

Abstract
Background
Stroke is a major cause of adult disability. Selective serotonin reuptake inhibitors (SSRIs) have been used for many years to manage depression and other mood disorders after stroke. The 2012 Cochrane Review of SSRIs for stroke recovery demonstrated positive effects on recovery, even in people who were not depressed at randomisation. A large trial of fluoxetine for stroke recovery (fluoxetine versus placebo under supervision) has recently been published, and it is now appropriate to update the evidence.
Objectives
To determine if SSRIs are more effective than placebo or usual care at improving outcomes in people less than 12 months post‐stroke, and to determine whether treatment with SSRIs is associated with adverse effects.
Search methods
For this update, we searched the Cochrane Stroke Group Trials Register (last searched 16 July 2018), the Cochrane Controlled Trials Register (CENTRAL, Issue 7 of 12, July 2018), MEDLINE (1946 to July 2018), Embase (1974 to July 2018), CINAHL (1982 July 2018), PsycINFO (1985 to July 2018), AMED (1985 to July 2018), and PsycBITE March 2012 to July 2018). We also searched grey literature and clinical trials registers.
Selection criteria
We included randomised controlled trials (RCTs) that recruited ischaemic or haemorrhagic stroke survivors at any time within the first year. The intervention was any SSRI, given at any dose, for any period, and for any indication. We excluded drugs with mixed pharmacological effects. The comparator was usual care or placebo. To be included, trials had to collect data on at least one of our primary (disability score or independence) or secondary outcomes (impairments, depression, anxiety, quality of life, fatigue, healthcare cost, death, adverse events and leaving the trial early).
Data collection and analysis
We extracted data on demographics, type of stroke, time since stroke, our primary and secondary outcomes, and sources of bias. Two review authors independently extracted data from each trial. We used standardised mean differences (SMDs) to estimate treatment effects for continuous variables, and risk ratios (RRs) for dichotomous effects, with their 95% confidence intervals (CIs). We assessed risks of bias and applied GRADE criteria.
Main results
We identified a total of 63 eligible trials recruiting 9168 participants, most of which provided data only at end of treatment and not at follow‐up. There was a wide age range. About half the trials required participants to have depression to enter the trial. The duration, drug, and dose varied between trials. Only three of the included trials were at low risk of bias across the key 'Risk of bias' domains. A meta‐analysis of these three trials found little or no effect of SSRI on either disability score: SMD −0.01 (95% CI −0.09 to 0.06; P = 0.75; 2 studies, 2829 participants; moderate‐quality evidence) or independence: RR 1.00 (95% CI 0.91 to 1.09; P = 0.99; 3 studies, 3249 participants; moderate‐quality evidence). We downgraded both these outcomes for imprecision.
SSRIs reduced the average depression score (SMD 0.11 lower, 0.19 lower to 0.04 lower; 2 trials, 2861 participants; moderate‐quality evidence), but there was a higher observed number of gastrointestinal side effects among participants treated with SSRIs compared to placebo (RR 2.19, 95% CI 1.00 to 4.76; P = 0.05; 2 studies, 148 participants; moderate‐quality evidence), with no evidence of heterogeneity (I2 = 0%). For seizures there was no evidence of a substantial difference. When we included all trials in a sensitivity analysis, irrespective of risk of bias, SSRIs appeared to reduce disability scores but not dependence. One large trial (FOCUS) dominated the results.
We identified several ongoing trials, including two large trials that together will recruit more than 3000 participants.
Authors' conclusions
We found no reliable evidence that SSRIs should be used routinely to promote recovery after stroke. Meta‐analysis of the trials at low risk of bias indicate that SSRIs do not improve recovery from stroke. We identified potential improvements in disability only in the analyses which included trials at high risk of bias. A further meta‐analysis of large ongoing trials will be required to determine the generalisability of these findings.
Plain language summary
Selective serotonin reuptake inhibitors for stroke recovery
Review question What are the effects of selective serotonin uptake inhibitor (SSRI) drugs on recovery from stroke?
Background Stroke is a major cause of disability. Stroke‐related disability can include difficulty with daily tasks such as toileting, washing, and walking. Sometimes disability is so severe that a person becomes dependent on others for performing basic activities (this is known as 'dependence'). Our previous Cochrane Review published in 2012 suggested that SSRI drugs (a class of drug usually used to treat mood problems, which work by changing the level of chemicals in the brain), might improve recovery after stroke, thereby reducing disability and increasing the chance of being independent after a stroke, However, when we looked at only the high‐quality trials, the effect was less convincing.
A large trial recruiting more than 3000 participants has now been completed and so it is necessary to update this review. In our main analyses we decided to include only high‐quality trials, that is those which used rigorous methods to avoid biases (such as the person assessing outcome being aware of whether the stroke survivor received the active drug or placebo). In this review, we refer to them as 'low risk of bias' trials.
If disability and dependency can be improved by a simple drug, this could have a major impact on quality of life for many stroke survivors.
We also wanted to find out whether SSRIs had other benefits, for example improving the severity of any arm or leg weakness, mood, anxiety, quality of life, and also whether SSRIs were associated with side effects such as bleeding or seizures.
Study characteristics In total we found 63 trials recruiting 9168 stroke survivors within one year of their stroke. There was a wide age range. About half the trials required participants to have depression to enter the trial. The duration, drug, and dose varied between trials. However, only three of these trials were at low risk of bias; the participants in these trials did not have to be depressed to enter the trial, and they were all recruited soon after the stroke.
Key results When we combined data from these three studies at low risk of bias, which recruited 3249 participants, SSRIs did not affect disability score or dependency. SSRIs reduced the risk of future depression but increased the risk of problems with the digestive system. There was no evidence of a substantial difference in seizures. When we combined data from all the studies, irrespective of risks of bias, there appeared to be a beneficial effect on recovery, but this was almost certainly because the studies at high risk of bias tended to give the positive results. The evidence is current until July 2018.
Quality of the evidence We are confident that the results are reliable when we included just the studies at low risk of bias. When we included all studies regardless of risk of bias we found that SSRIs reduced disability. When they become available, we will include the results from two large ongoing trials in a future update.
Summary of findings
Summary of findings for the main comparison. SSRI versus control at end of treatment, by SSRI for stroke recovery.
| SSRI versus control at end of treatment, by SSRI, for stroke recovery* | ||||||
|
Patient or population: people with stroke recovery
Settings: hospital
Intervention: SSRI versus control at end of treatment, by SSRI * Summary of Findings table based on studies with low risk of bias. | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of Participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Control | SSRI versus control at end of treatment, by SSRI | |||||
| Disability (primary analysis) | The mean disability (primary analysis) in the intervention groups was 0.01 standard deviations lower (0.09 lower to 0.06 higher) | 2829 (2 studies) | ⊕⊕⊕⊝ moderatea | Outcomes: Stroke Impact Scale (SIS) score at 6 months (FOCUS Trial Collaboration 2018); Barthel Index (BI) score on day 90 (Marquez Romero 2013) | ||
| Independent on modified Rankin score (mRS 0 to 2) (primary analysis) | Study population | RR 1.00 (0.91 to 1.09) | 3249 (3 studies) | ⊕⊕⊕⊝ moderateb,c | ‐ | |
| 367 per 1000 | 367 per 1000 (334 to 400) |
|||||
| Neurological deficit score | The mean neurological deficit score in the intervention groups was 0.3 standard deviations lower (0.63 lower to 0.04 higher) | 142 (2 studies) | ⊕⊕⊕⊝ moderated |
Outcomes: Fugl‐Meyer Assessment (FMA) score on day 90 (Marquez Romero 2013); National Institutes of Health Stroke Scale (NIHSS) score on day 90 (Chollet 2011) | ||
| Depression (continuous data) | The mean depression (continuous data) in the intervention groups was 0.11 standard deviations lower (0.19 to 0.04 lower) | 2861 (2 studies) | ⊕⊕⊕⊝ moderatee | Outcomes Mental Health Inventory 5 (MHI‐5) score at 6 months (FOCUS Trial Collaboration 2018); Montgomery‐Åsberg Depression Rating Scale (MADRS) score on day 90 (Chollet 2011) | ||
| Death | Study population | RR 0.99 (0.79 to 1.25) | 3254 (3 studies) | ⊕⊕⊕⊕ high | ‐ | |
| 80 per 1000 | 80 per 1000 (64 to 101) | |||||
| Number of seizures | Study population | RR 1.47 (0.99 to 2.18) | 3275 (3 studies) | ⊕⊕⊕⊝ moderatef | ‐ | |
| 24 per 1000 | 36 per 1000 (24 to 53) | |||||
| Gastrointestinal side effects | Study population | RR 2.19 (1.00 to 4.76) | 148 (2 studies) | ⊕⊕⊕⊝ moderateg | ‐ | |
| 107 per 1000 | 234 per 1000 (107 to 508) | |||||
| *The basis for the assumed risk (e.g. the mean control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio; SMD: standardised mean difference | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
aDisability is outcome is reported as a standardised mean difference. The sample size is large (> 400) and the 95% CI overlaps no effect and therefore we have downgraded for imprecision.(GRADE 2013). bHeterogeneity may be due to clinical variation ‐ Chollet 2011 and Marquez Romero 2013 give 20 mg fluoxetine for 90 days and FOCUS Trial Collaboration 2018 gives 20 mg for 180 days; methodological differences (when the outcomes are measured) ‐ Chollet 2011 and Marquez Romero 2013 measure at 90 days and FOCUS Trial Collaboration 2018 measures at 180 days; or population differences ‐ Chollet 2011 recruited participants only with ischaemic stroke with motor deficits, Marquez Romero 2013 recruited only participants with haemorrhagic stroke and FOCUS Trial Collaboration 2018 recruited all pathological subtypes. The trials were performed in different countries where other factors such as the amount of therapy might influence outcome. These are plausible explanations for the observed heterogeneity and we have therefore we have not downgraded the evidence (Schünemann 2017). cThe optimal information size criterion (FOCUS Trial Collaboration 2018) has been met and the 95% CI overlaps no effect; we have therefore downgraded for imprecision (GRADE 2013). dNeurological deficit outcomes reported as a SMD. Sample size is < 400 and we have therefore downgraded for imprecision (GRADE 2013). eStudies used different measures of depression. This variability in study design may have contributed to variability in intervention effects. fThe sample size is large (> 2000) but the 95% CI overlaps no effect; we have therefore downgraded for imprecision (GRADE 2013). gStudies are small with too few events and wide CIs; we have therefore downgraded for imprecision (Schünemann 2017).
Background
Description of the condition
Worldwide, stroke is the second leading cause of death, the third leading cause of disability (Johnson 2016), and results in 6.5 million years being lived with disability (GBD 2015). Although major advances in the early reperfusion of ischaemic stroke have been achieved in recent years (e.g. by intravenous thrombolysis, thrombectomy, and prevention of early recurrent stroke), effective, safe and widely accessible and affordable treatments that facilitate early and sustained recovery after stroke are urgently needed to further reduce the burdens of disability and dependency after stroke.
Description of the intervention
Selective serotonin reuptake inhibitors (SSRIs) are drugs that have been available for many years. They are widely used to treat mood disorders, including those that occur after stroke, such as depression and emotional lability (i.e. emotional behaviour that the patient reports as being outside normal control and that occurs in situations that previously would not have provoked such behaviour) (Allida 2019). Small trials suggest that fluoxetine, one of the SSRIs, might have a favourable effect on motor recovery after stroke (Chollet 2011; Yi 2010). Our 2012 Cochrane Review of SSRI for stroke recovery confirmed the positive effects seen in the small trials. Combining all SSRIs into a single review and meta‐analysis is justified because the mechanism of action for the different drugs are very similar.
How the intervention might work
In animal studies, multiple potentially beneficial effects of SSRIs have been demonstrated in both normal and diseased brains. First, SSRIs have a neurotrophic effect. Neurotrophins are a family of proteins that are involved in embryogenesis (formulation of an embryo) and organogenesis (development of organs). They control neural plasticity (ability to change, or easily changed or shaped) in adults, regulate synaptic activity and neurotransmitter synthesis, and are essential for the regeneration of nerves (Lang 2004). The development of new nerve cells in adults is generally restricted to specific areas of the brain, namely the subependymal cells of the ventricular system and the subgranular zone of the dentate gyrus in the hippocampus (Ming 2005). SSRIs increase neurogenesis and expression of neurotrophic or growth factors in the adult hippocampus (Schmidt 2007), and this is likely to account for the behavioural benefits of antidepressants in animals (Santarelli 2003). Importantly, several studies have shown that migration of new neurones to damaged areas of brain may occur (Wiltrout 2007), and that neurogenesis can also occur within areas of damaged brain, for example in people with Alzheimer's disease and in animal models of Alzheimer's disease (Taupin 2006). Second, fluoxetine may have a neuroprotective effect (i.e. protect nerve cells when the brain is damaged, e.g. by a stroke). In animals, there may be several mechanisms for neuroprotective effects of SSRIs, such as reducing inflammation (e.g. repression of microglia activation) (Lim 2009), and by enhancement of specific protein expression (hypoxia inducible factor–1 alpha, heme oxygenase‐1) (Shin 2009). Third, SSRIs can indirectly affect an important hormonal system in the body, the adrenergic system, through up‐regulation (i.e. increase a cellular component of a cell, such as ribonucleic acid (RNA) or protein, in response to an external variable) of beta1 receptors (Pälvimäki 1994).
In healthy humans, functional magnetic resonance imaging (fMRI) studies have demonstrated that fluoxetine can modulate cerebral motor activity (Loubinoux 1999). In eight chronic stroke participants in Zittel 2008, a single dose of citalopram 40 mg led to improvements in dexterity.
SSRIs may also improve recovery after stroke simply through their effect on preventing or treating depression and anxiety, and through improving sleep and alertness.
Why it is important to do this review
Our 2012 Cochrane Review of SSRIs for stroke recovery showed that SSRIs appeared to reduce dependence, disability, neurological impairment, anxiety, and depression after stroke, even in participants without depression. However, there was heterogeneity between trials and methodological limitations in many of the trials. When we included only those trials at low risk of bias, effect sizes were much smaller. The review generated the hypothesis that SSRIs might promote recovery after stroke, and the review authors recommended that well‐designed trials were needed to determine whether SSRIs given routinely to people early after stroke improved their recovery.
SSRIs interact with platelet function and clotting, and therefore may have adverse effects in people with stroke, particularly those with haemorrhagic stroke, and these adverse effects might outweigh any potential benefits.
Three large collaborative trials were designed based on the results of the 2012 Cochrane Review (Mead 2012), to test the hypothesis that fluoxetine given early after stroke would improve recovery, or in other words, lead to less dependency and less disability at follow‐up. The largest, the FOCUS Trial Collaboration 2018, recruited over 3000 participants and has now reported, but is not included in any systematic review or meta‐analysis.
Cochrane Reviews should be updated regularly, ideally every two years. In practice, this is not always possible but they should certainly be updated when substantial new evidence becomes available. Our review team knew that the results of FOCUS, a major trial, would be available in December 2018 and so we planned this current update to include FOCUS (FOCUS Trial Collaboration 2018). If a simple, inexpensive drug such as one of the SSRIs improves stroke recovery, this would have major implications for patients, carers, health services, social care services, and the economy.
Objectives
To determine if SSRIs are more effective than placebo or usual care at improving outcomes in people less than 12 months post‐stroke, and to determine whether treatment with SSRIs is associated with adverse effects.
Methods
Criteria for considering studies for this review
Types of studies
We included all randomised controlled trials (RCTs) in people with a clinical diagnosis of stroke (Hatano 1976), where an SSRI had been given within the first year of stroke onset, i.e.: 1) trials that stipulated that participants had to be recruited within 12 months of stroke onset, or 2) trials where the mean (or median) time since stroke was less than 12 months. Trials had to have measured at least one of our outcomes of interest in order to be included in the meta‐analysis. For those trials which did not report data in a form that we could use, we attempted to get the raw data from the authors, and if this was not possible, we retained the studies in the list of included studies.
We included trials:
with more than two arms (e.g. SSRI versus another active treatment versus placebo). We included data from the SSRI arm and the placebo arm (or usual‐care arm if a placebo was not used), and discarded data from the other active treatment arms;
in all languages.
We excluded trials:
using a quasi‐experimental design (i.e. where investigators describe a non‐random component in the sequence generation process, such as date of admission);
using a cross‐over design;
in which two or more active interventions were compared against each other rather than against a placebo or a standard care group;
combined an SSRI with another active intervention and compared it to the active treatment alone.
There was no restriction on the eligibility of RCTs on the basis of sample size or duration of follow‐up.
Types of participants
We included trials that had recruited survivors of a stroke, defined as a sudden‐onset focal neurological disturbance, assumed to be vascular in origin, and lasting more than 24 hours (Hatano 1976). Trials had to recruit participants within 12 months of stroke onset, or the mean time since stroke had to be less than 12 months. We intended to include trials in subarachnoid haemorrhage and perform a subgroup analysis but we did not find any such trials. We intended to exclude trials of mixed populations (e.g. stroke and head injury) unless separate results for those with stroke were available, but we found no such trials.
Types of interventions
We included any drug classified as a SSRI (e.g. fluvoxamine, fluoxetine, sertraline, citalopram and paroxetine). We included any dose or mode of delivery, given for any duration and for any reason (e.g. to aid neurological recovery, to treat depression or anxiety or emotionalism, or to prevent depression or anxiety or other mood disorders). We did not include drugs that have mixed effects that include SSRI actions.
The comparator arm could include usual care or a placebo. We excluded studies in which fluoxetine was compared with another active intervention (e.g. another type of drug or herb or acupuncture). In this update, we also excluded trials that combined an SSRI with another active treatment and compared with the active treatment alone, because of the potential for interaction between the SSRI and other active treatment.
Types of outcome measures
Primary outcomes
We had two primary outcomes:
independence at end of treatment. In stroke trials this is typically measured using the modified Rankin Scale (mRS), with a score of 0 to 2 conventionally considered to represent independence;
disability score at the end of treatment. Measures included, but were not limited to, Barthel index (BI) or Functional Independence Measure (FIM).
Although disability scores and independence (or not) are arguably measuring the same concept, disability scores provide a more detailed description of functional outcome than simply using a dichotomous outcome such as independence or not. In other words, we were interested in performance in personal activities of daily living (ADL)/disability (measured using disability scores) and independence in performance in personal ADL/disability measured using dichotomous outcome (independent or not).
Note that 'end of treatment' depends on the duration of treatment, and so the outcome might be measured at different time points in different trials. But we justified this approach provided that trials measured the outcome at the same time point in each group.
Secondary outcomes
Impairments (which can include neurological deficit scores, motor deficit scores)
Depression
Anxiety
Quality of life
Fatigue
Healthcare cost
Death
Adverse events including gastrointestinal (GI) side effects, bleeding, seizures, and any other side effect
Leaving the trial early (for any reason, including death)
We anticipated that most trials would assess these at the end of treatment and possibly at one or more time points. We did not stipulate a minimum follow‐up time. We did not stipulate in advance precisely how multiple time points would be handled (if they had been found); we will consider this for the next update.
Search methods for identification of studies
See the methods for the Cochrane Stroke Group Specialised register. We searched for trials in all languages and arranged for the translation of trials where necessary.
Electronic searches
We developed the MEDLINE search strategy with the help of the Cochrane Stroke Group Information Specialist and adapted it for the other databases.
We searched the following electronic bibliographic databases:
Cochrane Stroke Group Trials Register (16 July 2018);
Cochrane Central Register of Controlled Trials (CENTRAL) Issue 7 of 12, (July 2018) (Appendix 1);
MEDLINE (from 1948 to 16 July 2018) (Appendix 2);
Embase Ovid (from 1980 to 16 July 2018) (Appendix 3);
CINAHL EBSCO (Cumulative Index to Nursing and Allied Health Literature (1982 to July 2018) (Appendix 4);
AMED Ovid (Allied and Complementary Medicine) (from 1985 to 16 July 2018) (Appendix 5);
PsycINFO Ovid (from 1967 to 16 July 2018) (Appendix 6);
PsycBITE Pyschological Database for Brain Impairment Treatment Efficacy (www.psycbite.com/) (16 July 2018).
In addition, we searched the following ongoing trials registers (Appendix 7):
Stroke Trials Registry (www.strokecenter.org/trials) (26 June 2018);
US National Intitutes of Health ongoing Trials Register ClinicalTrials.gov (www.ClinicalTrials.gov) (16 July 2018);
ISRCTN Registry (www.isrctn.com) (26 June 2018);
EU Clinical Trials Register (www.clinicaltrialsregister.eu) (26 June 2018);
World Health Organization International Clinical Trials Registry Platform (apps.who.int/trialsearch/) (last searched 16 July 2018).
Evidence for this update included search results from the previous version of this review (Mead 2012), combined with results from the above searches. In Mead 2012 there were no date limits and searches were applied from inception of databases.
Searching other resources
In an effort to identify further published, unpublished and ongoing trials, we:
searched reference lists of included trials and relevant reviews when full texts were retrieved for detailed scrutiny;
contacted researchers in the field.
Data collection and analysis
Selection of studies
Joshua Cheyne (Cochrane Stroke Group Information Specialist), ran the searches of CENTRAL, MEDLINE, Embase, CINAHL, AMED, and PsycINFO, and downloaded the resulting references into Reference Manager. These were imported into Covidence, which automatically removed some, but not all, of the duplicates.
Any two review authors (from GM, EL, LL, MK, BS, SW, RT, A‐SR, or C‐FH) independently scrutinised the resulting titles and abstracts and excluded obviously irrelevant reports and duplicates. We obtained full texts of potentially eligible studies. Any two review authors (from GM, EL, LL, MK, BS, SW, RT, A‐SR, C‐FH) independently applied inclusion and exclusion criteria; if there was lack of consensus, a third review author (usually GM unless she had already scrutinised the paper) also applied inclusion and exclusion criteria.
For this update we include a study flow diagram that includes the number of unique references identified by the searches, the number of records excluded after preliminary screening of titles and abstracts, the number of records retrieved in full text, and the number fulfilling our inclusion criteria (see Figure 1).
Data extraction and management
For the new eligible trials we had identified, we created the necessary data fields in Covidence for each individual trial. Any two review authors (from GM, EL, LL, MK, MH, BS, SW, RT, LL, A‐SR, C‐FH) independently extracted data from each new trial.
We extracted the following data:
the report: author, year and source of publication;
the study: sample characteristics, social demography;
the participants: stroke sequence (first‐ever versus recurrent), social situation, time since stroke onset, prior history of psychiatric illness, current neurological status, stroke severity, whether people with aphasia were recruited, the proportion with depression at baseline (if recorded by trialists). We did not extract information on location or size of lesion as this was unlikely to have been recorded by the trialists, and brain imaging often does not show a visible infarct in people with minor strokes;
the research design and features: adherence, non‐response and length of follow‐up;
the intervention: type, duration, dose, timing and mode of delivery;
the effect size: sample size, nature of outcome, estimate and standard deviation (SD) (or standard error (SE));
Source of funding.
Assessment of risk of bias in included studies
We assessed risks of bias using the Cochrane 'Risk of bias' tool (Higgins 2017). We assessed the methods used in each study to control for the following potential sources of bias: sequence generation (selection bias); allocation concealment (selection bias); blinding of participants, personnel and outcome assessors (performance and detection bias); incomplete outcome data (attrition bias); selective outcome reporting (reporting bias); and other potential threats to validity.
For incomplete outcome data, we categorised as 'low risk' if missing data were imputed using appropriate methods or if missing outcome data overall were less than 5%.
We extracted data on source of funding, and listed this under 'Other sources of bias'. If the source of funding was not given, or if there were links with the pharmaceutical industry and no explicit statement that the funder had no input into the design or analysis of the study, we classified this as 'unclear risk'. We also recorded any other potential threats to validity.
We also extracted data on how adverse effects were reported, and listed these in the descriptions of the studies.
If a trial author was also one of the review authors, then a review author who was not involved in the trial extracted data and assessed quality.
Measures of treatment effect
For dichotomous data, we reported risk ratios (RRs). For ordinal scales, where there was a well‐recognised cut‐point in the scale (e.g. mRS) we analysed the data as a dichotomous outcome (dependent or independent).
For ordinal scales with no recognised cut‐point, we analysed the data as continuous data. The data required for meta‐analyses of continuous data in Review Manager 5 are means and standard deviations (SDs) (Review Manager 2014). When extracting continuous data from the study reports, we checked whether trials reported the SD or the standard error (SE). We had planned to use the SE or 95% confidence interval (CI) to compute the SD when SDs were missing, but this was not needed as all the trials reported SDs.
For ordinal scales and continuous data, we calculated standardised mean differences (SMDs), because different scales were used for the same outcomes (e.g. BI and FIM for disability score, the Beck Depression Inventory (BDI) or the Hamilton Rating Scale for Depression (HAMD) for depression). It should be noted that the SMD does not correct for differences in the direction of the scale. As some scales increase with disease severity and others decrease, we multiplied the mean value from one set of trials by −1. For example, in the National Institute of Health Stroke Scale a low score indicates a less severe stroke, whilst a low score in the Scandinavian Stroke Scale (SSS) indicates a more severe stroke.
If there was more than one outcome measure in the same domain (e.g. two different depression scales), we made a post hoc decision to select the one with the most complete data. We did not specify all 'acceptable' outcome measures in this review, but we will need to do this for future updates.
Unit of analysis issues
The number of observations in the analysis should match the number of 'units' that were randomised. We considered outcomes measured at the end of treatment and at the end of follow‐up in separate analyses. For side effects, we considered the number of participants developing a particularly side effect rather than the total number of side effects in each group.
Dealing with missing data
For this update, we contacted authors of new trials to obtain any data that we needed for our meta‐analysis that had not been included in a published full‐text article or an abstract.
Assessment of heterogeneity
We assessed whether there was evidence of inconsistency in our results by considering possible clinical, methodological, and statistical heterogeneity. We assessed clinical and methodological heterogeneity by comparing similarities in our included studies between study designs, participants, interventions, and outcomes.
We quantified the effect of heterogeneity using the I2 statistic. We assessed statistical heterogeneity by visually examining forest plots. We used the following cut‐offs from the Cochrane Handbook for Systematic Reviews of Interventions as a rough guide to interpretation (Section 9.5.2; Deeks 2017):
0% to 40% is not considered important;
30% to 60% suggests moderate heterogeneity;
50% to 90% suggests substantial heterogeneity;
75% to 100% is considerable heterogeneity
Assessment of reporting biases
We searched clinical trials registers to identify published protocols for each of our included studies. We checked for selective reporting of results by comparing the published protocol with the published full‐text article and by scrutinising the aims and methods of the trials and comparing these with outcomes reported. We found several papers by the same authors, and contacted the authors to check whether the publications were duplicates or to check if the included study populations were unique. If it was not possible to determine whether different publications reported overlapping groups of participants, we included just one of the papers and listed the others as awaiting assessment.
If we had identified a sufficient number of included studies at low risk of bias (i.e. more than 10 studies (Sterne 2017)), we would have generated a funnel plot to assess risk of publication bias in the review; an asymmetrical funnel plot might have suggested publication of only positive results (Egger 1997).
We deployed a comprehensive search strategy in an effort to avoid reporting biases in our review methodology. See Search methods for identification of studies.
We tried to avoid language bias by including all trials, irrespective of language: we sought translation where needed.
Data synthesis
We completed meta‐analysis of outcomes for which we had comparable effect measures from more than one study, and when measures of heterogeneity indicated that pooling of results was appropriate. We used the statistical calculator provided in Review Manager 5 to perform meta‐analysis (Review Manager 2014).
We used a fixed‐effect model (Mantel 1959), rather than a random‐effects model because of the dominance of the FOCUS trial (FOCUS Trial Collaboration 2018); random effects would have given too much weight to the smaller trials. The dominance of the FOCUS trial makes a fixed‐effect model a more reliable indicator of the effect than the average across the smaller trials. We assessed the robustness of the results to choice of model using a sensitivity analysis.
In the 2012 review, we performed multiple meta‐analyses of all outcomes, and included all trials irrespective of risk of bias. We then explored the influence of each aspect of bias on estimates of effects in a series of sensitivity analyses. This approach generated the hypothesis that SSRIs might improve stroke recovery, but also suggested that the apparently beneficial effects might simply have been due to bias, with trials at higher risk of bias tending to give positive results. This approach generated multiple forest plots.
For this update, we decided to limit our primary analysis to studies at low risk of bias (Higgins 2017), as we wanted to reliably find out whether SSRIs are more effective than placebo or usual care at improving disability or independence in people less than 12 months post‐stroke.
We reached decisions on overall risk of bias by study by consideration of six 'Risk of bias' domains: sequence generation, allocation concealment, blinding of participants and trial personnel, blinding of outcome assessment, incomplete outcome data, and selective outcome reporting. We required a study to have a judgement of low risk of bias in all six domains in order to categorise it as having an overall low risk of bias. We did, however, include the studies with an unclear or high risk of bias judgements in these six domains in a sensitivity analysis of our co‐primary outcomes of disability and independence.
Subgroup analysis and investigation of heterogeneity
If there had been at least two studies at low risk of bias, we would have explored variability in the participants, interventions, and outcomes among studies using the following subgroups.
Type of SSRI.
Trials with depression at baseline as an inclusion criterion and those where depression was not an inclusion criterion.
Time since stroke at recruitment. We categorised these as less than three months (0 ‐ 90 days), three to six months (91 to 108 days), six to nine months (181 to 271 days) or nine to 12 months (272 to 365 days).
If we found high statistical heterogeneity we still performed the subgroup analysis, but considered the reason for this heterogeneity.
Sensitivity analysis
We explored the potential effects of decisions made as part of the review process as follows.
We then included all studies regardless of 'Risk of bias' judgement for our primary outcomes of disability score and independence.
We conducted meta‐analysis using the alternate meta‐analytical effects model (fixed‐effect or random‐effects).
We conducted a meta‐analysis using the alternate 'last available follow‐up' time point.
We compared effect estimates from the above results with effect estimates from the main analysis. We reported differences that altered the interpretation of effects.
GRADE and 'Summary of findings'
We created a 'Summary of findings' table using the following outcomes: disability; dependent according to the mRS; neurological deficit score; depression (continuous data); death; seizures; and gastrointestinal side effects (Table 1). We used the five GRADE considerations (study limitations, consistency of effect, imprecision, indirectness and publication bias) to assess the quality of the body of evidence as it relates to the studies that contribute data to the meta‐analyses for the prespecified outcomes (Atkins 2004). We used methods and recommendations described in Chapter 11 of the Cochrane Handbook for Systematic Reviews of Interventions (Schünemann 2017), using GRADEproGDT software (GRADEproGDT 2015). We justified all decisions to downgrade the quality of studies using footnotes, and made comments to aid the reader's understanding of the review where necessary.
Results
Description of studies
For substantive descriptions of studies see: Characteristics of included studies, Characteristics of excluded studies, Characteristics of studies awaiting classification, and Characteristics of ongoing studies.
Results of the search
For this update, we screened 2988 references from database searches and accessed the available full‐text reports for 499 studies.
The previous version of this review (Mead 2012) identified 56 eligible completed studies recruiting 4109 participants, five ongoing studies (Anonymous 2005; Hankey 2011; Carda 2009; Kim 2011; FOCUS Trial Collaboration 2018), and two studies that were awaiting classification (Sitzer 2002; Whyte 2005).
The flow of search results for the previous version of the review are reported in Mead 2012. We report details of the search for this update in a PRISMA flow chart (Figure 1).
1.
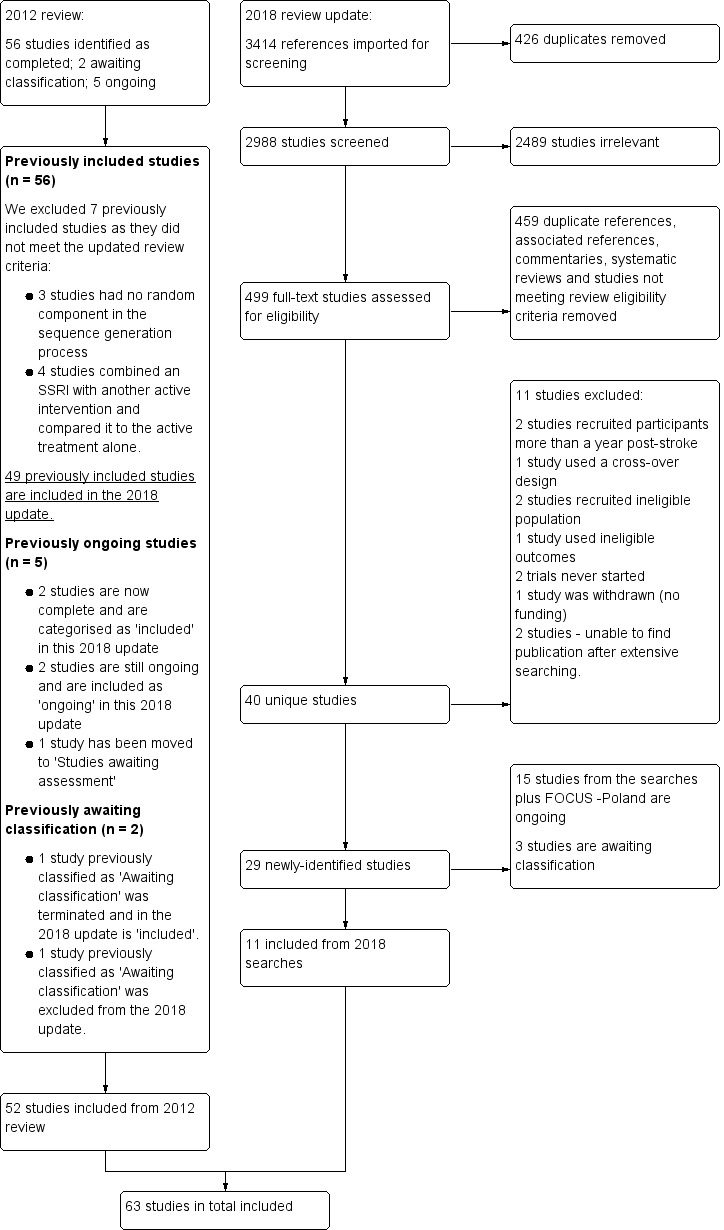
Flow diagram showing the searches for this update. FOCUS Poland identified through personal communication
Included studies
The previous version of this review identified 56 eligible completed studies recruiting 4109 participants (Acler 2009; Almeida 2006; Andersen 1994; Brown 1998; Burns 1999; Chen 2001; Chen 2002; Chen 2005a; Chen 2005b; Cheng 2003; Chollet 2011; Dam 1996; Feng 2004; Fruehwald 2003; GlaxoSmithKline 1998; Guo 2009; He 2004; He 2005; Hu 2002; Huang 2002; Jia 2005; Kong 2007; Lai 2006; Li 2004a; Li 2004b; Li 2005; Li 2006; Li 2008; Liu 2006; Meara 1998; Miao 2004; Murray 2005; Pariente 2001; Rasmussen 2003; Restifo 2001; Robinson 2000a; Robinson 2000b; Robinson 2008; Song 2006; Wang 2003; Wen 2006; Wiart 2000; Xie 2005; Xu 2001; Xu 2006; Yang 2002; Yang 2011; Ye 2004; Zhou 2008; Finkenzeller 2009; Ji 2000; Li 2002; Liang 2003; Liu 2004; Xu 2007; Zhou 2003).
