three weeks: standardised mean difference (SMD) −0.23, 95% CI −0.45 to −0.00; P = 0.05, I2 = 57%; 4 studies, 907 participants. See Analysis 1.8. This measure contains a large amount of heterogeneity, with an I2 of 57%. Visual examination of the forest plot shows that this is mainly contributed by Pope 1991. Pope 1991 is a small study with only 29 participants, conducted in 1991 and at significant risk of bias, and a clear outlier from the other studies. Removing the results from Pope 1991 reduces I2 to 0%, suggesting that this study alone is the source of the heterogeneity.
1.8. Analysis.
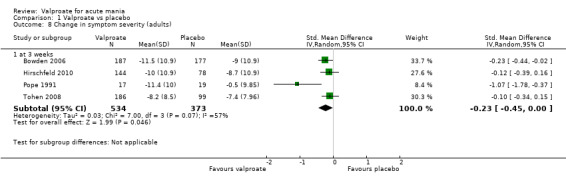
Comparison 1 Valproate vs placebo, Outcome 8 Change in symptom severity (adults).
1.9. Efficacy (continuous): we assessed the efficacy of valproate by assessing the change in symptom severity, using mean endpoint scores or mean change scores on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: children and adolescents
Very low‐quality evidence found that there was not strong evidence for a difference in mania rating scale scores between the two groups at four weeks: SMD −0.09, 95% CI −0.41 to 0.24; P = 0.60; 1 study, 144 participants. In addition, very low‐quality evidence found that there was not strong evidence of a difference in mania rating scale scores between the two groups at six weeks: SMD −0.51, 95% CI −1.38 to 0.36; P = 0.25; 1 study, 28 participants. See Analysis 1.9.
1.9. Analysis.
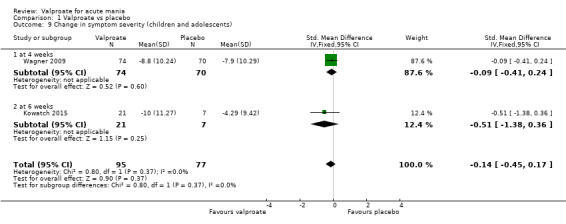
Comparison 1 Valproate vs placebo, Outcome 9 Change in symptom severity (children and adolescents).
1.10. Acceptability (dichotomous): information on dropouts due to 1. side effects 2. lack of efficacy 3. any other reasons 4. all causes, during the trial as a proportion of the total number of randomised participants: adults
There was evidence for an effect of valproate on dropouts due to adverse events: OR 2.59, 95% CI 1.33 to 5.05; P = 0.005, I2 = 0%; 5 studies, 931 participants, although the magnitude of this effect is uncertain. In addition, there was evidence for an effect of placebo on dropouts due to inefficacy: OR 0.50, 95% CI 0.36 to 0.69; P < 0.001, I2 = 0%; 6 studies, 1156 participants. There was not strong evidence for a difference in dropouts due to other reasons (OR 1.18, 95% CI 0.83 to 1.66; P = 0.35, I2 = 22%; 6 studies, 1156 participants) or dropouts due to all causes (OR 0.83, 95% CI 0.64 to 1.07; P = 0.16, I2 = 0%; 6 studies, 1156 participants) between the two groups. See Analysis 1.10.
1.10. Analysis.
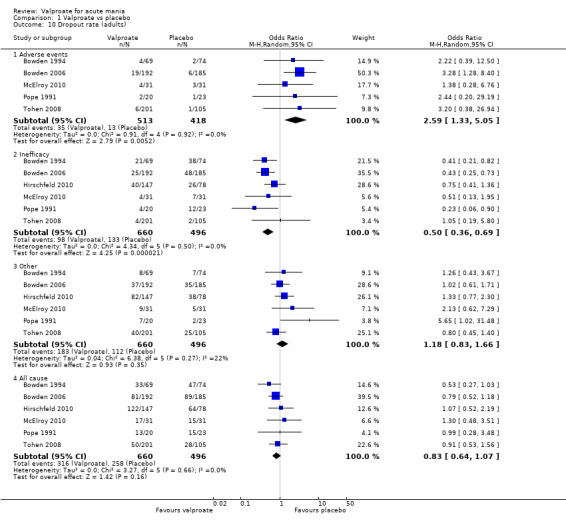
Comparison 1 Valproate vs placebo, Outcome 10 Dropout rate (adults).
1.11. Acceptability (dichotomous): information on dropouts due to 1. side effects 2. lack of efficacy 3. any other reasons 4. all causes, during the trial as a proportion of the total number of randomised participants: children and adolescents
There was not strong evidence for a difference in dropouts due to adverse events (OR 1.26, 95% CI 0.31 to 5.06; P = 0.75, I2 = 0%; 2 studies, 179 participants), dropouts due to inefficacy (OR 1.53, 95% CI 0.51 to 4.61; P = 0.45, I2 = 0%; 2 studies, 179 participants), dropouts due to other reasons (OR 1.44, 95% CI 0.51 to 4.08; P = 0.50, I2 = 0%; 2 studies, 179 participants), or dropouts due to all causes (OR 1.77, CI 0.83 to 3.78; P = 0.14, I2 = 0%; 2 studies, 179 participants) between the two groups. See Analysis 1.11.
1.11. Analysis.
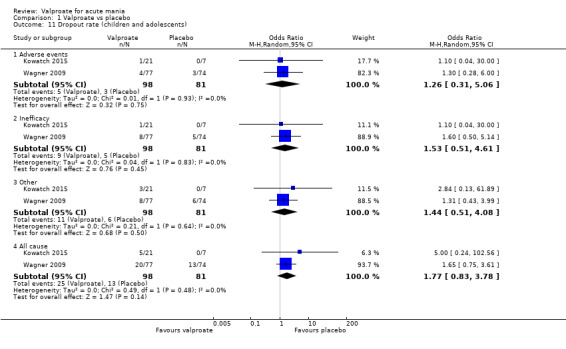
Comparison 1 Valproate vs placebo, Outcome 11 Dropout rate (children and adolescents).
1.12. Global functioning: the proportion of participants who improved based on the final scores of 1 ‐ 2 on the CGI‐I or CGI‐BP‐I: children and adolescents
There was not strong evidence for a difference in global functioning at four weeks (OR 0.83, 95% CI 0.42 to 1.66; P = 0.61; 1 study, 151 participants), nor at six weeks (OR 13.70, 95% CI 0.69 to 270.30; P = 0.09; 1 study, 28 participants) between the two groups. See Analysis 1.12.
1.12. Analysis.
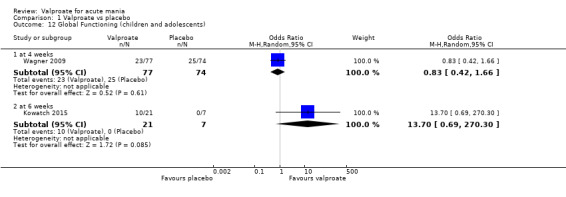
Comparison 1 Valproate vs placebo, Outcome 12 Global Functioning (children and adolescents).
Comparison 2: Valproate versus carbamazepine
One study contributed to this comparison (Vasudev 2000).
Primary outcomes
2.1. Efficacy (dichotomous): number of participants experiencing a 50% or greater reduction in mean score on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: adults
Very low‐quality evidence found that there was not strong evidence for a difference in response rates between the two groups at four weeks: OR 2.41, 95% CI 0.52 to 11.10; P = 0.26; 1 study, 30 participants. See Analysis 2.1.
2.1. Analysis.
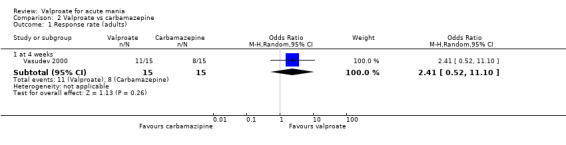
Comparison 2 Valproate vs carbamazepine, Outcome 1 Response rate (adults).
2.2. Tolerability (dichotomous): number with any adverse event: adults
There was low‐quality evidence for an effect of carbamazepine on experienced adverse events, although the magnitude of this effect is uncertain: OR 0.13, 95% CI 0.02 to 0.82; P = 0.03; 1 study, 30 participants. See Analysis 2.2.
2.2. Analysis.
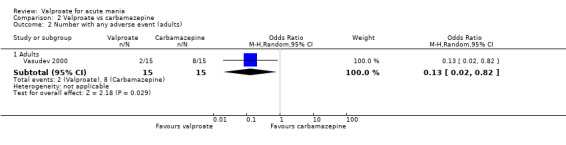
Comparison 2 Valproate vs carbamazepine, Outcome 2 Number with any adverse event (adults).
Secondary outcomes
2.3. Tolerability (dichotomous): individual adverse events: adults
There was evidence that fewer participants in the valproate group reported dizziness compared to participants in the carbamazepine group, although the magnitude of this effect is uncertain: OR 0.08, 95% CI 0.01 to 0.79; P = 0.03; 1 study, 30 participants. See Table 18 for a complete overview of all adverse events.
12. Adverse events ‐ valproate vs carbamazepine (adults).
| Adverse effect | Odds ratio; 95% CI; heterogeneity; N of studies, N of participants | P value |
| Ataxia/tremors | OR 0.29, 95% CI 0.03 to 3.12; 1 study, 30 participants | P = 0.30 |
| Dizziness | OR 0.08, 95% CI 0.01 to 0.79; 1 study, 30 participants | P = 0.03 |
| Lethargy | OR 0.14, 95% CI 0.01 to 1.42; 1 study, 30 participants | P = 0.10 |
| Nausea/vomiting | OR 0.18, 95% CI 0.03 to 1.07; 1 study, 30 participants | P = 0.06 |
| Rash | OR 0.31, 95% CI 0.01 to 8.28; 1 study, 30 participants | P = 0.49 |
| Raised liver enzyme | OR 1.00, 95% CI 0.06 to 17.62; 1 study, 30 participants | P = 1.00 |
2.4. Efficacy (continuous): we assessed the efficacy of valproate by assessing the change in symptom severity, using mean endpoint scores or mean change scores on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: adults
There was very low‐quality evidence for an effect of valproate on the reduction of mania rating scores at four weeks: mean difference (MD) −12.00, 95% CI −21.82 to −2.18; P = 0.02; 1 study, 30 participants. See Analysis 2.4.
2.4. Analysis.
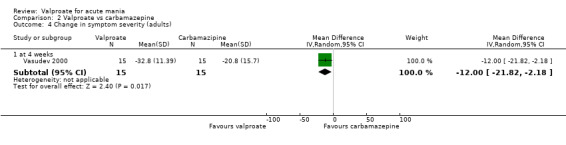
Comparison 2 Valproate vs carbamazepine, Outcome 4 Change in symptom severity (adults).
2.5. Acceptability (dichotomous): information on dropouts due to 1. side effects 2. lack of efficacy 3. any other reasons 4. all causes, during the trial as a proportion of the total number of randomised participants: adults
There was not strong evidence for a difference in dropout rates due to other reasons (OR 1.00, 95% CI 0.17 to 5.98; P = 1.00; 1 study, 30 participants) or as dropouts due to all causes (OR 1.00, 95% CI 0.17 to 5.98; P = 1.00; 1 study, 30 participants) between the two groups. See Analysis 2.5.
2.5. Analysis.
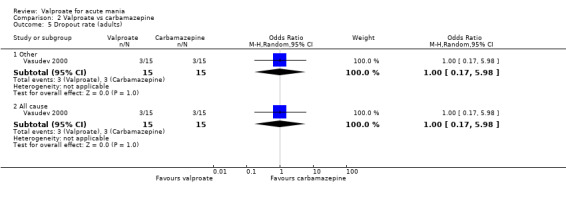
Comparison 2 Valproate vs carbamazepine, Outcome 5 Dropout rate (adults).
Comparison 3: Valproate versus endoxifen
One study contributed to this comparison (Ahmad 2016).
Primary outcomes
3.1. Efficacy (dichotomous): number of participants experiencing a 50% or greater reduction in mean score on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: adults
Very low‐quality evidence found that there was not strong evidence for a difference in response rate between the two groups at three weeks: OR 2.19, 95% CI 0.83 to 5.78; P = 0.11; 1 study, 84 participants. See Analysis 3.1.
3.1. Analysis.
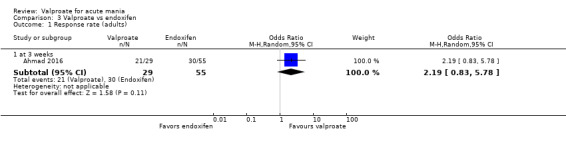
Comparison 3 Valproate vs endoxifen, Outcome 1 Response rate (adults).
3.2. Tolerability (dichotomous): number with any adverse event: adults
Very low‐quality evidence found that there was not strong evidence for a difference in experiencing any adverse events between the two groups: OR 1.88, 95% CI 0.73 to 4.86; P = 0.19; 1 study, 84 participants. See Analysis 3.2.
3.2. Analysis.
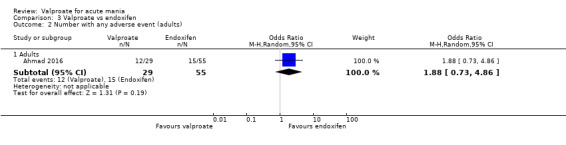
Comparison 3 Valproate vs endoxifen, Outcome 2 Number with any adverse event (adults).
Secondary outcomes
3.3. Tolerability (dichotomous): individual adverse events: adults
There was evidence that fewer participants in the valproate group reported nausea compared to participants in the endoxifen group, although the magnitude of this effect is uncertain: OR 5.52, 95% CI 1.00 to 30.50; P = 0.05; 1 study, 84 participants. See Analysis 3.3 and Table 19 for a complete overview of all adverse events.
3.3. Analysis.
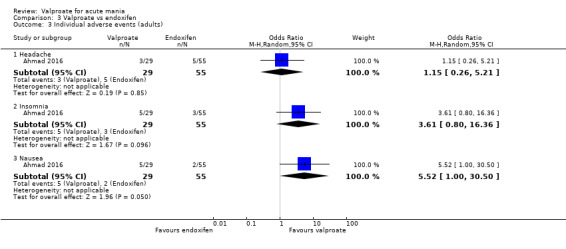
Comparison 3 Valproate vs endoxifen, Outcome 3 Individual adverse events (adults).
13. Adverse events ‐ valproate vs endoxifen (adults).
| Adverse effect | Odds ratio; 95% CI; heterogeneity; N of studies, N of participants | P value |
| Headache | OR 1.15, 95% CI 0.26 to 5.21; 1 study, 84 participants | P = 0.85 |
| Insomnia | OR 3.61, 95% CI 0.80 to 16.36; 1 study, 84 participants | P = 0.10 |
| Nausea | OR 5.52, 95% CI 1.00 to 30.50; 1 study, 84 participants | P = 0.05 |
Comparison 4: Valproate versus haloperidol
One study contributed to this comparison (McElroy 1996).
Primary outcomes
4.1. Efficacy (dichotomous): number of participants experiencing a 50% or greater reduction in mean score on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: adults
Very low‐quality evidence found that there was not strong evidence for a difference in response rate between the two groups at one week: OR 1.82, 95% CI 0.46 to 7.18; P = 0.39; 1 study, 36 participants. See Analysis 4.1.
4.1. Analysis.
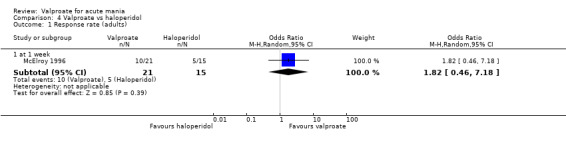
Comparison 4 Valproate vs haloperidol, Outcome 1 Response rate (adults).
Secondary outcomes
4.2. Tolerability (dichotomous): Individual adverse events: adults
There was low‐quality evidence that fewer participants in the valproate group reported extrapyramidal side effects compared to participants in the haloperidol group, although the magnitude of this effect is uncertain: OR 0.02, 95% CI 0.00 to 0.40; P = 0.01; 1 study, 36 participants. See Analysis 4.2. and Table 20 for a complete overview of all adverse events.
4.2. Analysis.
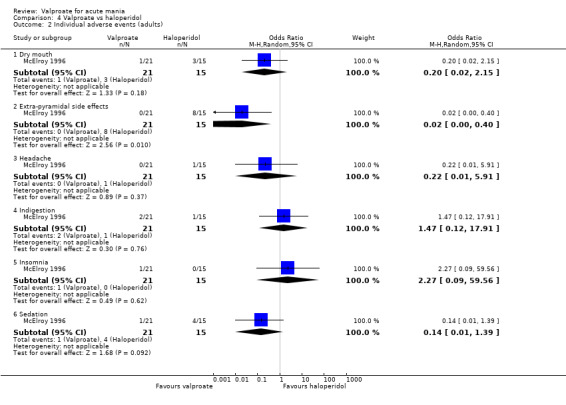
Comparison 4 Valproate vs haloperidol, Outcome 2 Individual adverse events (adults).
14. Adverse events ‐ valproate vs haloperidol (adults).
| Adverse effect | Odds ratio; 95% CI; heterogeneity; N of studies, N of participants | P value |
| Dry mouth | OR 0.20, 95% CI 0.02 to 2.15; 1 study, 36 participants | P = 0.18 |
| Extra‐pyramidal side effects | OR 0.02, 95% CI 0.00 to 0.40; 1 study, 36 participants | P = 0.01 |
| Headache | OR 0.22, 95% CI 0.01 to 5.91; 1 study, 36 participants | P = 0.37 |
| Indigestion | OR 1.47, 95% CI 0.12 to 17.91; 1 study, 36 participants | P = 0.76 |
| Insomnia | OR 2.27, 95% CI 0.09 to 59.56; 1 study, 36 participants | P = 0.62 |
| Sedation | OR 0.14, 95% CI 0.01 to 1.39; 1 study, 36 participants | P = 0.09 |
4.3. Efficacy (continuous): We assessed the efficacy of valproate by assessing the change in symptom severity, using mean endpoint scores or mean change scores on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: adults
Very low‐quality evidence found that there was not strong evidence for a difference in mania rating scores between the two groups at one week: MD −3.60, 95% CI −11.48 to 4.28; P = 0.37;1 study, 36 participants. See Analysis 4.3.
4.3. Analysis.
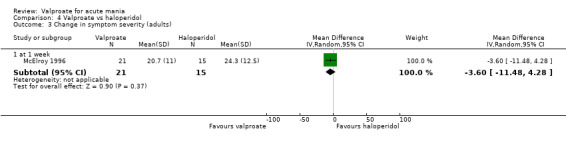
Comparison 4 Valproate vs haloperidol, Outcome 3 Change in symptom severity (adults).
Comparison 5: Valproate versus lithium
Six studies contributed to this comparison (Bowden 1994; Freeman 1992; Geller 2012; Hirschfeld 1999; Shafti 2008; Young 2017); One of these studies investigated the effects of valproate in children and adolescents (Geller 2012).
Primary outcomes
5.1. Efficacy (dichotomous): number of participants experiencing a 50% or greater reduction in mean score on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: adults
Moderate‐quality evidence found that there was not strong evidence for a difference in response rate between the two groups at three weeks: OR 0.80, 95% CI 0.48 to 1.35; P = 0.40, I2 = 16%; 3 studies, 356 participants. See Analysis 5.1.
5.1. Analysis.
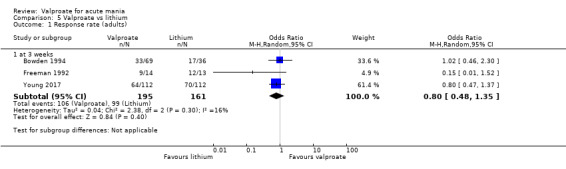
Comparison 5 Valproate vs lithium, Outcome 1 Response rate (adults).
5.2. Efficacy (dichotomous): number of participants experiencing a 50% or greater reduction in mean score on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: children and adolescents
Very low‐quality evidence found that there was no strong evidence for a difference in response rate between the two groups at eight weeks: OR 0.57, 95% CI 0.31 to 1.07; P = 0.08; 1 study, 197 participants. See Analysis 5.2.
5.2. Analysis.
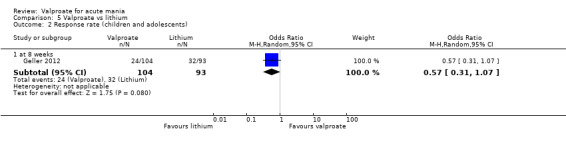
Comparison 5 Valproate vs lithium, Outcome 2 Response rate (children and adolescents).
5.3. Tolerability (dichotomous): number with any adverse event: adults
Low‐quality evidence found that there was not strong evidence for a difference in experiencing any adverse events between the two groups: OR 0.61, 95% CI 0.25 to 1.50; P = 0.28, I2 = 0%; 2 studies, 164 participants. See Analysis 5.3.
5.3. Analysis.
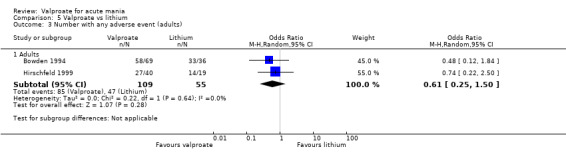
Comparison 5 Valproate vs lithium, Outcome 3 Number with any adverse event (adults).
Secondary outcomes
5.4. Tolerability (dichotomous): individual adverse events: adults
There was evidence that fewer participants in the valproate group reported fever compared to participants in the lithium group (OR 0.09, 95% CI 0.01 to 0.81; P = 0.03; 1 study, 105 participants; Analysis 5.4.5) and that more participants in the valproate group reported pain compared to participants in the lithium group (OR 8.12, 95% CI 1.02 to 64.86; P = 0.05; 1 study, 105 participants; Analysis 5.4.8), although the magnitude of these effects is uncertain. See Analysis 5.4 and Table 21 for a complete overview of all adverse events.
5.4. Analysis.
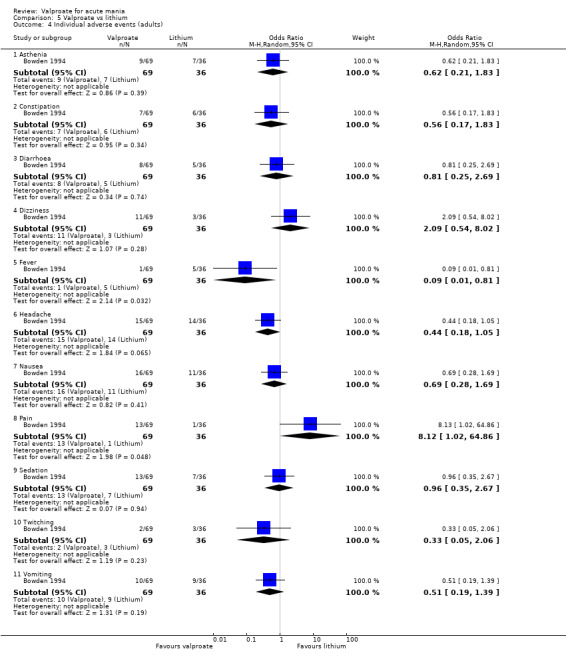
Comparison 5 Valproate vs lithium, Outcome 4 Individual adverse events (adults).
15. Adverse events ‐ valproate vs lithium (adults).
| Adverse effect | Odds ratio; 95% CI; heterogeneity; N of studies, N of participants | P value |
| Asthenia | OR 0.62, 95% CI 0.21 to 1.83; 1 study, 105 participants | P = 0.39 |
| Constipation | OR 0.56, 95% CI 0.17 to 1.83; 1 study, 105 participants | P = 0.34 |
| Diarrhoea | OR 0.81, 95% CI 0.25 to 2.69; 1 study, 105 participants | P = 0.74 |
| Dizziness | OR 2.09, 95% CI 0.54 to 8.02; 1 study, 105 participants | P = 0.28 |
| Fever | OR 0.09, 95% CI 0.01 to 0.81; 1 study, 105 participants | P = 0.03 |
| Headache | OR 0.44, 95% CI 0.18 to 1.05; 1 study, 105 participants | P = 0.07 |
| Nausea | OR 0.69, 95% CI 0.28 to 1.69; 1 study, 105 participants | P = 0.41 |
| Pain | OR 8.12, 95% CI 1.02 to 64.86; 1 study, 105 participants | P = 0.05 |
| Sedation | OR 0.96, 95% CI 0.35 to 2.67; 1 study, 105 participants | P = 0.94 |
| Twitching | OR 0.33, 95% CI 0.05 to 2.06; 1 study, 105 participants | P = 0.23 |
| Vomiting | OR 0.51, 95% CI 0.19 to 1.39; 1 study, 105 participants | P = 0.19 |
5.5. Tolerability (dichotomous): individual adverse events: children and adolescents
There was evidence that fewer participants in the valproate group reported dry mouth/excessive thirst (OR 0.34, 95% CI 0.17 to 0.65; P = 0.001; 1 study, 190 participants; Analysis 5.5.4) and frequent urination (OR 0.45, 95% CI 0.22 to 0.93; P = 0.03; 1 study, 190 participants; Analysis 5.5.8) compared to the lithium group. See Analysis 5.5 and Table 22 for a complete overview of all adverse events.
5.5. Analysis.
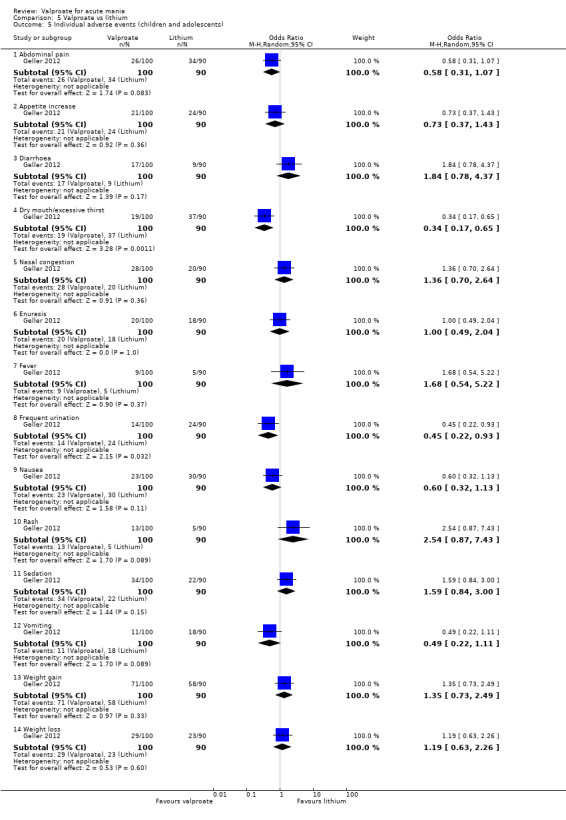
Comparison 5 Valproate vs lithium, Outcome 5 Individual adverse events (children and adolescents).
16. Adverse events ‐ valproate vs lithium (children and adolescents).
| Adverse effect | Odds ratio; 95% CI; heterogeneity; N of studies, N of participants | P value |
| Abdominal pain | OR 0.58, 95% CI 0.31 to 1.07; 1 study, 190 participants | P = 0.08 |
| Appetite increase | OR 0.73, 95% CI 0.37 to 1.43; 1 study, 190 participants | P = 0.36 |
| Diarrhoea | OR 1.84, 95% CI 0.78 to 4.37; 1 study, 190 participants | P = 0.17 |
| Dry mouth/excessive thirst | OR 0.34, 95% CI 0.17 to 0.65; 1 study, 190 participants | P = 0.001 |
| Nasal congestion | OR 1.36, 95% CI 0.70 to 2.64; 1 study, 190 participants | P = 0.36 |
| Enuresis | OR 1.00, 95% CI 0.49 to 2.04; 1 study, 190 participants | P = 1.00 |
| Fever | OR 1.68, 95% CI 0.54 to 5.22; 1 study, 190 participants | P = 0.37 |
| Frequent urination | OR 0.45, 95% CI 0.22 to 0.93; 1 study, 190 participants | P = 0.03 |
| Nausea | OR 0.60, 95% CI 0.32 to 1.13; 1 study, 190 participants | P = 0.11 |
| Rash | OR 2.54, 95% CI 0.87 to 7.43; 1 study, 190 participants | P = 0.09 |
| Sedation | OR 1.59, 95% CI 0.84 to 3.00; 1 study, 190 participants | P = 0.15 |
| Vomiting | OR 0.49, 95% CI 0.22 to 1.11; 1 study, 190 participants | P = 0.09 |
| Weight gain | OR 1.35, 95% CI 0.73 to 2.49; 1 study, 190 participants | P = 0.33 |
| Weight loss | OR 1.19, 95% CI 0.63 to 2.26; 1 study, 190 participants | P = 0.60 |
5.6. Efficacy (continuous): We assessed the efficacy of valproate by assessing the change in symptom severity, using mean endpoint scores or mean change scores on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: adults
There was very low‐quality evidence for an effect of lithium on mania rating scores at three weeks: SMD 0.69, 95% CI 0.14 to 1.25; P = 0.01, I2 = 6%; 2 studies, 57 participants. See Analysis 5.6.
5.6. Analysis.
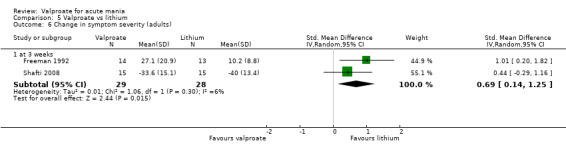
Comparison 5 Valproate vs lithium, Outcome 6 Change in symptom severity (adults).
5.7. Efficacy (continuous): We assessed the efficacy of valproate by assessing the change in symptom severity, using mean endpoint scores or mean change scores on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: adults
There was not strong evidence for a difference in MSRS scores (frequency) between the two groups at three weeks: MD 7.80, 95% CI −2.11 to 17.71; P = 0.12; 1 study, 30 participants. See Analysis 5.7.
5.7. Analysis.
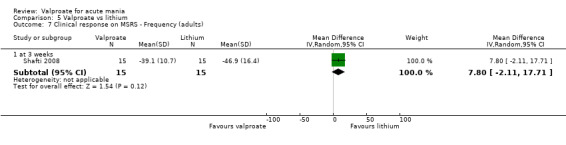
Comparison 5 Valproate vs lithium, Outcome 7 Clinical response on MSRS ‐ Frequency (adults).
5.8. Efficacy (continuous): We assessed the efficacy of valproate by assessing the change in symptom severity, using mean endpoint scores or mean change scores on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: children and adolescents
Very low‐quality evidence found that there was no strong evidence of a difference in mania rating scores between the two groups at eight weeks: MD 1.40, 95% CI −2.03 to 4.83; P = 0.42; 1 study, 190 participants. See Analysis 5.8.
5.8. Analysis.
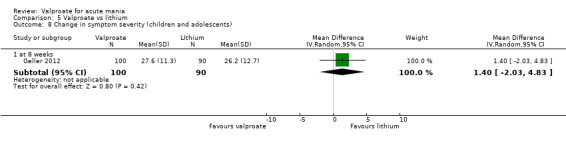
Comparison 5 Valproate vs lithium, Outcome 8 Change in symptom severity (children and adolescents).
5.9. Acceptability (dichotomous): adults
There was not strong evidence for a difference in dropout rates due to adverse events (OR 0.49, 95% CI 0.12 to 2.10; P = 0.34; 1 study, 105 participants), dropouts due to inefficacy (OR 0.89, 95% CI 0.42 to 1.88; P = 0.76, I2 = 0%; 2 studies, 164 participants), dropouts due to other reasons (OR 0.60, 95% CI 0.26 to 1.39; P = 0.23, I2 = 0%; 2 studies, 164 participants), and dropouts due to all causes (OR 0.83, 95% CI 0.47 to 1.45; P = 0.51, I2 = 22%; 3 studies, 388 participants) between the two groups. See Analysis 5.9.
5.9. Analysis.
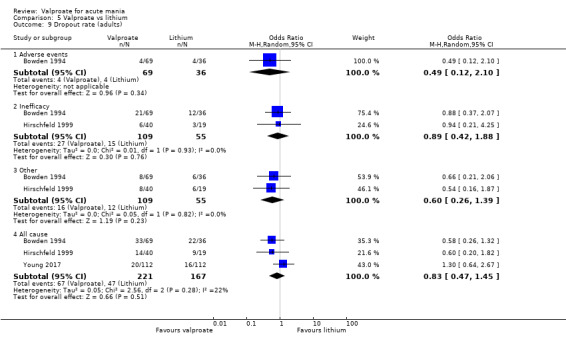
Comparison 5 Valproate vs lithium, Outcome 9 Dropout rate (adults).
5.10. Acceptability (dichotomous): children and adolescents
There was not strong evidence for a difference in dropout rates due to adverse events (OR 0.43, 95% CI 0.12 to 1.46; P = 0.17; 1 study, 197 participants), dropouts due to other reasons (OR 0.92, 95% CI 0.47 to 1.81; P = 0.81; 1 study, 197 participants), and dropouts due to all causes (OR 0.74, 95% CI 0.39 to 1.37; P = 0.33; 1 study, 197 participants) between the two groups. See Analysis 5.10.
5.10. Analysis.
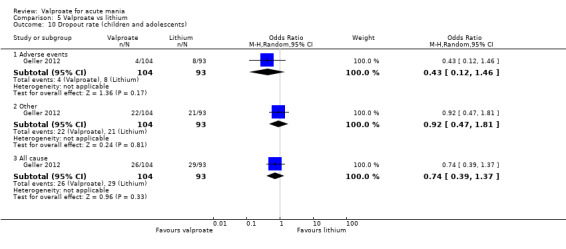
Comparison 5 Valproate vs lithium, Outcome 10 Dropout rate (children and adolescents).
Comparison 6: Valproate versus olanzapine
Four studies contributed to this comparison (Tohen 2002; Tohen 2008; Xu 2015; Zajecka 2002). In addition to the results on changes in mania rating scale scores at the time points presented below, Xu 2015 also provided further data which showed evidence for an effect of olanzapine on mania rating scale scores at two weeks (SMD 0.58, 95% CI 0.12 to 1.04; P = 0.01;1 study, 76 participants) and at four weeks (SMD 0.61, 95% CI 0.15 to 1.07; P = 0.01;1 study, 76 participants).
Primary outcomes
6.1. Efficacy (dichotomous): number of participants experiencing a 50% or greater reduction in score on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: adults
Low‐quality evidence found that there was not strong evidence for a difference in response rate between the two groups at three weeks: OR 0.77, 95% CI 0.48 to 1.25; P = 0.29, I2 = 57%; 2 studies, 667 participants; See Analysis 6.1. This measure contains a large amount of heterogeneity (I2 = 57%). This comparison contains two studies: Tohen 2002 and Tohen 2008. Differences in these studies may contribute to this heterogeneity. Tohen 2002 recruited anyone with a YMRS score of over 20, while Tohen 2008 only recruited those with scores between 20 and 30, excluding those with more serious mania. Additionally, while Tohen 2008 excluded all those with psychotic features, this was not an exclusion criterion in Tohen 2002. Indeed, Tohen 2002 found in a subgroup analysis that olanzapine was more efficacious in those with psychotic features but this difference was not present in those without any psychosis. These methodological difference may explain some of this heterogeneity.
6.1. Analysis.
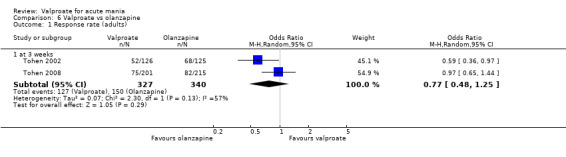
Comparison 6 Valproate vs olanzapine, Outcome 1 Response rate (adults).
Secondary outcomes
6.2. Tolerability (dichotomous): individual adverse events: adults
There was evidence that more participants in the valproate group experienced insomnia (OR 6.17, 95% CI 1.35 to 28.17; P = 0.02; 1 study, 416 participants; Analysis 6.2.11) and nausea (OR 4.12, 95% CI 2.22 to 7.62; P < 0.001, I2 = 0%; 3 studies, 747 participants; Analysis 6.2.12) compared to olanzapine, although the magnitude of these effects is unclear. In contrast, there was evidence that fewer participants in the valproate group reported neck rigidity (OR 0.21, 95% CI 0.04 to 0.98; P = 0.05; 1 study, 251 participants; Analysis 6.2.13), oedemas (OR 0.05, 95% CI 0.00 to 0.81; P = 0.04; 1 study, 120 participants; Analysis 6.2.15), sedation (OR 0.50, 95% CI 0.28 to 0.91; P = 0.02, I2 = 0%; 2 studies, 536 participants; Analysis 6.2.18), somnolence (OR 0.36, 95% CI 0.23 to 0.57; P < 0.001, I2 = 0%; 3 studies, 747 participants; Analysis 6.2.20), speech disorder (OR 0.09, 95% CI 0.02 to 0.50; P = 0.006, I2 = 0%; 2 studies, 371 participants; Analysis 6.2.21), tremor (OR 0.28, 95% CI 0.09 to 0.82; P = 0.02, I2 = 0%; 2 studies, 331 participants; Analysis 6.2.23), weight gain (OR 0.44, 95% CI 0.28 to 0.70; P < 0.001, I2 = 0%; 4 studies, 867 participants; Analysis 6.2.25), and xerostomia (OR 0.25, 95% CI 0.11 to 0.57; P = 0.001, I2 = 44%; 3 studies, 747 participants; Analysis 6.2.26) compared to participants in the olanzapine group, although the magnitude of these effects varies. See analysis Analysis 6.2 and Table 23 for a complete overview of all adverse events.
6.2. Analysis.
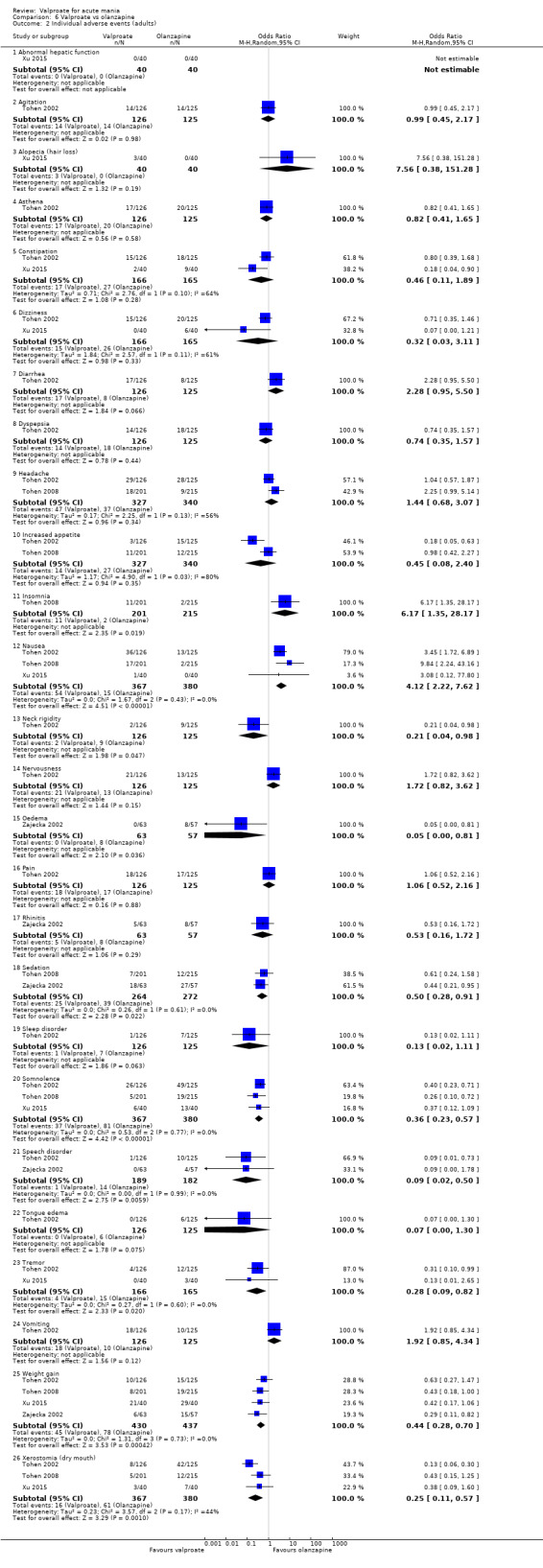
Comparison 6 Valproate vs olanzapine, Outcome 2 Individual adverse events (adults).
17. Adverse events ‐ valproate vs olanzapine (adults).
| Adverse effect | Odds ratio; 95% CI; heterogeneity; N of studies, N of participants | P value |
| Abnormal hepatic function | Not estimable | N/A |
| Agitation | OR 0.99, 95% CI 0.45 to 2.17; 1 study, 251 participants | P = 0.98 |
| Alopecia (hair loss) | OR 7.56, 95% CI 0.38 to 151.28; 1 study, 80 participants | P = 0.19 |
| Asthena | OR 0.82, 95% CI 0.41 to 1.65; 1 study, 251 participants | P = 0.58 |
| Constipation | OR 0.46, 95% CI 0.11 to 1.89; I2 = 64%; 2 studies, 331 participants | P = 0.28 |
| Dizziness | OR 0.32, 95% CI 0.03 to 3.11; I2 = 61%; 2 studies, 331 participants | P = 0.33 |
| Diarrhea | OR 2.28, 95% CI 0.95 to 5.50; 1 study, 251 participants | P = 0.07 |
| Dyspepsia | OR 0.74, 95% CI 0.35 to 1.57; studies = 1; participants = 251 | p = 0.44 |
| Headache | OR 1.44, 95% CI 0.68 to 3.07; I2 = 56%; 2 studies, 667 participants | P = 0.34 |
| Increased appetite | OR 0.45, 95% CI 0.08 to 2.40; I2 = 80%; 2 studies, 667 participants | P = 0.35 |
| Insomnia | OR 6.17, 95% CI 1.35 to 28.17; 1 study, 416 participants | P = 0.02 |
| Nausea | OR 4.12, 95% CI 2.22 to 7.62; I2 = 0%; 3 studies, 747 participants | P < 0.001 |
| Neck rigidity | OR 0.21, 95% CI 0.04 to 0.98; 1 study, 251 participants | P = 0.05 |
| Nervousness | OR 1.72, 95% CI 0.82 to 3.62; 1 study, 251 participants | P = 0.15 |
| Oedema | OR 0.05, 95% CI 0.00 to 0.81; 1 study, 120 participants | P = 0.04 |
| Pain | OR 1.06, 95% CI 0.52 to 2.16; 1 study, 251 participants | P = 0.88 |
| Rhinitis | OR 0.53, 95% CI 0.16 to 1.72; 1 study, 120 participants | P = 0.29 |
| Sedation | OR 0.50, 95% CI 0.28 to 0.91; I2 = 0%; 2 studies, 536 participants | P = 0.02 |
| Sleep disorder | OR 0.13, 95% CI 0.02 to 1.11; 1 study, 251 participants | P = 0.06 |
| Somnolence | OR 0.36, 95% CI 0.23 to 0.57; I2 = 0%; 3 studies, 747 participants | P < 0.001 |
| Speech disorder | OR 0.09, 95% CI 0.02 to 0.50; I2 = 0%; 2 studies, 371 participants | P = 0.006 |
| Tongue edema | OR 0.07, 95% CI 0.00 to 1.30; 1 study, 251 participants | P = 0.08 |
| Tremor | OR 0.28, 95% CI 0.09 to 0.82; I2 = 0%; 2 studies, 331 participants | P = 0.02 |
| Vomiting | OR 1.92, 95% CI 0.85 to 4.34; 1 study, 251 participants | P = 0.12 |
| Weight gain | OR 0.44, 95% CI 0.28 to 0.70; I2 = 0%; 4 studies, 867 participants | P < 0.001 |
| Xerostomia (dry mouth) | OR 0.25, 95% CI 0.11 to 0.57; I2 = 44%; 3 studies, 747 participants | P = 0.001 |
6.3. Remission (dichotomous): A score of 12 or less on the YMRS (or equivalent on other validated mania rating scales): adults
There was not strong evidence for a difference in remission rates between the two groups at three weeks: OR 0.73, 95% CI 0.46 to 1.15; P = 0.17, I2 = 50%; 2 studies, 667 participants. See Analysis 6.3. This measure contains a large amount of heterogeneity (I2 = 50%). This comparison covers the same studies as Analysis 6.1, with the same probable sources for the heterogeneity.
