

Abstract
Background
Poliomyelitis mainly affects unvaccinated children under five years of age, causing irreversible paralysis or even death. The oral polio vaccine (OPV) contains live attenuated virus, which can, in rare cases, cause a paralysis known as vaccine‐associated paralytic polio (VAPP), and also vaccine‐derived polioviruses (VDPVs) due to acquired neurovirulence after prolonged duration of replication. The incidence of poliomyelitis caused by wild polio virus (WPV) has declined dramatically since the introduction of OPV and later the inactivated polio vaccine (IPV), however, the cases of paralysis linked to the OPV are currently more frequent than those related to the WPV. Therefore, in 2016, the World Health Organization (WHO) recommended at least one IPV dose preceding routine immunisation with OPV to reduce VAPPs and VDPVs until polio could be eradicated.
Objectives
To assess the effectiveness, safety, and immunogenicity of sequential IPV‐OPV immunisation schemes compared to either OPV or IPV alone.
Search methods
In May 2019 we searched CENTRAL, MEDLINE, Embase, 14 other databases, three trials registers and reports of adverse effects on four web sites. We also searched the references of identified studies, relevant reviews and contacted authors to identify additional references.
Selection criteria
Randomised controlled trials (RCTs), quasi‐RCTs, controlled before‐after studies, nationwide uncontrolled before‐after studies (UBAs), interrupted time series (ITS) and controlled ITS comparing sequential IPV‐OPV schedules (one or more IPV doses followed by one or more OPV doses) with IPV alone, OPV alone or non‐sequential IPV‐OPV combinations.
Data collection and analysis
We used standard methodological procedures expected by Cochrane.
Main results
We included 21 studies: 16 RCTs involving 6407 healthy infants (age range 96 to 975 days, mean 382 days), one ITS with 28,330 infants and four nationwide studies (two ITS, two UBA). Ten RCTs were conducted in high‐income countries; five in the USA, two in the UK, and one each in Chile, Israel, and Oman. The remaining six RCTs were conducted in middle‐income countries; China, Bangladesh, Guatemala, India, and Thailand. We rated all included RCTs at low or unclear risk of bias for randomisation domains, most at high or unclear risk of attrition bias, and half at high or unclear risk for conflict of interests. Almost all RCTs were at low risk for the remaining domains. ITSs and UBAs were mainly considered at low risk of bias for most domains.
IPV‐OPV versus OPV
It is uncertain if an IPV followed by OPV schedule is better than OPV alone at reducing the number of WPV cases (very low‐certainty evidence); however, it may reduce VAPP cases by 54% to 100% (three nationwide studies; low‐certainty evidence). There is little or no difference in vaccination coverage between IPV‐OPV and OPV‐only schedules (risk ratio (RR) 1.01, 95% confidence interval (CI) 0.96 to 1.06; 1 ITS study; low‐certainty evidence). Similarly, there is little or no difference between the two schedule types for the number of serious adverse events (SAEs) (RR 0.88, 95% CI 0.46 to 1.70; 4 studies, 1948 participants; low‐certainty evidence); or the number of people with protective humoral response P1 (moderate‐certainty evidence), P2 (for the most studied schedule; two IPV doses followed by OPV; low‐certainty evidence), and P3 (low‐certainty evidence).
Two IPV doses followed by bivalent OPV (IIbO) may reduce P2 neutralising antibodies compared to trivalent OPV (moderate‐certainty evidence), but may make little or no difference to P1 or P2 neutralising antibodies following an IIO schedule or OPV alone (low‐certainty evidence). Both IIO and IIbO schedules may increase P3 neutralising antibodies compared to OPV (moderate‐certainty evidence). It may also lead to lower mucosal immunity given increased faecal excretion of P1 (low‐certainty evidence), P2 and P3 (moderate‐certainty evidence) after OPV challenge.
IPV‐OPV versus IPV
It is uncertain if IPV‐OPV is more effective than IPV alone at reducing the number of WPV cases (very low‐certainty evidence). There were no data regarding VAPP cases. There is no clear evidence of a difference between IPV‐OPV and OPV schedules for the number of people with protective humoral response (low‐ and moderate‐certainty evidence). IPV‐OPV schedules may increase mean titres of P1 neutralising antibodies compared to OPV alone (low‐ and moderate‐certainty evidence), but the effect on P2 and P3 titres is not clear (very low‐ and moderate‐certainty evidence).
IPV‐OPV probably reduces the number of people with P3 poliovirus faecal excretion after OPV challenge with IIO and IIOO sequences (moderate‐certainty evidence), and may reduce the number with P2 (low‐certainty evidence), but not with P1 (very low‐certainty evidence). There may be little or no difference between the schedules in number of SAEs (RR 0.92, 95% CI 0.60 to 1.43; 2 studies, 1063 participants, low‐certainty evidence).
The number of persons with P2 protective humoral immunity and P2 neutralising antibodies are probably lower with most sequential schemes without P2 components (i.e. bOPV) than with trivalent OPV or IVP alone (moderate‐certainty evidence).
IPV (3)‐OPV versus IPV (2)‐OPV
One study (137 participants) showed no clear evidence of a difference between three IPV doses followed by OPV and two IPV doses followed by OPV, on the number of people with P1 (RR 0.98, 95% CI 0.93 to 1.03), P2 (RR 1.00, 95% CI 0.97 to 1.03), or P3 (RR 1.01, 95% CI 0.97 to 1.05) protective humoral and intestinal immunity; all moderate‐certainty evidence. This study did not report on any other outcomes.
Authors' conclusions
IPV‐OPV compared to OPV may reduce VAPPs without affecting vaccination coverage, safety or humoral response, except P2 with sequential schemes without P2 components, but increase poliovirus faecal excretion after OPV challenge for some polio serotypes. Compared to IPV‐only schedules, IPV‐OPV may have little or no difference on SAEs, probably has little or no effect on persons with protective humoral response, may increase neutralising antibodies, and probably reduces faecal excretion after OPV challenge of certain polio serotypes.
Using three IPV doses as part of a IPV‐OPV schedule does not appear to be better than two IPV doses for protective humoral response.
Sequential schedules during the transition from OPV to IPV‐only immunisation schedules seems a reasonable option aligned with current WHO recommendations. Findings could help decision‐makers to optimise polio vaccination policies, reducing inequities between countries.
Plain language summary
Sequential inactivated (IPV) and live oral (OPV) poliovirus vaccines for preventing poliomyelitis
Background to the question
Poliomyelitis (most commonly called Polio) mainly affects children under the age of five who have not been vaccinated against it. Polio causes permanent paralysis and even death. Polio can be prevented by vaccines, which provide defence against the disease (antibodies) in body fluids (also called humoral immunity) and also gut mucosal immunity. Polio‐related paralysis is caused by wild polio virus (WPV) and also in rare cases by the weakened live vaccine virus in the oral polio vaccine (OPV). The number of wild polio cases has gone down dramatically since the introduction and widespread use of the OPV and inactivated polio vaccine (IPV). However, the cases of paralysis linked to the OPV are currently more frequent than those related to the WPV. Since 2016 the World Health Organization has started to recommend that before a child is given the OPV immunisation they must have had at least one dose of the IPV mainly to limit the incidence of cases of paralysis linked to the OPV until polio is wiped out worldwide.
Review question
Are polio vaccine schedules that include both IPV and OPV as effective and safe as either OPV or IPV alone?
Study characteristics
We searched databases of scientific studies and found 21 studies to include in this review. Studies included 16 randomised trials with 6407 infants, one additional study followed 28330 infants over time and another four were nationwide studies.
Certainty of the evidence
We assessed the included evidence for how certain we are that the effects are true and would not be altered with the addition of more evidence. In general, the certainty of the evidence was judged to be low to moderate but it was very low for some outcomes.
