

Abstract
Background
Epilepsy is a common neurological condition that affects up to 1% of the population. Nearly 30% of people with epilepsy are resistant to currently available antiepileptic drugs (AEDs) and require treatment with multiple antiepileptic drugs in combination. Tiagabine is one of the newer AEDs that can be used as an adjunct (add‐on) to standard AEDs.
Objectives
To evaluate the efficacy and tolerability of tiagabine when used as an add‐on treatment for people with drug‐resistant focal seizures.
Search methods
This is an updated Cochrane review, last published in 2014. For the latest update, we searched the following databases on 22 January 2019: Cochrane Register of Studies (CRS Web), which includes the Cochrane Epilepsy Group's Specialized Register and the Cochrane Central Register of Controlled Trials, MEDLINE (Ovid, 1946 to January 21, 2019), ClinicalTrials.gov, and the WHO International Clinical Trials Registry Platform. We imposed no language restrictions. We also contacted the manufacturers of tiagabine and experts in the field to identify any ongoing or unpublished studies.
Selection criteria
We included randomised placebo‐controlled add‐on trials conducted in people of any age with focal epilepsy. The studies could be double‐, single‐, or unblinded and of parallel or cross‐over design. They had to have a minimum treatment period of eight weeks. We also included trials using an active drug control group.
Data collection and analysis
Two review authors independently assessed trials for inclusion and extracted data according to the standard methodological procedures expected by the Cochrane Collaboration for this review update. We resolved disagreements by discussion. Outcomes investigated included 50% or greater reduction in seizure frequency, treatment withdrawal, adverse effects, effects on cognition and quality of life. The primary analyses were performed by intention‐to‐treat. We calculated worst‐case and best‐case analyses for seizure outcomes. We evaluated dose response using regression models. Two review authors assessed risk of bias in each study using the Cochrane 'Risk of bias' tool.
Main results
No further studies were added since the previous update in 2014. The review included six trials (four parallel‐group and two cross‐over group trials) consisting of 948 participants. For the main comparison, tiagabine versus placebo, all participants were aged between 12 and 77 years and the study treatment periods ranged from 12 to 22 weeks. The overall risk ratio (RR) with 95% confidence intervals (CIs) for a 50% or greater reduction in seizure frequency (tiagabine versus placebo) was 3.16 (95% CI 1.97 to 5.07; 3 trials; 769 participants; high‐certainty evidence). Because of differences in response rates among trials, regression models were unable to provide reliable estimates of response to individual doses. The RR for treatment withdrawal (tiagabine versus placebo) was 1.81 (95% CI 1.25 to 2.62; 3 trials, 769 participants; moderate‐certainty evidence). Dizziness and tremor were significantly associated with tiagabine therapy. For cognitive and quality‐of‐life outcomes, the limited available data suggested no significant effects on cognition, mood, or adjustment. One trial comparing tiagabine with an active drug control group (tiagabine versus topiramate) found no significant differences between the two add‐on drugs for a 50% or greater reduction in seizure frequency (RR 0.54, 95% CI 0.19 to 1.58; 1 trial; 41 participants) or for treatment withdrawal (RR 1.43, 95% CI 0.74 to 2.74; one trial; 41 participants). We judged two of the six included studies to have low risk of bias, three studies to have an unclear risk of bias, and one study to have a high risk of bias. Methods for randomisation sequence generation were the least reported trial design factor and generated the most concerns regarding risk of bias. We rated the overall certainty of the evidence as largely moderate to high using the GRADE approach. We rated the evidence for two of the adverse effect outcomes, nausea and tremor, as low certainty.
Authors' conclusions
Tiagabine reduced seizure frequency but was associated with some adverse effects when used as an add‐on treatment in people with drug‐resistant focal epilepsy. The findings of the current review are mainly applicable to adults and adolescents, and may not necessarily be applicable to children as none of the trials included participants aged under 12 years. We found no significant differences between tiagabine and topiramate as add‐on drugs; however, evidence was provided by a single trial and was therefore limited.
Plain language summary
Tiagabine add‐on therapy for drug‐resistant focal epilepsy
Background
Epilepsy is a disorder in which recurrent seizures are caused by abnormal electrical discharges from the brain. Most seizures can be controlled by a single antiepileptic drug. Unfortunately, some people require more than one antiepileptic drug to control their seizures (drug‐resistant epilepsy), especially if the seizures originate from one area of the brain (focal epilepsy), rather than affecting the entire brain (generalised epilepsy).
Tiagabine is a newer antiepileptic drug that can be used in conjunction with a person's normal antiepileptic drug regimen. This review assessed the evidence available for how effective tiagabine is in reducing seizures, as well as looking at the side effects that may be associated with its use when it is used alongside other antiepileptic medication(s) for people with drug‐resistant focal epilepsy.
Results
We found six trials that included 948 people with focal epilepsy. These trials were all randomised controlled trials (RCTs) that compared the antiepileptic drug tiagabine with a placebo (an inactive, dummy treatment which should not affect epilepsy) or with a different antiepileptic drug for a period of up to 24 weeks. We found that tiagabine, when used with another antiepileptic drug, was three times more effective than placebo at reducing the number of seizures in people with drug‐resistant focal epilepsy. Adding tiagabine to people's usual treatment was, however, associated with an increase in side effects, such as dizziness and tremor. People using tiagabine were over four times more likely to experience tremor than those using placebo; however, only one trial reported this adverse event so the evidence for this is limited. People taking tiagabine in addition to other drugs were nearly twice as likely to withdraw from treatment than those taking placebo. We found no significant differences between tiagabine and topiramate, another antiepileptic drug, as add‐on drugs.
Conclusions
Overall, there was high‐certainty evidence for the outcome of seizure reduction, which means that we are confident that the effect we have reported is accurate. The trials included in this review did not examine the long‐term effects of tiagabine as an add‐on treatment or the effects of tiagabine in children aged below the age of 12 years. Future research is needed to determine how tiagabine performs in comparison with other newer antiepileptic drugs.
The evidence is current to January 2019.
Summary of findings
Summary of findings for the main comparison. Tiagabine compared to placebo control for drug‐resistant focal epilepsy.
| Tiagabine compared to placebo control for drug‐resistant focal epilepsy | ||||||
| Patient or population: People with drug‐resistant focal epilepsy Setting: Hospital outpatient setting Intervention: Tiagabine Comparison: Placebo control | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with placebo control | Risk with Tiagabine | |||||
|
50% or greater reduction in seizure frequency (Intention‐to‐treat analysis) Follow‐up range: 12 to 22 weeks |
Study population | RR 3.16 (1.97 to 5.07) | 769 (3 RCTs) | ⊕⊕⊕⊕ HIGH1 4 | Tiagabine increases the number of participants achieving a 50% or greater reduction in seizure frequency | |
| 69 per 1,000 | 218 per 1,000 (136 to 350) | |||||
|
Treatment withdrawal Follow‐up range: 12 to 22 weeks |
Study population | RR 1.81 (1.25 to 2.62) | 769 (3 RCTs) | ⊕⊕⊕⊝ MODERATE1 | Tiagabine likely increases treatment withdrawal | |
| 113 per 1,000 | 204 per 1,000 (141 to 295) | |||||
|
Dizziness Follow‐up range: 12 to 22 weeks |
Study population | RR 1.69 (1.13 to 2.51) | 769 (3 RCTs) | ⊕⊕⊕⊝ MODERATE1 | Tiagabine likely increases the number of participants who experience dizziness | |
| 160 per 1,000 | 270 per 1,000 (181 to 402) | |||||
|
Fatigue Follow‐up range: 12 to 22 weeks |
Study population | RR 1.38 (0.89 to 2.14) | 769 (3 RCTs) | ⊕⊕⊕⊝ MODERATE1 | Tiagabine probably slightly increases the number of participants who experience dizziness | |
| 149 per 1,000 | 206 per 1,000 (133 to 319) | |||||
|
Nausea Follow‐up range: 12 to 22 weeks |
Study population | RR 1.24 (0.69 to 2.22) | 769 (3 RCTs) | ⊕⊕⊝⊝ LOW2 | Tiagabine may slightly increase the number of participants who experience nausea but we are uncertain | |
| 95 per 1,000 | 117 per 1,000 (65 to 210) | |||||
|
Somnolence Follow‐up range: 12 to 22 weeks |
Study population | RR 1.18 (0.76 to 1.83) | 769 (3 RCTs) | ⊕⊕⊕⊝ MODERATE1 | Tiagabine likely slightly increases the number of participants who experience somnolence | |
| 156 per 1,000 | 185 per 1,000 (119 to 286) | |||||
|
Tremor Follow‐up range: 12 to 22 weeks |
Study population | RR 4.56 (1.00 to 20.94) | 297 (1 RCT) | ⊕⊕⊝⊝ LOW2 3 4 | Tiagabine may cause a large increase the number of participants who experience nausea but we are uncertain | |
| 33 per 1,000 | 150 per 1,000 (33 to 690) | |||||
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI) CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: We are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: We are moderately confident in the effect estimate: The true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: Our confidence in the effect estimate is limited: The true effect may be substantially different from the estimate of the effect Very low certainty: We have very little confidence in the effect estimate: The true effect is likely to be substantially different from the estimate of effect | ||||||
1 Downgraded once for imprecision ‐ number of events was insufficient and did not satisfy the optimal information size.
2 Downgraded twice for imprecision ‐ very limited number of events was insufficient and did not satisfy the optimal information size.
3 Downgraded once for publication bias ‐ data were published by only one study.
4 Upgraded once for large effect ‐ RR > 2.00.
Background
Description of the condition
Epilepsy is a common neurological condition, with an estimated incidence of 50 per 100,000 people and a prevalence of five to 10 per 1000 in the developed world (Sander 1996). It has previously been estimated that between 2% and 3% of the population will be given a diagnosis of epilepsy at some time in their lives (Hauser 1993). A more recent systematic review has, however, highlighted that the lifetime prevalence of epilepsy is lower, with 7.6 people per 1000 estimated to be affected (95% confidence interval (CI) 6.17–9.38) (Fiest 2017).
Most people who receive a diagnosis of epilepsy will go into remission; however, up to 30% will continue to have seizures (i.e. become drug‐resistant), despite treatment with adequate doses of an antiepileptic drug (AED) as monotherapy or in combination with other AEDs (Cockerell 1995). Individuals with drug‐resistant epilepsy most commonly have focal‐onset seizures, which are divided into three types: simple focal, complex focal, and secondary generalised tonic‐clonic seizures (Commission 1989). Focal seizures originate from one area of the brain and are restricted to one hemisphere. During secondary generalised tonic‐clonic seizures, however, the initial focal‐onset seizure evolves such that the entire brain is then affected, incorporating both hemispheres. For the purposes of this review, people will be considered drug‐resistant if they have failed to respond to a minimum of two AEDs given as monotherapy or in combination, according to the definition of drug‐resistant epilepsy recommended by the International League Against Epilepsy (ILAE 2010).
Description of the intervention
Over the past 10 to 15 years, there has been renewed interest in the development of new AEDs as standard drugs (e.g. carbamazepine, phenytoin, valproate) do not leave all people with epilepsy seizure‐free, and are associated with adverse effects. New AEDs are initially tested in randomised controlled trials (RCTs) as an add‐on treatment for people with drug‐resistant focal epilepsy. Because they have demonstrated a therapeutic effect in these trials, new AEDs tend to be licensed for add‐on use before monotherapy trials have been undertaken in which new AEDs are compared with standard AEDs.
Tiagabine is a relatively new AED and was only approved by the US Food and Drug Administration (FDA) for use as an add‐on therapy for adults and children, over the age of 12 years, with focal‐onset seizures in September 1997 (Food and Drug Administration 1997). Pharmacokinetic studies in healthy volunteers have demonstrated that tiagabine is associated with good bioavailability after oral consumption (Gustavson 1995). Specifically, tiagabine displays linear pharmacokinetics with the maximum serum concentration (over 95% of the drug) being achieved within two hours after dosing (Gustavson 1995).