6.3. Analysis.
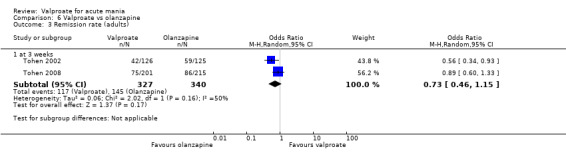
Comparison 6 Valproate vs olanzapine, Outcome 3 Remission rate (adults).
6.4. Efficacy (continuous): We assessed the efficacy of valproate by assessing the change in symptom severity, using mean endpoint scores or mean change scores on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: adults
Low‐quality evidence found that there was not strong evidence for a difference in mania rating scores between the two groups at one week: SMD 0.35, 95% CI −0.11 to 0.80; P = 0.13; 1 study, 76 participants. By contrast, there was high‐quality evidence for an effect of olanzapine on mania rating scale scores at three weeks: SMD 0.25, 95% CI 0.11 to 0.39; P < 0.001, I2 = 5%; 4 studies, 826 participants. See Analysis 6.4.
6.4. Analysis.
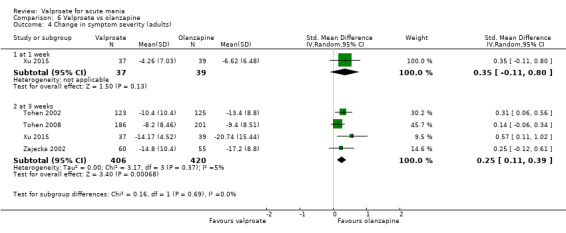
Comparison 6 Valproate vs olanzapine, Outcome 4 Change in symptom severity (adults).
6.5. Acceptability (dichotomous): information on dropouts due to 1. side effects 2. lack of efficacy 3. any other reasons 4. all causes, during the trial as a proportion of the total number of randomised participants: adults
There was not strong evidence for a difference in dropout rates due to adverse events (OR 0.61, 95% CI 0.25 to 1.49; P = 0.28, I2 = 22%; 3 studies, 616 participants), due to inefficacy (OR 1.36, 95% CI 0.65 to 2.86; P = 0.41, I2 = 0%; 3 studies, 616 participants), dropouts due to other reasons (OR 1.14, 95% CI 0.76 to 1.72; P = 0.52, I2 = 0%; 3 studies, 616 participants), and dropout due to all causes (OR 1.04, 95% CI 0.71 to 1.52; P = 0.84, I2 = 0%; 3 studies, 616 participants) between the two groups. See Analysis 6.5.
6.5. Analysis.
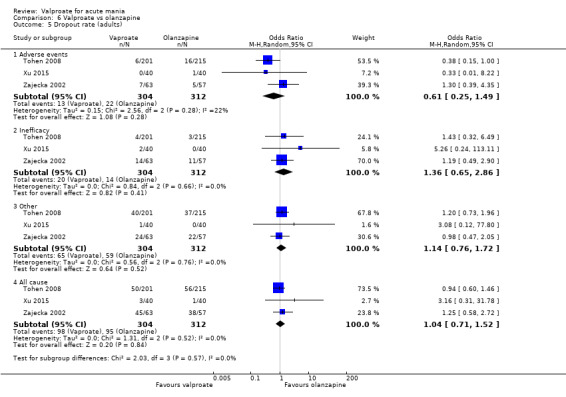
Comparison 6 Valproate vs olanzapine, Outcome 5 Dropout rate (adults).
Comparison 7: Valproate + olanzapine versus olanzapine monotherapy
One study contributed to this comparison (Xu 2015). In addition to the results on changes in mania rating scale scores at the time points presented below, Xu 2015 also provided further data which showed that there was not strong evidence for a difference in mania rating scores between the two groups at two weeks (MD −2.07, 95% CI −8.51 to 4.37; P = 0.53; 1 study, 76 participants) or at four weeks (MD −3.76, 95% CI −9.29 to 1.77; P = 0.18; 1 study, 76 participants).
Primary outcomes
No data on primary outcomes were available for this comparison.
Secondary outcomes
7.1. Tolerability (dichotomous): individual adverse events: adults
There were no differences between the study groups for any adverse events. See Table 24 for a complete overview of all adverse events.
18. Adverse events ‐ valproate + olanzapine vs olanzapine alone (adults).
| Adverse effect | Odds ratio; 95% CI; heterogeneity; N of studies, N of participants | P value |
| Alopecia (hair loss) | OR 5.26, 95% CI 0.24 to 113.11; 1 study, 80 participants | P = 0.29 |
| Constipation | OR 0.73, 95% CI 0.24 to 2.20; 1 study, 80 participants | P = 0.58 |
| Dizziness | OR 1.89, 95% CI 0.61 to 5.82; 1 study, 80 participants | P = 0.27 |
| Somnolence | OR 0.79, 95% CI 0.30 to 2.06; 1 study, 80 participants | P = 0.63 |
| Tremor | OR 0.65, 95% CI 0.10 to 4.11; 1 study, 80 participants | P = 0.65 |
| Weight gain | OR 1.31, 95% CI 0.47 to 3.61; 1 study, 80 participants | P = 0.61 |
| Xerostomia (dry mouth) | OR 0.67, 95% CI 0.19 to 2.33; 1 study, 80 participants | P = 0.53 |
7.2. Efficacy (continuous): We assessed the efficacy of valproate by assessing the change in symptom severity, using mean endpoint scores or mean change scores on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: adults
Low‐quality evidence showed that there was not strong evidence for a difference in mania rating scores between the two groups at one week: MD −0.87, 95% CI −5.16 to 3.42; P = 0.69; 1 study, 76 participants. In addition, low‐quality evidence found that there was no difference between the two groups at three weeks: MD −2.76, 95% CI −9.17 to 3.65; P = 0.40; 1 study, 76 participants. See Analysis 7.2.
7.2. Analysis.
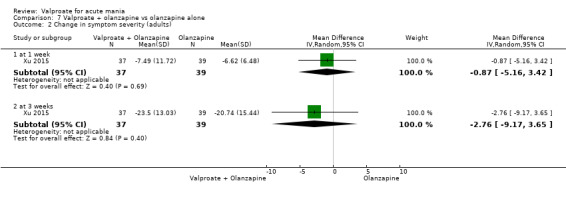
Comparison 7 Valproate + olanzapine vs olanzapine alone, Outcome 2 Change in symptom severity (adults).
7.3. Acceptability (dichotomous): information on dropouts due to 1. side effects 2. lack of efficacy 3. any other reasons 4. all causes, during the trial as a proportion of the total number of randomised participants: adults
There was not strong evidence for a difference in dropouts due to adverse events (OR 1.00, 95% CI 0.06 to 16.56; P = 1.00; 1 study, 80 participants), dropouts due to other reasons (OR 3.08, 95% CI 0.12 to 77.80; P = 0.50; 1 study, 80 participants), or dropouts due to all causes (OR 2.05, 95% CI 0.18 to 23.59; P = 0.56; 1 study, 80 participants) between the two groups. See Analysis 7.3.
7.3. Analysis.
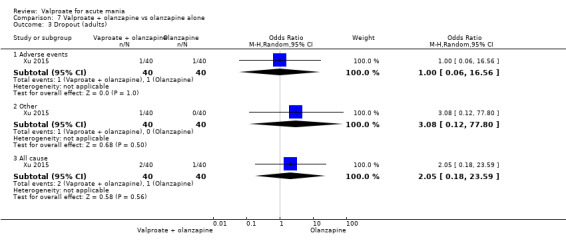
Comparison 7 Valproate + olanzapine vs olanzapine alone, Outcome 3 Dropout (adults).
Comparison 8: Valproate versus oxcarbazepine
One study contributed to this comparison (Kakkar 2009). In addition to the results on changes in mania rating scale scores at the time points presented below, Kakkar 2009 also provided further data which showed that there was not strong evidence for a difference between the two group at one day (MD 0.97, 95% CI −1.88 to 3.82; P = 0.50; 1 study, 60 participants), at two days (MD 2.50, 95% CI −0.82 to 5.82; P = 0.14; 1 study, 60 participants), at one week (MD 1.17, 95% CI −1.66 to 4.00; P = 0.42; 1 study, 60 participants), at two weeks (MD 0.37, 95% CI −2.33 to 3.07; P = 0.79; 1 study, 60 participants), at four weeks (MD 0.97, 95% CI −1.90 to 3.84; P = 0.51; 1 study, 60 participants), at five weeks (MD 1.10, 95% CI −1.75 to 3.95; P = 0.45;1 study, 60 participants), at six weeks (MD −0.16, 95% CI −2.77 to 2.45; P = 0.90; 1 study, 60 participants), at seven weeks (MD 0.00, 95% CI −2.56 to 2.56; P = 1.00; 1 study, 60 participants), at nine weeks (MD −0.63, 95% CI −2.84 to 1.58; P = 0.58; 1 study, 60 participants), at 10 weeks (MD −0.73, 95% CI −2.64 to 1.18; P = 0.45; 1 study, 60 participants), at 11 weeks (MD −1.66, 95% CI −3.41 to 0.09; P = 0.06; 1 study, 60 participants), or at 12 weeks (MD −1.10, 95% CI −2.66 to 0.46; P = 0.17; 1 study, 60 participants).
Primary outcomes
8.1. Tolerability (dichotomous): number with any adverse event: adults
There was low‐quality evidence for an effect of valproate on experiencing any adverse events, although the magnitude of this effect is uncertain: OR 4.67, 95% CI 1.57 to 13.87; P = 0.006; 1 study, 60 participants. See Analysis 8.1.
8.1. Analysis.
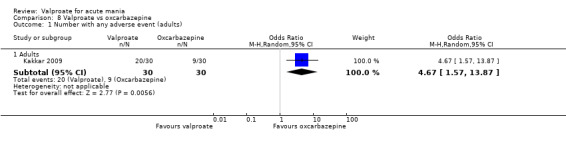
Comparison 8 Valproate vs oxcarbazepine, Outcome 1 Number with any adverse event (adults).
Secondary outcomes
8.2.Tolerability (dichotomous): individual adverse events: adults
There were no differences between the study groups in any adverse events. See Table 25 for a complete overview of all adverse events.
19. Adverse events ‐ valproate vs oxcarbazepine (adults).
| Adverse effect | Odds ratio; 95% CI; heterogeneity; N of studies, N of participants | P value |
| Alopecia | OR 3.10, 95% CI 0.12 to 79.23; 1 study, 60 participants | P = 0.49 |
| Constipation | OR 7.76, 95% CI 0.38 to 157.14; 1 study, 60 participants | P = 0.18 |
| Diarrhoea | OR 5.35, 95% CI 0.25 to 116.31; 1 study, 60 participants | P = 0.29 |
| Dizziness | OR 2.74, 95% CI 0.63 to 11.82; 1 study, 60 participants | P = 0.18 |
| Dry mouth | OR 2.07, 95% CI 0.18 to 24.15; 1 study, 60 participants | P = 0.56 |
| Dyspepsia | OR 1.00, 95% CI 0.19 to 5.40; 1 study, 60 participants | P = 1.00 |
| Headache | OR 2.25, 95% CI 0.51 to 9.99; 1 study, 60 participants | P = 0.29 |
| Increased appetite | OR 7.76, 95% CI 0.38 to 157.14; 1 study, 60 participants | P = 0.18 |
| Nausea | OR 1.52, 95% CI 0.42 to 5.47; 1 study, 60 participants | P = 0.52 |
| Pain in abdomen | OR 13.16, 95% CI 0.69 to 249.48; 1 study, 60 participants | P = 0.09 |
| Rash | OR 3.10, 95% CI 0.12 to 79.23; 1 study, 60 participants | P = 0.49 |
| Sedation | OR 1.80, 95% CI 0.39 to 8.32; 1 study, 60 participants | P = 0.45 |
| Thrombocytopenia | OR 3.10, 95% CI 0.12 to 79.23; 1 study, 60 participants | P = 0.49 |
| Vomiting | OR 1.63, 95% CI 0.41 to 6.47; 1 study, 60 participants | P = 0.49 |
| Weight gain | OR 10.36, 95% CI 0.53 to 201.45; 1 study, 60 participants | P = 0.12 |
8.3. Remission (dichotomous): A score of 12 or less on the YMRS (or equivalent on other validated mania rating scales): Adults
Very low‐quality evidence found that there was not strong evidence for a difference in response rate between the two groups at 12 weeks: OR 2.25, 95% CI 0.51 to 9.99; P = 0.29; 1 study, 60 participants. See Analysis 8.3.
8.3. Analysis.
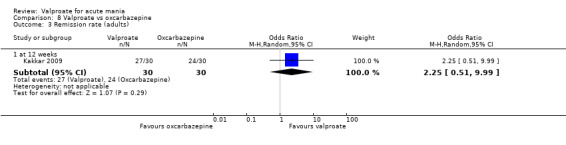
Comparison 8 Valproate vs oxcarbazepine, Outcome 3 Remission rate (adults).
8.4. Efficacy (continuous): We assessed the efficacy of valproate by assessing the change in symptom severity, using mean endpoint scores or mean change scores on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: adults
Very low‐quality evidence found that there was not strong evidence for a difference in mania rating scores at three days (MD 1.60, 95% CI −1.24 to 4.44; P = 0.29; 1 study, 60 participants), at three weeks (MD 0.73, 95% CI −2.17 to 3.63; P = 0.79; 1 study, 60 participants), and at eight weeks (MD −0.40, 95% CI −2.60 to 1.80; P = 0.72; 1 study, 60 participants). See Analysis 8.4.
8.4. Analysis.
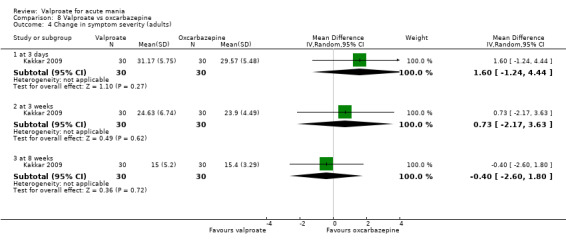
Comparison 8 Valproate vs oxcarbazepine, Outcome 4 Change in symptom severity (adults).
Comparison 9: Valproate versus quetiapine
Two studies contributed to this comparison (DelBello 2006; Feifel 2011). One of these studies aimed to investigate the effects of valproate within children (DelBello 2006).
Primary outcomes
No data on primary outcomes were available for this comparison.
Secondary outcomes
9.1. Tolerability (dichotomous): Individual adverse events: adults
There were no differences between the study groups in any adverse events. See Table 26 for a complete overview of all adverse events.
20. Adverse events ‐ valproate vs quetiapine (adults).
| Adverse effect | Odds ratio; 95% CI; heterogeneity; N of studies, N of participants | P value |
| Head, eyes, ears, nose, and throat | OR 0.17, 95% CI 0.02 to 1.67; 1 study, 30 participants | P = 0.13 |
| Gastrointestinal | OR 1.20, 95% CI 0.24 to 6.06; 1 study, 30 participants | P = 0.83 |
| Genito‐urinary | OR 1.15, 95% CI 0.07 to 20.34; 1 study, 30 participants | P = 0.92 |
| Musculo/skeletal | OR 1.15, 95% CI 0.07 to 20.34; 1 study, 30 participants | P = 0.92 |
| Pulmonary | OR 1.17, 95% CI 0.14 to 9.59; 1 study, 30 participants | P = 0.89 |
| Psychiatric | OR 0.51, 95% CI 0.11 to 2.36; 1 study, 30 participants | P = 0.39 |
9.2. Tolerability (dichotomous): Individual adverse events: children and adolescents
There were no differences between the study groups in any adverse events. See Table 27 for a complete overview of all adverse events.
21. Adverse events ‐ valproate vs quetiapine (children and adolescents).
| Adverse effect | Odds ratio; 95% CI; heterogeneity; N of studies, N of participants | P value |
| Dizziness | OR 1.00, 95% CI 0.32 to 3.17; 1 study, 50 participants | P = 1.00 |
| Dry mouth | OR 0.28, 95% CI 0.05 to 1.53; 1 study, 50 participants | P = 0.14 |
| Gastrointestinal upset | OR 0.81, 95% CI 0.23 to 2.88; 1 study, 50 participants | P = 0.75 |
| Increased appetite | OR 1.57, 95% CI 0.24 to 10.30; 1 study, 50 participants | P = 0.64 |
| Insomnia | OR 0.31, 95% CI 0.03 to 3.16; 1 study, 50 participants | P = 0.32 |
| Sedation or lethargy | OR 0.38, 95% CI 0.12 to 1.18; 1 study, 50 participants | P = 0.09 |
9.3. Remission (dichotomous): a score of 12 or less on the YMRS (or equivalent on other validated mania rating scales): children and adolescents
There was moderate‐quality evidence for an effect of quetiapine on remission at four weeks: OR 0.26, 95% CI 0.08 to 0.85; P = 0.03; 1 study, 50 participants. See Analysis 9.3.
9.3. Analysis.
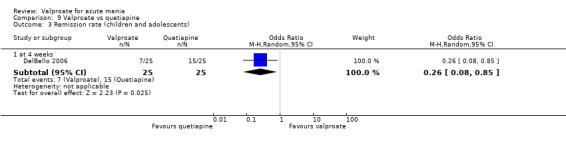
Comparison 9 Valproate vs quetiapine, Outcome 3 Remission rate (children and adolescents).
9.4. Efficacy (continuous): We assessed the efficacy of valproate by assessing the change in symptom severity, using mean endpoint scores or mean change scores on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: children and adolescents
Low‐quality evidence found that there was no strong evidence for a difference in mania rating scores between the two groups at four weeks: MD 4.00, 95% CI −2.10 to 10.10; P = 0.20; 1 study, 50 participants. See Analysis 9.4.
9.4. Analysis.
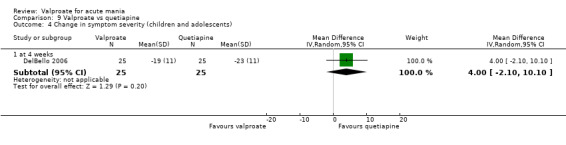
Comparison 9 Valproate vs quetiapine, Outcome 4 Change in symptom severity (children and adolescents).
9.5. Acceptability (dichotomous): Information on dropouts due to 1. side effects 2. lack of efficacy 3. any other reasons 4. all causes, during the trial as a proportion of the total number of randomised participants: adults
Very low‐quality evidence found that there was not strong evidence for a difference in dropouts due to all causes between the two groups: OR 1.73, 95% CI 0.31 to 9.57; P = 0.53; 1 study, 30 participants. See Analysis 9.5.
9.5. Analysis.
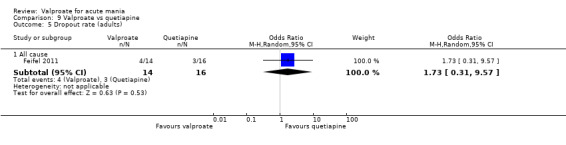
Comparison 9 Valproate vs quetiapine, Outcome 5 Dropout rate (adults).
9.6. Acceptability (dichotomous): information on dropouts due to 1. side effects 2. lack of efficacy 3. any other reasons 4. all causes, during the trial as a proportion of the total number of randomised participants: children and adolescents
There was not strong evidence for a difference in dropouts due to inefficacy (OR 1.00, 95% CI 0.13 to 7.72; P = 1.00; 1 study, 50 participants), dropouts due to other reasons (OR 1.00, 95% CI 0.22 to 4.54; P = 1.00; 1 study, 50 participants), or dropouts due to all causes (OR 1.00, 95% CI 0.27 to 3.66; P = 1.00; 1 study, 50 participants) between the two groups. See Analysis 9.6.
9.6. Analysis.
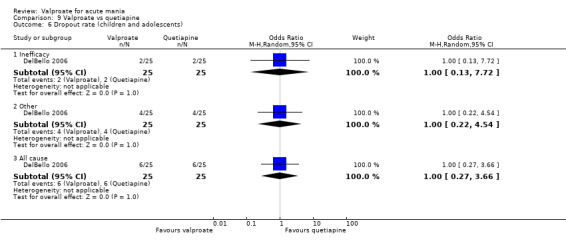
Comparison 9 Valproate vs quetiapine, Outcome 6 Dropout rate (children and adolescents).
9.7. Global functioning: the proportion of participants who improved at endpoint based on the final scores of 1 ‐ 2 on the CGI‐I or CGI‐BP‐I: children and adolescents
There was evidence for an effect of quetiapine on global functioning scores, although the magnitude of the effect is uncertain: OR 0.24, 95% CI 0.06 to 0.92; P = 0.04; 1 study, 50 participants. See Analysis 9.7.
9.7. Analysis.
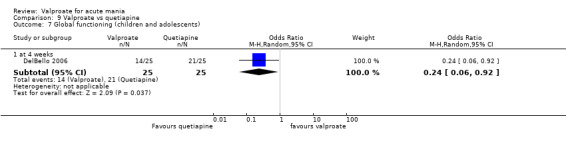
Comparison 9 Valproate vs quetiapine, Outcome 7 Global functioning (children and adolescents).
Comparison 10: Valproate versus risperidone
Two studies contributed to this comparison (Geller 2012, Kowatch 2015), both investigating the effects of valproate in children and adolescents. The results for continuous efficacy were reported at six weeks by Kowatch 2015 (SMD 0.92, 95% CI 0.25 to 1.58; P = 0.007; 1 study, 39 participants) and at eight weeks by Geller 2012 (SMD 1.03, 95% CI 0.73 to 1.34; P < 0.001; 1 study, 189 participants). We aggregated these results to reflect overall long‐term efficacy outcomes (5 ‐ 12 weeks).
Primary outcomes
10.1. Efficacy (dichotomous): number of participants experiencing a 50% or greater reduction in mean score on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: children and adolescents
There was low‐quality evidence for an effect of risperidone on response rate at eight weeks: OR 0.16, 95% CI 0.08 to 0.29; P < 0.001; 1 study, 197 participants. See Analysis 10.1.
10.1. Analysis.
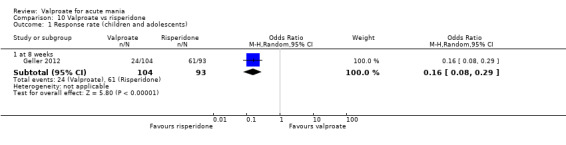
Comparison 10 Valproate vs risperidone, Outcome 1 Response rate (children and adolescents).
Secondary outcomes
10.2. Tolerability (dichotomous): individual adverse events: children and adolescents
There was evidence that fewer participants in the valproate group compared to risperidone reported appetite increase (OR 0.08, 95% CI 0.04 to 0.16; P < 0.001; 1 study, 189 participants; Analysis 10.2.2), dry mouth/excessive thirst (OR 0.44, 95% CI 0.23 to 0.85; P = 0.01; 1 study, 189 participants; Analysis 10.2.3), sedation (OR 0.50, 95% CI 0.28 to 0.91; P= 0.02; 1 study, 189 participants; Analysis 10.2.16), and weight gain (OR 0.12, 95% CI 0.04 to 0.34; P < 0.001; 1 study, 189 participants; Analysis 10.2.18). In contrast, there was evidence that more participants in the valproate group reported weight loss compared to risperidone: OR 3.63, 95% CI 1.61 to 8.19; P = 0.002; 1 study, 189 participants; Analysis 10.2.19. See Analysis 10.2 and Table 28 or a complete overview of all adverse events.
10.2. Analysis.
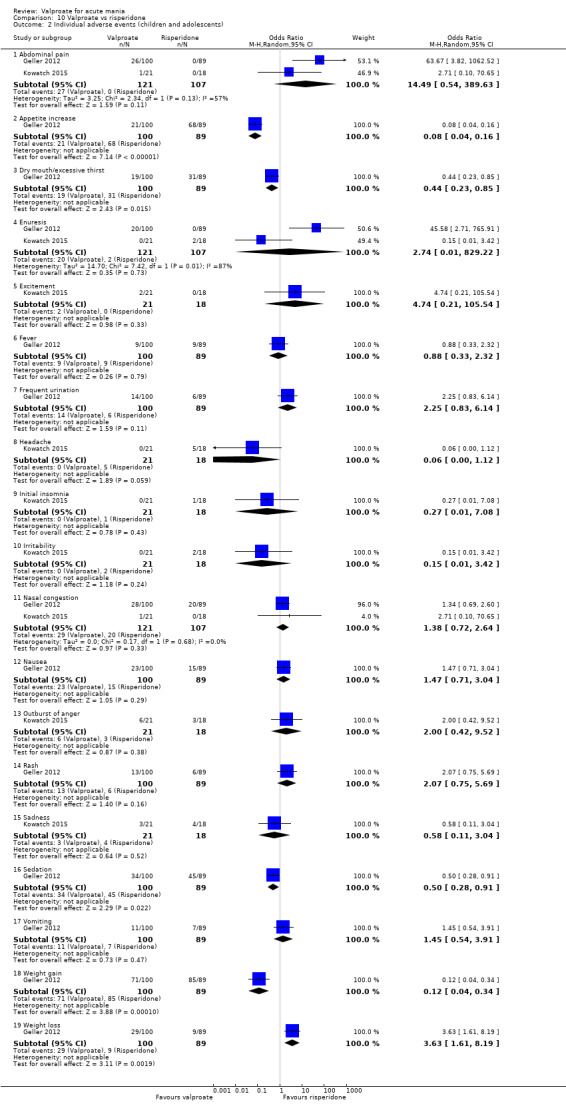
Comparison 10 Valproate vs risperidone, Outcome 2 Individual adverse events (children and adolescents).
22. Adverse events ‐ valproate vs risperidone (children and adolescents).
| Adverse effect | Odds ratio; 95% CI; heterogeneity; N of studies, N of participants | P value |
| Abdominal pain | OR 14.49, 95% CI 0.54 to 389.63; I2 = 57%; 2 studies, 228 participants | P = 0.11 |
| Appetite increase | OR 0.08, 95% CI 0.04 to 0.16; 1 study, 189 participants | P < 0.001 |
| Dry mouth/excessive thirst | OR 0.44, 95% CI 0.23 to 0.85; 1 study, 189 participants | P = 0.01 |
| Enuresis | OR 2.74, 95% CI 0.01 to 829.22; I2 = 87%; 2 studies, 228 participants | P = 0.73 |
| Excitement | OR 4.74, 95% CI 0.21 to 105.54; 1 study, 39 participants | P = 0.33 |
| Fever | OR 0.88, 95% CI 0.33 to 2.32; 1 study, 189 participants | P = 0.79 |
| Frequent urination | OR 2.25, 95% CI 0.83 to 6.14; 1 study, 189 participants | P = 0.11 |
| Headache | OR 0.06, 95% CI 0.00 to 1.12; 1 study, 39 participants | P = 0.06 |
| Initial insomnia | OR 0.27, 95% CI 0.01 to 7.08; 1 study, 39 participants | P = 0.43 |
| Irritability | OR 0.15, 95% CI 0.01 to 3.42; 1 study, 39 participants | P = 0.24 |
| Nasal congesion | OR 1.38, 95% CI 0.72 to 2.64; I2 = 0%; 2 studies, 228 participants | P = 0.33 |
| Nausea | OR 1.47, 95% CI 0.71 to 3.04; 1 study, 189 participants | P = 0.29 |
| Outburst of anger | OR 2.00, 95% CI 0.42 to 9.52; 1 study, 39 participants | P = 0.38 |
| Rash | OR 2.07, 95% CI 0.75 to 5.69; 1 study, 189 participants | P = 0.16 |
| Sadness | OR 0.58, 95% CI 0.11 to 3.04; 1 study, 39 participants | P = 0.52 |
| Sedation | OR 0.50, 95% CI 0.28 to 0.91; 1 study, 189 participants | P = 0.02 |
| Vomiting | OR 1.45, 95% CI 0.54 to 3.91; 1 study, 189 participants | P = 0.47 |
| Weight gain | OR 0.12, 95% CI 0.04 to 0.34; 1 study, 189 participants | P < 0.001 |
| Weight loss | OR 3.63, 95% CI 1.61 to 8.19; 1 study, 189 participants | P = 0.002 |
10.3. Efficacy (continuous): We assessed the efficacy of valproate by assessing the change in symptom severity, using mean endpoint scores or mean change scores on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: children and adolescents
There was low‐quality evidence for an effect of risperidone on mania rating scores between 5 and 12 weeks: SMD 1.01, 95% CI 0.74 to 1.29; P < 0.001 ,I2 = 0%; 2 studies, 228 participants. See Analysis 10.3.
10.3. Analysis.
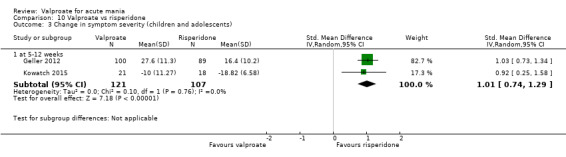
Comparison 10 Valproate vs risperidone, Outcome 3 Change in symptom severity (children and adolescents).
10.4. Acceptability (dichotomous): Information on dropouts due to 1. side effects 2. lack of efficacy 3. any other reasons 4. all causes, during the trial as a proportion of the total number of randomised participants: children and adolescents
Low‐quality evidence showed that more participants in the valproate group dropped out due to all possible causes compared to the risperidone group: OR 1.96, 95% CI 1.00 to 3.82; P = 0.05, I2 = 0%; 2 studies, 236 participants. There was not strong evidence for a difference in dropouts due to adverse events (OR 1.39, 95% CI 0.35 to 5.52; P = 0.05, I2 = 0%; 2 studies, 236 participants), dropouts due to inefficacy (OR 2.71, 95% CI 0.10 to 70.65; P = 0.55; 1 study, 39 participants), and dropouts due to other reasons (OR 1.89, 95% CI 0.91 to 3.90; P = 0.09, I2 = 0%; 2 studies, 236 participants). See Analysis 10.4.
10.4. Analysis.
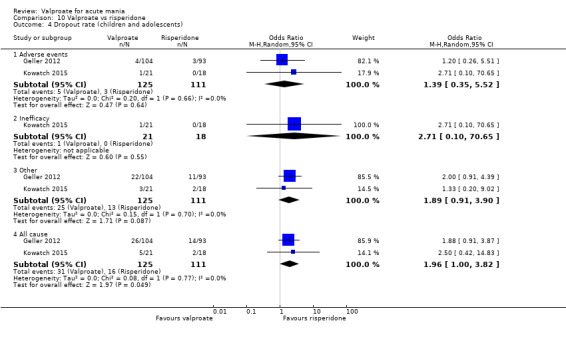
Comparison 10 Valproate vs risperidone, Outcome 4 Dropout rate (children and adolescents).
10.5. Global functioning: the proportion of participants who improved at endpoint based on the final scores of 1 ‐ 2 on the CGI‐I or CGI‐BP‐I: children and adolescents
There was evidence for an effect of risperidone on global functioning scores at six weeks: OR 0.14, 95% CI 0.03 to 0.75; P = 0.02; 1 study, 39 participants. See Analysis 10.5.
10.5. Analysis.
Comparison 10 Valproate vs risperidone, Outcome 5 Global functioning (children and adolescents).
Comparison 11: Valproate + risperidone versus risperidone monotherapy
One study contributed to this comparison (Moosavi 2014). In addition to the results on remission at the time points presented below, Moosavi 2014 also provided further data which showed there was not strong evidence for a difference in remission rate at two weeks (OR 0.73, 95% CI 0.23 to 2.28; P = 0.59; 1 study, 48 participants) nor for partial or full remission at two weeks (OR 0.71, 95% CI 0.18 to 2.74; P = 0.62; 1 study, 48 participants). See Analysis 11.2 and Analysis 11.3.
11.2. Analysis.
Comparison 11 Valproate + risperidone vs risperidone alone, Outcome 2 Remission (adults).
11.3. Analysis.
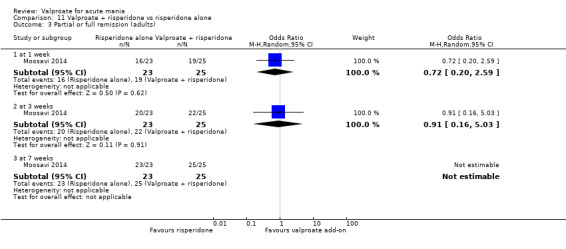
Comparison 11 Valproate + risperidone vs risperidone alone, Outcome 3 Partial or full remission (adults).
Primary outcomes
11.1. Tolerability (dichotomous): number with any adverse event: adults
Very low‐quality evidence found that there was not strong evidence of a difference in experiencing any adverse events between the two groups: OR 0.93, 95% CI 0.27 to 3.16; P = 0.91; 1 study, 48 participants. See Analysis 11.1.
11.1. Analysis.
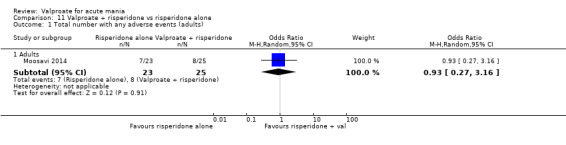
Comparison 11 Valproate + risperidone vs risperidone alone, Outcome 1 Total number with any adverse events (adults).
Secondary outcomes
11.2. Remission (dichotomous): a score of 12 or less on the YMRS ‐ or equivalent on other validated mania rating scale: adults
For this comparison, Moosavi 2014 classified a participant as being in 'full remission' if they displayed no DSM‐IV‐ TR criteria.
Very low‐quality evidence found that there was not strong evidence for a difference in remission rate at one week (OR 0.66, 95% CI 0.20 to 2.17; P = 0.49; 1 study, 48 participants), at three weeks (OR 0.91, 95% CI 0.16 to 5.03; P = 0.91; 1 study, 48 participants), and at seven weeks (OR 0.91, 95% CI 0.12 to 7.07; P = 0.93; 1 study, 48 participants) between the two groups. See Analysis 11.2.
11.3. Partial or full remission (dichotomous): a score of 12 or less on the YMRS (or equivalent on other validated mania rating scales): adults
For this comparison, Moosavi 2014 classified a participant as being in 'partial remission' if they displayed only one or two DSM‐IV‐TR criteria. There was not strong evidence for a difference in partial‐remission rate at one week (OR 0.72, 95% CI 0.20 to 2.59; P = 0.62; 1 study, 48 participants), or at three weeks (OR 0.91, 95% CI 0.16 to 5.03; P = 0.91; 1 study, 48 participants) between the two groups. The results at seven weeks were not estimable. See Analysis 11.3.
Comparison 12: Valproate versus topiramate
Two studies contributed to this comparison (Hebrani 2009; Mahmoudi‐Gharaei 2012). Both studies investigated the effects of valproate in children and adolescents. The results for continuous efficacy were reported at six weeks by Mahmoudi‐Gharaei 2012 (OR 0.05, 95% CI −0.68 to 0.78; P = 0.89; 1 study, 29 participants) and at eight weeks by Hebrani 2009 (OR −0.73, 95% CI −1.10 to −0.36; P < 0.001; 1 study, 120 participants). We aggregated these results to reflect overall long‐term efficacy outcomes (5 ‐ 12 weeks).
Primary outcomes
No data on primary outcomes were available for this comparison.
Secondary outcomes
12.1. Tolerability (dichotomous): individual adverse events: children and adolescents
There was evidence that more participants in the valproate group experienced drowsiness (OR 0.34, 95% CI 0.15 to 0.79; P = 0.01; 1 study, 142 participants; Analysis 12.1.3) and nausea (OR 0.40, 95% CI 0.17 to 0.95; P = 0.04; 1 study, 142 participants; Analysis 12.1.7) compared to topiramate, although the magnitude of these effects is unclear. See Analysis 12.1 and Table 29 for a complete overview of all adverse events.
12.1. Analysis.
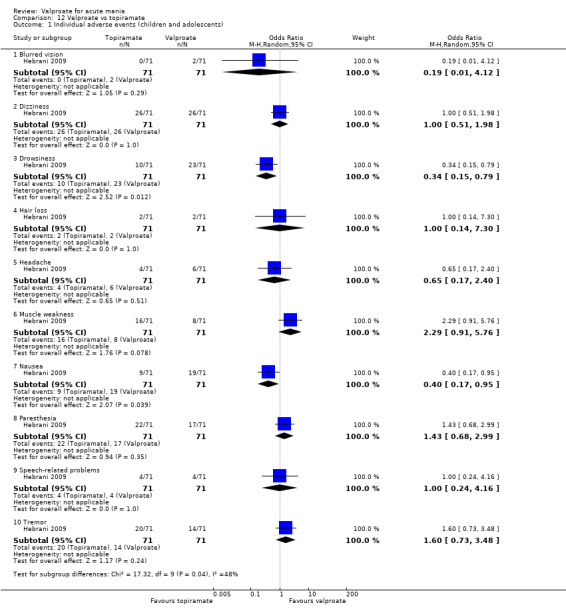
Comparison 12 Valproate vs topiramate, Outcome 1 Individual adverse events (children and adolescents).
23. Adverse events ‐ valproate vs topiramate (children and adolescents).
| Adverse effect | Odds ratio; 95% CI; heterogeneity; N of studies, N of participants | P value |
| Blurred vision | OR 0.19, 95% CI 0.01 to 4.12; 1 study, 142 participants | P = 0.29 |
| Dizziness | OR 1.00, 95% CI 0.51 to 1.98; 1 study, 142 participants | P = 1.00 |
| Drowsiness | OR 0.34, 95% CI 0.15 to 0.79; 1 study, 142 participants | P = 0.01 |
| Hair loss | OR 1.00, 95% CI 0.14 to 7.30; 1 study, 142 participants | P = 1.00 |
| Headache | OR 0.65, 95% CI 0.17 to 2.40; 1 study, 142 participants | P = 0.51 |
| Muscle weakness | OR 2.29, 95% CI 0.91 to 5.76; 1 study, 142 participants | P = 0.08 |
| Nausea | OR 0.40, 95% CI 0.17 to 0.95; 1 study, 142 participants | P = 0.04 |
| Paraesthesia | OR 1.43, 95% CI 0.68 to 2.99; 1 study, 142 participants | P = 0.35 |
| Speech‐related problems | OR 1.00, 95% CI 0.24 to 4.16; 1 study, 142 participants | P = 1.00 |
| Tremor | OR 1.60, 95% CI 0.73 to 3.48; 1 study, 142 participants | P = 0.24 |
12.2. Efficacy (continuous): We assessed the efficacy of valproate by assessing the change in symptom severity, using mean endpoint scores or mean change scores on the YMRS ‐ or any other equivalent standardised rating scale ‐ from baseline: children and adolescents
Very low‐quality evidence found that there was no evidence for a difference in mania rating scores between the two groups between 5 and 12 weeks: SMD −0.41, 95% CI −1.16 to 0.35; P = 0.29, I2 = 72%; 2 studies, 149 participants. See Analysis 12.2.1. This measure contains a large amount of heterogeneity (I2 = 72%). This is probably due to the differences between the studies in this outcome. Hebrani 2009 compared the use of valproate and topiramate as monotherapies and found that valproate was significantly more effective. In contrast, Mahmoudi‐Gharaei 2012 compared the use of valproate and topiramate as a third add‐on medication to lithium and risperidone. These different scenarios may explain the substantially different results in these two studies.
12.2. Analysis.
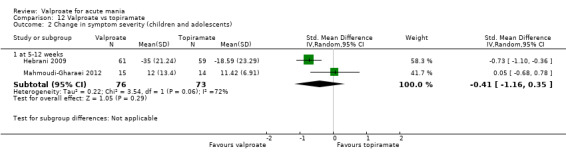
Comparison 12 Valproate vs topiramate, Outcome 2 Change in symptom severity (children and adolescents).
12.3. Acceptability (dichotomous): Information on dropouts due to 1. side effects 2. lack of efficacy 3. any other reasons 4. all causes, during the trial as a proportion of the total number of randomised participants: children and adolescents
There was not strong evidence for a difference in dropout rates due to adverse events (OR 0.31, 95% CI 0.01 to 8.28; P = 0.49; 1 study, 30 participants) or dropouts due to all causes (OR 0.31, 95% CI 0.01 to 8.28; P = 0.49; 1 study, 30 participants) between the two groups. See Analysis 12.3.
12.3. Analysis.
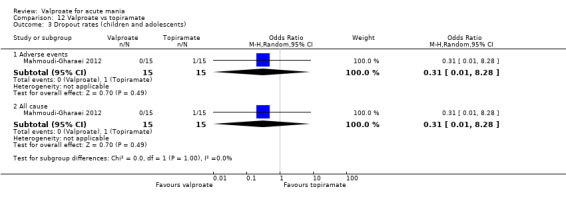
Comparison 12 Valproate vs topiramate, Outcome 3 Dropout rates (children and adolescents).
Sensitivity analyses
1. Excluding treatment‐resistant mania
Only three studies examined treatment‐resistant mania (Freeman 1992; Mahmoudi‐Gharaei 2012; Pope 1991).
The sensitivity analyses run on the placebo comparison response rate for adults excluding Pope 1991 did not affect the results, and the confidence interval and the P value remained significant (Before: OR 2.05, 95% CI 1.32 to 3.20; P = 0.001, I2 = 48%; 4 studies, 869 participants; versus After: OR 1.83, 95% CI 1.31 to 2.55; P < 0.001, I2 = 18%; 3 studies, 826 participants; Analysis 13.1). We did not conduct an analysis of the placebo comparison tolerability primary outcome (number with any adverse effect), as none of the above studies contributed to these results.
13.1. Analysis.
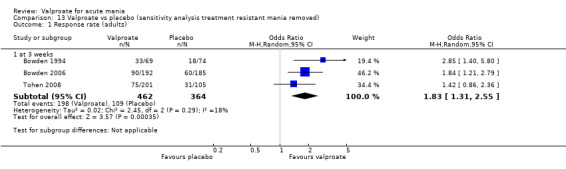
Comparison 13 Valproate vs placebo (sensitivity analysis treatment resistant mania removed), Outcome 1 Response rate (adults).
The sensitivity analyses run on the lithium comparison response rate for adults excluding Freeman 1992 did not materially affect the results, and the confidence interval and the P value remained non‐significant (Before: OR 0.80, 95% CI 0.48 to 1.35; P = 0.40, I2 = 16%; 3 studies, 356 participants; versus After: OR 0.86, 95% CI 0.55 to 1.35; P = 0.52, I2 = 0%; 2 studies, 329 participants; Analysis 14.1). We did not conduct an analysis on the lithium comparison tolerability primary outcome (number with any adverse effect), as none of the above studies contributed to these results.
14.1. Analysis.
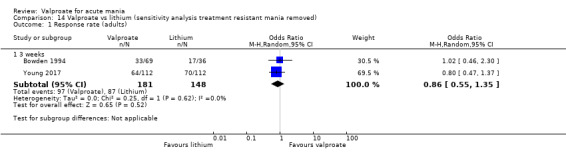
Comparison 14 Valproate vs lithium (sensitivity analysis treatment resistant mania removed), Outcome 1 Response rate (adults).
We could not run an analysis excluding Mahmoudi‐Gharaei 2012 which examined topiramate and valproate, as it was the only study in its section.