Key results
IPV‐OPV compared to OPV may reduce the cases of paralysis linked to OPV by 54% to 100% without affecting vaccination coverage, the number of serious adverse events, and humoral immunity. However, it may worsen mucosal immunity for some types of polio.
IPV‐OPV compared to IPV may make little or no difference on serious adverse events, probably makes little or no difference in the number of persons with protective humoral immunity, may increase neutralising antibodies and probably improves intestinal mucosal immunity of vaccinated people.
Three doses of IPV followed by OPV appears to be no different than two IPV doses followed by OPV on the number of people with protective humoral and gut immunity.
Authors' conclusions
The main potential benefit of IPV‐OPV, compared to OPV, is that may reduce cases of paralysis linked to OPV. It could be a more affordable option to IPV during the final stages of polio eradication, hence reducing inequities between countries.
Summary of findings
Background
Please see the Glossary in Appendix 1 for a list of acronyms used in this review.
Description of the condition
Poliomyelitis is a communicable disease in humans that mainly affects unimmunised children under five years of age. Wild poliovirus (WPV), which has three strains (serotype 1, 2 and 3), causes paralysis. The paralysis is also caused, albeit rarely, by the oral polio vaccine (OPV), which involves the same three serotypes.
WPV spreads primarily by faecal‐to‐oral transmission in poor sanitary conditions. It can also spread through pharyngea‐to‐oral secretions. The virus enters the body through the oral and nasal cavities, replicates in the gastrointestinal tract, and is then shed, through faeces, into the environment. Initial symptoms of polio infection include fever, fatigue, headache, vomiting, stiffness in the neck, and pain in the limbs. One in 200 infections leads to irreversible paralysis. Five to 10 per cent of those paralysed die when their breathing muscles become immobilised (WHO 2015b). In the pre‐vaccination era, most cases of paralysis were caused by serotype 1.
Since the introduction of OPV, four of the six regions of the World Health Organization (WHO) have been certified free of WPV: the Americas in 1994; Western Pacific in 2000; Europe in 2002; and South East Asia in 2014. Approximately 80% of the world’s people now live in polio‐free areas (CDC 2015). Only two countries remain polio endemic in 2019 (Afghanistan and Pakistan), compared to three countries in 2018 (Afghanistan, Pakistan and Nigeria) and 125 countries in 1988 (GPEI 2018). The virus continues to circulate unabated in these less‐developed countries because of poor seroconversion rates among the recipients of OPV (Faden 1993), coupled with programmatic issues such as reaching vulnerable children in remote areas and ensuring sustainable financial and political support for the programmes. Failure to eradicate polio from these few countries could result in as many as 200,000 new cases every year, within 10 years, all over the world. Thus, as long as a single child remains infected, children in all countries are at risk of contracting polio.
While there is no cure for polio, polio vaccines can protect a child for life (WHO 2015b). However, continued use of OPV has been linked to vaccine‐associated paralytic poliomyelitis (VAPP) and vaccine‐derived polioviruses (VDPVs) (Platt 2014). VAPP is defined as an event of paralysis that occurs in a vaccinee between seven and 60 days after receiving a dose of OPV, with the neurological deficit remaining 60 days after onset. It is caused by the OPV virus. Using risk estimates of VAPP from previously published studies from different countries, Platt 2014 calculated the risk of VAPP as 4.7 cases per million births (range = 2.4 to 9.7 cases), leading to a global annual burden estimate of 498 cases (range = 255 to 1018 cases). When the analysis was restricted to estimates from countries that currently use OPV, the VAPP risk was 3.8 cases per million births (range = 2.9 to 4.7 cases) and a burden of 399 cases (range = 306 to 490) (Platt 2014). The incidence of recipient VAPP in different countries has been estimated to range from 0.33 to 19.08 cases per million births (Minor 2009).
VDPVs are Sabin viruses whose genetic sequence have mutated, diverging from the original OPV strain as a result of prolonged replication or transmission and reacquired neurovirulence. VDPV can be further subcategorised into circulating vaccine‐derived poliovirus (cVDPV), which circulates in populations that are seriously under‐vaccinated; immunodeficiency‐related vaccine‐derived poliovirus (iVDPV), which occurs in people who are unable to develop an immune response (i.e. those with rare immunodeficiency disorders); and ambiguous vaccine‐derived poliovirus (aVDPV), isolated cases of which very little is known (WHO 2015c). Unlike serotypes 1 and 3, serotype 2 continues to cause a number of cases of cVPDP (cVPDP‐2; 65 cases in 2013, 56 in 2014, 30 in 2015 and 5 in 2016), thus complicating the epidemiology of polio as well as vaccine selection and scheduling for supplementary immunisation activities in many countries (WHO 2017). Fewer than 90 cVDPV cases per year were reported to the WHO between 2000 and 2016, with the exception of the year 2009 when the number of cases peaked, mainly due to a large outbreak in Nigeria. Considering that time period, more than 94% of cVDPV cases and 66% of iVDPV cases identified since the introduction of OPV were due to serotype 2; cVDPV due to serotype 1 represented 4% but in 2015 to 2016 it went up to 66% (Jorba 2016). cVDPV‐2 is the most represented strain, accounting for 85.8% of all reported cases (Lopalco 2017). Using the WHO and United Nations databases, we conducted a proportion meta‐analysis (using the random‐effects model), to determine the global incidence of cases of cVDPV by WHO region and by polio vaccination scheme. Between 2000 and 2016, 798 cases of cVDPV were reported in 25 countries around the world. None of these cases occurred with the inactivated polio vaccine (IPV), and only two cases occurred with IPV‐OPV. The remaining cases (99.7%) occurred with OPV or OPV‐IPV, representing a combined annual incidence of 14 cVDPV/million (95% confidence interval (CI) 13 to 15), ranging annually from 3 to 26 cVDPV/million. Probably the most remarkable finding is that there is no evidence that cVDPV tends to disappear and it is virtually only associated with exclusive use of OPV (Ciapponi 2017).
Description of the intervention
Immunisation against poliovirus infection represents one of the world's greatest medical achievements. The incidence of poliomyelitis has declined dramatically since the introduction and widespread use of live oral polio vaccine (OPV) and inactivated polio vaccine (IPV). It is now recommended that all children should receive four doses of vaccine before entering school. Regimens of IPV only, OPV only, sequential IPV and OPV (IPV‐OPV), the inverse (OPV‐IPV) or simultaneous IPV + OPV are acceptable. Each regimen has advantages and disadvantages. In special circumstances, one of the regimens is preferred or recommended.
OPV contains a live, attenuated (weakened) vaccine‐virus (Sabin vaccine‐virus). Low‐ and middle‐income countries (LMICs) rely on OPV to control WPV transmission because of its low cost, ease of administration, as well as induction of mucosal (gut) and herd (i.e. the indirect protection of unvaccinated children) immunity that limit the spread of the virus. There are four different types of OPV: trivalent OPV (tOPV); monovalent OPV serotype 1 (mOPV1); monovalent OPV serotype 3 (mOPV3); and bivalent OPV (bOPV), which contains serotypes 1 and 3.
tOPV has a simplified immunisation schedule, and consequently, has been the vaccine of choice for routine immunisation and achieving global polio eradication. Three doses given at least two months apart are sufficient to develop an optimal immune response; antibody prevalence to all three serotypes approximates 96% after the third dose (McBean 1988), with 84% to 98% of those vaccinated showing detectable serum antibodies to all three serotypes five years after primary immunisation (Krugman 1977).
In developing countries, however, multiple doses of tOPV given at 6, 10, and 14 weeks of age, as recommended by the WHO Expanded Program for Immunization (EPI) (Sutter 2000), have been shown to produce active immunity in only a small proportion of infants. Following this three‐dose regimen, low seroconversion rates have been documented in many locations, averaging 73% for serotype 1, 90% for serotype 2 and 70% for serotype 3 (Patriarca 1991). Diarrhoeal disease and co‐infection with other enteroviruses at the time of immunisation are major factors that affect immunity following immunisation.