How the intervention might work
Tiagabine is derived from the gamma‐aminobutyric acid (GABA) uptake inhibitor, nipecotic acid. It exerts its antiepileptic effect by inhibiting presynaptic and glial uptake of GABA, the main inhibitory neurotransmitter of the brain (Ostergaard 1995; Morimoto 1997). This action increases the availability of extracellular GABA in the brain, thereby increasing inhibition of neuronal electrical activity. Tiagabine thus produces an overall reduction in cortical excitability (Fink‐Jensen 1992).
Why it is important to do this review
This Cochrane Review is part of a series of reviews conducted to investigate the newer AEDs. Early reviews of tiagabine reported its effects on seizure frequency and adverse effects (Marson 1996; Marson 1997). These reviews investigated the most common adverse effects associated with tiagabine; however, they did not assess any potential cognitive effects that tiagabine might have. Specifically, there is concern that AEDs may impair cognitive abilities (Eddy 2011). As a consequence, we have included outcomes that assess cognitive effects and we have also chosen to include quality‐of‐life outcomes, so as to assess the global impact of this drug on well‐being. In this review, we assessed the effects of tiagabine on seizures, adverse effects, cognition, and quality of life, when used as add‐on treatment for people with drug‐resistant focal seizures.
Objectives
To evaluate the efficacy and tolerability of tiagabine when used as an add‐on treatment for people with drug‐resistant focal seizures.
Methods
Criteria for considering studies for this review
Types of studies
RCTs.
Double‐blind, single‐blind, or unblinded trials.
Placebo‐controlled or active drug control group.
Parallel‐group or cross‐over studies.
Minimum treatment period of eight weeks.
Types of participants
People of any age with drug‐resistant localisation‐related (focal‐onset) seizures (i.e. experiencing simple focal, complex focal, or secondary generalised tonic‐clonic seizures).
Types of interventions
The active treatment group received treatment with tiagabine, in addition to conventional AED treatment; the control group received matched placebo or a different add‐on AED, in addition to conventional AED treatment.
Types of outcome measures
Primary outcomes
50% or greater reduction in seizure frequency
We chose the proportion of people with a 50% or greater reduction in seizure frequency during the treatment period compared with the pre‐randomisation baseline period as the primary outcome. We chose this outcome as it is commonly reported in this type of study, and can be calculated for studies that do not report it directly, provided that baseline seizure data have been reported.
Secondary outcomes
Treatment withdrawal
We used the proportion of people who had treatment withdrawn during the course of the treatment period as a measure of global effectiveness. Treatment is likely to be withdrawn because of adverse effects, lack of efficacy, or a combination of the two, and this is an outcome to which individuals make a direct contribution. In trials of short duration, it is likely that adverse effects will be the most common reason for withdrawal.
Adverse effects
1. The proportion of people experiencing any of the following five adverse effects, considered by the review authors to be common and important adverse effects of AEDs. (a) Ataxia. (b) Dizziness. (c) Fatigue. (d) Nausea. (e) Somnolence.
2. The proportion of people experiencing the five most common adverse effects if different from point 1 above.
Cognitive effects
To date, no consensus has been reached on which instruments should be used to assess the effects of AEDs on cognition. As a result, the assessment of cognitive effects has been approached in a heterogeneous way (Cochrane 1998). In view of this difficulty, we tabulated results, but made no attempt to combine results in a meta‐analysis.
Quality of life
Once again, no consensus has been reached on which instruments should be used to assess quality of life (Baker 2000); we therefore tabulated results, but made no attempt to combine results in a meta‐analysis.
Search methods for identification of studies
Electronic searches
This review is an update of a Cochrane Review first published in 1999. We ran searches for the original review in 1999, and ran subsequent searches in June 2003, March 2005, January 2008, June 2010, December 2011, and November 2013. For the latest update, we searched the following databases on 22 January 2019, with no language restrictions.
The Cochrane Register of Studies (CRS Web; includes the Cochrane Epilepsy Group's Specialized Register and the Cochrane Central Register of Controlled Trials CENTRAL), using the search strategy outlined in Appendix 1.
MEDLINE (Ovid, 1946 to January 21, 2019), using the search strategy outlined in Appendix 2.
ClinicalTrials.gov, using the search strategy outlined in Appendix 3.
WHO International Clinical Trials Registry Platform (ICTRP), using the search terms: tiagabine AND epilepsy.
For the search conducted 4 August 2016, we searched the following databases, with no language restrictions.
The Cochrane Epilepsy Group Specialized Register (4 August 2016), using the search strategy outlined in Appendix 4.
The Cochrane Central Register of Controlled Trials (CENTRAL) via the Cochrane Register of Studies Online (CRSO; 4 August 2016), using the search strategy outlined in Appendix 5.
MEDLINE Ovid (1946 to 4 August 2016), using the search strategy outlined in Appendix 6.
ClinicalTrials.gov (4 August 2016), using the search terms: Tiagabine AND epilepsy.
WHO International Clinical Trials Registry Platform (ICTRP; 4 August 2016), using the search terms: Tiagabine AND epilepsy.
Searching other resources
We reviewed the reference lists of retrieved studies to search for additional reports of relevant studies.
We contacted Sanofi‐Synthelabo (makers of tiagabine) and experts in the field to ask for information about unpublished or ongoing studies.
Data collection and analysis
Selection of studies
In the previous versions of the review, two review authors (RB and KMM) had independently extracted data and assessed risk of bias for the studies included (Pulman 2012; Pulman 2014). For the current review update, two new review authors (RB and KMM) independently assessed the eligibility of studies for inclusion from the most recent searches. We resolved disagreements by mutual discussion.
Data extraction and management
We extracted the following information for each trial, using a data extraction form.
Trial design
Methods of sequence generation and randomisation concealment.
Method of blinding.
Whether any people had been excluded from reported analyses.
Duration of baseline period.
Duration of treatment period.
Dose(s) of tiagabine tested.
Participant demographic information
Total number of people allocated to each treatment group.
Age and sex of participants.
Number with focal and generalised seizures.
Seizure frequency during baseline period.
Number of background drugs.
We also contacted study sponsors to confirm the following information, if it was not available in the published text:
Method of randomisation.
Total number randomly assigned to each group.
Number of people in each group achieving a 50% or greater reduction in seizure frequency per treatment group.
Number of people having treatment withdrawn post‐randomisation per treatment group.
-
For those excluded:
the reason for exclusion;
whether any of those excluded completed the treatment phase;
whether any of those excluded had a 50% or greater reduction in seizure frequency during the treatment phase.
Outcomes
We recorded the number of people who experienced each outcome according to randomly assigned group.
Assessment of risk of bias in included studies
Two review authors (RB and KMM) independently assessed the risk of bias for each trial, using the Cochrane 'Risk of bias' tool, as described in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011). We discussed and resolved disagreements. We rated included studies as adequate, inadequate, or unclear on six domains applicable to RCTs: randomisation method, allocation concealment, blinding methods, incomplete outcome data, selective outcome reporting, and other sources of bias.
Measures of treatment effect
We presented the primary outcome of seizure reduction as a risk ratio (RR). We also presented the secondary outcomes, including seizure freedom, treatment withdrawal, and adverse effects, as RRs.
For continuous outcome data (i.e. cognition and quality of life), we expected that different measures may have been used for these outcomes. If we had found this to be the case, we intended to use the standardised mean difference to present these data if this was deemed appropriate, and if the data were available.
Unit of analysis issues
The inclusion of cross‐over studies in meta‐analyses introduces unit of analysis issues. We had intended to include data from the first treatment period of each study in the meta‐analysis, essentially regarding the first treatment period as a parallel study, and thus, avoiding any issues of carry‐over effect. Two cross‐over studies were included in this review; however, both trial publications contained insufficient information to allow our planned analyses.
Dealing with missing data
We sought missing data from the study authors. We carried out intention‐to‐treat (ITT), best‐case and worst‐case analyses on the primary outcome to account for missing data. We presented all analyses in the main report.
Assessment of heterogeneity
We assessed clinical heterogeneity by comparing the distribution of important individual participant factors among trials (e.g. age, seizure type, duration of epilepsy, number of AEDs taken at the time of randomisation) and trial factors (e.g. randomisation concealment, blinding, losses to follow‐up). We examined statistical heterogeneity with a Chi² test and the I² statistic for heterogeneity; when we found no significant heterogeneity (P > 0.10), we used a fixed‐effect model. Had we found significant heterogeneity, we intended to use a random‐effects model analysis, using the inverse variance method.
Assessment of reporting biases
We requested all protocols from study authors to enable a comparison of outcomes of interest. We investigated outcome reporting bias with the ORBIT matrix system (Kirkham 2010). We had planned to create and examine funnel plots; however, we could not undertake this because of the small number of trials included.
Data synthesis
We used a fixed‐effect model meta‐analysis to synthesise the data. Our preferred estimator was the Mantel‐Haenszel RR. For the outcomes of 50% or greater reduction in seizure frequency and treatment withdrawal, we used 95% CIs. For individual adverse effects, we used 99% CIs to make an allowance for multiple testing.
We had expected to carry out the following analyses.
Intervention group versus controls on seizure reduction.
Intervention group versus controls on treatment withdrawal.
Intervention group versus controls on adverse effects.
Intervention group versus controls on cognitive effects.
Intervention group versus controls on quality of life.
We had planned to stratify each analysis by type of control group (i.e. placebo or active control) and by study characteristics to ensure the appropriate combination of study data. However, because of the limited number of studies eligible for inclusion, the analyses could not be executed as planned. Instead, we conducted two separate comparisons, 'tiagabine versus placebo control' and 'tiagabine versus topiramate', due to the availability of data, and then addressed each outcome separately for the two comparisons.
Our analyses included all participants in the treatment group to which they had been allocated. For the efficacy outcome (50% or greater reduction in seizure frequency), we undertook three analyses, two of which were sensitivity analyses:
-
Primary (ITT) analysis: participants not completing follow‐up or with inadequate seizure data were assumed to be non‐responders. Analysis by ITT was reported by all of the included studies.
Worst‐case analysis (sensitivity analysis): participants not completing follow‐up or with inadequate seizure data were assumed to be non‐responders in the intervention group, and responders in the placebo group.
Best‐case analysis (sensitivity analysis): participants not completing follow‐up or with inadequate seizure data were assumed to be responders in the intervention group, and non‐responders in the placebo group.
The purpose of the best‐case and worst‐case analyses is to test the whether the assumption made during ITT analysis, that all participants not completing follow‐up or with inadequate seizure data are non‐responders, affects the estimated effect size.
Dose regression analysis
We examined the dose‐response relationship using logistic regression in the framework of generalised linear models (McCullagh 1989), and Generalized Linear Interactive Modelling (GLIM) statistical software (Smith 2015). For this model, we defined a binary variable with a value of 0 if the response was less than 50%, and a value of 1 if it was 50% or higher. Models included an indicator variable for trials. To examine the effect of dose, we considered the following as possible explanatory variables: dose levels, dose as a continuous variable, and logarithmic transformation of dose. We also considered interactions between dose and trials. We calculated odds ratios (ORs) and response rates.
Cognitive and quality of life data
As stated under Secondary outcomes, we tabulated data for these outcomes, but made no attempt to undertake a meta‐analysis. We found that trials had not used similar outcome measures, so we did not undertake a meta‐analysis, as it was deemed inappropriate to do so.
Subgroup analysis and investigation of heterogeneity
We did not plan to undertake any subgroup analyses. Instead, we had intended to investigate heterogeneity using sensitivity analysis, if it had been deemed appropriate.
Sensitivity analysis
We had intended to carry out sensitivity analysis if we detected irregularities between study quality, characteristics of participants, interventions, and outcomes. Upon examining the data collected, sensitivity analyses were not deemed necessary.