2. Excluding studies with unclear blinding or allocation bias
Only two studies have completely unbiased blinding or allocation (Bowden 2006; Bowden 1994).
The sensitivity analyses run on the placebo comparison response in adults included both Bowden 2006 and Bowden 1994. The analysis on the response rate did not affect the results, and the confidence interval and the P value remained significant (Before: OR 2.05, 95% CI 1.32 to 3.20; P = 0.001, I2 = 48%; 4 studies, 869 participants; versus After: OR 2.08, 95% CI 1.41 to 3.05; P < 0.001, I2 = 8%; 2 studies, 520 participants; Analysis 15.1). The sensitivity analyses run on the placebo comparison tolerability primary outcome (number with any adverse effect) did not affect the results, and the confidence interval and the P value remained significant (Before: OR 1.63, 95% CI 1.13 to 2.36; P = 0.009, I2 = 0%; 3 studies, 745 participants; versus After: OR 1.88, 95% CI 1.22 to 2.90; P = 0.004, I2 = 0%; 2 studies, 520 participants; Analysis 15.2).
15.1. Analysis.
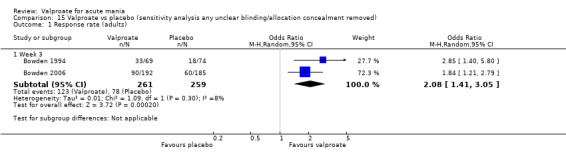
Comparison 15 Valproate vs placebo (sensitivity analysis any unclear blinding/allocation concealment removed), Outcome 1 Response rate (adults).
15.2. Analysis.
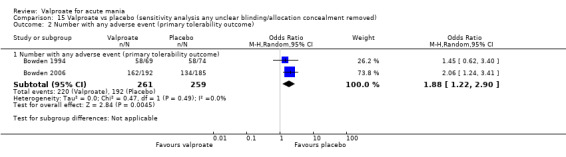
Comparison 15 Valproate vs placebo (sensitivity analysis any unclear blinding/allocation concealment removed), Outcome 2 Number with any adverse event (primary tolerability outcome).
The sensitivity analyses run on the lithium comparison included just Bowden 1994. The sensitivity analysis run on the response rate did not affect the results, and the confidence interval and the P value remained non‐significant (Before: OR 0.80, 95% CI 0.48 to 1.35; P = 0.40, I2 = 16%; 3 studies, 356 participants; versus After: OR 1.02, 95% CI 0.46 to 2.30; P = 0.95; 1 study, 105 participants; Analysis 16.1).
16.1. Analysis.
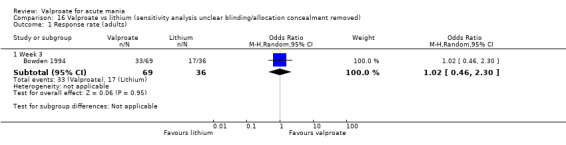
Comparison 16 Valproate vs lithium (sensitivity analysis unclear blinding/allocation concealment removed), Outcome 1 Response rate (adults).
The sensitivity analysis run on the tolerability primary outcome (number with any adverse effect) did not affect the results, and the confidence interval and the P value remained non‐significant (Before: OR 0.61, 95% CI 0.25 to 1.50; P = 0.28, I2 = 0%; 2 studies, 164 participants; versus After: OR 0.48, 95% CI 0.12 to 1.84; P = 0.28; 1 study, 105 participants; Analysis 16.2).
16.2. Analysis.
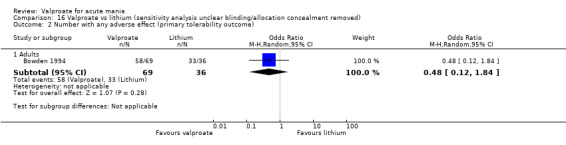
Comparison 16 Valproate vs lithium (sensitivity analysis unclear blinding/allocation concealment removed), Outcome 2 Number with any adverse effect (primary tolerability outcome).
3. Excluding add‐on medication
Only the lithium versus topiramate comparison contained both add‐on (Mahmoudi‐Gharaei 2012) and monotherapy studies (Hebrani 2009). However these studies did not report data about primary outcomes, so no sensitivity analyses were possible.
4. Excluding studies with estimated standard deviations
We did not estimate any standard deviations for any data in primary outcomes. We estimated the standard deviations for secondary outcomes in one study (Hirschfeld 2010).
We used a standard deviation estimate for Hirschfeld 2010 in our analysis of change in symptom severity for valproate versus placebo (Analysis 1.8). We decided to use the suggested tactic of preferring the most conservative standard deviation from similar studies. For Hirschfeld 2010 we have therefore used 10.9 as the most conservative standard deviation in other comparable studies. Removing Hirschfeld 2010 for the sensitivity analysis did not materially change the results, although they did become marginally statically non‐significant (Before: SMD −0.23, 95% CI −0.45 to −0.00; P = 0.05, I2 = 57%; 4 studies, 907 participants; versus After: SMD −0.31, 95% CI −0.63 to 0.02; P = 0.07, I2 = 70%; 3 studies, 685 participants; Analysis 17.1).
17.1. Analysis.
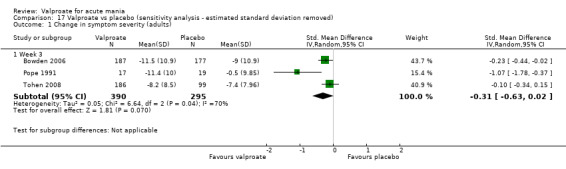
Comparison 17 Valproate vs placebo (sensitivity analysis ‐ estimated standard deviation removed), Outcome 1 Change in symptom severity (adults).
5. Comparing random‐effects analyses to fixed‐effect analyses
We checked the results using both random‐effects and fixed‐effect models for all primary outcomes on all comparisons. Exchanging fixed‐effect analyses for random‐effects analyses made no material difference to any primary outcome.
6. Excluding studies with psychotic features
Only one study included participants with psychotic features (McElroy 1996), whereas three studies excluded patients with psychotic features (McElroy 2010; Moosavi 2014; Tohen 2008). There are not enough trials with enriched numbers of participants with psychotic features to run a sensitivity analysis on these. We have therefore run this sensitivity analysis by excluding all trials with any participants with psychotic features in our primary outcomes.
None of the studies contributing to the valproate versus lithium or risperidone comparisons presented primary outcomes, so we did not conduct a sensitivity analysis for these.
For the valproate versus placebo comparison, McElroy 2010 and Tohen 2008 excluded people with psychotic features for the primary outcomes. Of these, only Tohen 2008 reported response rates in participants before four weeks. Including only Tohen 2008 in the analysis did not change the pattern of results, although they did become statically non‐significant by virtue of having fewer studies (Before: OR 2.05, 95% CI 1.32 to 3.20; P = 0.001, I2 = 48%; 4 studies, 869 participants; versus After: OR 1.42, 95% CI 0.86 to 2.36; P = 0.17; 1 study, 306 participants; Analysis 18.1)
18.1. Analysis.
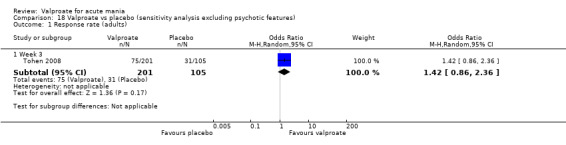
Comparison 18 Valproate vs placebo (sensitivity analysis excluding psychotic features), Outcome 1 Response rate (adults).
For the valproate versus olanzapine comparison, Tohen 2008 was the only study with no participants with psychotic features. Due to the difference in our findings in this comparison between our response rate and change in symptom severity, we decided also to run sensitivity analyses on our secondary measure of the change in symptom severity, as well as the primary outcomes. The response rate trended away from significance in this sensitivity analysis (Before: OR 0.77, 95% CI 0.48 to 1.25; P = 0.29, I2 = 57%; 2 studies, 667 participants; versus After: OR 0.97, 95% CI 0.65 to 1.44; P = 0.86; 1 study, 416 participants; Analysis 19.1). This may be in line with the hypothesis that mania without psychotic features may respond less well to antipsychotics. The change in mania rating scale effect became non‐significant in this sensitivity analysis, although the direction of effect did not materially change (Before: SMD 0.26, 95% CI 0.10 to 0.42; P = 0.001, I2 = 19%; 4 studies, 826 participants; versus After: SMD 0.14, 95% CI −0.06 to 0.34; P = 0.17; 1 study, 387 participants; Analysis 19.2).
19.1. Analysis.
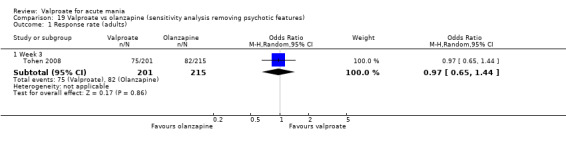
Comparison 19 Valproate vs olanzapine (sensitivity analysis removing psychotic features), Outcome 1 Response rate (adults).
19.2. Analysis.
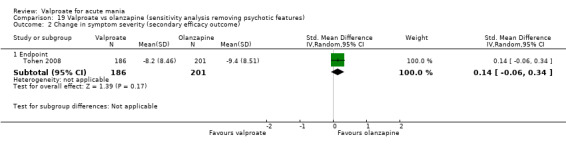
Comparison 19 Valproate vs olanzapine (sensitivity analysis removing psychotic features), Outcome 2 Change in symptom severity (secondary efficacy outcome).
Discussion
Summary of main results
A summary of the results for each comparison can be found in the 'Summary of findings' tables. A full accounting of all comparisons can be found in the forest plot section of the results (Data and analyses).
This review included 25 trials, 15 more than the previous update (Macritchie 2003), and expanded comparisons of valproate, most notably to risperidone in children and adolescents. It is worth noting, however, that there were only six studies in the paediatric population and even among our adult comparisons only the placebo versus valproate comparison had five or more trials. This means that the data are not yet at the stage where the meta‐analysis allows for a comprehensive aggregation of effects and therefore increases uncertainty about the estimates made about outcomes in this review.
The emerging body of research on the effects of valproate in children and adolescents failed to establish efficacy for the use of valproate. There was very low‐quality evidence that failed to find a difference in the response rate of valproate compared to placebo at four weeks. Furthermore, low‐quality evidence in this review suggested a higher response rate of risperidone over valproate at eight weeks. The response rate was not reported in the valproate‐quetiapine comparisons; there was also moderate‐quality evidence that quetiapine resulted in higher rates of remission (secondary outcome) than valproate at four weeks. This suggests that younger populations with bipolar may have a substantially different response profile from older patient populations, which should be considered when prescribing for this patient group (Findling 2018).
For adults, there is good evidence for the use of valproate over placebo. Specifically, high‐quality evidence showed higher response rates in participants treated with valproate compared to placebo at three weeks, and moderate‐quality evidence showed that valproate is more effective in reducing manic rating scores at three weeks.
The evidence was mixed for olanzapine's efficacy compared to valproate in adults. Two studies addressed the primary outcome of response rate, with low‐quality evidence not supporting a difference between the treatments at three weeks. It is important, however, to note that both these were conducted by the same group and differed substantially in their outcomes, which makes the conclusion of non‐superiority difficult to interpret. Their different findings could be due to a difference in the inclusion of participants with psychotic symptoms and severity of mania. The study that did not find a difference had excluded people with more severe mania (YMRS higher than 30) and with psychotic symptoms. The more sensitive quantitative analysis of mania rating scale changes, a secondary outcome, which could encompass results from four studies, showed high‐quality evidence for a superior effect of olanzapine at three weeks, although this difference was small. Overall, given the higher quality of this evidence, we think that olanzapine may be more efficacious than valproate in the acute setting at controlling mania. This conclusion is in line with evidence from Cipriani 2011. However, as expected this review was unable to be as definitive in its findings as the network meta‐analysis, which allows for increased precision through multiple comparisons.
The results concerning lithium and valproate were similarly mixed (McKnight 2019). While moderate‐quality evidence showed no evidence for a difference in response rates between the two treatments at three weeks, very low‐quality evidence examining changes in mania scores at three weeks (secondary efficacy measure) suggested lithium was more efficacious than valproate. These quantitative results are from two small and poor‐quality studies, while three larger, better‐designed trials indicated no difference in response rates between the two groups. There is, however, some evidence to suggest any superiority of lithium in the acute setting (Duffy 2018).
For the other comparisons, there was very low‐quality evidence which found no difference in response rates between valproate and carbamazepine, endoxifen or haloperidol in adults or between valproate and lithium in children and adolescents. Very low‐quality evidence found no difference in remission rates (secondary measure) between valproate and oxcarbazepine in adults at 12 weeks. There was very low‐quality evidence for a difference in mania rating scale changes (secondary measure) between valproate as an add‐on therapy to olanzapine or compared to topiramate in adults. We were unable to extract any useful efficacy data for the comparison of quetiapine versus valproate in adults. These comparisons contained low numbers of small‐scale studies, which limit the conclusions that can be drawn from them.
For tolerability, there was moderate‐quality evidence that valproate causes side effects in more people than placebo in adults. There was low‐quality evidence that valproate causes side effects in fewer people compared to carbamazepine, and that oxcarbazepine causes fewer side effects then valproate. There is very low‐quality evidence for no difference in the number of people with side effects when comparing valproate to endoxifen, or as an add‐on therapy to risperidone. Moderate‐quality evidence did not find a difference in the numbers with side effects between lithium and valproate. The main tolerability outcome was not reported in comparisons of valproate versus haloperidol, quetiapine, risperidone or topiramate. Individual side effects varied, including high‐quality evidence that olanzapine causes significantly more weight gain and sedation than valproate in adults, while very low‐quality evidence shows that risperidone causes more sedation and weight gain than valproate in children.
Overall completeness and applicability of evidence
We included all trials that were in previous network meta‐analysis, suggesting our search was thorough and robust. Furthermore, we searched the grey literature and should, therefore, have found other data that would have been applicable to this review if present. Remission and response to valproate were not reported for many of the studies, even though they are important in terms of clinical practice and prognosis, which affects the completeness of the evidence base. The lack of clear reporting of response rates has a substantial negative effect on the ability of this review to come to a conclusion on key questions such as 'Is olanzapine more effective than valproate for the treatment of acute mania?'. Furthermore, several published papers contain data without standard deviations, which renders the data more difficult to interpret. Standard efficacy measures that are reported across trials in this area are sorely needed, to enable data aggregation strategies to be conducted more effectively. Finally, most trials only reported results at the endpoint, which limited the temporal data we could extract which might be able to show certain treatments acting more rapidly.
The studies in this review included participants with a range of severity of manic symptoms, as shown by the different entrance scores on the Young Mania Rating Scale (YMRS). This heterogeneity contributes to that seen in our results. It may also limit the applicability of results to individual patients, depending on the severity of that patient’s presentation. However, most studies using YMRS used a cut‐off of 17 to 20 to recruit participants. Other potential sources of heterogeneity include studies that include or exclude patients with psychotic features (e.g. Tohen 2002 versus Tohen 2008) and analyses that include old trials with very small numbers of participants (e.g. Pope 1991). It is important to note that, due to ethical and practical considerations, severely ill and psychotic patients may not have been well represented in the available trials. The applicability of the findings of this review to the spectrum of bipolar patients seen in clinical practice, therefore, has its limits, and this should be borne in mind when using the results. This is a well‐known problem with the available literature on mania (Licht 2002). Furthermore, mania is only one element of bipolar disorder, although the most externally disruptive element. Depressive episodes which predominate in bipolar disorder are not considered in this review. Furthermore, there is an increasing understanding that mood instability in inter‐episode periods is a major contributor to morbidity in bipolar disorder (Altshuler 2006; Bonsall 2015). Future reviews may wish to consider the effects that medication such as valproate can have on this inter‐episode variation that can contribute much to morbidity.
It is worth noting that only six studies were carried out in children and adolescents. This reflects the recency of research on bipolar disorder in children and adolescents, and also the increased difficulty of consenting for true blinding and randomisation in a younger population. The paucity of research in this field means that we should be cautious in reaching categorical conclusions. Indeed, even among our adult comparisons only the placebo versus valproate comparison had five or more trials. This means that the data are not yet at the stage where meta‐analysis allows for a completely comprehensive aggregation of effects and increases uncertainty about the outcomes of this review.
Finally, the review deals with the acute setting of mania, with the most common length of endpoints being three weeks. Most people who experience a manic episode will need to be on long‐term treatment; the implication of side effects over three weeks is very different from any longer‐term effects. For example, any metabolic effects of second‐generation antipsychotics (SGAs), while only a minor inconvenience over three weeks, contribute significantly to morbidity if continued over the long term (Alvarez‐Jiménez 2008; De Hert 2011). Additionally, the effectiveness of SGAs when examined over the short term is probably helped by their more sedative profile, but this may not translate into efficacy in the longer term. Thus, while this review is helpful for the acute management of mania, its ability to help guide longer‐term treatment is limited.
Quality of the evidence
We assessed the quality of evidence by looking at the risks of bias of the trials in each comparison, imprecision suggested by wide confidence intervals or small numbers of trials/participants, inconsistency as assessed by I2, indirectness if outcomes were not directly generated from comparisons, publication bias, and large effect sizes. The full methods used to assess quality of evidence of studies is detailed in the Methods section under the 'Summary of findings' table section (Sensitivity analysis). The full summary of assessment of risk of bias can also be found in the Methods (Assessment of risk of bias in included studies).
Due to the large scope of this review, the quality of evidence is very mixed. Most of the evidence is classified as low‐ or very low‐quality. This reflects the relatively high rates of bias as seen in the 'Risk of bias summary' (Figure 3). Notable exceptions to this include the comparisons of valproate versus placebo, lithium and olanzapine, where some of the comparisons achieve ratings of moderate or high quality. These three comparisons as a whole involve more trials than the others and involve hundreds of participants. Additionally, they involve trials that are better‐designed with a lower risk of bias than other comparisons.
Particular issues that came up in assessing bias were that randomisation and allocation bias remained hard to assess due to unclear reporting in study reports. Furthermore, a continuing issue remains that most studies do not publish protocols before the study begins. This is reflected in the fact that studies did poorly on the assessment of reporting bias. Pre‐published protocols are a key tool in the fight against post hoc P‐hacking (Simmons 2011), which also enables better tracking of planned studies and therefore any publication bias that might be present. Pre‐published protocols should, therefore, be included as standard operating procedures when planning a study (De Angelis 2004). These are starting to become more common, and all three studies in our review that had low selective reporting bias were published post‐2008. Another issue in our review was that due to the large scope of the question several comparisons between valproate and alternative treatments draw on evidence from only one study, eliminating any benefit from data aggregation and decreasing the quality of evidence in these comparisons.
Potential biases in the review process
Even though we did our utmost to retrieve all relevant studies by searching electronic databases and grey literature, handsearching, and checking reference lists, we cannot rule out the possibility that some relevant data are missing from this review. One potential limitation of this review is that we could not obtain all the relevant data, despite efforts to contact the authors. This was due to a combination of non‐response to our inquires, and to authors who had published historical studies often moving on to later positions and being unable to access the relevant data. This is a common problem in meta‐analyses (Selph 2014). Unfortunately, we could not check statistically for publication bias, as there were not enough data for any single comparison to be able to produce a funnel plot analysis. This means that we cannot rule out publication bias affecting the studies within this meta‐analysis, as we know that this is a problem that has historically affected clinical data (Easterbrook 1991).
Agreements and disagreements with other studies or reviews
This review is an update of the 2003 Cochrane Review on valproate in acute mood episodes in bipolar disorder (Macritchie 2003). The previous review contained ten trials examining five comparisons, whereas the current review examines 25 trials with 12 comparisons. The current update concurred with its previous iteration that there is consistent evidence that valproate is an efficacious treatment for acute mania in adults compared with placebo. However, this review suggests that valproate may be less efficacious than olanzapine, as although low‐quality evidence found no difference in response rates, high‐quality evidence found that olanzapine causes a greater decrease in manic symptoms than valproate. This finding would align with recently‐published evidence that establishes valproate as being a treatment for mania superior to placebo but inferior to SGAs such as olanzapine (Cipriani 2011; Yildiz 2015). Recent changes to NICE guidelines reflect this gradual shift in the evidence base, with valproate no longer considered a first‐line option (NICE 2018a).
The two most recent reviews of treatments for acute mania (Cipriani 2011; Yildiz 2015) are both network meta‐analyses that concur with our broad findings. We included all the included studies in Yildiz 2015 and Cipriani 2011 that used valproate as a comparison. In terms of standardised mean difference on manic scores (the main outcome in these network meta‐analyses), our review found a similar magnitude of effect comparing placebo and valproate (SMD −0.23, 95% CI −0.45 to −0.00; P = 0.05) as Cipriani 2011 (SMD −0.20, 95% CI −0.37 to −0.04) and Yildiz 2015 (SMD −0.32, 95% CI −0.15 to −0.5). Similarly, our results comparing olanzapine and valproate (SMD 0.26, 95% CI 0.10 to 0.42; P = 0.001) showed few differences from Cipriani 2011 (SMD 0.23, 95% CI 0.06 to 0.40) or from Yildiz 2015 (SMD 0.16, 95% CI −0.04 to 0.34), although it is important to note that Yildiz 2015 (unlike our review) and Cipriani 2011 did not find a statistically significant positive difference. A possible reason for this result may be the inclusion of idiosyncratic drugs such as tamoxifen in the Yildiz 2015 network meta‐analysis. For example, the Yildiz 2015 network meta‐analysis concludes that tamoxifen is significantly more effective then every other antimanic drug, with a standardised mean difference of more than two for every comparison. This is despite Yildiz 2015 only including two trials that tested tamoxifen, both of which were comparing it to placebo as opposed to other active agents. Careful attention needs to be paid to checking consistency and transitivity when doing network meta‐analyses, especially when interventions with only a few comparisons are included.
These recent reviews and the original meta‐analysis (Cipriani 2011; Macritchie 2003; Yildiz 2015) did not examine mania in children and adolescents. Very‐low quality evidence in this review found no evidence of valproate's efficacy compared with placebo, while low‐quality evidence suggested that SGAs outperform valproate in controlling manic symptoms. Hazell 2012 was also unable to establish valproate's efficacy in paediatric and adolescent mania, although (unlike our review) that study is a systematic review without a meta‐analysis. This suggests that paediatric mania may have a different response profile from its adult counterpart , which is similar to what has been found recently for major depressive disorder (Cipriani 2016).
The new CANMAT guidelines (CANMAT 2018) disagree somewhat with our review. The guidelines place multiple treatments on the same level of efficacy but relegate olanzapine to a second‐line drug in light of tolerability concerns. They quote Yildiz 2015 as the source for their efficacy judgements, and as discussed above this meta‐analysis found no difference between olanzapine and valproate. This differs from Cipriani 2011 and from our analysis, which found effects in quantitative measures of mania. The decision to relegate olanzapine to a second‐line choice on the basis of poor tolerability is in contrast with Cipriani 2011, who found the tolerability of olanzapine to be superior to valproate; their primary outcome was dropout rate (OR 0.78, 95% CI 0.52 to 1.17). Our review did not find any reporting of our primary tolerability outcome, but on dropout rates found no difference in acute tolerability of valproate and olanzapine (OR 1.04, 95% CI 0.71 to 1.52). Furthermore, the Yildiz 2015 network meta‐analysis cited as the basis for efficacy also found a non‐significant difference in favour of olanzapine over valproate (OR 0.69, 95% CI 0.47 to 1.02). It is therefore not clear to us where this conclusion in the CANMAT guidelines has come from. Perhaps this decision may be due to long‐term metabolic effects that olanzapine causes (Alvarez‐Jiménez 2008), but this would fit better under the separate safety rating which CANMAT has, as this is not an issue of lack of tolerability. Furthermore, there is a separate column for safety risks as a maintenance treatment, under which repercussions for long‐term health would go; metabolic poor health is not an acute but a longer‐term safety risk. It is, however, important to take into account long‐term safety risks when prescribing in the acute setting. We find it odd that valproate is listed as having no safety concerns in mania, given its large teratogenic risk as discussed below.
Authors' conclusions
Implications for practice.
There is consistent evidence that valproate is an efficacious treatment for acute mania in adults, with high‐quality evidence showing superiority over placebo in response rate. Moderate‐quality evidence did not find a difference between lithium and valproate for response rate; use of these medications should be decided on their side‐effects profiles and long‐term effects. Comparisons with most other medications are of insufficient quality and conclusiveness to be used as a basis for practice.
Our findings are varied with regards to olanzapine with low‐quality evidence finding no evidence of difference in response rate but high quality evidence showing a small effect difference in favour of olanzapine with regards to reducing manic symptoms. This suggest that olanzapine could be a slightly more effective treatment for mania compared to valproate in the acute setting. High‐quality evidence suggests that valproate causes less sedation and weight gain than olanzapine and avoidance of sedation or metabolic side effects may represent a reason to prescribe valproate over olanzapine. This is especially true when considering long‐term management when starting acute therapy that is likely to be continued.
The emerging data on acute mania in children suggest the possibility of a substantially different response profile compared to adults. Low‐quality evidence in this review suggests that risperidone may have increased efficacy over valproate, while very low‐quality evidence fails to establish the efficacy of valproate over placebo. However, these data were from only one or two trials that used an open approach, and were thus at a high risk of bias. This makes the overall quality of the evidence poor, limiting confidence in these conclusions and thus the extent to which it can be trusted to make clinical decisions. However, it is worth noting that currently there is a lack of evidence showing valproate's efficacy in this age group, if not any strong evidence of a lack of effect. The use of valproate to augment monotherapy in treating paediatric and adolescent mania is not something for which this review was able to find evidence of any quality.
The choice of antimanic drug in clinical practice must also be influenced by other situational concerns such as previous response to any medications, the need for prophylactic treatment, comorbid physical illness, specific adverse effect profiles of the drug, and patients’ choice (Vitiello 2018). With regard to adverse events, the teratogenicity of valproate is an important event which cannot be analysed in the short three‐week studies that this meta‐analysis encompassed. However, the risks that valproate poses need to be weighed up against the risks inherent in a manic condition which the valproate could potentially ameliorate.
Implications for research.
This review has highlighted that too many published studies lack complete information on study conduct, and entail a plethora of biases (Barbui 2004). Future researchers must focus on avoiding common biases detected by this and other systematic reviews to ensure the validity of their research results. One way to achieve this goal would be to emphasize the importance of publishing detailed study protocols in which concrete research plans are outlined and fellow researchers' input on trial design is sought before the trial begins. Future researchers should aim to increase the transparency of a trial's characteristics in published articles, allowing fellow researchers to immediately assess the quality of the data and to facilitate good scientific conduct for follow‐up studies. Investigators should start using online data‐sharing to further strengthen this transparency. Specifically, at the point of trial completion, the whole data set should be archived online and be made readily available for fellow researchers (Furukawa 2016a). One recurring problem in the published reports used for this review was the lack of reporting on data points beyond baseline and endpoint measures. These data, however, would be extremely useful to establish the time course of any medication, especially when focusing on the efficacy of short, acute episodes, and we would like to encourage future researchers not to neglect the reporting of all intervals.
In view of the practical and ethical difficulties surrounding the inclusion of a placebo group, especially for severely ill patients, future trials should focus on the comparison of valproate with other medications (Geddes 2015). The lack of evidence on valproate as an add‐on treatment could help identify directions for future research about valproate. Only two trials here addressed the question of the use of valproate as an add on agent (Xu 2015, Moosavi 2014). However monotherapy with anticonvulsants for adults is increasingly rare, decreasing from 10% to 4% from 2005 ‐ 2012 (Kleimann 2016) and most medication prescriptions in bipolar disorder are dual prescriptions, usually of a SGA combined with a mood stabiliser (Kleimann 2016, Karanti 2016). It would, therefore, seem prudent to compare valproate versus other interventions as add‐on therapies in a more systematic way in randomised controlled trials. This would also help with clinical decision‐making when patients who are already on a long‐term relapse‐prevention treatment become manic (Miura 2014). As discussed previously, these trials should be sure to report both response rates and change in manic symptom scoring scales at successive time points, as well as full lists of adverse events and dropout rates. Future Cochrane Reviews might focus on guiding clinical practice in dual therapy.
Outcome measures of relevance to both patients and clinicians, such as length of hospital stay, occupational and social assessments and reports of patient satisfaction, should be included (Harrison 2016). Trials in this area of research should focus on applying rating scales and outcome measures that can facilitate strong and meaningful research results and also be easily translated into clinical practice. In order to achieve this goal, we would advise researchers to use information from existing systematic reviews as practical guidance to carefully plan study design, and to seek input from fellow experts in the field (Mulder 2018). In addition, good reporting of outcomes measures at all time points is necessary, in a form that is easily extractable into meta‐analysis, in any good‐quality study and using remote technology (Goodday 2019).
One key area in which trials fell short was in the consistency with which they measured valproate levels. Research has shown that the serum concentration of valproate is directly related to efficacy (Allen 2006). However, trials have no consistent way of establishing serum valproate levels, measuring this inconsistently if at all. In addition, several of the trials had a significant proportion of participants below the recommended valproate serum levels, although this did not clearly seem to influence results. In order to allow comparison, it is advisable that a standardised method for assessing valproate levels in medicated patients is established and followed, in order to identify the exact link between valproate concentrations and efficacy.
Finally, although exploration of mania in a paediatric setting has begun, it is still missing a large double‐blinded trial. Now that the baseline efficacy of some of the drugs involved has been established, good‐quality blinded evidence would help consolidate the basis for medication choice in this population.
What's new
| Date | Event | Description |
|---|---|---|
| 7 October 2019 | New citation required and conclusions have changed | Review updated. 15 new studies added. |
| 7 October 2019 | New search has been performed | Updated searches on 28 September 2018. |
History
Protocol first published: Issue 1, 1999 Review first published: Issue 1, 2003
| Date | Event | Description |
|---|---|---|
| 8 September 2002 | New citation required and conclusions have changed | Substantive amendment |
Acknowledgements
The authors are grateful to Cochrane Common Mental Disorders Group for help in constructing and implementing the search strategy. We also acknowledge the contribution of Keith Reid, Karine Macritchie and Allan Young for their work on the previous version of the review. We finally thank Jan Fischer for his help with writing the protocol for this updated review.
CRG Funding Acknowledgement: The National Institute for Health Research (NIHR) is the largest single funder of the Cochrane Common Mental Disorders Group.
Andrea Cipriani is supported by the National Institute for Health Research (NIHR) Oxford Cognitive Health Clinical Research Facility, by an NIHR Research Professorship (grant RP‐2017‐08‐ST2‐006) and by the NIHR Oxford Health Biomedical Research Centre (grant BRC‐1215‐20005).
Disclaimer: The views and opinions expressed herein are those of the authors and do not necessarily reflect those of the UK National Health Service, the NIHR, or the UK Department of Health and Social Care.
Appendices
Appendix 1. Cochrane Specialised Register (CCMDCTR) ‐ core MEDLINE search strategy
Core search strategy used to inform the Cochrane Common Mental Disorders Controlled Trials Register (CCMDCTR): OVID MEDLINE A weekly search alert based on condition + RCT filter only 1. [MeSH Headings]: eating disorders/ or anorexia nervosa/ or binge‐eating disorder/ or bulimia nervosa/ or female athlete triad syndrome/ or pica/ or hyperphagia/ or bulimia/ or self‐injurious behavior/ or self mutilation/ or suicide/ or suicidal ideation/ or suicide, attempted/ or mood disorders/ or affective disorders, psychotic/ or bipolar disorder/ or cyclothymic disorder/ or depressive disorder/ or depression, postpartum/ or depressive disorder, major/ or depressive disorder, treatment‐resistant/ or dysthymic disorder/ or seasonal affective disorder/ or neurotic disorders/ or depression/ or adjustment disorders/ or exp antidepressive agents/ or anxiety disorders/ or agoraphobia/ or neurocirculatory asthenia/ or obsessive‐compulsive disorder/ or obsessive hoarding/ or panic disorder/ or phobic disorders/ or stress disorders, traumatic/ or combat disorders/ or stress disorders, post‐traumatic/ or stress disorders, traumatic, acute/ or anxiety/ or anxiety, castration/ or koro/ or anxiety, separation/ or panic/ or exp anti‐anxiety agents/ or somatoform disorders/ or body dysmorphic disorders/ or conversion disorder/ or hypochondriasis/ or neurasthenia/ or hysteria/ or munchausen syndrome by proxy/ or munchausen syndrome/ or fatigue syndrome, chronic/ or obsessive behavior/ or compulsive behavior/ or behavior, addictive/ or impulse control disorders/ or firesetting behavior/ or gambling/ or trichotillomania/ or stress, psychological/ or burnout, professional/ or sexual dysfunctions, psychological/ or vaginismus/ or Anhedonia/ or Affective Symptoms/ or *Mental Disorders/
2. [Title/ Author Keywords]: (eating disorder* or anorexia nervosa or bulimi* or binge eat* or (self adj (injur* or mutilat*)) or suicide* or suicidal or parasuicid* or mood disorder* or affective disorder* or bipolar i or bipolar ii or (bipolar and (affective or disorder*)) or mania or manic or cyclothymic* or depression or depressive or dysthymi* or neurotic or neurosis or adjustment disorder* or antidepress* or anxiety disorder* or agoraphobia or obsess* or compulsi* or panic or phobi* or ptsd or posttrauma* or post trauma* or combat or somatoform or somati#ation or medical* unexplained or body dysmorphi* or conversion disorder or hypochondria* or neurastheni* or hysteria or munchausen or chronic fatigue* or gambling or trichotillomania or vaginismus or anhedoni* or affective symptoms or mental disorder* or mental health).ti,kf.
3. [RCT filter]: (controlled clinical trial.pt. or randomized controlled trial.pt. or (randomi#ed or randomi#ation).ab,ti. or randomly.ab. or (random* adj3 (administ* or allocat* or assign* or class* or control* or determine* or divide* or distribut* or expose* or fashion or number* or place* or recruit* or subsitut* or treat*)).ab. or placebo*.ab,ti. or drug therapy.fs. or trial.ab,ti. or groups.ab. or (control* adj3 (trial* or study or studies)).ab,ti. or ((singl* or doubl* or tripl* or trebl*) adj3 (blind* or mask* or dummy*)).mp. or clinical trial, phase ii/ or clinical trial, phase iii/ or clinical trial, phase iv/ or randomized controlled trial/ or pragmatic clinical trial/ or (quasi adj (experimental or random*)).ti,ab. or ((waitlist* or wait* list* or treatment as usual or TAU) adj3 (control or group)).ab.)
4. (1 and 2 and 3)
Records were screened for reports of RCTs within the scope of the Cochrane Common Mental Disorders Group. Secondary reports of RCTs were tagged to the appropriate study record. Similar weekly search alerts were also conducted on OVID Embase and PsycINFO, using relevant subject headings (controlled vocabularies) and search syntax, appropriate to each resource.
Appendix 2. Other database searches
Update search ‐ 2016 to 28 September 2018
Cochrane Library Trials database, n = 197
Ovid MEDLINE, n = 80
Ovid Embase, n = 177
Ovid PsycINFO, n = 34
WHO ICTRP, (all years) n = 56
ClinicalTrials.gov (all years), n = 53
TOTAL = 597 De‐dup‐1 = 389 De‐dup‐2 (against 2016 search results (where n = 683)) = 325 to screen (222 from bibliographic databases, 103 trial registry records)
Search Strategies CLib:Trials ℅ Cochrane Register of Studies Online (CRSO) (valpro* or divalpro*) and (bipolar or mania or manic or hypomani* or "affective psycho*" or "rapid cycling" or schizoaffective) AND 01/01/2016 TO 06/09/2018:CD n=197 ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ Database: Ovid MEDLINE(R) and Epub Ahead of Print, In‐Process & Other Non‐Indexed Citations and Daily <1946 to September 27, 2018> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 Valproic Acid/ (11861) 2 (valproate or valproic acid or divalpro*).mp. (18589) 3 1 or 2 (18589) 4 "bipolar and related disorders"/ or bipolar disorder/ (37574) 5 (mania or manic or hypomani*).mp. (18355) 6 affective psychosis.mp. (834) 7 (rapid cycling or schizoaffective).mp. (6448) 8 (psychos* or psychotic or anti‐psycho* or antipsycho*).ti,kf. (86661) 9 or/4‐8 (128201) 10 3 and 9 (2389) 11 controlled clinical trial.pt. (92662) 12 randomized controlled trial.pt. (468815) 13 (randomi#ed or randomi#ation or randomi#ing).ti,ab,kf. (555318) 14 (RCT or "at random" or (random* adj3 (administ* or allocat* or assign* or class* or control* or determine* or divide* or division or distribut* or expose* or fashion or number* or place* or recruit* or split or subsitut* or treat*))).ti,ab,kf. (459916) 15 placebo*.ab,ti,kf. (199065) 16 trial.ab,ti,kf. (520800) 17 groups.ab. (1835758) 18 (control* and (trial or study or group*) and (placebo or waitlist* or wait* list* or ((treatment or care) adj2 usual))).ti,ab,kf,hw. (181135) 19 ((single or double or triple or treble) adj2 (blind* or mask* or dummy)).ti,ab,kf. (159960) 20 double‐blind method/ or random allocation/ or single‐blind method/ (258249) 21 or/11‐20 (2722159) 22 exp animals/ not humans.sh. (4498736) 23 21 not 22 (2295697) 24 10 and 23 (723) 25 (2016* or 2017* or 2018*).yr,dp,dt,ep,ez. (3506290) 26 24 and 25 (80) *************************** Database: Embase <1974 to 2018 Week 39> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 valproic acid/ (59043) 2 (valproate or valproic acid or divalpro*).mp. (64263) 3 1 or 2 (64263) 4 mania/ or hypomania/ or manic psychosis/ (19397) 5 (mania or manic or hypomani*).mp. (37429) 6 bipolar mania/ or "mixed mania and depression"/ or rapid cycling bipolar disorder/ (2936) 7 *bipolar disorder/ (22870) 8 bipolar disorder/dt [Drug Therapy] (11057) 9 (bipolar and (psychos* or psychot*)).mp. (23835) 10 affective psychosis.mp. (2381) 11 (rapid cycling or mixed state?).mp. (2920) 12 or/4‐11 (66961) 13 3 and 12 (8698) 14 randomized controlled trial/ (512565) 15 randomization.de. (79288) 16 controlled clinical trial/ and (Disease Management or Drug Therapy or Prevention or Rehabilitation or Therapy).fs. (251998) 17 *clinical trial/ (17539) 18 placebo.de. (322751) 19 placebo.ti,ab. (275171) 20 trial.ti. (250746) 21 (randomi#ed or randomi#ation or randomi#ing).ti,ab,kw. (783459) 22 (RCT or "at random" or (random* adj3 (administ* or allocat* or assign* or class* or control* or determine* or divide* or division or distribut* or expose* or fashion or number* or place* or recruit* or split or subsitut* or treat*))).ti,ab,kw. (622098) 23 ((singl$ or doubl$ or trebl$ or tripl$) adj3 (blind$ or mask$ or dummy)).mp. (277510) 24 (control* and (trial or study or group) and (placebo or waitlist* or wait* list* or ((treatment or care) adj2 usual))).ti,ab,kw,hw. (315426) 25 or/14‐24 (1522513) 26 ((animal or nonhuman) not (human and (animal or nonhuman))).de. (5201323) 27 25 not 26 (1386891) 28 13 and 27 (2235) 29 (2016* or 2017* or 2018*).yr,dp,dc. (4429602) 30 28 and 29 (177) *************************** Database: PsycINFO <1806 to September Week 4 2018> Search Strategy: ‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐‐ 1 valproic acid/ (1685) 2 (valproate or valproic acid or divalpro*).ti,ot,ab,id. (4817) 3 1 or 2 (4832) 4 mania/ or hypomania/ (5921) 5 (mania or manic or hypomani*).ti,ot,ab,id. (20298) 6 bipolar disorder/ or affective psychosis/ (25603) 7 (bipolar and (psychos* or psychot*)).ti,ot,ab,id. (8197) 8 affective psychosis.ti,ot,ab,id. (913) 9 (rapid cycling or mixed state?).ti,ot,ab,id. (1586) 10 or/4‐9 (39456) 11 clinical trials.sh. (11069) 12 (randomi#ed or randomi#ation or randomi#ing).ti,ab,id. (75573) 13 (RCT or at random or (random* adj3 (administ* or allocat* or assign* or class* or control* or determine* or divide* or division or distribut* or expose* or fashion or number* or place* or recruit* or split or subsitut* or treat*))).ti,ab,id. (90175) 14 (control* and (trial or study or group) and (placebo or waitlist* or wait* list* or ((treatment or care) adj2 usual))).ti,ab,id,hw. (26437) 15 ((single or double or triple or treble) adj2 (blind* or mask* or dummy)).ti,ab,id. (24554) 16 trial.ti. (26642) 17 placebo.ti,ab,id,hw. (37936) 18 treatment outcome.md. (19117) 19 treatment effectiveness evaluation.sh. (22334) 20 mental health program evaluation.sh. (2049) 21 or/11‐20 (178423) 22 3 and 10 and 21 (474) 23 (2016* or 2017* or 2018*).yr,an. (504582) 24 22 and 23 (34) *************************** ICTRP (28‐Sept‐2018) valproate AND mania OR valproate AND manic OR valproic AND mania OR valproic AND manic OR divalproate AND mania OR divalproate AND manic OR divalproex AND mania OR divalproex AND manic ClinicalTrials.gov (28‐Sept‐2018) Condition: mania OR manic Other terms: valproate OR valproic OR divalproate OR divalproex | *****************************************************************************************************
Appendix 3. List of interactions with authors
We contacted Dr Ahmad to request a complete list of experienced side effects by the participants as well as information on baseline characteristics of the study population. We also enquired additional data for the efficacy measures beyond taken at day 4, day 7, day 14, and day 21, because only baseline and endpoint measures were provided in the text (Ahmad 2016). We did not receive a reply.
We contacted Dr Chopra about questions on the biases in Kakkar 2009. Although we received a reply, the authors did not answer our questions.
We liaised with Dr Delbello who helped us to clarify our questions on allocation concealment for DelBello 2006. However, the author was not able to provide us with additional data for time points which were measured but not reported in the text (i.e. all time points except baseline and endpoint).
We contacted Mr Toktam Faghihi to clarify the method of randomisation, concealment, and blinding procedures for Mahmoudi‐Gharaei 2012. However, we did not receive a reply.
We contacted Dr Feifel, to request the standard deviations of the presented results as these were crucial for the analyses. We also requested results for the YMRS, CGI‐S, CGI‐G, BARS, and MADRS which had been taken on day 1, day 3, day 7, day 14, and day 21 according to the study report (Feifel 2011), but had not been reported on in the Results section. We did not receive a reply.
We contacted Dr Freeman to clarify which method of randomisation was used in the study, as well as the procedures in place to ensure that allocation was not predictable. We also intended to confirm with Dr Freeman who conducted the outcome assessments and if these raters were blind to treatment allocation (Freeman 1992). We did not receive a reply.
We contacted Dr Hebrani to clarify the method of sequence generation, blinding in the study, confirm the number of participants randomised, ask about data on efficacy assessments on all reported datapoints in Hebrani 2009. However, we did not receive a response.
We liaised with Professor Hirschfeld to request the original data as well as on biases from his two studies (Hirschfeld 1999; Hirschfeld 2010). He was, however, not able to retrieve any information on these two studies because he had discontinued his work at the University of Texas.
We liaised with Dr Kowatch and the lead pharmacist of Kowatch 2015. Dr Kowatch and the pharmacist explained that they used a random‐number generator to produce a random sequence in the study. Dr Kowatch provided us with additional data which had been outlined in the pre‐published protocol and in the publication ("Subjects were assessed weekly for efficacy"), but were not reported in the text. However, we did not receive the accompanying standard deviations or standard errors of the data and so were unable to use these additional data in the analyses for this review.