Of these we excluded seven (439 participants) from this update (Finkenzeller 2009; Ji 2000; Li 2002; Liang 2003; Liu 2004; Xu 2007; Zhou 2003), as they did not fulfil our more stringent inclusion criteria. See Excluded studies.
Two previously ongoing studies are now complete (FOCUS Trial Collaboration 2018; Kim 2011), two studies are still ongoing (Anonymous 2005; Hankey 2011), and one study has been moved to studies awaiting assessment, as we were unable to make contact with the investigators (Carda 2009). One study previously classified as 'Awaiting assessment' was terminated due to difficulties meeting recruitment goals; it did not state the number of participants but has been retained in our narrative review (Whyte 2005).
We identified a further 11 eligible studies from the 2018 search (Andersen 2013; Black‐Schaffer 2012; Birchenall 2019; Gao 2016; He 2016; Marquez Romero 2013; Pan 2018; Razazian 2014; Savadi Oskouie 2012; Shah 2016; Zhao 2011). Thus, in addition to FOCUS Trial Collaboration 2018, Kim 2011 and Whyte 2005 there is a total of 14 new studies for this update, recruiting a further 5498 participants.
There are now 63 included studies recruiting a total of 9168 randomised participants. Not all the studies provided data that could be used in the meta‐analysis (thus the denominator in the forest plots does not equal 9168). Two trials did not state the number of participants (Meara 1998; Whyte 2005).
Of the 63 included studies:
32 trials used fluoxetine (Birchenall 2019; Black‐Schaffer 2012; Brown 1998; Chen 2001; Cheng 2003; Chollet 2011; Dam 1996; Feng 2004; FOCUS Trial Collaboration 2018; Fruehwald 2003; He 2004; He 2016; Hu 2002; Huang 2002; Kong 2007; Li 2004a; Li 2004b; Li 2008; Marquez Romero 2013; Pariente 2001; Razazian 2014; Restifo 2001; Robinson 2000a; Robinson 2000b; Shah 2016; Song 2006; Wang 2003; Wen 2006; Wiart 2000; Xu 2001; Zhao 2011; Zhou 2008);
8 trials used sertraline (Almeida 2006; Burns 1999; Guo 2009; Meara 1998; Murray 2005; Rasmussen 2003; Whyte 2005; Xie 2005);
11 used paroxetine (Chen 2002; Chen 2005b; GlaxoSmithKline 1998; He 2005; Lai 2006; Li 2005; Pan 2018; Xu 2006; Yang 2002; Yang 2011; Ye 2004);
8 used citalopram (Acler 2009; Andersen 1994; Andersen 2013; Gao 2016; Li 2006; Liu 2006; Miao 2004; Savadi Oskouie 2012);
2 used escitalopram (Kim 2011; Robinson 2008);
1 used either sertraline or fluoxetine (Jia 2005)
1 used citalopram or fluoxetine (Chen 2005a)
Baseline sociodemographic and clinical characteristics
Six trials do not present baseline demographic and clinical characteristics for each group, but rather the baseline demographic and clinical characteristics for only those completing the trial are presented (He 2016; Kim 2011; Pan 2018; Razazian 2014; Savadi Oskouie 2012; Shah 2016).
The mean age of participants ranged from 51 ± 7 years (Song 2006) to 75.6 years (Wang 2003), with most trials recruiting participants in their 60s (data from 48/63studies).
Mean time since stroke
Of the 63 included studies:
38 report recruiting participants between 0 and 90 days after stroke onset: (Acler 2009; Almeida 2006; Andersen 1994; Andersen 2013; Birchenall 2019; Chen 2001; Chen 2005b; Cheng 2003; Chollet 2011; Feng 2004; FOCUS Trial Collaboration 2018; Fruehwald 2003; Gao 2016; He 2004; He 2016; Hu 2002; Huang 2002; Kim 2011; Kong 2007; Li 2004a; Li 2004b; Li 2008; Marquez Romero 2013; Pan 2018; Rasmussen 2003; Robinson 2008; Savadi Oskouie 2012; Shah 2016; Song 2006; Wen 2006; Wiart 2000; Xie 2005; Xu 2001; Xu 2006; Yang 2011; Ye 2004; Zhao 2011; Zhou 2008). A further three trials described participants as having an 'acute stroke'; we assume this meant zero to three months, so have included these in the zero‐to‐three‐month group (He 2005; Lai 2006; Li 2006).Two further studies reported that the mean time since stroke was between five and 16 weeks, so we included these in the zero‐to three‐month group (Robinson 2000a; Robinson 2000b). One trial, which did not recruit any participants, had an inclusion criterion of less than 15 days before stroke (Black‐Schaffer 2012);
four trials report recruiting participants three to six months (91 to 108 days) after stroke onset: Dam 1996 (described as participants being one to six months); Miao 2004, Murray 2005, and Yang 2002 ('recovery phase of stroke' two to six months);
two trials report recruiting participants at six to nine months (181 to 271 days) after stroke onset (Guo 2009; Liu 2006);
no trials reported recruiting participants between nine and 12 months after stroke;
one trial reported the experimental and control group being median 10.5 months and 5.5 months after stroke, respectively (Burns 1999).
12 trials did not report the precise time (Brown 1998; Chen 2002; Chen 2005a; GlaxoSmithKline 1998 (less than 12 months); Jia 2005; Li 2005; Meara 1998; Pariente 2001; Razazian 2014; Restifo 2001; Wang 2003; Whyte 2005).
Depression as an inclusion criterion
Thirty‐three studies included participants affected by depression (i.e. depression used as an inclusion criterion): Andersen 1994; Chen 2001; Chen 2002; Chen 2005a; Chen 2005b; Cheng 2003; Feng 2004; Fruehwald 2003; GlaxoSmithKline 1998; Guo 2009; He 2005; Hu 2002; Huang 2002; Jia 2005; Lai 2006; Li 2004a; Li 2004b; Li 2005; Li 2006; Li 2008; Liu 2006; Meara 1998; Miao 2004; Murray 2005; Robinson 2000a; Song 2006; Wang 2003; Wiart 2000; Xie 2005; Xu 2001; Yang 2002; Yang 2011; Ye 2004.
Thirty studies did not use depression as an inclusion criterion: Acler 2009; Almeida 2006; Andersen 2013; Birchenall 2019; Black‐Schaffer 2012; Brown 1998; Burns 1999; Chollet 2011; Dam 1996; FOCUS Trial Collaboration 2018; Gao 2016; He 2004; He 2016; Kim 2011; Kong 2007; Marquez Romero 2013; Pan 2018; Pariente 2001; Rasmussen 2003; Razazian 2014; Restifo 2001; Robinson 2000b; Robinson 2008; Savadi Oskouie 2012; Shah 2016; Wen 2006; Whyte 2005; Xu 2006; Zhao 2011; Zhou 2008.
Criteria for diagnosing depression varied between trials.
Excluded studies
In line with the guidance for Cochrane Reviews, which states that the list of excluded studies should be as brief as possible and should not list studies that obviously do not fulfil the inclusion criteria, we have now listed only 20 of these in the Characteristics of excluded studies'. Of these 20 studies, the reasons for exclusion are as follows.
We excluded three studies that we had previously included but were no longer eligible for this review, as there was no random component in the sequence generation process (Li 2002; Liang 2003; Zhou 2003). We excluded four studies that combined an SSRI with another active intervention and compared it to the active treatment alone (Finkenzeller 2009; Ji 2000; Liu 2004; Xu 2007).
We excluded two studies listed as 'Awaiting classification' in the previous version of this review because we could find no published results, and when we sought further information from the authors we received no responses (Graffagnino 2002; Sitzer 2002). Given the insufficient information to assess eligibility and, owing to the length of time since the study abstract was published, we have now excluded these studies.
We excluded four studies because they recruited participants more than one year post‐stroke (Berends 2009; Choi Kwon 2008; Gourab 2015; Sun 2015).
Other reasons for exclusion were: cross‐over design (Andersen 1993), ineligible outcomes (Robinson 2011), trial never started (Andersen 2012; Anderson 2002), study withdrawn (no funding) (University of Alabama 2013), and unable to find publication after extensive searching (Anonymous 2012; Anonymous 2012b).
See Characteristics of excluded studies for studies excluded during this update. Studies excluded in previous searches are listed in Mead 2012.
Ongoing studies
In addition to one study identified as ongoing in the previous review (Anonymous 2005), we identified 14 new ongoing studies from the clinical trials register searches (Chollet 2016; Cocho 2015; Dike 2019; Farokhi 2017; Fregni 2014; Hankey 2011; Karimialavijeh 2017; Leibovitch 2018; Levitt 2019; Lundström 2014; Pastore‐Wapp 2016; Pirzeh 2012; Sadaat 2012; Sahin 2016), and one from personal contact (FOCUS‐Poland 2014). See Characteristics of ongoing studies.
Studies awaiting classification
We were unable to assess review eligibility for four studies (Carda 2009; Guo 2015; He 2012; Jurcau 2016). Carda 2009 has been moved from 'Ongoing studies' in the previous review (Mead 2012), to 'Studies awaiting classification' in this update, as we were unable to make contact with the investigators. Jurcau 2016 was published as an abstract and included insufficient detail; the author did not respond to requests for information. From a combination of trial registration details and published information, and after contacting authors, we were uncertain whether three studies (Guo 2015; He 2012; He 2016) included unique study populations and we therefore decided to include the data from one publication (He 2016), and classified the remaining two as 'Awaiting classification' (Guo 2015; He 2012). See Characteristics of studies awaiting classification.
Risk of bias in included studies
All 63 studies were RCTs.
2.

Risk of bias summary: review authors' judgements about each risk of bias item for each included study.
3.
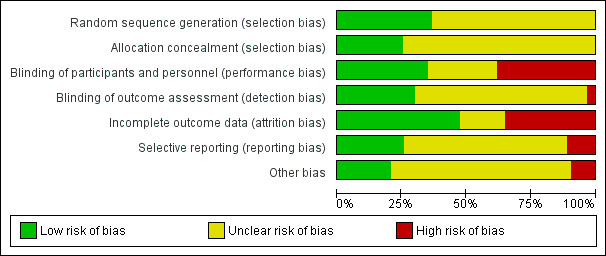
'Risk of bias' graph: review authors' judgements about each 'risk of bias' item presented as percentages across all included studies.
Allocation
We judged the random sequence generation to be adequately presented (i.e. there was a low risk of bias) in 22 studies. For the remaining 41 studies, the risk of selection bias was unclear. There was no study in which the random sequence generation was inadequate (i.e. there was a high risk of bias).
Sixteen studies had adequate (low risk) allocation concealment. In the other 47 studies, the details provided did not allow an assessment of the methods used to prevent investigators and participants from foreseeing the assignment (unclear risk of bias).
Blinding
We judged the blinding of participants and personnel (performance bias) as low risk in 22 studies, as high risk in 24 studies, and as unclear risk in the remaining 17 studies.
We judged the blinding of outcome assessment (detection bias) to be at low risk in 19 studies, at high risk in two studies, and at unclear risk in the remaining 42 studies.
Incomplete outcome data
The risk of incomplete outcome data (attrition bias) was at low risk in 30 studies, at high risk in 22 studies, and at unclear risk in the remaining 11 studies.
Selective reporting
We judged missing data to be at low risk in 16 studies, high risk in seven studies, and as having unclear risk in the other 40 studies.
Other potential sources of bias
We judged 13 studies to be at low risk of bias from other potential sources of bias, at high risk in six studies, and as having unclear risk in the other 44 studies.
Overall risk of bias
The FOCUS Trial Collaboration 2018, Chollet 2011, and Marquez Romero 2013 studies had an overall low risk of bias (i.e. low risk of bias in each of six domains: sequence generation, allocation concealment, blinding of participants and trial personnel, blinding of outcome assessment, incomplete outcome data and selective outcome reporting) (Figure 2).
Effects of interventions
See: Table 1
Primary outcomes
Disability score at the end of treatment
We combined data for studies with an overall low risk of bias for the outcome of disability score, using a standardised mean difference (SMD) with a fixed‐effect model (SMD −0.01, 95% CI −0.09 to 0.06; P = 0.75; 2 studies, 2829 participants; moderate‐quality evidence) with no important heterogeneity There was no difference in measures of disability score between SSRI intervention and placebo (Analysis 1.1).
1.1. Analysis.
Comparison 1 SSRI versus control at end of treatment, by SSRI, Outcome 1 Disability (primary analysis).
Independent on modified Rankin score (mRS 0 to 2) at the end of treatment
We combined data for studies with an overall low risk of bias for the outcome of independent on mRS 0 to 2 using a risk ratio with a fixed‐effect model (RR 1.00, 95% CI 0.91 to 1.09; P = 0.99; 3 studies, 3249 participants; moderate‐quality evidence). There was no difference in mRS (independence) between SSRI intervention and placebo (Analysis 1.2), but substantial heterogeneity between studies with an I2 of 78%.
1.2. Analysis.
Comparison 1 SSRI versus control at end of treatment, by SSRI, Outcome 2 Independent on modified Rankin score (mRS 0 to 2) (primary analysis).
Secondary outcomes
Neurological deficit score at the end of treatment
We combined data for studies with an overall low risk of bias for the outcome of neurological deficit score, using the SMD with a fixed‐effect model. The analysis found no difference in neurological scores between SSRI and placebo (SMD −0.30, 95% CI −0.63 to 0.04; P = 0.08; 2 studies, 142 participants; moderate‐quality evidence), with no important heterogeneity (I2 = 0%) (Analysis 1.3).
1.3. Analysis.
Comparison 1 SSRI versus control at end of treatment, by SSRI, Outcome 3 Neurological deficit score.
Depression severity at end of treatment (continuous data)
We combined data for studies with an overall low risk of bias for the outcome of depression using the SMD with a fixed‐effect model (SMD −0.11, 95% CI −0.19 to −0.04; P = 0.002; 2 studies, 2861 participants; moderate‐quality evidence). Participants who received an SSRI intervention had significantly lower end‐of‐treatment scores on measures of depression than those participants receiving placebo (Analysis 1.4). However, there was substantial heterogeneity between trials (I2 = 69%).
1.4. Analysis.
Comparison 1 SSRI versus control at end of treatment, by SSRI, Outcome 4 Depression (continuous data).
Depression at the end of treatment (dichotomous data)
Data were not available for more than one study at low risk of bias for any analysis (Analysis 1.5). This high‐quality study demonstrated that fluoxetine reduced the risk of depression at the end of treatment (RR 0.78, 95% CI 0.66 to 0.92; 1 study, 3127 participants).
1.5. Analysis.
Comparison 1 SSRI versus control at end of treatment, by SSRI, Outcome 5 Depression (dichotomous data).
Anxiety severity at end of treatment (continuous data)
No studies at low risk of bias reported measures of anxiety.
Anxiety severity at end of treatment (dichotomous data)
No studies at low risk of bias reported number of diagnoses of anxiety.
Cognition at end of treatment (continuous data)
No studies at low risk of bias reported cognition at the end of treatment.
Death at end of treatment
We combined data for studies with an overall low risk of bias for the outcome of death, using a risk ratio with a fixed‐effect model. The analysis found no difference in the total number of deaths between SSRI and placebo (RR 0.99, 95% CI 0.79 to 1.25; P = 0.95; 3 studies, 3254 participants; high‐quality evidence), with no evidence of heterogeneity (I2 = 0%) (Analysis 1.6).
1.6. Analysis.
Comparison 1 SSRI versus control at end of treatment, by SSRI, Outcome 6 Death.
Side effects: seizures at end of treatment
We combined data for studies with an overall low risk of bias for the outcome of seizures, using a risk ratio with a fixed‐effect model. The analysis found no evidence of a substantial difference in the total number of seizures between SSRI and placebo (RR 1.47, 95% CI 0.99 to 2.18; 3 studies, 3275 participants; moderate‐quality evidence), with no evidence of heterogeneity (I2 = 0%) (Analysis 1.7).
1.7. Analysis.
Comparison 1 SSRI versus control at end of treatment, by SSRI, Outcome 7 Seizures.
Side effects: gastrointestinal side effects at end of treatment
We combined data for studies with an overall low risk of bias for the outcome of gastrointestinal side effects, using a risk ratio with a fixed‐effect model. There was a higher number of gastrointestinal side effects (P = 0.05) among participants treated with SSRIs compared to placebo (RR 2.19, 95% CI 1.00 to 4.76; 2 studies, 148 participants; moderate‐quality evidence), with no evidence of heterogeneity (I2 = 0%) (Analysis 1.8).
1.8. Analysis.
Comparison 1 SSRI versus control at end of treatment, by SSRI, Outcome 8 Gastrointestinal side effects.
Side effects: bleeding at end of treatment
Data were not available for more than one study at low risk of bias for any analysis (Analysis 1.9). The risk ratio was 0.96, 95% CI 0.56 to 1.66.
1.9. Analysis.
Comparison 1 SSRI versus control at end of treatment, by SSRI, Outcome 9 Bleeding.
Change in depression score between baseline and follow‐up
No studies at low risk of bias reported change in depression score between baseline and follow‐up.
Change in cognition between baseline and follow‐up
No studies at low risk of bias reported change in cognition between baseline and follow‐up.
Leaving the study early (before the end of scheduled follow‐up)
We combined data for studies with an overall low risk of bias for the outcome of leaving the study before the end of scheduled follow‐up, using a risk ratio with a fixed‐effect model. The analysis found no difference between SSRI and placebo, with no evidence of heterogeneity (RR 1.01, 95% CI 0.48 to 2.10; P = 0.98; 3 studies, 3277 participants) with no heterogeneity (I2 = 0%) (Analysis 1.10).
1.10. Analysis.
Comparison 1 SSRI versus control at end of treatment, by SSRI, Outcome 10 Leaving the trial before the end of scheduled follow‐up.
Motor deficits
We combined data for studies with an overall low risk of bias for the outcome of motor deficit score, using the SMD with a fixed‐effect model. The analysis found no difference between SSRI and placebo (SMD 0.02, 95% CI −0.05 to 0.09; P = 0.58; 3 studies, 2936 participants) with considerable evidence of heterogeneity (I2 = 88%) (Analysis 1.11).
1.11. Analysis.
Comparison 1 SSRI versus control at end of treatment, by SSRI, Outcome 11 Motor deficits.
Note that data from Chollet 2011 are adjusted means, and data from Marquez Romero 2013 are means and SDs estimated from reported medians and interquartile ranges (Wan 2014).
Quality of life
Of the high‐quality trials, only FOCUS reported quality of life (FOCUS Trial Collaboration 2018). There was no difference between groups in the Euroquol 5D‐5L.
Fatigue
Of the high‐quality trials, only FOCUS reported fatigue (FOCUS Trial Collaboration 2018). This was measured using the SF‐36 vitality score. There was no difference in fatigue between the groups.
Healthcare costs
No trial reported healthcare costs.
We have included those outcomes in the Table 1 which we decided were key to decision‐making.
Subgroup analyses by intervention characteristics and subsets of participant
We did not perform preplanned subgroup analyses by intervention characteristics and subsets of participant (including with or without depression) as there were insufficient studies at low risk of bias. All the trials at low risk of bias stipulated that the participants did not have to have depression to enter the trial.
Sensitivity analysis
Inclusion of all studies regardless of 'Risk of bias' judgement for our primary outcomes
We included all studies regardless of 'Risk of bias' judgement for the co‐primary outcome of disability at the end of treatment using a SMD and a fixed‐effect model. Participants who received an SSRI intervention had significantly lower end‐of‐treatment scores on measures of disability than those participants receiving placebo or standard care/practice (SMD 0.23, 95% CI 0.18 to 0.29; P < 0.001; 26 studies, 5334 participants) with considerable heterogeneity between trials (Chi2 = 328.10, df = 25 (P < 0.001); I2 = 92%) (Analysis 1.12).
1.12. Analysis.
Comparison 1 SSRI versus control at end of treatment, by SSRI, Outcome 12 Disability (sensitivity analyses all studies regardless of RoB).
Re‐analysis included all studies regardless of 'Risk of bias' judgement for the outcome of independence. Modified Rankin score (mRS 0 to 2) at the end of treatment did not alter the result (RR 0.97, 95% CI 0.91 to 1.03; P = 0.35, I2 = 74%; 5 studies, 4002 participants) (Chi2 = 15.57, df = 4 (P = 0.004) (Analysis 1.13).
1.13. Analysis.
Comparison 1 SSRI versus control at end of treatment, by SSRI, Outcome 13 Independent on modified Rankin score (mRS 0 to 2) (sensitivity analysis).
Meta‐analysis using the alternate meta‐analytical effects model (fixed‐effect or random‐effects)
We re‐analysed the data for our primary outcomes (disability score and independence on modified Rankin score 0 to 2) using the random‐effects analysis for the three high‐quality trials. For mRs 0 to 2, this altered the effect estimate to 1.83 (95% CI 0.74 to 4.56). Note that the random‐effects model gives more weight to smaller trials; this explains the implausibly large effect size. For disability, the same effect size was obtained irrespective of whether fixed or random effects were used.
Meta‐analysis using the alternative end of follow‐up time point
Two of the studies at low risk of bias (Chollet 2011; Marquez Romero 2013) reported outcome data only at the end of treatment. FOCUS Trial Collaboration 2018 reported outcomes at the end of treatment and also six months after the end of treatment. With just one high‐quality trial reporting results at an alternative end point, it was therefore not possible to perform a meta‐analysis.
Discussion
Summary of main results
For this update we included 63 studies with 9168 participants.
Of the 63 included studies, 32 trials used fluoxetine, eight trials used sertraline, 11 used paroxetine, eight used citalopram, two used escitalopram, one used either sertraline or fluoxetine, and one used citalopram or fluoxetine.
We assessed only three of the 63 included studies to be at low risk of bias across the key domains. The three trials at low risk of bias compared fluoxetine to placebo. We included these three trials in our meta‐analysis.
Comparing fluoxetine to placebo, we found moderate‐quality evidence of no beneficial effects of fluoxetine on our two primary outcomes (disability and independence). We found moderate‐quality evidence that fluoxetine reduced the severity of depression evaluated using a continuous outcome. We found moderate‐quality evidence that fluoxetine increased gastrointestinal side effects compared to placebo. We found a non‐significant excess of seizures in those allocated to fluoxetine. We found no difference for other outcomes.
Overall completeness and applicability of evidence
This review includes studies from different settings (e.g. countries; high‐, middle‐ and low‐income settings; healthcare systems), with different criteria for selecting participants (e.g. methods of pre‐randomisation diagnosis and investigation, inclusion and exclusion criteria), that may reflect differences between the trial protocol and routine clinical practice (e.g. inclusion of participants based on a diagnosis of stroke made using brain imaging: brain imaging is unlikely to be either available or affordable in routine clinical care in many low‐ and middle‐income country settings); and different characteristics of randomised participants (e.g. baseline demographic and clinical characteristics, stroke severity, time since stroke onset, presence or absence of depression, severity of depression). These trial characteristics may in part explain the heterogeneity of results, but we know from our previous review that the most probable cause of heterogeneity is trial quality.
Six published studies did not present the baseline demographic and clinical characteristics for each group, but rather they reported the baseline demographic and clinical characteristics for those completing the trial (i.e. a subset of all those randomised). This makes it very difficult to compare the study groups at baseline.
There is a discordance between the results for disability (one of our co‐primary outcomes) between the trials at low risk of bias, which showed no effect, and all trials (a positive effect). This is because trials at high risk of bias tended to be positive.
The results of the meta‐analysis of the three trials at low risk of bias are applicable to clinical practice, although the meta‐analysis is dominated by the UK FOCUS trial which recruited participants from the National Health Service (FOCUS Trial Collaboration 2018). Nevertheless, FOCUS had broad inclusion criteria, and the demographics of those recruited are similar to UK patients with stroke. Chollet 2011 recruited participants from France and Marquez Romero 2013 recruited participants from Mexico. FOCUS Trial Collaboration 2018 included participants with both haemorrhagic and ischaemic stroke, Chollet 2011 recruited participants with ischaemic stroke, and Marquez Romero 2013 recruited participants with haemorrhagic stroke.
There is a theoretical risk that SSRIs might carry particular risks in people with haemorrhagic stroke, due to their effects on platelet aggregation and bleeding. An individual patient meta‐analysis is needed to explore this. This might be possible when the AFFINITY (Hankey 2011), and EFFECTS (Lundström 2014) trials are published.
We were unable to explore the influence of the type of SSRI, as all three high‐quality trials used fluoxetine.
The searches were performed in July 2018. Had we had sufficient resources, we would have updated the searches again immediately prior to publication of the review. Instead, we aim to update the review soon after two large key trials are published in 2020. We are not aware of any studies that have been published since July 2018.
Quality of the evidence
For the evaluation of quality of the evidence, we contacted authors of primary studies as yet unpublished for data on outcomes. We did not contact authors of primary studies for supplementary information on features of the study design that were unclear or omitted from published trial reports; rather, we assessed the study based on the information available in the published report. We did contact authors of primary studies for clarification when there were multiple publications indicating separate studies with unique populations.
We used the Cochrane 'Risk of bias' tool to assess study methodology. In this update we decided to restrict meta‐analyses to studies at low risk of bias, because in the previous review there was evidence that the apparently beneficial effects of SSRI on recovery might have been simply due to methodological limitations of the included trials. For this update, only three of the 63 included studies met our criteria for overall low risk of bias.
The meta‐analysis of the high‐quality studies is dominated by FOCUS Trial Collaboration 2018, which provided more than 90% of the data, and so it is not surprising that the results of the meta‐analysis are very similar to the results of FOCUS. FOCUS was neutral for its primary outcome (mRS at six months), unlike the other two high‐quality but smaller studies, which were both positive.
We performed sensitivity analyses of our two co‐primary outcomes (disability and independence) by including all the available outcome data, irrespective of risk of bias. Like our initial (hypothesis‐generating) Cochrane Review, we found that SSRIs reduced disability at the end of treatment. However, it is highly likely that this positive effect is due to biases in trial quality. It will therefore be important to update this review again when further data become available, to increase the generalisability of the findings.
We found a high I2 measure for independence. This might reflect different settings, different stroke types, and different durations of treatment.
Potential biases in the review process
We conducted the review using robust Cochrane methodology, with two review authors independently assessing studies for eligibility, extracting data, and carrying out 'Risk of bias' assessment. Five review authors were also authors of the FOCUS Trial Collaboration 2018 (MD, GM, EL, GH, MH) and so review authors who were independent of the trial extracted the data from FOCUS, and performed quality assessment and the meta‐analysis.
We made some changes to the review during this update. We used Covidence for screening and data extraction. We excluded trials which combined an SSRI with another active intervention and compared it to the active treatment alone. We restricted the criteria for considering studies for this review to randomised controlled trials and excluded studies where investigators described a non‐random component in the sequence‐generation process. We incorporated 'Risk of bias' assessments in analyses by restricting the primary analysis to studies at low risk of bias. We performed sensitivity analyses to determine how the conclusions were affected by including studies at unclear and high risk of bias; we found evidence of a beneficial effect of SSRIs on measures of disability with inclusion of studies at unclear and high risk of bias with considerable heterogeneity. For the outcome 'independence', the results were unchanged. We made these changes to the review in order to increase the robustness of our evidence.
Comparing the effect estimates in the this update with the effect estimates from only the high‐quality trials in the original review suggest that our decisions have not introduced bias.
Agreements and disagreements with other studies or reviews
This review has demonstrated that SSRIs do not improve recovery after stroke. This is in contrast with other meta‐analyses, including our own 2012 Cochrane Review, which showed a positive effect of SSRIs on recovery.
This difference is almost certainly because previous meta‐analyses included trials at higher risk of bias.
Authors' conclusions
Implications for practice.
Based on our meta‐analysis of the trials at low risk of bias, there is currently no indication for the routine prescription of SSRIs to promote stroke recovery. Fluoxetine reduces the risk of depression, but this is probably not a sufficiently strong rationale to give all people with stroke a six‐month course of the drug. Nevertheless, the data in this review have provided further information about the risks of SSRIs in stroke, which will enable those clinicians who may wish to give prophylactic SSRI to patients at high risk of depression to weigh up the risks and the benefits.
Implications for research.
A meta‐analysis is generally more robust if it includes several high‐quality trials. We will therefore update this review when two further large trials using fluoxetine have been published. There are also smaller ongoing trials which we will include in a future update. In the meantime, we recommend that further new trials exploring whether fluoxetine improves recovery after stroke are not established.
What's new
| Date | Event | Description |
|---|---|---|
| 14 March 2019 | New citation required and conclusions have changed | We include 2 new high‐quality trials. Meta‐analysis of all the high‐quality trials shows no effect on either of the co‐primary outcomes of independence and disability. Meta‐analysis of all trials irrespective of trial quality showed that SSRIs reduced disability at the end of treatment. |
| 14 March 2019 | New search has been performed | We have clarified that there are 2 primary outcomes: independence and disability. For modified Rankin Score (mRS) in advance of starting this update, we decided to report the proportion of independent participants compared with the proportion dead or dependent which is the usual convention in stroke trials. In the previous version we had reported the proportion dependent and had excluded the dead participants from the analysis. We checked the total number of participants included in the 2012 review. We had stated that the trials included 4060 participants; there were errors in the arithmetic (due to counting number allocated rather than those recruited, and omitting to count data from 2 small trials). When we recalculated the figures, there were 4109 recruited. We excluded 7 of these trials (439 participants) which had combined an SSRI with another active intervention and compared it to the active treatment alone or where there was a non‐random component to sequence generation process (see list of excluded studies in text). We added 14 new completed trials, recruiting 5498 participants. There are now a total of 63 trials recruiting a total of 9168 participants. We decided to restrict our primary analyses only to those trials at low risk of bias. We did this because we wished to provide a clear answer about the risks and benefits of SSRIs, which was not influenced by trial quality and because it would have been impractical, given the resources for this update, to perform analyses including all the low‐quality trials. We made this decision before we knew the results of the largest trial in this review (FOCUS). We have, however, performed a sensitivity analysis for dependence and disability (our primary outcomes) using data from all trials; as in the first version of the review, this sensitivity analysis showed that when low‐quality trials are included, results tend to be in favour of SSRIs. We adhered to the MECIR standards for conduct and reporting. We shortened our list of excluded studies in line with the Cochrane Handbook, by not listing those studies that obviously did not fulfil inclusion criteria, including those studies which clearly had an ineligible comparator, intervention or study design. |
History
Protocol first published: Issue 11, 2011 Review first published: Issue 11, 2012
| Date | Event | Description |
|---|---|---|
| 26 August 2013 | Amended | The review authors identified minor errors following publication of the previous version. These errors have now been corrected and have resulted in very minor changes in SMD for disability and some I2 values. The changes have not materially changed the results or conclusions of the review. Changes made: (1) the total number of participants has been changed from 4059 to 4060; (2) Almeida 2006 recruited people without depression; this has been corrected in the 'Characteristics of included studies' table, and data have been moved to 'did not have to have depression' in the relevant subgroup analyses; (3) disability data for Acler 2009 had been entered incorrectly; this has now been corrected. |
Acknowledgements
For this update, we are grateful to the following people.
Joshua Cheyne, Cochrane Stroke Information Specialist, ran the literature searches.
Maureen Harding obtained articles for full‐text review.
Dr Juan Marquez Romero provided unpublished data from the FMRICH trial on 8 September 2018 (Professor Mead was the recipient of this information).
Professor Chollet responded to requests for information about other new trials.
Dr Jan Bembenek and Professor Anna Czlonkowska provided further information about FOCUS‐Poland.
Professor Peter Sandercock commented on the analysis of the fluoxetine data.
Dr Pavel Lindberg kindly provided information from Birchenall 2019.