With the eradication of WPV serotype 2 by 1999, and continued circulation of WPV serotypes 1 and 3 in select geographic areas, bOPV was introduced in endemic areas. Like monovalent OPV (either serotype 1 or serotype 3), bOPV was developed to support global polio eradication efforts in resource‐limited settings (Cáceres 2001; Grassly 2006; Sutter 2010). Schemes containing bOPV are attractive approaches because these formulations, like monovalents formulations, have superior immunogenicity to tOPV for the corresponding serotype (Cáceres 2001; Grassly 2006; Sutter 2010), and without the interference from the serotype 2 component of the trivalent formulation, they may be more effective in treating outbreaks caused by a single WPV serotype (Grassly 2009).
IPV is an inactivated (dead) form of WPV, which is produced from the three serotypes. Since 1978, a new method of production resulted in higher potency per dose and significantly greater immunogenicity than the original IPV (Gold 1994). In this review, when we use the term IPV we are referring to enhanced potency IPV, because the original one is obsolete. Unlike OPV, IPV never causes VAPP, and hence is the vaccine of choice for routine immunisation in high‐income countries (HICs).
Sequential immunisation schedules of IPV‐OPV of at least three doses, starting with one or more doses of IPV and followed by one or more doses of OPV, could offer the same benefits of both vaccines and avoid the risks of VAPP and VDPV, which are associated with OPV alone.
The World Health Organization (WHO), the United Nations Children's Fund (UNICEF), the Rotary Foundation, the US Centers for Disease Control and Prevention (CDC) and The Gates Foundation have spearheaded the campaign to end polio through the Global Polio Eradication Initiative (GPEI), which helps to co‐ordinate vaccination campaigns worldwide, as well as environmental monitoring and evaluation of the cause of incident cases of paralysis associated to the wild or vaccinal poliovirus.
How the intervention might work
The introduction of IPV has stimulated the prospects of using IPV and OPV simultaneously. The combined vaccine schedule could reduce the frequency of VAPP and provide good mucosal immunity. Several studies have suggested that two doses of IPV followed by two doses of OPV provide excellent systemic and local immunity against polioviruses serotypes 1, 2, and 3 (Asturias 2007; Faden 1993; McBean 1988). At least two doses of IPV are necessary to induce more than 90% of protective antibodies against polioviruses before the first dose of OPV is administered. A non‐Cochrane systematic review found that IPV did not induce sufficient intestinal mucosal immunity to reduce the prevalence of faecal virus shedding after challenge with OPV, although there was some evidence that it could reduce the quantity of virus shed (Hird 2012).
Why it is important to do this review
In January 1997, the Advisory Committee on Immunization Practices (ACIP) recommended the adoption of a sequential IPV‐OPV immunisation schedule for the USA (CDC 1997). The schedule of IPV at two months and four months of age, followed by OPV at 12 to 18 months and again at four to six years, was intended to minimise the risk for VAPP while maintaining population immunity to the potential introduction of WPV. This switch to a schedule containing IPV represented one of the most significant changes to US vaccine policy (Faden 1993; Kew 2004). It resulted in considerable concern that parents might not want their children to receive numerous, simultaneous injections; that physicians might be reluctant to administer multiple injections at a single visit; and that the change to a sequential IPV‐OPV vaccination schedule would lead to reduced vaccination coverage of children. However, a study that followed children in two large, west coast, US health maintenance organisations (HMO) and evaluated a number of different measures of their immunisation coverage at one and two years of age showed that the changeover from an OPV‐only schedule to one containing IPV had little, if any, negative impact on vaccine coverage (Davis 2001).
In 2012, the Strategic Advisory Group of Experts on Immunization (WHO 2012), the body responsible for advising the WHO on global vaccination policy, recommended the replacement of tOPV with bOPV in all countries by 2016, preceded by the introduction of at least one dose of IPV in routine immunisation programmes. This schedule was implemented in April 2016 (PAHO 2017).
At present, 49 countries use sequential IPV‐OPV vaccination schedules (Table 4), and 17 countries have used it in the past (Table 5); 86 countries use an OPV‐IPV schedule; four countries use OPV + IPV; 51 countries use IPV exclusively; and five use OPV exclusively. Although the WHO strategy aims to stop OPV vaccination completely and replace it with IPV vaccination, this goal is not yet close, and the sequential IPV‐OPV vaccination schedule could have an important role during the transition. A systematic review of the evidence on sequential IPV‐OPV vaccination schedules could facilitate an evidence‐based, decision‐making process.
1. Countries currently using sequential IPV‐OPV schemes.
| Country | Year of introduction in entire country | IPV | OPV | ||
| Doses | Schedule | Doses | Schedule | ||
| Antigua and Barbuda | 2015 | 2 | 2, 4 months | 2 | 6, 18 months |
| Albania | 2014 | 2 | 2, 4 months | 3 | 6 months; 2, 6 years |
| Argentina | 2017 | 2 | 2, 4 months | 3 | 6, 15‐18 months; 6 years |
| Bahamas (the) | 2017 | 2 | 2, 4 months | 1 | 6 months |
| Bahrain | 2008 | 5 | 2, 4, 6, 18 months; 5 years | 4 | 4, 6, 18 months; 5 years |
| Barbados | 2017 | 6 | 2, 4, 6, 18 months; 4.5, 10‐11 years | 4 | 6, 18 months; 4.5, 10‐11 years |
| Belize | 2017 | 4 | 2, 4, 6, 4 years | 4 | 4, 6, 18 months; 4 years |
| Bolivia (Plurinational State of) | 2017 | 1 | 2 months | 4 | 4, 6, 18 months; 4 years |
| Bosnia and Herzegovina | 2008 | 4 | 2, 4, 5‐6 months; 5 years | 2 | 18 months; 14 years |
| Brazil | 2012 | 2 | 2, 4 months | 3 | 6, 15 months; 4 years |
| Chile | 2017 | 1 | 2 months | 2 | 6, 18 months |
| China | 2017 | 1 | 2 months | 3 | 3, 4, months; 4 years |
| Colombia | 2017 | 1 | 2 months | 3 | 6, 18 months; 5 years |
| Dominica | 2017 | 2 | 2, 4 months | 4 | 6, 18 months; 3, 10‐12 years |
| Dominican Republic (the) | 2017 | 1 | 2 months | 4 | 4, 6, 18 months; 4 years |
| Ecuador | 2017 | 1 | 2 months | 3 | 4, 6, > 1 year |
| Georgia | 2017 | 3 | 2, 3, 4 months | 5 | 5, 7, 18 months; 4‐5, ≥ 14 years |
| Grenada | 2015 | 1 | 2 months | 2 | 6, 18 months |
| Guatemala | 2017 | 1 | 2 months | 4 | 4, 6, 18 months; 4 years |