We contacted Dr McElroy to request the method of randomisation, randomisation concealment, and blinding procedures for McElroy 1996. We also asked for additional data measures on data points other than baseline and endpoint, as the study report states that data were "collected at baseline and 6 times during the active treatment". We did not receive a reply.
We contacted Dr Moosavi to ask about the method of sequence generation and allocation concealment in Moosavi 2014. We further attempted to clarify which parties were blinded and how the blinding was maintained. Lastly, we tried to confirm that 65 participants were randomised and the reasons for study dropout. We did not receive a response.
We contacted Dr Shafti to request the method of sequence generation and allocation concealment in Shafti 2008. We further requested an overview of participants' baseline characteristics and the titration schedule. We did not receive a reply.
We successfully liaised with Dr Tohen to clarify our question about randomisation techniques and blindness of outcome assessors in his two studies (Tohen 2002; Tohen 2008).
We contacted Dr Wagner to request the method of sequence generation and allocation concealment in Wagner 2009. We further enquired if the outcome assessors were blind and wanted to confirm that the adjunctive medication was balanced between the treatment groups. We did not receive a reply.
We contacted Dr. Young to request the means and standard deviations for the YMRS scores, as well as information on tolerability that had not been provided in Young 2017. However, we did not receive a reply.
We contacted Dr Zajecka to clarify the method of randomisation used, whether or not the raters of the outcome assessment were blind, and if the adjunctive medication was balanced between the study groups (Zajecka 2002). However, we did not receive a reply.
Data and analyses
Comparison 1. Valproate vs placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response rate (adults) | 5 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 at 3 weeks | 4 | 869 | Odds Ratio (M‐H, Random, 95% CI) | 2.05 [1.32, 3.20] |
| 1.2 at 8 weeks | 1 | 62 | Odds Ratio (M‐H, Random, 95% CI) | 1.50 [0.54, 4.15] |
| 2 Response rate (children and adolescents) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 at 4 weeks | 1 | 151 | Odds Ratio (M‐H, Random, 95% CI) | 1.11 [0.51, 2.38] |
| 3 Number with any adverse event (adults; children and adolescents) | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Adults | 3 | 745 | Odds Ratio (M‐H, Random, 95% CI) | 1.63 [1.13, 2.36] |
| 3.2 Children and adolescents | 1 | 150 | Odds Ratio (M‐H, Random, 95% CI) | 1.39 [0.71, 2.71] |
| 4 Individual adverse events (adults) | 6 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Abdominal pain | 2 | 439 | Odds Ratio (M‐H, Random, 95% CI) | 2.62 [1.18, 5.82] |
| 4.2 Abnormal dreams | 1 | 62 | Odds Ratio (M‐H, Random, 95% CI) | 10.31 [0.53, 200.18] |
| 4.3 Agitation | 1 | 43 | Odds Ratio (M‐H, Random, 95% CI) | 3.62 [0.14, 93.84] |
| 4.4 Anorexia | 2 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 4.44 [0.47, 41.59] |
| 4.5 Arthralgia | 1 | 62 | Odds Ratio (M‐H, Random, 95% CI) | 5.34 [0.25, 115.89] |
| 4.6 Asthenia | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 1.44 [0.50, 4.09] |
| 4.7 Ataxia | 1 | 43 | Odds Ratio (M‐H, Random, 95% CI) | 6.35 [0.29, 140.55] |
| 4.8 Chest tightness | 1 | 43 | Odds Ratio (M‐H, Random, 95% CI) | 3.62 [0.14, 93.84] |
| 4.9 Back pain | 2 | 287 | Odds Ratio (M‐H, Random, 95% CI) | 0.63 [0.03, 14.86] |
| 4.10 Constipation | 4 | 473 | Odds Ratio (M‐H, Random, 95% CI) | 0.59 [0.23, 1.52] |
| 4.11 Diarrhoea | 5 | 850 | Odds Ratio (M‐H, Random, 95% CI) | 1.20 [0.61, 2.39] |
| 4.12 Diplopia | 1 | 43 | Odds Ratio (M‐H, Random, 95% CI) | 1.16 [0.07, 19.80] |
| 4.13 Dizziness | 2 | 520 | Odds Ratio (M‐H, Random, 95% CI) | 2.76 [1.57, 4.85] |
| 4.14 Dry eyes | 1 | 43 | Odds Ratio (M‐H, Random, 95% CI) | 3.62 [0.14, 93.84] |
| 4.15 Dry mouth | 2 | 368 | Odds Ratio (M‐H, Random, 95% CI) | 1.08 [0.34, 3.40] |
| 4.16 Dysarthria | 1 | 43 | Odds Ratio (M‐H, Random, 95% CI) | 3.62 [0.14, 93.84] |
| 4.17 Dyspepsia | 3 | 664 | Odds Ratio (M‐H, Random, 95% CI) | 2.17 [1.10, 4.28] |
| 4.18 Dysuria | 2 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 1.17 [0.06, 22.96] |
| 4.19 Fever | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 0.35 [0.04, 3.43] |
| 4.20 Headache | 6 | 1156 | Odds Ratio (M‐H, Random, 95% CI) | 0.91 [0.66, 1.25] |
| 4.21 Hot flashes | 1 | 62 | Odds Ratio (M‐H, Random, 95% CI) | 7.74 [0.38, 156.36] |
| 4.22 Hypertension | 1 | 62 | Odds Ratio (M‐H, Random, 95% CI) | 5.34 [0.25, 115.89] |
| 4.23 Increased appetite | 2 | 368 | Odds Ratio (M‐H, Random, 95% CI) | 1.55 [0.54, 4.48] |
| 4.24 Insomnia | 2 | 368 | Odds Ratio (M‐H, Random, 95% CI) | 1.56 [0.54, 4.48] |
| 4.25 Nausea | 5 | 931 | Odds Ratio (M‐H, Random, 95% CI) | 2.00 [1.38, 2.90] |
| 4.26 Oedema | 1 | 62 | Odds Ratio (M‐H, Random, 95% CI) | 5.34 [0.25, 115.89] |
| 4.27 Pain | 3 | 563 | Odds Ratio (M‐H, Random, 95% CI) | 1.16 [0.70, 1.94] |
| 4.28 Palpitations | 1 | 43 | Odds Ratio (M‐H, Random, 95% CI) | 1.16 [0.07, 19.80] |
| 4.29 Photophobia | 1 | 62 | Odds Ratio (M‐H, Random, 95% CI) | 5.34 [0.25, 115.89] |
| 4.30 Rash | 1 | 62 | Odds Ratio (M‐H, Random, 95% CI) | 3.21 [0.32, 32.74] |
| 4.31 Sedation | 5 | 931 | Odds Ratio (M‐H, Random, 95% CI) | 1.76 [0.95, 3.24] |
| 4.32 Somnolence | 3 | 593 | Odds Ratio (M‐H, Random, 95% CI) | 1.76 [0.80, 3.88] |
| 4.33 Tremor | 1 | 62 | Odds Ratio (M‐H, Random, 95% CI) | 7.74 [0.38, 156.36] |
| 4.34 Twitching | 1 | 143 | Odds Ratio (M‐H, Random, 95% CI) | 5.52 [0.26, 117.01] |
| 4.35 Upper respiratory tract infection | 2 | 439 | Odds Ratio (M‐H, Random, 95% CI) | 3.24 [1.30, 8.09] |
| 4.36 Vomiting | 4 | 625 | Odds Ratio (M‐H, Random, 95% CI) | 3.18 [1.77, 5.70] |
| 4.37 Weight increase | 2 | 368 | Odds Ratio (M‐H, Random, 95% CI) | 1.89 [0.20, 18.26] |
| 5 Individual adverse events (children and adolescents) | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Abdominal pain | 2 | 178 | Odds Ratio (M‐H, Random, 95% CI) | 3.74 [0.62, 22.59] |
| 5.2 Ammonia increased | 1 | 150 | Odds Ratio (M‐H, Random, 95% CI) | 9.25 [0.49, 174.87] |
| 5.3 Excitement | 1 | 28 | Odds Ratio (M‐H, Random, 95% CI) | 1.92 [0.08, 44.92] |
| 5.4 Difficulty concentrating | 1 | 28 | Odds Ratio (M‐H, Random, 95% CI) | 0.01 [0.00, 0.15] |
| 5.5 Difficulty waking in the morning | 1 | 28 | Odds Ratio (M‐H, Random, 95% CI) | 0.10 [0.00, 2.78] |
| 5.6 Dyspepsia (Indigestion) | 1 | 150 | Odds Ratio (M‐H, Random, 95% CI) | 5.00 [0.24, 105.93] |
| 5.7 Enuresis | 1 | 28 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.8 Gastritis | 1 | 150 | Odds Ratio (M‐H, Random, 95% CI) | 9.25 [0.49, 174.87] |
| 5.9 Headache | 2 | 178 | Odds Ratio (M‐H, Random, 95% CI) | 1.07 [0.44, 2.61] |
| 5.10 Initial Insomnia | 1 | 28 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.11 Irritability | 1 | 28 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 5.12 Nausea | 1 | 150 | Odds Ratio (M‐H, Random, 95% CI) | 0.13 [0.02, 1.06] |
| 5.13 Outburst of anger | 1 | 28 | Odds Ratio (M‐H, Random, 95% CI) | 6.29 [0.31, 127.06] |
| 5.14 Pharyngitis streptococcal | 1 | 150 | Odds Ratio (M‐H, Random, 95% CI) | 7.10 [0.36, 139.78] |
| 5.15 Pharyngolaryngeal pain | 1 | 150 | Odds Ratio (M‐H, Random, 95% CI) | 0.48 [0.04, 5.41] |
| 5.16 Rash | 1 | 150 | Odds Ratio (M‐H, Random, 95% CI) | 4.06 [0.44, 37.17] |
| 5.17 Nasal congestion | 2 | 178 | Odds Ratio (M‐H, Random, 95% CI) | 0.38 [0.06, 2.37] |
| 5.18 Sedation | 1 | 150 | Odds Ratio (M‐H, Random, 95% CI) | 0.40 [0.12, 1.37] |
| 5.19 Sadness | 1 | 28 | Odds Ratio (M‐H, Random, 95% CI) | 0.42 [0.05, 3.22] |
| 5.20 Somnolence | 1 | 150 | Odds Ratio (M‐H, Random, 95% CI) | 5.14 [0.59, 45.10] |
| 5.21 Upper respiratory tract infection | 1 | 150 | Odds Ratio (M‐H, Random, 95% CI) | 0.97 [0.06, 15.85] |
| 5.22 Vomiting | 1 | 150 | Odds Ratio (M‐H, Random, 95% CI) | 1.72 [0.59, 4.99] |
| 5.23 Weight increase | 1 | 150 | Odds Ratio (M‐H, Random, 95% CI) | 1.97 [0.18, 22.24] |
| 6 Remission rate (adults) | 2 | 683 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.61 [1.17, 2.22] |
| 6.1 at 3 weeks | 2 | 683 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.61 [1.17, 2.22] |
| 7 Remission rate (children and adolescents) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 at 4 weeks | 1 | 151 | Odds Ratio (M‐H, Random, 95% CI) | 0.87 [0.37, 2.04] |
| 8 Change in symptom severity (adults) | 4 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 8.1 at 3 weeks | 4 | 907 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.23 [‐0.45, ‐0.00] |
| 9 Change in symptom severity (children and adolescents) | 2 | 172 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.14 [‐0.45, 0.17] |
| 9.1 at 4 weeks | 1 | 144 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.09 [‐0.41, 0.24] |
| 9.2 at 6 weeks | 1 | 28 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.51 [‐1.38, 0.36] |
| 10 Dropout rate (adults) | 6 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 Adverse events | 5 | 931 | Odds Ratio (M‐H, Random, 95% CI) | 2.59 [1.33, 5.05] |
| 10.2 Inefficacy | 6 | 1156 | Odds Ratio (M‐H, Random, 95% CI) | 0.50 [0.36, 0.69] |
| 10.3 Other | 6 | 1156 | Odds Ratio (M‐H, Random, 95% CI) | 1.18 [0.83, 1.66] |
| 10.4 All cause | 6 | 1156 | Odds Ratio (M‐H, Random, 95% CI) | 0.83 [0.64, 1.07] |
| 11 Dropout rate (children and adolescents) | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 11.1 Adverse events | 2 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 1.26 [0.31, 5.06] |
| 11.2 Inefficacy | 2 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 1.53 [0.51, 4.61] |
| 11.3 Other | 2 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 1.44 [0.51, 4.08] |
| 11.4 All cause | 2 | 179 | Odds Ratio (M‐H, Random, 95% CI) | 1.77 [0.83, 3.78] |
| 12 Global Functioning (children and adolescents) | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 12.1 at 4 weeks | 1 | 151 | Odds Ratio (M‐H, Random, 95% CI) | 0.83 [0.42, 1.66] |
| 12.2 at 6 weeks | 1 | 28 | Odds Ratio (M‐H, Random, 95% CI) | 13.70 [0.69, 270.30] |
Comparison 2. Valproate vs carbamazepine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response rate (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 at 4 weeks | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 2.41 [0.52, 11.10] |
| 2 Number with any adverse event (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Adults | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 0.13 [0.02, 0.82] |
| 3 Individual adverse events (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Ataxia/tremors | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 0.29 [0.03, 3.12] |
| 3.2 Dizziness | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 0.08 [0.01, 0.79] |
| 3.3 Lethargy | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 1.42] |
| 3.4 Nausea/vomiting | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 0.18 [0.03, 1.07] |
| 3.5 Rash | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 0.31 [0.01, 8.28] |
| 3.6 Raised liver enzyme | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.06, 17.62] |
| 4 Change in symptom severity (adults) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 at 4 weeks | 1 | 30 | Mean Difference (IV, Random, 95% CI) | ‐10.00 [‐21.82, ‐2.18] |
| 5 Dropout rate (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Other | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.17, 5.98] |
| 5.2 All cause | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.17, 5.98] |
2.3. Analysis.
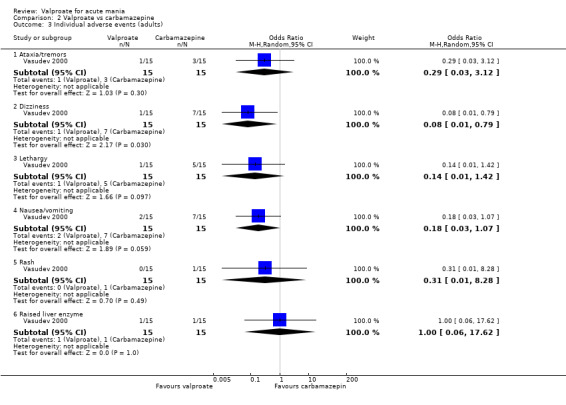
Comparison 2 Valproate vs carbamazepine, Outcome 3 Individual adverse events (adults).
Comparison 3. Valproate vs endoxifen.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response rate (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 at 3 weeks | 1 | 84 | Odds Ratio (M‐H, Random, 95% CI) | 2.19 [0.83, 5.78] |
| 2 Number with any adverse event (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Adults | 1 | 84 | Odds Ratio (M‐H, Random, 95% CI) | 1.88 [0.73, 4.86] |
| 3 Individual adverse events (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Headache | 1 | 84 | Odds Ratio (M‐H, Random, 95% CI) | 1.15 [0.26, 5.21] |
| 3.2 Insomnia | 1 | 84 | Odds Ratio (M‐H, Random, 95% CI) | 3.61 [0.80, 16.36] |
| 3.3 Nausea | 1 | 84 | Odds Ratio (M‐H, Random, 95% CI) | 5.52 [1.00, 30.50] |
Comparison 4. Valproate vs haloperidol.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response rate (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 at 1 week | 1 | 36 | Odds Ratio (M‐H, Random, 95% CI) | 1.82 [0.46, 7.18] |
| 2 Individual adverse events (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dry mouth | 1 | 36 | Odds Ratio (M‐H, Random, 95% CI) | 0.2 [0.02, 2.15] |
| 2.2 Extra‐pyramidal side effects | 1 | 36 | Odds Ratio (M‐H, Random, 95% CI) | 0.02 [0.00, 0.40] |
| 2.3 Headache | 1 | 36 | Odds Ratio (M‐H, Random, 95% CI) | 0.22 [0.01, 5.91] |
| 2.4 Indigestion | 1 | 36 | Odds Ratio (M‐H, Random, 95% CI) | 1.47 [0.12, 17.91] |
| 2.5 Insomnia | 1 | 36 | Odds Ratio (M‐H, Random, 95% CI) | 2.27 [0.09, 59.56] |
| 2.6 Sedation | 1 | 36 | Odds Ratio (M‐H, Random, 95% CI) | 0.14 [0.01, 1.39] |
| 3 Change in symptom severity (adults) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 at 1 week | 1 | 36 | Mean Difference (IV, Random, 95% CI) | ‐3.60 [‐11.48, 4.28] |
Comparison 5. Valproate vs lithium.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response rate (adults) | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 at 3 weeks | 3 | 356 | Odds Ratio (M‐H, Random, 95% CI) | 0.80 [0.48, 1.35] |
| 2 Response rate (children and adolescents) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 at 8 weeks | 1 | 197 | Odds Ratio (M‐H, Random, 95% CI) | 0.57 [0.31, 1.07] |
| 3 Number with any adverse event (adults) | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Adults | 2 | 164 | Odds Ratio (M‐H, Random, 95% CI) | 0.61 [0.25, 1.50] |
| 4 Individual adverse events (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Asthenia | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 0.62 [0.21, 1.83] |
| 4.2 Constipation | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 0.56 [0.17, 1.83] |
| 4.3 Diarrhoea | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 0.81 [0.25, 2.69] |
| 4.4 Dizziness | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 2.09 [0.54, 8.02] |
| 4.5 Fever | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 0.09 [0.01, 0.81] |
| 4.6 Headache | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 0.44 [0.18, 1.05] |
| 4.7 Nausea | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 0.69 [0.28, 1.69] |
| 4.8 Pain | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 8.12 [1.02, 64.86] |
| 4.9 Sedation | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 0.96 [0.35, 2.67] |
| 4.10 Twitching | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 0.33 [0.05, 2.06] |
| 4.11 Vomiting | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 0.51 [0.19, 1.39] |
| 5 Individual adverse events (children and adolescents) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Abdominal pain | 1 | 190 | Odds Ratio (M‐H, Random, 95% CI) | 0.58 [0.31, 1.07] |
| 5.2 Appetite increase | 1 | 190 | Odds Ratio (M‐H, Random, 95% CI) | 0.73 [0.37, 1.43] |
| 5.3 Diarrhoea | 1 | 190 | Odds Ratio (M‐H, Random, 95% CI) | 1.84 [0.78, 4.37] |
| 5.4 Dry mouth/excessive thirst | 1 | 190 | Odds Ratio (M‐H, Random, 95% CI) | 0.34 [0.17, 0.65] |
| 5.5 Nasal congestion | 1 | 190 | Odds Ratio (M‐H, Random, 95% CI) | 1.36 [0.70, 2.64] |
| 5.6 Enuresis | 1 | 190 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.49, 2.04] |
| 5.7 Fever | 1 | 190 | Odds Ratio (M‐H, Random, 95% CI) | 1.68 [0.54, 5.22] |
| 5.8 Frequent urination | 1 | 190 | Odds Ratio (M‐H, Random, 95% CI) | 0.45 [0.22, 0.93] |
| 5.9 Nausea | 1 | 190 | Odds Ratio (M‐H, Random, 95% CI) | 0.60 [0.32, 1.13] |
| 5.10 Rash | 1 | 190 | Odds Ratio (M‐H, Random, 95% CI) | 2.54 [0.87, 7.43] |
| 5.11 Sedation | 1 | 190 | Odds Ratio (M‐H, Random, 95% CI) | 1.59 [0.84, 3.00] |
| 5.12 Vomiting | 1 | 190 | Odds Ratio (M‐H, Random, 95% CI) | 0.49 [0.22, 1.11] |
| 5.13 Weight gain | 1 | 190 | Odds Ratio (M‐H, Random, 95% CI) | 1.35 [0.73, 2.49] |
| 5.14 Weight loss | 1 | 190 | Odds Ratio (M‐H, Random, 95% CI) | 1.19 [0.63, 2.26] |
| 6 Change in symptom severity (adults) | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.1 at 3 weeks | 2 | 57 | Std. Mean Difference (IV, Random, 95% CI) | 0.69 [0.14, 1.25] |
| 7 Clinical response on MSRS ‐ Frequency (adults) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 7.1 at 3 weeks | 1 | 30 | Mean Difference (IV, Random, 95% CI) | 7.80 [‐2.11, 17.71] |
| 8 Change in symptom severity (children and adolescents) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 8.1 at 8 weeks | 1 | 190 | Mean Difference (IV, Random, 95% CI) | 1.40 [‐2.03, 4.83] |
| 9 Dropout rate (adults) | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 9.1 Adverse events | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 0.49 [0.12, 2.10] |
| 9.2 Inefficacy | 2 | 164 | Odds Ratio (M‐H, Random, 95% CI) | 0.89 [0.42, 1.88] |
| 9.3 Other | 2 | 164 | Odds Ratio (M‐H, Random, 95% CI) | 0.60 [0.26, 1.39] |
| 9.4 All cause | 3 | 388 | Odds Ratio (M‐H, Random, 95% CI) | 0.83 [0.47, 1.45] |
| 10 Dropout rate (children and adolescents) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 Adverse events | 1 | 197 | Odds Ratio (M‐H, Random, 95% CI) | 0.43 [0.12, 1.46] |
| 10.2 Other | 1 | 197 | Odds Ratio (M‐H, Random, 95% CI) | 0.92 [0.47, 1.81] |
| 10.3 All cause | 1 | 197 | Odds Ratio (M‐H, Random, 95% CI) | 0.74 [0.39, 1.37] |
Comparison 6. Valproate vs olanzapine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response rate (adults) | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 at 3 weeks | 2 | 667 | Odds Ratio (M‐H, Random, 95% CI) | 0.77 [0.48, 1.25] |
| 2 Individual adverse events (adults) | 4 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Abnormal hepatic function | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
| 2.2 Agitation | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 0.99 [0.45, 2.17] |
| 2.3 Alopecia (hair loss) | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 7.56 [0.38, 151.28] |
| 2.4 Asthena | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 0.82 [0.41, 1.65] |
| 2.5 Constipation | 2 | 331 | Odds Ratio (M‐H, Random, 95% CI) | 0.46 [0.11, 1.89] |
| 2.6 Dizziness | 2 | 331 | Odds Ratio (M‐H, Random, 95% CI) | 0.32 [0.03, 3.11] |
| 2.7 Diarrhea | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 2.28 [0.95, 5.50] |
| 2.8 Dyspepsia | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 0.74 [0.35, 1.57] |
| 2.9 Headache | 2 | 667 | Odds Ratio (M‐H, Random, 95% CI) | 1.44 [0.68, 3.07] |
| 2.10 Increased appetite | 2 | 667 | Odds Ratio (M‐H, Random, 95% CI) | 0.45 [0.08, 2.40] |
| 2.11 Insomnia | 1 | 416 | Odds Ratio (M‐H, Random, 95% CI) | 6.17 [1.35, 28.17] |
| 2.12 Nausea | 3 | 747 | Odds Ratio (M‐H, Random, 95% CI) | 4.12 [2.22, 7.62] |
| 2.13 Neck rigidity | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 0.21 [0.04, 0.98] |
| 2.14 Nervousness | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 1.72 [0.82, 3.62] |
| 2.15 Oedema | 1 | 120 | Odds Ratio (M‐H, Random, 95% CI) | 0.05 [0.00, 0.81] |
| 2.16 Pain | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 1.06 [0.52, 2.16] |
| 2.17 Rhinitis | 1 | 120 | Odds Ratio (M‐H, Random, 95% CI) | 0.53 [0.16, 1.72] |
| 2.18 Sedation | 2 | 536 | Odds Ratio (M‐H, Random, 95% CI) | 0.50 [0.28, 0.91] |
| 2.19 Sleep disorder | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 0.13 [0.02, 1.11] |
| 2.20 Somnolence | 3 | 747 | Odds Ratio (M‐H, Random, 95% CI) | 0.36 [0.23, 0.57] |
| 2.21 Speech disorder | 2 | 371 | Odds Ratio (M‐H, Random, 95% CI) | 0.09 [0.02, 0.50] |
| 2.22 Tongue edema | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 0.07 [0.00, 1.30] |
| 2.23 Tremor | 2 | 331 | Odds Ratio (M‐H, Random, 95% CI) | 0.28 [0.09, 0.82] |
| 2.24 Vomiting | 1 | 251 | Odds Ratio (M‐H, Random, 95% CI) | 1.92 [0.85, 4.34] |
| 2.25 Weight gain | 4 | 867 | Odds Ratio (M‐H, Random, 95% CI) | 0.44 [0.28, 0.70] |
| 2.26 Xerostomia (dry mouth) | 3 | 747 | Odds Ratio (M‐H, Random, 95% CI) | 0.25 [0.11, 0.57] |
| 3 Remission rate (adults) | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 at 3 weeks | 2 | 667 | Odds Ratio (M‐H, Random, 95% CI) | 0.73 [0.46, 1.15] |
| 4 Change in symptom severity (adults) | 4 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 at 1 week | 1 | 76 | Std. Mean Difference (IV, Random, 95% CI) | 0.35 [‐0.11, 0.80] |
| 4.2 at 3 weeks | 4 | 826 | Std. Mean Difference (IV, Random, 95% CI) | 0.25 [0.11, 0.39] |
| 5 Dropout rate (adults) | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 Adverse events | 3 | 616 | Odds Ratio (M‐H, Random, 95% CI) | 0.61 [0.25, 1.49] |
| 5.2 Inefficacy | 3 | 616 | Odds Ratio (M‐H, Random, 95% CI) | 1.36 [0.65, 2.86] |
| 5.3 Other | 3 | 616 | Odds Ratio (M‐H, Random, 95% CI) | 1.14 [0.76, 1.72] |
| 5.4 All cause | 3 | 616 | Odds Ratio (M‐H, Random, 95% CI) | 1.04 [0.71, 1.52] |
Comparison 7. Valproate + olanzapine vs olanzapine alone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Individual adverse events (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Alopecia (hair loss) | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 5.26 [0.24, 113.11] |
| 1.2 Constipation | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 0.73 [0.24, 2.20] |
| 1.3 Dizziness | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 1.89 [0.61, 5.82] |
| 1.4 Somnolence | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 0.79 [0.30, 2.06] |
| 1.5 Tremor | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 0.65 [0.10, 4.11] |
| 1.6 Weight gain | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 1.31 [0.47, 3.61] |
| 1.7 Xerostomia (dry mouth) | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 0.67 [0.19, 2.33] |
| 2 Change in symptom severity (adults) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 at 1 week | 1 | 76 | Mean Difference (IV, Random, 95% CI) | ‐0.87 [‐5.16, 3.42] |
| 2.2 at 3 weeks | 1 | 76 | Mean Difference (IV, Random, 95% CI) | ‐2.76 [‐9.17, 3.65] |
| 3 Dropout (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Adverse events | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.06, 16.56] |
| 3.2 Other | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 3.08 [0.12, 77.80] |
| 3.3 All cause | 1 | 80 | Odds Ratio (M‐H, Random, 95% CI) | 2.05 [0.18, 23.59] |
7.1. Analysis.
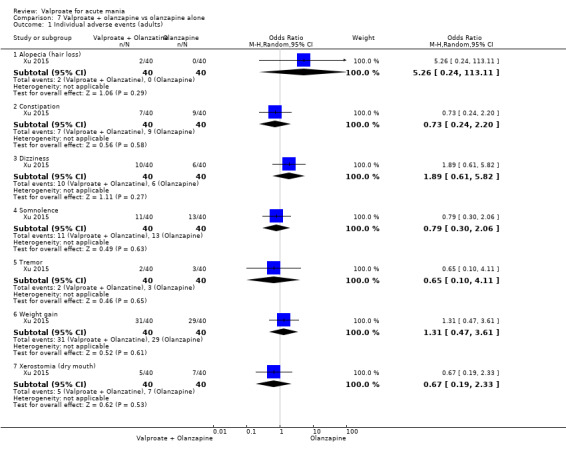
Comparison 7 Valproate + olanzapine vs olanzapine alone, Outcome 1 Individual adverse events (adults).
Comparison 8. Valproate vs oxcarbazepine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Number with any adverse event (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Adults | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 4.67 [1.57, 13.87] |
| 2 Individual adverse events (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Alopecia | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 3.10 [0.12, 79.23] |
| 2.2 Constipation | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 7.76 [0.38, 157.14] |
| 2.3 Diarrhoea | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 5.35 [0.25, 116.31] |
| 2.4 Dizziness | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 2.74 [0.63, 11.82] |
| 2.5 Dry mouth | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 2.07 [0.18, 24.15] |
| 2.6 Dyspepsia | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.19, 5.40] |
| 2.7 Headache | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 2.25 [0.51, 9.99] |
| 2.8 Increased appetite | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 7.76 [0.38, 157.14] |
| 2.9 Nausea | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 1.52 [0.42, 5.47] |
| 2.10 Pain in abdomen | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 13.16 [0.69, 249.48] |
| 2.11 Rash | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 3.10 [0.12, 79.23] |
| 2.12 Sedation | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 1.8 [0.39, 8.32] |
| 2.13 Thrombocytopenia | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 3.10 [0.12, 79.23] |
| 2.14 Vomiting | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 1.63 [0.41, 6.47] |
| 2.15 Weight gain | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 10.36 [0.53, 201.45] |
| 3 Remission rate (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 at 12 weeks | 1 | 60 | Odds Ratio (M‐H, Random, 95% CI) | 2.25 [0.51, 9.99] |
| 4 Change in symptom severity (adults) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 at 3 days | 1 | 60 | Mean Difference (IV, Random, 95% CI) | 1.60 [‐1.24, 4.44] |
| 4.2 at 3 weeks | 1 | 60 | Mean Difference (IV, Random, 95% CI) | 0.73 [‐2.17, 3.63] |
| 4.3 at 8 weeks | 1 | 60 | Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐2.60, 1.80] |
8.2. Analysis.
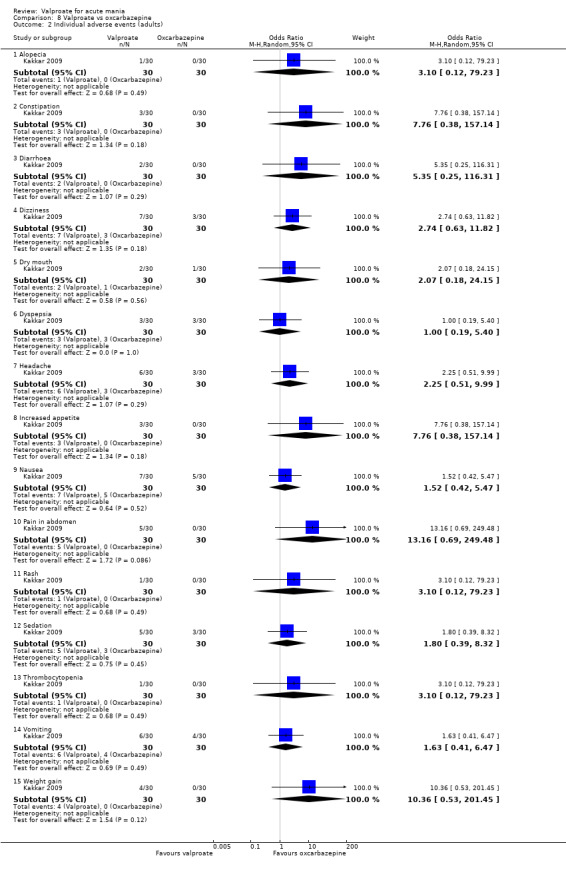
Comparison 8 Valproate vs oxcarbazepine, Outcome 2 Individual adverse events (adults).
Comparison 9. Valproate vs quetiapine.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Individual adverse events (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Head,eyes, ears, nose, and throat | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 0.17 [0.02, 1.67] |
| 1.2 Gastrointestinal | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 1.2 [0.24, 6.06] |
| 1.3 Genito‐urinary | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 1.15 [0.07, 20.34] |
| 1.4 Musculo/skeletal | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 1.15 [0.07, 20.34] |
| 1.5 Pulmonary | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 1.17 [0.14, 9.59] |
| 1.6 Psychiatric | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 0.51 [0.11, 2.36] |
| 2 Individual adverse events (children and adolescents) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Dizziness | 1 | 50 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.32, 3.17] |
| 2.2 Dry mouth | 1 | 50 | Odds Ratio (M‐H, Random, 95% CI) | 0.28 [0.05, 1.53] |
| 2.3 Gastrointestinal upset | 1 | 50 | Odds Ratio (M‐H, Random, 95% CI) | 0.81 [0.23, 2.88] |
| 2.4 Increased appetite | 1 | 50 | Odds Ratio (M‐H, Random, 95% CI) | 1.57 [0.24, 10.30] |
| 2.5 Insomnia | 1 | 50 | Odds Ratio (M‐H, Random, 95% CI) | 0.31 [0.03, 3.16] |
| 2.6 Sedation or lethargy | 1 | 50 | Odds Ratio (M‐H, Random, 95% CI) | 0.38 [0.12, 1.18] |
| 3 Remission rate (children and adolescents) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 at 4 weeks | 1 | 50 | Odds Ratio (M‐H, Random, 95% CI) | 0.26 [0.08, 0.85] |
| 4 Change in symptom severity (children and adolescents) | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 at 4 weeks | 1 | 50 | Mean Difference (IV, Random, 95% CI) | 4.0 [‐2.10, 10.10] |
| 5 Dropout rate (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 All cause | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 1.73 [0.31, 9.57] |
| 6 Dropout rate (children and adolescents) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 6.1 Inefficacy | 1 | 50 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.13, 7.72] |
| 6.2 Other | 1 | 50 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.22, 4.54] |
| 6.3 All cause | 1 | 50 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.27, 3.66] |
| 7 Global functioning (children and adolescents) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 at 4 weeks | 1 | 50 | Odds Ratio (M‐H, Random, 95% CI) | 0.24 [0.06, 0.92] |
9.1. Analysis.
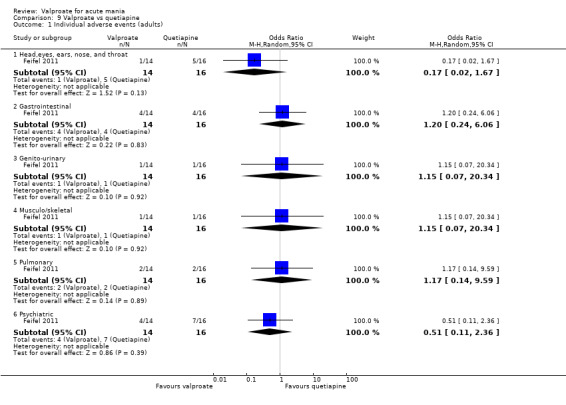
Comparison 9 Valproate vs quetiapine, Outcome 1 Individual adverse events (adults).
9.2. Analysis.
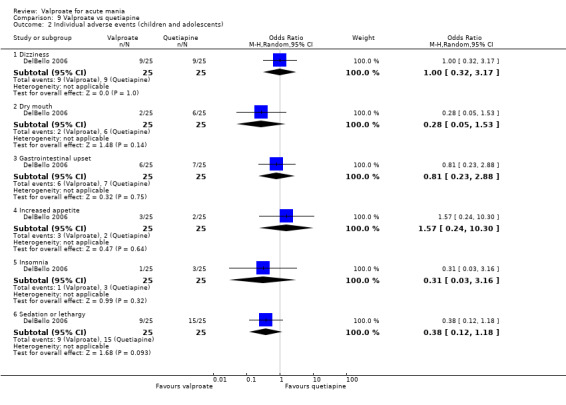
Comparison 9 Valproate vs quetiapine, Outcome 2 Individual adverse events (children and adolescents).
Comparison 10. Valproate vs risperidone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response rate (children and adolescents) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 at 8 weeks | 1 | 197 | Odds Ratio (M‐H, Random, 95% CI) | 0.16 [0.08, 0.29] |
| 2 Individual adverse events (children and adolescents) | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Abdominal pain | 2 | 228 | Odds Ratio (M‐H, Random, 95% CI) | 14.49 [0.54, 389.63] |
| 2.2 Appetite increase | 1 | 189 | Odds Ratio (M‐H, Random, 95% CI) | 0.08 [0.04, 0.16] |
| 2.3 Dry mouth/excessive thirst | 1 | 189 | Odds Ratio (M‐H, Random, 95% CI) | 0.44 [0.23, 0.85] |
| 2.4 Enuresis | 2 | 228 | Odds Ratio (M‐H, Random, 95% CI) | 2.74 [0.01, 829.22] |
| 2.5 Excitement | 1 | 39 | Odds Ratio (M‐H, Random, 95% CI) | 4.74 [0.21, 105.54] |
| 2.6 Fever | 1 | 189 | Odds Ratio (M‐H, Random, 95% CI) | 0.88 [0.33, 2.32] |
| 2.7 Frequent urination | 1 | 189 | Odds Ratio (M‐H, Random, 95% CI) | 2.25 [0.83, 6.14] |
| 2.8 Headache | 1 | 39 | Odds Ratio (M‐H, Random, 95% CI) | 0.06 [0.00, 1.12] |
| 2.9 Initial insomnia | 1 | 39 | Odds Ratio (M‐H, Random, 95% CI) | 0.27 [0.01, 7.08] |
| 2.10 Irritability | 1 | 39 | Odds Ratio (M‐H, Random, 95% CI) | 0.15 [0.01, 3.42] |
| 2.11 Nasal congestion | 2 | 228 | Odds Ratio (M‐H, Random, 95% CI) | 1.38 [0.72, 2.64] |
| 2.12 Nausea | 1 | 189 | Odds Ratio (M‐H, Random, 95% CI) | 1.47 [0.71, 3.04] |
| 2.13 Outburst of anger | 1 | 39 | Odds Ratio (M‐H, Random, 95% CI) | 2.0 [0.42, 9.52] |
| 2.14 Rash | 1 | 189 | Odds Ratio (M‐H, Random, 95% CI) | 2.07 [0.75, 5.69] |
| 2.15 Sadness | 1 | 39 | Odds Ratio (M‐H, Random, 95% CI) | 0.58 [0.11, 3.04] |
| 2.16 Sedation | 1 | 189 | Odds Ratio (M‐H, Random, 95% CI) | 0.50 [0.28, 0.91] |
| 2.17 Vomiting | 1 | 189 | Odds Ratio (M‐H, Random, 95% CI) | 1.45 [0.54, 3.91] |
| 2.18 Weight gain | 1 | 189 | Odds Ratio (M‐H, Random, 95% CI) | 0.12 [0.04, 0.34] |
| 2.19 Weight loss | 1 | 189 | Odds Ratio (M‐H, Random, 95% CI) | 3.63 [1.61, 8.19] |
| 3 Change in symptom severity (children and adolescents) | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 3.1 at 5‐12 weeks | 2 | 228 | Std. Mean Difference (IV, Random, 95% CI) | 1.01 [0.74, 1.29] |
| 4 Dropout rate (children and adolescents) | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 4.1 Adverse events | 2 | 236 | Odds Ratio (M‐H, Random, 95% CI) | 1.39 [0.35, 5.52] |
| 4.2 Inefficacy | 1 | 39 | Odds Ratio (M‐H, Random, 95% CI) | 2.71 [0.10, 70.65] |
| 4.3 Other | 2 | 236 | Odds Ratio (M‐H, Random, 95% CI) | 1.89 [0.91, 3.90] |
| 4.4 All cause | 2 | 236 | Odds Ratio (M‐H, Random, 95% CI) | 1.96 [1.00, 3.82] |
| 5 Global functioning (children and adolescents) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 at 6 weeks | 1 | 39 | Odds Ratio (M‐H, Random, 95% CI) | 0.14 [0.03, 0.75] |
Comparison 11. Valproate + risperidone vs risperidone alone.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Total number with any adverse events (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Adults | 1 | 48 | Odds Ratio (M‐H, Random, 95% CI) | 0.93 [0.27, 3.16] |
| 2 Remission (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 at 1 week | 1 | 48 | Odds Ratio (M‐H, Random, 95% CI) | 0.66 [0.20, 2.17] |
| 2.2 at 3 weeks | 1 | 48 | Odds Ratio (M‐H, Random, 95% CI) | 0.91 [0.16, 5.03] |
| 2.3 at 7 weeks | 1 | 48 | Odds Ratio (M‐H, Random, 95% CI) | 0.91 [0.12, 7.07] |
| 3 Partial or full remission (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 at 1 week | 1 | 48 | Odds Ratio (M‐H, Random, 95% CI) | 0.72 [0.20, 2.59] |
| 3.2 at 3 weeks | 1 | 48 | Odds Ratio (M‐H, Random, 95% CI) | 0.91 [0.16, 5.03] |
| 3.3 at 7 weeks | 1 | 48 | Odds Ratio (M‐H, Random, 95% CI) | 0.0 [0.0, 0.0] |
Comparison 12. Valproate vs topiramate.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Individual adverse events (children and adolescents) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Blurred vision | 1 | 142 | Odds Ratio (M‐H, Random, 95% CI) | 0.19 [0.01, 4.12] |
| 1.2 Dizziness | 1 | 142 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.51, 1.98] |
| 1.3 Drowsiness | 1 | 142 | Odds Ratio (M‐H, Random, 95% CI) | 0.34 [0.15, 0.79] |
| 1.4 Hair loss | 1 | 142 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.14, 7.30] |
| 1.5 Headache | 1 | 142 | Odds Ratio (M‐H, Random, 95% CI) | 0.65 [0.17, 2.40] |
| 1.6 Muscle weakness | 1 | 142 | Odds Ratio (M‐H, Random, 95% CI) | 2.29 [0.91, 5.76] |
| 1.7 Nausea | 1 | 142 | Odds Ratio (M‐H, Random, 95% CI) | 0.40 [0.17, 0.95] |
| 1.8 Paresthesia | 1 | 142 | Odds Ratio (M‐H, Random, 95% CI) | 1.43 [0.68, 2.99] |
| 1.9 Speech‐related problems | 1 | 142 | Odds Ratio (M‐H, Random, 95% CI) | 1.0 [0.24, 4.16] |
| 1.10 Tremor | 1 | 142 | Odds Ratio (M‐H, Random, 95% CI) | 1.60 [0.73, 3.48] |
| 2 Change in symptom severity (children and adolescents) | 2 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 at 5‐12 weeks | 2 | 149 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.41 [‐1.16, 0.35] |
| 3 Dropout rates (children and adolescents) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 3.1 Adverse events | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 0.31 [0.01, 8.28] |
| 3.2 All cause | 1 | 30 | Odds Ratio (M‐H, Random, 95% CI) | 0.31 [0.01, 8.28] |
Comparison 13. Valproate vs placebo (sensitivity analysis treatment resistant mania removed).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response rate (adults) | 3 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 at 3 weeks | 3 | 826 | Odds Ratio (M‐H, Random, 95% CI) | 1.83 [1.31, 2.55] |
Comparison 14. Valproate vs lithium (sensitivity analysis treatment resistant mania removed).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response rate (adults) | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 3 weeks | 2 | 329 | Odds Ratio (M‐H, Random, 95% CI) | 0.86 [0.55, 1.35] |
Comparison 15. Valproate vs placebo (sensitivity analysis any unclear blinding/allocation concealment removed).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response rate (adults) | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Week 3 | 2 | 520 | Odds Ratio (M‐H, Random, 95% CI) | 2.08 [1.41, 3.05] |
| 2 Number with any adverse event (primary tolerability outcome) | 2 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Number with any adverse event (primary tolerability outcome) | 2 | 520 | Odds Ratio (M‐H, Random, 95% CI) | 1.88 [1.22, 2.90] |
Comparison 16. Valproate vs lithium (sensitivity analysis unclear blinding/allocation concealment removed).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response rate (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Week 3 | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 1.02 [0.46, 2.30] |
| 2 Number with any adverse effect (primary tolerability outcome) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 2.1 Adults | 1 | 105 | Odds Ratio (M‐H, Random, 95% CI) | 0.48 [0.12, 1.84] |
Comparison 17. Valproate vs placebo (sensitivity analysis ‐ estimated standard deviation removed).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Change in symptom severity (adults) | 3 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 1.1 Week 3 | 3 | 685 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.31 [‐0.63, 0.02] |
Comparison 18. Valproate vs placebo (sensitivity analysis excluding psychotic features).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response rate (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Week 3 | 1 | 306 | Odds Ratio (M‐H, Random, 95% CI) | 1.42 [0.86, 2.36] |
Comparison 19. Valproate vs olanzapine (sensitivity analysis removing psychotic features).