Appendices
Appendix 1. Cochrane Central Register of Controlled Trials (CENTRAL) search strategy
#1. MeSH descriptor Cerebrovascular Disorders explode all trees #2. (stroke in Title, Abstract or Keywords or poststroke in Title, Abstract or Keywords or post‐stroke in Title, Abstract or Keywords or cerebrovasc* in Title, Abstract or Keywords or (brain in Title, Abstract or Keywords and vasc* in Title, Abstract or Keywords) or (cerebral in Title, Abstract or Keywords and vasc* in Title, Abstract or Keywords) or cva* in Title, Abstract or Keywords or apoplex* in Title, Abstract or Keywords or SAH in Title, Abstract or Keywords) #3. ( (brain* in Title, Abstract or Keywords or cerebr* in Title, Abstract or Keywords or cerebell* in Title, Abstract or Keywords or intracran* in Title, Abstract or Keywords or intracerebral in Title, Abstract or Keywords) and (ischemi* in Title, Abstract or Keywords or ischaemi* in Title, Abstract or Keywords or infarct* in Title, Abstract or Keywords or thrombo* in Title, Abstract or Keywords or emboli* in Title, Abstract or Keywords or occlus* in Title, Abstract or Keywords) ) #4. ( (brain* in Title, Abstract or Keywords or cerebr* in Title, Abstract or Keywords or cerebell* in Title, Abstract or Keywords or intracerebral in Title, Abstract or Keywords or intracranial in Title, Abstract or Keywords or subarachnoid in Title, Abstract or Keywords) and (haemorrhage* in Title, Abstract or Keywords or hemorrhage* in Title, Abstract or Keywords or haematoma* in Title, Abstract or Keywords or hematoma* in Title, Abstract or Keywords or bleed* in Title, Abstract or Keywords) ) #5. MeSH descriptor hemiplegia this term only #6. MeSH descriptor paresis explode all trees #7. MeSH descriptor Gait Disorders, Neurologic explode all trees #8. (hemipleg* in Title, Abstract or Keywords or hemipar* in Title, Abstract or Keywords or paresis in Title, Abstract or Keywords or paretic in Title, Abstract or Keywords) #9. (#1 or #2 or #3 or #4 or #5 or #6 or #7 or #8) #10. MeSH descriptor Serotonin Uptake Inhibitors explode all trees #11. (serotonin in Title, Abstract or Keywords or 5‐HT in Title, Abstract or Keywords or “5 HT” in Title, Abstract or Keywords or 5‐hydroxytryptamine in Title, Abstract or Keywords or “5 hydroxytryptamine” in Title, Abstract or Keywords) #12. (uptake in Title, Abstract or Keywords or reuptake in Title, Abstract or Keywords or re‐uptake in Title, Abstract or Keywords) #13. inhib* in Title, Abstract or Keywords #14. (#11 and #12 and #13) #15. SSRI* in Title, Abstract or Keywords #16. (alaproclat* in Title, Abstract or Keywords or cericlamin* in Title, Abstract or Keywords or citalopram in Title, Abstract or Keywords or dapoxetin* in Title, Abstract or Keywords or escitalopram in Title, Abstract or Keywords or femoxetin* in Title, Abstract or Keywords or fluoxetin* in Title, Abstract or Keywords or fluvoxamin* in Title, Abstract or Keywords or paroxetin* in Title, Abstract or Keywords or sertralin* in Title, Abstract or Keywords or trazodone in Title, Abstract or Keywords or vilazodone in Title, Abstract or Keywords or zimelidine in Title, Abstract or Keywords) #17. (Celexa in Title, Abstract or Keywords or Cipramil in Title, Abstract or Keywords or Cipram in Title, Abstract or Keywords or Recital in Title, Abstract or Keywords or Emocal in Title, Abstract or Keywords or Dalsan in Title, Abstract or Keywords or Sepram in Title, Abstract or Keywords or Seropram in Title, Abstract or Keywords or Citox in Title, Abstract or Keywords or Priligy in Title, Abstract or Keywords or Lexapro in Title, Abstract or Keywords or Cipralex in Title, Abstract or Keywords or Seroplex in Title, Abstract or Keywords or Esertia in Title, Abstract or Keywords or Prozac in Title, Abstract or Keywords or Fontex in Title, Abstract or Keywords or Seromex in Title, Abstract or Keywords or Seronil in Title, Abstract or Keywords or Sarafem in Title, Abstract or Keywords or Ladose in Title, Abstract or Keywords or Motivest in Title, Abstract or Keywords or Fluctin in Title, Abstract or Keywords or fluox in Title, Abstract or Keywords or Lovan in Title, Abstract or Keywords or Luvox in Title, Abstract or Keywords or Fevarin in Title, Abstract or Keywords or Faverin in Title, Abstract or Keywords or Favoxil in Title, Abstract or Keywords or Movox in Title, Abstract or Keywords or Paxil in Title, Abstract or Keywords or Seroxat in Title, Abstract or Keywords or Sereupin in Title, Abstract or Keywords or Aropax in Title, Abstract or Keywords or Deroxat in Title, Abstract or Keywords or Divarius in Title, Abstract or Keywords or Rexetin in Title, Abstract or Keywords or Xetanor in Title, Abstract or Keywords or Paroxat in Title, Abstract or Keywords or Loxamine in Title, Abstract or Keywords or Zoloft in Title, Abstract or Keywords or Lustral in Title, Abstract or Keywords or Serlain in Title, Abstract or Keywords or Asentra in Title, Abstract or Keywords) #18. (#10 or #14 or #15 or #16 or #17) #19. (#9 and #18)
Appendix 2. MEDLINE (Ovid) search strategy
1. cerebrovascular disorders/ or exp basal ganglia cerebrovascular disease/ or exp brain ischemia/ or exp carotid artery diseases/ or exp intracranial arterial diseases/ or exp "intracranial embolism and thrombosis"/ or exp intracranial hemorrhages/ or stroke/ or exp brain infarction/ or vertebral artery dissection/ 2. (stroke or poststroke or post‐stroke or cerebrovasc$ or brain vasc$ or cerebral vasc$ or cva$ or apoplex$ or SAH).tw. 3. ((brain$ or cerebr$ or cerebell$ or intracran$ or intracerebral) adj5 (isch?emi$ or infarct$ or thrombo$ or emboli$ or occlus$)).tw. 4. ((brain$ or cerebr$ or cerebell$ or intracerebral or intracranial or subarachnoid) adj5 (haemorrhage$ or hemorrhage$ or haematoma$ or hematoma$ or bleed$)).tw. 5. hemiplegia/ or exp paresis/ 6. (hemipleg$ or hemipar$ or paresis or paretic).tw. 7. exp Gait Disorders, Neurologic/ 8. or/1‐7 9. exp Serotonin Uptake Inhibitors/ 10. ((serotonin or 5‐HT or 5 HT or 5‐hydroxytryptamine or 5 hydroxytryptamine) adj5 (uptake or reuptake or re‐uptake) adj5 inhib$).tw. 11. SSRI$1.tw. 12. (alaproclat$ or cericlamin$ or citalopram or dapoxetin$ or escitalopram or femoxetin$ or fluoxetin$ or fluvoxamin$ or paroxetin$ or sertralin$ or trazodone or vilazodone or zimelidine).tw,nm. 13. (Celexa or Cipramil or Cipram or Recital or Emocal or Dalsan or Sepram or Seropram or Citox or Priligy or Lexapro or Cipralex or Seroplex or Esertia or Prozac or Fontex or Seromex or Seronil or Sarafem or Ladose or Motivest or Fluctin or fluox or Lovan or Luvox or Fevarin or Faverin or Favoxil or Movox or Paxil or Seroxat or Sereupin or Aropax or Deroxat or Divarius or Rexetin or Xetanor or Paroxat or Loxamine or Zoloft or Lustral or Serlain or Asentra).tw,nm. 14. 9 or 10 or 11 or 12 or 13 15. 8 and 14 16. exp animals/ not humans.sh. 17. 15 not 16 18. Randomized Controlled Trials as Topic/ 19. random allocation/ 20. Controlled Clinical Trials as Topic/ 21. control groups/ 22. clinical trials as topic/ or clinical trials, phase i as topic/ or clinical trials, phase ii as topic/ or clinical trials, phase iii as topic/ or clinical trials, phase iv as topic/ 23. Clinical Trials Data Monitoring Committees/ 24. double‐blind method/ 25. single‐blind method/ 26. Placebos/ 27. placebo effect/ 28. cross‐over studies/ 29. Multicenter Studies as Topic/ 30. Therapies, Investigational/ 31. Drug Evaluation/ 32. Research Design/ 33. Program Evaluation/ 34. evaluation studies as topic/ 35. randomized controlled trial.pt. 36. controlled clinical trial.pt. 37. (clinical trial or clinical trial phase i or clinical trial phase ii or clinical trial phase iii or clinical trial phase iv).pt. 38. multicenter study.pt. 39. (evaluation studies or comparative study).pt. 40. meta analysis.pt. 41. meta‐analysis as topic/ 42. random$.tw. 43. (controlled adj5 (trial$ or stud$)).tw. 44. (clinical$ adj5 trial$).tw. 45. ((control or treatment or experiment$ or intervention) adj5 (group$ or subject$ or patient$)).tw. 46. (quasi‐random$ or quasi random$ or pseudo‐random$ or pseudo random$).tw. 47. ((multicenter or multicentre or therapeutic) adj5 (trial$ or stud$)).tw. 48. ((control or experiment$ or conservative) adj5 (treatment or therapy or procedure or manage$)).tw. 49. ((singl$ or doubl$ or tripl$ or trebl$) adj5 (blind$ or mask$)).tw. 50. (coin adj5 (flip or flipped or toss$)).tw. 51. latin square.tw. 52. versus.tw. 53. (cross‐over or cross over or crossover).tw. 54. placebo$.tw. 55. sham.tw. 56. (assign$ or alternate or allocat$ or counterbalance$ or multiple baseline).tw. 57. controls.tw. 58. (treatment$ adj6 order).tw. 59. (meta‐analy$ or metaanaly$ or meta analy$ or systematic review or systematic overview).tw. 60. or/18‐59 61. 17 and 60
Appendix 3. EMBASE (Ovid) search strategy
1. cerebrovascular disease/ or basal ganglion hemorrhage/ or exp brain hematoma/ or exp brain hemorrhage/ or exp brain infarction/ or exp brain ischemia/ or exp carotid artery disease/ or cerebral artery disease/ or cerebrovascular accident/ or exp intracranial aneurysm/ or exp occlusive cerebrovascular disease/ or stroke/ 2. stroke unit/ or stroke patient/ 3. (stroke or poststroke or post‐stroke or cerebrovasc$ or brain vasc$ or cerebral vasc$ or cva$ or apoplex$ or SAH).tw. 4. ((brain$ or cerebr$ or cerebell$ or intracran$ or intracerebral) adj5 (isch?emi$ or infarct$ or thrombo$ or emboli$ or occlus$)).tw. 5. ((brain$ or cerebr$ or cerebell$ or intracerebral or intracranial or subarachnoid) adj5 (haemorrhage$ or hemorrhage$ or haematoma$ or hematoma$ or bleed$)).tw. 6. hemiparesis/ or hemiplegia/ or paresis/ 7. (hemipleg$ or hemipar$ or paresis or paretic).tw. 8. or/1‐7 9. exp serotonin uptake inhibitor/ 10. ((serotonin or 5‐HT or 5 HT or 5‐hydroxytryptamine or 5 hydroxytryptamine) adj5 (uptake or reuptake or re‐uptake) adj5 inhib$).tw. 11. SSRI$1.tw. 12. (alaproclat$ or cericlamin$ or citalopram or dapoxetin$ or escitalopram or femoxetin$ or fluoxetin$ or fluvoxamin$ or paroxetin$ or sertralin$ or trazodone or vilazodone or zimelidine).tw. 13. (Celexa or Cipramil or Cipram or Recital or Emocal or Dalsan or Sepram or Seropram or Citox or Priligy or Lexapro or Cipralex or Seroplex or Esertia or Prozac or Fontex or Seromex or Seronil or Sarafem or Ladose or Motivest or Fluctin or fluox or Lovan or Luvox or Fevarin or Faverin or Favoxil or Movox or Paxil or Seroxat or Sereupin or Aropax or Deroxat or Divarius or Rexetin or Xetanor or Paroxat or Loxamine or Zoloft or Lustral or Serlain or Asentra).tw,tn. 14. 9 or 10 or 11 or 12 or 13 15. 8 and 14 16. limit 15 to human 17. Randomized Controlled Trial/ 18. Randomization/ 19. Controlled Study/ 20. control group/ 21. clinical trial/ or phase 1 clinical trial/ or phase 2 clinical trial/ or phase 3 clinical trial/ or phase 4 clinical trial/ or controlled clinical trial/ 22. Double Blind Procedure/ 23. Single Blind Procedure/ or triple blind procedure/ 24. placebo/ 25. "types of study"/ 26. research subject/ 27. random$.tw. 28. (controlled adj5 (trial$ or stud$)).tw. 29. (clinical$ adj5 trial$).tw. 30. ((control or treatment or experiment$ or intervention) adj5 (group$ or subject$ or patient$)).tw. 31. (quasi‐random$ or quasi random$ or pseudo‐random$ or pseudo random$).tw. 32. ((singl$ or doubl$ or tripl$ or trebl$) adj5 (blind$ or mask$)).tw. 33. (coin adj5 (flip or flipped or toss$)).tw. 34. versus.tw. 35. placebo$.tw. 36. controls.tw. 37. or/17‐36 38. 16 and 37
Appendix 4. CINAHL (Ebsco) search strategy
S23. S12 and S22 S22. S13 or S17 or S18 or S19 or S20 or S21 S21. AB Celexa or Cipramil or Cipram or Recital or Emocal or Dalsan or Sepram or Seropram or Citox or Priligy or Lexapro or Cipralex or Seroplex or Esertia or Prozac or Fontex or Seromex or Seronil or Sarafem or Ladose or Motivest or Fluctin or fluox or Lovan or Luvox or Fevarin or Faverin or Favoxil or Movox or Paxil or Seroxat or Sereupin or Aropax or Deroxat or Divarius or Rexetin or Xetanor or Paroxat or Loxamine or Zoloft or Lustral or Serlain or Asentra S20. TI Celexa or Cipramil or Cipram or Recital or Emocal or Dalsan or Sepram or Seropram or Citox or Priligy or Lexapro or Cipralex or Seroplex or Esertia or Prozac or Fontex or Seromex or Seronil or Sarafem or Ladose or Motivest or Fluctin or fluox or Lovan or Luvox or Fevarin or Faverin or Favoxil or Movox or Paxil or Seroxat or Sereupin or Aropax or Deroxat or Divarius or Rexetin or Xetanor or Paroxat or Loxamine or Zoloft or Lustral or Serlain or Asentra S19. TI ( alaproclat* or cericlamin* or citalopram or dapoxetin* or escitalopram or femoxetin* or fluoxetin* or fluvoxamin* or paroxetin* or sertralin* or trazodone or vilazodone or zimelidine ) OR AB ( alaproclat* or cericlamin* or citalopram or dapoxetin* or escitalopram or femoxetin* or fluoxetin* or fluvoxamin* or paroxetin* or sertralin* or trazodone or vilazodone or zimelidine ) S18. TI SSRI* OR AB SSRI* S17. S14 and S15 and S16 S16. TI inhib* OR AB inhib* S15. TI ( uptake or reuptake or re‐uptake ) OR AB ( uptake or reuptake or re‐uptake ) S14. TI ( serotonin or 5‐HT or 5 HT or 5‐hydroxytryptamine or 5 hydroxytryptamine ) OR AB ( serotonin or 5‐HT or 5 HT or 5‐hydroxytryptamine or 5 hydroxytryptamine ) S13. (MH "Serotonin Uptake Inhibitors+") S12. S1 or S2 or S3 or S6 or S9 or S10 or S11 S11. TI ( hemipleg* or hemipar* or paresis or paretic ) or AB ( hemipleg* or hemipar* or paresis or paretic ) S10. (MH "Hemiplegia") S9. S7 and S8 S8. TI ( haemorrhage* or hemorrhage* or haematoma* or hematoma* or bleed* ) or AB ( haemorrhage* or hemorrhage* or haematoma* or hematoma* or bleed* ) S7. TI ( brain* or cerebr* or cerebell* or intracerebral or intracranial or subarachnoid ) or AB ( brain* or cerebr* or cerebell* or intracerebral or intracranial or subarachnoid ) S6. S4 and S5 S5. TI ( ischemi* or ischaemi* or infarct* or thrombo* or emboli* or occlus* ) or AB ( ischemi* or ischaemi* or infarct* or thrombo* or emboli* or occlus* ) S4. TI ( brain* or cerebr* or cerebell* or intracran* or intracerebral ) or AB ( brain* or cerebr* or cerebell* or intracran* or intracerebral ) S3. TI ( stroke or poststroke or post‐stroke or cerebrovasc* or brain vasc* or cerebral vasc or cva or apoplex or SAH ) or AB ( stroke or poststroke or post‐stroke or cerebrovasc* or brain vasc* or cerebral vasc or cva or apoplex or SAH ) S2. (MH "Stroke Patients") OR (MH "Stroke Units") S1. (MH "Cerebrovascular Disorders") OR (MH "Basal Ganglia Cerebrovascular Disease+") OR (MH "Carotid Artery Diseases+") OR (MH "Cerebral Ischemia+") OR (MH "Cerebral Vasospasm") OR (MH "Intracranial Arterial Diseases+") OR (MH "Intracranial Embolism and Thrombosis") OR (MH "Intracranial Hemorrhage+") OR (MH "Stroke") OR (MH "Vertebral Artery Dissections")
Appendix 5. AMED (Ovid) search strategy
1. cerebrovascular disorders/ or cerebral hemorrhage/ or cerebral infarction/ or cerebral ischemia/ or cerebrovascular accident/ or stroke/ 2. (stroke or poststroke or post‐stroke or cerebrovasc$ or brain vasc$ or cerebral vasc$ or cva$ or apoplex$ or SAH).tw. 3. ((brain$ or cerebr$ or cerebell$ or intracran$ or intracerebral) adj5 (isch?emi$ or infarct$ or thrombo$ or emboli$ or occlus$)).tw. 4. ((brain$ or cerebr$ or cerebell$ or intracerebral or intracranial or subarachnoid) adj5 (haemorrhage$ or hemorrhage$ or haematoma$ or hematoma$ or bleed$)).tw. 5. hemiplegia/ 6. (hemipleg$ or hemipar$ or paresis or paretic).tw. 7. or/1‐6 8. antidepressive agents/ 9. ((serotonin or 5‐HT or 5 HT or 5‐hydroxytryptamine or 5 hydroxytryptamine) adj5 (uptake or reuptake or re‐uptake) adj5 inhib$).tw. 10. SSRI$1.tw. 11. (alaproclat$ or cericlamin$ or citalopram or dapoxetin$ or escitalopram or femoxetin$ or fluoxetin$ or fluvoxamin$ or paroxetin$ or sertralin$ or trazodone or vilazodone or zimelidine).tw. 12. (Celexa or Cipramil or Cipram or Recital or Emocal or Dalsan or Sepram or Seropram or Citox or Priligy or Lexapro or Cipralex or Seroplex or Esertia or Prozac or Fontex or Seromex or Seronil or Sarafem or Ladose or Motivest or Fluctin or fluox or Lovan or Luvox or Fevarin or Faverin or Favoxil or Movox or Paxil or Seroxat or Sereupin or Aropax or Deroxat or Divarius or Rexetin or Xetanor or Paroxat or Loxamine or Zoloft or Lustral or Serlain or Asentra).tw. 13. 8 or 9 or 10 or 11 or 12 14. 7 and 13
Appendix 6. PsycINFO (Ovid) search strategy
1. cerebrovascular disorders/ or cerebral hemorrhage/ or exp cerebral ischemia/ or cerebral small vessel disease/ or cerebrovascular accidents/ or subarachnoid hemorrhage/ 2. (stroke or poststroke or post‐stroke or cerebrovasc$ or brain vasc$ or cerebral vasc$ or cva$ or apoplex$ or SAH).tw. 3. ((brain$ or cerebr$ or cerebell$ or intracran$ or intracerebral) adj5 (isch?emi$ or infarct$ or thrombo$ or emboli$ or occlus$)).tw. 4. ((brain$ or cerebr$ or cerebell$ or intracerebral or intracranial or subarachnoid) adj5 (haemorrhage$ or hemorrhage$ or haematoma$ or hematoma$ or bleed$)).tw. 5. (hemipleg$ or hemipar$ or paresis or paretic).tw. 6. hemiparesis/ or hemiplegia/ 7. or/1‐6 8. exp serotonin reuptake inhibitors/ 9. ((serotonin or 5‐HT or 5 HT or 5‐hydroxytryptamine or 5 hydroxytryptamine) adj5 (uptake or reuptake or re‐uptake) adj5 inhib$).tw. 10. SSRI$1.tw. 11. (alaproclat$ or cericlamin$ or citalopram or dapoxetin$ or escitalopram or femoxetin$ or fluoxetin$ or fluvoxamin$ or paroxetin$ or sertralin$ or trazodone or vilazodone or zimelidine).tw. 12. (Celexa or Cipramil or Cipram or Recital or Emocal or Dalsan or Sepram or Seropram or Citox or Priligy or Lexapro or Cipralex or Seroplex or Esertia or Prozac or Fontex or Seromex or Seronil or Sarafem or Ladose or Motivest or Fluctin or fluox or Lovan or Luvox or Fevarin or Faverin or Favoxil or Movox or Paxil or Seroxat or Sereupin or Aropax or Deroxat or Divarius or Rexetin or Xetanor or Paroxat or Loxamine or Zoloft or Lustral or Serlain or Asentra).tw. 13. 8 or 9 or 10 or 11 or 12 14. 7 and 13
Appendix 7. Search strategy for the trial registers
Patient: stroke
Intervention: alaproclate OR cericlamineOR citalopram OR clomipramine OR dapoxetine OR etoperidone OR femoxetine OR fenfluramine OR fluoxetine OR fluvoxamine OR norfenfluramine OR paroxetine OR sertraline OR trazodone OR vilazodone OR zimelidine
Comparison: placebo
Trial status: ongoing OR Recruiting OR Not yet recruiting OR Active
Age: adult OR older adult
Methods: Randomised Controlled Study
Data and analyses
Comparison 1. SSRI versus control at end of treatment, by SSRI.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Disability (primary analysis) | 2 | 2829 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.01 [‐0.09, 0.06] |
| 1.1 Fluoxetine | 2 | 2829 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.01 [‐0.09, 0.06] |
| 2 Independent on modified Rankin score (mRS 0 to 2) (primary analysis) | 3 | 3249 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.91, 1.09] |
| 3 Neurological deficit score | 2 | 142 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.30 [‐0.63, 0.04] |
| 3.1 Fluoxetine | 2 | 142 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.30 [‐0.63, 0.04] |
| 4 Depression (continuous data) | 2 | 2861 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.11 [‐0.19, ‐0.04] |
| 4.1 Fluoxetine | 2 | 2861 | Std. Mean Difference (IV, Fixed, 95% CI) | ‐0.11 [‐0.19, ‐0.04] |
| 5 Depression (dichotomous data) | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 5.1 Fluoxetine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 6 Death | 3 | 3254 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.79, 1.25] |
| 6.1 Fluoxetine | 3 | 3254 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.99 [0.79, 1.25] |
| 7 Seizures | 3 | 3275 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.47 [0.99, 2.18] |
| 7.1 Fluoxetine | 3 | 3275 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.47 [0.99, 2.18] |
| 8 Gastrointestinal side effects | 2 | 148 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.19 [1.00, 4.76] |
| 8.1 Fluoxetine | 2 | 148 | Risk Ratio (M‐H, Fixed, 95% CI) | 2.19 [1.00, 4.76] |
| 9 Bleeding | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 9.1 Fluoxetine | 1 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.0 [0.0, 0.0] | |
| 10 Leaving the trial before the end of scheduled follow‐up | 3 | 3277 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.48, 2.10] |
| 10.1 Fluoxetine | 3 | 3277 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.01 [0.48, 2.10] |
| 11 Motor deficits | 3 | 2936 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.02 [‐0.05, 0.09] |
| 11.1 Fluoxetine | 3 | 2936 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.02 [‐0.05, 0.09] |
| 12 Disability (sensitivity analyses all studies regardless of RoB) | 26 | 5334 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.23 [0.18, 0.29] |
| 12.1 Fluoxetine | 15 | 3919 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.14 [0.08, 0.20] |
| 12.2 Sertraline | 1 | 130 | Std. Mean Difference (IV, Fixed, 95% CI) | 1.38 [0.99, 1.76] |
| 12.3 Paroxetine | 5 | 293 | Std. Mean Difference (IV, Fixed, 95% CI) | 1.29 [1.03, 1.55] |
| 12.4 Citalopram | 5 | 992 | Std. Mean Difference (IV, Fixed, 95% CI) | 0.24 [0.11, 0.37] |
| 13 Independent on modified Rankin score (mRS 0 to 2) (sensitivity analysis) | 5 | 4002 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.97 [0.91, 1.03] |
| 13.1 Fluoxetine | 3 | 3249 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.00 [0.91, 1.09] |
| 13.2 Sertraline | 1 | 111 | Risk Ratio (M‐H, Fixed, 95% CI) | 1.0 [0.97, 1.04] |
| 13.3 Citalopram | 1 | 642 | Risk Ratio (M‐H, Fixed, 95% CI) | 0.90 [0.82, 0.98] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Acler 2009.
| Methods | Study type: interventional (clinical trial) Intervention model: parallel assignment Primary purpose: treatment |
|
| Participants | 20 participants Location: Italy Setting: inpatient Inclusion criteria: first‐ever ischaemic stroke, CT or MRI documenting a single monohemispheric lesion, age below 80 years, within 3 months of onset Exclusion criteria: major affective disorders, alcohol abuse and dementia leading to unco‐operative behaviour, pacemakers, metal in the head, concomitant neuropathies, systemic vasculopathies, major affective disorders Treatment: 10 people, mean age 68 ± 7 years, 6 men Control: 10 people, mean age 65 ± 7 years, 6 men |
|
| Interventions | Citalopram 10 mg daily Placebo: identical pill daily Duration of treatment: at least 4 months Duration of follow‐up: not stated |
|
| Outcomes | Motor cortex excitability NIHSS Lindmark Scale BI HDRS BDI No data on death, GI upset, bleeds or seizures |
|
| Funding source | Source of funding not stated; unclear whether or not a drug company was involved in the study | |
| Notes | Dates of study not stated. Any conflicts of interest not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random numbers |
| Allocation concealment (selection bias) | Unclear risk | Method of allocation concealment not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Stated blinded, placebo was 'an identical pill' |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Stated blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | It is not stated whether data from all recruited participants are reported |
| Selective reporting (reporting bias) | Unclear risk | Side effects were not reported although they were assessed |
| Other bias | Unclear risk | − |
Almeida 2006.
| Methods | Parallel design Analysis: ITT (last observation carried forward), withdrawn owing to becoming depressed, AE, treating practitioner started antidepressant, medical advice, no reason given, not contactable ‐ numbers not included |
|
| Participants | Location: Australia Setting: inpatient Treatment: 55 people, mean ± SD age 68 ± 13 years, 67% men Control: 56 people, mean ± SD age 67 ± 13 years, 62% men Stroke criteria: acute ischaemic or haemorrhagic stroke, diagnosis by clinical signs (ICD‐10) and CT (100% imaged, 10/111 CT scan did not show acute ischaemia); stroke on average < 2 weeks prior to randomisation Not depressed (HADS‐D had to be over 7) Other entry criteria: not stated Comparability of treatment groups: more participants in treatment group with previous heart attack and stroke, also higher levels of hypertension Exclusion criteria: severe communication difficulties, unstable medical condition, severe cognitive impairment and depression (MMSE < 10), taking antidepressants within 4 weeks of stroke, contraindication to sertraline, previous reaction to sertraline, could not speak English |
|
| Interventions | Treatment: sertraline 50 mg daily (night) Control: matched placebo Duration: treatment continued for 24 weeks Duration of follow‐up (post‐treatment to study end): 28 weeks |
|
| Outcomes | Depression: change in scores from baseline to end of treatment on HDRS, proportion depressed Change in MMSE scores mRS Death Leaving the trial early Check list of possible AEs read out to participant by a research nurse |
|
| Funding source | Funded by an unrestricted grant from Rotary Health Research Fund | |
| Notes | Recruitment June 2004 to June 2006 Conflicts of interest not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random numbers |
| Allocation concealment (selection bias) | Low risk | Centralised |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Stated in paper, matched placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated in paper |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Performed last observation carried forward |
| Selective reporting (reporting bias) | Low risk | Trial protocol published on www.strokecentre.org/trials |
| Other bias | Low risk | No other obvious biases |
Andersen 1994.
| Methods | Parallel design Analysis (ITT) last observation carried forward and per protocol: death (1 treatment, 1 control) withdrawn owing to AE (6 treatment, 1 control), all excluded from analysis |
|
| Participants | Location: Denmark Setting: mixed Treatment: 33 people, mean ± SD age 68 ± 4 years, 36% men Control: 33 people, mean ± SD age 66 ± 9 years, 66% men Stroke criteria: ischaemic stroke and PICH; diagnosis via clinical signs and CT (100%); stroke 2 to 52 weeks prior to randomisation (average time 12 weeks) Depression criteria: HDRS score > 12 (score transformed to appropriate DSM‐III‐R criteria) Other entry criteria: none stated Comparability of treatment groups: balanced Exclusion criteria: depression within last year, receiving current treatment for depression, severe dementia or communication problems, degenerative or expansive neurological disease, decreased consciousness |
|
| Interventions | Treatment: citalopram 10 mg in participants > 66 years, 20 mg in participants < 67 years daily; dose doubled if no response to treatment within 3 weeks Control: matched placebo Duration: treatment continued for 6 weeks Duration of follow‐up (post‐treatment to study end): 0 Note that although the protocol on www.strokecentre.org/trials states that mood scores were measured up to 1 year post‐stroke, this probably refers to the time since stroke at the time of randomisation |
|
| Outcomes | Depression: change in scores from baseline to end of treatment on HDRS Melancholia scale Proportion no longer meeting entry criteria (< 13 on HDRS) 50% reduction in HDRS score Additional: leaving the study early Death AEs (unwanted drug effects were registered and evaluated at the same intervals using a side effect scale) Unable to use: BI, Social Activities Index, MMSE (data not presented) |
|
| Funding source | Funded by Lundbeck Foundation, Medical Research Foundation for North Jutland County, The Aalgorg Diocese Research Foundation, Consultant Otorhinolaryngologist Kopp's Foundation and Stine and Martinus Sorensen's Foundation. Lundbeck Pharma A/S provided the citalopram and placebo; thus we have classified this as 'unclear risk'. | |
| Notes | Recruitment 1 February 1991 to 29 February 1992. Conflicts of interest not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Blocks of 4 used |
| Allocation concealment (selection bias) | Low risk | Centralised opaque envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Matched placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Those who were blinded were not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Although there were dropouts, analysis performed both per protocol and using last observation carried forward |
| Selective reporting (reporting bias) | Low risk | Trial published on www.strokecentre.org/trials The primary outcome was reported |
| Other bias | Unclear risk | − |
Andersen 2013.
| Methods | Multicentre Study type: interventional (clinical trial) Intervention model: parallel assignment Primary purpose: treatment |
|
| Participants | 642 participants Country: Denmark Setting: inpatient At randomisation number allocated: citalopram n = 319; placebo n = 323 % male at baseline: citalopram n = 199/319 (62%); placebo n = 222/323 (69%) Age at baseline: mean age, citalopram 68 ± 13 (n = 319); placebo 68 ± 13 (n = 323) Subtype of stroke at baseline: not available Severity of stroke at baseline: NIHSS, citalopram 5.3 ± 5.6; placebo 4.8 ± 4.8 Time since stroke onset: mean time from last known 'well' to first treatment 1.7 days (median 1, IQR 0 to 6) Inclusion criteria:
Exclusion Criteria:
|
|
| Interventions | Experimental: citalopram 20 mg (10 mg if aged ≥ 65 years or having reduced liver/kidney function) or placebo once daily for 6 months Comparator: ½ to 2 tablets with no intrinsic drug activity per day for 6 months |
|
| Outcomes | Primary outcomes
Secondary outcomes within or at 6 months
|
|
| Funding source | TrygFonden, the Danish Council for Independent Research, the Regional Medicine Fund, and the Aarhus University Research Foundation | |
| Notes | Dates study conducted: September 2013 to December 2016 Declarations of Interest: Dr Kraglund received speaker honoraria from Bristol‐Meyers Squibb and Pfizer. Dr Iversen received speaker honoraria from Boehringer Ingelheim, Bristol‐Myers Squibb, Bayer, AstraZeneca, and Pfizer and has previously participated in advisory board meetings for Boehringer Ingelheim, Bristol‐Myers Squibb, Pfizer, Bayer, AstraZeneca, and Amgen. Dr Grove has received speaker honoraria from AstraZeneca, Baxter, Bayer, Boehringer Ingelheim, Bristol‐Myers Squibb, MSD, and Pfizer and has previously participated in advisory board meetings for AstraZeneca, Bayer, Boehringer Ingelheim, and Bristol‐Myers Squibb. Dr Andersen reports other from MSD, personal fees from AstraZeneca, outside the submitted work |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A computer‐generated randomization code was used to randomize patients in blocks of 10." |
| Allocation concealment (selection bias) | Low risk | Quote: "Citalopram was commercially available (Sandoz, Denmark) and production of the placebo and randomization was prepared by a pharmacy independently of the investigators (Glostrup Pharmacy, Denmark). The tablets were indistinguishable and were supplied in numbered containers." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Blinding of participant, care provider, and investigator assured and unlikely that the blinding could have been broken |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of outcome assessor assured and unlikely that the blinding could have been broken |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition/exclusions reported with reasons provided (including did not start on study medication, consent withdrawn, side effects, indication for open label, other reasons (no detail provided)). At < 31 days of study medication, twice as many participants in the citalopram group withdrew consent (n = 29/318 (9%)) compared to the placebo group (n = 14/320 (4%)). However, at < 31 days twice as many participants in the placebo group (n = 12/318(4%)) compared to the citalopram group (n = 6/320 (2%) were switched to open label. Attrition/exclusions: 51/319 (16%) in the citalopram group and 39/319 (11%) in the placebo group. The investigators use LOCF in their intention‐to‐treat analysis. LOCF assumes that missing values are missing completely at random and ignores improvements or deteriorations in the participants condition since dropout and therefore stops improvements or declines in outcome measures. LOCF introduces risk of false or biased conclusions (Molnar 2008) |
| Selective reporting (reporting bias) | Low risk | The study protocol is available and all of the study's prespecified (primary and secondary) outcomes that are of interest to the review have been reported in the prespecified way |
| Other bias | Low risk | The study appears to be free of other sources of bias |
Birchenall 2019.
| Methods | Study type: interventional (clinical trial) Intervention model: parallel assignment Primary Purpose: treatment |
|
| Participants | 6 participants Country: France Setting: inpatient At randomisation number allocated: 6 although unclear as to which group % male: not available Age: not available Subtype of stroke: not available Severity of stroke: not available Time since stroke onset: not available Inclusion criteria:
Exclusion criteria:
Withdrawal criteria: not stated |
|
| Interventions | Experimental: fluoxetine; 1 pill of 20 mg/day, during 3 months Comparator: placebo of fluoxetine; 1 pill of 20 mg/day, during 3 months |
|
| Outcomes | Primary outcome:
Secondary outcomes recorded at 3 and 6 months:
|
|
| Funding source | Not stated | |
| Notes | No published data, unpublished data say 6 patients, none of whom died, so we have used this information Dates study conducted: February 2014 to August 2015 Declarations of Interest: none reported. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No information available |
| Allocation concealment (selection bias) | Unclear risk | No information available |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | No information available |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No information available |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | No information available |
| Selective reporting (reporting bias) | Unclear risk | No information available |
| Other bias | Unclear risk | No information available |
Black‐Schaffer 2012.