Summarising and interpreting results
We used the GRADE approach, as outlined in the GRADE Handbook (Schünemann 2013), to assess the certainty of the evidence and to interpret findings. We created 'Summary of findings' tables using GRADEpro GDT software (which imports data from Review Manager 5 software (GRADEpro GDT 2015)) for the outcomes 50% reduction in seizure frequency and treatment withdrawal, and for the following adverse effects: dizziness, fatigue, nausea, somnolence, and tremor.
Results
Description of studies
Results of the search
The two most recent searches, conducted August 2016 and January 2019, identified 29 records from the databases outlined in Electronic searches. We removed six duplicates, leaving 23 records, which we screened for inclusion in the review. All 23 records were excluded at this point because of irrelevance; no new studies were included in this review update. We included a total of six studies in this review, which had been identified during previous searches, three of which were included in meta‐analyses. See Figure 1 for a diagram of study flow for the two most recent searches.
1.
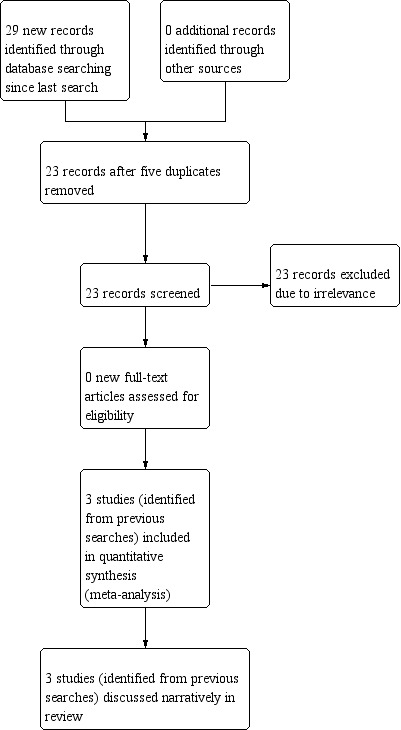
Study flow diagram for searches conducted August 2016 and January 2019
Included studies
We found six studies that were eligible for inclusion in this review. All trials were initially sponsored by Novo Nordisk as part of a pre‐licensing programme; however, the drug patent is now owned by Sanofi‐Synthelabo. Three of the four parallel‐group studies identified, plus the two cross‐over studies, specifically compared tiagabine with placebo for people with drug‐resistant focal seizures (Crawford 2001; Kalviainen 1998; Richens 1995; Sachdeo 1997; Uthman 1998). The remaining parallel‐group study (Fritz 2005) compared tiagabine with an alternative AED, topiramate, in people with drug‐resistant focal seizures.
The trials largely shared the same inclusion and exclusion criteria. People were excluded from these studies if they had a history of non‐epileptic attacks; any active progressive disease of the central nervous system (e.g. brain tumour); any significant illness within the previous three months; any medical or neurological disorder requiring frequent changes in medication or dosage; abnormal laboratory findings that were not attributable to their concomitant AEDs; a history of drug abuse or addiction (including alcohol); or poor compliance with past medication or medical advice. Women who were pregnant or at risk of pregnancy and those who were lactating were also excluded. Valproate monotherapy was not allowed in two studies (Sachdeo 1997; Uthman 1998).
One parallel trial had a 12‐week pre‐randomisation baseline period. People who had eight or more focal seizures during this period were eligible to be randomly assigned (Kalviainen 1998). The treatment period lasted 22 weeks, and 154 individuals were randomly assigned ‐ 77 to placebo and 77 to tiagabine 30 mg per day. Data on cognitive and quality‐of‐life effects on a subset of 43 people were reported separately (Kalviainen 1996).
Another parallel trial had an eight‐week screening period, a titration phase of three months and a three‐month maintenance phase. Twenty‐one participants were randomly assigned to tiagabine (final dose at least 20 mg per day) and 20 were randomly assigned to topiramate treatment (final dose at least 200 mg per day). This study reported seizure outcome, cognitive outcome, and quality‐of‐life effects (Fritz 2005). Notably, the study did not include a placebo control arm and, consequently, could not contribute data to the main comparison (tiagabine versus placebo). Instead, the data from the study was incorporated into a separate comparison, tiagabine versus topiramate.
Sachdeo 1997, a parallel‐group trial, included an eight‐week pre‐randomisation baseline period, and people who had eight or more seizures during this period were eligible to be randomly assigned. The treatment period lasted 12 weeks, and 318 individuals were randomly assigned ‐ 107 to placebo and 211 to tiagabine 32 mg per day. Of those randomly assigned to tiagabine, 106 were allocated to receive 16 mg twice a day and 105 were allocated to receive 8 mg four times a day. No effects on cognition or quality of life were reported for this study.
Another parallel trial had a 12‐week pre‐randomisation baseline period. People who had eight or more focal seizures during this period were eligible to be randomly assigned. The treatment period lasted 20 weeks, and 297 individuals were randomly assigned ‐ 91 to placebo, 61 to 16 mg, 88 to 32 mg and 57 to 56 mg tiagabine per day (Uthman 1998). Data on cognitive and quality‐of‐life effects were reported separately, for a subset of 162 individuals (Dodrill 1997).
The cross‐over trials were similar in design, and used what has been called a response‐conditional design (Crawford 2001; Richens 1995). People who had six or more seizures in the eight weeks before the study were entered into a 12‐week screening phase. During the screening phase, all participants were given tiagabine, the dose of which was titrated up with a target dose of 52 mg per day in Richens 1995, and 64 mg per day in Crawford 2001. Participants with a reduction in seizure frequency of 25% or more during the screening phase were eligible to be randomly assigned (hence, response was conditional). Eligible individuals were randomly assigned in a two‐by‐two cross‐over trial, to a sequence of placebo‐tiagabine or tiagabine‐placebo. Treatment periods lasted seven weeks and the cross‐over periods three weeks. Richens 1995 screened 94 individuals, 46 of whom were randomly assigned; Crawford 2001 screened 88 individuals, 44 of whom were randomly assigned. The reports for these two studies contained insufficient information to allow our planned analyses using data from the first treatment period. For the cohort recruited in Richens 1995, the cognitive effects of tiagabine were reported separately, for a subset of 22 individuals (Sveinbjornsdottir 1994).
In total, the studies assessing seizure outcomes and treatment withdrawal randomly assigned 948 individuals. Studies assessing adverse effects randomly assigned 859 participants. Data on cognitive effects were available for 251 individuals, and data on quality of life were available for 229 individuals. Further details are provided in the Characteristics of included studies,
Excluded studies
Four studies were previously excluded (Arroyo 2005; Bauer 1995; Gustavson 1997; Uldall 1995). See Characteristics of excluded studies tables for reasons for exclusion.
Risk of bias in included studies
Overall, two studies were judged to be at low risk of bias (Kalviainen 1998; Sachdeo 1997), three studies at unclear risk of bias (Crawford 2001; Richens 1995; Uthman 1998), and one study at high risk of bias (Fritz 2005). For the outcome of 50% reduction in seizure frequency, the three trials that contributed to the meta‐analysis were judged to be at low risk of bias, overall. The reasons for the 'Risk of bias' judgements for each domain for each study are summarised in the 'Risk of bias' tables within the Characteristics of included studies tables. See Figure 2 and Figure 3 for a visual representation of the risk of bias in all included studies.
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
3.
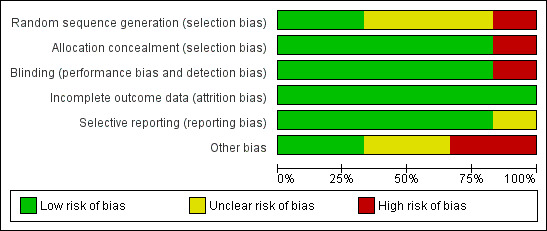
Risk of bias graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
Allocation
All included studies randomly assigned participants to treatment arms. Of the six trials included, only two used computer‐generated sequences to allocate participants (Fritz 2005; Kalviainen 1998). The other studies did not provide details on how their sequence allocation was generated. All but Fritz 2005 used sequentially allocated sealed packages to conceal group allocation. Fritz 2005 used no methods of concealment. Overall for sequence generation, we rated risk of bias as unclear for three studies (Crawford 2001; Richens 1995; Uthman 1998), low for two studies (Kalviainen 1998; Sachdeo 1997), and high for one study (Fritz 2005). For allocation concealment, we rated risk of bias to be low for five studies and high for one study (Fritz 2005).
Blinding
All studies reported that they were double‐blinded, except for Fritz 2005, which was an open‐label study, and thus used no blinding techniques. In the five other included studies (Crawford 2001; Kalviainen 1998; Richens 1995; Sachdeo 1997; Uthman 1998), identical packaging and medication were used to maintain blinding. No specific details regarding who was blinded were provided in the papers (i.e. participants, study personnel, or outcome assessors). Overall, for blinding, we rated risk of bias to be low for five studies and high for one study.
Incomplete outcome data
All studies reported attrition rates and reasons for dropout. An ITT analysis was used by all studies to deal with missing data. Overall, for incomplete outcome data, we rated risk of bias to be low for all six studies.
Selective reporting
Most of the studies detailed outcomes within the methods of the paper and reported the data; however, no protocols were available to allow detailed assessment of this. Fritz 2005 selected a subset of randomly assigned participants, and reported baseline measures only for this subset. Overall, for selective reporting, we rated risk of bias to be low for five studies and unclear for one study.
Other potential sources of bias
When assessing the trials for other sources of bias, we were unable to detect any other potential sources of bias for two of the included trials (Kalviainen 1998; Sachdeo 1997). We hence awarded these two trials a low 'Risk of bias' judgement. We judged that the remaining four trials were at risk of other potential sources of bias.
The two cross‐over trials (Crawford 2001; Richens 1995) shared a response‐conditional study design that required participants to attain a 25% or greater reduction in seizure frequency during the screening phase so as to be eligible for randomisation into the treatment phase of the studies. As a result, both trials preselected participants who were expected to have a clinical response to tiagabine, thus generating bias. Notably, trials which use this specific type of study design likely overestimate the therapeutic effectiveness of a given treatment and are assumed to generate biased findings. We thus assessed both trials to be at a high risk of other potential sources of bias.
We further judged that the trial by Fritz 2005 was at unclear risk of other potential sources of bias. There was incomplete reporting of baseline demographic data for participants. Specifically, baseline data were not provided for participants who discontinued from the study. It was, therefore, not possible to determine whether there were any baseline imbalances in the trial.
Similarly, we assessed that the trial by Uthman 1998 was at unclear risk of other potential sources of bias. This arose from the unequal randomisation ratio (3:2:3:2 to placebo, 16 mg, 32 mg, and 56 mg tiagabine, respectively) used in the study design, and the lack of reasoning provided to justify its use. Generally, unequal randomisation ratios tend to favour allocation to intervention. Such randomisation ratios are associated with a greater placebo effect as more participants infer that they have been allocated to the active treatment (Hey 2014). In contrast, in this study, the randomisation ratio favoured allocation to placebo and to tiagabine 32 mg/day. This could, however, have similarly influenced participants' perception of their treatment allocation and could thus have biased their responsiveness. Due to the unclear impact that this might have on the trial data, we awarded the trial an unclear risk of bias judgement for this domain.
Effects of interventions
See: Table 1
As already outlined in the description of studies, we found four parallel‐group and two cross‐over studies. The cross‐over studies used a response‐conditional design, in which participants were given tiagabine during a screening period, and those with a 25% or greater reduction in seizure frequency were eligible to be randomly assigned. The people randomly assigned were, therefore, highly selected and were biased towards a response to tiagabine. We chose to report the effects on seizure frequency from the parallel‐group and cross‐over trials separately. In addition, reports of the cross‐over studies did not provide detailed adverse effect data or data on treatment withdrawal, and the parallel‐group studies only contributed results for these outcomes.