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Response rate (adults) | 1 | Odds Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 Week 3 | 1 | 416 | Odds Ratio (M‐H, Random, 95% CI) | 0.97 [0.65, 1.44] |
| 2 Change in symptom severity (secondary efficacy outcome) | 1 | Std. Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 2.1 Endpoint | 1 | 387 | Std. Mean Difference (IV, Random, 95% CI) | 0.14 [‐0.06, 0.34] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Ahmad 2016.
| Methods | Randomised double‐blind trial | |
| Participants |
Number of total participants/type of patients:
84 participants were randomised. Inpatient study
Inclusion criteria:
Sex: male and female
Age: 18 ‐ 65 years
Diagnosis: diagnosed with BPD I and having displayed an acute manic or mixed episode (with or without psychotic features) according to DSM‐IV‐TR. YMRS total score of ≥ 20 and a score of ≥ 4 on the CGI–S Scale at the time of screening and at randomisation. Other: willing to give written informed consent along with at least 1 first‐degree relative/legally acceptable representative (LAR), who were capable of understanding the purposes and risks of the trial and had given written informed consent, which included compliance with the study requirements and restrictions listed in the consent form. The participants were previously treated with at least one of the drugs, viz., lithium, valproate, carbamazepine, or an atypical (except for clozapine) or typical antipsychotic at some time during the course of their bipolar illness. Their last intake of the medication(s) for BPD I was within 2 – 7 days prior to randomisation, depending on the individual drug’s plasma half‐life. The male patients of child‐begetting potential and female patients of child‐bearing potential,who were practising adequate contraception, were enrolled in the study. Female participants were not pregnant or lactating and had a negative serum pregnancy test at the time of screening and negative urine pregnancy test at the time of randomisation Exclusion criteria: Newly‐diagnosed and not having any suitable treatment exposure in the past for their bipolar mood disorder; clinically significant suicidal or homicidal ideation; serious, unstable illnesses including hepatic, renal, gastroenterologic, respiratory, cardiovascular (including Ischaemic heart disease), endocrinologic, neurologic, immunologic, or haematologic disease as per history and medical examination. |
|
| Interventions |
Location: India, various hospital sites (cities not stated) Study duration: Not stated Treatment groups: 1. Valproate extended‐release: 1000 mg 2. Endoxifen: stage I: 4 mg/day Stage II: 8 mg/day Concomitant medications: All psychotropic medications except benzodiazepines (lorazepam/diazepam only) were discontinued at least 2 days before randomisation. Benzodiazepines (lorazepam/diazepam only) (up to 5 mg/day, preferably in divided doses) were allowed as adjunctive medication as needed at the discretion of the investigator from 2 days prior to randomisation, but not beyond the first 10 days of investigational medicinal product dosing. Benzodiazepines were avoided within 12 hours of scheduled mania ratings. The use of 2 benzodiazepines was permitted to reduce undue excitement by using these adjuvants in an appropriate manner while avoiding efficacy and safety overlap with the endoxifen or valproate Length of study: 21 days Randomisation: The study was conducted in a two‐stage parallel assignment. Participants entering the study were randomly assigned 2:1 (endoxifen: valproate) Stage 1: 42 participants were randomised (endoxifen: 27; valproate: 15) Stage 2: 42 were randomised (endoxifen: 28; valproate: 14) |
|
| Outcomes |
Primary outcome: change in the YMRS total score (≥ 50% decrease from baseline) Secondary outcome: change in the MADRS total score, CGI‐S score, CGI‐I, and C‐SSRS score Safety/tolerability measures included adverse events. Study withdrawals were reported. Plasma concentrations of endoxifen were reported |
|
| Funding | Not stated | |
| Conflict of interest | "The authors declare no conflicts of interest" | |
| Notes | Despite no declaration of conflict of interest by the authors, the lead author's corresponding address is the pharmaceutical company that owns the patent on endoxifen. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A randomisation schedule prepared at Lambda Therapeutic Research, India. The randomisation schedule was generated using SAS v. 9. 3 (Cary, NC) by an unblended biostatistician before commencement of the study." |
| Allocation concealment (selection bias) | Low risk | Quote: "The randomisation schedule was kept under controlled access, which was handled only by the pharmacy custodian or designate, until the blind was broken. Study medication for each individual patient was prepackaged and pre‐numbered and provided to each participating site according to the randomisation schedule. A designed pharmacy employee dispensed the study medication serially at the site." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The blind was to be broken only if knowledge of the treatment regimen assisted medical management of the patient in an acute emergency" Quote: "On breaking the treatment randomisation code in case of an emergency, the patient was to be withdrawn from the study." Quote: "A designated pharmacy employee dispensed the study medication serially at the site." |
| Blinding of outcome assessment (detection bias) Efficacy | Unclear risk | Quote: "All psychiatrists participating in this double‐blind trial at different sites were well trained and had experience in using DSM‐IV." Comment: Although it is implied that outcome assessors are blinded, it is not made explicit in the text who is assessing outcomes. It is also not clear if these assessors are blind and no description of any procedures to maintain the blind of the assessors is described. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: No group had more than 30% dropout rate. |
| Selective reporting (reporting bias) | Unclear risk | Comment: No pre‐published protocol found. We are therefore unable to assess the risk. |
| Other bias | High risk | Comment: Two‐stage design where if lower endoxifen dose failed against valproate, a higher endoxifen dose was trialled. This is a biased approach and this trial should have been run in parallel design. Additionally, baseline characteristics of groups are not reported. Upon requesting the baseline characteristics from the authors, we did not receive a reply. The authors do include no information on whether or not the concomitant medication was balanced between the two groups. |
Bowden 1994.
| Methods | Randomised double‐blind trial | |
| Participants |
Number of total participants/type of patients: 179 participants randomised. Inpatient study Inclusion criteria: Sex: male and female Age: 18 ‐ 65 years Diagnosis: Research Diagnostic Criteria for manic disorder, based on the structured interview and rating scale of the Schedule for Affective Disorders and Schizophrenia (SADS). Patients also had MRS scores of ≥ 14 on the last washout day with scores of 2 or higher on at least 4 items and had undetectable serum lithium concentrations prior to randomisation. Exclusion criteria:
|
|
| Interventions |
Location: USA, hospital sites in San Antonio, Houston, Indianapolis, Atlanta, Cleveland, Dallas, Miami, and Chicago Study duration: Not stated Treatment groups: 1. Valproate: was administered at an initial dose of 750 mg/d. On day 3, the total daily dosages of valproate were increased to 1000 mg, and trough serum concentrations of both drugs were determined. On day 5, an unblinded physician at each centre reviewed the serum concentration and adjusted the dosage of active medication. 2. Lithium carbonate was administered at an initial dose of 900 mg/d. On day 3, the total daily dosages of lithium were increased to 1200 mg and trough serum concentrations of both drugs were determined. On day 5, an unblinded physician at each centre reviewed the serum concentration and adjusted the dosage of active medication. 3. Placebo adjusted according to blinded protocol specified dosing schedules. Concomitant medications: The protocol allowed the use of adjunctive chloral hydrate or lorazepam as needed for control of agitation, irritability, restlessness, insomnia, and hostile behaviours. The maximum daily dosages of chloral hydrate and lorazepam were 4 g and 2 mg, respectively, through treatment day 4, then 2 g and 1 mg, respectively, through day 10. These medications were not permitted during the 8 hours before behavioural assessments. Neuroleptic drugs were not allowed in the protocol. Duration of trial: 21 days Randomisation: 69 patients randomised to valproate 36 randomised to lithium 74 randomised to placebo |
|
| Outcomes |
Primary outcome: changes in the MRS derived from Schedule for Affective Disorders and Schizophrenia (SADS) Secondary outcomes: GAS and ADRS Safety measures included adverse events and platelet counts Study withdrawals were reported Outcomes were clinician assessed |
|
| Funding | "This study was funded in part by a grant from Abbott Laboratories, North Chicago III." | |
| Conflict of interest | "Drs Brugger and Morris purchase Abbott Laboratories stock on a regular basis as part of their retirement plans; Drs Bowden, Swann, Calabrese, and Janicak have received honoraria from Abbott for educational programs; Dr Janicak owns equities in Abbott Laboratories through an investment plan for retirement funds. Drs Davis and Goodnick have received remuneration from Abbott as speakers. Dr Small owns stock in Abbott Laboratories and has received honoraria for participation in educational programs. Dr Garza‐Trevi\l=n˜\ohas received remuneration from Abbott for participation in continuing medical education activities. Dr Risch has received remuneration from Abbott as a speaker and a consultant." | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned to divalproex, lithium, or placebo in a 2:1:2 ratio. A separate randomisation schedule for each centre was generated prior to the study start." |
| Allocation concealment (selection bias) | Low risk | Quote: "Centers were sent patient numbers in blocks of 10; unknown to the investigators, treatment group assignments were randomised in blocks of five within each set of 10 numbers." Comment: Although block randomisation is subject to a higher risk of selection bias, the sub‐block method reduced the chance of the investigators being able to predict participant allocation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "On day 5, an unblinded physician at each centre reviewed the serum concentration and adjusted the dosage of active medication" Quote: "Drug dosage was raised on each adjustment day unless precluded by an adverse event or a serum concentration of valproate or lithium exceeding 1041 µ/L (150 µg/mL) or 1.5 mmol/L (conventionally expressed as milliequivalents per litre), respectively. Comparable adjustments were made in the dosage of placebo according to blinded protocol specified dosing schedules." Quote: "Study medication (active drug or placebo) was dispensed in divided doses three times daily, 30 min after meals, in identical‐appearing capsules." |
| Blinding of outcome assessment (detection bias) Efficacy | Low risk | Quote: "Whenever possible, the same blinded investigator rated the patient throughout the study." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Dropout rates in placebo‐group were 64%, in lithium‐group 61% and valproate group 48%. All groups were over 30% dropout rates, but dropout rates were comparable (less than two‐fold difference) and overall dropout rate below 75%. |
| Selective reporting (reporting bias) | Unclear risk | Comment: No pre‐published protocol found. We were therefore unable to assess risk of bias. |
| Other bias | Low risk | Comment: None identified |
Bowden 2006.
| Methods | Randomised double‐blind trial | |
| Participants |
Number of total participants/type of patients: 377 participants randomised. Inpatient study initially but discharge from hospital allowed if:
Inclusion criteria: Sex: male and female Age: 18 ‐ 65 years Diagnosis: Participants had a current DSM‐IV‐TR primary diagnosis of bipolar I disorder (manic or mixed type) confirmed with SCID assessment. Participants were hospitalised for a current acute manic exacerbation of illness. The MRS score had to be ≥ 18 with at least 4 item scores > 1 at screening and day 1 before randomisation. Other:Participants were hospitalised no more than 7 days immediately before screening/washout or were in the process of being hospitalised at the time of screening. At least 1 prior manic or mixed episode within the past 3 years was required. Exclusion criteria: History of schizophrenia or schizoaffective disorder Current axis II or I disorder that would affect compliance of confounds study interpretation. Current episode secondary to antidepressant use, drug use or medical disorder. Receiving protocol banned psychotropic medication within the lesser of 5 half‐lives or 10 days before randomisation Receipt of a depot neuroleptic within 1 inter‐injection period before randomisation; History of clozapine use; history of active substance abuse within last 3 months before screening, evidence of drug and alcohol withdrawal; positive urine screen for phencyclidine, opiates, cocaine, or amphetamines; history of intolerance or failure to respond to valproate therapy; use of valproate regularly in the 30 days before study entry |
|
| Interventions |
Location: USA, 33 hospital sites (cities not described) Study duration: April 2003 ‐ May 2004 Treatment groups: 1. Valproate extended‐release: Dose initiated at 25 mg/kg/day rounded up to the nearest 500 mg dose. On day 3 all participants' doses were increased by 500 mg. Additional dose adjustments could occur on days 7, 12, 17, depending on investigator discretion Target serum valproate was 85 ‐ 125 ug/mL 2. Placebo Concomitant medications: No other psychotropic medication other than lorazepam was allowed during the washout and treatment period. The maximum single dose of lorazepam allowed was 2 mg/dose with 6 mg/day in screening, 4 mg/day on days 1 ‐ 7 and 2 mg/day on days 8 ‐ 10. None allowed after day 10 or within 8 hours of efficacy evaluations Length of study: 21 days Randomisation: 192 participants randomised to valproate extended‐release 185 participants randomised to placebo |
|
| Outcomes |
Primary outcome: Change in MRS Scores (response: ≥ 50%; remission ≤ 12) Secondary outcomes: Change from baseline to each scheduled visit for the MRS and its subscales, the MSS, changes on the BIS, changes on the DSS and change on the GAS Safety measures included adverse events and platelet counts Metabolic changes (e.g. weight gain) were reported Study withdrawals were reported |
|
| Funding | "This study was supported by Abbott Laboratories, Abbott Park, III." | |
| Conflict of interest | "Dr. Bowden has been a consultant for Abbott Laboratories, GlaxoSmithKline, Janssen, Lilly Research, Sanofi‐Synthelabo, and UCB Pharma; has received grant/research support from Abbott Laboratories, Bristol‐Myers Squibb, Elan Pharmaceuticals, GlaxoSmithKline, Janssen, Lilly Research, National Institute of Mental Health, Parke Davis, R. W. Johnson Pharmaceutical Institute, SmithKline Beecham, and Stanley Medical Research Foundation; and has participated in speakers bureaus for Abbott Laboratories, AstraZeneca, GlaxoSmithKline, Janssen, Lilly Research, and Pfizer. Dr. Swann has been a consultant for Abbott, GlaxoSmithKline, Janssen, Shire, Novartis, Ortho‐McNeil, and AstraZeneca; has received grant/research support from Abbott, Pfizer, Janssen, Ciba, Eli Lilly, GlaxoSmithKLine, Parke Davis, and Ortho‐McNeil. Dr. Calabrese has received funding from Abbott, AstraZeneca, Merck, GlaxoSmithKline, Janssen, Eli Lily, and Pfizer; and has had consulting agreements/served on advisory boards for Abbott, AstraZeneca, Bristol‐Myers Squibb/Otsuka, Eli Lilly, GlaxoSmithKline, Janssen, and Teva. Drs. Rubenfaer, Collins, and Abi‐Saab report no additional financial or other relationships relevant to the subject of this article" | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Eligible patients were randomly assigned on day 1 in a 1:1 ratio" Comment: Although little information is provided here as to the method of randomisation, previous studies by Bowden (e.g. Bowden 1994) in the same area have used robust randomisation techniques. We have therefore decided to assign this a low risk of bias. |
| Allocation concealment (selection bias) | Low risk | Comment: Although little information is provided here as to the method of allocation concealment, previous studies by Bowden (Bowden 1994) in the same area have used robust allocation concealment techniques. We have therefore decided to assign this a low risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double blind" Quote: "Blinded medication was administered once daily in the morning" Comment: A system of central valproate‐serum analysis, with matched sham‐calls if medication dosage needed to be changed was employed to maintain blind. |
| Blinding of outcome assessment (detection bias) Efficacy | Low risk | Quote: "The same rater performed all evaluations for a patient where possible" Comment: Although it is not made explicit that the raters were blind, a previous Bowden study (Bowden 1994) used a similar system with blind raters. We have therefore decided to assign a low risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Completion rates were comparable between the divalproex ER (58%) and placebo (52%) treatment groups" Comment: Although both groups had a greater than 30% dropout rate, the rates were comparable (less than two‐fold difference). |
| Selective reporting (reporting bias) | Unclear risk | Comment: Trial was completed in May 2004. Protocol was updated with primary outcome measures in February 2006, and therefore cannot be considered a pre‐published protocol. We are therefore unable to assess bias. |
| Other bias | Low risk | None identified |
DelBello 2006.
| Methods | Randomised double‐blind trial | |
| Participants |
Number of total participants/type of patients: 50 participants randomised. Inpatient study Inclusion criteria: Sex: male and female Age: adolescents (12 ‐ 18 years) Diagnosis: Bipolar I disorder, manic or mixed episode, according to the DSM‐IV‐TR criteria and determined by the WASH‐U‐KSADS interview. In addition, to be included in this study, patients were required to have a YMRS score of ≥ 20 at baseline. Exclusion criteria: Patients were excluded by having a substance use disorder (other than nicotine use disorder) within the previous 3 months; an unstable medical or neurological illness; a history of intolerance or non‐response to quetiapine or valproate monotherapy; or treatment with an antidepressant, an anticonvulsant (other than as noted below), or atomoxetine within 7 days (fluoxetine within 4 weeks) or an antipsychotic or psychostimulant within 48 weeks from baseline. Patients who had received treatment with lithium, valproate, or carbamazepine previously were required to have undetectable serum concentrations ( < 0.4 mEq/L, < 30 µg/mL, and < 4 µg/mL, respectively). |
|
| Interventions |
Location: USA, children's hospital site in Cincinnati Study duration: July 2002 ‐ January 2004 Treatment groups: 1. Valproate: participants randomised to valproate received an initial dose of 20 mg/kg/day in the evening (5 ‐7 p.m.) on day 0 and matching placebo pills of quetiapine. Doses of valproate administered at bedtime on successive days were adjusted to achieve serum valproic acid levels of 80 to 120 µg/mL. Within the valproate group, the mean valproic acid level at endpoint was 101 µg/mL; 96% of participants achieved a therapeutic valproic acid level (> µg/ mL) by day 7. 2. Quetiapine: participants randomised to quetiapine received a dose of 100 mg on day 0, which was increased to 400 mg/day by days 4 to 7, and up to a maximum of 600 mg/day, thereafter in 1 or 2 divided doses, with matching placebo pills. Within the quetiapine group, the mean (SD) quetiapine dose at endpoint was 412 (SD 83) mg/day in responders. Concomitant medications: The use of lorazepam (a maximum of 4 mg/day for days 0 ‐ 7 and 2 mg/day for days 8 ‐ 14) was permitted during the study. 3 participants (12%) in the quetiapine group received a total of 5 doses of lorazepam for agitation during the first week of their study participation. 2 participants (7%) in the valproate group received a total of 3 doses of lorazepam for agitation during the first week of their study participation. Length of study: 28 days Randomisation: Randomisation was stratified by age group 25 patients randomised to valproate 25 patients randomised to quetiapine |
|
| Outcomes |
Primary outcome: Change in YMRS score Secondary outcomes: Clinical Global Impression‐Bipolar Disorder Version Improvement scores for overall illness and manic symptoms CGI‐BP‐I overall and CGI‐BP‐I mania). Response was defined by an endpoint CGI‐BP‐I score ≤ 2 (much or very much improved) for overall bipolar disorder and mania at endpoint. Remission rates, defined by an endpoint YMRS score ≤ 12. Change from baseline to endpoint in the Positive and Negative Syndrome Scale, Positive Subscale (PANSS‐P), the CDRS‐R. Tolerability evaluations included assessments of adverse events, vital signs, and movement scales Study withdrawals were reported |
|
| Funding | "The study was supported by a grant from AstraZeneca" | |
| Conflict of interest | "Dr. DelBello has received research support, speaker honoraria, and/or consulting fees from Abbott Laboratories, AstraZeneca, Bristol‐Myers Squibb, Eli Lilly, GlaxoSmithKline, Janssen, OrthoMcNeil, Pfizer, and Shire. Dr. Kowatch has received research support, speaking honoraria, and/or consulting fees from Bristol‐Myers Squibb, GlaxoSmithKline, Janssen, and AstraZeneca. Dr. Adler has received research support, speaker honoraria, and/or consulting fees from Abbott Laboratories, AstraZeneca, Eli Lilly, Janssen, and Pfizer. Dr. Welge has received research support from Abbott Laboratories. Dr. Barzman has received research support from AstraZeneca and Pfizer. Dr. Nelson has received research support, speaker honoraria, and/or consulting fees from Cephalon Pharmaceutical, Eli Lilly, Forest Laboratories, Merck, Pfizer, and Wyeth‐Ayerst Pharmaceuticals. Dr. Strakowski has received research support, speaker honoraria, and/or consulting fees from Abbott Laboratories, AstraZeneca, BristolMyers Squibb, Eli Lilly, Forest Laboratories, Janssen, Ortho‐McNeil, and Pfizer. Mr. Stanford has no financial relationships to disclose." | |
| Notes | Study focused on teenagers between 12 – 18 years of age | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: “Randomization was assigned by investigational pharmacists and was stratified by gender and the presence of psychosis using a random number generator.” |
| Allocation concealment (selection bias) | Unclear risk | Quote: “Randomization was assigned by investigational pharmacists and was stratified by gender and the presence of psychosis using a random number generator.” Comment: We contacted the lead investigator who told us that randomisation was done by investigational pharmacist who prepared the medication. However, it is still unclear in what way the randomisation sequence was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The study medications were administered in a double‐dummy, double‐blinded manner" Quote: "All patients received placebo pills for the medication to which they were not randomised." Quote: "Two psychiatrists who were not blinded to the treatment status of patients (R.A.K., S.M.S.) and who did not perform efficacy or tolerability ratings on any patient monitored valproate levels on all of the patients and adjusted the valproate dose to achieve a therapeutic valproic acid serum level. Some patients receiving quetiapine had their placebo doses adjusted to avoid breaking the blind (i.e., yoking)." |
| Blinding of outcome assessment (detection bias) Efficacy | Low risk | Quote: “Specifically, all patients, caregivers, and investigational staff who performed efficacy and tolerability ratings were blinded to patient treatment group.” |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: In all groups dropout rate was below 30%. |
| Selective reporting (reporting bias) | Unclear risk | Comment: No pre‐published protocol found. We are therefore unable to assess the risk. |
| Other bias | Low risk | Comment: None identified |
Feifel 2011.
| Methods | Randomised single‐blind trial | |
| Participants |
Number of total participants/type of patients: 30 participants randomised. Inpatient study Inclusion criteria: Sex: male and female Age: 18 ‐ 65 years Diagnosis: Bipolar I disorder, most recent episode manic, or bipolar I disorder, most recent episode mixed, with or without psychotic features, as defined by DSM‐IV were recruited. Participants were recruited from among those presenting to the University of California, San Diego (UCSD) Medical Center’s emergency department and deemed needing hospitalisation for their mania, and from those recently admitted to the inpatient psychiatry unit for treatment of acute mania. Consenting participants were enrolled if they scored > 17 on the YMRS as well as receiving a score of 4 (moderate) or higher on the CGI‐S. Exclusion criteria: Treatment with a depot antipsychotic was within 1 treatment cycle. |
|
| Interventions |
Location: USA, San Diego Medical Center Study duration: Not stated Treatment groups: 1. Valproate extended‐release: Dose initiated at 30 mg/kg/day orally taken at night, rounded up to the nearest 500 mg dose, with adjustments made through the trial to obtain optimal serum valproic acid levels between 85 and 125 mug/mL 2. Quetiapine: Dose given orally at an initial dose of 200 mg/day and titrated up to a target dose of 600 to 800 mg/day, based upon published rapid‐loading regimens Concomitant medications: Lorazepam was provided for agitation and insomnia as needed for rescue only. The maximum dose of lorazepam was 6 mg in the first 7 days, 4 mg for the next 3 days, and 2 mg/day for the remainder of the study. Those who required a greater amount of lorazepam were excluded. Nonpsychotropic medications were allowed as deemed necessary by the participant’s treated physician. Length of study: 21 days Randomisation: 14 patients randomised to valproate extended‐release 16 patients randomised to quetiapine |
|
| Outcomes |
Primary outcome: Change from baseline to endpoint on the YMRS Secondary outcomes: CGI‐S, CGI‐I, Extra Pyramidal Symptoms Rating Scale (ESRS), MADRS, Behavioural Activity Rating Scale (BARS) Adverse events were reported Study withdrawals were reported |
|
| Funding | "The study was supported by a research grant from Abbott Laboratories" | |
| Conflict of interest | "Dr. Feifel has received funding for research, consulting, or speaking from Abbott Labs, Alexa, AstraZeneca, Bristol‐Myers Squibb, Eli Lilly, Forest, Janssen, Merck, Otsuka, Pfizer, Sanofi, Shionogi, Shire, and Sunovion; Dr. MacDonald has received funding for research, consulting, or speaking from Cypress Bioscience, Eli Lilly, Onu Pharmaceuticals, and Pfizer; Ms. Galangue, Cobb, and Dinca and Drs. Becker, Cooper, and Hadley report no conflicts of interest relevant to the content of this article." | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: No comment on randomisation procedure in text. We contacted the authors but received no reply. |
| Allocation concealment (selection bias) | Unclear risk | Comment: No comment on randomisation concealment in text. We contacted the authors but received no reply. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Raters but not patients or treating physicians were blinded." |
| Blinding of outcome assessment (detection bias) Efficacy | Low risk | Quote: "Raters but not patients or treating physicians were blinded." Quote: "Independet raters, blind to the subjects' treatments used the following scales to assess efficacy." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: In all groups dropout rate was below 30%. |
| Selective reporting (reporting bias) | High risk | Comment: Mean change in YMRS scores from baseline to endpoint reported without standard deviations. All other data represented only in graphical form. This leaves us unable to conduct any analysis on any of these data. |
| Other bias | Low risk | Comment: None identified |
Freeman 1992.
| Methods | Randomised double‐blind trial | |
| Participants |
Number of total participants/type of patients: 27 participants were randomised. Inpatient study Inclusion criteria: Sex: male and female Age: no information provided Diagnosis: meeting DSM‐III‐R criteria for a manic episode Other: able to give informed consent Exclusion criteria: 1. Abnormal EEGs, LFTs, TFTs, haematological findings or positive urine drug screens 2. History of drug or alcohol abuse within the previous 12 months or before the onset of major affective episode 3. Focal neurological abnormalities |
|
| Interventions |
Location: USA, Houston Study duration: Not stated Treatment groups: 1. Valproate group: 1500 mg/day for first week, 2250 mg/day for second week and 3000 mg/day for third week 2. Lithium started at 0.5 mEq/kg a day, increased to maximum of 1800 mg/day or 1.5 mmol/L Concomitant medications: Rescue medication, including chloral hydrate and lorazepam, was allowed for extreme behavioural problems not responding to non‐pharmacological interventions Duration of trial: 21 days Randomisation: 14 patients randomised to valproate 13 patients randomised to lithium |
|
| Outcomes |
Outcomes: 1. Changes on the YMRS scores as assessed by SADS‐C 2. GAS 3. BPRS 4. Study withdrawals were reported |
|
| Funding | Not stated | |
| Conflict of interest | Not stated | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were assigned randomly to treatment with lithium or valproate in a double‐blind fashion" Comment: Although participants were randomly assigned, there is no detail on the method used in the text. We were therefore unable to evaluate bias. Comment: We contacted the authors but did not receive a reply. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Patients were assigned randomly to treatment with lithium or valproate in a double‐blind fashion" Comment: There is no detail on any procedures present to conceal allocation. We were therefore unable to evaluate bias. Comment: We contacted the authors but did not receive a reply. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double‐blind" Quote: "Plasma drug levels were monitored on days 5‐7 of each week, the results were sent to the research pharmacist, and the dose was adjusted by a non‐blinded, non‐treating physician." Comment: Medication was given in liquid form and always diluted to a total volume of 30 ml. Procedures in place to ensure blinding seem reasonable. |
| Blinding of outcome assessment (detection bias) Efficacy | Unclear risk | Quote: "Double‐blind" Quote: "the dose was adjusted by a non‐blinded, non‐treating physician. The SADS‐C, the GAS and the BPRS were administered at the time of dose adjustments." Comment: The dose was adjusted by a non‐blinded physician and the rating scales were administered at the time of dose adjustments. This implies that the outcomes may have been evaluated by an unblinded clinician. However, there is no detail in text about who administered the rating scales. On contacting the authors for clarification, we received no reply. We are therefore unable to assess bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: In all groups dropout rate was below 30%. |
| Selective reporting (reporting bias) | Unclear risk | Comment: No pre‐published protocol found. We are therefore unable to assess the risk. |
| Other bias | Low risk | Comment: None identified. |
Geller 2012.
| Methods | Randomised single‐blind trial | |
| Participants |
Number of total participants/type of patients: 290 participants randomised. Outpatient study Inclusion criteria: Sex: male and female Age: children and adolescents (6 ‐ 15 years) Diagnosis: DSM‐IV diagnosis of bipolar I disorder, manic or mixed episode, for at least 4 consecutive weeks immediately preceding baseline, with a CGAS score of 60 or less at baseline and in good physical health. Co‐occurring attention‐deficit hyperactivity, oppositional defiant, and conduct disorder were allowed because these are common comorbidities in childhood mania. Suicidal ideation was allowed if there was no imminent risk. Participants required no history of receiving study psychotropics or their equivalents. All medication histories were verified by physician or pharmacy records, or both, to enhance interview accuracy. Exclusion criteria: An IQ of less than 70, a lifetime history of schizophrenia, pervasive developmental disorder or major medical or neurological disease, substance use dependency, alcohol or drug abuse within the past 4 weeks, pregnancy, sexually active and not using contraceptives, or nursing. Other psychotropics and medications associated with psychiatric symptoms were not permitted. |
|
| Interventions |
Location: USA, hospital sites in Washington DC, Baltimore, Pittsburgh, Dallas, and St. Louis Study duration: 2003‐2008 Treatment groups: Valproate: started at 125 mg or 250 mg (depending on participant's weight) for 2 days, then 125/250 mg twice a day. This dose was adjusted based on the participant's reaction to up to 111 ‐ 135 µg/ml Risperidone: started at 0.25 mg or 0.5 mg for 2 days (depending on participant's weight), then 0.5 mg twice a day. This dose was adjusted based on the participant's reaction to up to 2.0 ‐ 3.0 mEq/L Lithium: started at 150 mg or 300 mg (depending on participant's weight) for 2 days, then 150/300 mg twice a day. This dose was adjusted based on the participant's reaction to up to 1.1 ‐ 1.3 mEq/L Concomitant medications: Maintenance methylphenidate and amphetamine preparations (total daily dose equivalent to < 60 mg methylphenidate), verified by pharmacy/physician records, and allergy/asthma medications were allowed, to mimic usual clinical practice. No stimulant dose adjustment was allowed during protocol. Antidepressants were tapered during the first week of study to avoid risk of increased mania symptoms. Participants required no history of receiving study psychotropics or their equivalents. All medication histories were verified by physician or pharmacy records, or both, to enhance interview accuracy Length of study: 8 weeks Randomisation: Randomisation was stratified by age group. 104 patients randomised to valproate 93 patients randomised to risperidone 93 patients randomised to lithium |
|
| Outcomes |
Primary outcome: CGI‐BP‐I. Secondary outcome: K‐SADS Mania Rating Scale (KMRS). Adverse events were reported. Study withdrawals were reported. |
|
| Funding | "This work was supported by the National Institute of Mental Health" | |
| Conflict of interest | "Dr Geller reports the following for the work under consideration: a grant from NIMH; support for travel to meetings from NIMH; payment for writing or reviewing the manuscript from NIMH; and provision of writing assistance, equipment, or administrative support from NIMH. Dr Geller also reports the following from outside the submitted work: consultancy for NIMH and the US Food and Drug Administration (FDA) Federal Advisory Committees; employment at Washington University in St Louis, Missouri; grants from NIMH; payment for lectures from Vanderbilt University and the International Review of Bipolar Disorder; payment for manuscript preparation from NIMH; royalties from Guilford Press; travel, accommodations, and meeting expenses from NIMH and FDA for service on Federal Advisory Committees; payment from Massachusetts Medical Society for Journal Watch in Psychiatry Associate Editorship. Dr Luby reports the following for the work under consideration: grant from NIMH and provision of medicines from Abbott. Dr Luby also reports the following from outside the submitted work: employment at Washington University School of Medicine in St Louis, Missouri; grants/grants pending fromNIMH, National Alliance for Research on Schizophrenia and Depression, and CHADS; and royalties from Guilford Press. Dr Joshi reports the following from the work under consideration: a grant from NIMH; support for travel to meetings from NIMH; provision of medicines from Abbott. Dr Joshi also reports the following from outside the submitted work: employment at Children’s National Medical Center in Washington, DC. Dr Wagner reports the following from the work under consideration: grant from NIMH and provision of medicines from Abbott. Dr Wagner also reports the following from outside the submitted work: consultancy for Forest, American Institute of BiologicalSciences, Krog and Partners, and National Institutes of Health; employment at University of Texas Medical Branch in Galveston; payment for lectures from American Psychiatric Association, Letters and Sciences, American Society of Clinical Psychopharmacology, Toledo Hospital, American Academy of Child and Adolescent Psychiatry, Madison Institute of Medicine, Mexican Psychiatric Association, Contemporary Forums, Doctors Hospital at Renaissance, CME LLC, Nevada Psychiatric Association, and Quantia Communications; payment for manuscript preparation from Guilford Publications, Health and Wellness Education Partners, American Psychiatric Publishing Inc, Springer Publishing, CMP Medica, UBM Medica, and Wolters Kluver Health; payment from Physician’s Postgraduate Press, Inc, for serving as deputy editor of the Journal of Clinical Psychiatry. Dr Wagner also sits on the Scientific Advisory Board of the Child and Adolescent Bipolar Foundation and on the Scientific Advisory Board of the Depression and Bipolar Support Alliance. Dr Emslie reports the following from the work under consideration: a grant from NIMH and provision of medicines from Abbott. Dr Emslie also reports the following from outside the submitted work: consultancy for Biobehavioral Diagnostics, Inc, Eli Lilly, Forest, GlaxoSmithKline, Pfizer, Shire, Validus, and Wyeth; employment at University of Texas Southwestern Medical Center; grants/grants pending from NIMH, Biobehavioral Diagnostics, Inc, Eli Lilly, Forest, GlaxoSmithKline, Shire, and Somerset; payment for lectures, including service on speakers bureaus from Forest; and receiving payment for manuscript preparation from British Medical Journal Online. Dr Walkup reports the following for the work under consideration: a grant from NIMH; support for travel to meetings from NIMH; and provision of medicines from Abbott. Dr Axelson reports the following for the work under consideration: a grant from NIMH. Ms Bolhofner reports the following for the work under consideration: a grant from NIMH. Ms Bolhofner also reports the following from outside the submitted work: employment at Washington University in St Louis, Missouri, and grants from NIMH. Dr Robb reports the following for the work under consideration: a grant from NIMH; support for travel to meetings from NIMH; provision of medicines from Abbott; and payment for serving as clinical pharmacologist for the Eunice Kennedy Shriver National Institute of Child Health and Human Development. Dr Robb also reports the following from outside the submitted work: board membership at Lilly, Bristol Myers Squibb, Otsuka, Shinogi, and McNeil Pediatrics; consultancy for Lundbeck; employment at Children’s National Medical Center; expert testimony for a case on antipsychotic use; grants/grants pending from Bristol Meyers Squibb, McNeil Pediatrics, Merck Scherring Plough, GlaxoSmithKline, Janssen, Sepracor, Supernus, Otsuka, Pfizer, Johnson and Johnson, and Forest; payment for service on speakers bureaus from Bristol Myers Squibb, Lilly, and McNeil Pediatrics; royalties from Epocrates; payment for development of education presentations from University of Minnesota, American Academy of Child& Adolescent Psychiatry, and American Academy of Pediatrics; stock/stock options from Lilly, Pfizer, Johnson and Johnson, GlaxoSmithKline, and 3M. Dr Robb also sits on the Children and Adults with Attention‐Deficit/Hyperactivity Disorder professional advisory board and program committee for the American Psychiatric Association annual meeting, and her husband sits on American Epilepsy Society Board and Scientific Committee for Child Neurology Society. Dr Wolf reports the following for the work under consideration: a grant from NIMH. Dr Riddle reports the following for the work under consideration: a grant from NIMH and provision of medicines from Abbott. Dr Riddle also reports the following from outside the submitted work: employment at Johns Hopkins University; expert testimony for Teva Canada; and receiving aripiprazole for an NIMH study. Dr Birmaher reports the following for the work under consideration: a grant from NIMH. Dr Birmaher also reports the following from outside the submitted work: consultancy for Schering Plough, Dey Pharma, Forest, and Jazz Pharmaceuticals; and royalties from Random House and Lippincott Williams and Wilkins. Dr Nusrat reports the following for the work under consideration: a grant from NIMH; support for travel to meetings from NIMH; and provision of medicines from Abbott. Dr Nusrat also reports the following from outside the submitted work: employment at Children’s National Medical Center and grants/grants pending from Merck/Scherring Plough, GlaxoSmithKline, Janssen, Sepracor, Supernus, Otsuka, Pfizer, Johnson and Johnson, and Forest. Dr Ryan reports the following for the work under consideration: a grant from NIMH; support for travel to meetings from NIMH; and provision of medicines from Abbott. Dr Ryan also reports the following from outside the submitted work: employment at the University of Pittsburgh and the University of Pittsburgh Medical Center. Ms Tillman reports the following for the work under consideration: a grant from NIMH and payment for writing or reviewing the manuscript from NIMH. Ms Tillman also reports the following from outside the submitted work: employment at Washington University in St Louis and receiving travel/accommodations/meeting expenses from NIMH. Dr Lavori reports the following for the work under consideration: a grant from NIMH." | |
| Notes | Study focused on children/teenagers between 6 – 15.11 years of age. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The ranuni function in the SAS version 8.1 statistical software package (SAS Institute Inc) was used to create random lists of the 3 medications for each combination of the stratifying variables at each site." |
| Allocation concealment (selection bias) | Low risk | Quote: "When subjects were randomly assigned, the randomised medication was determined by selecting the next available entry in the list corresponding to the subject’s stratifying variables and site. Randomization was performed at the coordinating site, and a form identifying the randomised medication was e‐mailed to the site’s non‐blinded staff members.” |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Patients, family members, and treating clinicians were aware of treatment assignment." |
| Blinding of outcome assessment (detection bias) Efficacy | Low risk | Quote: "Independent evaluators (IEs) who were blinded to medication status administered baseline and endpoint assessments.Masking of the treatment assignment to the IEs was strictly enforced by using staff who were totally uninvolved with the subjects’ treatment. Families were instructed not to reveal either the medication or adverse events to the blinded end‐point raters.Separate,non‐blinded interviewers conducted the weekly assessments." Comment: Baseline and endpoint measures are our primary outcome, and these are low risk. Midpoint scores are high risk, as raters were not blinded. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "The discontinuation rate was significantly higher for subjects randomly assigned to the lithium group than for subjects randomly assigned to the risperidone group (32.2 % vs 15.5%)." Comment: Dropout rates above 30% in at least 1 group. Rates two‐fold higher in lithium group compared to risperidone. |
| Selective reporting (reporting bias) | High risk | Quote: "Adverse effects were considered present if the severity score was a 2 or 3 on a scale of 0 to 3. The only adverse effects presented are those that occurred in at least 5% of subjects who were treated with the given medication and that had at least a twofold increase or decrease during the study" Comment: Efficacy outcomes reported in line with pre‐published protocol. Comment: Adverse events reported in a way that makes analysis of the data impossible. We contacted the authors to ask for a full list of experienced side effects but they did not reply. |
| Other bias | Low risk | Comment: None identified. |
Hebrani 2009.
| Methods | Randomised double‐blind trial | |
| Participants |
Number of total participants/type of patients: 142 participants randomised. Inpatient study Inclusion criteria: Sex: male and female Age: adolescents (12 ‐ 18 years) Diagnosis: bipolar I disorder based on DSM‐IV‐TR, manic or mixed episode, YMRS ≥ 20 at baseline Exclusion criteria: Substance use disorder (other than nicotine) within the previous 3 months; unstable medical or neurological illness; history of intolerance or no response to topiramate or valproate monotherapy; ongoing treatment with an antidepressant or an anticonvulsant (other than as noted below) within 4 weeks or an antipsychotic or psychostimulant within at least 48 hours. Patients who were receiving treatment with lithium, valproate, or carbamazepine were required to have undetectable serum concentrations ( = 0.4 mEq/L = 30 µg/mL, and = 4 µg/mL, respectively) before entering the study, to ensure an adequate washout period. |
|
| Interventions |
Location: Iran, Mashad Study duration: September 2 ‐ September 2007 Treatment groups: 1. Valproate: participants in valproate group received an initial dose of 200 – 400 mg in the evening ( 5 – 7 p.m.) on day 0. Every 2 days the dose was increased by 200 – 400 mg; based on tolerability, the maximum dose was increased to 1200 mg by day 7 2. Topiramate: topiramate group participants received a dose of 50 mg on day 0; every 2 days the dose was increased by 50 mg, and based on tolerability, the maximum dose (400 mg/days) was reached by day 7. After complete titration, participants were maintained on a stable dose through 8 weeks Concomitant medications: The use of antidepressants/anticonvulsant or antipsychotics was not permitted. The use of lorazepam was permitted to reduce agitation or insomnia during the study (a maximum of 4 mg/day for days 0 ‐ 7 and 2 mg/day for days 8 ‐ 14). Length of study: 8 weeks Randomisation: 71 patients randomised to valproate 71 patients randomised to topiramate |
|
| Outcomes |
Primary outcome: Changes in scores on the YMRS Secondary outcome: CGAS Adverse events were reported Study withdrawals were reported |
|
| Funding | Not stated | |
| Conflict of interest | Not stated | |
| Notes | Study focused on teenagers between 12 – 18 years of age | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: No comment on randomisation procedure in text. We contacted the authors but received no reply. |
| Allocation concealment (selection bias) | Unclear risk | Comment: No comment on randomisation concealment in text. We contacted the authors but received no reply. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Medications were prescribed in a double‐blind manner." Comment: No more information than this given on blinding procedures, so it is difficult to exactly establish whether it was blind. |
| Blinding of outcome assessment (detection bias) Efficacy | Low risk | Quote: "All investigational staff members who performed efficacy and tolerability rating scales were blind to the patient treatment group" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: In all groups dropout rate was below 30%. |
| Selective reporting (reporting bias) | Unclear risk | Comment: No pre‐published protocol found. We are therefore unable to assess the risk. |
| Other bias | Low risk | Comment: None identified |
Hirschfeld 1999.