| Guyana | 2017 | 1 | 2 months | 4 | 4, 6, 18, months; 4 years |
| Haiti | 2017 | 1 | 1.5 months | 3 | 2.5, 3.5, 9 months |
| Honduras | 2017 | 1 | 2 months | 4 | 4, 6, 18 months; 4 years |
| Israel | 2017 | 4 | 2, 4, 6, 12 months | 2 | 6, 18 months |
| Jamaica | 2017 | 5 | 1.5, 3, 5‐6, 18 months; 4‐6 years | 5 | 3, 5‐6, 18 months; 4‐6 years |
| Jordan | 2005 | 3 | 3, 4, 5 months | 5 | 4, 5, 9, 18 months; 6 years |
| Kazakhstan | 2013 | 2 | 2, 4 months OR 3, 18 months | 1 | 12 months |
| Kuwait | 2010 | 1 | 2 months | 4 | 4, 6, 12, 18 months |
| Lebanon | 2012 | 1 | 2 months | 6 | 4, 6, 18 months; 4‐5, 10‐12, 16‐18 years |
| Mexico | 2008 | 4 | 2, 4, 6, 18 months | 2 | > 6 months; < 5 Years |
| Montenegro | 2011 | 3 | 9, 17, 23 weeks | 3 | 18 months; 6, 14 years |
| Nicaragua | 2017 | 1 | 2 months | 2 | 4, 6 months |
| Oman | 2010 | 1 | 9 weeks‐2 months | 5 | 4, 6, 18 months; 6, 18 years |
| Paraguay | 2017 | 1 | 2 months | 4 | 4, 6, 18 months; 4 years |
| Peru | 2013 | 2 | 2, 4 months | 3 | 6, 18 months; 4 years |
| Qatar | 2010 | 2 | 2, 4 months | 4 | 4, 6, 18 months; 4‐6 years |
| Russian Federation (the) | 2008 | 3 | 3, 4.5 months | 4 | 6, 18, 20 months; 14 years |
| Saint Kitts and Nevis | 2017 | 1 | 2 months | 5 | 4, 6, 18 months; 4.5‐5, 15 years |
| Saint Lucia | 2017 | 1 | 2 months | 5 | 4, 6, 18 months; 5, 11‐12 years |
| Saint Vincent and the Grenadines | 2017 | 2 | 2, 4 months | 2 | 6, 18 months |
| Saudi Arabia | 2008 | 3 | 2, 4, 6 months | 4 | 6, 12, 18 months; 6 years |
| Serbia | 2015 | 4 | 2, 4, 3.5, 18 months | 2 | 7, 14 years |
| Suriname | 2017 | 1 | 2 months | 4 | 4, 6, 18 months; 4 years |
| The former Yugoslav Republic of Macedonia | 2015 | 2 | 2‐3.5, 6‐18 months | 2 | 7, 14 years |
| Trinidad and Tobago | 2017 | 3 | 2, 3, 6 months | 4 | 4, 6, 18 months; 4‐5 years |
| Tunisia | 2014 | 2 | 2, 3 months | 5 | 6, 18 months; 6,12, 18 years |
| Turkey | 2008 | 5 | 2, 4, 6, 18 months; 6 years | 2 | 6, 18 months |
| Ukraine | 1959 | 2 | 3, 4 months | 4 | 5, 18 months; 6, 14 years |
| United Arab Emirates | 2014 | 1 | 2 months | 4 | 4, 6, 18 months; 5‐6 years |
| Venezuela (Bolivarian Republic of) | 2017 | 1 | 2 months | 4 | 4, 6, 18 months; 4‐5 years |
| IPV: inactivated poliovirus vaccine; OPV: oral poliovirus vaccine. | |||||
Sources: WHO vaccine‐preventable diseases: monitoring system. 2018 global summary, available at: apps.who.int/immunization_monitoring/globalsummary/schedules (accessed 6 June 2018) and www.who.int/entity/immunization/monitoring_surveillance/data/year_vaccine_introduction.xls?ua=1 (accessed 6 June 2018)
In summary, 86 countries use OPV‐IPV, 3 OPV + IPV, 51 exclusively IPV and 5 exclusively OPV.
The number of countries using use a single IPV dose varies according the scheme used: with IPV‐OPV scheme, 21/49 countries (43%) and with OPV‐IPV or OPV+IPV 82/89 (92%).
2. Countries that have used sequential IPV‐OPV schemes.
| Country | Schemes | Years |
| Belarus | 2 IPV, 3 OPV | 2014‐2016 |
| Bermuda | 2 IPV, 2 OPV | 2007 |
| Canada | 2 IPV, 1 OPV | 1978‐1979 |
| Cyprus | 2 IPV, 3 OPV | 2003‐2008 |
| Croatia | 1 IPV, 6 OPV | 2003‐2008 |
| Denmark | 3 IPV, 3 OPV | 1968‐1997 |
| Gaza | 1 OPV, 2 IPV/OPV, 2 OPV | 1978‐1988 |
| Hungary | 1 IPV, 5 OPV | 1992‐2006 |
| Italy | 2 IPV, 2 OPV | 1999‐2002 |
| Latvia | 3 IPV, 3 OPV | 2001‐2007 |
| Lithuania | 4 IPV, 2 OPV | 2004‐2007 |
| Malaysia | 4 IPV, 1 OPV | 2009‐2016 |
| Poland | 3 IPV, 1 OPV | 1958‐2016 |
| Romania | 1 IPV, 5 OPV | 2008‐2009 |
| Syrian Arab Republic (The) | 2 IPV, 4 OPV | 2008‐2016 |
| USA | 2 IPV, 1 OPV | 1997‐1999 |
| West Bank | 5 IPV, 2 OPV | 1978‐1988 |
| IPV: inactivated poliovirus vaccine; OPV: oral poliovirus vaccine. | ||
Objectives
To assess the effectiveness, safety, and immunogenicity of sequential inactivated poliovirus vaccine‐oral poliovirus vaccine (IPV‐OPV) immunisation schemes compared to either OPV or IPV alone.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs), quasi‐RCTS, controlled before‐and‐after studies (CBAs), uncontrolled before‐and‐after studies (UBAs), interrupted time series studies (ITSs) and controlled interrupted time series studies (CITSs) that meet the inclusion criteria listed in the EPOC 2011a data collection checklist (See Appendix 2).
For vaccine‐associated paralytic polio (VAPP) only, we also accepted nationwide UBA studies evaluating the impact of changing the vaccination policy to sequential IPV‐OPV vaccination schemes (See Appendix 2).
Types of participants
People entitled to receive IPV‐OPV vaccination schemes.
Types of interventions
Experimental intervention
Sequential IPV‐OPV schedule: one or more doses of IPV followed by one or more doses of OPV.
Comparator intervention
IPV alone.
OPV alone.
Non‐sequential combinations of IPV‐OPV.
Types of outcome measures
Primary outcomes
Paralytic polio, measured as change in level and change in slope by ITS.
Secondary outcomes
Vaccine‐associated paralytic polio (VAPP), measured as number of VAPPs or VAPP per million of OPV doses.
Vaccine‐derived poliovirus (VDPV) shedding in stool, measured as number of VDPVs or VDPV per million of OPV doses.
Protective immune responses, measured as risk ratio (RR) of protective humoral response and mean titres of neutralising antibody by serotype (humoral).
Intestinal immunity, measured as RR of polio faecal excretion after OPV challenge by serotype.
Vaccination coverage in children, measured as average proportion of vaccine coverage.
Safety, measured as RR of serious adverse events (SAEs), and proportion of SAEs.
Search methods for identification of studies
Electronic searches
We did not use any methods filters or limit the searches by date or language. We first searched the following databases in August 2014, We updated the searches in August 2015, July 2016, August 2018 and May 2019, Exact search dates for each search are reported in Appendix 3.
Cochrane Central Register of Controlled Trials (CENTRAL; 2019, Issue 5) in the Cochrane Library and which includes the Cochrane Developmental, Psychosocial and Learning Problems Specialised Register (searched 14 May 2019).
MEDLINE Ovid (1946 to April Week 3 2019).
Embase Ovid (1980 to 2019 Week 18).
Science Citation Index Web of Science (1970 to 14 May 2019).
Conference Proceedings Citation Index ‐ Science Web of Science (1990 to 23 July 2019).
Scopus Elsevier (searched 23 July 2019).
LILACS (Latin American and Caribbean Health Science Information database; regional.bvsalud.org/php/index.php?lang=en; searched 14 May 2019).
Cochrane Database of Systematic Reviews (CDSR; 2019, Issue 7), part of the Cochrane Library (searched 31 July 2019).