Of the parallel‐group studies, three were placebo‐controlled. Data extracted from these studies thus contributed to the main meta‐analysis comparison, 'tiagabine versus placebo control'. We assessed the active‐controlled parallel study that examined tiagabine and topiramate (Fritz 2005), separately, in a second meta‐analysis comparison, 'tiagabine versus topiramate'.
Tiagabine versus placebo control
50% or greater reduction in seizure frequency: parallel trials
A Chi² test for heterogeneity for the response to tiagabine indicated no significant heterogeneity between trials (ITT analysis: Chi² = 0.81, degrees of freedom (df) = 2, P = 0.67; worst‐case analysis: Chi² = 2.64, df = 2, P = 0.27; best‐case analysis: Chi² = 0.97, df = 2, P = 0.61). The overall RR for a response to tiagabine was 3.16 (95% CI 1.97 to 5.07), indicating that participants were significantly more likely to respond to tiagabine than to placebo. The RRs for worst‐case and best‐case analyses were 1.19 (95% CI 0.88 to 1.62) and RR 6.14 (95% CI 3.92 to 9.63), respectively (Analysis 1.1; Figure 4).
1.1. Analysis.
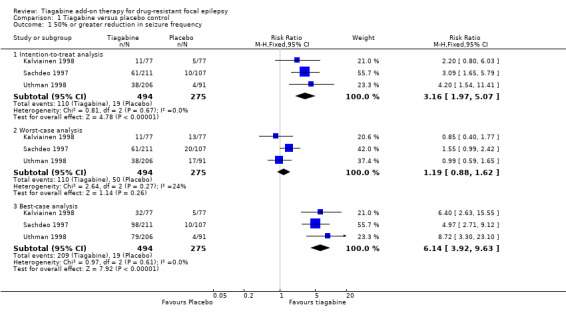
Comparison 1 Tiagabine versus placebo control, Outcome 1 50% or greater reduction in seizure frequency.
4.
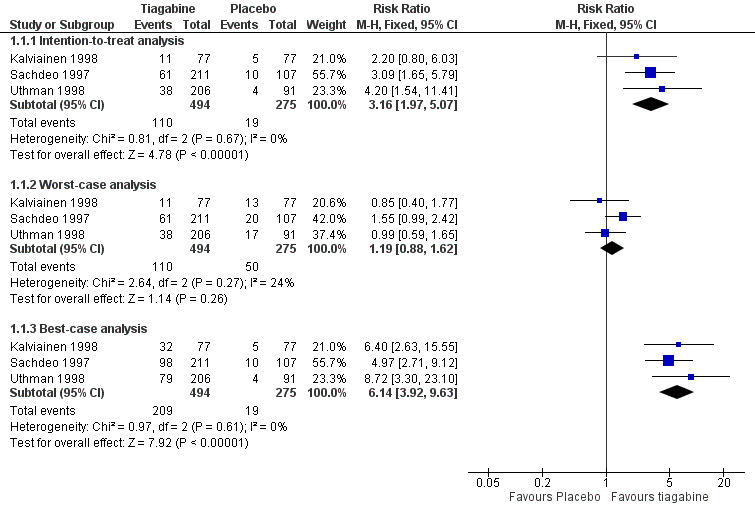
Forest plot of comparison: 1 Tiagabine versus placebo control—50% or greater reduction in seizure frequency, outcome: 1.1 50% or greater reduction in seizure frequency.
Dose regression analysis
Due to differences in response rates in individual trials, it was not possible to give valid estimates of precise responses to individual doses. Instead, the model with the best summary of the log odds compared Kalviainen 1998 and Uthman 1998 with Sachdeo 1997. After adjustments for trial effects, this model included two contrasting dose groups: one group comparing placebo and tiagabine 16 mg per day, and the other group comparing tiagabine 30 mg, 32 mg, and 56 mg per day. The reduction in deviance due to dose at these two levels, with adjustment for trial effects, was 40.6 on 1 df (P = 0.001). Upon contrast of trials included, the residual deviance was 4.8 on 5 df.
The estimated OR relative to placebo was 3.67 (95% CI 2.30 to 5.86). The OR for Uthman 1998 and Kalviainen 1998 versus Sachdeo 1997 was 1.65 (95% CI 1.11 to 2.44).
Actual and estimated response rates are given in Table 2. Although we were unable to give precise overall estimates for the proportion of participants responding to individual doses, evidence suggests an effect for daily doses of tiagabine 30 mg to 56 mg or more. As an indication of the possible effects we can say that with placebo rates in the range of 6% to 10%, at least an additional 13%, and possibly 20%, of people similar to those in these trials, would experience a 50% or greater reduction in seizure frequency when taking a dose of tiagabine 30 mg or more per day.
1. Percentage of 50% responders (95% CI), intention‐to‐treat regression analysis.
| Trial | Placebo, proportion | 16 mg/d | 30 to 32 mg/d | 56 mg/d |
| Uthman 1998actual | 4.4 | 8.2 | 19.3 | 18.1 |
| Kalviainen 1998actual | 6.5 | 14.3 | ||
| Uthman 1998;Kalviainen 1998fitted | 6.3 (4.1 to 9.4) | 6.3 (4.1 to 9.4) | 19.7 (15.2 to 25.0) | 19.7 (15.2 to 25.0) |
| Sachdeo 1997actual | 10.2 | 28.6 | ||
| Sachdeo 1997fitted | 9.8 (6.4 to 14.9) | 28.7 (23.3 to 34.9) |
50% or greater reduction in seizure frequency: cross‐over trials
Reports of the cross‐over trials contained insufficient data from the first treatment period to allow the analyses that were planned. We therefore summarised the reported results from the two cross‐over trials below.
Of the 46 people randomly assigned in Richens 1995, 11 (24%) had a 50% or greater reduction in seizure frequency of complex focal seizures in the tiagabine phase versus the placebo phase. Of the 44 people randomly assigned in Crawford 2001, 12 (27%) had a 50% reduction in seizure frequency in the tiagabine versus the placebo phase. The proportion of responders was higher than that seen in the parallel‐group trials; this is not surprising, given that people in the cross‐over trials had to have an apparent response to tiagabine before they were randomly assigned.
Treatment withdrawal: parallel trials
Treatment withdrawal data were available only for the parallel‐group trials. A Chi² test for heterogeneity suggested no significant statistical heterogeneity (Chi² = 1.75, df = 2, P = 0.42). The overall RR for discontinuation for any reason was 1.81 (95% CI 1.25 to 2.62), indicating that people were significantly more likely to withdraw from tiagabine than from placebo (Analysis 1.2).
1.2. Analysis.
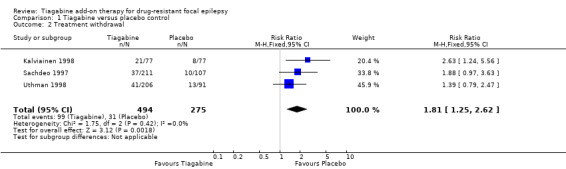
Comparison 1 Tiagabine versus placebo control, Outcome 2 Treatment withdrawal.
Adverse effects: parallel‐group trials
For the parallel‐group trials, in addition to the five adverse effects specified in the protocol for this review, headache, infection, nervousness, and tremor were among the five most commonly reported adverse effects. A Chi² test for heterogeneity of adverse effects indicated no significant heterogeneity between trials. The following adverse effects were significantly associated with tiagabine: dizziness (RR 1.69, 99% CI 1.13 to 2.51) and tremor (4.56, 99% CI 1.00 to 20.94). In contrast, the following were not significantly associated with tiagabine: fatigue (RR 1.38, 99% CI 0.89 to 2.14), nervousness (10.65, 99% CI 0.78 to 146.08), ataxia (1.24, 99% CI 0.34 to 4.55), nausea (1.24, 99% CI 0.69 to 2.22), somnolence (1.18, 99% CI 0.76 to 1.83), headache (1.15, 99% CI 0.48 to 2.79), and infection (1.00, 99% CI 0.36 to 2.76) (Analysis 1.3).
1.3. Analysis.

Comparison 1 Tiagabine versus placebo control, Outcome 3 Adverse effects.
Adverse effects: cross‐over trials
For the cross‐over trials, Crawford 2001 reported that eight people experienced adverse effects while taking tiagabine and 10 people reported adverse effects while taking placebo. While taking tiagabine, two people reported dizziness and another two reported a lack of coordination. While taking placebo, three people reported accidental injury. All other adverse effects were reported by one individual only. Richens 1995 did not report a detailed breakdown of adverse effects occurring with tiagabine or placebo.
Cognitive effects: parallel‐group and cross‐over trials
Reviewing the cognitive effects of tiagabine is complicated by the lack of a uniform of approach to the assessment of such effects (Dodrill 1997; Kalviainen 1996; Sveinbjornsdottir 1994). In total, the included studies used a total of 24 neuropsychological tests, only two of which ‐ the Stroop test and the Rey auditory verbal learning test ‐ were used in all three studies that reported cognitive effects. Three tests were used by two of the three studies, and the remaining 19 tests were used in just one study. Even when the same test had been used in two or more studies, it was not always clear that the same aspects of the test had been used. The tests used in the individual studies are outlined in Table 3.
2. Neuropsychological tests used.
| Study | Test |
| Dodrill 1997 | Stroop test |
| Rey auditory verbal learning test | |
| Controlled oral word association test | |
| Lafayette grooved pegboard | |
| Benton visual retention test | |
| Symbol digit modalities test | |
| Wonderlic personnel test | |
| Digit cancellation | |
| Fritz 2005 | Edinburgh inventory |
| Mehrfachwahl‐Wortschatz‐Intelligenztest (MWT‐B) | |
| Kurztest zur cerebralen Insuffizienz (c.I. test) | |
| Trailmaking test | |
| Weiner test system | |
| HAWIE‐R (verbal memory) | |
| Corsi block test | |
| Verbal learning and memory test (VLMT) | |
| Diagnosticum fur cerebralschaden (DSC‐R) (visual memory) | |
| Boston naming test | |
| Word fluency test (LPS) | |
| Token test | |
| Kalviainen 1996 | Stroop test |
| Rey auditory verbal learning test | |
| Controlled oral word association test | |
| Modified finger tapping test | |
| Binary choice reaction | |
| Full‐scale I.Q. (WAIS) | |
| Logical prose, story A from the Wechsler memory scale | |
| Forward digit span | |
| Corsi block span | |
| Alternating S‐task | |
| Letter cancellation task | |
| The WMS visual reproduction subtest | |
| Auditory and visual reaction times | |
| Sveinbjornsdottir 1994 | Stroop test |
| Rey auditory verbal learning test | |
| Binary choice reaction | |
| Modified finger tapping | |
| Semantic processing | |
| Information processing speed | |
| Bimanual hand movements | |
| Simple reaction time | |
| Tapping rate | |
| Verbal memory |
No statistically significant differences were noted at the 0.01 level of confidence for any test, and statistically significant differences at the 0.05 level were seen for only one test (Benton visual retention test, form F) (Table 4). It is worth noting that the results from another two tests were close to reaching statistical significance but did not (P = 0.051). Consequently, the evidence was insufficient to conclude that tiagabine as an add‐on treatment had an effect on cognition compared with placebo.
3. Neuropsychological tests with statistically significant differences.
| Test | Study | Result (P) | Treatment favoured |
| Benton visual retention test, form F | Dodrill 1997 | 0.049 | Placebo |
| Benton visual retention test, form G | Dodrill 1997 | 0.051 | Placebo |
| Symbol digit modalities test | Dodrill 1997 | 0.051 | Placebo |
Quality‐of‐life measures
Two reports addressed quality‐of‐life outcomes (Dodrill 1997; Kalviainen 1996). Both reported effects on mood and adjustment, but used different tests to assess them (Table 5). Neither study found a significant difference between tiagabine and placebo; hence, evidence was insufficient to conclude that tiagabine had an effect on quality of life compared with placebo.