| Methods | Study type: interventional (clinical trial) Intervention model: parallel assignment Primary purpose: treatment |
|
| Participants | 0 participants (aimed to recruit 25 participants) Country: USA Setting: inpatient At randomisation number allocated: 0 % male: not available Age: not available Subtype of stroke: not available Severity of stroke: not available Time since stroke onset: not available Inclusion criteria:
Exclusion criteria:
|
|
| Interventions | Experimental: fluoxetine 20 mg daily for 90 days starting day 5 to10 after stroke Comparator: placebo participants will take 1 placebo pill daily for 90 days |
|
| Outcomes | Primary outcome measures:
Secondary Outcome Measures
|
|
| Funding source | Not stated | |
| Notes | clinicaltrials.gov/ct2/show/NCT01674868 Withdrawn ‐ unable to find patients meeting inclusion/exclusion criteria Dates study conducted: April 2013 to December 2015 (estimated completion date) Declarations of Interest: none reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Withdrawn: unable to find patients meeting inclusion/exclusion criteria |
| Allocation concealment (selection bias) | Unclear risk | Withdrawn: unable to find patients meeting inclusion/exclusion criteria |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Withdrawn: unable to find patients meeting inclusion/exclusion criteria |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Withdrawn: unable to find patients meeting inclusion/exclusion criteria |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Withdrawn: unable to find patients meeting inclusion/exclusion criteria |
| Selective reporting (reporting bias) | Unclear risk | Withdrawn: unable to find patients meeting inclusion/exclusion criteria |
| Other bias | Unclear risk | Withdrawn: unable to find patients meeting inclusion/exclusion criteria |
Brown 1998.
| Methods | Parallel design Analysis: per protocol: 1 withdrawn (treatment), excluded from analysis |
|
| Participants | Diagnosis: stroke, time from stroke to randomisation not reported Randomised 10 to treatment and 10 to control Treatment: 9 completed treatment, mean ± SD age 61.4 ± 8.6 years, 55% men Control: 10 people completed placebo, mean ± SD age 63.7 ± 5.4 years, 60% men Emotionalism criteria: emotionalism of at least 4 weeks' duration assessed during semi‐structured interview using a modified Lawson and MacLeod rating scale, in addition to frequency of outbursts Exclusion criteria: cognitive impairment, dysphasia, major depressive disorder |
|
| Interventions | Treatment: fluoxetine 20 mg daily Control: matched placebo Duration: 10 days Duration of follow‐up: (end of treatment to end of study) 0 |
|
| Outcomes | Used leaving the study early Unable to use data from HDRS, Lawson and MacLeod Scale, self‐rating scales (mean and SD not presented) Also reported emotional outbursts; we have not used these in our analyses AEs: not presented |
|
| Funding source | Funder not stated | |
| Notes | Dates of study not stated; conflicts of interest not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation method not stated |
| Allocation concealment (selection bias) | Unclear risk | Randomised by independent statistician |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | States blinding, matched placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | States blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Only 1 withdrawn (5% of participants) ‐ we categorised this as low risk |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to make judgement |
| Other bias | Unclear risk | No other obvious biases, baseline balanced |
Burns 1999.
| Methods | Parallel design Analysis: ITT: 2 withdrawn and 1 death (treatment), 1 death (placebo), last value carried forward |
|
| Participants | Diagnosis: stroke. Months from stroke: median (range) 10.5 months (1 ± 156) in sertraline group and 5.5 months (1.5 ± 48) in the control group Treatment: 14 people Control: 14 people Exclusion criteria: less than 1 month since stroke, depression or dementia using the DSM III‐R criteria |
|
| Interventions | Treatment: sertraline 50 mg daily Control: matched placebo Duration: treatment continued for 8 weeks Duration of follow‐up: 2 weeks off treatment. All scores became non‐significant (though data not reported so could not be used in the analysis) |
|
| Outcomes | Able to use:
Method of collecting AEs was not stated Unable to use: MADRS, BI, MMSE (data not presented) |
|
| Funding source | Funded by an unrestricted personal grant from Pfizer, the manufacturers of sertraline | |
| Notes | Dates of study not stated, conflicts of interest not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Blocks of 4 using list produced by medical statistics department |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Matched placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Run‐out was single‐blind, treatment was double‐blind, but unclear whether outcome assessors were blind |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Analysis: ITT, LOCF |
| Selective reporting (reporting bias) | Low risk | Trial details published on www.strokecentre.org/trials, although unable to use data from MADRS Given that the main aim was to explore effect on emotionalism, this is unlikely to have biased results |
| Other bias | Unclear risk | Placebo group younger, uncertain influence on bias Statistical analysis was carried out independently by the Applied Statistics Research Unit in Canterbury |
Chen 2005a.
| Methods | To observe the changes of neurotransmitter in people with post‐stroke depression by using Encephalofluctuography Technology, and observe the effect of antidepression treatment on the activity of neurotransmitter | |
| Participants | 48 participants with post‐stroke depression | |
| Interventions | Treatment: 24 people received citalopram 20 mg plus usual care, or fluoxetine if side effects such as nausea, emesis Control: 24 people usual care alone |
|
| Outcomes | Encephalofluctuography technology Level of sympathin and 5‐hydroxytryptamine at 4 weeks and 3 months after treatment started |
|
| Funding source | Not stated | |
| Notes | No data from our endpoints of interest, so data not included in a meta‐analysis Recruitment March 2001 to December 2001 Conflicts of interest not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Randomly divided" but method not stated |
| Allocation concealment (selection bias) | Unclear risk | Method not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | Unclear |
| Other bias | Unclear risk | Unclear |
Chen 2005b.
| Methods | Parallel group Analysis: according to allocated treatment group |
|
| Participants | Country: China Setting: inpatient Stroke criteria: first ever stroke, onset time ≤ 7 days, haemorrhagic and ischaemic, clinical diagnosis plus confirmation by imaging (though not clear whether a stroke lesion had to be present), at least 1 limb with muscle power grade 3 or less, BI ≤ 50, no consciousness disturbance Mood criteria: HAMD > 16 Treatment: 40 people, mean age 63.5 years, 29 men Control: 38 people, mean age 65.8 years, 25 men No difference in baseline depression and BI between treatment and control group Excluded: severe cardiac, hepatic and renal organic diseases, psychiatric disorders |
|
| Interventions | Treatment: paroxetine 20 mg daily plus routine stroke medication, nerve nutritional agents, acupuncture and rehabilitation Control: routine stroke medication, nerve nutritional agents, acupuncture and rehabilitation Duration of treatment: 12 weeks Duration of follow‐up (post‐treatment to study end): 0 weeks |
|
| Outcomes | HAMD BI Death Number completing the trial AEs not reported |
|
| Funding source | No description of funding | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropouts: none |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | No obvious risks, baseline similar |
Chen 2001.
| Methods | Randomised trial Aim: to observe effects of integrative Chinese herb YuLeShu and fluoxetine on the depressive symptoms and rehabilitation of neurological impairment in patients with post‐stroke depression |
|
| Participants | Country: China Setting: not described Participants: internal carotid system cerebral infarction or haemorrhage within previous 2 months Fluoxetine: 19 people, mean age 61.71 ± 8.13 years, 8 men Control: 18 people, mean age 62.85 ± 7.32 years, 7 men Depression: diagnosis of depression according to DSM‐IV Inclusion criteria: HDRS ≥ 20 but < 35 and/or Zung SDS ≥ 41 Exclusion criteria: HDRS > 35, previous depression, aphasia, severe cardiac, pulmonary, hepatic and renal diseases, previous stroke |
|
| Interventions | 3 groups: fluoxetine plus usual care versus YuLeShu plus usual care versus usual care. We are using the fluoxetine plus usual care versus usual care alone in the comparison | |
| Outcomes | HDRS Zung SDS BI Scandinavian Neurological Stroke Scale (also known as CSS) Stated no side effects, but not clear which side effects were sought, or how they were sought. They were reported at 4, 8 and 12 weeks after treatment |
|
| Funding source | Funded by a local scientific academic fund, drug company not involved | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "using a computer", but method not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Dropouts: 2 people dropped out of the fluoxetine group, 1 dropped out of the YuLeShu group and 2 dropped out of the control group |
| Selective reporting (reporting bias) | Unclear risk | Protocol not published |
| Other bias | Unclear risk | Reported that of the people who completed the tests, there were no differences in baseline No comment on whether there were differences in baseline for the entire group |
Chen 2002.
| Methods | Parallel group (3 groups: doxepin, paroxetine, placebo; we used the paroxetine and placebo data in our review) Aim: treat depression and determine effect on neurological function |
|
| Participants | Country: China Setting: unclear Stroke diagnosis: diagnostic criteria of the 4th National Meeting of the Cerebrovascular Diseases proved by CT or MRI Time since stroke: not known Depression diagnosis: Classification and Diagnosis of Psychosis in China (2nd edition) Treatment: 24 people, age and gender not given Control: 24 people, age and gender not given Exclusion: pre‐stroke mental disease, cognition disorder (MMSE < 24), marked deterioration in depression during treatment (HAMD > 24) or suicide mood, intolerance to drug |
|
| Interventions | Treatment: paroxetine 20 mg 3 times per day Control: placebo guvitamine once per day Duration of treatment: 8 weeks Duration of follow‐up (post‐treatment to study end): unclear: follow‐up is performed 'after treatment' so we assume this is at 8 weeks (so post‐treatment to study end = 0) |
|
| Outcomes | HAMD BI CSS Death/side effects/leaving the trial early Method of reporting side effects not stated |
|
| Funding source | Funder not stated, unclear if there was drug company involvement | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Placebo was used, but unclear if this was matching |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Dropouts: 4 in placebo and 0 in paroxetine |
| Selective reporting (reporting bias) | Unclear risk | No protocol available |
| Other bias | Unclear risk | Demographic data not provided, so we cannot determine whether the baseline was balanced |
Cheng 2003.
| Methods | Parallel design Aim: to treat depression and augment rehabilitation Analysis: according to allocated treatment group |
|
| Participants | Location: China Setting: inpatient Treatment: 25 people Control: 32 people Whole group (including non‐depression group, depression control group and depression treatment group): 132 (mean age 62 ± 12 years, 79 men) Stroke: ischaemic stroke or PICH, clinical diagnosis plus confirmation on brain imaging (not clear that a stroke lesion had to be present), clear consciousness Depression diagnosis (at 2 weeks after stroke onset): psychiatric interview, DSM IV criteria Excluded: major psychological trauma history in previous 1 year, severe mental retardation, severe impairment of lingual expression or comprehension, major complicated medical event in previous 1 year |
|
| Interventions | Treatment: fluoxetine 20 mg daily Control: no fluoxetine Duration of treatment: 6 months Duration of follow‐up (post‐treatment to study end): 6 months |
|
| Outcomes | SSS ADL HAMD Zung SDS Zung SAS No deaths, none left trial early No data on AEs |
|
| Funding source | No description of funding | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 59 participants were diagnosed to have depression by symptoms but only 57 were included in the results table |
| Selective reporting (reporting bias) | High risk | No protocol, no report of the results of the self‐rating anxiety scale |
| Other bias | Unclear risk | No clear description of differences between the treatment and control group |
Chollet 2011.
| Methods | Randomised parallel‐group trial | |
| Participants | Location: France Setting: stroke units Inclusion criteria: aged 18 to 85 years with FMMS of 55 or less, acute ischaemic stroke with hemiparesis or hemiplegia, 5 to 10 days after stroke onset, unclear if there had to be a visible lesion on brain imaging Treatment: 59 people, mean ± SD age 66.4 ± 11.7 years; 63% men Control: 59 people, mean ± SD age 62.9 ± 13.4 years; 59% men Comparability of treatment groups: total FMMS score fluoxetine 17.1 compared with 13.4 in placebo Previous stroke more common in the fluoxetine group; fluoxetine group had more diabetes Exclusions: clinical depression or treatment with antidepressants, MADRS > 19, aphasia severe enough to mask detection/assessment of depression, pregnancy, patient on neuroleptics/benzodiazepines, owing to undergo carotid endarterectomy, other major diseases that would prevent follow‐up |
|
| Interventions | Treatment: fluoxetine 20 mg daily for 90 days Control: identical capsules to active drug Duration of treatment: 90 days Duration of follow‐up (treatment end to study end): 0 days |
|
| Outcomes | Primary outcome: the mean change of FMMS score between inclusion (day 0) and day 90 after the start of the study drug Secondary endpoints were NIHSS, mRS and MADRS measured at days 0, 30 and 90 |
|
| Funding source | Funded by French national programme for clinical research: the sponsor had no involvement in study design, data collection, data analysis, data interpretation or writing the report | |
| Notes | Recruitment 14 March 2005 to 9 June 2009. Authors state "no conflicts of interest" | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Balanced by centre with an allocation based on a block size of 4 generated with a computer random‐number generator |
| Allocation concealment (selection bias) | Low risk | Sequentially‐numbered opaque envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Identical capsules for control arm |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | All study site investigators and all investigators were masked to treatment allocation |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Dropouts: 2 participants died (1 in each group) and 3 dropped out ‐ not stated how missing outcome data were dealt with |
| Selective reporting (reporting bias) | Low risk | Trial protocol published on www.strokecentre.org/trials, all outcomes were reported |
| Other bias | Unclear risk | Note difference in baseline: it is not clear what effect this had on results, so we have classified this as 'unclear risk' |
Dam 1996.
| Methods | Parallel design Analysis: per protocol: withdrawn because of AEs (2 treatment), all excluded from analysis |
|
| Participants | Location: Italy Setting: unclear Treatment: 18 people, mean ± SD age 68 ± 9 years, 44% men Control: 17 people, mean ± SD age 68 ± 5.5 years, 44% men Stroke criteria: ischaemic, unilateral MCA territory stroke, diagnosis via clinical signs and CT (100%), stroke 1 to 6 months prior to randomisation (average time 3 months) Other inclusion criteria: unable to walk Comparability of treatment groups: balanced Exclusion: history of major affective disorders; alcohol abuse; or a history or evidence or both of severe heart, lung, kidney or liver diseases or mental deterioration |
|
| Interventions | Treatment: fluoxetine 20 mg daily Control: matched placebo Duration: treatment continued on average 74 ± 6 days, duration not reported for control group Duration of follow‐up (treatment end to study end): 0 |
|
| Outcomes | Depression: change in scores from baseline to end of treatment on HDRS Additional: graded neurological scale (HSS), BI Leaving the study early Death AEs including seizures ‐ unclear if these were reported systematically |
|
| Funding source | Funding source not stated | |
| Notes | Dates of recruitment and conflicts of interest not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No description |
| Allocation concealment (selection bias) | Unclear risk | No description |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Examining neurologists blind to treatment". Comment: Unclear if this refers to outcome assessors or the neurologist caring for the participant. However, placebo was 'matched' so this is low risk |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | See above |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 2/35 dropouts, per‐protocol analysis |
| Selective reporting (reporting bias) | Low risk | Trial available, including results on www.strokecentre.org/trials ‐ all specified outcome measures were reported |
| Other bias | Unclear risk | Baseline characteristics similar in the 2 groups |
Feng 2004.
| Methods | Aim: to study the influence of Jieyu Huoxue decoction on rehabilitation of patients with depression after cerebral infarction | |
| Participants | Country: China 4 groups: fluoxetine plus usual care, Jieyu Huoxue decoction plus usual care, usual care in people with depression, usual care in people with no depression We are using data from 'fluoxetine plus usual care' versus 'usual care in people with depression' Setting: mixed inpatient and outpatient Stroke criteria: ischaemic stroke within 1 month of stroke onset, clinical diagnosis plus confirmation by imaging. Did not state whether a visible lesion was needed to make a diagnosis Depression: psychiatric interview using DSM IV, Zung SDS ≥ 41 Included those with no previous psychiatric history 54 participants with post‐stroke depression were randomised 18 received fluoxetine plus usual care, 18 received usual care only and 18 received Jieyu Huoxue decoction Of the 54 participants with depression randomised, mean age: 71.5 ± 6.7 years, 24 men Excluded: previous stroke, previous depression, and severe cardiac, pulmonary, hepatic and renal diseases |
|
| Interventions | Treatment: fluoxetine 20 mg daily plus usual stroke care Control: usual stroke care Duration of treatment: 60 days Duration of follow‐up (post‐treatment to study end): 0 weeks |
|
| Outcomes | Zung SDS ADL ‐ although score not referenced, so not used in analysis MESSS Reported side effects in fluoxetine group but not in the control group Unclear how side effects were collected |
|
| Funding source | Funding source not stated | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not stated |
| Allocation concealment (selection bias) | Unclear risk | No description |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | No blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 8 participants dropped out (2 in fluoxetine group, 2 in the depression control group, 1 in the Jieyu Huoxue decoction, 3 in no‐depression control) |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | Baseline balanced |
FOCUS Trial Collaboration 2018.
| Methods | Multicentre RCT Study type: interventional (clinical trial) Primary purpose: treatment |
|
| Participants | 3127 participants Country: UK Setting: inpatient At randomisation number allocated: N = 3127: fluoxetine (n = 1564); placebo (n = 1563) % male: fluoxetine (62%); placebo (61%) Age: mean age: fluoxetine = 71·2 ± 12.4; placebo = 71·5 ± 12.1 Subtype of stroke:
Severity of stroke: NIHSS, Median (IQR) fluoxetine (6 (3 to 11)); placebo (6 (3 to 11)) Time since stroke onset: mean days: fluoxetine 6.9 ± 3.6; placebo 7.0 ± 3.6 Inclusion criteria
Exclusion criteria
|
|
| Interventions | Experimental: 20 mg orally once daily for 6 months Comparator: matching placebo orally once daily for 6 months |
|
| Outcomes | Primary outcome:
Secondary outcome measures:
|
|
| Funding source | MHRA approval granted. Start‐up phase funded by The Stroke Association. Main phase funded by NIHR | |
| Notes | ISRCTN83290762. Recruitment 10 September 2012 to 31 March 2017. Authors declared no conflicts of interest | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Patients were randomly assigned in a 1:1 ratio to receive fluoxetine or placebo, by use of a centralised randomization system." |
| Allocation concealment (selection bias) | Low risk | Quote: "Patients were randomly assigned in a 1:1 ratio to receive fluoxetine or placebo, by use of a centralised randomization system." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Patients, their families, and the health‐care team including the pharmacist, staff in the coordinating centre, and anyone involved in outcome assessments were all masked to treatment allocation by use of a placebo capsule that was visually identical to the fluoxetine capsules even when broken open." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Patients, their families, and the health‐care team including the pharmacist, staff in the coordinating centre, and anyone involved in outcome assessments were all masked to treatment allocation by use of a placebo capsule that was visually identical to the fluoxetine capsules even when broken open." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | For the primary outcome of mRS at 6 months data were available for fluoxetine n = 1553/1564 (99.3%) and placebo n = 1553/1563 (99.3%) |
| Selective reporting (reporting bias) | Low risk | The study protocol is available and all of the study's prespecified (primary and secondary) outcomes that are of interest in the review have been reported |
| Other bias | Low risk | The study appears to be free of other sources of bias |
Fruehwald 2003.
| Methods | Parallel design Analysis: per protocol: Withdrawals: death (1 treatment), withdrawn owing to AEs (1 treatment, 2 control), all excluded from analysis |
|
| Participants | Location: Austria Setting: inpatients Treatment: 28 people, mean ± SD age 65 ± 14 years, 46% men Control: 26 people, mean ± SD age 64 ± 14 years, 71% men Stroke criteria: ischaemic stroke and PICH; diagnosis via clinical signs and CT (100%); stroke on average 11 days prior to randomisation Depression criteria: psychiatric interviews, HDRS score > 15 Other entry criteria: not stated Comparability of treatment groups: non‐significant trend towards more women and right‐sided strokes in treatment group Exclusion criteria: MMSE < 20, more than mild communication deficit, diseases of the central nervous system and previous neurodegenerative or expansive neurological disorders |
|
| Interventions | Treatment: fluoxetine 20 mg daily, dose escalation at 4 weeks if HDRS score > 13 Control: matched placebo Duration of treatment: 12 weeks Duration of follow‐up (end of treatment to study end): 15 months |
|
| Outcomes | Depression: change in scores from baseline to end of treatment of HDRS, BDI and CGI (item 1) Proportion of responders (< 13 HDRS) Additional: SSS Death AEs (selected data) Unable to use: RS, BI, MMSE (data not presented at follow‐up) AEs data on dizziness, nausea and cephalalgia (data not presented by group) |
|
| Funding source | The medication was supplied by Lannacher Heilmittel, Lannach, Austria | |
| Notes | Recruitment 1 June 1998 to 31 Decmeber 1998. Conflicts of interest not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computerised randomisation, using random permutated block design |
| Allocation concealment (selection bias) | Low risk | Centralised concealment |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | States blinded, used matching placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | States blinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 4/54, per protocol analysis |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | Baseline balanced All participants were randomly assigned to either fluoxetine or placebo treatment by the drug company independently of the research teams and the study centres |
Gao 2016.
| Methods | Study type: interventional (clinical trial) Primary purpose: treatment |
|
| Participants | 274 participants Country: China Setting: outpatient At randomisation number allocated: N = 274, citalopram (n = 91); placebo (n = 91); cognitive behavioural therapy (n = 92) % male: 51.8% Age: mean age, citalopram 66.0 ± 7.3 (n = 91); placebo 67.2 ± 9.6 (n = 91); cognitive behavioural therapy 64.9 ± 8.0 (n = 92) Subtype of stroke: not available Severity of stroke: not available Time since stroke onset: acute ischaemic stroke within the previous 7 days Inclusion criteria:
Exclusion criteria:
|
|
| Interventions | Experimental: citalopram 20 mg per day for a minimum of 3 months + general discussions Comparator 1: placebo + general discussions Comparator 2: placebo + cognitive behavioural therapy |
|
| Outcomes |
|
|
| Funding source | Natural Science Foundation of China [81100243, 81171131, 81272564, 81272795, 81100893, 81172197, and 81372484], the Natural Science Foundation of Liaoning Province in China [No. L2013296], and Liaoning Science and Technology Plan Projects [No. 2011225020] | |
| Notes | This trial was particular in that recruitment happened at 4 different time points: at 0 months, 3 months, 6 months and 9 months from discharge. Inclusion criteria required that participants suffered from post‐stroke depression. Participants were invited to complete the BDI and those with a score > 10 were included, provided other criteria were met Group 'placebo + general discussions' and 'citalopram + general discussions were included. No significant differences observed in the 2 included groups Dates study conducted: Participants enrolled between October 2011 and June 2013 Declarations of Interest: none reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Randomization into one of three intervention groups was undertaken by an independent researcher using computer‐generated random number sequences..." |
| Allocation concealment (selection bias) | Low risk | Quote: "....that were prepared in advance and placed in consecutively numbered, sealed, opaque envelopes." |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | Study described as "single blind". Quote: "The researcher successively opened the envelopes corresponding to different time periods and determined the intervention by patient number." Quote: "The study therapists acted as clinical evaluators." Quote: "The study therapists were asked not to divulge any treatment information to their patients." Comment: Care providers, investigator and outcome assessors were all aware of allocation. |
| Blinding of outcome assessment (detection bias) All outcomes | High risk | Quote: "The study therapists acted as clinical evaluators." Quote: "The study therapists were asked not to divulge any treatment information to their patients." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Quote: "in Group A [placebo + general discussions] , one patient violated protocol in the second time period, one could no longer be reached, and one left the study owing to stroke recurrence in the third time period; in Group B [citalopram + general discussions], persistent side‐effects from the drugs led five patients to leave the study (two owing to orthostatic dizziness, one owing to palpitation, and two owing to constipation)" Comment: Attrition reported for each intervention group and reasons given Group A (placebo + general discussions) 3/91 = 3% attrition Group B (citalopram + general discussions) 5/91 = 5% attrition Overall = 4% attrition |
| Selective reporting (reporting bias) | Unclear risk | There is no study protocol available. Therefore insufficient information to judge yes or no |
| Other bias | Low risk | The study appears to be free from other sources of bias |
GlaxoSmithKline 1998.
| Methods | Parallel group Analysis: according to treatment group |
|
| Participants | Location: not stated Setting: not stated Stroke criteria: "documented diagnosis of stroke within 12 months prior to screening" Mood: MADRS score > 17 Treatment: 112 people, age 64.3 ± 11.4 years, 61 men Control: 117 people, 65.6 ± 10.5 years, 64 men Excluded: concurrent psychiatric disorders, concurrent psychotropic pharmacotherapy, patients who posed a suicidal risk, patients with substance abuse/dependence, concurrent psychotropic pharmacotherapy, MMSE < 24, participating in another clinical trial, serious medical condition or clinically significant finding on screening or baseline evaluation that would preclude the administration of paroxetine and an intolerance to paroxetine |
|
| Interventions | Treatment: paroxetine 20 to 50 mg daily Control: placebo (not stated whether matching) Duration of treatment: 8 weeks Duration of follow‐up (treatment to study end): 0 weeks |
|
| Outcomes | Change from baseline to endpoint in MADRS Proportion of participants scoring < 8 on the MADRS total score at the endpoint (we used this in our analysis) Changes from baseline to endpoint on the BI Change from baseline to endpoint on RS score Change from baseline to endpoint on the Clinical Global Improvement Severity of Illness Score (CGI‐S Proportion of responders based on CGI‐Global Improvement (CGI‐G) score (score of < 4) at endpoint GI side effects reported, but unclear whether these are 'events' or 'participants', so we cannot use these data. It is not clear how the side effects were collected Withdrawal from study |
|
| Funding source | Source of funding not stated, but we assume it was funded by GlaxoSmithKline | |
| Notes | Study period 29 August 1998 to 15 October 1999. Conflicts of interest not stated. Study number PAR625. Date updated: 11 March 2005 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation method not described |
| Allocation concealment (selection bias) | Unclear risk | Allocation concealment: not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Blinding not described, used placebo but not stated whether identical |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Blinding not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 20 in each group dropped out |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | Insufficient information to make clear judgement |
Guo 2009.
| Methods | Parallel group, 3‐arm trial, comparing sertraline plus routine care versus routine care versus acupuncture plus routine care. We are using the sertraline plus routine care versus routine care in this review Aim: to treat depression Analysis: according to allocated treatment |
|
| Participants | Country: China Setting: unknown Stroke criteria: first ever stroke, clinical diagnosis plus relevant lesion on imaging, age ≥ 60 years old Depression criteria: HAMD score ≥ 8, no depression prior to stroke Treatment: 40 people, mean age 67.6 ± 12.43 years, 23 men Control: 40 people, mean age 64.5 ± 12.07 years, 22 men Exclusions: psychiatric disorders or family psychiatric disorders, severe cognitive impairment, global aphasia, sensory aphasia, apraxia, severe cardiac, hepatic, renal, lung or other severe somatic disorder, consciousness disturbance, severe deafness, family or patient unable to comply |
|
| Interventions | Treatment: sertraline 50 mg daily plus stroke care (acute, secondary prevention, rehabilitation and psychotherapy) Control: stroke care (acute, secondary prevention, rehabilitation and psychotherapy) Duration of treatment: 6 weeks Duration of follow‐up: (treatment end to study end): 6 months |
|
| Outcomes | HAMD NIHSS FIM (reported cognition and mobility scores only) SF‐36 AEs not reported |
|
| Funding source | Funded by a local scientific academic fund | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random‐number table |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessor blind |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts, analysed by allocated treatment |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Low risk | No obvious risk, balance baseline |
He 2004.
| Methods | Parallel group Analysis: according to treatment allocation |
|
| Participants | Location: China Setting: inpatient Inclusion criteria: all pathological types of stroke, clinical diagnosis plus confirmation by imaging (did not state that a visible lesion was needed to make the diagnosis), first ever stroke Depression diagnosis: 'HAMD scores'. Translation of paper: did not have to have depression at recruitment Treatment: 36 people, mean age 70.8 ± 6.7 years, 25 men Control: 35 people, mean age 70.4 ± 6.8 years, 23 men Exclusion: psychiatric disorders, dysphasia, consciousness disturbance, agnosia, severe dementia |
|
| Interventions | Treatment: fluoxetine 20 mg daily plus usual stroke care Control: usual stroke care Duration of treatment: 8 weeks Duration of follow‐up (treatment end to study end): 0 |
|
| Outcomes | HAMD SSS No description of how side effects were collected |
|
| Funding source | Funded by local scientific academic fund | |
| Notes | Reported that there were no AEs, so we have assumed no seizures or GI side effects | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Outcome assessors blind" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 13 dropped out after randomisation |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Low risk | Balanced baseline, no obvious risks |
He 2005.
| Methods | Parallel design. 3 groups: paroxetine, paroxetine plus psychotherapy, control. We are using paroxetine and control data in this review Analysis: according to treatment group |
|
| Participants | Location: China Setting: inpatient Stroke criteria: first ever stroke; ischaemic and haemorrhagic, timing: "acute", clinical diagnosis plus confirmation by imaging (though not clear whether a stroke lesion had to be present or not) Mood criteria: meets ICD‐10 organic depression and organic anxiety diagnostic criteria on psychiatric interview, HAMD score ≥ 17 and HAMA score ≥ 14 Treatment: 27 people, mean age 62.4 ± 6.1 years, 14 men Control: 27 people, mean age 63.2 ± 5.7 years, 16 men Exclusion: previous psychiatric disorder, antidepressants and "nerve block agents" in recent 3 months, severe cognitive impairment, aphasia, severe cardiac, hepatic and renal function impairment, allergy to paroxetine, severe suicidal behaviour |
|
| Interventions | Treatment: paroxetine 20 mg plus routine stroke treatment Control: routine stroke treatment Duration of treatment: 6 weeks Duration of follow‐up: end of treatment to study end: 0 |
|
| Outcomes | SSS BI HAMD HAMA TESS Also reported GI upset and dizziness. They did not list any seizures in the list of AEs, so we are assuming no seizures in either groups Unclear how side effects were collected |
|
| Funding source | Funded by a local scientific academic fund | |
| Notes | The authors mentioned using the SDS and the SAS for evaluation, but they did not report the results of SDS and SAS | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts, analysed according to treatment group |
| Selective reporting (reporting bias) | High risk | No protocol, the authors mentioned using the SDS and the SAS for evaluation but they did not report the results |
| Other bias | Low risk | Balanced baseline |
He 2016.
| Methods | Study type: interventional (clinical trial) Primary purpose: prevention |
|
| Participants | 374 participants Country: China Setting: inpatient At randomisation numbers allocated: N = 300 Experimental group 1: fluoxetine immediately after enrolment n = 100; comparator group 1: fluoxetine 7 days after enrolment n = 100; comparator group 2: no fluoxetine n = 100 % male: unclear Age: experimental, unclear; comparator 1, unclear; comparator 2, unclear Subtype of stroke: unclear Severity of stroke NIHSS score at baseline: unclear Experimental: unclear Comparator 1: unclear Comparator 2: unclear Time from stroke onset: within 1 week after onset of cerebral infarction Inclusion criteria:
Exclusion criteria:
Withdrawal criteria:
|
|
| Interventions | Experimental: 20 mg of fluoxetine a day for 90 days and conventional therapy Comparator: conventional therapy |
|
| Outcomes | Primary outcome at days 15, 90 and 180
Secondary outcome at days 90 and 180
|
|
| Funding source | This study was funded by Science and Technology Department of Guangdong, China (grant number: 2011B031800130), Science and Technology Innovation Committee of Shenzhen, China (grant number: 201101020), and Health and Family Planning Committee of Shenzhen, China (grant number: 201501009). It was registered on the Chinese Clinical Trial Registry (number: ChiCTR‐TRC‐12002078) | |
| Notes | Dates study conducted: Unclear. Either from June 2011 to December 2012 (ChiCTR‐TRC‐12002078) or from December 2015 to June 2016 (ChiCTR‐IPR ‐ 15007658) Declarations of Interest: none reported Trial registration detail (ChiCTR‐TRC‐12002078) does not match but rather matches ChiCTR‐IPR ‐ 15007658. Baseline demographic and clinical characteristics for each group not presented, but rather the baseline demographic and clinical characteristics for those completing the trial (i.e. a subset of all those randomised at baseline) are presented |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Table of random numbers" |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information on method of allocation concealment to judge yes or no |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement yes or no |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "The evaluator was banned from participation in the treatment or from querying of the randomisation data." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | For the primary outcome of NIHSS score at 15, 90 and 180 days there was 8/187 (4%) lost to follow‐up in the experimental group; 16/187 (15%) in the comparator group. Twice as many participants in the comparator group (16/187(9%)) compared to the fluoxetine group (8/187 (4%)) were lost to follow‐up. Attrition and exclusions were not fully reported > 5% lost to follow‐up |
| Selective reporting (reporting bias) | High risk | The trial registration number/protocol does not match the study design presented, but rather matches ChiCTR‐IPR ‐ 15007658 |
| Other bias | High risk | The baseline data presented in table 1: comparison of data at baseline between control group and the treatment group are not true baseline characteristics (i.e. at randomisation). The data presented in table 1 are the baseline characteristics of all those completing the trial which is a subgroup of all participants randomised. We cannot tell if there is whether there was any baseline imbalance in important demographic or clinical characteristics |
Hu 2002.
| Methods | Parallel design Aim: to study effect of antidepressants on depressive symptoms and nervous function |
|
| Participants | Country: China Setting: inpatient Stroke criteria: all pathological stroke types, clinical diagnosis plus confirmation by imaging (though unclear whether a relevant lesion had to be visible), onset of stroke 0.5 to 2 months, no obvious aphasia Depression: according to CCMD‐II‐R Treatment: 42 people, mean age 61.4 ± 3.6 years, 32 men Control: 30 people, mean age 60 ± 4.8 years, 23 men |
|
| Interventions | Treatment: fluoxetine 20 mg daily Control: no other antidepressant Duration of treatment: 8 weeks Duration of follow‐up (end of treatment to study end): 0 |
|
| Outcomes | HAMD MESSS However, these data were not usable, as they were reported as proportions above or below "decrement levels" Reported side effects but unclear how this was done None left the trial early |
|
| Funding source | Source of funding not stated | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation method not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | Balanced baseline, no other obvious risks |
Huang 2002.