Database of Abstracts of Reviews of Effects (DARE; 2015, Issue 2, final issue of DARE), part of the Cochrane Library (searched 11 August 2015).
IndMED (Indian Medical Journals; indmed.nic.in; last searched 11 May 2018; access attempted 31 July 2019 but website could not be reached).
IBECS (Spanish Bibliographical Index in Health Sciences; regional.bvsaud.org/php/index.php?lang=en; searched 14 May 2019).
PAHO HQ Library Catalog (Pan American Health Organization Headquarters Library Catelogue; regional.bvsalud.org/php/index.php?lang=en; searched 2 June 2019).
WHOLIS (World Health Organization Library Information System; regional.bvsalud.org/php/index.php?lang=en; searched 23 July 2019).
IMSEAR (Index Medicus for South‐East Asia Region; www.globalindexmedicus.net/; searched 31 July 2019).
SciELO (Scientific Electronic Library Online; regional.bvsalud.org/php/index.php?lang=en; searched 1 June 2019).
African Index Medicus (AIM; indexmedicus.afro.who.int; searched 31 July 2019).
IMEMR (Index Medicus for the Eastern Mediterranean Region;www.globalindexmedicus.net/; searched 31 July 2019).
ICTRP (International Clinical Trials Registry Platform; apps.who.int/trialsearch; searched 31 July 2019).
ClinicalTrials.gov (clinicaltrials.gov; searched 2 June 2019).
ISRCTN registry (previously mRCT; www.isrctn.com/; searched 2 June 2019).
The search strategies for each database are reported in Appendix 4.
Searching other resources
We checked the reference lists of relevant studies, and contacted authors of included studies to identify any additional published or unpublished data. In addition, we searched for reports of adverse effects on the websites of the following organisations.
Current Problems in Pharmacovigilance (www.mhra.gov.uk).
Australian Adverse Drug Reactions Bulletin (from 1995 to 2009), which was replaced by the Advisory Committee on the Safety of Medicines (ACSOM) in January 2010.
European Public Assessment Reports from the European Medicines Evaluation Agency (www.emea.eu).
Food and Drug Administration (FDA) Medwatch (www.fda.gov/medwatch).
Data collection and analysis
We performed the systematic review following the guidance in the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011a). We report here only the methods that we were able to use. Methods that we had planned to use (Ciapponi 2014), but could not be performed, are reported in Table 6.
3. Planned methods that could not be performed.
| Section | Unused methods | Reason for nonuse |
| Measures of treatment effect |
Dichotomous data We planned to use the Peto odds ratio (OR) for very infrequent outcomes. We planned to report the risk difference (RD) if the result obtained by this measure was different from the RR. Continuous We planned to calculate the standardised mean difference (SMD) with 95% CI if the same outcomes were measured by different scales. |
It was not necessary. |
| Unit of analysis issues | For studies in which clusters of individuals were randomised (cluster‐RCTs, cluster quasi‐RCTs) or allocated (cluster CBAs) to intervention groups, but where inference is intended at the level of the individual, we planned to analyse them appropriately to account for correlation of observations within clusters. Standard statistical methods assume independence of observations, and their use in these types of studies will generally result in artificially small P values and overly narrow CIs for the effect estimates (Ukoumunne 1999). These studies can generally be re‐analysed by making assumptions about the intra‐cluster correlation (ICC). We planned to obtain estimates of the ICC by contacting study authors or imputing them from data presented in the study. Where this was not feasible, we planned to use external estimates from similar studies, if available (Campbell 2000). If this was not possible, we planned not to combine the findings of these studies in a meta‐analysis, but to present the results in an additional table. We also planned to combine the adjusted measures of effects of cluster‐RCTs with the results of non‐cluster trials, and to perform a Sensitivity analysis on meta‐analyses, including cluster‐RCTs, in which we would compare the effect estimates with and without the inclusion of the cluster trials. | We did not include cluster trials. |
| Dealing with missing data | We planned to impute missing dichotomous data through an intention‐to‐treat (ITT) analysis based on the available data (all participants analysed in the group to which they were allocated, whether they received the allocated intervention or not). We also planned to perform a Sensitivity analysis, assigning the worst outcome to those lost to follow‐up, to assess the impact of a 'worse case' scenario. | We did not impute dichotomous data. |
| Subgroup analysis and investigation of heterogeneity | We planned to conduct the following subgroup analyses.
|
We did not have enough data to perform these subgroup analyses. |
| Data synthesis | Where there were multiple, appropriate outcomes, we planned to selected the median effect. Where there were only two, we planned to selected the more conservative result. | This scenario did not arise. |
| We planned to use the generic inverse‐variance model of analysis if we needed to combine continuous and dichotomous data or clustered and non‐clustered data. | This scenario did not arise. | |
| We planned to estimate a regression coefficient (with its standard error) that describes the effect of the interventions. We planned to standardise the direction of effect (for example, positive or negative) so that a negative coefficient planned to describe an improvement in outcome attributable to the intervention. | We performed an ARIMA analysis. | |
It may not be appropriate to combine the results quantitatively when the assessed outcomes, settings, and interventions are diverse. For these results, we planned to provide a descriptive summary of the data using one of the following methods:
|
In these cases, we presented the results of the subgroup analysis. | |
| Sensitivity analysis | We planned to conduct sensitivity analyses based on the following characteristics.
We also planned to conduct additional sensitivity analyses:
|
We could not perform the first analysis because no study was at high risk of bias for allocation concealment. We did not include cluster trials so did not perform the third analysis. The last three scenarios did not arise. |
ARIMA: autoregressive integrated moving average; CBAs: controlled before and after study; CI: confidence interval; RCT: randomised controlled trial.
Selection of studies
Pairs of review authors independently considered studies for possible inclusion using EROS (Early Review Organizing Software) (Glujovsky 2011; Ciapponi 2011; Glujovsky 2010). First, the review authors independently screened the titles and abstracts of all records identified by the searches, discarding any that were clearly irrelevant. Second, they independently examined the full texts of all potentially relevant records, or those for which more information was needed to determine eligibility, to identify those that met the inclusion criteria (Criteria for considering studies for this review). Any disagreements were discussed amongst all review authors until a consensus was reached. We presented our selection process in a PRISMA diagram (Liberati 2009; Moher 2009).
Data extraction and management
We used an adapted, electronic version of a data collection checklist developed by Cochrane Effective Practice and Organisation of Care (EPOC 2011a). We piloted the form prior to use.
Pairs of review authors independently extracted descriptive, risk of bias, and numerical outcome data from each study using EROS. For descriptive data, a review author extracted the data onto a Google spreadsheet, which a second review author verified. Discrepancies were resolved by discussion with the entire team. We collected the following information from each study.
Study references
Name of author(s) and year of publication
Start and stop dates for study
Location of the study (for example, country, region or district, and city)
Study characteristics
Study design (RCT, quasi‐RCT, CBA, UBA, ITS and CITS)
Intervention and comparisons: description and characteristics
Number and type of vaccines
OPV poliovirus types
Timing (frequency of intervention, and duration of intervention)
Schedule of IPV (I) or OPV (O) sequence (for example, IOO, IOOO, IIO, IIOO)
Setting of the intervention (rural or urban)
Participants
Age
Sex
Socioeconomic status
Baseline health problems
In addition, for quasi‐RCTs, we also recorded whether the study restricted participant selection, or demonstrated balance or matching between intervention and control groups on possible confounders (such as age, sex), or both.