4. Tests of mood and adjustment.
| Study | Test | Domain |
| Dodrill 1997 | Profile of Mood States (POMS) | Tension‐anxiety |
| Depression‐dejection | ||
| Anger‐hostility | ||
| Vigour‐activity | ||
| Fatigue‐inertia | ||
| Confusion‐bewilderment | ||
| Total mood disturbance | ||
| Washington Psychosocial Seizure Inventory (WPSI) | Family background | |
| Emotional adjustment | ||
| Interpersonal adjustment | ||
| Vocational adjustment | ||
| Financial status | ||
| Adjustment to seizures | ||
| Medicine and medical management | ||
| Overall functioning | ||
| Mood Rating Scale | Average score | |
| Fritz 2005 | Befindlichkeits‐Skala (BFS) | Dysphoria |
| Beck Depression Inventory | Depression | |
| Self‐rating Anxiety Scale (SAS) | Anxiety | |
| QOLIE‐31 | Health‐related quality of life | |
| Sveinbjornsdottir 1994 | The Mood Adjective Check List (MACL) | Depression |
| Anxiety | ||
| Fatigue | ||
| Activity | ||
| Aggression | ||
| Rating Scale Adapted from Brooks and McKinlay | Individual's behaviour |
Tiagabine versus topiramate
Only Fritz 2005 compared tiagabine with an active control drug (topiramate); therefore, we did not pool the data for this comparison.
50% or greater reduction in seizure frequency
Within Fritz 2005, we did not note any significant differences between the two add‐on drugs (RR 0.54, 95% CI 0.19 to 1.58; Analysis 2.1). According to the study authors' analysis of 37 participants, three participants (one tiagabine, two topiramate) became seizure‐free; 11 participants (four tiagabine, seven topiramate) reduced their seizure frequency by 50% or more.
2.1. Analysis.

Comparison 2 Tiagabine versus topiramate, Outcome 1 50% or greater reduction in seizure frequency (ITT).
Treatment withdrawal
We found no significant differences between tiagabine and topiramate for withdrawal from the study (RR 1.43, 95% CI 0.74 to 2.74; Analysis 2.2) (Fritz 2005).
2.2. Analysis.
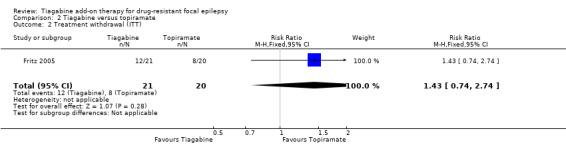
Comparison 2 Tiagabine versus topiramate, Outcome 2 Treatment withdrawal (ITT).
Cognitive effects
For this outcome, the authors of Fritz 2005 did not compare the two add‐on AEDs. Both add‐on drugs were examined over separate time points: baseline to post‐titration phase and post‐titration to maintenance phase. Within the first evaluation, baseline to post‐titration, significant deterioration was found within the topiramate group in phonemic verbal fluency (P = 0.001), semantic verbal fluency (P = 0.006), language comprehension (P = 0.002), working memory (P < 0.05), and visual block tapping (P = 0.032). A significant deterioration in verbal memory was also found in the tiagabine group (P = 0.039).
Within the post‐titration to maintenance phase, one significant improvement was found in mental flexibility in the topiramate group (P = 0.045). No changes were found in the tiagabine group for any of the cognitive outcomes.
Quality‐of‐life measures
Again, Fritz 2005 did not use appropriate analysis; hence, we were unable to compare the two add‐on AEDs. Within the baseline to post‐titration phase, significantly more complaints about medication were reported within the tiagabine group (P = 0.048). Participants taking topiramate reported significantly higher depression scores (P = 0.011), lower cognitive functioning (P = 0.024), and increased medication adverse effects (P = 0.008).
For the post‐titration to maintenance phase evaluation, the tiagabine group was significantly more fatigued and had less energy (P = 0.025). No other differences were found from post‐titration to maintenance.
Discussion
Summary of main results
This review assessed the efficacy and tolerability of tiagabine as an add‐on treatment for people with drug‐resistant focal epilepsy. Six trials were included in this review and all trials were initially sponsored by Novo Nordisk, as part of a pre‐licensing programme. The drug patent is now owned by Sanofi‐Synthelaboe.
Five of the six included trials used adequate methods of allocation concealment. However, we rated risk of bias for the method of randomisation sequence generation to be low in only two of the trials. All trials, except one (Fritz 2005), were double‐blinded and used identical packaging and medication to maintain this. We rated risk of bias for missing data as low in all studies. Investigators fully reported attrition and utilised ITT analyses. We had no access to the protocols for any of the studies; however, we did not consider selective outcome reporting bias to be an issue for the included studies. Despite these limitations, this review provided high‐certainty evidence that tiagabine reduced seizure frequency for people with drug‐resistant focal seizures compared with placebo. Specifically, a significantly larger proportion of people who were allocated to tiagabine achieved a clinically relevant reduction in seizure frequency (50% or greater) and were considered to be responders to treatment, compared with those who were allocated to placebo. As a result of the differences in response rates amongst the trials, the regression model was unable to provide valid estimates for the proportion of participants who would be expected to respond to specific doses of tiagabine. We can, however, suggest that for people who are similar to those recruited into the trials reviewed, we might expect that at least an additional 13%, possibly 20%, of people will experience a 50% or greater reduction in seizure frequency when taking a dose of tiagabine 30 mg or more per day, compared with placebo.
Results for the outcome of treatment withdrawal showed that tiagabine was more likely to be withdrawn than placebo. In trials of relatively short duration, such as those reviewed here, this is likely to represent problems with tolerability rather than poor seizure control. Of the adverse effects investigated, dizziness and tremor were significantly more likely to occur with tiagabine than with placebo. It is, however, important to recognise that the evidence for tremor is very limited, with the outcome being reported only by a single study. With regards to cognitive and quality‐of‐life effects, due to a lack of uniformity in the approach used to test for cognitive effects and quality of life, as well as the relatively small numbers of participants tested, evidence was insufficient to conclude whether tiagabine had an effect on these outcomes.
Given that only one trial included in this review compared add‐on tiagabine with another add‐on AED, the results from this study must be interpreted with caution. Authors of this study compared the two drugs for only two of the outcomes included in this review. Given the increasing number of licensed AEDs, this is an important issue that must be addressed in trials that compare one drug to another.
We should emphasise that the results of this review do not provide information about the effects of tiagabine when used as monotherapy, and that the results apply only to focal seizures.
Overall completeness and applicability of evidence
All of the included studies were conducted in adult populations. Notably, one study (Sachdeo 1997) also included adolescents aged over 12 years, but none of the studies included children under 12 years old. Consequently, this review is unable to predict the effectiveness or tolerability of tiagabine in children. Furthermore, as discussed above, only one study (Fritz 2005) investigated the use of add‐on tiagabine compared with that of an alternative AED, topiramate. As a result, this review cannot be considered informative about the effectiveness or tolerability of tiagabine compared with other AEDs.
With regards to applicability of the evidence, two of the trials (Crawford 2001; Richens 1995) were associated with limitations that could impact the applicability of the review findings (and were, hence, excluded from the meta‐analyses). Both trials included the use of a response‐conditional design, the aim of which was to select and randomly assign a group of people likely to respond favourably to tiagabine. In theory, this would maximise the effects observed, and should lead to fewer individuals needing to be recruited into trials before a significant treatment effect is observed. This is not a process that mimics clinical practice and the results of these studies are difficult to translate into everyday clinical practice.
The length of the treatment period was another limitation for all included trials: seven weeks in the cross‐over studies and 12 to 22 weeks in the parallel studies. Clinical practice would be better informed by trials of longer duration, especially for a chronic condition such as epilepsy.
Certainty of the evidence
Evidence for the main comparison, 'tiagabine versus placebo control', was GRADE assessed and a 'Summary of findings' table was constructed (See Table 1).
For all three studies that contributed data to the 'tiagabine versus placebo control' comparison, we rated the overall risk of bias as low and considered the evidence to be methodologically sound. As a result, we did not downgrade the evidence for risk of bias for any of the outcomes. We did, however, downgrade the evidence for five of the outcomes (50% or greater seizure reduction, treatment withdrawal, dizziness, fatigue and somnolence) once to moderate certainty due to imprecision. Specifically, the number of events extracted for each of the outcomes was insufficient and did not satisfy the optimal information size. We then upgraded the evidence for the outcome 50% or greater reduction in seizure frequency due to the large effect size detected, providing an overall rating of high‐certainty evidence for this outcome. The evidence for the outcomes treatment withdrawal, dizziness, fatigue and somnolence remained rated as moderate certainty.
For the outcomes nausea and tremor we downgraded the evidence twice for imprecision, as the number of events was exceptionally low (< 100 events). We rated the evidence as low certainty for the outcome nausea.We downgraded the evidence for tremor further, due to suspected publication bias. Data for the outcome tremor were provided only by one study (Uthman 1998). The other two studies (Kalviainen 1998; Sachdeo 1997) did not report tremor. Kalviainen 1998 specified in their publication that an adverse effect was reported in the journal article only if it had been reported by more than 5% of participants. Moreover, Sachdeo 1997 stated that adverse effects were reported only if significantly more participants in one, or both, of the two tiagabine treatment groups experienced an adverse event compared with those receiving placebo. This therefore implies that data on tremor were not included in the publications because the adverse event was not commonly reported by study participant, thus contradicting the data extracted from Uthman 1998. This subsequently led to the suspicion of publication bias. We then, however, upgraded the evidence for tremor back to low certainty of evidence due to the large effect size recognised by Uthman 1998.
Potential biases in the review process
We are not aware of any biases in the review process. We successfully conducted an extensive search of the available literature. We consulted the reference lists of retrieved studies to ensure that there were no additional references that had not been identified by the search strategies. We made all attempts to obtain any additional information from the manufacturer of tiagabine, Sanofi‐Synthelabo, and we contacted experts in the field to enquire about unpublished or ongoing studies. We therefore feel that we exhausted all possible sources of data relevant to the review.
Agreements and disagreements with other studies or reviews
Our findings and conclusions for the current review update are consistent with those of previous versions of this review (Pereira 2002; Pulman 2012; Pulman 2014). This is largely because no further studies have been identified for inclusion since the addition of Fritz 2005 in the review update completed in September 2010 (Pereira 2002).
We were able to identify another systematic review that similarly investigated the efficacy of add‐on tiagabine (Adkins 1998). In this review, the authors identified five studies for inclusion. Notably, all five studies were included in this current review, namely: Crawford 2001; Kalviainen 1998; Richens 1995; Sachdeo 1997; Uthman 1998. Data from all five studies, including the two cross‐over studies (Crawford 2001; Richens 1995), were combined into a single meta‐analysis. Notably, in our review, we only included data from the two cross‐over studies (Crawford 2001; Richens 1995) in a narrative synthesis and did not combine the data, from these studies, into the meta‐analysis conducted. Despite the fact that the meta‐analysis conducted by Adkins 1998 included additional data, not combined into our meta‐analysis, the findings of the review remained in agreement with our current findings, regarding 50% or greater seizure reduction. The pooled analysis, by Adkins 1998, indicated that 23% of participants randomised to receive tiagabine attained a 50% or greater reduction in seizure frequency, opposed to only 9% of the participants who were randomised to placebo. Here, we calculated a RR of 3.16 (95% CI 1.97 to 5.07). This RR predicts that participants randomised to tiagabine are three times more likely to achieve a 50% or greater reduction in seizure frequency than those randomised to placebo. This is consistent with the statistics reported in the Adkins 1998 review.
A separate review by Leppik 1995 investigated the tolerability of add‐on tiagabine in people with drug‐resistant focal epilepsy by reviewing data from five short‐term RCTs and six open‐label long‐term extension studies. The review described dizziness, fatigue, nervousness, tremor, diarrhoea, and depressed mood as the most commonly reported adverse effects during short‐term RCTs. Similarly, in this current Cochrane Review update, we observed that dizziness, tremor, and nervousness were reported by significantly more participants randomised to tiagabine than by those randomised to placebo. The number of participants reporting fatigue, however, did not reach significance (P = 0.06). We did not address diarrhoea and depressed mood in this review update; however, it would advisable to incorporate these adverse effects into future review updates.