| Methods | Randomised double‐blind trial | |
| Participants |
Number of total participants/type of patients: 59 participants randomised. Inpatient study Inclusion criteria: Sex: male and female Age: 18 ‐ 60 years Diagnosis: DSM‐IV criteria for bipolar disorder currently manic or mixed episode and a score of ≥ 14 on the YMRS Exclusion criteria: Intolerance to valproate or lithium Concurrent medical disorders Substance dependence Serious risk of suicide Pregnancy Screened positive for amphetamines or phencyclidine Depot antipsychotic drugs Investigational drug within previous 4 weeks Any drug that may interfere with safety or efficacy of trial medications |
|
| Interventions |
Location: USA, hospital site(s) (cities not stated) Date of study: Not stated Treatment groups: 1. Valproate ‐ non‐loading: 750 mg 3 times a day (days 1 and 2) with gradual dose titration (days 3 ‐ 10) Valproate ‐ loading: 30 mg/kg a day (days 1 and 2) then 20 mg/kg per day (days 3 ‐ 10) 2. Lithium: 300 mg 3 times a day (days 1 and 2); gradual dose titration (days 3 ‐ 10) Concomitant medications: Lorazepam was allowed to manage agitation, insomnia, restlessness, irritation, and hostility (4 mg/day on days 1 ‐ 4 and 2 mg/day on days 5 ‐ 7) Length of study: 10 days Randomisation: 20 patients randomised to valproate non‐loading 20 patients randomised to valproate loading 19 randomised to lithium |
|
| Outcomes |
Outcomes: 1. Changes on the YMRS scores as assessed by the Schedule for Affective Disorders and Schizophrenia Change Version (SADS‐C) 2. GAS Adverse events were reported Study withdrawal was reported |
|
| Funding | "Sponsored by Abbott Laboratories (M95‐305)." | |
| Conflict of interest | Not stated | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomised" Comment: No other information about method of randomisation, so we were unable to assess risk of bias. We established contact with the lead author who no longer has access to the data or protocols. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Randomised" Comment: No other information about method of allocation concealment, so we were unable to assess risk of bias.We established contact with the lead author who no longer has access to the data or protocols. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double blind" Quote: "Blinded medication was provided in identical‐appearing grey tablets so that all patients received the same total number of capsules." |
| Blinding of outcome assessment (detection bias) Efficacy | Low risk | Quote: "Blinded raters evaluated the patients" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "Seven patients (35%) in each of the valproate‐treated groups and 9 (47%) in the lithium standard‐titration group discontinued the study medication before the conclusion of the trial." Comment: Both groups have a > 30% dropout rate. However, dropout rates are comparable (less than a two‐fold difference). |
| Selective reporting (reporting bias) | High risk | Comment: Means and standard deviations of all outcomes are not reported. We established contact with the lead author who no longer has access to these data. |
| Other bias | Low risk | Comment: None identified |
Hirschfeld 2010.
| Methods | Randomised double‐blind trial | |
| Participants |
Number of total participants/type of patients: 225 participants randomised. Inpatient study Inclusion criteria: Sex: male and female Age: 18 ‐ 65 years Diagnosis: a DSM‐IV diagnosis of bipolar I disorder (manic or mixed episode) confirmed by Structured Clinical Interview for DSM‐IV (SCID). Participants had to have a MRS score ≥ 25 with at least 4 items having a score ≥ 3 on the final day of the screening/washout period. Exclusion criteria: Having one of the 5 schizophrenia‐like symptoms listed in the SCID as excluding a person for a diagnosis of manic syndrome while not manic or if their first manic episode occurred when older then 60 years. History of schizophrenia or schizoaffective disorder axis I (e.g. anxiety disorder), or axis II (e.g. personality disorder) that would interfere with compliance or confound interpretation of study results. Current manic or mixed episode is drug‐induced or secondary to a medical disorder (e.g. AIDS, corticosteroids). Current manic or mixed episode is believed to be caused by antidepressant use (i.e. antidepressant‐induced mania) Had first manic episode after age 60 years Has ever taken clozapine Has received depot neuroleptic medication within 1 inter‐injection interval of first dose of study drug Urine toxicology screen is positive for phencyclidine (PCP), opiates, cocaine or amphetamines History of active alcohol or substance dependence within past 3 months History of failed treatment on adequate valproate therapy for bipolar disorder Has taken depakote (DR or ER) regularly over the last 30 days Has serious violent, homicidal, or suicidal ideation Women of childbearing potential were allowed on condition that they were not pregnant and agreed to use effective contraception |
|
| Interventions |
Location: USA, hospital site(s) (cities not stated) Study duration: May 1998 ‐ July 1999 Treatment groups: 1. Valproate extended‐release: dosing initiated at 20 mg/kg once daily with increases allowed at days 5,10 and 15 if manic symptomology persisted 2. Placebo Concomitant medications: chloral hydrate and lorazepam allowed as rescue medications Length of study: 21 days Randomisation: 147 patients randomised to valproate extended‐release 78 patients randomised to placebo |
|
| Outcomes |
Primary outcome: change in MRS Secondary outcomes: Manic Syndrome Score, Behavioural and Ideation Score, Brief Agitation Rating Scale, Overt Aggression Scale, BPRS Safety/tolerability measures included adverse events and several laboratory evaluations (e.g. platelet count) Study withdrawals were reported |
|
| Funding | "Funding/support: Financial support for the study was provided by Abbott" | |
| Conflict of interest | "Potential conflict of interest: Dr Hirschfeld is a consultant for or a member of the advisory boards for Dainippon Sumitomo, Forest, Health and Wellness Partners, Pfizer, and Takeda; and receives royalties from Compact Clinicals, Taylor and Francis Group, Epocrates, Ogilvy Healthworld, Merck Manual, and Jones and Bartless. Dr Bowden is a consultant for Pfizer, sanofi‐aventis, and Schering; receives grant/research support from Repligen, Bristol‐Myers Squibb, and Janssen; and receives honoraria from Physicians Postgraduate Press (J Clin Psychopedia) and American College of Clinical Psychiatry. Dr Wozniak was employee of, is a stockholder of, has received a pension from, and has a spouse employed by Abbott. Drs Vigna and Collins are employees of Abbott." | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients randomly assigned in a 2:1 ratio" Comment: No other information about method of randomisation, so we were unable to assess risk of bias. We established contact with the lead author who no longer has access to the data or protocols. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Patients randomly assigned in a 2:1 ratio." Comment: No other information about method of allocation concealment, so we were unable to assess risk of bias. We established contact with the lead author who no longer has access to the data or protocols. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: “Double‐blind” Quote: “Blinded serum valproate levels were obtained prior to dosing on days 5, 10 and 15 and on last day of treatment period” Comment: No mention of who was blind or any procedures to keep blinding in place. We established contact with the lead author who no longer has access to the data or protocols. |
| Blinding of outcome assessment (detection bias) Efficacy | Unclear risk | Quote: “All ratings were performed by the same individual at approximately the same time of day for each subject within each site” Comment: No mention of any blinding, so we were unable to assess risk of bias. We established contact with the lead author who no longer has access to the data or protocols. |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: Participants were discharged from the study if they reached 2 improvement criteria: 1. “MRS score reduced by 50% or more from the last day of the washout period” and 2. “no MRS item was >3 at the time of the last rating of the treatment period” This meant that 40% of participants were enrolled in the study for less then 7 days. The valproate group had a dropout rate of 83%, the placebo group 82%. As the combined dropout rates were higher than 75% there is a high risk of bias. |
| Selective reporting (reporting bias) | High risk | Comment: Published protocol where primary outcome is the same as in the paper. However protocol was first uploaded to clinicaltrials.gov in 2003, but the trial was completed in 1999. This therefore cannot be considered a pre‐published protocol, and thus bias risk is uncertain. Standard deviations were not reported, so therefore rated high risk. |
| Other bias | Low risk | Comment: None identified |
Kakkar 2009.
| Methods | Randomised double‐blind trial | |
| Participants |
Number of total participants/type of patients: 60 participants randomised. Inpatient study Inclusion Criteria: Sex: male and female Age: 18 – 50 years Diagnosis: meet DSM‐IV diagnostic criteria for a manic episode of bipolar disorder. Participants were required to have a baseline YMRS score of ≥ 20 for study entry Exclusion Criteria: Serious and unstable medical illness. DSM‐IV substance dependence within the past 30 days (except nicotine and caffeine); documented history of intolerance to oxcarbazepine or valproate; treatment with lithium, an anticonvulsant, or an antipsychotic medication within 24 hours of randomisation, serious suicidal risk, hypomania, bipolar depression, mixed states, rapid cycling disorder states and patients exhibiting severe excitement, and pregnant or lactating women |
|
| Interventions |
Location: India, hospital site in New Delhi Date of study: Not stated Treatment groups: 1. Valproate: this group received oral valproate starting from 750 mg/day up to a maximum of 2000 mg/day for 12 weeks. It was decided that the dose would be reduced if any participant develops excessive sedation, ataxia, tremors, or any other intolerable side effects.The mean dose was 1425 mg/day (range 750 – 2000 mg) in the valproate group 2. Oxcarbazepine: this group received oral oxcarbazepine starting from 300 mg/day and gradually increasing it to a maximum of 2400 mg/day for 12 weeks. Dose was to be reduced if any treatment‐emergent intolerable side effects including features of hyponatraemia. The mean dose was 1280 mg/day (range 1000 – 2400 mg) in the oxcarbazepine group Concomitant medications: The study protocol allowed the use of adjunctive lorazepam (maximum dose of 2 mg/day) as needed for the control of agitation, irritation, restless, insomnia, and hostile behaviour. However, lorazepam was not permitted during the 8 hours before behavioural assessment. The use of antipsychotics was considered an exclusion criterion Length of study: 12 weeks Randomisation: 30 patients randomised to valproate 30 patients randomised to oxcarbazepine |
|
| Outcomes |
Primary outcome: Change in YMRS from baseline to endpoint. An adequate response in the YMRS was taken as a decrease in scores by at least 50%. Adverse events were reported |
|
| Funding | Not stated | |
| Conflict of interest | Not stated | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: No comment on randomisation procedure in text. We contacted the authors but received no reply. |
| Allocation concealment (selection bias) | Unclear risk | Comment: No comment on randomisation concealment in text. We contacted the authors but received no reply. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double‐blind" Comment: No discussion of who is blind or procedures in place to ensure blinding, which makes bias unassessable. |
| Blinding of outcome assessment (detection bias) Efficacy | Unclear risk | Quote: "Double‐blind" Comment: No discussion of who is blind or procedures in place to ensure blinding, which makes bias unassessable. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "In general, both the treatments were well tolerated as evidenced by high study completion rates." Comment: No dropouts mentioned. In combination with the comment above and reporting on all participants we can assume < 30% dropout rates in any group. |
| Selective reporting (reporting bias) | Unclear risk | Comment: No pre‐published protocol found. We are therefore unable to assess the risk. |
| Other bias | Unclear risk | Quote: "The study protocol allows the use of adjunctive lorazepam [...] lorazepam was not permitted during the eight hours before behavioural assessment." Comment: No information about whether lorazepam was balanced between the two groups. |
Kowatch 2015.
| Methods | Randomised double‐blind trial | |
| Participants |
Number of total participants/type of patients: 46 participants were randomised. Outpatient study Inclusion criteria: Sex: male and female Age: children (3 ‐ 7 years) Diagnosis: bipolar I diagnosis according to the DSM IV‐TR criteria, manic or mixed episode (with or without psychotic features) with a score ≥ 20 on the YMRS at the time of randomisation Exclusion criteria: Clinically significant or unstable hepatic, renal, gastroenterological, respiratory, cardiovascular, endocrine, immunological, haematological, or other systemic medical conditions; neurological disorders including epilepsy, stroke, or severe head trauma; clinically significant laboratory abnormalities on complete blood count (CBC) with differential, electrolytes, blood urea nitrogen (BUN), creatinine, hepatic transaminases, urinalysis, thyroid indices (T3, total T4, tree T4, thyroid‐stimulating hormone (TSH)) and electrocardiogram (ECG); mania caused by a general medical condition or substance‐induced mania; mental retardation (intelligence quotient (IQ) < 70); evidence of foetal alcohol syndrome or an alcohol‐related neurodevelopmental disorder; or schizophrenia or other psychotic disorders (including schizophreniform disorder, schizoaffective disorder, delusional disorder, brief psychotic disorder, shared psychotic disorder, psychotic disorder caused by a general medical condition, substance induced psychotic disorder, psychotic disorder not otherwise specified) as defined in the DSM‐IV |
|
| Interventions |
Location: USA, hospital sites in Columbus, Cincinnati, and Wichita Study duration: Not stated Treatment groups: 1. Valproate (VPA): initial dose of 10 mg/kg/day on a twice‐daily schedule beginning on day 0. VPA levels were adjusted to achieve a blood level of 80 – 100 µg/mL. The mean dose of VPA at endpoint was 300 mg/day. The mean level of VPA at study endpoint was 81 ± µg/mL 2. Risperidone: the mean dose of risperidone at endpoint was 0.5 mg/day (range 0.5 – 0.75 mg/day). Medications were administered in a double‐blind manner on a twice‐daily basis 3. Placebo: active medication and placebo were administered in liquid form matched for taste and colour Concomitant medications: the concurrent use of antipsychotics, antidepressants, and mood stabiliser/anticonvulsant medication other than the study drug was not allowed during study participation. The adjunctive use of oral chlorpromazine in low doses, 10 – 20 mg/day, 2 to 3 times a week, was allowed for sleep disturbance or agitation during the first 2 weeks of this trial Length of study: 6 weeks of intervention in addition to 3 ‐ 7 days screening period. There was a 1‐week washout period prior to study entry, except for those who took aripiprazole or fluoxetine who needed 4 weeks. Other psychotropic medication required 2 weeks Randomisation: 21 patients randomised to valproate 18 patients randomised to risperidone 7 patients randomised to placebo |
|
| Outcomes |
Outcomes: 1. Change in YMRS scores from baseline 2. Change in CDRS‐R scores from baseline 3. Clinical Global Impression ‐ Bipolar Illness ‐ Severity Scale (CGI‐BP‐S) 4. Clinical Global Impression ‐ Bipolar Illness ‐ Improvement Scale (CGI‐BP‐I) Safety/Tolerability: Adverse events, changes in body measures (e.g., BMI) Study withdrawals were reported |
|
| Funding | "The Stanley Medical Research Foundation funded study was designed to test the efficacy of risperidone versus VPA in children and adolescents with symptomatic bipolar I or II disorder during a mixed, manic, or hypomanic episode." | |
| Conflict of interest | "Dr. Kowatch is a consultant and faculty for the Resource for Advancing Children’s Health (REACH) Institute. He receives research support from the National Institute of Mental Health (NIMH). He is employed by Ohio State University and is an editor for Current Psychiatry. Drs. Scheffer, Monroe, Delgado, and Altaye, and Ms. Lagory disclosed no conflicts of interest." | |
| Notes | Study focused on children between 3 and 7 years of age | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Comment: Randomisation procedure not specified in text. We contacted the authors who informed that a random‐number generator was used to generate a random sequence. |
| Allocation concealment (selection bias) | Low risk | Comment: Procedures to conceal allocation not specified in text. We contacted the authors who informed us that the random sequences were stored in the pharmacy department and were only accessible to the pharmacy staff. Therefore, low risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Active medication and placebo were administered in liquid form matched for taste and colour. Medication were administered in a double‐blinded manner on a twice daily basis." Quote: "An independent, unblinded study psychiatrist adjusted the VPA dosis to achieve a therapeutic level." Quote: "An unblinded study coordinator [...] coordinated dose increases with the unblinded medical monitor at each site." Comment: Blinding procedures seem robust. |
| Blinding of outcome assessment (detection bias) Efficacy | High risk | Quote: "Subjects were assessed weekly for efficacy during the acute phase by one of the site principal investigators, who was blind to medication status and adverse events (AEs)." Quote: "An unblinded study coordinator performed the weekly side‐effect ratings using the Side Effects Form for Children and Adolescents." Comment: Efficacy measures properly blinded. However, adverse events are not. Therefore, overall high risk of bias. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: In all groups dropout rate was below 30%. |
| Selective reporting (reporting bias) | Low risk | Comment: Pre‐published protocol found. This protocol reports on all primary outcomes. |
| Other bias | Unclear risk | Comment: Chlorpromazine allowed as adjunctive medication but no comment on doses in either group is made and no comment on any group differences either. |
Mahmoudi‐Gharaei 2012.
| Methods | Randomised single‐blind trial | |
| Participants |
Number of total participants/type of patients: 30 participants randomised. Inpatient study Inclusion criteria: Sex: male and female Age‐group : children and adolescents (11 ‐ 18 years) Diagnosis: diagnosis of current bipolar disorder I (BD) according to the DSM‐IV‐TR in the manic or mixed phase (with or without psychotic features), eligible for adding adjunctive therapy, who were treated with lithium + risperidone at therapeutic doses for at least 4 weeks were selected. Other: in the context of the study centre, children and adolescents with BD are regularly initiated on two agents (lithium + risperidone) together. Therefore, those patients who received the above combination for at least 4 weeks and met the criteria for adjunctive therapy entered the study. Indications for receiving adjunctive therapy included experiencing a recurrent episode, or relapse evaluation by 1 child and adolescent psychiatrist. Exclusion criteria: (i) Comorbidity of mental retardation, pervasive developmental disorder, seizure disorder, anorexia nervosa according to clinical assessment; (ii) a severe mental illness requiring ECT or other treatment modalities during trial; (iii) any contraindication to the study drugs; (iv) current substance abuse or dependence within 3 months; (v) pregnancy; (vi) clinically significant medical illness; (vii) body weight under 30 kg; (viii) positive personal or family history of nephrolithiasis |
|
| Interventions |
Location: Iran, hospital site in Tehran Study duration: Not stated Treatment groups: 1. Lithium + risperidone + valproate: valproate was initiated at a dose of 10 mg/kg in divided doses with increase of 5 mg/kg every 3 days to a maximum daily dose of 20 – 30 mg/kg or as tolerated. Mean dose: 927.27 (SD 134.83) Maximum dose: 1200; lithium and risperidone were kept stable. 2.Lithium + risperidone + topiramate: topiramate was initiated at a dose of 25 mg/day; dosage was increased by 25 mg every 3 days to a maximum dose of 200 mg/day or increased as tolerated. Mean dose: 177.08 (SD 31.00) Maximum dose: 250; lithium and risperidone were kept stable Concomitant medications: Concomitant treatments with haloperidol injections (as needed for severe agitation) and biperiden (in the event of facing EPS symptoms) were allowed. Participants were not allowed to have any other concomitant treatments, including medications for their comorbid illnesses Length of study: 6 weeks Randomisation: 15 patients randomised to lithium, risperidone, and valproate 15 patients randomised to lithium, risperidone, and topiramate |
|
| Outcomes |
Outcomes: 1. Changes on YMRS scale 2. Changes on the CGI scale Study withdrawals were reported |
|
| Funding | "Funding sources supported by Teheran University of Medical Scienes" | |
| Conflict of interest | Not stated | |
| Notes | Study focused on children/teenagers between 11 – 18 years of age | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: No comment on randomisation procedure in text. We contacted the authors but received no reply. |
| Allocation concealment (selection bias) | Unclear risk | Comment: No comment on randomisation concealment in text. We contacted the authors but received no reply. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Single‐blind" Comment: No comment on the nature of the single blind (who exactly was blind). No comment on procedures to maintain blinding. We contacted the authors but received no reply. However, as a single‐blind study, either the participants or medical staff must be unblinded. |
| Blinding of outcome assessment (detection bias) Efficacy | Unclear risk | Quote: "Single‐blind" Comment: No comment on the nature of the single blind (who exactly was blind). No comment on procedures to maintain blinding. We contacted the authors but received no reply. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: In all groups dropout rate was below 30%. |
| Selective reporting (reporting bias) | Unclear risk | Comment: No pre‐published protocol found. We are therefore unable to assess the bias. |
| Other bias | High risk | Comment: Group receiving adjunctive topiramate had significantly different baseline characteristics (more hospitalisations). |
McElroy 1996.
| Methods | Randomised single‐blind trial | |
| Participants |
Number of total participants/type of patients: 36 participants randomised. Inpatient study Inclusion criteria: Sex: male and female Age: 18 ‐ 65 years Diagnosis: DSM‐III‐R diagnosed bipolar disorder, manic or mixed episode with psychotic features Exclusion criteria: Those unable to provide informed consent Prior treatment with valproate Unstable medical condition History of seizures or neurological disorders Psychoactive substance dependence |
|
| Interventions |
Location: USA, hospital site in Cincinnati Study duration: Not stated Treatment groups: 1. Valproate oral loading dose of 20 mg/day adjusted to achieve serum level of 50 micrograms/L 2. Haloperidol: 0.2 mg/kg/day in divided doses Concomitant medications: in participants receiving psychotropic medications prior to admission, these medications (other than lorazepam up to 4 mg/day) were discontinued upon admission to the unit Benztropine was administered as needed for treatment of extrapyramidal side effects Length of study: 6 days Randomisation: 21 patients randomised to valproate 15 patients randomised to haloperidol |
|
| Outcomes |
Primary outcome: Changes on the YMRS Secondary outcomes: Changes in the global scores of the Scale for Assessment of Positive Symptoms (SAPS) and SAPS subscale scores, total dose of adjunctive lorazepam received per participant and length of hospital stay Adverse events were reported Study withdrawals were reported |
|
| Funding | "Support in part by grants from Abbott Laboratories and the Theorode and Vada Stanley Foundation" | |
| Conflict of interest | Not stated | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: No comment on randomisation procedure in text. We contacted the authors but received no reply. |
| Allocation concealment (selection bias) | Unclear risk | Comment: No comment on randomisation concealment in text. We contacted the authors but received no reply. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Although the rater was blind to treatment, patients were not." |
| Blinding of outcome assessment (detection bias) Efficacy | High risk | Quote: "Although the rater was blind to treatment, patients were not." Quote: "The substantially higher rate of extrapyramidal side effects in the haloperidol treated patients may have compromised the raters blindness." Comment: 53.3% (n = 8) of the haloperidol participants had extrapyramidal side effects compared to none in the other group. This is likely to have compromised the rater blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: All participants completed the study. |
| Selective reporting (reporting bias) | Unclear risk | Comment: No pre‐published protocol found. We are therefore unable to assess the bias. |
| Other bias | High risk | Comment: To deal with extrapyramidal side effects, participants in the haloperidol group received benztropine. No participants in the valproate group received this medication. Differences between the 2 groups may potentially have been affected by this difference |
McElroy 2010.
| Methods | Randomized double‐blind trial | |
| Participants |
Number of total participants/type of patients: 62 participants randomised. Outpatient study Inclusion criteria: Sex: male and female Age: 18 years and over Diagnosis: met DSM‐IV‐TR criteria for bipolar I or II disorder or bipolar disorder not otherwise specified and currently experiencing a hypomanic, manic, or mixed episode (as determined by the Structured Clinical Interview for DSM‐IV‐TR33); have a YMRS score ≥ 10 and < 21 at the baseline assessment and at least 1 prior study screening visit at least 3 days, but no longer than 2 weeks, before baseline; had an overall CGI‐BP score ≥ 2 and < 5 Exclusion criteria: Considered severely psychiatrically ill or in need of psychiatric hospitalisation in the judgment of the clinical investigator Baseline YMRS score ≥ 21, CGI‐BP score ≥ 5, or Inventory of Depressive Symptoms (IDS) 35 score ≥ 39 Experiencing clinically significant suicidal ideation, homicidal ideation, or psychotic features Current DSM‐IV‐TR diagnosis of delirium, dementia, or other cognitive disorder or a lifetime DSM‐IV‐TR psychotic disorder DSM‐IV‐TR substance dependence disorder (except for nicotine dependence) within 3 months of study entry, a current DSM‐IV‐TR diagnosis of cocaine, stimulant, or hallucinogen abuse, or a urine drug screen positive for cocaine, stimulants, or hallucinogens Clinically significant finding on medical history, physical examination, electrocardiogram, or laboratory testing History of allergy or hypersensitivity to any valproate or valproate preparation Women were excluded if they were pregnant, lactating, or, if fertile, not practising a form of medically accepted contraception Must be receiving no psychotropics for the 1 week (4 weeks for fluoxetine or depot antipsychotics) before the baseline assessment, except as needed lorazepam (up to 2 mg/d) or zaleplon (up to 10 mg/d) |
|
| Interventions |
Location: USA, hospital site in Cincinnati Study duration: October 2003 ‐ November 2007 Treatment groups: 1. Valproate ER administered at an initial dose of 15 mg/kg/day, rounded up or down to the nearest 500 mg, and subsequently adjusted to a dose considered optimal based on the participant’s clinical response and side effects, but not to exceed 30 mg/kg/day 2. Placebo Concomitant medications: as needed use of lorazepam 0.5 – 2.0 mg/day was allowed for the management of affective symptoms for the first 2 weeks of the study; as needed lorazepam 0.5 – 1.0 mg/d was allowed for the next 2 weeks. No lorazepam was permitted for the final 4 weeks. As needed zaleplon (10 – 20 mg/day at bedtime) was allowed for management of insomnia throughout the study Length of study: 8 weeks Randomisation: 31 patients randomised to valproate 31 patients randomised to placebo |
|
| Outcomes |
Primary outcome: Changes in YMRS Other outcomes: Adverse events, withdrawal from study and associated reasons, IDS, CGI‐BP , HARS, and GAF scales also collected |
|
| Funding | "This investigator‐initiated study was funded in part by a grant from Abbott Laboratories. Abbott Laboratories also partially funded poster production costs and the first author’s travel expenses to present the poster." | |
| Conflict of interest | "Dr McElroy is a consultant to or member of the scientific advisory boards of AstraZeneca, Eli Lilly, Jazz, and Schering‐Plough; is a principal or co‐investigator on research studies sponsored by Abbott, AstraZeneca, Bristol‐Myers Squibb, Cephalon, Eli Lilly, Forest, GlaxoSmithKline, Jazz, National Institute of Mental Health (NIMH), Marriot Foundation, Orexigen, Shire, and Takeda; is inventor on United States Patent No. 6,323,236 B2: Use of Sulfamate Derivatives for Treating Impulse Control Disorders; and has received payments from Johnson & Johnson. Dr Jefferson has received grant/research support from The Program for Minority Research Training in Psychiatry and The American Psychiatric Institute for Research and Education (5T32 MH 19126‐18). Dr Keck is presently or has been in the past year a principal or co‐investigator on research studies sponsored by AstraZeneca, Cephalon, GlaxoSmithKline, Eli Lilly, Epi‐Q, Jazz, Marriot Foundation, NIMH, Orexigen, and Pfizer; has been reimbursed for consulting, in the past year, to GlaxoSmithKline, Bristol‐Myers Squibb, Pfizer, Quantia MD, Shering‐Plough, and Sepracor; and is inventor on United States Patent No. 6,387,956: Methods of Treating Obsessive‐Compulsive Spectrum Disorder Comprises the Step of Administering an Effective Amount of Tramadol to an Individual. Drs Welge and Guerdjikova and Messrs Martens and Creech have no personal affiliations or financial relationships with any commercial interest to disclose relative to the article." | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned to receive valproate ER or placebo in a 1:1 ratio according to computer‐generated coding. Randomization was balanced by use of permuted blocks." Comment: Computer‐generated coding is a low‐risk strategy |
| Allocation concealment (selection bias) | Low risk | Quote: "Allocation concealment was achieved by having the research pharmacy perform the randomisation, package the study medication, and maintain the integrity of the blinded information throughout the trial" Comment: Pharmacy ensured randomisation separate from investigators |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double blind" Quote: "Unblinded investigators were to be notified only of concentrations ≥150 μg/mL. To maintain the blind, similar notifications were to be given for a placebo patient who was at the same point in the study. No serum valproate levels, however, exceeded 150 μg/mL" Quote: "All study medication was in identical 500‐mg tablets supplied in numbered containers and dispensed to patients according to a predetermined randomisation schedule" Comment: Blinding procedures seem effective |
| Blinding of outcome assessment (detection bias) Efficacy | Unclear risk | Comment: No information on blinding of outcome assessors |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: 17 participants (57%) in the valproate ER group and 15 participants (50%) in the placebo group did not complete all 8 weeks of treatment. This was not a significant difference between group (Fisher exact test, P = 0.796). Thus even though both groups' dropout rates are above 30% they are at low risk of bias |
| Selective reporting (reporting bias) | Unclear risk | Comment: Pre‐published trial protocol, however the publication of primary outcome was only made on 27 December 2007. This is after the study finished recruiting all its participants. This makes it unclear whether the primary outcome was selected before or after some of the results were known, making this outcome at unclear risk. |
| Other bias | Low risk | Comment: None identified |
Moosavi 2014.
| Methods | Randomised single‐blind trial | |
| Participants |
Number of total participants/type of patients: 65 participants appear to have been randomised in this study. We contacted the authors to clarify this matter but did not receive a reply. Inpatient study Inclusion criteria: Sex: male and female Age: 20 ‐ 60 years (We suspect the authors made an error in the text where they state that "age older than 20 or less than 60 (...) were exclusion criteria") Diagnosis: diagnosis of bipolar I disorder (manic phase without psychotic features) based on DSM‐IV‐TR criteria were included Exclusion criteria: Substance dependency, comorbidity with other psychiatric disorders, general medical diseases (hepatic, kidney, respiratory, etc.), age older than 20 or less than 60 and pregnancy were exclusion criteria (but see above for a specification on age). |
|
| Interventions |
Location: Iran, hospital site in Sari City Date of study: 2012 ‐ 2013 Treatment groups: 1. Risperidone: participants received risperidone with a starting dose of 6 ‐ 8 mg a day in divided dose 2. Risperidone + valproate: participants received valproate 800 ‐ 1200 mg a day in divided dose plus risperidone with the same dose as the risperidone‐only group Concomitant medications: Clonazepam (2 ‐ 3 mg a day) and trihexyphenidyl (4 ‐ 6 mg a day) started in divided dose in both groups. All participants in both groups have received prophylactic anticholinergic drugs (trihexyphenidyl) 6 mg daily in divided dose) and benzodiazepine (clonazepam) 2 ‐ 3 mg daily Length of study: 7 weeks Randomisation: It is unclear how many people were randomised |
|
| Outcomes |
Outcomes: 1. Full remission: without DSM‐IV‐TR criteria 2. Partial remission: 1 or 2 criteria 3. No remission: 3 or more criteria or no change Adverse events Study withdrawals |
|
| Funding | Not stated | |
| Conflict of interest | The authors reported no conflict of interest | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: No comment on randomisation procedure in text. We contacted the authors but received no reply. |
| Allocation concealment (selection bias) | Unclear risk | Comment: No comment on randomisation concealment in text. We contacted the authors but received no reply. |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Quote: "Single‐blind" Comment: No comment on the nature of the single blind (who exactly was blind). No comment on procedures to maintain blinding. We contacted the authors but received no reply. However, as a single‐blind study, either the participants or medical staff must be unblinded. |
| Blinding of outcome assessment (detection bias) Efficacy | Unclear risk | Quote: "Single‐blind" Comment: No comment on the nature of the single blind (who exactly was blind). No comment on procedures to maintain blinding. We contacted the authors but received no reply. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: 17 out of 65 participants dropped out (26.2%) during the course of the study. There is no information on which groups these dropouts came from. Dropouts were not included in the final analysis. It is therefore impossible to comment on whether attrition bias was present. |
| Selective reporting (reporting bias) | Unclear risk | Comment: No pre‐published protocol found. We are therefore unable to assess the bias. |
| Other bias | Low risk | Comment: None identified |
Pope 1991.
| Methods | Randomised double‐blind trial | |
| Participants |
Number of total participants/type of patients: 43 participants randomised. Inpatient study Inclusion criteria: Sex: male and female Age 18 ‐ 65 years Diagnosis: bipolar disorder, manic phase, according to DSM‐III‐R criteria. Previously resistant or intolerant to lithium Other: beginning at the 4th month of the study, an entry criterion was added requiring women to be postmenopausal or surgically sterilised Exclusion criteria: If person had previously received more than one 250 mg dose of valproate Significant medical disorder History of neurological disease or focal neurological signs on examination Paroxysmal activity on EEG Comorbid psychoactive substance dependence Female patients were post‐menopausal or surgically sterilised |
|
| Interventions |
Location: USA, hospital site in Belmont, Massachusetts Study duration: Not stated Treatment groups: 1. Valproate: starting on day 0, participants received 3 tablets a day of study medication. Each tablet contained either 250 mg of valproate or a matching inert placebo 2. Placebo: Concomitant medications: No other psychotropic medication was allowed during the course of the study except lorazepam Duration of trial: 21 days Randomisation: 20 patients randomised to valproate 23 patients randomised to placebo |
|
| Outcomes |
Outcomes: 1. YMRS ‐ response defined as 50% reduction 2. GAS 3. BPRS Adverse events were reported Study withdrawals were reported |
|
| Funding | "This investigation was supported by Clinical Research Center grant MH‐362224 from the National Institute of Mental Health, Bethesda, Md, by a grant from the Philipp S. Weld, Jr Memorial Fund, McLean Hospital, by a grant from Abbott Laboratories, Chicago, III" | |
| Conflict of interest | Not stated | |
| Notes | 7 participants have dropped out during the first 7 days and have not been included in the analyses. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Tablets were supplied in numbered bottles containing drug or placebo as determined by a random number sequence; each patient recruited was assigned the next numbered bottle" Comment: Medications were assigned using a random‐number sequence, so low risk of bias. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Tablets were supplied in numbered bottles containing drug or placebo as determined by a random number sequence; each patient recruited was assigned the next numbered bottle." Comment: It appears that the numbered bottles are given to participants sequentially, so that the allocation group is potentially predictable. However, it is unclear if this was done centrally by the pharmacist or the study staff, so unclear risk of bias. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double blind" Quote: "No communication regarding the status of patients' under study was permitted between the unblinded investigator and the other investigators, save that the unblinded investigator was informed if patients complained of any side effects from the study medications" Comment: Valproate serum levels were released to 1 unblinded investigator who adjusted dosage and also made sham‐adjustments to placebos. |
| Blinding of outcome assessment (detection bias) Efficacy | Low risk | Quote: "The investigator who performed ratings (P.E.K.) remained blind throughout the study." Quote: "No communication regarding the status of patients' under study was permitted between the unblinded investigator and the other investigators, save that the unblinded investigator was informed if patients complained of any side effects from the study medications." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: 16/20 (80%) of the valproate group and 19/23 (82.6%) of the placebo group did not complete the entire study. Therefore, as the average dropout rate is above 75%, we consider this study to be at a high risk of bias. |
| Selective reporting (reporting bias) | Unclear risk | Comment: No pre‐published protocol found. We are therefore unable to assess bias. |
| Other bias | High risk | Quote: "The 17 patients randomised to valproate received a mean total dose of 5.8 ± 7.0 mg of lorazepam during the study, the 19 patients randomised to placebo received a mean total dose of 13.9 ± 10.3 mg ‐ a significantly greater amount." |
Shafti 2008.
| Methods | Randomised double‐blind trial | |
| Participants |
Number of total participants/type of patients: 30 female participants randomised. Inpatient study Inclusion criteria: Sex: only female patients Age: no information provided Diagnosis: DSM‐IV‐TR diagnosis of bipolar I disorder who had been admitted in the hospital because of recent appearance of a new episode of manic symptoms Exclusion criteria: None |
|
| Interventions |
Location: Iran, Tehran Date of study: Not stated Treatment groups: 1. Valproate: 200 mg uncoated tablets. The mean (SD) dosage of valproate prescribed in this trial was 1026 ± 148.64 mg 2. Lithium carbonate: 300 mg uncoated tablets. The mean (SD) dosage of lithium was 1240 ± 222.96 mg/day Mean serum level of lithium also was 0.873 ± 01486 mEq/L Concomitant medications: Although benzodiazepine (lorazepam) and typical antipsychotic (haloperidol) as adjunctive agents were permissible during trial, neither combining anticonvulsant nor atypical antipsychotic was prescribed during the aforesaid assessment. Mean (SD) dosage of haloperidol used in this trial was 5.87 ± 1.09 mg in the lithium group and 6.02 ± 0.84 mg in the valproate group. Mean (SD) dosage of adjunctive lorazepam also was 3.38 ± 1.06 in the first group and 3.46 ± 0.94 in the second one. There were no significant differences in this regard. Length of study: 21 days Randomisation: 15 patients randomised to valproate 15 patients randomised to lithium |
|
| Outcomes |
Primary outcomes: Changes in the Manic State Rating Scale (MSRS); measuring frequency and intensity; Changes in the CGI‐S scale Study withdrawals were reported |
|
| Funding | "The authors declare no funding or conflicts of interest." | |
| Conflict of interest | "The authors declare no funding or conflicts of interest." | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: No comment on randomisation procedure in text. We contacted the authors but received no reply. |
| Allocation concealment (selection bias) | Unclear risk | Comment: No comment on allocation concealment in text. We contacted the authors but received no reply. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double‐blind" Quote: "The tablets were prescribed while previously inserted into empty and similar capsules, which were prepared in this regard to make patients blind with respect to the procedure.The evaluators were also unaware concerning the aforesaid partition and the type of medications arranged for each group." Comment: The procedures to maintain the blind seem reasonable. |
| Blinding of outcome assessment (detection bias) Efficacy | Low risk | Quote: "The evaluators were also unaware concerning the aforesaid partition and the type of medications arranged for each group." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "There was no premature discontinuation in neither of groups" |
| Selective reporting (reporting bias) | Unclear risk | Comment: No pre‐published protocol found. We are therefore unable to assess bias. |
| Other bias | Low risk | Comment: None identified |
Tohen 2002.
| Methods | Randomised double‐blind trial | |
| Participants |
Number of total participants/type of patients: 251 participants randomised. Inpatient study Inclusion criteria: Sex: male and female Age: 18 ‐ 75 years Diagnosis: diagnosis of bipolar I disorder, manic or mixed episode, with or without psychotic features. Clinical diagnoses were confirmed with the Structured Clinical Interview for DSM‐IV. A score of ≥ 20 on the YMRS was required at both the screening visit and on the day of random assignment to study groups (baseline) Exclusion criteria: Serious and unstable medical illness, DSM‐IV substance dependence within the past 30 days (except nicotine or caffeine), documented history of intolerance to olanzapine or valproate, and treatment with lithium, an anticonvulsant, or an antipsychotic medication within 24 hours of random assignment to study groups |
|
| Interventions |
Location: USA, 48 sites (cities not stated) Study duration: Not stated Treatment groups: 1. Olanzapine: 5 – 20 mg/day. The initial daily dose was 15 mg/day of olanzapine consistent with the recommendations of the manufacturers. Investigators made dose adjustments primarily on the basis of clinical response but also on plasma levels and adverse events. Participants who did not tolerate the minimum dose level for treatment (5 mg/day olanzapine or 500 mg/day valproate) were discontinued from participation in the study. Mean modal doses for olanzapine were 17.4 mg/day 2. Valproate: 500 – 2500 mg/day. The initial daily doses was 750 mg/day of valproate, consistent with the recommendations of the manufacturers. Investigators made dose adjustments primarily on the basis of clinical response but also on plasma levels (valproate levels of 50 ‐ 125 ug/ml were aimed for) and adverse events. Participants who did not tolerate the minimum dose level for treatment (5 mg/day olanzapine or 500 mg/day valproate) were discontinued from participation in the study. Mean modal dose of 1401.2 mg/day. Concomitant medication: Concomitant lorazepam use was restricted to a maximum dose of 2 mg/day, and administration was not allowed within 8 hours of the administration of a symptom rating scale. Benztropine was permitted to treat extrapyramidal symptoms up to a maximum of 2 mg/day throughout the course of the study. Benztropine was not allowed as prophylaxis for extrapyramidal symptoms. Length of study: 21 days Randomisation: 126 randomised to valproate 125 randomised to olanzapine |
|
| Outcomes |
Primary outcome: Changes on YMRS from baseline to endpoint. In addition, clinical response was defined as ≥ 50% improvement in the YMRS score at endpoint Secondary outcome: HDRS Safety was assessed using adverse events measures (using Abnormal Involuntary Movement Scale, Barnes Akaesthesia Rating Scale, Simpson Angus Rating Scale) and by monitoring other laboratory test values (e.g. ECG results, weight changes) Study withdrawals were reported |
|
| Funding | "Sponsored by Lilly Research Laboratories" | |
| Conflict of interest | Not stated | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomly assigned" Comment: We contacted the lead author, who confirmed that random numbers were computer‐generated. |
| Allocation concealment (selection bias) | Unclear risk | Comment: Not mentioned in text. We contacted the lead author who confirmed that the random allocation was determined centrally (not on site). However, it is still unclear in what way the randomisation sequence was concealed. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double‐blind" Quote: "To maintain the blind all patients randomly assigned to receive olanzapine had blood drawn and sham 'divalproex' plasma level results were reported. [...] All investigators at the clinical sites and at Lily Research Laboratories remained blind to subjects' treatment assignment" Quote: "Blood was [...] shipped to an independent reference laboratory" |
| Blinding of outcome assessment (detection bias) Efficacy | Low risk | Comment: There is no mention in text of who conducted outcome assessments and whether the assessors were blind. However, we contacted the lead author who confirmed that the rater was blind. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Dropout rates in valproate group were 35.7% and in the olanzapine group 31.2%. All groups were over 30% dropout rates, but dropout rates were comparable (less than two‐fold difference) and overall dropout rate below 75%. |
| Selective reporting (reporting bias) | Unclear risk | Comment: No pre‐published protocol found. We are therefore unable to assess the bias. |
| Other bias | Low risk | Comment: None identified |
Tohen 2008.
| Methods | Randomised double‐blind trial | |
| Participants |
Number of total participants/type of patients: 521 participants randomised. Both inpatients and outpatients Inclusion criteria Sex: male and female Age: 18 ‐ 65 years Diagnosis: a diagnosis of DSM‐IV‐TR acute bipolar manic or mixed episode without psychotic features. In addition, participants were required to score ≥ 20 on the YMRS and ≤ 30 (mild to moderate) and a Clinical Global Impression for Bipolar Disorder‐Severity of Illness scale (CGI‐BP‐S) mania subscore of 3 or 4 at screening (week 1) and at randomisation (week 0). Women had to test negative for pregnancy and be using effective contraception. Exclusion criteria Rapid cycling course or Psychotic features in DSM‐IV‐TR |
|
| Interventions |
Location: USA, Lithuania, Pueto Rico, Romania, and Russia Date of study: October 2004 ‐ December 2006 Treatment groups: 1. Olanzapine (5 ‐ 20 mg) administered orally once in the evening. Mean dose 11.4 g (SD 2.49 g) 2. Valproate (500 ‐ 2500 mg) administered orally 3 times a day if ≥ 750 mg, twice a day if not Mean dose 848.4 mg (SD 135.62 mg)/Plasma levels 61.3 mg/L (SD 32.04 mg/L) 3. Placebo Placebo capsules used to balance all medications out to 3 doses a day. Concomitant medication: Lorazepam ≤ 2 mg a day was allowed as long as > 8 hours before psychiatric assessment. Anticholinergics and ongoing thyroids supplementation therapy were permitted Length of study: 21 days Randomisation: 215 randomised to olanzapine 201 randomised to valproate 105 randomised to placebo |
|
| Outcomes |
Primary outcome: Change in scores on the YMRS from baseline to endpoint Secondary outcome: CGI‐BP, MADRS Safety was assessed using adverse events measures (using Abnormal Involuntary Movement Scale, Barnes Akaesthesia Rating Scale, Simpson Angus Rating Scale) and by monitoring other laboratory test values (e.g. ECG results, weight changes) Study withdrawals were reported |
|
| Funding | "This study was supported by Eli Lilly and Co., Indianapolis, Ind." | |
| Conflict of interest | Not stated | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A computer generated random sequence randomly assigned patients to treatment group within each study site." |
| Allocation concealment (selection bias) | Unclear risk | Quote: "A computer generated random sequence randomly assigned patients to treatment group within each study site." Comment: We contacted the lead author who confirmed that the random allocation was determined centrally (not on site). However, the method of concealment is still unclear and there is a chance that involved participants and staff would be able to anticipate allocation. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double‐blind" Quote: "To keep investigators blind to treatment‐assignment, all study drugs were dispensed by an interactive voice response/web/fax tool [...] dose adjustments were conducted via the interactive voice response/web/fax tool. To maintain blinding, every time the interactive voice response/web/fax tool send a message to alter the dose of a valproate treated patient, a dummy message was send to alter the dose of an olanzapine or placebo treated patient." Quote: "To maintain blinding, all patients had blood collected for assessing valproate concentration, irrespective of whether they received valproate." Quote: "All study medication appeared identical." |
| Blinding of outcome assessment (detection bias) Efficacy | Low risk | Quote: "To keep investigators blind to treatment‐assignment, all study drugs were dispensed by an interactive voice response/web/fax tool [...] dose adjustments were conducted via the interactive voice response/web/fax tool." Comment: Although blinding procedures appear robust, there is no explicit mention of who conducted outcome assessments and if they were blind. We contacted the author who confirmed that the rating was conducted by site raters who were blind. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: In all groups dropout rate was below 30%. |
| Selective reporting (reporting bias) | Low risk | Comment: Protocol found. Primary outcome as stated on protocol. |
| Other bias | Low risk | Comment: None identified |
Vasudev 2000.