| Methods | Parallel design Aim: efficacy and tolerance of fluoxetine in early post‐stroke depression Analysis: according to treatment group |
|
| Participants | Country: China Setting: inpatient Stroke criteria: first ever stroke, with single unilateral lesion, clinical diagnosis with imaging consistent with stroke, both ischaemic and haemorrhagic, recruited 2 weeks after stroke onset Depression criteria: CCMD II‐R depression diagnosis Treatment: 40 people, age and gender not stated Control: 40 people, age and gender not stated Participants in the treatment and control groups were selected from a group of 168 first‐ever acute stroke patients with average age of 62 ± 8.1 years, 76 men |
|
| Interventions | Treatment: fluoxetine 20 mg daily Control: placebo Duration of treatment: 4 weeks Duration of follow‐up (treatment end to study end): 0 |
|
| Outcomes | HAMD CSS Did not report death Unclear how AEs were reported: no obvious AEs were found, but they did not specifically report seizures |
|
| Funding source | Source of funding not stated | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not stated |
| Allocation concealment (selection bias) | Unclear risk | Method not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Placebo used, but unclear if identical |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts, analysed according to treatment group |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | No description of the differences between treatment and control group in baseline characteristics |
Jia 2005.
| Methods | Parallel design Aim: to determine the effect of early intervention for post stroke depression on movement after 3 months of stroke |
|
| Participants | Country: China Setting: inpatient Inclusion: aged 40 to 75 years, all pathological types of stroke, clinical diagnosis plus confirmation by imaging (did not state whether a relevant lesion had to be present to make a diagnosis), able to give informed consent Depression diagnosis: Zung SDS > 41 for screening for depression, HDRS for evaluation of the depression severity level Treatment: 92 people randomised, 90 accepted allocation, mean age 55.6 ± 6.5 years, 60 men Control: 92 people randomised, 90 accepted allocation, mean age 55.1 ± 6.8, 55 men Excluded: organic psychiatric disorders such as Alzheimer's disease or degenerative disease, functional disorders such as schizophrenia and affective disorders |
|
| Interventions | Treatment: either fluoxetine or sertraline (given sertraline if also had anxiety) plus routine stroke care Control: routine stroke care Duration of treatment: 3 months Duration of follow‐up: 3 years but the authors did not describe the extent of neurological function damage and HAMD scores in the third year |
|
| Outcomes | HAMD Extent of neurological damage Recurrent stroke Death Did not report AEs |
|
| Funding source | Source of funding not stated | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Dropouts: 6 in treatment group (2 refused allocation), 4 in control group (2 refused allocation) |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | Balanced baseline |
Kim 2011.
| Methods | Multicentre Study type: interventional (clinical trial) Intervention model: parallel assignment Primary purpose: prevention |
|
| Participants | 478 participants Country: South Korea Setting: inpatient. At neurology departments in 17 university hospitals throughout South Korea At randomisation number allocated: N = 478, escitalopram (n = 241); placebo (n = 237) % male at baseline: unclear Age at baseline: unclear Subtype of stroke at baseline: unclear Severity of stroke at baseline: unclear Time since stroke onset: acute ischaemic stroke or intracerebral haemorrhage within the previous 21 days Inclusion criteria:
Exclusion criteria
Withdrawal criteria: not stated |
|
| Interventions | Experimental: escitalopram: first week 5 mg, 2nd week ˜ 12 week: 10 mg Comparator: "sugar pill". First week 5 mg, 2nd week ˜ 12 week: 10 mg |
|
| Outcomes | Primary outcomes collected at 3 months
Secondary outcomes:
|
|
| Funding source | Dong‐A Pharmaceutical Company, grants from the Ministry for Health, Welfare, and Family Affairs, South Korea | |
| Notes |
NCT01278498 Baseline demographic and clinical characteristics for each group not presented, but rather the baseline demographic and clinical characteristics for those completing the trial (i.e., a subset of all those randomised at baseline) are presented. Dates study conducted: January 2011 to December 2015. Declarations of Interest: none reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Eligible patients were enrolled by investigators at each centre, and randomly assigned in a 1:1 ratio using a web‐based system to the escitalopram group or the placebo group after being assigned a subject number. Randomisation was done with random permuted blocks of sizes four to six, and was stratified by centre. The placebo was identical in appearance to escitalopram" |
| Allocation concealment (selection bias) | Low risk | Quote: "Eligible patients were enrolled by investigators at each centre, and randomly assigned in a 1:1 ratio using a web‐based system to the escitalopram group or the placebo group after being assigned a subject number. Randomisation was done with random permuted blocks of sizes four to six, and was stratified by centre" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "The placebo was identical in appearance to escitalopram" Quote: "The individual treatment code was stored separately by the main medical statistician (E‐JL) and two designated statisticians. All investigators including interviewers and assessors of the outcome, participants, and care providers were masked to treatment assignment throughout the study. The code could be unblinded only with the approval of the steering committee." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "All investigators including interviewers and assessors of the outcome, participants, and care providers were masked to treatment assignment throughout the study. The code could be unblinded only with the approval of the steering committee." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | The following participants were excluded from the 'full analysis set' post‐randomisation from both escitalopram group and placebo group:
Reasons for attrition were reported (withdrew consent, violated protocol, considered for treatment for depression, death). Numbers were similar in both groups At 12 weeks, escitalopram group 67/241 (28%) attrition and placebo 73/237(31%) attrition Attrition greater than 5% It is not clear how missing data were imputed for the intention‐to‐treat analysis; Quote: "we used latest available records for analysis." |
| Selective reporting (reporting bias) | Low risk | The study protocol is available and all the study's prespecified (primary outcomes and secondary outcomes) that are of interest in the review have been reported in the prespecified way.= |
| Other bias | High risk | The baseline data presented in table 1: comparison of data at baseline between control group and the treatment group are not true baseline characteristics (i.e. at randomisation). The data presented in table 1 are the baseline characteristics of all those completing the trial which is a subgroup of all participants randomised. We cannot tell if there is whether there was any baseline imbalance in important demographic or clinical characteristics |
Kong 2007.
| Methods | Parallel Aim: to study whether fluoxetine could prevent post‐stroke depression and improve neurological function |
|
| Participants | Country: China Setting: inpatient Stroke: met diagnostic criteria of various cerebrovascular diseases formulated in the 4th National Cerebrovascular Disease conference and confirmed as stroke by CT or MRI, all hemiplegic, within 7 days of onset HAMD score of no depression Treatment: 48 people, mean age 64 ± 7 years, 60% men Control: 42 people, mean age 62 ± 7 years, 57% men Exclusion: major depression, current antidepressants, allergy to fluoxetine, substance abuse, bipolar disorder, schizophrenia, MMSE ≤ 23/30, substance abuse, obvious liver and renal deficit |
|
| Interventions | Treatment: fluoxetine 20 mg daily Control: matching placebo capsules Duration of treatment: 8 weeks Duration of follow‐up (end of treatment to end of study): 0 |
|
| Outcomes | HAMD BI NIHSS Reported "somatic side effects and hyponatraemia" but not death or other side effects Authors state that "side effect rating was assessed at each visit" but unclear how this was done |
|
| Funding source | Source of funding not stated. Fluoxetine and placebo were supplied by Lilly Pharmaceutical Company | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated table of random digits |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Identical capsules, participants blinded |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | States that researchers were blinded |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 17/90 dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | Balanced baseline |
Lai 2006.
| Methods | Parallel design Analysis: analysed according to allocated treatment groups |
|
| Participants | Location China Setting: inpatients Treatment: 40 people Control: 40 people Total: mean age 60 ± 14 years, 43 men Stroke criteria: unclear stroke types, clinical diagnosis plus brain imaging (though not clear that stroke lesion had to be present), acute stroke Depression criteria: HAMD at least 7, or Zung SDS > 53, but no clear description about using which scale for inclusion criteria Other entry criteria: none stated Comparability of treatment groups: unclear Exclusion criteria: unclear |
|
| Interventions | Treatment: paroxetine 20 mg daily Control: placebo Duration: treatment continued for 2 months Duration of follow‐up (end of treatment to end of study): 0 |
|
| Outcomes | Depression: HAMD, Zung SDS (abnormal if the score is > 53) Additional: Zung SAS (abnormal is the score is > 50) Death The author described that they recorded AEs but they did not report any AEs |
|
| Funding source | Source of funding not stated | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not stated |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Placebo used, not stated if matching |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No participant dropped out |
| Selective reporting (reporting bias) | High risk | No protocol, stated that they would evaluate side effects but these were not reported |
| Other bias | Unclear risk | Demographic details at baseline not described |
Li 2004a.
| Methods | Parallel group Aim: to study effects of fluoxetine on neurological impairment and post‐stroke depression |
|
| Participants | Location: China Setting: inpatient Stroke: inclusion: all pathological types, clinical diagnosis plus confirmation by imaging that relevant lesion visible, CSS 16 to 30 Depression criteria: HAMD scores ≥ 17 and DSM IV diagnostic criteria Treatment: 33 people, mean age 60.33 years, 24 men Control: 34 people, mean age 60.44 years, 23 men Excluded severe psychiatric disorders, severe cardiac, pulmonary, hepatic and renal disease |
|
| Interventions | Treatment: fluoxetine 20 mg daily plus routine acute stroke care Control: routine acute stroke care Duration of treatment: 4 weeks Duration of follow‐up (end of treatment to end of study): 0 |
|
| Outcomes | CSS Depression incidence Laboratory monitoring parameters AEs (method of reporting not stated) |
|
| Funding source | Source of funding not stated | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer random numbers |
| Allocation concealment (selection bias) | Low risk | Opaque sealed envelopes |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | Balanced baseline |
Li 2004b.
| Methods | Parallel design Aim: to treat depression |
|
| Participants | Country: China Setting: inpatient Stroke criteria: ischaemic stroke, clinical diagnosis plus imaging confirmation (though not clear that a relevant lesion had to be seen), stroke onset time ≤ 7 days Depression criteria: HAMD score ≥ 8 Treatment: 37 people, age 48 to 87 years, 17 men Control: 36 people, age 53 to 82 years, 15 men Exclusion: previous depression or psychiatric interview, dementia (according to MMSE scores), aphasia, severe cardiac, pulmonary, hepatic, renal function impairment, consciousness disturbance |
|
| Interventions | Treatment: fluoxetine 20 mg daily plus usual stroke care Control: usual stroke care Duration: 8 weeks Duration of follow‐up (treatment end to study end): 0 |
|
| Outcomes | HAMD CSS (cannot use as reported as a categorical variable) MMSE (reported as a dichotomous variable) BI (reported as a dichotomous variable) Data for continuous variables not provided Death reported Side effects in treatment group only reported, not control group. Method of reporting side effects not stated |
|
| Funding source | Source of funding not stated | |
| Notes | Note that the sum of numbers in each category of HAMD at 8 weeks in the control group adds up to 30, not 32 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not described |
| Allocation concealment (selection bias) | Unclear risk | Method not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Dropouts: 6 in treatment and 4 in control group |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | Baseline balanced |
Li 2005.
| Methods | Parallel design Improvement of post‐stroke depression and augmentation of rehabilitation |
|
| Participants | Country: China Setting: inpatient Stroke criteria: all stroke, clinical diagnosis plus confirmation on imaging (though not clear whether a relevant lesion had to be present) Depression according to CCMD‐II‐R Treatment: 74 participants Control: 74 participants Participaients in the treatment and control groups were selected from a group of 368 stroke patients with an average age of 57 ± 11.8 years, age range 33 to 84 years, 240 men Excluded: previous psychiatric disorders, severe dementia, aphasia, consciousness disturbance |
|
| Interventions | Treatment: paroxetine 20 mg daily plus routine stroke treatment Control: routine stroke treatment Duration of treatment: 4 weeks Duration of follow‐up (end of treatment to study end): 0 |
|
| Outcomes | HAMD SSS Deaths Side effects not recorded |
|
| Funding source | Source of funding not stated | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Randomisation method not described |
| Allocation concealment (selection bias) | Unclear risk | No description |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated whether blinded |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Analysed according to allocated treatment group, no participant dropped out |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | No description of differences between treatment and control group |
Li 2006.
| Methods | Parallel group | |
| Participants | All pathological types of stroke, CT or MRI needed for diagnosis Inclusion criteria: depression diagnosed by Chinese Classification of Mental Disorders 3 and HAMD ≥ 18, no previous organic brain disorder, and no previous psychiatric history, clear consciousness, no comprehension problems, normal language, first acute stroke, first episode of depression Treatment: 52 people, mean ± SD age 61.12 ± 10.25, 32 men Control: 53 people, mean ± SD age 60.89 ± 9.12, 35 men |
|
| Interventions | Treatment: citalopram 20 mg daily plus usual care Control: usual care Duration of treatment: 12 weeks Duration of follow‐up (end of treatment to end of study): 0 |
|
| Outcomes | HDRS (also known as HAMD) BI CSS MMSE Side effects reported according to the participant's complaints and observation, no description of who recorded AEs; and reported only for the treatment group |
|
| Funding source | Source of funding not stated | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | No description |
| Allocation concealment (selection bias) | Unclear risk | No description |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | No description |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 2 dropouts in treatment group, 4 in control group. 1 in treatment group died, and 2 in the control group died (i.e. > 5%) |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | Baseline balanced |
Li 2008.
| Methods | Parallel trial, 3 (fluoxetine versus "free and easy wandering" versus placebo), we are using the fluoxetine versus placebo comparison in this review | |
| Participants | Country: China Setting: unclear Stroke criteria: by neuroimaging, ischaemic or PICH Depression diagnosis: "each patient was evaluated by a psychiatrist", HAMD > 20 included Fluoxetine group: 60 people, mean age 69.2 ± 3.5 years, men 41.6% Control: 30 people, mean age 67.8 ± 3.9 years, men 56.7% Excluded psychiatric illness other than depression, antidepressants within previous 2 weeks, MMSE < 23, severe aphasia |
|
| Interventions | Treatment: fluoxetine 20 to 40 mg daily Control: placebo Duration of treatment: 8 weeks Duration of follow‐up (treatment end to study end): 0 |
|
| Outcomes | HAMD BI Description of why participants left the trial early AEs (reported by participant or observed/elicited by physician at each visit) |
|
| Funding source | Funded by the Natural Science Foundation of Shandong Province, People's Republic of China. None of authors had financial ties with the companies producing the medications in this study | |
| Notes | Note twice as many in fluoxetine as in control group study conducted between March 2006 to September 2007. None of the authors or departments involved in the study had financial ties with the companies producing the medications used in this study |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Computer‐generated random numbers |
| Allocation concealment (selection bias) | Unclear risk | Unclear |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Paper states blinded, used placebo (though unclear if matching) |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | 4/90 dropped out (< 5%) |
| Selective reporting (reporting bias) | Unclear risk | No placebo |
| Other bias | Low risk | Balanced baseline |
Liu 2006.
| Methods | Parallel design Aim: to study effect of citalopram on post‐stroke depression and neurological functional rehabilitation |
|
| Participants | Country: China Setting: inpatient Stroke criteria: stroke during "recovery phase" at 6 to 9 months, NIHSS score ≥ 13, HAMD score ≥ 17 60 people randomised, of whom 38 were men, mean age 60.7 ± 8.6 years. Demographics for treatment and control groups were not provided Treatment: 30 people, age and gender not stated Control: 30 people, age and gender not stated Exclusion criteria: previous psychiatric disorder, dementia, aphasia, consciousness disturbance |
|
| Interventions | Treatment: citalopram 20 mg daily plus routine stroke care Control: routine stroke care Duration of treatment: 6 weeks Duration of follow‐up (treatment end to study end): 0 |
|
| Outcomes | HAMD NIHSS BI Death |
|
| Funding source | Source of funding not stated | |
| Notes | AEs not reported | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | Baseline balance reported by authors |
Marquez Romero 2013.
| Methods | Multicentre Study type: interventional (clinical trial) Primary purpose: supportive care |
|
| Participants | 32 participants Country: Mexico Setting: inpatient At randomisation number allocated: N = 32: fluoxetine (n = 15); placebo (n = 17) % men: 50% Age: mean age 55.1 ± 12.2 Subtype of stroke: not available Severity of stroke: NIHSS, Median (IQR): fluoxetine (12 (5)); placebo (14 (5)) Time since stroke onset: within 10 days Inclusion criteria:
Exclusion criteria:
Withdrawal criteria:
|
|
| Interventions | Experimental: fluoxetine 20 mg orally once daily for 90 days Comparator: matching placebo orally once daily for 90 days |
|
| Outcomes | Primary outcome
Secondary outcomes
|
|
| Funding source | Psicofarma S.A. de C.V. | |
| Notes |
NCT01737541 Terminated (study recruitment was suspended due to lack of funding) Dates study conducted: November 2012 to August 2014 Declarations of Interest: none reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A pharmaceutical laboratory (Psicofarma™ S.A. de C.V.) will be responsible for the manufacture and randomization of the investigational product, which will be achieved using a web‐based randomization program. This program will be set to assign participants equally to each site at a ratio of 1:1." |
| Allocation concealment (selection bias) | Low risk | Quote: "Each of the sites will be assigned 22 participants. The manufacturer will then deliver the pre‐randomized bottles containing the investigational product to each recruiting center. Study subjects who satisfy the eligibility criteria at each recruiting center will receive the investigational product corresponding to a consecutive number assigned according to their entrance to the study." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Fluoxetine and placebo tablets will be identical in form, color, odor and packaging." "Both the investigator and the subject will be blinded to the assignment of the study drugs. The manufacturer of the tablets will label the investigational drugs by the randomization code number. The labeled experimental products will be provided to the recruiting centers by the manufacturer. An envelope containing all randomization codes will be delivered to the principal investigator and will be kept sealed until the conclusion of the trial." |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding of participants and key study personnel ensured, and unlikely that blinding could have been broken |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Aimed to recruit 44 per group (total of 88) 35 in each group + 20% to allow for predicted 20% loss to follow‐up Actual enrolment N = 32. Quote: "Two patients (one in each group) did not take any medication returning the unopened bottles at visit 1 and had to be excluded from analysis." Comment: Report includes data from 30 participants (14 participants in the fluoxetine group and 16 in the placebo group) |
| Selective reporting (reporting bias) | Low risk | The study protocol is available and all of the study's prespecified (primary and secondary) outcomes that are of interest in the review have been reported |
| Other bias | Low risk | The study appears to be free of other sources of bias |
Meara 1998.
| Methods | Parallel design Analysis: unclear |
|
| Participants | Location: Wales, UK
Setting: inpatient Treatment: unclear Control: unclear Stroke criteria: ischaemic stroke > 11 weeks prior to randomisation Depression criteria: GDS (15‐item) score > 4 Other entry criteria not stated Exclusion criteria: moderate to severe dementia, severe aphasia, communication difficulties, poorly controlled epilepsy |
|
| Interventions | Treatment: sertraline 50 mg daily, dose escalation to 100 mg for non‐responders at 2 weeks Control: matched placebo Duration: treatment continued for 6 weeks |
|
| Outcomes | Depression: change in scores from baseline to end of treatment on GDS
Unable to use GDS, BI, MMSE, FAI, FAST Leaving trial early Death AEs |
|
| Funding source | Source of funding not stated | |
| Notes | Contacted author for more details but no response We could not use the data in our meta‐analysis Dates of study not stated. Conflicts not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Double‐blind reported, those who were blind not described |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Double‐blind reported, those who were blind not described |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Not described |
| Selective reporting (reporting bias) | Unclear risk | Insufficient data to make a judgement |
| Other bias | Unclear risk | Insufficient data to make a judgement |
Miao 2004.
| Methods | Parallel group 9 not allocated (5 in treatment group refused allocation, 4 in the control group refused allocation) |
|
| Participants | Country: China Setting: mixed inpatient and outpatient All stroke pathological types, clinical diagnosis plus confirmation by imaging that a relevant lesion was visible, 2 to 8 months after stroke, clear consciousness, no comprehension problem, 1 lesion in 1 hemisphere, normal language comprehension Mood: depression after stroke onset, HAMD score ≥ 20 Participants: 90 randomised, 34 in each group at treatment end Treatment: 34 people, age 58.16 ± 8.49 years, 19 men Control: 34 people, age 62.45 ± 8.24 years, 18 men Exclusion criteria: other organic brain disorders and other aetiologies‐related depression |
|
| Interventions | Treatment: citalopram 20 mg daily plus usual stroke care Control: usual stroke care Duration of treatment: 6 weeks Duration of follow‐up (treatment end to study end): 0 |
|
| Outcomes | HAMD SDS Efficacy Death AEs (only in the citalopram group) Method of recording AEs was not stated |
|
| Funding source | Source of funding not stated | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Simple random sampling" Comment: no further description given |
| Allocation concealment (selection bias) | Unclear risk | Allocation not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Blinding described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 9 not allocated after randomisation, 13 dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | Baseline balanced |
Murray 2005.
| Methods | Parallel design Analysis: ITT (last observation carried forward) and per‐protocol: death (2 control), no efficacy (16 treatment, 22 control), withdrawn owing to AE (8 treatment, 5 control), withdrew consent (1 control), all excluded from analysis |
|
| Participants | Location: Sweden Setting: mixed Treatment: 62 people, mean ± SD age 71 ± 10 years, 52% men Control: 61 people, mean ± SD age 71 ± 10 years, 44% men Stroke criteria: all subtypes, diagnosis by WHO criteria and CT (100%); stroke 3 to 367 days prior to randomisation (average time 128 days) Depression criteria: psychiatric interview (DSM‐IV, major and minor) and MADRS > 9 Other entry criteria: > 17 years of age, stroke within the previous 12 months Comparability of treatment groups: significant trend towards more left‐hemisphere lesion strokes in treatment group Exclusion criteria: under 18 years of age, severely impaired communication, apparent difficulties adhering to study protocol, acute myocardial infarction, other psychiatric illnesses other than depression, significant risk of suicide, antidepressants during the month after randomisation, current use of psychotropic medication or opiate analgesic drugs Participants with < 20% reduction in MADRS score at 6 weeks were excluded |
|
| Interventions | Treatment: sertraline 50 mg daily; possible dose escalation to 100 mg after 4 weeks Control: matching placebo Duration of treatment: 26 weeks Duration of follow‐up: (treatment end to study end): 0 |
|
| Outcomes | Depression: change in scores from baseline to end of treatment on MADRS Additional: leaving the study early Death Unable to use: Scandinavian Supervision Stroke Scale, BI, Stroke Unit Mental Status, Examination social performance, treatment costs, mortality, relative's situation, neuropsychological performance, neurological recovery (data not presented) AEs (selected data presented) using a modified version of the Udvalg for Kliniske Undersogelser side effect rating scale |
|
| Funding source | Funded by an unrestricted grant, study drug and placebo from Pfizer AG Sweden and grants from the AFA Insurances and Marianne and Marcus Wallenberg Foundation | |
| Notes | Recruitment September 1998 to January 2001. Conflicts stated; some of the authors have received grants from pharmaceutical companies | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Block randomisation |
| Allocation concealment (selection bias) | Low risk | Centralised randomisation |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | States blinding and used matching placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | States blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT as well as per protocol |
| Selective reporting (reporting bias) | High risk | No protocol, paper stated that ADL data and SSS data were collected, but these were not reported |
| Other bias | Unclear risk | Balanced baseline except that more participants had left hemisphere brain lesion in sertraline group than in placebo group (statistically significant) |
Pan 2018.
| Methods | Study type: interventional (clinical trial) Primary purpose: treatment |
|
| Participants | 170 participants Country: China Setting: inpatient At randomisation number allocated: 170, paroxetine (n = 85); usual care (n = 85) % male: paroxetine (71.8); usual care (unclear) Age: mean age paroxetine = 65.6 ± 7.56; placebo = unclear Subtype of stroke: not stated. Severity of stroke: NIHSS, Median (IQR): paroxetine 8 (6 – 10); usual care (unclear) Time since stroke onset: within 1 week Inclusion criteria:
Exclusion criteria
Withdrawal criteria: not stated |
|
| Interventions | Experimental: orally administrated paroxetine at dosages of 10 mg/day during week 1 and 20 mg/day thereafter, for a total treatment duration of 3 months Comparator: usual care |
|
| Outcomes | Outcomes were collected at 15, 90 and 180 days
|
|
| Funding source | No grant funding from any grant funding agency, commercial or not‐for‐profit organisations. | |
| Notes | There is no study protocol/trial register reference. Baseline sociodemographic and clinical characteristics are provided only for those who completed the study. The authors state that one of the inclusion criteria is Montreal Cognitive Assessment (MOCA) score of < 26 points. In the Results section they state that there were "72 cases of cognitive impairment" (i.e. a MoCA score of < 26 points) in the comparator group and 82 in the experimental group at days 15, 90 and 180. This suggests that either that the inclusion criteria were not strictly adhered to or if 100% of participants had a MoCA score of < 26 points at baseline then 10/82 participants in the comparator group and 3/85 in the experimental group have improved on the MoCA between days 0 and 15 Dates study conducted: participants recruited between January 2012 and June 2014 Declarations of Interest: none reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Random number table" |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to judge yes or no |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to judge yes or no |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "All scale evaluators were trained and tested by the main investigator and were blind to the group assignment." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | All data available for all participants in the experimental group (n = 85/85) and data available for (n = 82/85) participants in the comparison group for the Fugl–Meyer Motor Scale and the HAMD score For the MOCA (see 'Other bias' below) < 5% overall loss to follow‐up |
| Selective reporting (reporting bias) | Unclear risk | There is no study protocol/trial register reference, so insufficient information to judge yes or no. |
| Other bias | High risk | The authors state that one of the inclusion criteria is Montreal Cognitive Assessment (MoCA) score of < 26 points. In the Results section they state that there were "72 cases of cognitive impairment" (i.e., a MoCA score of < 26 points) in the comparator group and 82 in the experimental group at days 15, 90 and 180. This suggests that either that the inclusion criteria were not strictly adhered to or, if 100% of participants had a MoCA score of < 26 points at baseline then 10/82 participants in the comparator group and 3/85 in the experimental group have improved on the MoCA between days 0 and 15. The results 'Comparison of MoCA scores' and table 3 suggests otherwise |
Pariente 2001.
| Methods | Prospective double‐blind cross‐over placebo‐controlled study of 8 people with pure motor hemiparesis | |
| Participants | Lacunar ischaemic stroke, assessed by brain CT Quote: "Early phase of recovery" |
|
| Interventions | Single dose of fluoxetine | |
| Outcomes | fMRI (raw data provided) Finger tapping (presented as a graph, no raw data) NIHSS, motricity index, BI, trunk control test, Ashworth scale, somatosensory scale (no data) |
|
| Funding source | Source of funding not stated | |
| Notes | We could not use these data in our meta‐analyses. The authors reported that fluoxetine led to hyperactivation in the ipsi‐lesional (i.e. on the same side as the stroke lesion) primary motor cortex during a motor task; moreover, fluoxetine significantly improved motor skills of the affected side Dates of recruitment not given. Conflicts not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomisation code kept at the centre and broken at the end of the study |
| Allocation concealment (selection bias) | Low risk | Randomisation code kept at the centre and broken at the end of the study |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Double‐blind, placebo given |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Double‐blind |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Data on fMRI appears complete |
| Selective reporting (reporting bias) | Unclear risk | Data on clinical outcomes were not reported |
| Other bias | Unclear risk | Balanced baseline |
Rasmussen 2003.
| Methods | Parallel design Analysis: ITT (last observation carried forward) and per‐protocol: details of those excluded from analyses (35 treatment, 35 control) unclear |
|
| Participants | Location: Denmark Setting: unclear Treatment: 70 people, mean ± SD age 72 ± 9, 50% men Control: 67 people, mean ± SD age 68 ± 11, 51% men Stroke criteria: ischaemic and PICH; diagnosis by clinical signs and symptoms; stroke 0 to 4 weeks prior to randomisation Other entry criteria: not stated Comparability of treatment groups: participants in treatment group older on average |
|
| Interventions | Treatment: sertraline 50 mg daily; at any time after 2 weeks dose could be increased in 50 mg increments up to 150 mg daily; average dose 62.9 mg daily Control: matched placebo Duration of treatment: 12 months Duration of follow‐up (end of treatment to end of study): 0 |
|
| Outcomes | Depression: change in scores from baseline to end of treatment on HDRS Proportion scoring > 2 on the CGI or > 16 on the GDS at end of treatment Additional: leaving the study early. Did not report death Unable to use: HDRS, GDS, aphasia severity rating scale, European Stroke Scale, MMSE, Cambridge Cognitive Examination, SF‐36, BI (data not presented) AEs (detailed data not presented) evaluated by using the Udvalg for Kliniske Undersogelser Side Effect Rating Scale Did not report death |
|
| Funding source | Funding from Pfizer A/S, Gert Jorgensen legat and the Brain Cause. It is unclear whether the drug companies had input into the design and analysis of the study | |
| Notes | Recruitment January 1996 to May 1998. Conflicts not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not stated |
| Allocation concealment (selection bias) | Unclear risk | Method not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Matched placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Used ITT analysis and last observation carried forward |
| Selective reporting (reporting bias) | Low risk | Trial details published on www.strokecentre.org/trials |
| Other bias | Unclear risk | Those given sertraline were slightly older (by 4 years) but this is unlikely to introduce bias There was no significant difference between groups |
Razazian 2014.
| Methods | Study type: interventional (clinical trial) Primary purpose: treatment |
|
| Participants | 172 participants Country: Iran (Islamic Republic of) Setting: inpatient At randomisation number allocated: fluoxetine n = 86; placebo n = 86 % male: unclear Age: fluoxetine group = unclear; placebo = unclear Subtype of stroke: not available Severity of stroke: not available Time since stroke onset: not available Inclusion criteria
Exclusion criteria
Withdrawal criteria: not stated |
|
| Interventions | Experimental: fluoxetine, 20 mg once a day for 90 days Comparator: placebo fluoxetine for 90 days All participants received 30 sessions of routine physiotherapy during the rehabilitation period |
|
| Outcomes | Primary outcomes collected at day 45 and day 90
|
|
| Funding source | Kermanshah University of Medical Sciences. | |
| Notes | IRCT201312088323N7 Baseline demographic and clinical characteristics for each group not presented, but rather the baseline demographic and clinical characteristics for those completing the trial (i.e. a subset of all those randomised at baseline) are presented Dates study conducted: participants recruited between June 2013 and September 2014 Declarations of Interest: none reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "random permuted blocks". Comment: Insufficient information about the block randomisation to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of yes or no |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "placebo that was identical to the active drug in appearance and packaging" |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of yes or no |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 13% attrition at 90 days. 13% (n = 11/86) from the experimental group and 13% (n = 11/86) from the comparator group were excluded form the full set analysis at 90 days follow‐up. Reasons for attrition reported |
| Selective reporting (reporting bias) | Low risk | Protocol available and all the study's prespecified outcomes that are of interest to the review have been reported in a prespecified way |
| Other bias | High risk | The baseline data presented in table 1: patients demographic characteristics and risk factors and not true baseline characteristics (i.e. at randomisation). The data presented in table 1 are the characteristics of the full analysis set which is a subgroup of all participants randomised. We cannot tell if there is whether there was any baseline imbalance in important demographic or clinical characteristics |
Restifo 2001.
| Methods | Double‐blind study | |
| Participants | 10 participants with disabling hemiplegia owing to hemispheric ischaemic stroke in territory of left MCA | |
| Interventions | Treatment: fluoxetine 20 mg daily for 3 months plus usual care (including Bobath rehabilitation) Control: usual care including Bobath rehabilitation |
|
| Outcomes | Transmagnetic stimulation to establish motor reorganisation The authors reported that fluoxetine might modulate the primary motor cortex reorganisation |
|
| Funding source | Source of funding not stated | |
| Notes | Abstract only, full paper could not be found by our searches. Dates of study and conflicts of interest not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Random allocation", method not described |
| Allocation concealment (selection bias) | Unclear risk | Quote: "Random allocation", method not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | A placebo was used, not clear if it was matching |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Unclear from abstract |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Unclear from abstract |
| Selective reporting (reporting bias) | Unclear risk | Unclear from abstract |
| Other bias | Unclear risk | Unclear from abstract |
Robinson 2000a.
| Methods | Parallel design Comparison of fluoxetine, nortriptyline and placebo. We are using the fluoxetine and placebo data Analysis: per protocol, number excluded from analyses varied Data provided for depressed and non‐depressed separately. We are labelling the depressed group as Robinson 2000a (this trial), and the non‐depressed group as Robinson 2000b |
|
| Participants | Location: USA and Argentina Setting: mixed Treatment: 23 people with depression, mean ± SD age 65 ± 14 years; 17 men Control: 17 people with depression, mean ± SD age 73 ± 10 years; 9 men Stroke criteria: all subtypes, diagnosis by clinical signs and CT (100%), stroke within 6 months of recruitment, 18 to 85 years of age Stroke on average 16 weeks (fluoxetine) and 6 weeks (placebo) prior to randomisation Exclusion criteria: other significant medical illness, severe comprehension deficit, prior history of head injury, prior history of other brain disease (with the exception of stroke), participants on antidepressants (other than fluoxetine) were allowed to stop their antidepressant for a 2‐week washout period |
|
| Interventions | Treatment: fluoxetine 10 mg daily (3 weeks), 20 mg daily (3 weeks), 30 mg daily (3 weeks), 40 mg daily (3 weeks) Control: matched placebo Duration: treatment continued for 12 weeks Duration of follow‐up (end of treatment to end of study): 0 |
|
| Outcomes | Depression: change in scores from baseline to end of treatment on HDRS Additional: MMSE, JHFI Death AEs (method of reporting these was not stated) |
|
| Funding source | Funded by NIMH grants and grants from the Raul Carrea Institute of Neurological Research and Fundacion Perez Companc. Eli Lilly and company supplied the fluoxetine and placebo | |
| Notes | Note difference in time since stroke between treatment groups Dates of recruitment not stated. Conflicts not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random‐number table |
| Allocation concealment (selection bias) | Low risk | Concealment held by independent person |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Matched placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Per‐protocol and ITT analyses |
| Selective reporting (reporting bias) | Low risk | Protocol published www.strokecentre.org/trials |
| Other bias | Unclear risk | Imbalance in treatment groups for time since stroke and gender |
Robinson 2000b.