The review by Leppik 1995 emphasised that a large majority of the adverse effects associated with tiagabine were mild to moderate in severity and that they mostly occurred during dose titration. Importantly, the long‐term tolerability profile of tiagabine remained comparable to that observed during short‐term use, with dizziness continuing to be the most commonly reported adverse event. Likewise tremor, fatigue, and nervousness all continued to feature among the most frequently reported adverse effects associated with tiagabine, but remained mild to moderate in severity. Accidental injury and infection were also significantly more likely with tiagabine than with placebo during long‐term use. Notably, the majority of adverse effects were associated with the titration period, even during longer periods of use, and most resolved within a month of commencing treatment.
Authors' conclusions
Implications for practice.
Add‐on tiagabine reduced the frequency of seizures for people with drug‐resistant focal seizures. Doses of between 30 mg and 56 mg per day were likely to reduce the frequency of seizures by 50% or more, for between 13% and 20% of people. Doses higher than 56 mg per day were not tested in the trials included in this review. Decision‐makers should, however, be made aware that withdrawal of treatment is predicted to occur in twice as many people with add‐on tiagabine than with add‐on placebo, and this likely represents issues with tolerability. Specifically, the evidence presented here suggests that the adverse effects dizziness and tremor are significantly associated with add‐on tiagabine.
We found no significant differences in seizure frequency and treatment withdrawal between tiagabine and topiramate as add‐on drugs.
Implications for research.
To further evaluate the place of tiagabine in the armamentarium of available antiepileptic drugs, further studies are required to address the following:
The long‐term effects of add‐on tiagabine.
How tiagabine compares with other add‐on treatments in drug‐resistant focal epilepsy.
The role of tiagabine in childhood and generalised epilepsies.
Economic aspects of tiagabine therapy.
What's new
| Date | Event | Description |
|---|---|---|
| 22 January 2019 | New citation required but conclusions have not changed | Conclusions are unchanged. |
| 22 January 2019 | New search has been performed | Searches updated 22 January 2019; no new trials identified. |
History
Protocol first published: Issue 1, 2000 Review first published: Issue 3, 2002
| Date | Event | Description |
|---|---|---|
| 4 August 2016 | New search has been performed | Searches updated 04 August 2016; no new trials identified. |
| 4 August 2016 | New citation required but conclusions have not changed | Conclusions remain unchanged. |
| 11 November 2013 | New citation required but conclusions have not changed | Conclusions remain unchanged. |
| 11 November 2013 | New search has been performed | Searches updated 11 November 2013; no new trials identified. |
| 15 December 2011 | New citation required but conclusions have not changed | Searches updated 15 December 2011; no new trials identified. |
| 11 June 2010 | New search has been performed | Searches updated 11 June 2010; no new trials identified. |
| 21 August 2008 | Amended | Converted to new review format. |
| 15 February 2008 | New search has been performed | Searches updated 15 February 2008; no new trials identified. |
Acknowledgements
We thank Jennifer Pulman for her contributions to the two previous review updates (Pulman 2012; Pulman 2014).
Appendices
Appendix 1. Cochrane Register of Studies (CRS Web) search strategy
1. tiagabin* OR gabitril AND CENTRAL:TARGET
2. MESH DESCRIPTOR Epilepsy EXPLODE ALL AND CENTRAL:TARGET
3. MESH DESCRIPTOR Seizures EXPLODE ALL AND CENTRAL:TARGET
4. (epilep* OR seizure* OR convuls*):AB,KW,MC,MH,TI AND CENTRAL:TARGET
5. #2 OR #3 OR #4 AND CENTRAL:TARGET
6. #1 AND #5
7. #6 AND >04/08/2016:CRSCREATED
Appendix 2. MEDLINE (Ovid) search strategy
This search strategy is based on the Cochrane Highly Sensitive Search Strategy for identifying randomized trials published in Lefebvre 2011.
1. (Tiagabin* or Gabitril).tw.
2. exp Epilepsy/
3. exp Seizures/
4. (epilep$ or seizure$ or convuls$).tw.
5. 2 or 3 or 4
6. exp *Pre‐Eclampsia/ or exp *Eclampsia/
7. 5 not 6
8. (randomized controlled trial or controlled clinical trial or pragmatic clinical trial).pt. or (randomi?ed or placebo or randomly).ab.
9. clinical trials as topic.sh.
10. trial.ti.
11. 8 or 9 or 10
12. exp animals/ not humans.sh.
13. 11 not 12
14. 1 and 7 and 13
15. limit 14 to ed=20160804‐20190121
16. 14 not (1$ or 2$).ed.
17. 16 and (2016$ or 2017$ or 2018$ or 2019$).dt.
18. 15 or 17
19. remove duplicates from 18
Appendix 3. ClinicalTrials.gov search strategy
Interventional Studies | Epilepsy | Tiagabine | First posted on or after 08/04/2016
Appendix 4. Cochrane Epilepsy Group Specialized Register search strategy
#1 tiagabin* OR gabitril
#2 INREGISTER AND >11/11/2013:CRSCREATED
#3 #1 AND #2
Appendix 5. CENTRAL via CRSO search strategy
#1 gabitril OR tiagabina OR tiagabine
#2 (epilep* OR seizure* OR convuls*):TI,AB,KY
#3 MESH DESCRIPTOR Epilepsy EXPLODE ALL TREES
#4 MESH DESCRIPTOR Seizures EXPLODE ALL TREES
#5 #2 OR #3 OR #4
#6 #1 AND #5
#7 31/10/2013 TO 31/08/2016:DL
#8 #6 AND #7
#9 ("Conference Abstract"):PT AND INEMBASE
#10 #8 NOT #9
Appendix 6. MEDLINE (Ovid) search strategy
This search strategy is based on the Cochrane Highly Sensitive Search Strategy for identifying randomized trials published in Lefebvre 2011.
1. (Tiagabin* or Gabitril).tw.
2. exp Epilepsy/
3. exp Seizures/
4. (epilep$ or seizure$ or convuls$).tw.
5. 2 or 3 or 4
6. exp *Pre‐Eclampsia/ or exp *Eclampsia/
7. 5 not 6
8. (randomized controlled trial or controlled clinical trial or pragmatic clinical trial).pt. or (randomi?ed or placebo or randomly).ab.
9. clinical trials as topic.sh.
10. trial.ti.
11. 8 or 9 or 10
12. exp animals/ not humans.sh.
13. 11 not 12
14. 1 and 7 and 13
15. limit 14 to ed=20131111‐20160804
16. remove duplicates from 15
Earlier versions of this review employed the following search strategy. It was combined with phases 1 and 2 of the earlier Cochrane highly sensitive search strategy for MEDLINE as set out in Appendix 5b of the Cochrane Handbook for Systematic Reviews of Interventions (version 4.2.4, updated March 2005) (Higgins 2011).
1. tiagabine.tw. 2. exp epilepsy/ OR epilep$.tw. 3. exp seizures/ OR seizure$.tw. 4. convulsion$.tw. 5. 2 OR 3 OR 4 6. 1 AND 5
Data and analyses
Comparison 1. Tiagabine versus placebo control.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 50% or greater reduction in seizure frequency | 3 | Risk Ratio (M‐H, Fixed, 95% CI) | Subtotals only | |
| 1.1 Intention‐to‐treat analysis | 3 | 769 | Risk Ratio (M‐H, Fixed, 95% CI) | 3.16 [1.97, 5.07] |
| 1.2 Worst‐case analysis | 3 | 769 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.19 [0.88, 1.62] |
| 1.3 Best‐case analysis | 3 | 769 | Risk Ratio (M‐H, Fixed, 95% CI) | 6.14 [3.92, 9.63] |
| 2 Treatment withdrawal | 3 | 769 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.81 [1.25, 2.62] |
| 3 Adverse effects | 3 | Risk Ratio (M‐H, Fixed, 99% CI) | Subtotals only | |
| 3.1 Dizziness | 3 | 769 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.69 [1.13, 2.51] |
| 3.2 Tremor | 1 | 297 | Risk Ratio (M‐H, Fixed, 99% CI) | 4.56 [1.00, 20.94] |
| 3.3 Fatigue | 3 | 769 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.38 [0.89, 2.14] |
| 3.4 Nervousness | 1 | 318 | Risk Ratio (M‐H, Fixed, 99% CI) | 10.65 [0.78, 146.08] |
| 3.5 Ataxia | 1 | 297 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.24 [0.34, 4.55] |
| 3.6 Nausea | 3 | 769 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.24 [0.69, 2.22] |
| 3.7 Somnolence | 3 | 769 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.18 [0.76, 1.83] |
| 3.8 Infection | 1 | 154 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.0 [0.36, 2.76] |
| 3.9 Headache | 1 | 154 | Risk Ratio (M‐H, Fixed, 99% CI) | 1.15 [0.48, 2.79] |
Comparison 2. Tiagabine versus topiramate.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 50% or greater reduction in seizure frequency (ITT) | 1 | 41 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.54 [0.19, 1.58] |
| 2 Treatment withdrawal (ITT) | 1 | 41 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.43 [0.74, 2.74] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Crawford 2001.
| Methods | Randomised, double‐blind, placebo‐controlled, cross‐over study, based on a response‐dependent design. Two treatment arms: one TGB, one PCB. Participants randomly assigned to one of two sequences. TGB started during screening phase at 12 mg/d QID. Seven‐week double‐blind treatment period during which participants continued on TGB or crossed over to PCB arm | |
| Participants | Multi‐centre study (five centres: two in UK, two in The Netherlands, and one in Denmark) 44 participants with drug‐resistant focal epilepsy were randomly assigned (30 male), aged 18 to 53 years Participants already taking one to three background AEDs Median baseline seizure frequency = 2.7 per week | |
| Interventions | Group one: PCB Group two: TGB (optimal dose 64 mg/d) Mean daily dose for all randomly assigned participants was 46 mg during the double‐blind phase |
|
| Outcomes |
|
|
| Notes | From the 44 people randomly assigned to the double‐blind phase, seven were excluded from the intention‐to‐treat analysis. All participants were evaluated for adverse effects. Trial originally sponsored by Novo Nordisk, now owned by Sanofi‐Synthelabo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "patients were randomly assigned in a 1:1 ratio at each centre to one of two sequences of treatment" Comment: No further details on how the sequences were generated |
| Allocation concealment (selection bias) | Low risk | Comment: Sequentially allocated sealed packages used |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Comment: Used identical packaging and medication |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Attrition rates reported and intention‐to‐treat analysis employed |
| Selective reporting (reporting bias) | Low risk | Comment: Although no protocol was available, all outcomes reported in methods section were fully reported in text |
| Other bias | High risk | Quote: "The design has ethical advantages because eligibility for randomization into the double‐blind phase is dependent on patients showing some therapeutic benefit from the study drug (a ≥25% reduction in seizure rate) during the screening phase. This preselection maximizes the chances of detecting a difference in clinical response between the study drug and placebo with a relatively low number of patients" Comment: Study design was biased to detect a therapeutic effect with tiagabine. The study design also produced a misleading high discontinuation rate as participants were withdrawn during the screening phase as a result of not achieving a ≥25% reduction in seizure frequency |
Fritz 2005.