Methods
Unit of allocation
Unit of analysis
-
Criteria for assessment of risk of bias for:
RCTs and quasi‐RCTs (random sequence generation, allocation concealment, blinding of participants and personnel, blinding of outcome assessment, incomplete outcome data, selective reporting, and other bias);
CBA studies (baseline measurement, characteristics of studies using second site as control, blinded assessment of primary outcome(s)* (protection against detection bias), protection against contamination (studies using second site as control), reliable primary outcome measure(s), follow‐up of professionals (protection against exclusion bias), and follow‐up of participants); and
Interrupted time series (ITS) studies (protection against secular changes (intervention is independent of other changes, data were analysed appropriately, reason for the number of points pre‐ and postintervention given, and shape of the intervention effect was specified), and protection against detection bias (intervention unlikely to affect data collection, blinded assessment of primary outcome(s), completeness of data set, reliable primary outcome measure(s)).
Outcomes
Primary outcome (paralytic polio)
Secondary outcomes (VAPP, VDPV shedding in stool, protective immune responses (humoral and intestinal immunity), vaccination coverage of children, and safety)
Assessment of risk of bias in included studies
In EROS, pairs of review authors independently assessed the risk of bias in each included study using the ‘Risk of bias’ tool, described in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2011b) for RCTs, with additional criteria developed by EPOC for non‐clinical studies (EPOC 2017).
We assessed the risk of bias as described below. All review authors discussed any disagreements until they reached a consensus.
We present the results of our 'Risk of bias' assessment in the 'Risk of bias' tables (beneath the Characteristics of included studies tables). We generated a 'Risk of bias' summary graph using RevMan 5 (Review Manager 2014). We considered our 'Risk of bias' judgements when evaluating study limitations while preparing 'Summary of findings’ tables when assessing the overall certainty of the evidence for each outcome (Guyatt 2011; Hultcrantz 2017).
RCTs
Using the criteria set out in Appendix 5, we assessed the risk of bias in RCTs as low, high or unclear, across each of the following domains from the Cochrane ‘Risk of bias’ tool: sequence generation; allocation concealment; blinding of participants and personnel; blinding of outcome assessors; incomplete outcome data; selective outcome reporting; and other potential threats to validity (Higgins 2011b). Where we scored a study at unclear risk of bias, one review author attempted to obtain further information from the authors of the trial.
CBA and UBA studies
For CBAs, we used the following criteria: baseline measurement; characteristics for studies using the second site as control; blinded assessment of primary outcome(s); reliable primary outcome measure(s); follow‐up of professionals (protection against exclusion bias); and follow‐up of patients.
For UBAs, we used the same criteria, with the exception of baseline measurement and characteristics for studies using the second site as control.
For each included study, we rated the risk of bias in each domain as low, high or unclear using the criteria set out in Appendix 6,
ITS and CITS studies
For ITS studies, we assessed the risk of bias associated with the following seven domains: intervention independent of other changes; shape of intervention effect pre‐specified; intervention unlikely to affect data collection; blinding of outcome assessors to intervention allocation; incomplete outcome data; selective outcome reporting; and other sources of bias (EPOC 2017). For each included study, we rated the risk of bias in each domain as low, high or unclear using the criteria set out in Appendix 6,
As for CBAs, for CITS studies we included three additional domains that assess design‐specific threats to validity: imbalance of outcome measures at baseline; comparability of intervention and control group characteristics at baseline; and protection against contamination (EPOC 2017). We rated the risk of bias in each domain as low, high or unclear using the criteria set out in Appendix 6.
Measures of treatment effect
RCTs
Dichotomous outcomes
We calculated the risk ratio (RR) with 95% confidence intervals (CIs).
Continuous outcomes
We calculated the mean difference (MD) with 95% CI when the same outcomes were measured using similar scales (such as immune responses).
CBA and UBA studies
For continuous variables, we reported, if possible, the relative change, adjusted for baseline differences in the outcome measures; that is, the absolute post‐intervention difference between the intervention and control groups minus the absolute pre‐intervention difference between the intervention and control groups divided by the post‐intervention level in the control group. We re‐analysed CBA studies as ITS studies, if possible, using the methods described below.
ITS and CITS studies
The most basic approach is to graph the time series, and look for trends and patterns. We present the results for outcomes as changes along two dimensions: 'change in level' and 'change in slope'.
'Change in level' is the immediate effect of the policy. It is measured as the difference between the fitted values for the first post‐intervention data point (one month after the intervention) minus the predicted outcome one month after the intervention based on the pre‐intervention slope only. We calculated the relative change in level by dividing the change in level by the predicted outcome one month after the intervention based on the pre‐intervention slope only and then multiplying by 100%.
'Change in slope' is the change in the trend from pre‐ to post‐intervention that reflects the long‐term effect of the intervention. Since the interpretation of change in slope can be difficult, we presented the long‐term effects in a similar way to how we calculated and presented the relative immediate effects.
Unit of analysis issues
We did not included cluster‐trials (see Table 6 for planned methods to address this issue should they arise in future updates of this review).
Dealing with missing data
If information was missing or unclear, we contacted the study investigators for additional information or clarification. To reduce the risk of overly positive answers, we used open‐ended questions (as recommended in Chapter 16 the Cochrane Handbook for Systematic Reviews of Interventions; Higgins 2011c).
We imputed missing continuous data when necessary (calculating standard deviations from standard errors or using standard deviations from other studies).
Assessment of heterogeneity
Statistical heterogeneity exists if the observed intervention effects are more different from each other than one would expect due to random error (chance) alone. We obtained an initial visual overview of heterogeneity through scrutinising the forest plots and looking at the overlap between CIs around the estimate for each included study. To quantify the inconsistency across studies, and thus the impact of heterogeneity on the meta‐analysis, we used the I2 statistic to detect heterogeneity (Higgins 2003). In the latter case, we defined an I2 of > 50% as revealing substantial heterogeneity. We also interpreted the significance of the I2 test in light of (i) the magnitude and direction of effects and (ii) the strength of evidence for heterogeneity (for example, a CI for the I2, or the P value for the Chi2 test).
We assessed observable heterogeneity among the study questions and methods, to determine whether a meta‐analysis was appropriate. We also looked at the study participants, settings, interventions, and reported outcomes. We paid particular attention to the homogeneity of the methodology (such as variances in blinding and concealment of allocation) within and across included studies.
If we found evidence of statistical heterogeneity, we examined it in a subgroup analysis and a sensitivity analysis, as outlined in the respective sections below (Subgroup analysis and investigation of heterogeneity; Sensitivity analysis).
Assessment of reporting biases
To reduce possible publication bias, we employed strategies to search for and include relevant unpublished studies. These strategies included searching the grey literature and prospective trial registration databases to overcome time‐lag bias.
To investigate the likelihood of overt publication bias, when we found more than eight studies of different sizes we drew a funnel plot, plotting trial effects against inverse standard errors of the effects. Funnel plot asymmetry is found through 'eyeballing' the funnel plot. We recognise that the funnel plot is not the most reliable method of investigating reporting biases, since asymmetry can also result from other sources of selection bias (delayed publication, location biases, selective outcome reporting), and methodological issues leading to spuriously inflated effects in smaller studies, true heterogeneity, artefactual, and chance (Sterne 2011).
Data synthesis
For each comparison, we reported summary statistics for each of the included studies (RCTs, quasi‐RCTs, CBAs, UBAs, and controlled or non‐controlled ITSs). We used forest plots to display the data graphically.
For dichotomous data, we used the Mantel‐Haenszel method, and for continuous data, we used the inverse variance method.
We pooled the results from individual studies in a meta‐analysis using the random‐effects model by DerSimonian and Laird (DerSimonian 1986). We chose this method because we could not assume a single, underlying (fixed) treatment effect. When the impact of the intervention was assessed in individual studies on more than one outcome measure, we selected the outcome that best reflects the targeted intervention for pooling data.
We analysed ITS and UBAs studies separately to RCTs.