| Methods | Parallel design Comparison of fluoxetine, nortriptyline and placebo. We are using the fluoxetine and placebo data Analysis: per protocol, number excluded from analyses varies Data provided for depressed and non‐depressed separately. We are labelling the depressed group as Robinson 2000a, and the non‐depressed group as Robinson 2000b (this trial) |
|
| Participants | Location: USA and Argentina Setting: mixed Treatment: 17 non‐depressed people, mean ± SD age 66 ± 13 years, 15 men Control: 16 non‐depressed people, mean ± SD age 67 9 years, 12 men Stroke criteria: all subtypes, diagnosis by clinical signs and CT (100%), stroke within 6 months of recruitment, aged 18 to 85 years of age Stroke on average 8 weeks (treatment) and 5 weeks (control) prior to randomisation Comparability of treatment groups: unclear Exclusion criteria: other significant medical illness, severe comprehension deficit, prior history of head injury, prior history of other brain disease (with the exception of stroke), participants on antidepressants (other than fluoxetine) were allowed to stop their antidepressant for a 2‐week washout period |
|
| Interventions | Treatment: fluoxetine 10 mg daily (3 weeks), 20 mg daily (3 weeks), 30 mg daily (3 weeks), 40 mg daily (3 weeks) Control: matched placebo Duration: treatment continued for 12 weeks Duration of follow‐up (end of treatment to end of study): 0 |
|
| Outcomes | Depression: change in scores from baseline to end of treatment on HDRS Additional: MMSE, JHFI Death AEs (method of reporting these was not stated) |
|
| Funding source | Funded by NIMH grants and grants from the Raul Carrea Institute of Neurological Research and Fundacion Perez Companc. Eli Lilly and company supplied the fluoxetine and placebo | |
| Notes | Note difference in time since stroke between groups Dates of recruitment not stated. Conflicts of interest not stated |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Random‐number table |
| Allocation concealment (selection bias) | Low risk | Concealment held by independent person |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Matched placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT and per‐protocol |
| Selective reporting (reporting bias) | Low risk | Trial on www.strokecentre.org/trials |
| Other bias | Unclear risk | Note imbalance in time since stroke and in gender. |
Robinson 2008.
| Methods | Parallel group, 3‐arm (escitalopram, placebo, problem‐solving therapy group). We are using the escitalopram versus placebo arm in this review Analysis: ITT |
|
| Participants | Country: USA Setting: mixed: neurology department and newspaper advertisements Stroke criteria: ischaemic or haemorrhagic stroke not because of complications of intracranial aneurysm or intracranial vascular malformation; within 3 months of index stroke Mood: excluded if DSM IV for major or minor depression or HAMD > 17 Treatment (escitalopram): 59 people, mean ± SD age 61.2 ± 13.7, 38 men Control (matched placebo): 58 people, mean ± SD age 63.9 ± 11.1, 37 men Exclusion: acute coronary syndrome, neurodegenerative disorders, DSM IV criteria for alcohol or substance abuse |
|
| Interventions | Treatment: escitalopram 5 to 10 mg (depending on age ‐ lower dose given to > 65 years old) Control: matched placebo Duration of treatment: 12 months Duration of follow‐up (treatment end to study end): 0 |
|
| Outcomes | Diagnosis of depression HAMD (dichotomised) FIM (though no raw data provided in the paper for meta‐analysis) Social functioning examination Repeatable Battery for Neuropyschological Status The Iowa subset provided detailed information about cognition Participants, family members and primary care physicians were asked about AEs at 3 monthly intervals or sooner if an individual reported an AE using a standardised checklist |
|
| Funding source | The initial report states that "This work was supported solely by National Institute of Mental Health Grant RO1MH‐65134. All the study medications were purchased using NIMH grant funds." In a subsequent letter to the Journal, the authors disclosed honoraria and expenses from pharmaceutical companies, and that 1 of the authors owned Pfizer stock. However, the authors stated that the design and analysis of any of the expenses of the study were supported by monies, materials or any intellectual input from Forest Laboratories | |
| Notes | The escitalopram group had significantly more diabetes than the placebo group Financial disclosures: see above Recruitment: 9 July 2003 to 1 October 2007 |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Randomised blocks of 3, 6 and 9 |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Identical placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Outcome assessors were blind |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | ITT analyses, all participants used in analysis Dropouts: 5 in placebo and 7 drop‐outs in escitalopram) |
| Selective reporting (reporting bias) | Low risk | All specified outcome data reported. Trial published on www.strokecentre.org/trials |
| Other bias | Unclear risk | There was more diabetes in the escitalopram group than placebo group |
Savadi Oskouie 2012.
| Methods | Study type: interventional (clinical trial) Primary purpose: treatment |
|
| Participants | 144 participants Country: Islamic Republic of Iran Setting: inpatient At randomisation number allocated: N = 144; citalopram (n = 72); placebo (n = 72) % male at baseline: citalopram n = unclear; placebo n = unclear Age at baseline: citalopram (n = unclear); placebo (n = unclear) Subtype of stroke at baseline: unclear Severity of stroke at baseline: unclear Time since stroke onset: within 7 days Inclusion criteria:
Exclusion criteria:
Withdrawal criteria: not stated |
|
| Interventions | Experimental: oral citalopram 20 mg once daily Comparator: placebo |
|
| Outcomes | Primary outcome
Secondary outcome
|
|
| Funding source | Neurosciences Research Center (NSRC) of Tabriz University of Medical Sciences | |
| Notes | IRCT201203192150N2 Baseline demographic and clinical characteristics for each group not presented, rather the baseline demographic and clinical characteristics for those completing the trial (i.e. a subset of all those randomised at baseline) are presented. Dates study conducted: May 2012 to January 2014 Declarations of Interest: none reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "A total of 144 patients were randomized through an allocation sequence based on 2 blocks with size of 72, generated with a computer random number generator." |
| Allocation concealment (selection bias) | Low risk | Quote: "Allocation was concealed using the sequentially numbered black envelopes." |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Not explicitly stated that key study personnel and care providers were blinded, although implied by Quote: "The blinding code remained confidential until the end of the study." Quote: "placebo of the same shape and full packaging during the first day after hospital admission." |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not explicitly stated that outcome assessors were blinded, although perhaps implied by the fact Quote: "The blinding code remained confidential until the end of the study." |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Primary outcome data were available for 58 (81%) of the citalopram group and 50 (69%) of the placebo group. Reasons for attrition are reported but there are differences between groups: number of participants in the placebo group (n = 11) dead compared to the citalopram group (n = 4). 3 times the number of participants in the placebo group were depressed (n = 6) compared to the citalopram group (n = 2). Did not want to continue (placebo group (n = 5), citalopram group (n = 8). Intention‐to‐treat analyses were carried out (suppl table) assuming that 1. those lost to follow‐up had a poor outcome (i.e. did not improve their NIHSS scores from baseline) and 2. those participants in the placebo group who did not want to continue had a good outcome. Overall loss of > 5% |
| Selective reporting (reporting bias) | Low risk | The study protocol is available and all the study's prespecified (primary outcomes and secondary outcomes) that are of interest in the review have been reported in the prespecified way |
| Other bias | High risk | The baseline data presented in table 1: comparison of demographic and baseline variables and not true baseline characteristics (i.e. at randomisation). The data presented in table 1 are the characteristics of the full analysis set at 3 months which is a subgroup of all participants randomised. We cannot tell if there is whether there were any baseline imbalance in important demographic or clinical characteristics. However, given that approximately 3 times the number of participants in the placebo group (n = 11) died compared to the citalopram (n = 4) and 3 times the number of participants in the placebo group were depressed (n = 6) compared to the citalopram group (n = 2), this suggests that there may have been important group differences in clinical characteristics at baseline |
Shah 2016.
| Methods | Study type: interventional (clinical trial) Primary purpose: supportive care |
|
| Participants | 89 participants Country: India Setting: inpatient At randomisation number allocated: N = 89: fluoxetine (n = 45); placebo (n = 44) % male: unclear Age: unclear Subtype of stroke: unclear Severity of stroke: unclear Time since stroke onset: within 5 to 10 days Inclusion criteria:
Exclusion criteria:
Withdrawal criteria: not stated |
|
| Interventions | Experimental: fluoxetine 20 mg orally once daily for 90 days Comparator: matching placebo orally once daily for 90 days |
|
| Outcomes | Primary outcome
|
|
| Funding source | Not stated | |
| Notes | Baseline demographic and clinical characteristics for each group not presented, rather the baseline demographic and clinical characteristics for those completing the trial (i.e. a subset of all those randomised at baseline) are presented Dates study conducted: January 2014 to January 2015 Declarations of Interest: none reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information about the sequence generation process to permit judgement of yes or no |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information about the method of allocation concealment to permit judgement of yes or no |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | "Patients, attendants, study staff and investigators were masked to treatment allocation." However, "matching was done on a 1:1 basis for age, sex, severity of stroke " which suggests that some key study personnel were not blinded and this non‐blinding is likely to introduce bias |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of yes or no |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 3/45 (7%) participants in the fluoxetine and 2/44 (5%) in the placebo group were lost to follow‐up. Reasons for attrition/exclusion not reported. 6% lost to follow‐up |
| Selective reporting (reporting bias) | Unclear risk | No study protocol available. Insufficient information to permit judgement of yes or no |
| Other bias | High risk | The use of matching suggests a matched case control design rather than an RCT design. We cannot tell whether there was any baseline imbalance in important demographic or clinical characteristics |
Song 2006.
| Methods | Aim: to evaluate changes in depression and cognitive impairment in people with post‐stroke depression treated with fluoxetine Parallel trial |
|
| Participants | Country: China Setting: inpatient Stroke diagnosed by clinical criteria and "proved on CT" (though not clear if lesion had to be visible) Depression: diagnosed in accordance with the CCMD‐II‐R Treatment: 41 people, mean age 51 ± 7 years, 25 men), time since stroke: 3.5 days Control: 41 people, mean age 50 ± 8 years, 24 men), time since stroke: 3.7 days Excluded: previous mental disorders, previous "neurological disorder", if other psychiatric drugs had been taken, these had to be stopped for 1 week before fluoxetine was administered |
|
| Interventions | Treatment: fluoxetine 20 mg daily Control: placebo (although not stated whether this was identical to fluoxetine) Duration of treatment: 6 weeks Duration of follow‐up (treatment end to study end): 0 Side effects not reported |
|
| Outcomes | SDS p300 (an event‐related potential) Although the stated aim was to assess cognitive impairment, it is not clear how this was measured |
|
| Funding source | Source of funding not stated | |
| Notes | Recruitment December 1999 to June 2003. Conflicts of interest not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Placebo ‐ but not clear whether identical |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | Unclear |
| Other bias | Unclear risk | Balanced baseline |
Wang 2003.
| Methods | Parallel design 3‐arm trial: routine care, fluoxetine plus routine care, amitriptyline plus routine care. We are using the routine care and fluoxetine plus routine care in this analysis Aim: to observe effects of antidepressant therapy on post‐stroke and neurological rehabilitation in the elderly |
|
| Participants | Country: China Setting: inpatient Stroke criteria: ischaemic stroke, clinical diagnosis plus confirmation by imaging (although not clear whether a stroke lesion had to be present) Depression diagnosed according to CCMD‐II‐R diagnostic criteria, HAMD ≥ 18 Treatment: 64 people, mean age 75.6 ± 19.7 years, 39 men Control: 56 people, mean age 74.9 ± 20.8 years, 29 men Excluded: psychiatric disorder history, severe cardiac, pulmonary, hepatic and renal diseases |
|
| Interventions | Treatment: fluoxetine 20 to 80 mg daily (start at 20 mg/day, increase dosage at 3 weeks if poor therapeutic effect and no AE), plus usual stroke care Control: usual stroke care Duration of treatment: 12 to 24 weeks Duration of follow‐up (treatment end to study end): 6 to 9 months |
|
| Outcomes | HAMD Neurological function impairment score BI AEs not recorded |
|
| Funding source | Source of funding not stated | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 13 dropped out of fluoxetine group, and 9 dropped out of control group |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | Baseline appeared balanced but no statistical comparison between groups |
Wen 2006.
| Methods | Parallel trial Aim: to explore effects of prophylactic antidepression therapy on nerve functional rehabilitation after stroke Analysis: according to treatment group |
|
| Participants | Country: China Setting: inpatient Stroke criteria: acute stroke of all pathological subtypes, clinical diagnosis plus confirmation by imaging (although not clear whether a stroke lesion had to be present) Treatment: 42 people, mean age 56.8 years, men 19 Control: 42 people, mean age 57.2 years, men 16 Excluded those with primary psychiatric impairment and premorbid mood disorders, pre‐existing neurological disease causing confusion, severe systematic diseases and pulmonary, hepatic and renal failure |
|
| Interventions | Treatment: fluoxetine 20 mg daily plus routine stroke care Control: routine stroke care Duration of treatment: 8 weeks Duration of follow‐up (end of treatment to end of study): 0 |
|
| Outcomes | HAMD MESSS AEs (method of obtaining data not stated) Death |
|
| Funding source | Source of funding not stated | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Analysed according to treatment group, no dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | Balanced baseline |
Whyte 2005.
| Methods | Study type: interventional (clinical trial) Primary purpose: prevention |
|
| Participants | Number of participants: unclear Country: USA Setting: inpatient At randomisation number allocated: unclear % male: unclear Age: unclear Subtype of stroke: unclear Severity of stroke: unclear Time since stroke onset: unclear Inclusion criteria:
Exclusion criteria
|
|
| Interventions | Experimental: sertraline 12.5 mg/d for 3 days, increased to 25 mg/d for 4 days, then 50 mg/d for 7 days, then increased to 75 mg/d. Target dose = 75 mg per day for the remainder of participation in the study Comparator: matched placebo |
|
| Outcomes | Primary outcome collected at 12 months:
Secondary outcomes collected at 12 months:
|
|
| Funding source | None stated | |
| Notes | Terminated (recruitment goals could not be met). Last update 27 June 27 2014 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Insufficient information to permit judgement |
| Selective reporting (reporting bias) | Unclear risk | Insufficient information to permit judgement |
| Other bias | Unclear risk | Insufficient information to permit judgement |
Wiart 2000.
| Methods | Purpose: to treat early depression Parallel design Analysis: ITT (last observation carried forward), withdrawn owing to AE (1 treatment), protocol violation (1 treatment) |
|
| Participants | Location: France Setting: unclear Treatment: 16 people, mean ± SD age 66 ± 7 years, 65% men Control: 15 people, mean ± SD age 66 ± 12 years, 40% men Stroke criteria: ischaemic stroke and PICH, diagnosis by clinical signs and CT (100%); stroke on average 47 ± 22 days (treatment group) and 48 ± 20 days (control group) Depression criteria: psychiatric interview (ICD‐10 criteria) and MADRS score > 19 Other entry criteria: all antidepressant or neuroleptic drugs stopped 10 days prior to enrolment Comparability of treatment groups: balanced Exclusion criteria: severe psychiatric problems which required hospitalisation, severe aphasia, previous stroke, severe cognitive impairment, chronic alcoholism, chronic associated handicapping pathology, contraindication to fluoxetine |
|
| Interventions | Treatment: fluoxetine 20 mg daily Control: matched placebo Duration of treatment: 45 days Duration of follow‐up (treatment end to study end): 0 |
|
| Outcomes | Depression: change in scores from baseline to end of treatment of MADRS, 50% reduction in MADRS score Additional: FIMs MMSE Motricity Index Leaving the study early Death AEs ("evaluated qualitatively and quantitatively". Complete blood count, liver test and renal function test were carried out at each assessment visit) |
|
| Funding source | Lilly France Laboratory provided methodological and financial support | |
| Notes | Dates of recruitment not stated. Conflicts of interest not stated | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method of randomisation not stated |
| Allocation concealment (selection bias) | Unclear risk | Method not stated |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Quote: "Identical white capsules" given |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Method of blinding not stated |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Used last observation carried forward |
| Selective reporting (reporting bias) | Low risk | Trial published on www.strokecentre.org/trials. The primary outcome was reported |
| Other bias | Unclear risk | Baseline balanced |
Xie 2005.
| Methods | Aim: to study the effect of treatment with sertraline in elderly patients with post‐stroke depression Parallel study |
|
| Participants | Country: China Setting: unclear Recruited "clinically stable stroke patients with post‐stroke depression" No other inclusion and exclusion criteria given Mood: Zung SDS score ≥ 40 or GDS score 5 to 10 Treatment: 65 people, mean age 69.8 years, 29 men Control: 65 people, mean age 70.7 years, 27 men Time since stroke: mean 87.8 days, range 48 to 142 days |
|
| Interventions | Treatment: sertraline 50 mg/day plus usual stroke care Control: usual stroke care Duration of treatment: 12 weeks Duration of follow‐up: 0 |
|
| Outcomes | Zung SDS, GDS, ADL score AEs were not reported |
|
| Funding source | Local scientific academic fund funded the study | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | No clear description between treatment and control |
Xu 2001.
| Methods | Parallel Aim: to study the effect of fluoxetine on depression in early recovery stage of cerebral infarction |
|
| Participants | Country: China Setting: outpatient in rehabilitation clinic Stroke: first acute cerebral infarction, no description of the diagnostic criteria and the need for imaging confirmation, excluded large cerebral infarction or lacunar infarction (clinical condition too severe or too mild); onset to recruitment time mean 30 days Zung SDS ≥ 40 Treatment: 32 people Control: 31 people (no details of participant characteristics) Excluded if previous antidepressants |
|
| Interventions | Treatment: fluoxetine 20 mg daily plus usual stroke care Control: usual stroke care Duration of treatment: 8 weeks Duration of follow‐up (treatment end to study end): 0 |
|
| Outcomes | Zung SDS ADL (BI) Neural function deficient Death AEs not reported |
|
| Funding source | Source of funding not stated | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 10/62 dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | No clear description of stroke criteria and imaging |
Xu 2006.
| Methods | Parallel group Aim: to test whether early prophylactic antidepressant treatment by paroxetine has any beneficial influence on the rate of post‐stroke depression and rehabilitation |
|
| Participants | Country: China Setting: inpatient Stroke criteria: stroke onset time ≤ 3 days, age ≤ 75 years old, no previous psychiatric disorders, no obvious cognitive impairment or aphasia Depression diagnosis was not mentioned as an inclusion criteria, so we assumed that patients did not have to have depression to enter the trial Treatment: 32 people, mean age 65 ± 12 years, 17 men Control: 32 people, mean age 63 ± 11 years, 16 men Exclusion: no severe hepatic or renal impairment, DSM IV depression not stated as an inclusion, but none met criteria for depression initially |
|
| Interventions | Treatment: paroxetine 20 mg daily Control: placebo Duration of treatment: 12 weeks Duration of follow‐up (treatment end to study end): 0 |
|
| Outcomes | MESSS ADL Post‐stroke diagnosis incidence of depression according to DSM IV AEs not recorded |
|
| Funding source | Study funded by local scientific academic fund | |
| Notes | The number of participants in Table 1 (p187) were wrong (paroxetine/placebo: N = 32/32 should be N = 28/29) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Sequence numbers" |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Placebo used, but unclear if it was matched |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 7 participants dropped out |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Low risk | Baseline balance |
Yang 2002.
| Methods | Parallel group Aim: to study effects of antidepressant in treatment of people with post‐stroke depression |
|
| Participants | Country: China Setting: inpatients and outpatients Stroke criteria: recovery phase of stroke (2 to 6 months after ischaemic stroke, and 1.5 to 6 months after haemorrhagic stroke). We included this in the 3 to 6 month group. Clinical diagnosis of stroke (not stated whether confirmation by imaging was needed) Depression: HAMD > 7 Treatment: 64 people, mean age 64 ± 3 years, 40 men Control: 57 people, mean age 63 ± 5 years, 32 men |
|
| Interventions | Treatment: paroxetine 20 mg daily plus stroke treatment and rehabilitation Control: stroke treatment and rehabilitation Duration of treatment: 4 months Duration of follow‐up: 0 |
|
| Outcomes | Death They collected data on HAMD and CSS but did not report these data ADL score: did not state which one, so not used AEs not reported |
|
| Funding source | Source of funding not reported | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | High risk | 11/121 (9%) dropouts |
| Selective reporting (reporting bias) | High risk | No protocol. The paper stated that ADL data and depression data were collected, but these data were not reported |
| Other bias | Unclear risk | No baseline differences between groups, no other obvious source of bias |
Yang 2011.
| Methods | Aim: to treat early post‐stroke depression | |
| Participants | Country: China Setting: inpatient Stroke: all pathological types, clinical diagnosis plus confirmation of lesion on imaging, no previous psychiatric and psychological disorders, age < 75 years old, stroke onset time < 72 hours, NIHSS score: 4 to 19 Mood: HAMD ≥ 8 Treatment: 20 people, mean age 64 ± 8 years, 8 men Control: 22 people, mean age 64 ± 10 years, 13 men Note inconsistency between abstract (20 in treatment and 22 in control, but in tables of results, there are 22 in treatment and 20 in control). We have used the data from the abstract Excluded: functional psychiatric disorder, functional depression, psychoactive substance and addictive substance induced psychiatric disorders, infectious disease, severe cognitive impairment to affect communication, severe aphasia to affect communication, severe cardiac, pulmonary, hepatic and renal function impairment, previous organic brain disease such as brain tumour, or symptomatic stroke, encephalitis |
|
| Interventions | Treatment: paroxetine 20 mg daily plus usual stroke care Control: usual stroke care Duration of treatment: at least 3 months Duration of follow‐up: 0 |
|
| Outcomes | HAMD score, IL‐1β and IL‐6 level Death AEs not reported |
|
| Funding source | Source of funding: local scientific academic fund | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Case sequence" randomisation |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Low risk | No difference in baseline |
Ye 2004.
| Methods | Aim: to investigate whether antidepressive therapy is needed for people with post‐stroke depression or not, and the effect of different antidepressive drugs on the rehabilitation of psychological and neurological function after stroke 3 groups: paroxetine, imipramine and control. We are using the paroxetine versus control arm in this review |
|
| Participants | Country: China Setting: inpatient Stroke: all pathological subtypes, clinical diagnosis plus confirmation by imaging (did not state whether a visible lesion was needed to make the diagnosis), no positive psychiatric disorders or family history, clear consciousness, no comprehension problem Mood: inclusion criteria: HAMD score > 21, HAMA scale > 14 Treatment: 30 people, age 58.04 ± 8.28 years, 22 men Control: 30 people, age 59.21 ± 9.52 years, 17 men Exclusion criteria: severe cardiac, hepatic and renal diseases, multiple infarcts or haemorrhage |
|
| Interventions | Treatment: paroxetine 20 mg/day plus acute stroke routine care and rehabilitation Control: acute stroke routine care plus rehabilitation Duration of treatment: 12 weeks Duration of follow‐up (end of treatment to end of study): 0 |
|
| Outcomes | Chinese Neurological Impairment Scale, modified BI, HAMD, HAMA, Therapeutic Effect for Depression and Neurologic Function Death, GI upset Method of recording side effects not stated |
|
| Funding source | Source of funding not stated | |
| Notes | Inconsistent description about the number of recruitment and randomisation between abstract (N = 90) and result part (N = 93) of the text. The number for final analysis is consistent in the text | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Used "number table" ‐ but unclear if this was a random number table |
| Allocation concealment (selection bias) | Low risk | The study designer did not involve in assessment and treatment, the assessors did not know the allocation |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | The participants were blinded. Not clear if those delivering the treatment were blind, but no placebo used |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Assessors were blinded |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Only 1 dropped out in paroxetine group |
| Selective reporting (reporting bias) | Unclear risk | No protocol |
| Other bias | Unclear risk | Different numbers reported to have been recruited and randomised, baseline similar |
Zhao 2011.
| Methods | Study type: interventional (clinical trial) Primary purpose: treatment |
|
| Participants | Country: People's Republic of China Setting: inpatient At randomisation number allocated: N = 82: fluoxetine (n = 41); placebo (n = 41) % male: 58.5 Age: mean age 65 ± 12 Subtype of stroke: Ischaemic stroke: 61/82 (74%); haemorrhagic stroke: 21/82 (26%) Severity of stroke: MESSS: fluoxetine 23.2 ± 6.2 (n = 37); placebo 22.8 ± 5.8 (n = 34) Time since stroke onset: within 10 days Inclusion criteria:
Exclusion criteria: none Withdrawal criteria: not stated |
|
| Interventions | Experimental:fluoxetine 20 mg daily for 12 weeks Comparator: no fluoxetine |
|
| Outcomes | Outcomes collected at 2nd, 4th and 12 week of treatment and 12 weeks after the end of treatment
|
|
| Funding source | Not available | |
| Notes | Dates study conducted: 2008 to 2010 Declarations of Interest: none reported |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | The participants were randomised into 2 groups (with fluoxetine or without fluoxetine) according to the sequence number and a block randomisation table |
| Allocation concealment (selection bias) | Unclear risk | Insufficient information to permit judgement of yes or no |
| Blinding of participants and personnel (performance bias) All outcomes | Unclear risk | Insufficient information to permit judgement of yes or no |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Insufficient information to permit judgement of yes or no |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Attrition rate of fluoxetine group vs control group was 4/41 (9.8%) vs 7/41 (17.1%) > 5% loss to follow‐up |
| Selective reporting (reporting bias) | Unclear risk | No trial protocol available. Insufficient information to permit judgement of yes or no |
| Other bias | Low risk | The study appears to be free from other sources of bias |
Zhou 2008.
| Methods | Aim: to study effect of early paroxetine on post‐stroke depression and rehabilitation Parallel design Analysis: according to treatment groups |
|
| Participants | Country: China Setting: inpatient Stroke criteria: all stroke, clinical diagnosis plus confirmation by imaging (though not clear if a relevant stroke lesion had to be visible), stroke onset time ≤ 7 days, no obvious cognitive impairment, no obvious aphasia HAMD score < 8 Treatment: 36 people, mean age 63 ± 9.3 years, 16 men Control: 40 people, mean age 61 ± 9.6 years, 19 men Excluded: previous psychiatric disorders, severe hepatic and renal impairment, taking agents with obvious interaction with fluoxetine in recent 1 month |
|
| Interventions | Treatment: fluoxetine 20 mg daily plus acute stroke routine medication Control: acute stroke routine medication Duration of treatment: 8 weeks Duration of follow‐up: 0 |
|
| Outcomes | No raw data provided for any of the following outcomes: diagnosis of depression (CCMD‐3, HAMD, ADL, MESSS) Reported no deaths in either group. Unclear how data on side effects were collected |
|
| Funding source | Source of funding not stated | |
| Notes | − | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Method not described |
| Allocation concealment (selection bias) | Unclear risk | Not described |
| Blinding of participants and personnel (performance bias) All outcomes | High risk | No placebo |
| Blinding of outcome assessment (detection bias) All outcomes | Unclear risk | Not described |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | No dropouts, analysed according to allocated treatment group |
| Selective reporting (reporting bias) | High risk | No protocol, no raw data provided for several of the outcomes |
| Other bias | Low risk | Baseline similar |
ADL: activities of daily livingAE: adverse events; AE: adverse event; BDI: Beck Depression Inventory; BI: Barthel Index; CCMD‐II‐R: Chinese Classification of Mental Disorders, second edition, revised; CCMD‐3: Chinese Classification of Mental Disorders‐3; CGI: Clinical Global Impressions Scale; CSS: Chinese Stroke Scale; CT: computerised tomography; CTIMP: Clinical Trial of an Investigational Medical Product; EEG: electroencephalogram; FAI: Frenchay Activities Index; FAST: Frenchay Aphasia Screening Test; FIM: Functional Independence Measure; FMMS: Fugl‐Meyer Motor Scale; fMRI: functional magnetic resonance imaging; GDS: Geriatric Depression Scale; GI: gastrointestinal; HADS: Hospital Anxiety and Depression Scale; HAMA: Hamilton Anxiety scales; HAMD/HDRS: Hamilton Depression Rating Scale; HSS: Hemispheric Stroke Scale; ICD: International Classification of Diseases; ICH: intracerebral haemorrhage; IL: interleukin; ITT: intention‐to‐treat; IQR: interquartile range; JHFI: Johns Hopkins Functioning Inventory; LOCF: last‐observation‐carried‐forward; MADRS: Montgomery‐Åsberg Depression Rating Scale; MAOI: mono‐amino‐oxidase inhibitor; MCA: middle cerebral artery; MEP: motor evoked potentials; MESSS: Modified Edinburgh‐Scandinavian Stroke Scale; MMSE: Mini‐Mental State Examination; MRI: magnetic resonance imaging; mRS: modified Rankin score; NIHSS: National Institutes of Health Stroke Scale; PASE: Physical Activity Scale for the Elderly; PICH: primary intracerebral haemorrhage; RS: Rankin score; SAH: subarachnoid haemorrhage; SAS: Zung Self‐rating Anxiety Scale; SD: standard deviation; SDS: Zung Self‐rating Depression Scale; SF‐36: Short Form‐36; SSS: Scandinavian Stroke Scale; TESS: Treatment Emergent Symptom Scale; TIA: transient ischaemic attack; WHO: World Health Organization
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Andersen 1993 | Cross‐over design: double‐blind placebo‐controlled cross‐over protocol as follows: 7 days initial baseline registration, 21 days citalopram or placebo (randomised), 7 days wash‐out, 7 days baseline registration, and cross‐over to second 21‐day treatment period |
| Andersen 2012 | The trial never started |
| Anderson 2002 | The trial never started |
| Anonymous 2012 | Unable to find publication after extensive searching |
| Anonymous 2012b | Unable to find publication after extensive searching |
| Berends 2009 | Mean time from stroke onset to fluoxetine was 39.1 months |
| Choi Kwon 2008 | Participants more than 1 year post‐stroke |
| Finkenzeller 2009 | SSRI plus active intervention (psychotherapy) versus active treatment (psychotherapy) alone. This trial had been included in the original 2012 review but due to the potential interaction between the SSRI and psychotherapy we decided to exclude it in this update |
| Gourab 2015 | Time of stroke onset > 12 months |
| Graffagnino 2002 | Previously listed in 'Studies awaiting classification' (Mead 2012). Unable to access any full publication and we received no response from the author. Given the insufficient information to assess eligibility and, owing to the length of time since the study abstract (2002) was published, we have now excluded this study. CRSREF: 3340767 |
| Ji 2000 | SSRI plus active intervention versus active treatment alone |
| Li 2002 | There is no random component in the sequence generation process |
| Liang 2003 | There is no random component in the sequence generation process. This had been included in the 2012 review but on review of the methodology the review authors decided to exclude this for the update |
| Liu 2004 | SSRI plus active intervention versus active treatment alone |
| Robinson 2011 | Ineligible outcomes: prevention of generalised anxiety disorder |
| Sitzer 2002 | Previously listed in 'Studies awaiting classification' (Mead 2012). Unable to access any full publication and we received no response from the author. Given the insufficient information to assess eligibility and, owing to the length of time since the study abstract (2002) was published, we have now excluded this study |
| Sun 2015 | Mean time since onset 19.2 ± 3.5 months. No placebo or usual‐care control group (prozac, acupuncture, and prozac plus acupuncture) |
| University of Alabama 2013 | Study withdrawn (not funded) |
| Xu 2007 | This had been included in the 2012 review but it compares fluoxetine plus wulung capsule vs wulung capsule alone. Wulung capsule is an active comparator so we have therefore excluded this trial for this update |
| Zhou 2003 | There is no random component in the sequence generation process. This trial had been included in the 2012 version of the review but for this update we excluded it |
RCT: randomised controlled trial SNR: serotonin–norepinephrine reuptake inhibitor SSRI: selective serotonin reuptake inhibitor
Characteristics of studies awaiting assessment [ordered by study ID]
Carda 2009.
| Methods | Study type: interventional (clinical trial) Estimated enrolment: 200 participants Allocation: randomised Intervention model: parallel assignment Masking: quadruple (participant, care provider, investigator, outcomes assessor) Primary purpose: treatment |
| Participants | Country: Italy Setting: inpatient Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: escitalopram and rehabilitation. Escitalopram given 5 mg once a day for the first week, 10 mg once a day from the second to fourth week, and 20 mg daily until the 6th month Comparator: placebo and rehabilitation |
| Outcomes | Primary outcome collected at 2 and 6 months
Secondary outcomes collected at 2 and 6 months:
|
| Notes | clinicaltrials.gov/ct2/show/NCT00967408 Contacted author Prof Cisari; response received; data being analysed |
Guo 2015.
| Methods | Study type: interventional (clinical trial) Actual enrolment: 300 Allocation: randomised Intervention model: parallel assignment Masking: single (outcomes assessor) Primary purpose: prevention |
| Participants | Country: China Setting: inpatient At randomisation numbers allocated: N = 300 Experimental group 1: fluoxetine immediately after enrolment n = 100 Comparator group 1: fluoxetine 7 days after enrolment n = 100 Comparator group 2: no fluoxetine n = 100 % male: unclear Age: Experimental, unclear; Comparator 1, unclear; Comparator 2, unclear Subtype of stroke: unclear Severity of stroke NIHSS score at baseline: unclear Experimental: unclear Comparator 1: unclear Comparator 2: unclear Time from stroke onset: within 1 week after onset of cerebral infarction Inclusion criteria:
Exclusion criteria:
Withdrawal criteria:
|
| Interventions | Experimental: 20 mg of fluoxetine per day for 90 days given immediately after enrolment and conventional therapy of cerebral infarction Comparator 1: 20 mg of fluoxetine a day for 90 days given 7 days after enrolment and conventional therapy of cerebral infarction Comparator 2: no fluoxetine and conventional therapy of cerebral infarction |
| Outcomes | Primary outcome at days 15, 90 and 180
Secondary outcome at days 90 and 180
|
| Notes | ChiCTR‐TRC‐15007658 xuanyi_guo@163.com Baseline demographic and clinical characteristics for each group not presented, but rather the baseline demographic and clinical characteristics for those completing the trial (i.e. a subset of all those randomised at baseline) are presented |
He 2012.
| Methods | Study type: interventional (clinical trial) Actual enrolment: 404 Allocation: randomised Intervention model: parallel assignment Masking: single (outcomes assessor) Primary purpose: prevention |
| Participants | Country: China Setting: inpatient At randomisation numbers allocated: N = 404 Experimental group: fluoxetine n = 202 Comparator group n = 202 % male: 70.5% Age: Experimental: 61.14 ± 10.48; Comparator 62.72 ± 11.86 Subtype of stroke: unclear Severity of stroke NIHSS score at baseline: Experimental: Median 6 (IQR 4, 8) Comparator: Median 5 (IQR 3, 8) Time since stroke onset: mean days, fluoxetine 4.28 ± 1.89; placebo 4.08 ± 2.15 Inclusion criteria:
Exclusion criteria:
Withdrawal criteria:
|
| Interventions | Experimental: 20 mg of fluoxetine a day for 90 days and conventional therapy Comparator: conventional therapy |
| Outcomes |
|
| Notes | ChiCTR‐TRC‐12002078 xuanyi_guo@163.com |
Jurcau 2016.