| Methods | Open‐label, head‐to‐head controlled, parallel study. Two treatment arms: one TGB, one TPM. Participants were randomly assigned using a non‐random component. Duration of screening period: eight weeks, followed by a titration phase of three months and a maintenance period of three months | |
| Participants | No information regarding study sites or countries 41 participants with drug‐resistant focal epilepsy randomly assigned, aged 18 to 65 years Baseline data reported for only 30 participants who completed the whole study (18 male) Participants already taking one to three background AEDs |
|
| Interventions | Group one: TGB at least 20 mg/d; mean 32 mg/d Group two: TPM at least 200 mg/d; mean 335 mg/d |
|
| Outcomes |
|
|
| Notes | Non‐accurate baseline data reported. Four participants excluded from ITT analysis for seizure reduction outcome. Trial originally sponsored by Novo Nordisk, now owned by Sanofi‐Synthelabo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Comment: Non‐randomisation component employed in the sequence generation process. Sequence generated by odd versus even number of week in which participant was seen |
| Allocation concealment (selection bias) | High risk | Comment: No concealment used |
| Blinding (performance bias and detection bias) All outcomes | High risk | Comment: Study was open‐label |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Attrition rates reported and intention‐to‐treat analysis employed |
| Selective reporting (reporting bias) | Unclear risk | Comment: Although no protocol was available, all outcomes reported in methods section were fully reported in text. Participant characteristics reported for only 30 participants who completed the whole study of the 41 randomly assigned |
| Other bias | Unclear risk | Comment: Due to the incomplete reporting of participant characteristics at baseline it was not possible to ascertain whether any baseline imbalances existed |
Kalviainen 1998.
| Methods | Randomised, double‐blind, placebo‐controlled, two‐treatment parallel‐group study. Two treatment arms: one PCB, one TGB. Participants randomly assigned using computer‐generated sequence. Treatment period: 22 weeks (six‐week run‐in period, 12‐week fixed‐dose period, four‐week termination period) | |
| Participants | Multi‐centre study (11 centres in Europe—one each in Denmark and Sweden, two in Finland and seven in UK 154 participants with drug‐resistant focal epilepsy were randomly assigned (90 male), aged 17 to 71 years 77 were randomly assigned to placebo and 77 to 30 mg/d tiagabine Participants already taking one to three background AEDs Median four‐week baseline seizure frequency: placebo = 10.5, tiagabine = 12.2 Cognitive and quality of life effects were assessed on a subset of 43 individuals | |
| Interventions | Group one: PCB Group two: TGB 30 mg/d |
|
| Outcomes |
|
|
| Notes | No participants were excluded from analysis. 29 people withdrew from the study: 21 receiving tiagabine and eight receiving placebo. Trial originally sponsored by Novo Nordisk, now owned by Sanofi‐Synthelabo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Eligible patients entering the double‐blind phase were randomly allocated, according to a computer‐generated list" |
| Allocation concealment (selection bias) | Low risk | Comment: Used sequentially allocated sealed packages |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Comment: Used identical packaging and medication |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Attriition rates reported and ITT analysis employed |
| Selective reporting (reporting bias) | Low risk | Comment: Although no protocol was available, all outcomes reported in methods section were fully reported in text |
| Other bias | Low risk | Comment: None detected |
Richens 1995.
| Methods | Randomised, double‐blind, placebo‐controlled, cross‐over study based on a response‐dependent design. Two treatment arms: one PCB, one TGB. Participants randomly assigned using 1:1 ratio. Treatment period: 23 weeks (three‐week run‐in period, seven‐week assessment period, three‐week cross‐over period, seven‐week assessment period, three‐week termination period) | |
| Participants | Multi‐centre study (five centres in UK and Denmark) 94 participants with drug‐resistant focal epilepsy were randomly assigned (61% male), aged 19 to 71 years 25 participants were randomly assigned to placebo‐tiagabine sequence and 21 to tiagabine‐placebo Participants already taking one to three AEDs Median complex focal seizure rate per four weeks = six Cognitive effects were reported for a subset of 22 individuals | |
| Interventions | Group one: PCB Group two: TGB 52 mg/d QID Mean daily dose was 33.4 mg (range 12 to 52 mg) |
|
| Outcomes |
|
|
| Notes | From the 46 people randomly assigned to the double‐blind phase, seven failed to complete the study. Of these, only four were excluded from the intent‐to‐treat analysis. All participants were evaluated for adverse effects. The characteristics of people ineligible for the double‐blind phase were similar to those of people qualifying, with regard to epilepsy history, seizure frequency, and concurrent treatment. Trial originally sponsored by Novo Nordisk, now owned by Sanofi‐Synthelabo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Qualifying patients were randomly assigned in a 1:1 ratio at each centre to one of two treatment sequences" Comment: No further details of sequence generation |
| Allocation concealment (selection bias) | Low risk | Comment: Used sequentially allocated sealed packages |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Comment: Used identical packaging and medication |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Attrition rates reported and ITT analysis employed |
| Selective reporting (reporting bias) | Low risk | Comment: Although no protocol was available, all outcomes reported in methods section were fully reported in text |
| Other bias | High risk | Quote: "Of the 74 patients who completed the screening phase, 28 were ineligible for the double‐blind phase because their total seizure rate was not reduced by 25% and therefore tiagabine was tapered off over a 3‐week period" Comment: Study design was biased to detect a therapeutic effect with tiagabine. The study design also produced a misleading high discontinuation rate as participants were withdrawn during the screening phase as a result of not achieving a ≥25% reduction in seizure frequency |
Sachdeo 1997.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group study. Three treatment arms: one PLC, two TGB. Participants randomly assigned using ratio 1:1:1 in blocks of six Treatment periods = 12 weeks | |
| Participants | Multi‐centre US study (26 centres) 318 participants (178 male) aged 12 to 71 years with drug‐resistant focal epilepsy were randomly assigned 107 to PBO, 106 to TGB 16 mg BID, 105 to TGB 8 mg QID Valproate allowed in combination with an enzyme‐inducing drug but not as monotherapy Median baseline four‐week complex focal seizures; frequency during baseline was as follows: PCB = 8.4; TGB 16 mg BID = 8.4; TGB 8 mg QID = 7.9 | |
| Interventions | Add‐on placebo, TGB 16 mg BID or TGB 8 mg QID | |
| Outcomes |
|
|
| Notes | From the 318 people randomly assigned to the double‐blind phase, four were excluded from the ITT analyses. All 318 people were evaluated for adverse effects. 47 people withdrew from the study: 10 receiving PLC; 16 receiving TGB 16 mg BID; 21 receiving TGB 8 mg QID. Trial originally sponsored by Novo Nordisk, now owned by Sanofi‐Synthelabo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "patients were randomized in a ratio of 1:1:1 in blocks of six per study center" |
| Allocation concealment (selection bias) | Low risk | Comment: Used sequentially allocated sealed packages |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Quote: "this was done by dispensing tiagabine (2 and 4 mg) and placebo as identical‐appearing tablets" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Attrition rates reported and ITT analysis employed |
| Selective reporting (reporting bias) | Low risk | Comment: Although no protocol was available, all outcomes reported in methods section were fully reported in text |
| Other bias | Low risk | Comment: None detected |
Uthman 1998.
| Methods | Randomised, double‐blind, placebo‐controlled, parallel‐group study. Four treatment arms: one PCB, three TGB. Participants randomly assigned using ratio of 3:2:3:2. Treatment period: 20 weeks (four‐week titration phase, 12‐week fixed‐dose treatment period, four‐week tapering period) | |
| Participants | Multi‐centre US study (21 centres) 297 participants with drug‐resistant focal epilepsy were randomly assigned (172 male), aged 12 to 77 years Participants already taking one to three AEDs, valproate allowed in combination with an enzyme‐inducing drug but not as monotherapy Median four‐week baseline complex focal seizure frequency was: PCB = 7.4; TGB 16 mg = 8.5; TGB 32 mg = 9.6; TGB 56 mg = 9.1 Cognitive effects were reported for a subset of 162 individuals | |
| Interventions | Group one: PCB Group two: TGB 16 mg/d QID Group three: TGB 32 mg/d QID Group 4: TGB 56 mg/d QID |
|
| Outcomes |
|
|
| Notes | From the 297 people randomly assigned to the double‐blind phase, five were excluded from the ITT analyses because no double‐blind assessments were done or their centres lacked participants in all treatment groups. All 297 people were evaluated for adverse effects 54 people withdrew from the study: 13 receiving PCB; six receiving 16 mg TGB; 18 receiving 32 mg TGB, and 17 receiving 56 mg TGB. Trial originally sponsored by Novo Nordisk, now owned by Sanofi‐Synthelabo | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "The randomization ratio to treatment groups was 3:2:3:2 for placebo and 16 mg, 32 mg, and 56 mg or tiagabine (Gabitril), respectively" Comment: No further details of sequence generation reported in text |
| Allocation concealment (selection bias) | Low risk | Comment: Used sequentially allocated sealed packages |
| Blinding (performance bias and detection bias) All outcomes | Low risk | Comment: Used identical packaging and medication |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: Attrition rates reported and ITT analysis employed |
| Selective reporting (reporting bias) | Low risk | Comment: Although no protocol was available, all outcomes reported in methods section were fully reported in text |
| Other bias | Unclear risk | Comment: No reasoning was given for the use of the unequal randomisation ratio |
Abbreviations
- AED = antiepileptic drug;
- BID = twice daily;
- ITT = intention‐to‐treat;
- PCB = placebo;
- QID = four times daily;
- TGB = tiagabine;
- TPM = topiramate.
.
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Arroyo 2005 | This is a non‐placebo controlled study |
| Bauer 1995 | This is an open add‐on study of tiagabine for the treatment of people with resistant focal, secondarily generalised seizures, or both. In this study, the results reported were part of an ongoing multi‐national, multi‐centre trial. It is not randomised |
| Gustavson 1997 | This is an open‐label, single‐dose study that was designed to examine the pharmacokinetics of tiagabine in children with epilepsy. It is not randomised |
| Uldall 1995 | This is a single‐blind study of safety, tolerability, and preliminary efficacy of tiagabine as adjunctive treatment of children with epilepsy. It is not randomised |
Differences between protocol and review
According to the original protocol, we had planned to only include trials using a placebo control group in the review, assuming they met all other inclusion criteria. We decided to extend this to include any other studies with an active add‐on drug, used as a control group versus tiagabine.
The title of the review has been changed from "Tiagabine add‐on for drug‐resistant partial epilepsy" to "Tiagabine add‐on therapy for drug‐resistant focal epilepsy" in accordance with the latest classification of epilepsies, released by the International League Against Epilepsy (ILAE) (Scheffer 2017). Any previous mention of "partial epilepsy" within the review text was subsequently changed to "focal epilepsy".
Contributions of authors
RB conducted the update of this review. RB and KMM dual assessed trials for inclusion in the current review update and GRADE‐assessed the outcomes for the Table 1. JH and AGM provided support for the review update.
Sources of support
Internal sources
No sources of support supplied
External sources
-
National Institute for Health Research, UK.
This review update was supported by the National Institute for Health Research, via Cochrane Programme Grant funding to the Epilepsy Group. The views and opinions expressed therein are those of the authors and do not necessarily reflect those of the Systematic Reviews Programme, NIHR, NHS or the Department of Health.
Declarations of interest
RB: none known. JH: none known. KMM: none known. AGM: a consortium of pharmaceutical companies (GSK, EISAI, UCB Pharma) funded the National Audit of Seizure Management in Hospitals (NASH) through grants paid to the University of Liverpool. Professor Tony Marson is part funded by National Institute for Health Research Collaboration for Leadership in Applied Health Research and Care North West Coast (NIHR CLAHRC NWC).