| Methods | Randomised single‐blind trial | |
| Participants |
Number of total participants/type of patients: 30 participants attending outpatient clinic (hospitalised for study) Inclusion criteria: Sex: male and female Age: 18 ‐ 65 years Diagnosis: DSM‐III‐R bipolar disorder acute manic episode and a score of ≥ 20 on the YMRS Other: participants had to be medication‐free for 6 months. Exclusion criteria: Seizure disorder, cerebrovascular disorder, neurological disorder, general medical disorder Pregnant women or those on the contraceptive pill. Medication for mania prior to inclusion Drug or alcohol dependence. Requirement for ECT or antipsychotic medication |
|
| Interventions |
Location: India, New Delhi Study duration: Not stated Treatment groups: 1. Carbamazepine 200 mg twice daily increased by 200 mg to 800 mg daily aiming for 20 mg/kg/day in 3 divided doses by day 3 (range 800 ‐ 1200 mg day) 2. Valproate initiated 20 mg/kg/day (range 800 ‐ 1400 mg daily) Further increase in dose, 200 ‐ 400 mg/day carried out weekly until clinical improvement occurred or serum level not exceeding upper therapeutic range (14 mcg/ml CBZ and 125 mcg/ml valproate) Maximum doses: carbamazepine 800 ‐ 1600 mg/day; valproate 1000 ‐ 2200 mg/day by the end of week 4 Concomitant medication: No other psychotropic medication was allowed during the study. Diazepam and promethazine were allowed in initial phase of study to treat agitation or insomnia Length of study: 28 days Randomisation: 15 patients randomised to carbamazepine 15 patients randomised to valproate |
|
| Outcomes |
Primary outcome: changes in scores on the YMRS from baseline to endpoint. In addition, clinical response was defined as ≥ 50% improvement in the YMRS score at endpoint Safety was assessed by recording adverse events Study withdrawal was recorded |
|
| Funding | Not stated | |
| Conflict of interest | Not stated | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "The consecutive patients attending the out‐patient clinic and qualifying for the study according to the inclusion and exclusion criteria, were randomised to the two treatment groups (n=15 for each) according to the table of random numbers." |
| Allocation concealment (selection bias) | High risk | Quote: "The consecutive patients attending the out‐patient clinic and qualifying for the study according to the inclusion and exclusion criteria, were randomised to the two treatment groups (n=15 for each) according to the table of random numbers." Comment: Participants were assigned consecutively according to an open table of random numbers, so that the allocation group is potentially predictable. |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "The dosage in any given patient was guided by the feedback received from the laboratory; the person who estimated the serum levels in the laboratory was blind to the patient identity, blood samples having been sent in coded test tubes. [...] Another psychiatrist from the regular clinical division altered the dosage regimen on the basis of his clinical assessment and serum levels reported from the laboratory. While the patients knew they were receiving an active treatment, they did not know which one it was." Comment: No mention of double‐blind procedure. Although participants are blind it is never established in the text if investigators are blind. We are therefore unable to assess risk of bias. |
| Blinding of outcome assessment (detection bias) Efficacy | Low risk | Quote: "The two raters who assessed the mania status weekly, were blind to the medication status or serum levels attained by a particular patient." Comment: Raters appear to be blind. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: In all groups dropout rate was below 30%. |
| Selective reporting (reporting bias) | Unclear risk | Comment: No pre‐published protocol found. We are therefore unable to assess the risk of bias. |
| Other bias | High risk | Comment: On average, participants in the carbamazepine group received more rescue medication than those in the valproate group. |
Wagner 2009.
| Methods | Randomised double‐blind trial | |
| Participants |
Number of total participants/type of patients: 151 participants randomised. Outpatient study Inclusion criteria: Sex: male and female Age: 10 ‐17 years Diagnosis: DSM‐IV‐TR diagnosis of bipolar I disorder, manic or mixed episode, with or without psychotic features. Participants were required to have a YMRS score of ≥ 20 at the time of screening and at randomisation Exclusion criteria: Current manic episode that was drug‐induced or secondary to a medical disorder; a current diagnosis of a DSM‐IV‐TR axis I disorder other than attention‐deficit/hyperactivity disorder (ADHD), obsessive‐compulsive disorder, oppositional defiant disorder, conduct disorder, panic disorder, enuresis, encopresis, parasomnias, agoraphobia, specific phobia, social phobia or separation anxiety disorder; or a current axis II disorder that would interfere with compliance or confound study results interpretation. Patients with a history of substance abuse within the month before screening, substance dependence within 3 months before screening, or evidence of drug or alcohol withdrawal/intoxication at the time of randomisation were excluded. Mental retardation or cognitive deficits severe enough to confound study interpretation or interfere with compliance were exclusion criteria. Patients who had current serious violent, homicidal, or suicidal ideation were excluded. Female patients who were pregnant or lactating were excluded. Other exclusion criteria included patients expected to require hospitalisation for their manic or mixed episode and patients with clinically significant abnormal laboratory data, unstable medical conditions, or an underlying condition that would confound the interpretation of the study results. |
|
| Interventions |
Location: USA, 24 sites (cities not stated) Study duration: Not stated Treatment groups: 1. Valproate extended‐release: Study drug was initiated at 15 mg/kg a day (not to exceed 750 mg) and titrated in 250‐mg increments every 1 to 3 days to clinical response and/or a serum valproate concentration within the target range of 80 to 125 2 g/mL, as deemed appropriate by the investigator, to a maximum dosage of 35 mg/kg a day 2. Placebo: matching placebo tablets. Concomitant medications: Concurrent use of antipsychotic, antidepressant, and mood stabiliser/anticonvulsant medication other than the study drug was not allowed during the study participation. Patients who were taking a protocol‐prohibited psychotropic medication within 5 elimination half‐lives before randomisation were excluded. The adjunctive use of zolpidem tartrate (up to 10 mg a day for insomnia) and lorazepam (up to 4 mg for severe agitation) was permitted, up to 3 times a week during the washout period and the first 14 days of double‐blind period, except during the 8 hours before efficacy ratings. There were no restrictions on zolpidem tartrate or lorazepam during the long‐term study. Treatment of ADHD with stimulant medications (with the exception of pemoline) was also allowed during both the double‐blind and long‐term studies for participants whose dosage had been stable for 3 months before day 1, and the investigator planned to maintain this stable dose throughout the study, and this medication was not exacerbating mood symptoms. Use of atomoxetine was not allowed. Length of study: 28 days Randomisation: 77 randomised to valproate 74 randomised to placebo |
|
| Outcomes |
Primary outcome: Change from baseline to final evaluation on the YMRS. The definition of response was 50% or greater improvement on the YMRS total score from baseline. Remission was defined as a YMRS score of < 12 at final evaluation Secondary outcome: Clinical GAS, CGI‐I, Clinical Global Assessment Scale ‐ Severity (CGI‐S), CDRS‐R, ADHD Rating Scale IV, and TheCaregiverStrain Questionnaire (CGSQ) Safety was assessed using adverse events and by monitoring other laboratory test values (e.g. weight changes) Study withdrawals were reported |
|
| Funding | "This study was supported by a grant from Abbott Laboratories" | |
| Conflict of interest | "Dr. Wagner is with the University of Texas Medical Branch; Dr. Redden is with the Abbott Laboratories; Dr. Kowatch is with the University of Cincinnati Medical Center; Dr. Wilens is with the Massachusetts General Hospital; Dr. Segal is with the Segal Institute for Clinical Research; Dr. Chang is with the Stanford University School of Medicine; Drs. Vigna, Abi‐Saab, and Saltarelli are with Abbott Laboratories and the Divalproex ER Pediatric Mania Group; and Dr. Wozniak is with Advanced Clinical Research Services." | |
| Notes | Study focused on children/teenagers between 10 ‐ 17 years of age | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Patients were randomised in a 1:1 ratio to receive divalproex ER or matching placebo tablets" Comment: No other information about method of randomisation, so we are unable to assess risk of bias. We contacted the authors but did not receive a reply. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Patients were randomised in a 1:1 ratio to receive divalproex ER or matching placebo tablets" Comment: No other information about method of allocation concealment, so we are unable to assess risk of bias. We contacted the authors but did not receive a reply. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Double‐blind" Comment: A system of centralised unblinded serum valproate analysis was used. Procedures to ensure blinding was maintained included the centralised location and matched sham‐calls to placebo‐receiving participants. We contacted the authors but did not receive a reply. |
| Blinding of outcome assessment (detection bias) Efficacy | Unclear risk | Quote: "Double‐blind" Comment: It is unclear if the outcomes assessors were blind. We contacted the authors but did not receive a reply. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: In all groups dropout rate was below 30%. |
| Selective reporting (reporting bias) | Low risk | Comment: The primary outcome in this study (YMRS) is the same as on the pre‐published protocol. |
| Other bias | Unclear risk | Quote: "The adjunctive use of zolpidem tartrate (up to 10 mg per day for insomnia) and lorazepam (up to 4 mg for severe agitation) was permitted, up to 3 times per week during the washout period and the first 14 days of double‐blind period, except during the 8 hours before efficacy ratings." Comment: There is no information about whether use of adjunctive medication was balanced between the treatment groups. |
Xu 2015.
| Methods | Randomised double‐blind trial | |
| Participants |
Number of total participants/type of patients: 120 participants randomised. Inpatient study Inclusion criteria: Sex: male and female Age: 19 ‐ 60 years Diagnosis: all participants were diagnosed with bipolar I by qualified psychiatrists according to the 4th edition of the DSM‐IV. Only patients with a YMRS score of ≥ 17 were recruited for this study. Participants also needed to experience their first acute manic or mixed episode when included in this study. Exclusion criteria: Female patients with pregnancy or lactation Severe and unstable diseases, including cardiovascular, respiratory, liver, kidney, gastrointestinal, neurological, endocrine, immune, blood‐system conditions, narrow‐angle glaucoma, and seizures Substance dependence (except tobacco) according to DSM‐IV standards History of untolerated use of olanzapine or valproate and History of use of any antipsychotics or mood stabilisers |
|
| Interventions |
Location: China, Hangzhou Study duration: Not stated Treatment groups: 1. Valproate: 0.6 g/day (2 to 3 times a day orally). The dose of valproate was gradually increased to 1.2 ‐ 1.8 g/day based on the participant's reaction. At the end of treatment, the average dose of valproate was 1.53 (SD 0.22) g/day 2. Olanzapine: 10 mg/day (once a day orally). The dose of olanzapine was adjusted by 5 ‐ 20 mg/day based on the participant's condition. At the end of treatment, the average dose of olanzapine was 16.3 (SD 2.1) g/day 3. Combined valproate and olanzapine: dosages were the same as for the monotherapy. At the end of treatment, the average dose of valproate was 1.08 (SD 0.45) g/day and of olanzapine was 13.1 (SD 3.2) g/day Concomitant medications: Aside from trial medications, no other drugs were permitted during the study Length of study: 28 days Randomisation: 40 patients randomised to valproate 40 patients randomised to olanzapine 40 patients randomised to combined valproate and olanzapine |
|
| Outcomes |
Primary outcome: Change from baseline to final evaluation on the YMRS Secondary outcome: Changes on the CGI‐BP scale Adverse events were reported Study withdrawals were reported |
|
| Funding | "This work was supported by grants from the Health Department Foundation of Zhejiang Province (2011KYA073), the Natural Science Foundation of Zhejiang Province (Y2100294), and the Science and Technology Department Foundation of Zhejiang Province (2010C33038)." | |
| Conflict of interest | "The authors report no conflict of interest in this work" | |
| Notes | Table 1 seems to have mislabelled the study groups. Specifically, throughout the text group A received valproate, group B received olanzapine, and group C was the combined group. This group allocation is reversed for Group A and Group C in Table 1 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "For randomisation, a random number table with sequentially numbered, opaque and sealed envelopes was used to conceal the allocation sequences." |
| Allocation concealment (selection bias) | Low risk | Quote: "For randomisation, a random number table with sequentially numbered, opaque and sealed envelopes was used to conceal the allocation sequences." |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Quote: "Double‐blind, randomised controlled study." Quote: "The physician kept the randomisation code and no rater became aware of treatment allocations before requesting unmasking at the end of the study." Quote: "Trial medications were administered by nurses." Comment: It is unclear whether the medical personnel were blinded. |
| Blinding of outcome assessment (detection bias) Efficacy | Low risk | Quote: "Patient assessments were conducted by a professional psychiatrist who was blind to the experimental condition." Quote: "No rater became aware of treatment allocations before requesting unmasking at the end of the study." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Over 90% completion rates in all group. No significant group differences |
| Selective reporting (reporting bias) | Unclear risk | Comment: Protocol found but published on 17 August 2013 and trial completed on 31 May 2011; hence, the protocol had been published retrospectively. We are therefore unable to assess risk of bias. |
| Other bias | Low risk | Comment: Although we did not find any other biases, we noticed inconsistencies regarding the labelling of study groups. Specifically, table 1 does not reflect the group allocation outlined in the remainder of the text where group A (n = 37) = Valproate monotherapy, group B (n = 39) = Olanzapine monotherapy, group C (n = 38) = combined valproate and olanzapine therapy. We contacted the authors but did not receive a reply. Nontheless, we did not consider this simple error to justify high risk of bias. |
Young 2017.
| Methods | Randomised double‐blind trial | |
| Participants |
Number of total participants/type of patients: 224 participants randomised. Inpatient study Inclusion criteria: Sex: male and female Age: ≥ 60 years Diagnosis: patients who meet DSM‐V criteria for bipolar disorder type I with current manic, mixed or hypomanic episode. Only patients with a YMRS score of ≥ 18 were recruited for this study. Exclusion criteria: Diagnosis of schizophrenia, schizoaffective disorder, or delusional disorder Contra‐indication to lithium or valproate History of intolerance to lithium, valproate, lorazepam, or risperidone Failure of current episode to respond to at least 4 weeks of treatment with lithium or valproate Active substance dependence or other substance‐related safety issues Mood disorder due to a general medical condition (e.g. recent stroke, hyperthyroidism, porphyria, HIV infection, connective tissue disease) or a substance Rapid cycling mania Diagnosis of delirium or dementia or other brain degenerative diseases Inability to communicate in English Sensory impairment preventing participation in research assessments Unstable medical condition High risk for suicide Requirement for other immediate pharmacological intervention |
|
| Interventions |
Location: USA, (no cities stated) Study duration: Not stated Treatment groups: 1. Valproate: The starting dose was 500 mg/day. Afterwards, the dose was titrated to achieve a target range of 80 9 9 mcg/ml (the acceptable range being 40 – 99 mcg/ml) 2. Lithium: The starting dose was 300 mg/day. Afterwards, the dose was titrated to achieve a target range of 80 – 99 mEq (the acceptable range being 40 – 99 mEq). Concomitant medications: Other psychotropics were not allowed but medications for comorbid physical conditions were continued. When non‐steroidal anti‐inflammatory agents or thiazide diuretics were required, mood stabiliser dosages were adjusted based on serum concentrations. Participants with inadequate response after 3 weeks received open adjunctive risperidone. Behavioural interventions or lorazepam or both may be added, if necessary Length of study: 63 days Randomisation: 112 patients randomised to valproate 112 patients randomised to lithium |
|
| Outcomes |
Primary outcomes: Primary clinical tolerability outcome: The Sedation Item score of the UKU (Norwegian for Committee of Clinical Investigations) Side Effect Rating Scale Primary pharmacologic tolerability: Proportion of participants reaching concentration of target range Primary outcome measure: Change from baseline to final evaluation on the YMRS Secondary outcomes: tremor, weight gain, nausea and vomiting, and change in MADRS depression scores from baseline Rates of remission are reported (YMRS score ≤ 9) Study withdrawals were reported |
|
| Funding | "Supported by [grant numbers] from the US Public Health Service." | |
| Conflict of interest | "Dr. Young has received support from the National Institutes of Health (NIH). Dr. Mulsant has received research support from Brain Canada, the CAMH Foundation, the Canadian Institutes of Health Research, the NIH, Bristol‐Myers Squibb (medications for an NIH‐funded clinical trial), and Pfizer (medications for an NIH‐funded clinical trial). Within the past five years, he has also received some travel support from Roche. Dr. Sajatovic has received research grants from Pfizer, Merck, Ortho‐McNeil Janssen, Reuter Foundation, Woodruff Foundation, Reinberger Foundation, NIH, and the Centers for Disease Control. She is a consultant to United BioSource Corporation (Bracket), Prophase, Otsuka, Pfizer, and Amgen and has received royalties from Springer Press, Johns Hopkins University Press, Oxford Press and Lexicomp. Dr. Gildengers has participated in scientific advisory board meetings for Shire Pharmaceuticals. Drs. Gyulai, Al Jurdi and Chen have no disclosures. Dr. Beyer has research support from Eli Lilly, Elan, Forest, Novartis, Sanofi: Astra‐Zeneca, and Takeda. Drs. Marino and Bruce and Ms. Greenberg have had support from NIH. Drs. Kunik, Banerjee and Schulberg and Ms. Barrett have no disclosures. Dr. Reynolds receives research support from the NIH, the Commonwealth of Pennsylvania, the American Foundation for Suicide Prevention. He receives pharmaceutical supplies for his NIH‐sponsored research from Pfizer, Forest Laboratories, and Lilly. Dr. Alexopoulos receives support from the NIH; he is on the speakers’ bureaus of Astra Zeneca, Forest, Novartis, and Sunovion. Janssen Scientific Affairs, LLC provided risperidone to some sites." | |
| Notes | Study focused on older adults aged 60 or more | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Permuted block randomisation employed block sizes ranging randomly from 4 to 8 consecutive patients by site." |
| Allocation concealment (selection bias) | Low risk | Quote: "Eligible patients were randomised under double‐blind conditions on a 1:1 basis to lithium or divalproex (...) Participants received monotherapy with lithium or divalproex in over‐encapsulated pills given twice daily." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Eligible patients were randomised under double‐blind conditions", Quote: "Concentrations were reported to a non‐blind clinician who created an equivalent “dummy concentration” for the other drug (e.g., 0.58 mEq/L and 58 mcg/ml); both concentrations were then provided to the blinded psychiatrist" |
| Blinding of outcome assessment (detection bias) Efficacy | Unclear risk | Comment: The treating psychiatrist was blinded. However it is unclear who performed the outcome assessments. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Dropout in both groups was higher than 30% at the end of the study. Specifically, 57/112 (51%) of the lithium group and 49/112 (44%) of the valproate group did not complete the entire study. However, dropout rates are comparable (less than a two‐ fold difference) and the average dropout rate is below 75%. At week 3 (before risperidone was introduced into the study), however, the rates of dropout between lithium (14%) and valproate (18%) were under 30%. We therefore rate the risk of biases as low. |
| Selective reporting (reporting bias) | Low risk | Comment: The primary outcomes are the same as in the pre‐published protocol. We therefore rate the risk of biases as low. |
| Other bias | Unclear risk | Quote: "In participants who did not respond to behavioral intervention and lorazepam, oral risperidone rescue 0.5–1 mg was used up to twice a day and for up to three days in any week. Participants who required more than these doses terminated the study. After three weeks of treatment, risperidone up to 4mg/day was used for an inadequate response to lithium or divalproex, defined as YMRS ≥ 16, and lorazepam was tapered off. Patients not receiving adjunct risperidone could receive lorazepam 0.5–1.0 mg/day for persistent anxiety or insomnia." Comment: The odds of needing rescue lorazepam or risperidone did not differ statistically between groups (lithium: 60.7% vs valproate: 50.9%; OR 1.49, CI: 0.88 to 2.5, P = 0.14). Similarly, the use of adjunctive risperidone (i.e. consistent use up to 4 mg/day after week 3) did not differ significantly (lithium: 17.0% vs valproate: 14.3%; OR 1.23, CI 0.59 to 2.5, P = 0.58). However, the 2 groups differed in the use of daily lorazepam after day 28 (lithium: 9.8% vs valproate: 19.6%; Chi2(1) = 4.3; P = 0.038). Due to the use of adjunctive risperidone after week 3 we used only results up to week 3 in our analysis. |
Zajecka 2002.
| Methods | Randomised double‐blind trial | |
| Participants |
Number of total participants/type of patients: 120 participants randomised. After an inpatient period of up to 21 days, participants were followed as outpatients. Inclusion criteria: Sex: male and female Age: 18 ‐ 65 years Diagnosis: participants had to have a DSM‐IV primary diagnosis of bipolar disorder type I, and be hospitalised for an acute manic episode (defined as a score ≥ 25 on the SADS‐C MRS, with at least 4 scale items rated ≥ 3) Exclusion criteria: Diagnosis of axis I or II disorder that would interfere with the evaluation of the compounds being studied Drug or alcohol withdrawal symptoms Platelet count of < 100.000 mm3 Women who were pregnant or planning to become pregnant Mood disorder secondary to a medical condition. Patients who had previously failed trials of either valproate or olanzapine (in the opinion of the investigator) were also excluded. |
|
| Interventions |
Location: USA, 21 study centres (no cities stated) Study duration: Not stated Treatment groups: 1. Olanzapine: starting dose 10 mg/day. Dose increased by 500 mg valproate (starting 20 mg/kg/day in 3 divided doses) on days 3 and 6 if clinical symptoms of mania persisted. Mean maximum dose 14.7 mg day (range 2 ‐ 25 mg/day) 2. Valproate: starting dose 20 mg/kg/day. Dose increased by 5 mg on days 3 and 6 if clinical symptoms of mania persisted. Mean maximum dose 2115 mg day (range 750 ‐ 3250 mg/day) Concomitant medications: Investigators could prescribe rescue medications, including lorazepam, benztropine mesylate, chloral hydrate, and zolpidem, as adjunctive therapy. Lorazepam was allowed in single doses up to 3 mg/dose, but not exceeding 4 mg/day from days 1 through 7, 3 mg/dose from days 8 through 14 and 2 mg/day from day 15 to the end of the study. Benzotropine mesylate was permitted in single doses up to 2 mg/dose but not exceeding 4 mg/day. Chloral hydrate was allowed in single doses up to 1 g/dose, but not exceeding 3 g/day. Zolpidem was permitted in doses up to 10 mg/day. Chloral hydrate and Zolzidem were not to be administered concurrently. Adjunctive therapy was not to be administered within 8 hours prior to efficacy ratings. Duration of trial: 12 weeks Randomisation: 63 patients randomised to valproate 57 patients randomised to olanzapine |
|
| Outcomes |
Primary outcome: change in MRS Secondary outcomes: MSS, Behaviour and Ideation Scale (BIS), BPRS , HDRS, CGI‐I and Quality of life enjoyment and satisfaction questionnaire (Q‐LES‐Q) Adverse events were reported Study withdrawals were reported |
|
| Funding | "This research was supported by Abbott Laboratories, Abbott Park, III" | |
| Conflict of interest | "Dr. Zajecka has received grant/research support from Bristol‐Myers Squibb, Lilly, Cephalon, Cyberonics, GlaxoWellcome, Lichtwer Pharma, MIICRO, Otuka, Parke‐Davis, Pfizer, and Wyeth‐Ayerst; has been a consultant/advisory board member of Abbott, Bristol‐Myers Squibb, and Lilly; and has been on the speakers bureau for Abbott, Bristol‐Myers Squibb, Lilly, Pfizer/Roerig, SmithKline Beecham, Pharmacia & Upjohn, and Wyeth‐Ayerst. Dr. Weisler has received grant/research support from Lilly, Glaxo, Pfizer, Janssen, AstraZeneca, Bristol‐Myers Squibb, and Merck. Dr. Sachs has received grant/research support from Abbott, GlaxoSmithKline, Lilly, and Janssen and has been on the advisory board or speakers bureau for Abbott,GlaxoSmithKline, Lilly, Janssen, Solvay, Sanofi, AstraZeneca, Novaris, Elan, and Bristol‐Myers Squibb. Dr. Swann has received grant/research support from Abbott, Glaxo, Robert Wood Johnson; and has received honoraria from and been on a speakers bureau/advisory board for Abbott, GLaxo, and Janssen. Drs. Woznia and Sommerville are employees of and major stock holders in Abbott." | |
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Randomized, double‐blind, parallel‐group study" Comment: No information in text on method of randomisation. |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Randomized, double‐blind, parallel‐group study" Comment: No information in text on any procedures that ensured allocation concealment. |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Randomized, double‐blind, parallel‐group study" Quote: "To assess total valproate levels, serum samples were obtained [...] results were reported to a qualified, unblinded associate who then advised an investigator to reduce the number of the divalproex tables taken by any subject with a serum valproate > 125 µg/mL. To preserve the study blind, the unblinded associate concurrently advised that the number of placebo tables taken by a subject randomly assigned to received olanzapine also be reduced." |
| Blinding of outcome assessment (detection bias) Efficacy | Unclear risk | Quote: "Raters were trained in the use of the MRS prior to the start of the study, and each subject was evaluated by the same rater throughout the study" Comment: It is not made explicit in the text whether the rater is blinded. |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: The dropout rates were 71% for the valproate group and 67% for the olanzapine group prior to day 84. 38% of the valproate group and 32% of the olanzapine group prematurely discontinued prior to day 21. However, the reasons for dropout rates were similar between the 2 study groups at both time points. |
| Selective reporting (reporting bias) | Unclear risk | Comment: No pre‐published protocol found. We are therefore unable to assess the bias. |
| Other bias | Unclear risk | Quote: "Investigators could prescribe rescue medications, including lorazepam, benztropine mesylate, chloral hydrate and zolpidem, as adjunctive therapy. [...] Adjunctive therapy was not to be administered within 8 hours prior to efficacy ratings." Comment: It is not clear whether the use of the wide range of adjunctive medication was balanced between groups. |
BARS: Behavioural Activity Rating Scale; BD: bipolar disorder; BIS: Behavioral Inhibition System; BMI: body mass index; BPD: borderline personality disorder; BPRS: Brief Psychiatric Rating Scale; CDRS: Children's Depression Rating Scale; CGAS: Children's Global Assessment Scale; CGI: Clinical Global Impression; CNS: central nervous system; C‐SSRS: Columbia‐Suicide Severity Rating Scale; DSM: Diagnostic and Statistical Manual of Mental Disorders; ECT: electroconvulsive therapy; EEG: electroencephalogram; EPS: extrapyramidal side effects; GAS: Global Assessment Scale; HDRS: Hamilton Rating Scale for Depression; IQ: intelligence quotient; kg: kilogram; K‐SADS: Kiddie Schedule for Affective Disorders and Schizophrenia; L: litre; LFT: liver function tests; MADRS: Montgomery–Åsberg Depression Rating Scale; mEq: milliequivalents; mg: milligram; min: minute; ml: millilitre; mmol: millimole; MRS: Modified Rankin Scale MSS: Manic Syndrome Score; n: number; SADS: Schedule for Affective Disorders and Schizophrenia; SCID: Structured Clinical Interview; SD: standard deviation; TFT: thyroid function tests; µ: micro; µg: microgram; WASH‐U‐KSADS: Washington University in St. Louis Kiddie Schedule for Affective Disorders and Schizophrenia; YMRS: Young Mania Rating Scale
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Bowden 2008 | A 12‐week, randomised trial comparing valproate to lithium. Was unblinded |
| Campos 2010 | Single‐site, parallel‐group, randomised, outcome assessor‐blinded trial. BD I patients according to the DSM‐IV‐TR, in depressive, manic/hypomanic or mixed episode, aged 18 to 35 years are eligible. Comparing lithium + carbamazepine vs lithium + valproate. However included all stages of bipolar, not specific to acute mania |
| Clothier 1992 | Double‐blind, randomised comparison of valproate and lithium. Mania was not acute |
| Emrich 1992 | Double‐blind, placebo‐controlled study of valproate in acute mania. Used an ABA design. Diagnostic criteria used were ICD 9 |
| Findling 2002 | A prospective study that looked at prophylactic dosing of high‐risk youths ‐ no acute mania |
| Jahangard 2012 | A trial where patients on valproate prior to having ECT either had their valproate stopped or continued on valproate during ECT in a blinded manner. In line with our protocol, we exclude trials where all participants are on open‐label valproate prior to discontinuation randomisation |
| Keck 2005 | Post hoc analysis of results obtained in a 12‐month, double‐blind, placebo‐controlled trial of valproate and lithium involving 372 participants. Original trial also ineligible as acutely manic phase was treated in an non‐randomised manner |
| Müller‐Oerlinghausen 2000 | 21‐day, randomised, double‐blind, parallel‐group, placebo‐controlled trial comparing valproate add‐on to neuroleptics with placebo add‐on. However they included a large number of participants without bipolar disorder (schizoaffective disorder) |
| Novartis 2007 | A randomised, double‐blind, placebo‐controlled study comparing add‐on carbamazepine to placebo when added to a mood stabiliser. Mood stabiliser allocation was neither randomised nor blinded |
| Pavuluri 2010 | Double‐blind, pseudo‐randomised, outpatient clinical trial with 66 children and adolescents. Participants were allocated to either risperidone or valproate. Randomisation used an alternating schedule, where each successive participant was allocated to the opposite treatment |
| Pavuluri 2012 | 6‐week, double‐blind, randomised trial of risperidone plus placebo versus valproate plus placebo MRI and pharmacological study. However participants were not acutely manic |
| Revicki 2005 | Randomised clinical trial, 201 adults hospitalised with bipolar I manic or mixed episodes were randomised to valproate or lithium, in addition to usual psychiatric care, and followed for 1 year. Assessors were not blinded while participants were acutely manic |
| Sachs 2002 | 3‐week randomised, double‐blind, placebo‐controlled study included 156 bipolar disorder patients with a current manic or mixed episode who received a mood stabiliser (lithium or valproate) and placebo, risperidone, or haloperidol. Mood stabiliser allocation was not randomised |
| Sachs 2004 | Double‐blind, randomised treatment with quetiapine plus lithium/valproate, or placebo plus lithium/valproate. Mood stabiliser allocation not randomised |
| Sidana 2012 | 3‐week, open‐label, randomised,comparative, parallel‐group study. Raters not blinded |
| Suppes 2007 | 30 hypomanic patients were randomised to receive oxcarbazepine or valproate as add‐on or monotherapy for 8 weeks. A rater blind to treatment assignment performed all symptom ratings. Treatment was otherwise open‐label |
| Walkup 2015 | Randomised, controlled trial of 379 individuals aged 6 to 15 years. Randomised to lithium, valproate or risperidone. Continuation of TEAM trials that used patients that failed treatment in an earlier study |
| West 2011 | Prospective 6‐week, double‐blind,placebo‐controlled, randomised outpatient medication treatment trial of risperidone versus valproate. Randomisation used an alternating schedule where each successive patient was allocated to the opposite treatment, therefore pseudo‐randomisation |
| Yatham 2004 | Double‐blind, randomised comparison of treatment with quetiapine plus lithium/valproate or placebo plus lithium/valproate. The mood stabiliser allocation was not randomised |
| Yatham 2007 | Double‐blind, randomised, 6 weeks trial of quetiapine (up to 800 mg/day) and lithium/valproate vs placebo and lithium/valproate. The mood stabiliser allocation was not randomised |
BD: bipolar disorder; DSM: Diagnostic and Statistical Manual of Mental Disorders; ICD: International Classification of Diseases; mg: milligram
Characteristics of studies awaiting assessment [ordered by study ID]
Aliev 2003.
| Methods | Randomised single‐blind trial |
| Participants | Male and female adults with bipolar disorder |
| Interventions | Treatment groups: 1.Valproate 2.Lithium |
| Outcomes | Not stated |
| Notes | Could not locate this study. The article was originally published in Russian. |
Goswami 2001.
| Methods | No information found |
| Participants | No information found |
| Interventions |
Treatment groups:
1.Valproate No further information found |
| Outcomes | No information found |
| Notes | Could not locate this study |
Lambert 1987.
| Methods | Unclear |
| Participants | Adults |
| Interventions |
Treatment groups:
1.Valproate No further information found |
| Outcomes | Adverse events |
| Notes | Could not locate |
Morinigo 1992.
| Methods | No information found |
| Participants | No information found |
| Interventions |
Treatment groups:
1.Valproate No further information found |
| Outcomes | No information found |
| Notes | Could not locate. The article was originally published in Spanish. |
Mosolov 1991.
| Methods | Controlled clinical trial |
| Participants | A total of 88 participants |
| Interventions |
Treatment groups:
1.Valproate (n = 28)
2.Carbamazepine (n = 30) 3.Lithium (n = 30) |
| Outcomes | No information found |
| Notes | Could not locate. The article was originally published in Russian. |
NCT00141505.
| Methods | Randomised double‐blind trial |
| Participants | Adults (aged 18 ‐ 65) with bipolar I disorder |
| Interventions |
Treatment groups: 1.Valproate 2.Bifeprunox 3.Placebo |
| Outcomes | Not stated |
| Notes | Could not locate |
Tiangin 1995.
| Methods | No information found |
| Participants | No information found |
| Interventions | Valproate No further information found |
| Outcomes | No information found |
| Notes | Could not locate. The article was originally published in Chinese. |
DSM: Diagnostic and Statistical Manual of Mental Disorders
GI: gastrointestinal
Characteristics of ongoing studies [ordered by study ID]
NCT01893229.
| Trial name or title | Comparative efficacy and acceptability of antimanic drugs in acute mania |
| Methods | Allocation: randomised Intervention model: parallel assignment Masking: single‐blind (participant) |
| Participants | 120 adults with a diagnosis of DSM‐IV bipolar I disorder |
| Interventions | Lithium Valproate Oxcarbazepine Quetiapine Olanzapine Ziprasidone |
| Outcomes | Primary outcome measures:
|
| Starting date | 2013 |
| Contact information | xuguiyun2908@hotmail.com |
| Notes |
DSM: Diagnostic and Statistical Manual of Mental Disorders; YMRS: Young Mania Rating Scale
Differences between protocol and review
We updated the protocol methods to reflect the most recent methodological standards expected by Cochrane, including implementation of the 'Risk of bias' tool and presentation of 'Summary of findings' tables.
We decided to include trials which had unbalanced rescue medications between groups. This is for two reasons. Firstly, in trials involving first generation antipsychotics it is unreasonable to expect balanced use of antiparkinsonian side‐effect medication. Secondly we decided that excluding these trials would potential exclude those with a positive result where the treatment group that benefited less from medication used more adjunctive medication but still showed improvement. We therefore decided to include these trials, but note in the 'Other potential bias' section in our bias assessment whether the use of medication was balanced. We also note the direction of medication imbalance compared to effect benefit.
Our original methods stated “In order to avoid missing any relatively rare or unexpected but important side effects, in the data extraction phase, we collected information on all side‐effects data reported in the studies and discussed ways to summarise them post hoc”. Accordingly we discussed that it would be useful to have data for each individual side effect for each comparison as different medications have different side effect profiles and this is vital in helping to guide treatment. Therefore, the decision was made to include all individual effects in our results as a secondary tolerability outcome.
The protocol noted that the 'Summary of findings' tables would include dropouts due to adverse events; we aimed to replace this with the number of participants with any adverse event. This is because the number of participants with any adverse event is our primary tolerability outcome. However, a large number of studies did not report this outcome (13 studies: DelBello 2006; Feifel 2011; Freeman 1992; Geller 2012; Hebrani 2009; Kowatch 2015; Mahmoudi‐Gharaei 2012; McElroy 1996; Pope 1991; Shafti 2008; Tohen 2002; Tohen 2008; Xu 2015). This meant that for some of our comparisons there were no data. We decided that to help interpretation we would additionally report an appropriate single adverse event, which we decided was sedation as a side effect common to all anti‐manic therapies, which is reported in all our main comparisons.
On the advice of a peer reviewer, we added a sensitivity analysis to assess different effects on psychotic versus non‐psychotic participants.
In our initial protocol there was some ambiguity, as we had stated we wanted to consider all “meaningful” time points for the outcomes, while in the protocol for this update we clearly stated we would extract data from three groups of time points; under 1 week, 1 to 2 weeks and 2 to 4 weeks. Some of the studies in this review reported useable data only for time points longer than this. We accept that data from time points after this are still meaningful and important to consider when answering the question of the usefulness of valproate in acute mania. We therefore thought it appropriate to add a fourth group of time points that we would extract from; 5 to 12 weeks, with eight weeks as the preferred week for comparison. We consider that beyond 12 weeks the question is no longer really about acute mania.
Contributions of authors
JJ*: Screened search results, retrieved papers against eligibility criteria, appraised quality of papers, extracted data from papers, wrote to authors of papers for additional information, entered data into Review Manager 5, analysed data, interpreted data, and wrote the review
RRZ*: Screened search results, screened and retrieved papers against eligibility criteria, appraised quality of papers, wrote to authors of papers for additional information, entered data into Review Manager 5, analysed data, interpreted data, and wrote the review.
JRG: Helped in designing search strategies and collecting supplemental data, interpreted review findings, provided a methodological and clinical perspective, and revised the manuscript.
AC: Co‐ordinated the update of the review, helped in designing search strategies and collecting supplemental data, interpreted review findings, provided a methodological and clinical perspective, and revised the manuscript.
* Equal contribution as authors.
Sources of support
Internal sources
-
University of Oxford, UK.
Salary for last authors, funding for applications used to make review
External sources
No sources of support supplied
Declarations of interest
Conflict of interests: Janina Jochim: no conflicts of interest Raphael Rifkin‐Zybutz: no conflicts of interest Andrea Cipriani: no conflicts of interest John Geddes: no conflicts of interest
These authors contributed equally to this work.