We analysed ITS data using the guidelines of the EPOC group (EPOC 2011b; EPOC 2011c), and reported outcomes in natural units. We reported pre‐intervention and post‐intervention means or proportions for both study and control groups, and calculated the unadjusted and adjusted (for any baseline imbalance) absolute change from baseline with 95% CIS. We used either a regression analysis with time trends before and after the intervention, which adjust for autocorrelation and any periodic changes, or an autoregressive, integrated, moving average (ARIMA) model to isolate the effect of the intervention from existing time trends (McCain 1979).
Subgroup analysis and investigation of heterogeneity
We performed the following subgroup analyses, where possible, to check if the intervention effect varied with different populations, interventions, or settings.
Timing of the first dose (at birth or at two months).
Type of dose sequence (IOO, IOOO, IIO, or IIOO).
Country (according to the current World Bank classification).
When we were not able to perform a meta‐analysis, we summarised the results for these subgroups within the text of the review.
We also used an I2 of > 50% to test for subgroup differences, since substantial heterogeneity would suggest differential interventions effects.
Due to a lack of studies, we were unable to conduct other preplanned subgroup analyses (Ciapponi 2014), which can be found in Table 6.
Sensitivity analysis
We performed a sensitivity analyses based on the following characteristic.
Method of meta‐analysis: we compared the results from the random‐effects and fixed‐effect models if there was unexplained heterogeneity between studies, to assess the robustness of the results.
Due to a lack of studies, we were unable to conduct our other preplanned analyses (Ciapponi 2014), which can be found in Table 6.
We conducted a post hoc sensitivity analysis of studies at low risk of bias for allocation concealment for persons with P1 protective humoral response.
Summarising and interpreting results
We imported data from RevMan 5 (Review Manager 2014) to GRADE profiler (GRADEpro GDT), and created 'Summary of findings' tables for the following comparisons:
IPV‐OPV compared to OPV alone;
IPV‐OPV compared to IPV alone; and
IPV(3)‐OPV compared to IPV(2)‐OPV.
The critical/important outcomes reported in all thee 'Summary of findings' tables are:
paralytic polio;
VAPP cases;
persons with protective humoral response;
neutralising antibodies;
persons with faecal excretion after OPV challenge;
vaccination coverage; and
serious adverse events.
We grouped, analysed and presented the results according to serotypes P1, P2 and P3 protective humoral and intestinal response (outcomes 3, 4, and 5), because the effect is partially independent of each other, and in that sense, many policies, like the replacement of tOPV by bOPV, are serotype specific. If we were not able to pool the data in a meta‐analysis due to considerable heterogeneity, we presented the scheme of two IPV doses (IIO) as the main subgroup for this outcome, since it is the most studied scheme.
Pairs of review authors independently graded the certainty of the evidence for each outcome using the GRADE approach (Guyatt 2011; Hultcrantz 2017; Schünemann 2011b, Schünemann 2013); discrepancies were resolved by consensus. For assessments of the overall certainty of the evidence for outcomes that included pooled data from RCTs, we initially graded the evidence as high certainty, downgrading the rating (by one level from high to moderate certainty, by two levels to low certainty, or three levels to very low‐certainty evidence) depending on the extent of accomplishment across the following criteria: study limitations (risk of bias); indirectness of evidence; inconsistency; imprecision of effect estimates; or publication bias. For certainty ratings for outcomes that included pooled data from ITS, CITS, CBA and UBA studies, we initially graded the evidence from as low certainty, upgrading the rating to moderate or high certainty if the pooled estimates revealed a large magnitude of effect, negligible concerns about confounders, or a strong dose‐response gradient. We used these assessments, along with the evidence (or lack thereof) for absolute benefit or harm of the interventions, and the sum of available data on all critical and important outcomes from each study included for each comparison, to draw conclusions about the effectiveness of sequential inactivated (IPV) and live oral (OPV) poliovirus vaccines for preventing poliomyelitis.
Results
Description of studies
Results of the search
We retrieved a total of 6960 records (6958 database records and 2 from other sources) and eliminated 3058 duplicates, We screened the remaining 3902 titles and abstracts, and from these, selected 205 full texts for further screening. We excluded 175 full‐text reports. The majority of these involved an ineligible intervention (n = 98) or ineligible study design (n = 44) and consequently are not described in the Excluded studies section. We do, however present the details of six full‐text reports that we excluded for less obvious reasons in the Excluded studies section.
We included 21 studies (from 27 reports; we considered the six additional reports as secondary references of Faden 1990; Ivanova 2018; O'Ryan 2015; Rennels 2000) in this review. See Included studies section for more information. We also identified three ongoing studies (NCT02412514; NCT03430349; NCT03614702).
See Figure 1.
1.

Study flow diagram.
Included studies
We included 16 RCTs (Anand 2015; Asturias 2007; Faden 1990; Halsey 1997; Jain 1997; Li 2016a; Linder 2000; Modlin 1997; O'Ryan 2015; Qiu 2017; Ramsay 1994; Rennels 2000; Simasathien 1994; Sutter 1997; West 2001; Yeh 2001), three ITS (Alexander 2004; Davis 2001; Von Magnus 1984), and two UBA (Ivanova 2018; Kapusinszky 2010). See Characteristics of included studies tables.
RCTs
Setting
The majority of RCTs (10/16) were conducted in high‐income countries: five in the USA (Faden 1990; Halsey 1997; Modlin 1997; West 2001; Yeh 2001); two in the UK (Ramsay 1994; Rennels 2000); and one apiece in Chile (O'Ryan 2015); Israel (Jain 1997); and Oman (Sutter 1997). The six remaining studies were conducted in middle‐income countries: two in China (Li 2016a; Qiu 2017); and one apiece in Bangladesh (Anand 2015), Guatemala (Asturias 2007), India (Linder 2000), and Thailand (Simasathien 1994).
The RCTs were published between 1990 and 2017, with only six of them published within the last 10 years (Anand 2015; Ivanova 2018; Kapusinszky 2010; Li 2016a; O'Ryan 2015; Qiu 2017). There date of patient recruitments were from 1986 (Faden 1990) to 2015 (Qiu 2017).
Participants and sample sizes
The 16 RCTs involved 6407 healthy infants (mean age = 382 days, range = 96 to 975 days, median = 365 days).
Comparisons
The RCTs compared a sequential IPV‐OPV schedule (one or more doses of IPV followed by one or more doses of OPV) with IPV alone (O'Ryan 2015; Rennels 2000), OPV alone (Li 2016a; Ramsay 1994; Simasathien 1994; Sutter 1997; West 2001), both IPV and OPV (Anand 2015; Asturias 2007; Faden 1990; Jain 1997; Modlin 1997; Qiu 2017; Yeh 2001), IPV and IPV + OPV (Halsey 1997), or different schemes of IPV‐OPV (Linder 2000).
Outcomes
All RCTs assessed serological immune response for each serotype as geometric mean antibody titres and protective humoral response (≥ 1:8 dilutions) from completing the vaccination schedule to 18 months. Faden 1990 also assessed mean antibody titres at five years. Five RCTs assessed poliovirus detection in stool or gastrointestinal mucosal immunity (Asturias 2007; Faden 1990; Modlin 1997; O'Ryan 2015; Ramsay 1994), and seven reported on safety outcomes (Asturias 2007; Li 2016a; O'Ryan 2015; Qiu 2017; Rennels 2000; West 2001; Yeh 2001).
ITS and UBA studies
We included two ITS studies (Davis 2001; Von Magnus 1984), two UBA studies (Ivanova 2018; Kapusinszky 2010), and one study that used a mixed design (ITS + UBA analysis for VAPP cases) (Alexander 2004).
Setting
Most (4/5) studies were conducted in high‐income countries: two in the USA (Alexander 2004; Davis 2001); one in Denmark (Von Magnus 1984); and one in Hungary (Kapusinszky 2010). The fifth study was conducted in the Russian Federation (Ivanova 2018); a middle‐income country.