| Methods | Study type: interventional (clinical trial) Actual enrolment: 89 Allocation: randomised Intervention model: parallel assignment Masking: unclear Primary purpose: treatment |
| Participants | Country: Romania Setting: inpatient At randomisation numbers allocated: N = 89 Experimental group: escitalopram = 43 Comparator group: ?secondary preventive treatment = 46 % male: unclear Age: unclear Subtype of stroke: unclear Severity of stroke: unclear Time since stroke onset: unclear Inclusion criteria: unclear Exclusion criteria: unclear withdrawal criteria: unclear |
| Interventions | Experimental: escitalopram 10 mg/day for 12 weeks Comparator: ?secondary preventive treatment = 46 |
| Outcomes | Outcomes collected at 3, 6 and 12 months post‐stroke
|
| Notes | Does not appear to be a clinical trial register number |
BDI: Beck Depression Inventory BI: Bathel Index FIM: Functional Independence Measure HAM‐D17: Hamilton Depression Scale MMSE: Mini‐Mental State Examination NIHSS: National Institutes of Health Stroke Scale od: once daily
Characteristics of ongoing studies [ordered by study ID]
Anonymous 2005.
| Trial name or title | Influence of escitalopram on the incidence of depression and dementia following acute middle cerebral artery territory infarction. A randomised, placebo‐controlled, double‐blind study |
| Methods | Study type: interventional (clinical trial) Estimated enrolment: 60 participants Allocation: randomised Intervention model: parallel assignment Masking: double (detail unclear) Primary purpose: prevention |
| Participants | Country: Germany Setting: inpatient Inclusion criteria
Exclusion criteria:
Aiming to recruit 60 participants |
| Interventions | Experimental: escitalopram Comparator: placebo |
| Outcomes | Depression (MADRS) after 180 days Incidence of dementia after 180 days (Clinical Dementia Rating scale) Severity of dementia Zarit Burden Interview Incidence of depression (Depression Visual Analogue Scale) Severity of post‐stroke depression Quality of life (SF‐36) Bayer Activities of Daily Living score NPI |
| Starting date | MHRA approval 7 April 2006; start date not known |
| Contact information | Not available. National Competent authority is Germany‐BFarm Sponsor Name: Central Institute for Mental Health, Mannheim, Division of Gerontopsychiatry |
| Notes | Details available on EudraCT website www.clinicaltrialsregister.eu/ctr‐search/trial/2005‐005266‐37/DE |
Chollet 2016.
| Trial name or title | Resting state MRI connectivity in acute ischemic stroke: Serotonin Selective Reuptake Inhibitor (SSRI) in enhancing motor recovery: a placebo controlled study |
| Methods | Study type: interventional (clinical trial) Estimated enrolment: 60 participants Allocation: randomised Intervention model: parallel assignment Masking: double (participant, investigator) Primary purpose: treatment |
| Participants | Country: France Setting: inpatient Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: 20 mg of fluoxetine capsule a day from day 0 to day 90 and have fMRI Comparator: cellulose placebo a day from day 0 to day 90 and have fMRI |
| Outcomes | Primary outcome at 90 days
Secondary outcome at 90 days
|
| Starting date | January 2016 |
| Contact information | Virginie Sattler, Dr sattler.v@chu‐toulouse.fr François Chollet, MD PhD chollet.f@chu‐toulouse.fr |
| Notes | NCT02767999 |
Cocho 2015.
| Trial name or title | Effect of serotonin and levodopa in ischemic stroke (SELEIS) |
| Methods | Study type: interventional (clinical trial) Estimated enrolment: 240 participants Allocation: randomised Intervention model: parallel assignment Masking: single (outcomes assessor) Primary purpose: treatment |
| Participants | Country: Spain Setting: inpatient Inclusion criteria:
Exclusion criteria:
Aiming to recruit 240 participants. |
| Interventions | Placebo comparator: placebo Active comparator: citalopram 20 mg Active comparator: sinemet plus 100 mg Sinemet plus + citalopram group |
| Outcomes | Rankin Scale at 12 months |
| Starting date | 1 January 2015 |
| Contact information | Dolores Cocho dcocho@fhag.es |
| Notes | NCT02386475 |
Dike 2019.
| Trial name or title | Pharmacological enhancement of motor function recovery in patients with ischaemic stroke: a trial of fluoxetine |
| Methods | Study type: interventional (clinical trial) Estimated enrolment : 40 participants Allocation: randomised Intervention model: parallel assignment Masking: double (participant, outcomes assessor) Primary purpose: treatment |
| Participants | Country: Nigeria Setting: inpatient Inclusion criteria:
Exclusion criteria:
Aiming to recruit 60 participants |
| Interventions | Experimental: 20 mg fluoxetine for 30 days plus standard treatment Comparator: standard treatment |
| Outcomes | Primary outcome:
Secondary outcome
|
| Starting date | 1 January 2015 |
| Contact information | Dike Franklin, Abak Road, Uyo, 530001, Nigeria frankincense4m@yahoo.com |
| Notes | PACTR201412000967245 |
Farokhi 2017.
| Trial name or title | Evaluation of fluoxetine and standard treatment efficacy on change to side effect of stroke of ischaemic strokes in both hemispheres in anterior circulation |
| Methods | Study type: interventional (clinical trial) Estimated enrolment: 60 participants Allocation: randomised Intervention model: parallel assignment Masking: unclear Primary purpose: treatment |
| Participants | Islamic Republic of Iran Inclusion criteria:
Exclusion criteria:
Aiming to recruit 60 participants |
| Interventions | Experimental: 20 milligram fluoxetine and standard treatment (antiplatelets, anticoagulant and statin) Comparator: placebo and standard treatment (antiplatelets, anticoagulant and statin) |
| Outcomes | Primary outcome collected at 1 and 2 months
|
| Starting date | 11 October 2017 |
| Contact information | Fariba Farokhi, Arak University of Medical Sciences Iran (Islamic Republic of Iran) f.farokhi@arakmu.ac.ir |
| Notes | www.irct.ir/trial/17976 |
FOCUS‐Poland 2014.
| Trial name or title | Fluoxetine Or Control Under Supervision ‐ Poland |
| Methods | Study type: interventional (clinical trial) Estimated enrolment: 200 participants Allocation: randomised Intervention model: parallel assignment Masking: double (participant, care provider) Primary purpose: treatment |
| Participants | Country: Poland Setting: inpatient Inclusion criteria:
Exclusion criteria:
Aiming to recruit 200 participants |
| Interventions | Fluoxetine 20 mg daily (1 capsule) for 6 months (180 capsules) vs placebo |
| Outcomes | The primary outcomes
Secondary endpoints
|
| Starting date | December 2014 |
| Contact information | jbemenek@o2.pl czlonkow@ipin.edu.pl |
| Notes |
Fregni 2014.
| Trial name or title | Effects rTMS combined with fluoxetine on motor recovery in stroke patients |
| Methods | Study type: interventional (clinical trial) Estimated enrolment: 45 participants Allocation: randomised Intervention model: parallel assignment Masking: triple (participant, investigator, outcomes assessor) Primary purpose: treatment |
| Participants | Country: USA Setting: inpatient Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental 1: active rTMS/active fluoxetine 10 daily 20‐minute sessions over 15 days of active low‐frequency rTMS, followed by 8 weekly 20‐minute sessions of active low‐frequency rTMS + active fluoxetine (20 mg) taken orally consecutively for 90 days Comparator 1: sham rTMS/active fluoxetine 10 daily 20‐minute sessions over 15 days of sham low‐frequency rTMS, followed by 8 weekly sessions of sham low‐frequency 20‐minute rTMS sessions + active fluoxetine (20 mg) taken orally consecutively for 90 days Comparator 2: sham rTMS/placebo fluoxetine 10 daily 20‐minute sessions over 15 days of sham low‐frequency rTMS. This will be followed by 8 weekly sessions of sham low‐frequency 20‐minute rTMS sessions + placebo fluoxetine (sugar pills) taken orally consecutively for 90 days |
| Outcomes | Primary outcome over 90‐day period
Secondary outcome over 90‐day period
|
| Starting date | 5 August 2014 |
| Contact information | Felipe Fregni, Principal Investigator, Spaulding Rehabilitation Hospital, Charlestown, Massachusetts, USA |
| Notes | NCT02208466 |
Hankey 2011.
| Trial name or title | Assessment oF FluoxetINe In sTroke recoverY (AFFINITY) trial |
| Methods | Multicentre Study type: interventional (clinical trial) Estimated enrolment: 1600 participants Allocation: randomised Intervention model: parallel assignment Masking: quadruple (participant, care provider, investigator, outcomes assessor) Primary purpose: treatment |
| Participants | Country: Australia Setting: inpatient Inclusion criteria
Exclusion criteria:
Aiming to recruit 1600 participants |
| Interventions | Fluoxetine 20 mg once daily or matching placebo capsules for 6 months |
| Outcomes | Primary outcome
Secondary outcomes at 180 and 365 days after randomisation
|
| Starting date | July 2012 |
| Contact information | graeme.hankey@health.wa.gov.au |
| Notes | ACTRN12611000774921 |
Karimialavijeh 2017.
| Trial name or title | Comparison of the effects of citalopram versus fluoxetine on motor recovery after stroke: a double‐blind placebo‐controlled randomised clinical trial |
| Methods | Study type: interventional (clinical trial) Estimated enrolment: 90 participants Allocation: randomised Intervention model: parallel assignment Masking: unclear Primary purpose: treatment |
| Participants | Iran (Islamic Republic of Iran) Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: 20 mg of fluoxetine orally, daily for 90 days, as well as physiotherapy sessions 1 hour a day, 5 days a week, for 12 weeks Comparator 1: 20 mg of citalopram orally, daily for 90 days, as well as physiotherapy sessions 1 hour a day, 5 days a week, for 12 weeks Comparator 2: capsules containing microcrystalline cellulose, orally, daily for 90 days, as well as physiotherapy sessions 1 hour a day, 5 days a week, for 12 weeks |
| Outcomes | Primary outcome collected at 90 days
|
| Starting date | 3 December 2017 |
| Contact information | Ehsan Karimialavijeh Dr Shariati hospital, North Karegar Ave1411713135 Tehran Iran (Islamic Republic of) drkarimi86@gmail.com |
| Notes | IRCT20141116019971N3 |
Leibovitch 2018.
| Trial name or title | FLuoxetine Opens Window to improve motor recovery after stroke (FLOW) |
| Methods | Study type: interventional (clinical trial) Estimated enrolment: 176 participants Allocation: randomised Intervention model: parallel assignment Intervention model description
Masking: quadruple (participant, care provider, investigator, outcomes assessor) Primary purpose: treatment |
| Participants | Country: Canada Setting: inpatient Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: fluoxetine hydrochloride (Prozac): 10 mg Prozac per day for 3 to 5 weeks and then 20 mg for 12 weeks (the duration of the exercise intervention) Comparator: an over‐encapsulated placebo (identical 'sugar pill'): 10 mg 'sugar pill' a day for 3 to 5 weeks and then 20 mg for 12 weeks (the duration of the exercise intervention) |
| Outcomes | Primary outcomes at 12 weeks
Secondary outcomes at 12 weeks and 6 months
|
| Starting date | 1 November 2018 |
| Contact information | Farrell Leibovitch farrell@canadianstroke.ca |
| Notes | NCT03448159 |
Levitt 2019.
| Trial name or title | Depression in haemorrhagic stroke |
| Methods | Study type: interventional (clinical trial) Estimated enrolment: 224 participants Allocation: randomised Intervention model: parallel assignment Intervention model description: double‐blinded placebo‐controlled randomised trial Masking: triple (participant, care provider, investigator) Primary purpose: prevention |
| Participants | Country: USA Setting: inpatient Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: fluoxetine 20 mg/day for a period of 1 year Comparator: placebo 20 mg/day for a period of 1 year |
| Outcomes | Primary outcomes at 1 year
Secondary outcomes at 1 year:
|
| Starting date | 1 March 2019 |
| Contact information | Cory M Kelly 206‐685‐3043 kellycm@neurosurgery.washington.edu |
| Notes | NCT03826875 |
Lundström 2014.
| Trial name or title | Efficacy oF Fluoxetine – a randomisEd Controlled Trial in Stroke (EFFECTS) |
| Methods | Multicentre RCT Study type: interventional (clinical trial) Estimated enrolment: 1500 participants Allocation: randomised Intervention model: parallel assignment Masking: quadruple (participant, care provider, investigator, outcomes assessor) Primary purpose: treatment |
| Participants | Sweden Inclusion criteria
Exclusion criteria
Aiming to recruit 1500 participants |
| Interventions | Fluoxetine (20 mg once daily) for 6 months with oral capsules |
| Outcomes | Outcomes collected at 6 months and 12 months Primary outcome
Secondary outcomes
|
| Starting date | 20 October 2014 |
| Contact information | Associate Professor Erik Lundström, Department of Neuroscience, Neurology, Uppsala University, Akademiska sjukhuset, 751 85 Uppsala, Sweden. Email: erik.lundstrom@neuro.uu.se. |
| Notes | clinicaltrials.gov/ct2/show/NCT02683213 |
Pastore‐Wapp 2016.
| Trial name or title | Cortical Ischemic Stroke and Serotonin (CISS) |
| Methods | Study type: interventional (clinical trial) Estimated enrolment: 90 participants Allocation: randomised Intervention model: parallel assignment Masking: quadruple (participant, care provider, investigator, outcomes assessor) Primary purpose: supportive care |
| Participants | Country: Switzerland Setting: inpatient Inclusion criteria:
Exclusion criteria:
|
| Interventions | Experimental: escitalopram 5 mg/day at the baseline visit (day 14 (±7) post‐stroke) for 7 days followed by a weekly dosage increase of 5 mg/day till target dose of escitalopram 20 mg/day. Participants remain on escitalopram 20 mg/day until visit 3 (day 90 (±14) post‐stroke) followed by dosage reduction of escitalopram 10 mg/day for 1 week Comparator: placebo 5 mg/day at the baseline visit (day 14 (±7) post‐stroke) for 7 days followed by a weekly dosage of 5 mg/day until target dose of placebo 20 mg/day. Participants remain on placebo 20 mg/day until visit 3 (day 90 (±14) post‐stroke) followed by placebo 10 mg/day for 1 week |
| Outcomes | Primary outcome
Secondary outcomes:
Other outcomes:
|
| Starting date | August 2016 |
| Contact information | Manuela Pastore‐Wapp manuela.pastore‐wapp@insel.ch |
| Notes | NCT02865642 |
Pirzeh 2012.
| Trial name or title | A study of sertraline effect on quality of life in stroke inpatients |
| Methods | Study type: interventional (clinical trial) Estimated enrolment: 80 participants Allocation: randomised Intervention model: parallel assignment Masking: unclear Primary purpose: prevention |
| Participants | Iran (Islamic Republic of Iran) Inclusion criteria:
Exclusion criteria:
Aiming to recruit 80 participants |
| Interventions | Experimental: 3 weeks after stroke sertraline 50 mg a day for 12 months versus Comparator: 3 weeks after stroke a placebo tablet every day |
| Outcomes | Primary outcomes collected at 3 months, 6 months, 9 months
Secondary outcomes collected at 3 months, 6 months, 9 months
|
| Starting date | 28 November 2012 |
| Contact information | Reza Pirzeh, Tabriz University of Medical Sciences, Iran (Islamic Republic of) pirzehr@tbzmed.ac.ir |
| Notes | IRCT2012101011062N1 |
Sadaat 2012.
| Trial name or title | Effect of fluoxetine on functional recovery of patients with cerebrovascular accident following middle cerebral artery trunk obstruction: a randomised clinical trial |
| Methods | Study type: interventional (clinical trial) Estimated enrolment: 60 participants Allocation: randomised Intervention model: parallel assignment Masking: unclear Primary purpose: treatment |
| Participants | Iran (Islamic Republic of Iran) Inclusion criteria
Exclusion criteria
|
| Interventions | Intervention: fluoxetine, 15 mg oral pill for the first month and 20 mg for the next 2 months Comparator: placebo, 15 mg oral pill for the first month and 20 mg for the next 2 months |
| Outcomes | Primary outcomes collected at discharge, 1 and 3 months
Secondary outcomes collected at discharge
|
| Starting date | 5 April 2012 |
| Contact information | Dr Babak Bakhshayesh Eghbali Poorsina hospital, Guilan University of Medical Sciences bakhshayesh@gums.ac.ir |
| Notes | IRCT201112228490N1 Contacted 7 February 2019 |
Sahin 2016.
| Trial name or title | Fluoxetine for visual recovery after ischemic stroke (FLUORESCE) |
| Methods | Study type: interventional (clinical trial) Estimated enrolment : 40 participants Allocation: randomised Intervention model: parallel assignment Masking: quadruple (participant, care provider, investigator, outcomes assessor) Primary purpose: treatment |
| Participants | USA Inclusion criteria 18 to 85 years Inclusion criteria:
Exclusion criteria:
Aiming to recruit 40 participants |
| Interventions | Experimental: 20 mg fluoxetine capsule by mouth once daily for 90 days Comparator: matching placebo |
| Outcomes | Outcomes collected at 6 months Primary outcome
Secondary outcomes
|
| Starting date | May 2016 |
| Contact information | Bogachan Sahin bogachan_sahin@rocheter.edu |
| Notes | NCT02737930 |
BDI: Beck Depression Inventory; BDNF: brain‐derived neurotrophic factor; BI: Barthel Index; CT: computerised tomography; CTIMP: Clinical Trial of an Investigational Medicinal Product; DWI: diffusion‐weighted imaging; FAI: Frenchay Activities Index; FLAIR: fluid‐attenuated inversion recovery; FMMS: Fugl Meyr Motor Score; fMRI: functional magnetic resonance imaging; HAM‐D: Hamilton Depression Rating Scale; HRQoL: Health‐Related Quality of Life; JTT: Jebsen Taylor Test; MADRS: Montgomery‐Åsberg Depression Rating Scale; MAOI: mono‐amino‐oxidase inhibitor; MCA: middle cerebral artery; MEP: motor evoked potential; MHI 5: Mental Health Inventory; MHRA: Medicines and Healthcare products Regulatory Agency MRA: magnetic resonance angiography; MRI: magnetic resonance imaging; mRS: modified Rankin score; NHP: Nottingham Health Profile; NIHSS: National Institute of Health Stroke Scale NPI: Neuropsychiatric Inventory Scale; PHQ‐9: Patient Health Questionnaire; rTMS: repetitive transcranial magnetic stimulation; SF‐36: Short Form‐36; SIS: Stroke Impact Scale; smRSq: simplified modified Rankin Scale questionnaire; SSRI: selective serotonin reuptake inhibitor; TCD: transcranial Doppler; TIA: transient ischaemic attack; TICSm: telephone interview for cognitive status ‐ modified; TMS: transcranial magnetic stimulation
Differences between protocol and review
Changes to 'Criteria for considering studies for this review'
We edited the Types of studies section: we excluded trials which combined an SSRI with another active intervention and compared it to the active treatment alone. We restricted the criteria for considering studies for this review to randomised controlled trials and excluded studies where investigators described a non‐random component in the sequence generation process.
Changes to 'Data collection and analysis'
We edited the Methods section of the review to reflect current MECIR standards, adding information for Assessment of risk of bias in included studies; Measures of treatment effect; Unit of analysis issues; Dealing with missing data; Assessment of heterogeneity; Assessment of reporting biases; and Subgroup analysis and investigation of heterogeneity. We also restricted the meta‐analyses to studies at low risk of bias.
Changes to Results
We excluded three studies that were previously included but were no longer eligible for this review, as there was a non‐random component in the sequence generation process (Li 2002; Liang 2003; Zhou 2003). We excluded four studies that combined an SSRI with another active intervention and compared it to the active treatment alone (Finkenzeller 2009; Ji 2000; Liu 2004; Xu 2007). We excluded two studies (Graffagnino 2002; Sitzer 2002), listed as 'Awaiting classification' in the previous version of this review (Mead 2012). We could find no published results and when we sought further information from the authors, we received no responses.
We renamed previously included studies to match current Cochrane standards: EMOTION 2011 is now Kim 2011; FOCUS 2011 is now FOCUS Trial Collaboration 2018; AFFINITY is now Hankey 2011; EFFECTS is now Lundström 2014.
Contributions of authors
Gillian Mead conceived the study, screened references, extracted data, assessed risk of bias, performed the analyses and wrote the first draft.
Lynn Legg searched for studies selected studies for inclusion, collected data, assessed risk of bias, managed studies through the review process, contributed to the final version.
Russel Tilney screened citations, retrieved potentially relevant papers and screened their eligibility, assisted with data extraction, performed 'Risk of bias' assessments and drafted an initial version of a manuscript for the fluoxetine trials.
Cheng Fang Hsieh screened citations, retrieved potentially relevant papers and screened their eligibility, assisted with data extraction, performed 'Risk of bias' assessments and approved the final version.
Simiao Wu screened citations, retrieved potentially relevant papers and screened their eligibility, assisted with data extraction , performed 'Risk of bias' assessments and approved the final version.
Erik Lundström screened citations, retrieved potentially relevant papers and screened their eligibility, assisted with data extraction performed 'Risk of bias' assessments and approved the final version.
Ann‐Sofie Rudberg screened citations, retrieved potentially relevant papers and screened their eligibility, assisted with data extraction, performed 'Risk of bias' assessments and approved the final version.
Mansur Kutlubaev screened citations, retrieved potentially relevant papers and screened their eligibility, assisted with data extraction, performed 'Risk of bias' assessments and approved the final version.
Babak Soleimani screened citations, retrieved potentially relevant papers and screened their eligibility, assisted with data extraction, performed 'Risk of bias' assessments and approved the final version.
Amanda Barugh screened citations, retrieved potentially relevant papers and screened their eligibility, assisted with data extraction, drafted the manuscript for submission, performed 'Risk of bias' assessments and approved the final version.
Maree Hackett screened citations, retrieved potentially relevant papers and screened their eligibility, assisted with data extraction, performed 'Risk of bias' assessments and approved the final version.
Graeme Hankey conceived the review, provided expertise in relation to analysis methods, checked the list of excluded studies and approved the final version of the review.
Martin Dennis provided topic expertise, advised on methods of analysis and approved the final version.
Sources of support
Internal sources
None, Other.
External sources
-
Stroke Research Network, UK.
Stroke Research Network in England provided some financial support to the Cochrane Stroke Group for assistance with the searches
-
Scotland, Other.
Scottish Stroke Research Network provided some funding to the Cochrane Stroke group for assistance with the searches
-
Incentive grant from National Institute of Health Research, UK.
£5000 incentive grant to support an honorarium to Lynn Legg
Declarations of interest
Lynn A Legg: none known.
Russel Tilney: none known.
Cheng‐Fang Hsieh: none known.
Simiao Wu: none known.
Erik Lundström: none known.
Ann‐Sofie Rudberg: none known.
Mansur A Kutlubaev: none known.
Martin Dennis: none known.
Babak Soleimani: none known.
Amanda Barugh: none known.
Maree L Hackett: during the completion of this work Maree Hackett was supported by a National Health and Medical Research Council of Australia Career Development Fellowship, Population Health (Level 2), APP1141328 (1/1/18‐31/12/21)
Graeme J Hankey: in the past three years, GJH has a project grant from the National Health and Medical Research Council of Australia to lead a trial of fluoxetine for stroke recovery (AFFINITY trial). He has also received honoraria from the American Heart Assocaition for serving as an associated editor of the journal CIrculation, and from AC Immune for chairing the data safety monitoring committee of two clinical trials of vaccines for Alzheimer's disease.
Gillian E Mead: has developed a course on exercise after stroke which was licensed to Later Life Training, who pay royalties for the course. These royalties are used to support further research in this area. She has received expenses for speaking at conferences on exercise and fatigue after stroke.
Gillian Mead, Martin Dennis, Maree Hackett, Erik Lundstrom and Graeme Hankey are investigators on the FOCUS trial (Fluoxetine or control under supervision) in the UK, the AFFINITY (Assessment of fluoxetine in stroke recovery) trial in Australia (Hankey 2011), and the EFFECTs trial in Sweden designed to assess the impact of fluoxetine on disability and dependency after stroke (Lundström 2014). None of these review authors extracted data from FOCUS Trial Collaboration 2018.
New search for studies and content updated (conclusions changed)
References
References to studies included in this review
Acler 2009 {published data only}
- Acler M, Avesani A, Fiaschi A, Manganotti P. Serotonergic modulation of brain excitability and motor recovery in patients affected by stroke. A double blind placebo RCT. International Journal of Stroke 2008;3:336. [Google Scholar]
- Acler M, Robol E, Fiaschi A, Manganotti P. A double blind placebo RCT to investigate the effects of serotonergic modulation on brain excitability and motor recovery in stroke patients. Journal of Neurology 2009;256(7):1152‐8. [DOI] [PubMed] [Google Scholar]
Almeida 2006 {published data only}
- Almeida OP, Waterreus A, Hankey GJ. Preventing depression after stroke: results from a randomized placebo‐controlled trial. Journal of Clinical Psychiatry 2006;67(7):1104‐9. [DOI] [PubMed] [Google Scholar]
Andersen 1994 {published data only}
- Andersen G, Vestergaard K, Lauritzen L. Effective treatment of post‐stroke depression with selective serotonin reuptake inhibitors. Journal of Neurology 1994;241:S42. [DOI] [PubMed] [Google Scholar]
- Andersen G, Vestergaard K, Lauritzen L. Effective treatment of post‐stroke depression with the selective serotonin reuptake inhibitor citalopram. Stroke 1994;25(6):1099‐104. [DOI] [PubMed] [Google Scholar]
- Andersen G, Vestergaard K, Lauritzen L. Post‐stroke depression treated with citalopram. Acta Neurologica Scandinavica 1994;89 Suppl 155:20. [Google Scholar]
- Andersen G, Vestergaard K, Lauritzen L. Post‐stroke depression treated with citalopram ‐ a selective serotonin reuptake inhibitor. Canadian Journal of Neurological Sciences 1993;20:S115. [Google Scholar]
- Andersen G, Vestergaard K, Lauritzen L. Post‐stroke depression treated with citalopram a selective serotonin reuptake inhibitor. 7th Scandinavian Meeting on Cerebrovascular Disease. 1993:54.
- Andersen G, Vestergaard K, Lauritzen LU. Effective treatment of depression following apoplexy with citalopram. Ugeskrift for Laeger 1995;157(14):2000‐3. [PubMed] [Google Scholar]
- Flicker C, Andersen G, Bayer L. A placebo controlled study of citalopram treatment for post‐stroke depression. 11th Annual Meeting of the American Association for Geriatric Psychiatry. 1998.
Andersen 2013 {published data only}
- *Kraglund KL, Mortensen JK, Damsbo AG, Modrau B, Simonsen SA, Iversen HK, et al. Neuroregeneration and vascular protection by citalopramin in acute ischemic stroke (TALOS). Stroke 2018;49(11):2568‐76. [DOI] [PubMed] [Google Scholar]
- Andersen 2013. The efficacy of citalopram treatment in acute stroke (TALOS). www.clinicaltrials.gov/ct2/show/NCT01937182 (first received 9 September 2013).
- Kraglund K, Mortensen JK, Grove EL, Johnsen SP, Andersen G. TALOS: a multicenter, randomised, double blind, placebo controlled trial to test the effects of citalopram in patients with acute stroke; Protocol. International Journal of Stroke 2015;10:985‐987. [DOI] [PubMed] [Google Scholar]
Birchenall 2019 {published and unpublished data}
- Birchenall J, Térémetz M, Roca P, Lamy JC, Oppenheim C, Maier MA, et al. Individual recovery profiles of manual dexterity, and relation to corticospinal lesion load and excitability after stroke ‐ a longitudinal pilot study. Neurophysiologie Clinique 2019;49(2):149‐64. [DOI] [PubMed] [Google Scholar]
- Centre Hospitalier St Anne. Evaluation by Transcranial Magnetic Stimulation of the Benefit of Fluoxetine on Motor Recovery After Stroke (EFLUSTIM). clinicaltrials.gov/ct2/show/NCT02063425 2014.
Black‐Schaffer 2012 {published data only}
- Black‐Schaffer R. Fluoxetine for motor, aphasia, and neglect recovery after ischemic stroke (FLAN). www.clinicaltrials.gov/ct2/show/NCT01674868 (first received 29 August 2012).
Brown 1998 {published data only}
- Brown KW, Sloan RL, Pentland B. Fluoxetine as a treatment for post‐stroke emotionalism. Acta Psychiatrica Scandinavica 1998;98(6):455‐8. [DOI] [PubMed] [Google Scholar]
Burns 1999 {published data only}
- Burns A, Russell E, Stratton‐Powell H, Tyrell P, O'Neill P, Baldwin R. Sertraline in stroke‐associated lability of mood. International Journal of Geriatric Psychiatry 1999;14(8):681‐5. [PubMed] [Google Scholar]
Chen 2001 {published data only}
- Chen WY, Liu FY, Yang AP. Study of effect of integrative Chinese herbs with fluoxetine on rehabilitation of neurological impairment in patients with post‐stroke depression. Journal of Chengdu University of Traditional Chinese Medicine 2001;24(4):20‐3. [Google Scholar]
Chen 2002 {published data only}
- Chen W, Wang G‐F, Chen X‐H, Sheng Y‐L, Zhu H. Effects of paroxetine on function recovery in patients with post‐stroke depression. Chinese Journal of Clinical Rehabilitation 2002;6(13):2014‐5. [Google Scholar]
Chen 2005a {published data only}
- Chen KN. Changes of neurotransmitter in patients with post‐stroke depression observed with encephalofluctuography technology. Chinese Journal of Clinical Rehabilitation 2005;9(16):118‐9. [Google Scholar]
Chen 2005b {published data only}
- Chen T, Li J, Han M. A study on paroxetine in the treatment for post‐stroke depression. Jiangxi Medicine 2005;40(7):382‐4. [Google Scholar]
Cheng 2003 {published data only}
- Cheng F, Shao G, Bao S. Study of effect on neurologic function rehabilitation in patient with post‐stroke depression. Chinese Journal of Clinical Rehabilitation 2003;7(1):108‐9. [Google Scholar]
Chollet 2011 {published data only}
- Chollet F, Tardy J, Albucher J‐F, Thalamas E, Berard E, Lamy C, et al. Fluoxetine for motor recovery after acute ischaemic stroke (FLAME): a randomised placebo‐controlled trial. Lancet Neurology 2011;10(2):123‐30. [DOI] [PubMed] [Google Scholar]
Dam 1996 {published data only}
- Dam M, Tonin P, Boni A, Pizzolato G, Casson S, Ermani M, et al. Effects of fluoxetine and maprotiline on functional recovery in poststroke hemiplegic patients undergoing rehabilitation therapy. Stroke 1996;27(7):1211‐4. [DOI] [PubMed] [Google Scholar]
Feng 2004 {published data only}
- Feng B‐L, Wang Q‐C, Zheng‐Yuan LI. Influence of Jieyu Huoxue decoction on rehabilitation of patients with depression after cerebral infarction. Journal of Chinese Integrated Medicine 2004;2(3):182‐4. [DOI] [PubMed] [Google Scholar]
FOCUS Trial Collaboration 2018 {published data only}
- FOCUS Trial Collaboration. Effects of fluoxetine on functional outcomes after acute stroke (FOCUS): a pragmatic, double‐blind, randomised, controlled trial. Lancet 2018;393(10168):265‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mead G. Fluoxetine Or Control Under Supervision (FOCUS) trial: to establish the effect(s) of routine administration of Fluoxetine in patients with a recent stroke. www.isrctn.com/ISRCTN83290762 ( first received 23 May 2012).
- Mead GE, Dennis MS, Innes K, MacLeod M, Sandercock PA, House A, et al. A multicentre randomised trial to establish the effect(s) of routine administration of fluoxetine in patients with a recent stroke (Fluoxetine Or Control Under Supervision, FOCUS). 21st European Stroke Conference. 2012:Abst OAID 26.
Fruehwald 2003 {published data only}
- Fruehwald S, Gatterbauer. Early fluoxetine treatment of post‐stroke depression: a three‐month double‐blind placebo‐controlled study with an open‐label long‐term follow up. Journal of Neurology 2003;250(3):347‐51. [DOI] [PubMed] [Google Scholar]
Gao 2016 {published data only}
- Gao J, Lin M, Zhao J, Bi S, Ni Z, Shang X. Different interventions for post‐ischaemic stroke depression in different time periods: a single‐blind randomized controlled trial with stratification by time after stroke. Clinical Rehabilitation 2017;31(7):71‐81. [DOI] [PubMed] [Google Scholar]
GlaxoSmithKline 1998 {published data only}
- GlaxoSmithKline. An 8‐week double‐blind placebo controlled parallel group study to assess the efficacy and tolerability of paroxetine in patients suffering from depression following stroke. GSK Clinical Study Register www.gsk clinicalstudyregister.com (accessed 31 August 2012).
Guo 2009 {published data only}
- Guo R‐Y, Su L, Wang CX. Effects of Linggui Bafa on the therapeutic effect and quality of life in patients of post‐stroke depression. Chinese Acupuncture & Moxibustion 2009;29(10):785‐90. [PubMed] [Google Scholar]
He 2004 {published data only}
- He P. Randomized controlled observation on the effect of early application of fluoxetine in preventing depression after stroke. Chinese Journal of Clinical Rehabilitation 2004;8(28):6016‐7. [Google Scholar]
He 2005 {published data only}
- He Y, Wang X, Xiao C. Prospective study of effects of paroxetine with mental intervention on depression and anxiety after stroke. Nervous Diseases and Mental Health 2005;5(1):6‐13. [Google Scholar]
- Wang X, He Y, Xiao C‐L. A clinical trial of paroxetine and psychotherapy in patients with post‐stroke depression and anxiety. Chinese Mental Health Journal 2005;19:564‐6. [Google Scholar]
He 2016 {published data only}
- Guo 2012. Multi‐center randomized clinical study of antidepressant treatment (fluoxetine) on secondary prevention of ischemic stroke [ChiCTR‐TRC‐12002078]. www.chictr.org.cn/showprojen.aspx?proj=7471 (first received 3 April 2012).