These authors contributed equally to this work
New search for studies and content updated (conclusions changed)
References
References to studies included in this review
Ahmad 2016 {published data only}
- Ahmad A, Sheikh S, Shah T, Reddy MS, Prasad BSV, Verma KK, et al. Endoxifen, a new treatment option for mania: A double‐blind, active controlled trial demonstrates the antimanic efficacy of endoxifen. Clinical and Translational Science 2016;9(5):252‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Bowden 1994 {published data only}
- Bowden CL, Brugger AM, Swann AC, Calabrese JR, Janicak PG, Petty F, et al. Efficacy of divalproex vs lithium and placebo in the treatment of mania. JAMA 1994;271(12):918‐24. [PubMed] [Google Scholar]
- Bowden CL, Davis J, Morris D, Swann A, Calabrese J, Lambert M, et al. Effect size of efficacy measures comparing divalproex, lithium and placebo in acute mania. Depression and Anxiety 1997;6(1):26‐30. [DOI] [PubMed] [Google Scholar]
- Bowden CL, Janicak PG, Orsulak P, Swann AC, Davis JM, Calabrese JR, et al. Relation of serum valproate concentration to response in mania. American Journal of Psychiatry 1996;153(6):765‐70. [DOI] [PubMed] [Google Scholar]
- Petty F, Rush J, Davis JM, Calabrese JR, Kimmel SE, Kramer GL, et al. Plasma GABA predicts acute response to divalproex in mania. Biological Psychiatry 1996;39(4):278‐84. [DOI] [PubMed] [Google Scholar]
- Swann AC, Bowden CL, Morris D, Calabrese JR, Petty F, Small J, et al. Depression during mania: treatment response to lithium or divalproex. Archives of General Psychiatry 1997;54(1):37‐42. [DOI] [PubMed] [Google Scholar]
Bowden 2006 {published data only}
- Bowden CL, Swann AC, Calabrese JR, Rubenfaer LM, Wozniak PJ, Collins MA, et al. A randomized, placebo‐controlled, multicenter study of divalproex sodium extended release in the treatment of acute mania. Journal of Clinical Psychiatry 2006;67(10):1501‐10. [PUBMED: 17107240] [DOI] [PubMed] [Google Scholar]
DelBello 2006 {published data only}
- DelBello MP, Kowatch RA, Adler CM, Stanford KE, Welge JA, Barzman DH, et al. A double‐blind randomized pilot study comparing quetiapine and divalproex for adolescent mania. Journal of the American Academy of Child and Adolescent Psychiatry 2006;45(3):305‐13. [PUBMED: 16540815] [DOI] [PubMed] [Google Scholar]
Feifel 2011 {published data only}
- Feifel D, Galangue B, Macdonald K, Cobb P, Dinca A, Becker O, et al. A naturalistic, single‐blind comparison of rapid dose administration of divalproex ER versus quetiapine in patients with acute bipolar mania. Innovations in Clinical Neuroscience 2011;8(1):29‐35. [PMC free article] [PubMed] [Google Scholar]
Freeman 1992 {published data only}
- Clothier J, Swann AC, Freeman T. Dysphoric mania. Journal of Clinical Psychopharmacology 1996;12(1 Suppl):13S‐16S. [DOI] [PubMed] [Google Scholar]
- Freeman TW, Clothier JL, Pazzaglia P, Lesem MD, Swann AC. A double‐blind comparison of valproate and lithium in the treatment of acute mania. American Journal of Psychiatry 1992;149(1):108‐11. [DOI] [PubMed] [Google Scholar]
Geller 2012 {published data only}
- Geller B, Luby JL, Joshi P, Wagner KD, Emslie G, Walkup JT, et al. A randomized controlled trial of risperidone, lithium, or divalproex sodium for initial treatment of bipolar I disorder, manic or mixed phase, in children and adolescents. Archives of General Psychiatry 2012;69(5):515‐28. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hebrani 2009 {published data only}
- Hebrani P, Behdani F, Manteghi AA. Double‐blind, randomized, clinical trial of topiramate versus sodium valproate for the treatment of bipolar disorder in adolescents. Pakistan Journal of Medical Sciences 2009;25(2):247‐52. [Google Scholar]
Hirschfeld 1999 {published data only}
- Hirschfeld RM, Allen MH, McEvoy JP, Keck PE, Russell JM. Safety and tolerability of oral loading divalproex sodium in acutely manic bipolar patients. Journal of Clinical Psychiatry 1999;60(12):815‐8. [DOI] [PubMed] [Google Scholar]
Hirschfeld 2010 {published data only}
- Hirschfeld RM, Bowden CL, Vigna NV, Wozniak P, Collins M. A randomized, placebo‐controlled, multicentre study of divalproex sodium extended‐release in the acute treatment of mania. Journal of Clinical Psychiatry 2010;71(4):426‐32. [DOI] [PubMed] [Google Scholar]
Kakkar 2009 {published data only}
- Kakkar AK, Rehan HS, Unni KES, Gupta NK, Chopra D, Kataria D. Comparative efficacy and safety of oxcarbazepine versus divalproex sodium in the treatment of acute mania: A pilot study. European Psychiatry 2009;24:178‐82. [DOI] [PubMed] [Google Scholar]
Kowatch 2015 {published data only}
- Kowatch RA, Scheffer RE, Monroe E, Delgado S, Altaye M, Lagory D. Placebo‐controlled trial of valproic acid versus risperidone in children 3 ‐ 7 years of age with bipolar I disorder. Journal of Child and Adolescent Psychopharmacology 2015;25(4):306‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mahmoudi‐Gharaei 2012 {published data only}
- Mahmoudi‐Gharaei J, Shahrivar Z, Faghihi T, Mohammadi MR, Tehrani‐Doost M, Alaghband‐Rad J, et al. Topiramate versus valproate sodium as adjunctive therapies to a combination of lithium and risperidone for adolescents with bipolar I disorder: Effects on weight and serum lipid profiles. Iranian Journal of Psychiatry 2012;7:1‐10. [PMC free article] [PubMed] [Google Scholar]
McElroy 1996 {published data only}
- McElroy SL, Keck PE, Stanton SP, Tugrul KC, Bennett JA, Strakowski SM. A randomised comparison of divalproex loading versus haloperidol in the initial treatment of acute psychotic mania. Journal of Clinical Psychiatry 1996;57:142‐6. [PubMed] [Google Scholar]
McElroy 2010 {published data only}
- McElroy SL, Martens BE, Creech RS, Welge JA, Jefferson L, Guerdjikova AI, et al. Randomized, double‐blind, placebo‐controlled study of divalproex extended release loading monotherapy in ambulatory bipolar spectrum disorder patients with moderate‐to‐severe hypomania or mild mania. Journal of Clinical Psychiatry 2010;71(5):557‐65. [DOI: 10.4088/JCP.08m04854yel] [DOI] [PubMed] [Google Scholar]
Moosavi 2014 {published data only}
- Moosavi SM, Ahmadi M, Monajemi MB. Risperidone versus risperidone plus sodium valproate for treatment of bipolar disorders: A randomized, double‐blind clinical‐trial. Global Journal of Health Science 2014;6(6):163‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pope 1991 {published data only}
- Pope HG, McElroy SL, Keck PE, Hudson JI. Valproate in the treatment of acute mania: A placebo controlled study. Archives of General Psychiatry 1991;48(1):62‐8. [DOI] [PubMed] [Google Scholar]
Shafti 2008 {published data only}
- Shafti SS, Shahveisi B. Comparison between lithium and valproate in the treatment of acute mania. Journal of Clinical Psychopharmacology 2008;28(6):718‐20. [DOI] [PubMed] [Google Scholar]
Tohen 2002 {published data only}
- Tohen M, Baker RW, Altshuler LL, Zarate CA, Suppes T, Ketter TA, et al. Olanzapine versus divalproex in the treatment of acute mania. American Journal of Psychiatry 2002;159(6):1011‐7. [PUBMED: 12042191] [DOI] [PubMed] [Google Scholar]
Tohen 2008 {published data only}
- Tohen M, Vieta E, Goodwin GM, Sun B, Amsterdam JD, Banov M, et al. Olanzapine versus divalproex versus placebo in the treatment of mild to moderate mania: a randomized, 12‐week, double‐blind study. Journal of Clinical Psychiatry 2008;69(11):1776‐89. [PUBMED: 19014751] [DOI] [PubMed] [Google Scholar]
Vasudev 2000 {published data only}
- Vasudev K, Goswami U, Kohli K. Carbamazepine and valproate monotherapy: feasibility, relative safety and efficacy, and therapeutic drug monitoring in manic disorder. Psychopharmacology 2000;150(1):15‐23. [DOI] [PubMed] [Google Scholar]
Wagner 2009 {published data only}
- Wagner KD, Redden R, Kowatch RA, Wilens TE, Segal S, Chang K, et al. A double‐blind, randomised, placebo‐controlled trial of divalproex extended‐release in the treatment of bipolar disorder in children and adolescents. Journal of the American Academy of Child and Adolescent Psychiatry 2009;48(5):519‐32. [DOI] [PubMed] [Google Scholar]
Xu 2015 {published data only}
- Xu L, Lu Y, Yang Y, Zheng Y, Chen F, Lin Z. Olanzapine‐valproate combination versus olanzapine or valproate monotherapy in the treatment of bipolar mania: A randomized controlled study in a Chinese population group. Neuropsychiatric Disease and Treatment 2015;11:1265‐71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Young 2017 {published data only}
- Young RC, Mulsant BH, Sajatovic M, Gildengers AG, Gyulai L, Al Jurdi RK, et al. Geri‐BD: A randomized double‐blind controlled trial of lithium and divalproex in the treatment of mania in older patients with bipolar disorder. American Journal of Psychiatry 2017;174(11):1086‐93. [DOI] [PMC free article] [PubMed] [Google Scholar]
Zajecka 2002 {unpublished data only}
- Zajecka JM, Weisler R, Sachs G, Swann AC, Wozniak P, Sommerville KW. A comparison of the efficacy, safety and tolerability of divalproex sodium and olanzapine in the treatment of bipolar disorder. Journal of Clinical Psychiatry 2002;63(12):1148‐55. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Bowden 2008 {published data only}
- Bowden C, Göğüş A, Grunze H, Häggström L, Rybakowski J, Viet E. A 12‐week, open, randomized trial comparing sodium valproate to lithium in patients with bipolar I disorder suffering from a manic episode. International Clinical Psychopharmacology 2008;23(5):254‐62. [DOI] [PubMed] [Google Scholar]
Campos 2010 {published data only}
- Campos RN, Costa LF, Bio DS, Soeiro de Souza MG, Garcia CR, Demétrio FN, et al. LICAVAL: combination therapy in acute and maintenance treatment of bipolar disorder. Trials 2010;11(72):1‐5. [DOI] [PMC free article] [PubMed] [Google Scholar]
Clothier 1992 {published data only}
- Clothier J, Swann AC, Freeman T. Dsyphoric mania. Journal of Clinical Psychopharmacology 1992;12(1):13S‐16S. [DOI] [PubMed] [Google Scholar]
Emrich 1992 {published data only}
- Emrich HM, Wolf R. Valproate treatment of mania. Progress in Neuro‐psychopharmacology & Biological Pyschiatry 1992;16(5):691‐701. [DOI] [PubMed] [Google Scholar]
Findling 2002 {published data only}
- Findling BL, Gracious NK, McNamara RJ, Stansbrey JR, Calabrese JR. The prevention of bipolar disorder in genetically at‐risk youths. International Journal of Neuropsychopharmacology 2002;5:S57. [Poster session at conference] [Google Scholar]
Jahangard 2012 {published data only}
- Jahangard L, Haghighi M, Bigdelou G, Bajoghli H, Brand S. Comparing efficacy of ECT with and without concurrent sodium valproate therapy in manic patients. Journal of ECT 2012;28(2):118‐23. [DOI] [PubMed] [Google Scholar]
Keck 2005 {published data only}
- Keck PE Jr, Bowden CL, Meinhold JM, Gyulai L, Prihoda TJ, Baker JD, et al. Relationship between serum valproate and lithium levels and efficacy and tolerability in bipolar maintenance therapy. International Journal of Psychiatry in Clinical Practice 2005;9(4):271‐7. [DOI] [PubMed] [Google Scholar]
Müller‐Oerlinghausen 2000 {published data only}
- Müller‐Oerlinghausen B, Retzow A, Henn FA, Giedke H, Walden J. Valproate as an adjunct to neuroleptic medication for the treatment of acute episodes of mania: a prospective, randomized, double‐blind, placebo‐controlled, multicenter study. Journal of Clinical Psychopharmacology 2000;20(2):195‐203. [DOI] [PubMed] [Google Scholar]
Novartis 2007 {unpublished data only}
- Novartis. Study of licarbazepine in the treatment of manic episodes of bipolar disorder. www.novctrd.com.
Pavuluri 2010 {published data only}
- Pavuluri MN, Henrya DB, Findling RL, Parnesa S, Carbraya JA, Mohammed T, et al. Double‐blind randomized trial of risperidone versus divalproex in paediatric bipolar disorder. Bipolar Disorders 2010;12:593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pavuluri 2012 {published data only}
- Pavuluri MN, Passarotti AM, Fitzgerald JM, Wegbreit E, Sweeney JA. Risperidone and divalproex differentially engage the fronto‐striato‐temporal circuitry in paediatric mania: A pharmacological functional magnetic resonance imaging study. Journal of the American Academy of Child & Adolescent Psychiatry 2012;51(2):157‐70. [DOI] [PMC free article] [PubMed] [Google Scholar]
Revicki 2005 {published data only}
- Revicki DA, Hirschfeld RM, Ahearn EP, Weisler RH, Palmer C, Keck PE Jr. Effectiveness and medical costs of divalproex versus lithium in the treatment of bipolar disorder: results of a naturalistic clinical trial. Journal of Affective Disorders 2005;86(2‐3):183–93. [DOI] [PubMed] [Google Scholar]
Sachs 2002 {published data only}
- Sachs GS, Grossman F, Ghaemi N, Okamoto A, Bowden CL. Combination of a mood stabilizer with risperidone or haloperidol for treatment of acute mania: a double‐blind, placebo‐controlled comparison of efficacy and safety. American Journal of Psychiatry 2002;159(7):1146–54. [DOI] [PubMed] [Google Scholar]
Sachs 2004 {published data only}
- Sachs G, Chengappa KN, Suppes T, Mullen JA, Brecher M, Devinedan NA, et al. Quetiapine with lithium or divalproex for the treatment of bipolar mania: a randomized, double‐blind, placebo‐controlled study. Bipolar Disorders 2004;6(3):213–23. [DOI] [PubMed] [Google Scholar]
Sidana 2012 {published data only}
- Sidana A, Verma P, Chavan BS. Comparative study of olanzapine versus divalproex in acute treatment of bipolar disorder : a 3‐week prospective study. Journal of Mental Health and Human Behavior 2012;17(1):5‐14. [Google Scholar]
Suppes 2007 {published data only}
- Suppes T, Kelly DI, Hynan LS, Snow DE, Sureddi S, Foster B, et al. Comparison of two anticonvulsants in a randomized, single‐blind treatment of hypomanic symptoms in patients with bipolar disorder. Australian and New Zealand Journal of Psychiatry 2007;41(5):397‐402. [DOI] [PubMed] [Google Scholar]
Walkup 2015 {published data only}
- Walkup JT, Wagner KD, Miller L, Yenokyan G, Luby JL, Joshi PT, et al. Treatment of early‐age mania: outcomes for partial and non responders to initial treatment. Journal of the American Academy of Child & Adolescent Psychiatry 2015;54(12):1008–19. [DOI] [PubMed] [Google Scholar]
West 2011 {published data only}
- West AE, Celio CI, Henry DB, Pavuluri MN. Child mania rating scale‐parent version: a valid measure of symptom change due to pharmacotherapy. Journal of Affective Disorders 2011;128(1‐2):112‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yatham 2004 {published data only}
- Yatham LN, Paulsson B, Mullen J, Vågerö AM. Quetiapine versus placebo in combination with lithium or divalproex for the treatment of bipolar mania. Journal of Clinical Psychopharmacology 2004;24(6):599–606. [DOI] [PubMed] [Google Scholar]
Yatham 2007 {published data only}
- Yatham LN, Vieta E, Young AH, Mö̈ller HJ, Paulsson B, Vågerö M. A double blind, randomized, placebo‐controlled trial of quetiapine as an add‐on therapy to lithium or divalproex for the treatment of bipolar mania. International Clinical Psychopharmacology 2007;22(4):212–20. [DOI] [PubMed] [Google Scholar]
References to studies awaiting assessment
Aliev 2003 {published data only}
- Aliev NA. Efficacy of depakine chrono and lithium combination in the treatment of manic states. Zh Nevrol Psikhiatr Im S S Korsakova. 2003;103(12):49‐51. [PUBMED: https://www.ncbi.nlm.nih.gov/pubmed/14763259] [PubMed] [Google Scholar]
Goswami 2001 {published data only}
- Goswami U, Ahuja J, Kohli K. Comparative efficacy and tolerability of lithium, carbamazepine and valproate in acute mania. Journal of Psychopharmacology 2001;15(Suppl 3):A19; B4. [Google Scholar]
Lambert 1987 {published data only}
- Lambert PA, Venaud G. Utilisation de valpromide en therapeutique psychiatrique [The use of valpromide in psychiatry]. Encephale 1987;13:367‐73. [PUBMED: https://www.ncbi.nlm.nih.gov/pubmed/3131112] [PubMed] [Google Scholar]
Morinigo 1992 {published data only}
- Morinigo A, Mateo L, Martin J, Noval D, Gonzalez S. The therapeutic effect of sodium valproate and carbamazepine in mania. [Efectos terapeuticos del valproato sodico y la carbamacepina en la mania]. Psiquis 1992;13(8):335‐8. [Google Scholar]
Mosolov 1991 {published data only}
- Mosolov SN. Comparative efficacy of preventive use of lithium carbonate, carbamazepine, and sodium valproate in affective and schizoaffective psychoses. Zhurnal Nevropatologii 1991;91(4):78‐83. [PUBMED: https://www.ncbi.nlm.nih.gov/pubmed/1650105] [PubMed] [Google Scholar]
NCT00141505 {unpublished data only}
- NCT00141505. Solvay Pharmaceuticals: PK effects of bifeprunox and valproate in bipolar I. clinicaltrials.gov/ct2/show/NCT00141505 (first received 1 September 2005).
Tiangin 1995 {published data only}
- Tiangin. Double‐blind placebo controlled study of valproate in treating lithium‐resistant mania. Chinese Journal of Psychiatry 1995;21:86‐7. [Google Scholar]
References to ongoing studies
NCT01893229 {unpublished data only}
- NCT01893229. Xu G, Lin K: Comparative efficacy and acceptability of antimanic drugs in acute mania. clinicaltrials.gov/ct2/show/NCT01893229 (first received 8 July 2013).
Additional references
Allen 2006
- Allen MH, Hirschfeld RM, Wozniak PJ, Baker JD, Bowden CL. Linear relationship of valproate serum concentration to response and optimal serum levels for acute mania.. American Journal of Psychiatry Feb;163(2):272‐5. [DOI] [PubMed] [Google Scholar]
Alonso 2011
- Alonso J, Petukhova M, Vilagut G, Chatterji S, Heeringa S, Üstün TB, et al. Days out of role due to common physical and mental conditions: results from the WHO World Mental Health surveys. Molecular Psychiatry 2011;16(12):1234‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
Altshuler 2006
- Altshuler LL, Post RM, Black DO, Keck PE Jr, Nolen WA, Frye MA, et al. Subsyndromal depressive symptoms are associated with functional impairment in patients with bipolar disorder: results of a large, multisite study. Journal of Clinical Psychiatry 2006;67(10):1551‐60. [DOI] [PubMed] [Google Scholar]
Alvarez‐Jiménez 2008
- Alvarez‐Jiménez M, González‐Blanch C, Crespo‐Facorro B, Hetrick S, Rodríguez‐Sánchez JM, Pérez‐Iglesias R, et al. Antipsychotic‐induced weight gain in chronic and first‐episode psychotic disorders: a systematic critical reappraisal.. CNS Drugs 2008;22(7):547‐62.. [PUBMED: 18547125] [DOI] [PubMed] [Google Scholar]
APA 1980
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM‐III). 3rd Edition. Washington DC: American Psychiatric Association, 1980. [Google Scholar]
APA 1987
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM‐III). 3rd Edition. Washington D.C: American Psychiatric Association, 1987. [Google Scholar]
APA 1994
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM‐IV). 4th Edition. Washington DC: American Psychiatric Association, 1994. [Google Scholar]
APA 2000
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders ‐ Text Revision (DSM‐IV‐TR). 4th Edition. Washington DC: American Psychiatric Association, 2000. [Google Scholar]
APA 2013
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM‐V). 5th Edition. Washington DC: American Psychiatric Association, 2013. [Google Scholar]
Atkins 2004
- Atkins D, Best D, Briss PA, Eccles M, Falck‐Ytter Y, Flottorp S, et al. GRADE Working Group. Grading quality of evidence and strength of recommendations. BMJ 2004;328(7454):1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
Barbui 2004
- Barbui C, Cipriani A, Brambilla P, Hotopf M. "Wish bias" in antidepressant drug trials?. Journal of Clinical Psychopharmacology 2004;24(2):126‐30. [DOI] [PubMed] [Google Scholar]
Bazinet 2006
- Bazinet RP, Weis MT, Rapoport SI, Rosenberger TA. Valproic acid selectively inhibits conversion of arachidonic acid to arachidonoyl‐CoA by brain microsomal long‐chain fatty acyl‐CoA synthetases: relevance to bipolar disorder. Psychopharmacology 2006;184(1):122‐9. [DOI] [PubMed] [Google Scholar]
Bonsall 2015
- Bonsall, Geddes JR, Goodwin, Holmes EA. Bipolar disorder dynamics: affective instabilities, relaxation oscillations and noise. Journal of the Royal Society Interface 2015;12(112):20150670. [DOI: 10.1098/rsif.2015.0670] [DOI] [PMC free article] [PubMed] [Google Scholar]
Bosetti 2005
- Bosetti F, Bell JM, Manickam P. Microarray analysis of rat brain gene expression after chronic administration of sodium valproate. Brain Research Bulletin 2005;65(4):331‐8. [DOI] [PubMed] [Google Scholar]
Bowden 2010
- Bowden CL, Mosolov S, Hranov L, Chen E, Habil H, Kongsakon R, et al. Efficacy of valproate versus lithium in mania or mixed mania: a randomized, open 12‐week trial. International Clinical Psychopharmacology 2010;25(2):60‐7. [DOI] [PubMed] [Google Scholar]
Bromley 2014
- Bromley R, Weston J, Adab N, Greenhalgh J, Sanniti A, McKay AJ, et al. Treatment for epilepsy in pregnancy: neurodevelopmental outcomes in the child. Cochrane Database of Systematic Reviews 2014, Issue 10. [DOI: 10.1002/14651858.CD010236.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Caddy 2015
- Caddy C, Amit BH, McCloud TL, Rendell JM, Furukawa T, McShane R, et al. Ketamine and other glutamate receptor modulators for depression in adults. Cochrane Database of Systematic Reviews 2015, Issue 9. [DOI: 10.1002/14651858.CD011612.pub2] [DOI] [PubMed] [Google Scholar]
CANMAT 2018
- Yatham LM, Kennedy SH, Parikh SV, Schaffer A, Bond DJ, Frey BN, et al. Canadian network for mood an anxiety treatments (CANMAT) and International society for Bipolar Disorders (ISBD) 2018 guidelines for the management of patients with bipolar disorder. Bipolar Disorders 2018;20(2):97‐170. [DOI] [PMC free article] [PubMed] [Google Scholar]
Carr 2018
- Carr O, Vos M, Saunders KE. Heart rate variability in bipolar disorder and borderline personality disorder: a clinical review. Evidence‐based Mental Health 2018;21(1):23‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
Chateauvieux 2010
- Chateauvieux S, Morceau F, Dicato M, Diederich M. Molecular and therapeutic potential and toxicity of valproic acid. Journal of Biomedicine & Biotechnology 2010;2010:479364. [DOI] [PMC free article] [PubMed] [Google Scholar]
Christensen 2013
- Christensen J, Grønborg TK, Sørensen MJ, Schendel D, Parner ET, Pedersen LH, et al. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA 2013;309(16):1696‐703. [DOI] [PMC free article] [PubMed] [Google Scholar]
Cipriani 2011
- Cipriani A, Barbui C, Salanti G, Rendell J, Brown R, Stockton S, et al. Comparative efficacy and acceptability of antimanic drugs in acute mania: a multiple‐treatments meta‐analysis. Lancet 2011;378(9799):1306‐15. [DOI] [PubMed] [Google Scholar]
Cipriani 2016
- Cipriani A, Zhou X, Giovane C, Hetrick SE, Qin B, Whittington C, et al. Comparative efficacy and tolerability of antidepressants for major depressive disorder in children and adolescents: a network meta‐analysis. Lancet 2016;388(10047):881‐90. [DOI] [PubMed] [Google Scholar]
Cummings 2011
- Cummings C, Stewart M, Stevenson M, Morrow J, Nelson J. Neurodevelopment of children exposed in utero to lamotrigine, sodium valproate and carbamazepine. Journal Arch Dis Child 2011;96(7):643‐7. [DOI] [PubMed] [Google Scholar]
De Angelis 2004
- Angelis C, Drazen JM, Frizelle FA, Haug C, Hoey J, Horton R, et al. Clinical trial registration: a atatement from the International Committee of Medical Journal Editors. New England Journal of Medicine 2004;351(12):1250‐1. [DOI: 10.1056/NEJMe048225] [DOI] [PubMed] [Google Scholar]
De Hert 2011
- De Hert, Dobbelaere M, Sheridan EM, Cohen D, Correll CU. Metabolic and endocrine adverse effects of second‐generation antipsychotics in children and adolescents: a systematic review of randomized, placebo controlled trials and guidelines for clinical practice.. European Psychiatry 2011;26(3):144‐58. [PUBMED: 21295450] [DOI] [PubMed] [Google Scholar]
Del Grande 2014
- Grande C, Muti M, Musetti L, Corsi M, Pergentini I, Turri M, et al. Lithium and valproate in manic and mixed states: a naturalistic prospective study. Journal of Psychopathology 2014;20:6‐10. [Google Scholar]
Doherty 2018
- Doherty J, Cooper M, Thapar A. Advances in our understanding of the genetics of childhood neurodevelopmental disorders. Evidence‐Based Mental Health 2018;21(4):171‐2. [DOI] [PMC free article] [PubMed] [Google Scholar]
Duffy 2018
- Duffy A, Heffer N, Goodday SM, Weir A, Patten S, Malhi GS, Cipriani A. Efficacy and tolerability of lithium for the treatment of acute mania in children with bipolar disorder: A systematic review: A report from the ISBD‐IGSLi joint task force on lithium treatment. Bipolar Disord 2018;20(7):583‐593. [DOI] [PubMed] [Google Scholar]
Easterbrook 1991
- Easterbrook PJ, Gopalan R, Berlin JA, Matthews DR. Publication bias in clinical research.. Lancet 1991;337(8746):867‐72. [DOI: 10.1016/0140-6736(91)90201-Y] [DOI] [PubMed] [Google Scholar]
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Biasin meta‐analysis detected by a simple, graphical test. BMJ 1997;315(7109):629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
Emrich 1980
- Emrich HM, Zerssen D, Kissling W, Möller HJ, Windorfer A. Effect of sodium valproate on mania ‐ The GABA‐hypothesis of affective disorders. Archiv für Psychiatrie und Nervenkrankheiten 1980;229(1):1‐16. [DOI] [PubMed] [Google Scholar]
FDA 2015
- Food, Drug Administration. Valproate sodium ‐ Labelling Revision (09/23/2015). www.accessdata.fda.gov/drugsatfda_docs/label/2015/020593s033lbl.pdf 2015.
Feighner 1972
- Feighner JP, Robins E, Guze SB, Woodruff RA, Winokur G, Munoz R. Diagnostic criteria for use in psychiatric research. Archives of General Psychiatry 1972;26(1):57‐63. [DOI] [PubMed] [Google Scholar]
Findling 2018
- Findling RL, Stepanova E, Youngstrom EA, Young AS. Progress in diagnosis and treatment of bipolar disorder among children and adolescents: an international perspective.. Evidence‐Based Mental Health 2018;21(4):177‐81. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fountoulakis 2012
- Fountoulakis KN, Kontis D, Gonda X, Siamouli M, Yatham LN. Treatment of mixed bipolar states. International Journal of Neuropsychopharmacology 2012;15(7):1015‐26. [DOI] [PubMed] [Google Scholar]
Freeman 2002
- Freeman MP, Freeman SA, McElroy SL. The comorbidity of bipolar and anxiety disorders: prevalence, psychobiology, and treatment issues. Journal of Affective Disorders 2002;68(1):1‐23. [DOI] [PubMed] [Google Scholar]
Furukawa 2002
- Furukawa TA, Guyatt GH, Griffith LE. An empirical study of summary effect measures in meta‐analyses. International Journal of Epidemiology 2002;31(1):72‐6. [DOI] [PubMed] [Google Scholar]
Furukawa 2006
- Furukawa TA, Barbui C, Cipriani A, Brambilla P, Watanabe N. Imputing missing standard deviations in meta‐analyses can provide accurate results. Journal of Clinical Epidemiology 2006;59(1):7–10. [DOI] [PubMed] [Google Scholar]
Furukawa 2016a
- Furukawa TA, Cipriani A, Atkinson LZ, Leucht S, Ogawa Y, Takeshima N, et al. Placebo response rates in antidepressant trials: a systematic review of published and unpublished double‐blind randomised controlled studies. Lancet Psychiatry 2016;3(11):1059‐66. [DOI] [PubMed] [Google Scholar]
Furukawa 2016b
- Furukawa TA, Salanti G, Atkinson LZ, Leucht S, Ruhe HG, Turner EH, et al. Comparative efficacy and acceptability of first‐generation and second‐generation antidepressants in the acute treatment of major depression: protocol for a network meta‐analysis. BMJ Open 2016;6(7):e010919. [DOI] [PMC free article] [PubMed] [Google Scholar]
GBD 2013 Risk Factors Collaborators 2015
- GBD 2013 Risk Factors Collaborators 2015. Global, regional, and national comparative risk assessment of 79 behavioural, environmental and occupational, and metabolic risks or clusters of risks in 188 countries, 1990‐2013: a systematic analysis for the Global Burden of Disease Study 2013. Lancet 2015;386(10010):2287‐323. [DOI] [PMC free article] [PubMed] [Google Scholar]
Geddes 2013
- Geddes JR, Miklowitz DJ. Treatment of bipolar disorder. Lancet 2013;381(9878):1672‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Geddes 2015
- Geddes JR, Cipriani A. Time to abandon placebo control in pivotal phase III trials?. World Psychiatry 2015;14(3):306‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Gerstner 2007
- Gerstner T, Büsing D, Bell N, Longin E, Kasper JM, Klostermann W, et al. Valproic acid‐induced pancreatitis: 16 new cases and a review of the literature. Journal of Gastroenterology 2007;42(1):39‐48. [DOI] [PubMed] [Google Scholar]
Goodday 2019
- Goodday SM, Cipriani A. Challenges in identifying behavioural markers of bipolar disorder through objective smartphone data. Aust N Z J Psychiatry 2019;53(2):168‐169. [DOI] [PubMed] [Google Scholar]
Goodwin 2016
- Goodwin GM, Haddad PM, Ferrier IN, Aronson JK, Barnes T, Cipriani A, et al. Evidence‐based guidelines for treating bipolar disorder: revised third edition recommendations from the British Association for Psychopharmacology. Journal of Psychopharmacology 2016;30(6):495‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADEpro GDT 2015 [Computer program]
- McMaster University (developed by Evidence Prime). GRADEpro GDT. Version Accessed 26 June 2019. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015.
Grande 2016
- Grande I, Berk M, Birmaher B, Vieta E. Bipolar disorder. Lancet 2016;387(10027):1561‐72. [DOI: 10.1016/S0140-6736(15)00241-X] [DOI] [PubMed] [Google Scholar]
Hahn 2005
- Hahn CG, Umapathy, Wang HY, Koneru R, Levinson DF, Friedman E. Lithium and valproic acid treatments reduce PKC activation and receptor‐G protein coupling in platelets of bipolar manic patients. Journal of Psychiatric Research 2005;39(4):355‐63. [DOI] [PubMed] [Google Scholar]
Harrison 2016
- Harrison PJ, Cipriani A, Harmer CJ, Nobre AC, Saunders K, Goodwin GM, et al. Innovative approaches to bipolar disorder and its treatment. Annals of the New York Academy of Sciences 2016;1366(1):674‐82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hazell 2012
- Hazell R, Jairam P. Acute treatment of mania in children and adolescents. Current Opinion in Psychiatry 2012;25(4):264‐70. [DOI] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003;327(7414):557‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JP, Deeks JJ, editor(s). Chapter 7: Selecting studies and collecting data. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions. Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from www.cochrane‐handbook.org.
Higgins 2017
- Higgins JP, Altman DG, Sterne JA, editor(s). Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Churchill R, Chandler J, Cumpston MS, editor(s), Cochrane Handbook for Systematic Reviews of Interventions version 5.2.0 (updated June 2017). The Cochrane Collaboration, 2017. Available from www.training.cochrane.org/handbook.
Hooshmand 2014
- Hooshmand F, Miller S, Dore J, Wang PW, Hill SJ, Portillo N, et al. Trends in pharmacotherapy in patients referred to a bipolar specialty clinic, 2000–2011. Journal of Affective Disorders 2014;155:283–7. [DOI] [PubMed] [Google Scholar]
Johannessen 2000
- Johannessen CU. Mechanisms of action of valproate: A commentatory. Neurochemistry International 2000;37(2‐3):103‐10. [DOI] [PubMed] [Google Scholar]
Joint Formulary Committee 2015
- Joint Formulary Committee. British National Formulary. 69. London: BMJ Group and Pharmaceutical Press, 2015. [Google Scholar]
Judd 2005
- Judd LL, Akiskal HS, Schettler PJ, Endicott J, Leon AC, Solomon DA, et al. Psychosocial disability in the course of bipolar I and bipolar II disorders: a prospective comparative, longitudinal study. Archives of General Psychiatry 2005;62(12):1322‐30. [DOI] [PubMed] [Google Scholar]
Karanti 2016
- Karanti A, Kardell M, Lundberg U, Landén M. Changes in mood stabilizer prescription patterns in bipolar disorder. Journal of Affective Disorders 2016;195:50‐6. [DOI] [PubMed] [Google Scholar]
Kendall 2014
- Kendall T, Morriss R, Mayo‐Wilson E, Marcus E, Guideline Development Group of the National Institute for Health and Care Excellence. Assessment and management of bipolar disorder: summary of updated NICE guidance. BMJ 2014;349:g5673. [DOI] [PubMed] [Google Scholar]
Kleimann 2016
- Kleimann A, Schrader V, Stübner S, Greil W, Kahl KG, Bleich S, Grohmann R, Frieling H, Toto S. Psychopharmacological treatment of 1650 in‐patients with acute mania‐data from the AMSP study.. Journal of Affective Disorders 2016;191(1):64‐71. [DOI] [PubMed] [Google Scholar]
Lambert 1966
- Lambert PA, Carraz G, Borselli S, Carrel S. Neuropsychotropic action of a new antiepileptic: depramide [Action neuropsychotrope d'un novel antiepileptique: le depamide]. Annales Medico‐Psychologiques 1966;1(5):707‐10. [PubMed] [Google Scholar]
Laursen 2011
- Laursen TM. Life expectancy among persons with schizophrenia or bipolar affective disorder. Schizophrenia Research 2011;131(1‐3):101‐4. [DOI] [PubMed] [Google Scholar]
Leibenluft 2000
- Leibenluft E. Women and bipolar disorder: An update. Bulletin of the Menninger Clinic 2000;64(1):5‐17. [PubMed] [Google Scholar]
Leo 1999
- Leo RJ, Narendran R. Anticonvulsant use in the treatment of bipolar disorder: a primer for primary care physicians. Primary Care Companion to the Journal of Clinical Psychiatry 1999;1(3):74‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
Li 2002
- Li X, Ketter TA, Frye MA. Synaptic, intracellular, and neuroprotective mechanisms of anticonvulsants: Are they relevant for the treatment and course of bipolar disorders?. Journal of Affective Disorders 2002;69(1‐3):1‐14. [DOI] [PubMed] [Google Scholar]
Licht 2002
- Licht RW. Limits of the applicability and generalizability of drug trials in mania. Bipolar Disorder 2002;4 Suppl 1:66–8. [DOI: 10.1034/j.1399-5618.4.s1.27.x] [DOI] [PubMed] [Google Scholar]
Malhi 2015
- Malhi GS, Bassett D, Boyce P, Bryant R, Fitzgerald PB, Fritz K, et al. Royal Australian and New Zealand College of Psychiatrists clinical practice guidelines for mood disorders. Australian and New Zealand Journal of Psychiatry 2015;49(12):1087‐206. [DOI] [PubMed] [Google Scholar]
Marson 2015
- Marson A, Sills G. Valproate. In: Shorvon S, Perucca E, Engel J editor(s). The Treatment of Epilepsy. 3rd Edition. Chichester, UK: John Wiley & Sons Ltd, 2015:652‐666. [Google Scholar]
McCloud 2015
- McCloud TL, Caddy C, Jochim J, Rendell JM, Diamond PR, Shuttleworth C, et al. Ketamine and other glutamate receptor modulators for depression in bipolar disorder in adults. Cochrane Database of Systematic Reviews 2015, Issue 9. [DOI: 10.1002/14651858.CD011611] [DOI] [PubMed] [Google Scholar]
McKnight 2019
- McKnight RF, Motte de Broöns de Vauvert SJGN, Chesney E, Amit BH, Geddes J, Cipriani A. Lithium for acute mania. Cochrane Database of Systematic Reviews 2019, Issue 6. [DOI: 10.1002/14651858.CD004048.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Meador 2008
- Meador K, Reynolds MW, Crean S, Fahrbach K, Probst C. Pregnancy outcomes in women with epilepsy: a systematic review and meta‐analysis of published pregnancy registries and cohorts.. Epilepsy Research 2008;81(1):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
Merikangas 2011
- Merikangas KA, Jin R, He J‐P, Kessler RC, Lee S, Sampson NA, et al. Prevalence and correlates of bipolar spectrum disorder in the World Mental Health Survey Initiative. Archives of General Psychiatry 2011;68(3):241‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
MHRA 2018
- Drug Safety Update ‐MHRA. https://assets.publishing.service.gov.uk/government/uploads/system/uploads/attachment_data/file/701831/DSU‐April‐2018‐PDF.pdf April 2018; Vol. 11, issue 9.
Miura 2014
- Miura T, Noma H, Furukawa TA, Mitsuyasu H, Tanaka S, Stockton S, et al. Comparative efficacy and tolerability of pharmacological treatments in the maintenance treatment of bipolar disorder: a systematic review and network meta‐analysis. Lancet Psychiatry 2014;1(5):351‐9. [DOI] [PubMed] [Google Scholar]
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, The PRISMA Group. Preferred reporting items for systematic reviews and meta‐analyses: The PRISMA Statement. BJM 2009;339:2535. [PMC free article] [PubMed] [Google Scholar]
Morrow 2006
- Morrow J, Russell A, Guthrie E, Parsons L, Robertson I, Waddell R, et al. Malformation risks of antiepileptic drugs in pregnancy: a prospective study from the UK Epilepsy and Pregnancy Register. Journal of Neurology 2006;77(2):193‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Mulder 2018
- Mulder R, Singh AB, Hamilton A, Das P, Outhred T, Morris G, et al. The limitations of using randomised controlled trials as a basis for developing treatment guidelines. Evidence‐Based Mental Health 2018;21(1):4‐6. [DOI: 10.1136/eb-2017-102701] [DOI] [PMC free article] [PubMed] [Google Scholar]
Murphy 2016
- Murphy S, Bennett K, Doherty CP. Prescribing trends for sodium valproate in Ireland. Seizure 2016;36:44–8. [DOI] [PubMed] [Google Scholar]
Nanau 2013
- Nanau RM, Neuman MG. Adverse drug reactions induced by valproic acid. Clinical Biochemistry 2013;46(15):1323‐38. [DOI] [PubMed] [Google Scholar]
NICE 2014
- National Institute for Health and Care Excellence (NICE). Bipolar disorder: the assessment and management of bipolar disorder in adults, children and young people in primary and secondary care (updated edition) (CG185). National Collaborating Centre for Mental Health; The British Psychological Society & The Royal College of Psychiatrists 2014.
NICE 2018a
- Ntional Institute for Health and Care Excellence (NICE). Bipolar disorder: assessment and management; Clinical guideline [CG185]. www.nice.org.uk/guidance/cg185/chapter/1‐Recommendations#managing‐mania‐or‐hypomania‐in‐adults‐in‐secondary‐care‐2) April 2018.
NICE 2018b
- National Collaborating Centre for Mental Health. Antenatal and postnatal mental health (National clinical guideline 192). www.nice.org.uk/guidance/cg192/evidence/full‐guideline‐pdf‐4840896925 2018.
Perruca 2002
- Perucca E. Pharmacological and therapeutic properties of valproate: A summary of 35 years of clinical experience. CNS Drugs 2002;16(10):695‐714. [DOI] [PubMed] [Google Scholar]
Philips 2013
- Phillips ML, Kupfer DJ. Bipolar disorder diagnosis: challenges and future directions. Lancet 2013;381(9878):1663–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
Poon 2012
- Poon SH, Sim K, Sum MY, Kuswanto CN, Baldessarini RJ. Evidence‐based options for treatment‐resistant adult bipolar disorder patients. Bipolar Disorders 2012;14(6):573–84. [DOI: 10.1111/j.1399-5618.2012.01042.x] [DOI] [PubMed] [Google Scholar]
Rho 1999
- Rho JM, Sankar R. The pharmacologic basis of antiepileptic drug action. Epilepsia 1999;40(11):1471‐83. [DOI] [PubMed] [Google Scholar]
Rosenberg 2007
- Rosenberg G. The mechanisms of action of valproate in neuropsychiatric disorders: can we see the forest for the trees?. Cellular and Molecular Life Sciences 2007;64(16):2090‐103. [DOI] [PMC free article] [PubMed] [Google Scholar]
Selph 2014
- Selph SS, Ginsburg AD, Chou R. Impact of contacting study authors to obtain additional data for systematic reviews: diagnostic accuracy studies for hepatic fibrosis. Systematic Reviews 2014;3:107. [DOI: 10.1186/2046-4053-3-107.] [DOI] [PMC free article] [PubMed] [Google Scholar]
Simmons 2011
- Simmons JP, Nelson LD, Simonsohn U. False‐positive psychology: undisclosed flexibility in data collection and analysis allows presenting anything as significant. Psychological Science 2011;22(11):1359–66. [DOI: 10.1177/0956797611417632] [DOI] [PubMed] [Google Scholar]
Spitzer 1978
- Spitzer RL, Endicott J, Robins E. Research diagnostic criteria: rationale and reliability. Archives of General Psychiatry 1978;35(6):773‐82. [DOI] [PubMed] [Google Scholar]
Sterne 2000
- Sterne JA, Gavaghan D, Egger M. Publication and related bias in meta‐analysis: power of statistical tests and prevalence in the literature. Journal of Clinical Epidemiology 2000;53(11):1119‐29. [DOI] [PubMed] [Google Scholar]
Sterne 2017
- Sterne JA, Egger M, Moher D, Boutron I, editor(s). Chapter 10: Addressing reporting biases. In: Higgins JP, Churchill R, Chandler J, Cumpston MS (editors), Cochrane Handbook for Systematic Reviews of Interventions version 5.2.0 (updated June 2017). The Cochrane Collaboration, 2017. Available from www.training.cochrane.org/handbook.
Swann 1997
- Swann AC, Bowden CL, Morris D, Calabrese JR, Petty F, Small J, et al. Depression during mania: treatment response to lithium or divalproex. Archives of General Psychiatry 1997;54(1):37‐42. [DOI] [PubMed] [Google Scholar]
Swann 2007
- Swann AC, Steinberg JL, Schneider L, Barratt ES, Dougherty DM. Manic symptoms and impulsivity during bipolar depressive episodes. Bipolar Disorders 2007;9(3):206‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
Tang 2004
- Tang Y, Glauser TA, Gilbert DL, Hershey AD, Privitera MD, Ficker DM, et al. Valproic acid blood genomic expression patterns in children with epilepsy – a pilot study. Acta Neurologica Scandinavica 2004;109(3):159‐68. [DOI] [PubMed] [Google Scholar]
Tansella 2006
- Tansella M, Thornicroft G, Barbui C, Cipriani A, Saraceno B. Seven criteria for improving effectiveness trials in psychiatry. Psychological Medicine 2006;36(5):711‐20. [DOI] [PubMed] [Google Scholar]
Taylor 2009
- Taylor D, Paton C, Kapur S. The South London and Maudsley NHS Foundation Trust & Oxleas NHS Foundation Trust Prescribing Guidelines. 10th Edition. Informa Healthcare, 2009. [Google Scholar]
Telles‐Correia 2017
- Telles‐Correia D, Barbosa A, Cortez‐Pinto H, Compos C, Rocha NB, Machado S. Psychotropic drugs and liver disease: A critical review of pharmacokinetics and liver toxicity. World Journal of Gastrointestinal Pharmacology and Therapeutics 2017;8(1):26‐38. [DOI] [PMC free article] [PubMed] [Google Scholar]
Vitiello 2018
- Vitiello B, Davico C. Twenty years of progress in paediatric psychopharmacology: accomplishments and unmet needs. Evid Based Ment Health 2018;21(4):e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
Vojta 2001
- Vojta C, Kinosian B, Glick H, Altshuler L, Bauer MS. Self‐reported quality of life across mood states in bipolar disorder. Comprehensive Psychiatry 2001;42(3):190‐5. [DOI] [PubMed] [Google Scholar]
Wandel 2016
- Wandel S, Roychoudhury S. Designing and analysing clinical trials in mental health: an evidence synthesis approach. Evidence‐Based Mental Health 2016;19(4):114‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wang 1996
- Wang HY, Friedman E. Enhanced protein kinase C activity and translocation in bipolar affective disorder brains. Biological Psychiatry 1996;40(7):568‐75. [DOI] [PubMed] [Google Scholar]
Wang 1999
- Wang HY, Markowitz P, Levinson D, Undie AS, Friedman E. Increased membrane‐associated protein kinase C activity and translocation in blood platelets from bipolar affective disorder patients. Journal of Psychiatric Research 1999;33(2):171‐9. [DOI] [PubMed] [Google Scholar]
Wang 2001
- Wang H, Friedman E. Increased association of brain protein kinase C with the receptor for activated C kinase‐1 (RACK1) in bipolar affective disorder. Biological Psychiatry 2001;50(5):364‐70. [DOI] [PubMed] [Google Scholar]
WHO 1992
- World Health Organization. The ICD‐10 classification of mental and behavioural disorders: clinical descriptions and diagnostic guidelines. Geneva: World Health Organization, 1992. [Google Scholar]
Williams 2018
- Williams T, Stein DJ, Ipser J. A systematic review of network meta‐analyses for pharmacological treatment of common mental disorders. Evidence‐Based Mental Health 2018;21(1):7‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Yatham 2002
- Yatham LN, Kusumakar LN, Kutcher SP. Bipolar Disorder: A Clinician's Guide to Biological Treatments. New York: Brunner‐Routledge, 2002. [Google Scholar]
Yildiz 2011
- Yildiz A, Vieta E, Leucht S, Baldessarini RJ. Efficacy of antimanic treatments: meta‐analysis of randomized, controlled trials. Neuropsychopharmacology 2011;36(2):75‐89. [PUBMED: 20980991] [DOI] [PMC free article] [PubMed] [Google Scholar]
Yildiz 2015
- Yildiz A, Nikodem M, Vieta E, Correll U, Baldessarini RJ. A network meta‐analysis on comparative efficacy and all‐cause discontinuation of antimanic treatments in acute bipolar mania. Psychological Medicine 2015;45:299‐317. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Macritchie 2003
- Macritchie K, Geddes J, Scott J, Haslam DR, Silva de Lima M, Goodwin G. Valproate for acute mood episodes in bipolar disorder. Cochrane Database of Systematic Reviews 2003, Issue 1. [DOI: 10.1002/14651858.CD004052] [DOI] [PubMed] [Google Scholar]