New search for studies and content updated (no change to conclusions)
References
References to studies included in this review
Crawford 2001 {published and unpublished data}
- Crawford PM, Meinardi H, Brown S, Rentmeester TW, Pedersen PC, Lassen LC. Tiagabine: efficacy and safety in adjunctive treatment of partial seizures. Epilepsia 2001;42(4):531‐8. [DOI] [PubMed] [Google Scholar]
Fritz 2005 {published data only}
- Fritz N, Glogau S, Hoffmann J, Rademacher M, Elger CE, Helmstaedter C. Efficacy and cognitive side effects of tiagabine and topiramate in patients with epilepsy. Epilepsy & Behavior 2005;6(3):373‐81. [DOI] [PubMed] [Google Scholar]
Kalviainen 1998 {published and unpublished data}
- Kalviainen R, Aikia M, Mervaala E, Saukkonen AM, Pitkanen A, Riekkinen PJ Sr. Long‐term cognitive and EEG effects of tiagabine in drug‐resistant partial epilepsy. Epilepsy Research 1996;25(3):291‐7. [DOI] [PubMed] [Google Scholar]
- Kalviainen R, Brodie MJ, Duncan J, Chadwick D, Edwards D, Lyby K. A double‐blind, placebo‐controlled trial of tiagabine given three‐times daily as add‐on therapy for refractory partial seizures. Epilepsy Research 1998;30:31‐40. [DOI] [PubMed] [Google Scholar]
Richens 1995 {published and unpublished data}
- Richens A, Chadwick DW, Duncan JS, Dam M, Gram L, Mikkelsen M, et al. Adjunctive treatment of partial seizures with tiagabine: a placebo‐controlled trial. Epilepsy Research 1995;21(1):37‐42. [PUBMED: 7641674] [DOI] [PubMed] [Google Scholar]
- Sveinbjornsdottir S, Sander JW, Patsalos PN, Upton D, Thompson PJ, Duncan JS. Neuropsychological effects of tiagabine, a potential new antiepileptic drug. Seizure 1994;3(1):29‐35. [DOI] [PubMed] [Google Scholar]
Sachdeo 1997 {published and unpublished data}
- Sachdeo RC, Leroy RF, Krauss GL, Drake ME Jr, Green PM, Leppik IE, et al. Tiagabine therapy for complex partial seizures: a dose‐frequency study. Archives of Neurology 1997;54:595‐601. [DOI] [PubMed] [Google Scholar]
Uthman 1998 {published and unpublished data}
- Dodrill CB, Arnett JL, Sommerville KW, Shu V. Cognitive and quality of life effects of differing dosages of tiagabine in epilepsy. Neurology 1997;48(4):1025‐31. [DOI] [PubMed] [Google Scholar]
- Uthman BM, Rowan AJ, Ahmann PA, Leppik IE, Schachter SC, Sommerville KW, et al. Tiagabine for complex partial seizures: a randomized, add‐on, dose‐response trial. Archives of Neurology 1998;55:56‐62. [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Arroyo 2005 {published data only}
- Arroyo S, Bootham BR, Brodie MJ, Duncan JS, Duncan R, Nieto M. A randomised open‐label study of tiagabine given two or three times daily in refractory epilepsy. Seizure 2005;14(2):81‐4. [DOI] [PubMed] [Google Scholar]
Bauer 1995 {published data only}
- Bauer J, Stawowy B, Lenders T, Bettig U, Elger CE. Efficacy and tolerability of tiagabine: results of an add‐on study in patients with refractory partial seizures. Journal of Epilepsy 1995;8(1):83‐6. [Google Scholar]
Gustavson 1997 {published data only}
- Gustavson LE, Boellner SW, Granneman GR, Qian JX, Guenther HJ, El‐Shourbagy T, et al. A single‐dose study to define tiagabine pharmacokinetics in pediatric patients with complex partial seizures. Neurology 1997;48(4):1032‐7. [DOI] [PubMed] [Google Scholar]
Uldall 1995 {published data only}
- Uldall P, Bulteau C, Pedersen SA, Dulac O, Meinild H, Lassen LC. Single‐blind study of safety, tolerability and preliminary efficacy of tiagabine as adjunctive treatment of children with epilepsy. Epilepsia 1995;36(Suppl 3):S147‐8. [Google Scholar]
Additional references
Adkins 1998
- Adkins JC, Noble S. Tiagabine: a review of its pharmacodynamic and pharmacokinetic properties and therapeutic potential in the management of epilepsy. Drugs 1998;55(3):437‐60. [PUBMED: 9530548] [DOI] [PubMed] [Google Scholar]
Baker 2000
- Baker GA, Hesdon M, Marson AG. Quality of life and behavioural outcome measures in randomized controlled trials of antiepileptic drugs: a systematic review of reporting standards. Epilepsia 2000;41(11):1357‐63. [DOI] [PubMed] [Google Scholar]
Cochrane 1998
- Cochrane HC, Baker GA, Chadwick DW. Neuropsychological outcomes in randomized controlled trials of antiepileptic drugs: a systematic review of methodology and reporting standards. Epilepsia 1998;39(10):1088‐97. [DOI] [PubMed] [Google Scholar]
Cockerell 1995
- Cockerell OC, Johnson AL, Sander JW, Hart YM, Shorvon SD. Remission of epilepsy: results from the national general practice study of epilepsy. Lancet 1995;346:140‐4. [DOI] [PubMed] [Google Scholar]
Commission 1989
- Commission on Classification and Terminology of the International League Against Epilepsy. Proposal for revised classification of epilepsies and epileptic syndromes. Epilepsia 1989;30(4):389‐99. [DOI] [PubMed] [Google Scholar]
Dodrill 1997
- Dodrill CB, Arnett JL, Sommerville KW, Shu V. Cognitive and quality of life effects of differing dosages of tiagabine in epilepsy. Neurology 1997;48(4):1025‐31. [DOI] [PubMed] [Google Scholar]
Eddy 2011
- Eddy CM, Rickards HE, Cavanna AE. The cognitive impact of antiepileptic drugs. Therapeutic advances in neurological disorders 2011;4(6):385‐407. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fiest 2017
- Fiest KM, Sauro KM, Wiebe S, Patten SB, Kwon CS, Dykeman J, et al. Prevalence and incidence of epilepsy: a systematic review and meta‐analysis of international studies. Neurology 2017;88(3):296‐303. [PUBMED: 27986877] [DOI] [PMC free article] [PubMed] [Google Scholar]
Fink‐Jensen 1992
- Fink‐Jensen A, Suzdak PD, Swedberg MD, Judge ME, Hansen L, Nielsen PG. The gamma‐aminobutyric acid (GABA) uptake inhibitor, tiagabine, increases extracellular brain levels of GABA in awake rats. European Journal of Pharmacology 1992;220(2‐3):197‐201. [PUBMED: 1425991] [DOI] [PubMed] [Google Scholar]
Food and Drug Administration 1997
- Food, Drug Administration. Approval Letter. www.accessdata.fda.gov/drugsatfda_docs/nda/97/020646ap_gabitril_apltr.pdf (accessed 01 March 2019).
GRADEpro GDT 2015 [Computer program]
- McMaster University (developed by Evidence Prime). GRADEpro Guideline Development Tool. Version Accessed 29 January 2019. Hamilton (ON): McMaster University (developed by Evidence Prime), 2015.
Gustavson 1995
- Gustavson LE, Mengel HB. Pharmacokinetics of tiagabine, a gamma‐aminobutyric acid‐uptake inhibitor, in healthy subjects after single and multiple doses. Epilepsia 1995;36(6):605‐11. [PUBMED: 7555975] [DOI] [PubMed] [Google Scholar]
Hauser 1993
- Hauser WA, Annegers JF, Kurland LT. Incidence of epilepsy and unprovoked seizures in Rochester, Minnesota: 1935‐1984. Epilepsia 1993;34(3):453‐68. [DOI] [PubMed] [Google Scholar]
Hey 2014
- Hey SP, Kimmelman J. The questionable use of unequal allocation in confirmatory trials. Neurology 2014;82(1):77‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2011
- Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
ILAE 2010
- Kwan P, Arzimanoglou A, Berg AT, Brodie MJ, Hauser WA, Mathern G, et al. Definition of drug resistant epilepsy. Consensus proposal by the ad hoc Task Force of the ILAE Commission on Therapeutic Strategies. Epilepsia 2010;51(9):1922. [DOI] [PubMed] [Google Scholar]
Kalviainen 1996
- Kalviainen R, Aikia M, Mervaala E, Saukkonen AM, Pitkanen A, Riekkinen PJ Sr. Long‐term cognitive and EEG effects of tiagabine in drug‐resistant partial epilepsy. Epilepsy Research 1996;25(3):291‐7. [DOI] [PubMed] [Google Scholar]
Kirkham 2010
- Kirkham J, Dwan K, Altman D, Gamble C, Dodd S, Smyth R, et al. The impact of outcome reporting bias in randomised controlled trials on a cohort of systematic reviews. BMJ 2010;340:365. [DOI] [PubMed] [Google Scholar]
Lefebvre 2011
- Lefebvre C, Manheimer E, Glanville J. Chapter 6: Searching for studies. In: Higgins JPT, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Leppik 1995
- Leppik IE. Tiagabine: the safety landscape. Epilepsia 1995;36 Suppl 6:S10‐3. [PUBMED: 8595787] [DOI] [PubMed] [Google Scholar]
Marson 1996
- Marson AG, Kadir ZA, Chadwick DW. New antiepileptic drugs: a systematic review of their efficacy and tolerability. BMJ 1996;313(7066):1169‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
Marson 1997
- Marson AG, Kadir ZA, Hutton JL, Chadwick DW. The new antiepileptic drugs: a systematic review of their efficacy and tolerability. Epilepsia 1997;38(8):859‐80. [DOI] [PubMed] [Google Scholar]
McCullagh 1989
- McCullagh P, Nelder JA. Generalized Linear Models. 2nd Edition. London: Chapman and Hall, 1989. [Google Scholar]
Morimoto 1997
- Morimoto K, Sato H, Yamamoto Y, Watanabe T, Suwaki H. Antiepileptic effects of tiagabine, a selective GABA uptake inhibitor, in the rat kindling model of temporal lobe epilepsy. Epilepsia 1997;38(9):966‐74. [PUBMED: 9579934] [DOI] [PubMed] [Google Scholar]
Ostergaard 1995
- Ostergaard LH, Gram L, Dam M. Tiagabine. In: Levy RH, Mattson RH, Medlrum BS editor(s). Mechanisms of Action in Antiepileptic Drugs. 4th Edition. New York: Raven Press, 1995. [Google Scholar]
Sander 1996
- Sander JW, Shorvon SD. Epidemiology of the epilepsies. Journal of Neurology, Neurosurgery, & Psychiatry 1996;61(5):433‐43. [DOI] [PMC free article] [PubMed] [Google Scholar]
Scheffer 2017
- Scheffer IE, Berkovic S, Capovilla G, Connolly MB, French J, Guilhoto L, et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017;58(4):512‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2013
- Schünemann H, Brożek J, Guyatt G, Oxman A, editor(s). GRADE handbook for grading quality of evidence and strength of recommendations (updated October 2013). Grade Working Group, 2013. Available from www.guidelinedevelopment.org/handbook.
Smith 2015
- Smith MJ. Generalised linear models. Statistical Analysis Handbook 2018 edition www.statsref.com/HTML/index.html?glim.html (accessed 27 February 2019).
Sveinbjornsdottir 1994
- Sveinbjornsdottir S, Sander JW, Patsalos PN, Upton D, Thompson PJ, Duncan JS. Neuropsychological effects of tiagabine, a potential new antiepileptic drug. Seizure 1994;3(1):29‐35. [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Pereira 2002
- Pereira J, Marson AG, Hutton JL. Tiagabine add‐on for drug‐resistant partial epilepsy. Cochrane Database of Systematic Reviews 2002, Issue 3. [DOI: 10.1002/14651858.CD001908] [DOI] [PubMed] [Google Scholar]
Pulman 2012
- Pulman J, Hutton J, Marson A. Tiagabine add‐on for drug‐resistant partial epilepsy. Cochrane Database of Systematic Reviews 2012, Issue 5. [DOI: 10.1002/14651858.CD001908.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]
Pulman 2014
- Pulman J, Hutton JL, Marson AG. Tiagabine add‐on for drug‐resistant partial epilepsy. Cochrane Database of Systematic Reviews 2014, Issue 2. [DOI: 10.1002/14651858.CD001908.pub3] [DOI] [PMC free article] [PubMed] [Google Scholar]