- He Y‐T, Tang B‐S, Cai Z‐L, Zeng S‐L, Jiang X, Guo Yi. Effects of fluoxetine on neural functional prognosis after ischemic stroke: a randomized controlled study in China. Journal of Stroke and Cerebrovascular Diseases 2016;25(4):761‐70. [DOI] [PubMed] [Google Scholar]
Hu 2002 {published data only}
- Hu Y, Suo A, Xiang L. The comparative study of the effectiveness of fluoxetine on the stroke patients with depressive symptoms. Shanghai Archives of Psychiatry 2002;14:149‐50. [Google Scholar]
Huang 2002 {published data only}
- Huang X‐H. The clinical correlation study and the effect of fluoxetine intervention on poststroke depression. Chinese Journal of Clinical Rehabilitation 2002;6(15):2296‐7. [Google Scholar]
Jia 2005 {published data only}
- Jia W. Effect of early intervention on recovery of motor function and recurrent stroke in patients with post‐stroke depression. Chinese Journal of Clinical Rehabilitation 2005;9(12):4‐5. [Google Scholar]
Kim 2011 {published data only}
- Kim JS. The preventative effect of escitalopram on depression and related emotional disorders in acute stroke patients, 2011. clinicaltrials.gov/ct2/show/NCT01278498 (first received 19 January 2011). [NCT01278498]
- Kim JS, Lee E‐J, Chang D‐ll, Park J‐H, Ahn SH, Cha J‐K, et al. Efficacy of early administration of escitalopram on depressive and emotional symptoms and neurological dysfunction after stroke: a multicentre, double‐blind, randomised, placebo‐controlled study. Lancet Psychiatry 2017;4(1):33‐41. [DOI] [PubMed] [Google Scholar]
Kong 2007 {published data only}
- Kong Y. Fluoxetine for poststroke depression: a randomized placebo controlled clinical trial. Neural Regeneration Research 2007;2(3):162‐5. [Google Scholar]
Lai 2006 {published data only}
- Lai J. The effect of using paroxetine to treat post stroke depression. Journal of Guangdong Medical College 2006;24(6):585‐6. [Google Scholar]
Li 2004a {published data only}
- Li J, He Q‐Y, Han M‐F. Recent effect of fluoxetine in improving neurologic impairment and preventing post‐stroke depression in the early stage. Chinese Journal of Clinical Rehabilitation 2004;8(7):1208‐9. [Google Scholar]
Li 2004b {published data only}
- Li C‐M, Jiang X‐D, Liao G, Lei J‐M, Lan S, Ni F‐W. Effect of antidepressant drugs in early period on the recovery of post‐stroke depression. Chinese Journal of Clinical Rehabilitation 2004;8(19):3713‐5. [Google Scholar]
Li 2005 {published data only}
- Li Y, Wang X, Qian FS. Related factors of post‐stroke depression and effect of paroxetine. Shandong Archives of Psychiatry 2005;18(4):209‐10. [Google Scholar]
Li 2006 {published data only}
- Li W‐Q, Li D‐X. The efficacy of citalopram for post‐stroke depression and its effects on stroke rehabilitation. International Journal of Cerebrovascular Diseases 2006;14(4):275‐8. [Google Scholar]
Li 2008 {published data only}
- Li L‐T, Wang S‐H, Ge H‐Y, Chen J, Yue S‐W, Yu M. The beneficial effects of the herbal medicine free and easy wanderer plus (FEWP) and fluoxetine on post‐stroke depression. Journal of Alternative and Complementary Medicine 2008;14(7):841‐6. [DOI] [PubMed] [Google Scholar]
Liu 2006 {published data only}
- Liu Y, Xu R. Effect of citalopram treatment on post‐stroke depression and neurological functional rehabilitation. Chinese Journal of Rehabilitation 2006;21(3):174‐5. [Google Scholar]
Marquez Romero 2013 {published data only}
- Marquez‐Romero JM. Fluoxetine for motor recovery after acute intracerebral hemorrhage (FMRICH). www.clinicaltrials.gov/ct2/show/NCT01737541 (first received 29 November 2012).
- Marquez‐Romero JM, Arauz A, Ruiz‐Sandoval JL, Cruz‐Estrada Ede L, Huerta‐Franco MR, Aguayo‐Leytte G, et al. Fluoxetine for motor recovery after acute intracerebral hemorrhage (FMRICH): study protocol for a randomized, double‐blind, placebo‐controlled, multicenter trial. Trials 2013;14:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marquez‐Romero JM, Reyes‐Martínez M, Huerta‐Franco MR, Ruiz‐Franco A, Silos H, Arauz A. Fluoxetine for motor recovery after acute intracerebral hemorrhage,the FMRICH Trial. Correspondence from Marquez‐Romero 2018. [DOI] [PubMed]
Meara 1998 {published data only}
- Meara RJ, Thalanany M, Balonwu V, Hobson P. The treatment of depression after stroke with the selective serotonin reuptake inhibitor sertraline. Cerebrovascular Diseases 1998;8 Suppl 4:90. [Google Scholar]
Miao 2004 {published data only}
- Miao S‐Y, Shi Y‐J. Related factors of post‐stroke depression and therapeutical effect of citalopram. Chinese Journal of Clinical Rehabilitation 2004;8(19):3718‐9. [Google Scholar]
Murray 2005 {published data only}
- Murray V, Arbin M, Bartfai A, Berggren A‐L, Landtblom A‐M, Lumdmark J, et al. Double‐blind comparison of sertraline and placebo in stroke patients with minor depression and less severe major depression. Journal of Clinical Psychiatry 2005;66(6):708‐16. [DOI] [PubMed] [Google Scholar]
Pan 2018 {published data only}
- Pan X‐L, Chen H‐F, Cheng X, Hu C‐C, Wang J‐W, Fu Y‐M, et al. Effects of paroxetine on motor and cognitive function recovery in patients with non‐depressed ischemic stroke: an open randomized controlled study. Brain Impairment Vol. doi: 10.1017/BrImp.2018.6.
Pariente 2001 {published data only}
- Guiraud‐Chaumeil B, Pariente J, Albucher J‐F, Loubinoux I, Chollet F. Rehabilitation after stroke [Recuperation neurologique post‐ischemique]. Bulletin de l'Académie Nationale de Médecine 2002;6:1015‐24. [PubMed] [Google Scholar]
- Pariente J, Loubinoux I, Carel C, Albucher JF, Leger A, Manelfe C, et al. Fluoxetine modulates motor performance and cerebral activation of patients recovering from stroke. Annals of Neurology 2001;50(6):718‐29. [DOI] [PubMed] [Google Scholar]
Rasmussen 2003 {published data only}
- Rasmussen A. Depression and stroke. Nordic Journal of Psychiatry 2001;55(4):288. [Google Scholar]
- Rasmussen A. Prophylactic treatment for post‐stroke depression and comorbidity. Journal of Psychosomatic Research 2000;48(3):66. [Google Scholar]
- Rasmussen A, Lunde M, Poulsen D, Sørensen K, Qvitzau S, Bech P. A double‐blind placebo controlled study of sertraline in the prevention of depression in stroke patients. European Neuropyschopharmacology 2002;12:231. [DOI] [PubMed] [Google Scholar]
- Rasmussen A, Lunde M, Poulsen DL, Sørensen K, Qvitzau S, Bech P. A double‐blind, placebo‐controlled study of sertraline in the prevention of depression in stroke patients. Psychosomatics 2003;44(3):216‐22. [DOI] [PubMed] [Google Scholar]
Razazian 2014 {published data only}
- Razazian N. A survey for assessment of effectiveness of fluoxetine on motor improvement in ischemic stroke patients. www.en.irct.ir/trial/8797 2014.
Restifo 2001 {published data only}
- Restifo DA, Lo Prest R, Lanza S, Giuffrida S, D'Aleo G, Rifici Di Bella C, et al. Motor cortex reorganization induced by fluoxetine in poststroke hemiplegic patients undergoing rehabilitation therapy: a study with transcranial magnetic stimulation. Neurorehabilitation and Neural Repair 2001;15(4):284. [Google Scholar]
Robinson 2000a {published data only}
- Jorge RE, Robinson RG, Arndt S, Starkstein S. Mortality and post‐stroke depression: a placebo controlled trial of antidepressants. American Journal of Psychiatry 2003;160:1823‐9. [DOI] [PubMed] [Google Scholar]
- Narushima K, Kosier JT, Robinson RG. Preventing post‐stroke depression: a 12 week double blind randomised treatment trial and 21 month follow‐up. Journal of Nervous and Mental Diseases 2002;190:296‐303. [DOI] [PubMed] [Google Scholar]
- Robinson RG, Schultz SK, Castillo C, Kopel T, Kosier JT, Newman RM, et al. Nortriptyline versus fluoxetine in the treatment of depression and in short‐term recovery after stroke: a placebo‐controlled, double‐blind study. American Journal of Psychiatry 2000;157(3):351‐9. [DOI] [PubMed] [Google Scholar]
Robinson 2000b {published data only}
- Jorge RE, Robinson RG, Arndt S, Starkstein S. Mortality and post‐stroke depression: a placebo controlled trial of antidepressants. American Journal of Psychiatry 2003;160:1823‐9. [DOI] [PubMed] [Google Scholar]
- Narushima K, Robinson RG. Preventing post‐stroke depression: a 12 week double blind randomised treatment trial and 21 month follow‐up. Journal of Nervous and Mental Diseases 2002;190:296‐303. [DOI] [PubMed] [Google Scholar]
- Robinson RG, Schultz SK, Castillo C, Kopel T, Kosier JT, Newman RM, et al. Nortriptyline versus fluoxetine in the treatment of depression and in short‐term recovery after stroke: a placebo‐controlled, double‐blind study. American Journal of Psychiatry 2000;157(3):351‐9. [DOI] [PubMed] [Google Scholar]
Robinson 2008 {published data only}
- Jorge RE, Acion L, Moser D, Adams HP, Robinson RG. Escitalopram and enhancement of cognitive recovery following stroke. Archives of General Psychiatry 2010;67(2):187‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robinson RG, Arndt S. Incomplete financial disclosure in a study of escitalopram and problem solving therapy for prevention of post‐stroke depression. JAMA 2009;301:1023‐4. [DOI] [PubMed] [Google Scholar]
- Robinson RG, Jorge RE, Moser DJ, Acion L, Solodkin A, Small SL, et al. Escitalopram and problem‐solving therapy for prevention of poststroke depression. JAMA 2008;299(20):2391‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
Savadi Oskouie 2012 {published data only}
- Savadi Oskouie D. Evaluation of the effect of citalopram on three months functional prognosis of acute ischemic stroke patients: a randomized clinical trial. www.en.irct.ir/trial/1766 (first received 22 April 2012).
- Savadi Oskouie D, Sharifipour E, Sadeghi Bazargani H, Hashemilar M, Nikanfar M, Ghazanfari Amlashi S, et al. Efficacy of citalopram on acute ischemic stroke outcome: a randomized clinical trial. Neurorehabilitation and Neural Repair 2017;31(7):638‐47. [DOI] [PubMed] [Google Scholar]
Shah 2016 {published data only}
- Shah IA, Asimi RP, Kawoos Y, Wani MA, Wani MA, Dar MA. Effect of fluoxetine on motor recovery after acute haemorrhagic stroke: a randomized trial. Journal of Neurology and Neurophysiology 2016; Vol. 7, issue 2. [DOI: 10.4172/2155-9562.1000364] [DOI]
Song 2006 {published data only}
- Song J‐G. Effects of fluoxetine hydrochloride on depressive symptoms and P300 after cerebral stroke. Chinese Journal of Clinical Rehabilitation 2006;10(14):160‐2. [Google Scholar]
Wang 2003 {published data only}
- Wang X, Tan Z, Wu Z, Gao J, Feng M. The effects of anti‐depression therapy on post‐stroke depression and neurologic rehabilitation in the elderly patients. Chinese Journal of Geriatrics 2003;22(5):270‐3. [Google Scholar]
Wen 2006 {published data only}
- Wen Z‐X. The influence of post‐stroke prophylactic anti‐depression treatment on nerve functional rehabilitation. Acta Academiae Medicinae Qingdao Universitatis 2006;42(3):253‐4. [Google Scholar]
Whyte 2005 {published data only}
- Whyte E. Setraline for prevention post stroke depression and improving rehabilitation outcomes. clinicaltrials.gov/ct2/show/NCT00177424 (first received 15 September 2005).
Wiart 2000 {published data only}
- Wiart L, Petit H, Joseph PA, Mazaux JM, Barat M. Fluoxetine in early poststroke depression: a double‐blind placebo‐controlled study. Stroke 2000;31(8):1829‐32. [DOI] [PubMed] [Google Scholar]
Xie 2005 {published data only}
- Xie R, Liu J, Quan H. A prospective random clinical contrast study of treatment with sertraline in elderly patients with post‐stroke depression. Chinese Journal of Clinical Neuroscience 2005;13(3):294‐7. [Google Scholar]
Xu 2001 {published data only}
- Xu J, Tan J, Ou L. A study on treatment of fluoxetine to depression in early recovery stage of cerebral infarction. Chinese Journal of Rehabilitation Medicine 2001;16(5):281‐3. [Google Scholar]
Xu 2006 {published data only}
- Xu J, Wang J, Liu J. Preventive effects of antidepressants on post‐stroke depression. Chinese Mental Health Journal 2006;20(3):186‐8. [Google Scholar]
Yang 2002 {published data only}
- Yang J, Zhao Y, Bai S. Controlled study on antidepressant treatment of patients with post‐stroke depression. Chinese Mental Health Journal 2002;16(12):871‐2. [Google Scholar]
Yang 2011 {published data only}
- Yang J. Therapeutic effect of paroxetine on patients with early poststroke depression and the serum interleukins. Chinese Journal of Cerebrovascular Diseases 2011;8(5):235‐8. [Google Scholar]
Ye 2004 {published data only}
- Ye L‐X, Wang H, Wang Y‐D, Zhong L, Liang D‐S, Guo Y. Effect of anti‐depressive therapy on the rehabilitation of psychological and neurological function after stroke. Chinese Journal of Clinical Rehabilitation 2004;8(31):6826‐8. [Google Scholar]
- Ye LX. Effect of Paxil and berhomine on post‐stroke anxiety‐depression and neurological recovery. Chinese Journal of Clinical Rehabilitation 2006;10(6):153‐5. [Google Scholar]
Zhao 2011 {published data only}
- Zhao P, Wang J‐P. Effects of antidepressants on neurofunctional recovery of post‐stroke patients with aphasia. Journal of Dalian Medical University 2011;33(1):55‐7. [Google Scholar]
Zhou 2008 {published data only}
- Zhou Z‐l, Liang L‐Z, Yan Y‐X. Preventive effects of fluoxetine on post‐stroke depression. Chinese Journal of Modern Applied Pharmacy 2008;25(3):263‐4. [Google Scholar]
References to studies excluded from this review
Andersen 1993 {published data only}
- Andersen G, Vestergaard K, Riis JO. Citalopram for post‐stroke pathological crying. Lancet 1993;342(8875):837‐9. [DOI] [PubMed] [Google Scholar]
Andersen 2012 {published data only}
- Andersen G. Efficacy of escitalopram treatment in acute stroke and the role of specific genotypes in stroke. www.clinicaltrialsregister.eu/ctr‐search/search?query=2011‐005541‐12 (first received 27 February 2012).
Anderson 2002 {published data only}
- Anderson C, Hackett M, Carter K, Ni MC. Maximising outcome by overcoming depression in stroke (MOODS) trial. Cerebrovascular Diseases 2002 2002;13(Suppl 3):79. [Google Scholar]
Anonymous 2012 {published data only}
- Anonymous 2012. Stroke and depression. Johns Hopkins Med Letters: Health After 50 2012;23(11):3. [Google Scholar]
Anonymous 2012b {published data only}
- Anonymous. Drug & Device News. Medical Malpractice Law & Strategy, New York Law Journal Newsletters 2012;30(2):5. [Google Scholar]
Berends 2009 {published data only}
- Berends HI, Nijlant JM, Putten MJ, Movig KL, IJzerman MJ. Single dose of fluoxetine increases muscle activation in chronic stroke patients. Clinical Neuropharmacology 2009; Vol. 32, issue 1:1‐5. [also known as Flu 2006] [PubMed]
Choi Kwon 2008 {published data only}
- Choi‐Kwon S, Choi J, Kwon SU, Kang DW, Kim JS. Fluoxetine improves the quality of life in patients with poststroke emotional disturbances. Cerebrovascular Diseases 2008;26(3):266‐71. [DOI] [PubMed] [Google Scholar]
Finkenzeller 2009 {published data only}
- Finkenzeller W, Zobel I, Rietz S, Schramm E, Berger M. Interpersonal psychotherapy and pharmacotherapy for post‐stroke depression. Feasibility and effectiveness. Der Nervenarzt 2009;80(7):805‐12. [DOI] [PubMed] [Google Scholar]
Gourab 2015 {published data only}
- Gourab K, Schmit BD, Hornby GT. Increased lower limb spasticity but not strength or function following a single‐dose serotonin reuptake inhibitor in chronic stroke. Archives of Physical Medicine and Rehabilitation 2015;96(12):2112‐9. [DOI] [PubMed] [Google Scholar]
Graffagnino 2002 {published data only}
- Graffagnino C. Poststroke depression and functional recovery (SADBRAIN). Duke University Medical Centre 2002.
Ji 2000 {published data only}
- Ji QM, Xie LP. Efficacy of fluoxetine in the treatment of 20 patients with depression after stroke. Herald of Medicine 2000;19(4):329. [Google Scholar]
Li 2002 {published data only}
- Li F, Gu DX, Ceng SH, Xu JW. Effect of paroxetine on prognosis of patients with post cerebral infarction depression. Chinese Journal of New Drugs and Clinical Remedies. 2002;21(1):11‐3. [Google Scholar]
Liang 2003 {published data only}
- Liang Z, Shuliang T. Clinical efficacy of fluoxetine in treatment of patients with depression after acute stroke. Chinese Journal of Clinical Rehabilitation 2003;7(13):1924‐5. [Google Scholar]
Liu 2004 {published data only}
- Liu L‐X. Recent effect of drug intervention on post‐stroke anxiety. Chinese Journal of Clinical Rehabilitation 2004;8(30):6600‐1. [Google Scholar]
Robinson 2011 {published data only}
- Robinson RG, Mikami K, Jang M, Jorge RE. Prevention of anxiety disorder after stroke. Journal of Neuropsychiatry and Clinical Neurosciences 2011;23(2):18. [Google Scholar]
Sitzer 2002 {published data only}
- Sitzer M, Huff W, Steckel R. Prevention of poststroke depression after acute ischemic stroke using the selective serotonin reuptake inhibitor sertraline (PreDis‐study). Stroke 2002;33(2):651‐2. [Google Scholar]
Sun 2015 {published data only}
- Sun YT, Bao YH, Wang SL, Chu JM, Li LP. Efficacy observation on the treatment of post‐stroke depression by acupuncture at the acupoints based on ziwuliuzhu and Prozac. Chinese Acupuncture and Moxibustion 2015;355(2):119‐22. [PubMed] [Google Scholar]
University of Alabama 2013 {published data only}
- University of Alabama. RCT of a neuroplasticity agent and CI therapy for severe arm paresis after stroke. clinicaltrials.gov/ct2/show/NCT01963832 (accessed 5th March 2019).
Xu 2007 {published data only}
- Xu B, Zhou WY, Zhang SJ. Observation of effect of Wulung capsule in treating post‐stroke depression. Chinese Journal of Integrated Traditional and Western Medicine 2007;27(7):640‐2. [PubMed] [Google Scholar]
Zhou 2003 {published data only}
- Zhou B. Effects of fluoxetine on neurofunctional recovery of non depressed patients after stroke. Chinese Journal of Clinical Rehabilitation 2003;7(3):374‐5. [Google Scholar]
References to studies awaiting assessment
Carda 2009 {published data only}
- Carda S, Cisari C. Effects of clinical and functional outcome of escitalopram in adult stroke patients. clinicaltrials.gov/ct2/show/NCT00967408 (first received 27 August 2009). [NCT00967408]
Guo 2015 {published data only}
- Guo Y. Effect of using fluoxetine at different time windows after ischemic stroke on neurological functional prognosis: a randomized controlled trial. www.chictr.org.cn/showprojen.aspx?proj=12925 (first received 27 December 2015).
- Guo Y, He Y, Tang B, Ma K, Cai Z, Zeng S, et al. Effect of using fluoxetine at different time windows on neurological functional prognosis after ischemic stroke. Restorative Neurology and Neuroscience 2016;34(2):177–87. [DOI] [PubMed] [Google Scholar]
He 2012 {published data only}
- Guo 2012. Multi‐center randomized clinical study of antidepressant treatment (fluoxetine) on secondary prevention of ischemic stroke [ChiCTR‐TRC‐12002078]. www.chictr.org.cn/showprojen.aspx?proj=7471 (first received 3 April 2012).
- He Y, Cai Z, Zeng S, Chen S, Tang B, Liang Y, Chang X, Guo Y. Effect of fluoxetine on three‐year recurrence in acute ischemic stroke: A randomized controlled clinical study. Clinical Neurology and Neurosurgery 2018;168:1‐6. [DOI] [PubMed] [Google Scholar]
Jurcau 2016 {published data only}
- Jurcau A, Simion A. Improved post‐ischaemic stroke recovery over 1 year with escitalopram for 3 months. European Journal of Neurology. Conference: 2nd Congress of the European Academy of Neurology 2016;23:614. [Google Scholar]
References to ongoing studies
Anonymous 2005 {published data only}
- Anonymous. Influence of escitalopram on the incidence of depression and dementia following acute middle cerebral artery territory infarction. A randomised placebo‐controlled double blind study. https://www.clinicaltrialsregister.eu/ctr‐search/search?query=2005‐005266‐37 (accessed prior to 12 May 2019).
Chollet 2016 {published data only}
- Sattler V, Chollet F. Serotonin Selective Reuptake Inhibitor (SSRI) effects on cerebral connectivity in acute ischemic stroke (RECONISE). www.clinicaltrials.gov/ct2/show/NCT02767999 (first received 11 May 2016).
Cocho 2015 {published data only}
- Cocho D. Effect of serotonin and levodopa in ischemic stroke. www.clinicaltrials.gov/ct2/show/NCT02386475 (first received 12 March 2015).
Dike 2019 {published data only}
- Dike FO, Ekeh BC, Ogun AS, Ekrikpo EU, Walshak P, Ogunniy AA. Pharmacological enhancement of motor function recovery in patients with ischaemic stroke: a trial of fluoxetine. Journal of Neurology and Stroke 2019;9(1):47‐51. [Google Scholar]
Farokhi 2017 {published data only}
- Farokhi F. Evaluation of fluoxetine and standard treatment efficacy on change to side effect of stroke of ischemic strokes in both hemispheres in anterior circulation. www.irct.ir/trial/17976 (first received 11 October 2017).
FOCUS‐Poland 2014 {published data only}
- Fluoxetine or Control under Supervision: Poland. Polish Office for Registration of Medicinal Products, Medical Devices and Biocidal Products ‐ EduraCT No: 2014‐001335‐37 2014.
Fregni 2014 {published data only}
- Fregni F. Effects rTMS combined with fluoxetine on motor recovery in stroke patients. www.clinicaltrials.gov/ct2/show/NCT02208466 (first received 5 August 2014).
Hankey 2011 {published data only}
- Hankey G. Assessment oF FluoxetINe In sTroke recoverY (AFFINITY). www.affinitytrial.org/ (accessed 19th February 2019).
- Hankey G. Assessment of fluoxetine in stroke recovery (AFFINITY) trial, 2011. www.anzctr.org.au/Trial/Registration/TrialReview.aspx?ACTRN=12611000774921 (first received 22 July 2011).
Karimialavijeh 2017 {published data only}
- Karimialavijeh E. Comparison of the effects of citalopram versus fluoxetine on motor recovery after stroke: A double‐blind placebo‐controlled randomized clinical trial. www.en.irct.ir/trial/17729 (first received 3 December 2017).
Leibovitch 2018 {published data only}
- Leibovitch F. Fluoxetine Opens Window to improve motor recovery after stroke (FLOW). www.clinicaltrials.gov/ct2/show/NCT03448159 (first received 27 February 2018).
Levitt 2019 {published data only}
- Levitt 2019. Depression in hemorrhagic stroke. www.clinicaltrials.gov/ct2/show/NCT03826875 (first received 1 February 2019).
Lundström 2014 {published data only}
- Lundström E. Establishing the effect(s) and safety of Fluoxetine initiated in the acute phase of stroke. www.isrctn.com/ISRCTN13020412 (first received 19 December 2014).
Pastore‐Wapp 2016 {published data only}
- Pastore‐Wapp M, Krammer W. Cortical ischemic stroke and serotonin (CISS). www.clinicaltrials.gov/ct2/show/NCT02865642 (first received 12 August 2016).
Pirzeh 2012 {published data only}
- Pirzeh R. A study of sertraline effect on quality of life in stroke inpatients. www.en.irct.ir/trial/11413 (first received 28 November 2012).
Sadaat 2012 {published data only}
- Saadat SM. Effect of fluoxetine on functional recovery of patients with cerebrovascular accident following middle cerebral artery trunk obstruction: a randomized clinical trial. www.en.irct.ir/trial/8954 (first received 25 January 2012).
Sahin 2016 {published data only}
- Sahin B, Leonhardt AM. Fluoxetine for visual recovery after ischemic stroke (FLUORESCE). www.clinicaltrials.gov/ct2/show/NCT02737930 (accessed 14 April 2016).
Additional references
Allida 2019
- Allida S, Patel K, House A, Hackett ML. Pharmaceutical interventions for emotionalism after stroke. Cochrane Database of Systematic Reviews 2019, Issue 3. [DOI: 10.1002/14651858.CD003690.pub4] [DOI] [PMC free article] [PubMed] [Google Scholar]
Deeks 2017
- Deeks JJ, Higgins JP, Altman DG, editor(s) on behalf of the Cochrane Statistical Methods Group. Chapter 9: Analysing data and undertaking metaanalyses. In: Higgins JP, Churchill R, Chandler J, Cumpston MS, editor(s), Cochrane Handbook for Systematic Reviews of Interventions version 5.2.0 (updated June 2017), The Cochrane Collaboration, 2017. Available from www.training.cochrane.org/handbook.
Egger 1997
- Egger M, Davey Smith G, Schneider M, Minder C. Bias in meta‐analysis detected by a simple, graphical test. BMJ 1997;315(7109):629‐34. [DOI] [PMC free article] [PubMed] [Google Scholar]
GBD 2015
- GBD 2015. Disease and Injury Incidence and Prevalence Collaborators. Global, regional, and national incidence, prevalence, and years lived with disability for 310 diseases and injuries, 1990–2015: a systematic analysis for the Global Burden of Disease Study. Lancet 2016;388(10053):1545–602. [DOI] [PMC free article] [PubMed] [Google Scholar]
GRADE 2013
- Schünemann H, Brożek J, Guyatt G, Oxman A, editor(s). Handbook for grading the quality of evidence and the strength of recommendations using the GRADE approach (updated October 2013). GRADE Working Group, 2013. Available from gdt.guidelinedevelopment.org/app/handbook/handbook.html.
GRADEproGDT 2015 [Computer program]
- McMaster University (developed by Evidence Prime, Inc.). GRADEpro Guideline Development Tool [Software]. McMaster University (developed by Evidence Prime, Inc.), 2015.
Hatano 1976
- Hatano S. Experience from a multicentre stroke register: a preliminary report. Bulletin of the World Health Organization 1976;54(5):541‐53. [PMC free article] [PubMed] [Google Scholar]
Higgins 2017
- Higgins JP, Altman DG, Sterne JA, editor(s). Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Churchill R, Chandler J, Cumpston MS, editor(s), Cochrane Handbook for Systematic Reviews of Interventions version 5.2.0 (updated June 2017), The Cochrane Collaboration, 2017. Available from www.training.cochrane.org/handbook.
Johnson 2016
- Johnson W, Onuma O, Owolabi M, Sachdev S. Stroke: a global response is needed. Bulletin of the World Health Organization 2016;94(9):634‐634A. [DOI] [PMC free article] [PubMed] [Google Scholar]
Lang 2004
- Lang UE, Jockers‐Scherübl MC, Hellweg R. State of the art of the neurotrophin hypothesis in psychiatric disorders: implications and limitations. Journal of Neural Transmission 2004;111(3):387‐411. [DOI] [PubMed] [Google Scholar]
Lim 2009
- Lim C‐M, Kim W‐W, Park J‐Y, Kim C, Yoon SH, Lee J‐K. Fluoxetine affords robust neuroprotection in the postischemic brain via its anti‐inflammatory effect. Journal of Neuroscience Research 2009;87(4):1037‐45. [DOI] [PubMed] [Google Scholar]
Loubinoux 1999
- Loubinoux I, Boulanouar K, Ranjeva J‐F, Carel C, Berry I, Rascol O, et al. Cerebral functional magnetic resonance imaging activation modulated by a single dose of the monoamine neurotransmission enhancers fluoxetine and fenozolone during hand sensorimotor tasks. Journal of Cerebral Blood Flow and Metabolism 1999;19(12):1365‐75. [DOI] [PubMed] [Google Scholar]
Mantel 1959
- Mantel N, Haenszel W. Statistical aspects of the analysis of data from retrospective studies of disease. Journal of the National Cancer Institute 1959;22(4):719‐748. [PubMed] [Google Scholar]
Ming 2005
- Ming GL, Song H. Adult neurogenesis in the mammalian central nervous system. Annual Review of Neuroscience 2005;28:223‐50. [DOI] [PubMed] [Google Scholar]
Molnar 2008
- Molnar FJ, Hutton B, Fergusson D. Does analysis using “last observation carried forward” introduce bias in dementia research?. CMAJ 2008;179(8):751–753. [DOI] [PMC free article] [PubMed] [Google Scholar]
Pälvimäki 1994
- Pälvimäki E‐P, Laakso A, Kuoppamäki M, Syvälahti E, Hietala J. Up‐regulation of beta l‐adrenergic receptors in rat brain after chronic citalopram and fluoxetine treatments. Psychopharmacology (Berl) 1994;115(4):543‐6. [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Santarelli 2003
- Santarelli L, Saxe M, Gross C, Surget A, Battaglia F, Dulawa S, et al. Requirement of hippocampal neurogenesis antidepressant treatments and animal models of depressive‐like behaviour. Behavioural Pharmacology 2003;301:805‐9. [DOI] [PubMed] [Google Scholar]
Schmidt 2007
- Schmidt HD, Duman RS. The role of neurotrophic factors in adult hippocampal neurogenesis, antidepressant treatments and animal models of depressive‐like behaviour. Behavioural Pharmacology 2007;18(5‐6):391‐418. [DOI] [PubMed] [Google Scholar]
Schünemann 2017
- Schünemann HJ, Oxman AD, Higgins JP, Vist GE, Glasziou P, Akl E, Guyatt GH on behalf of the Cochrane GRADEing Methods Group and the Cochrane Statistical Methods Group. Chapter 11: Completing ‘Summary of findings’ tables and grading the confidence in or quality of the evidence. In: Higgins JP, Churchill R, Chandler J, Cumpston MS, editor(s), Cochrane Handbook for Systematic Reviews of Interventions version 5.2.0 (updated June 2017). The Cochrane Collaboration, 2017. Available from www.training.cochrane.org/handbook.
Shin 2009
- Shin TK, Kang MS, Lee HY, Seo MS, Kim SG, Kim CD, et al. Fluoxetine and sertraline attenuate postischemic brain injury in mice. Korean Journal of Physiology and Pharmacology 2009;13(3):257‐63. [DOI] [PMC free article] [PubMed] [Google Scholar]
Sterne 2017
- Sterne JA, Egger M, Moher D, Boutron I, editor(s). . Chapter 10: Addressing reporting biases. In: Higgins JP, Churchill R, Chandler J, Cumpston MS, editor(s), Cochrane Handbook for Systematic Reviews of Interventions version 5.2.0 (updated June 2017). The Cochrane Collaboration, 2017. Available from www.training.cochrane.org/handbook.
Taupin 2006
- Taupin P. Adult neurogenesis and neuroplasticity. Restorative Neurology and Neuroscience 2006;24(1):9‐15. [PubMed] [Google Scholar]
Wan 2014
- Wan X, Wang W, Liu J, Tong T. Estimating the sample mean and standard deviation from the sample size, median, range and/or interquartile range. BMC Medical Research Methodology 2014;14:135. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wiltrout 2007
- Wiltrout C, Lang B, Yan Y, Dempsey RJ, Vemuganti R. Repairing brain after stroke: a review on post‐ischaemic neurogenesis. Neurochemistry International 2007;50(7‐8):1028‐41. [DOI] [PubMed] [Google Scholar]
Yi 2010
- Yi ZM, Liu F, Zhai SD. Fluoxetine for the prophylaxis of post‐stroke depression in patients with stroke: a meta‐analysis. International Journal of Clinical Practice 2010;64(9):1310‐7. [DOI] [PubMed] [Google Scholar]
Zittel 2008
- Zittel S, Weiller C, Liepert J. Citalopram improves dexterity in chronic stroke patients. Neurorehabilitation and Neural Repair 2008;22(3):311‐4. [DOI: 10.1177/1545968307312173] [DOI] [PubMed] [Google Scholar]
References to other published versions of this review
Mead 2011
- Mead GE, Hankey GJ, Kutlubaev MA, Lee R, Bailey M, Hackett ML. Selective serotonin reuptake inhibitors (SSRIs) for stroke. Cochrane Database of Systematic Reviews 2011, Issue 10. [DOI: 10.1002/14651858.CD009286] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mead 2012
- Mead GE, Hsieh CF, Lee R, Kutlubaev MA, Claxton A, Hankey GJ, et al. Selective serotonin reuptake inhibitors (SSRIs) for stroke recovery. Cochrane Database of Systematic Reviews 2012, Issue 11. [DOI: 10.1002/14651858.CD009286.pub2] [DOI] [PMC free article] [PubMed] [Google Scholar]