Participants and sample sizes
Four studies were conducted nationwide (Alexander 2004; Ivanova 2018; Kapusinszky 2010; Von Magnus 1984). The other study, Davis 2001, was conducted in two large, health maintenance organizations (HMO) in the USA and involved 28,330 infants aged one year old.
Comparisons
All but one study compared a sequential IPV‐OPV schedule with OPV. The exception was Von Magnus 1984, who compared an IPV‐OPV schedule with IPV.
Davis 2001 used an OPV‐only schedule in quarter four of 1995 to quarter three of 1996 (at ages two, four and 12 to 18 months and at four to six years). This changed to an IPV‐OPV schedule in quarter four of 1996 to quarter four of 1997 (IPV at ages two to four months, OPV at ages 12 to 18 months and at four to six years).
Outcomes
Both UBA studies (Ivanova 2018; Kapusinszky 2010) and the mixed‐design study (Alexander 2004) assessed the frequency of VAPP cases. Of the two ITS studies, one assessed the frequency of paralytic polio cases, percentage of persons with antibodies to type 1, 2 and 3 poliovirus, and acceptance rate (Von Magnus 1984). The other assessed immunisation status, immunisation up‐to‐date status and total number of missed‐opportunity visits (Davis 2001).
Excluded studies
We excluded six studies because they did not meet the inclusion criteria for this review (Criteria for considering studies for this review); four because they used an ineligible study design (McCollough 1969; Moǐseieva 2002; Swartz 1998; Wattigney 2001), and two because they used an ineligible intervention (Li 2016b; Ye 2018). See Characteristics of excluded studies tables.
There are no studies awaiting classification.
Ongoing studies
We identified three ongoing studies, which we describe in full in the Characteristics of ongoing studies tables. In brief:
NCT02412514: (I + O)OO (IPV + bOPV, bOPV and bOPV) versus IOO (IPV, bOPV and bOPV);
NCT03430349: IPV followed by novel OPV2 versus IPV followed by another novel OPV2; and
NCT03614702: 12 arms of one or two doses of cIPV/sIPV + one dose of tOPV versus one or two doses of cIPV/sIPV + one dose of bOPV.
Risk of bias in included studies
We assessed the risk of bias for RCTs separately to that of the ITS and UBA studies. We provide a summary of the results of our assessment below and graphically in Figure 2 and Figure 3. Further details can be found in the 'Risk of bias' tables (beneath the Characteristics of included studies tables).
2.

'Risk of bias' graph by experimental / quasi‐experimental design: review authors' judgements about each risk of bias item presented as percentages across all studies
3.

A summary table of review authors' judgements for each 'Risk of bias' item for each study
RCTs
Allocation (selection bias)
Randon sequence generation
We rated six studies at low risk of bias (Asturias 2007; Linder 2000; Modlin 1997; O'Ryan 2015; Qiu 2017; Ramsay 1994). We considered the 10 remaining studies to be at unclear risk of bias because they did not provide a complete description of the random sequence generation process.
Allocation concealment
We judged four studies at low risk of bias (Asturias 2007; Linder 2000; Qiu 2017; Simasathien 1994). We rated the 12 remaining studies at unclear risk of bias because they did not describe allocation concealment or the description was incomplete (Anand 2015; Faden 1990; Halsey 1997; Jain 1997; Li 2016a; Modlin 1997; O'Ryan 2015; Ramsay 1994; Rennels 2000; Sutter 1997; West 2001; Yeh 2001).
Blinding (performance bias and detection bias)
Blinding of participants and personnel
We rated all studies at low risk of performance bias since it is unlikely that the lack of blinding would have influenced the way in which the immunisation schedule was delivered.
Blinding of outcome assessment
We rated all studies at low risk of detection bias. Although the outcome assessment was not blinded, it is unlikely that objective outcomes (like antibody titres) would have been influenced by the lack of blinding.
Incomplete outcome data (attrition bias)
We judged four studies at low risk of attrition bias (Halsey 1997; Li 2016a; Linder 2000; Rennels 2000). We judged a further four studies at high risk of attrition bias because of attrition in the short term (from 14% to 30%) and long term (from 45% to 69%) (Faden 1990; Jain 1997; Modlin 1997; Yeh 2001). We rated the eight remaining studies at unclear risk of attrition bias because the description of attrition was incomplete, although we consider it unlikely to impact considerably on the results, since most outcomes had early presentation.
Selective reporting (reporting bias)
We considered all studies to be at of low risk of reporting bias.
We assessed reporting bias through visual inspection of funnel plots when there was more than eight studies included in a meta‐analysis. We found no significant asymmetries for the number of people with protective humoral response P1, P2 and P3 for the comparison IPV‐OPV versus OPV (Figure 4), or for the number of people with protective humoral response P1 for the comparison IPV‐OPV versus IPV (Figure 5).
4.
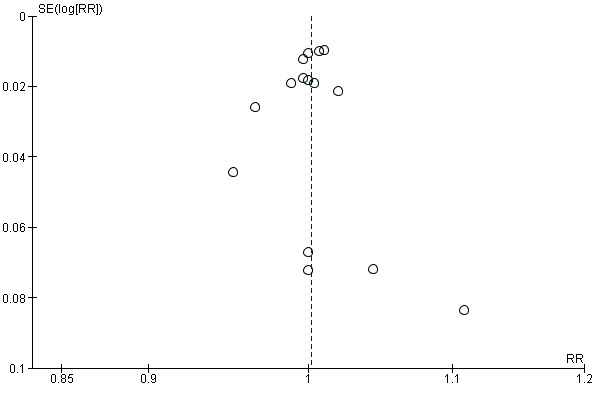
Funnel plot of comparison: 1 IPV‐OPV vs OPV, outcome: 1.1 Persons with P1 Protective humoral response.
5.

Funnel plot of comparison: 2 IPV‐OPV vs IPV, outcome: 2.1 Persons with P1 Protective humoral response.
Other potential sources of bias
All trials seems to be free of other potential sources of bias
Conflict of interest
Seven studies received no industry funding and we rated these studies at low risk of bias (Anand 2015; Li 2016a; O'Ryan 2015; Qiu 2017; Ramsay 1994; Simasathien 1994; Sutter 1997). We rated one study at high risk of bias because the director of the study was also the medical director of the sponsor (Li 2016a). We considered the eight remaining studies to be unclear risk of bias; six because they declared industry funding (Asturias 2007; Faden 1990; Halsey 1997; Rennels 2000; West 2001; Yeh 2001), and two because they did not describe funding (Jain 1997; Modlin 1997). There were no other potential sources of bias.
ITS and UBA studies
Protection against secular changes
Intervention is independent of other changes
We rated three studies at low risk of bias on this domain since the intervention appears to occur independently of other changes over time (Alexander 2004; Davis 2001; Von Magnus 1984).
Data were analysed appropriately
We considered two studies to be at unclear risk of bias on this domain because we re‐analysed the data as an ITS (Alexander 2004; Davis 2001).
We considered one study to be at high risk of bias because the available data did not allow us to re‐analyse it (Von Magnus 1984).
Reason for the number of points pre‐ and post‐intervention given
We rated three studies at low risk of bias on this domain because they provided many points pre‐ and post‐intervention (Alexander 2004; Davis 2001; Von Magnus 1984).
Shape of the intervention effect was specified
We rated the mixed study by Alexander 2004 at low risk of bias. We rated two studies at unclear risk of bias because the data were not designed as an ITS (Davis 2001, which we re‐analysed; and Von Magnus 1984, which did not specify the shape).
Protection against detection bias
Blinded assessment of primary outcome(s)
We rated three studies at low risk of bias because they used objective outcomes that are not likely to be influenced by the lack of blinding (Alexander 2004; Davis 2001; Von Magnus 1984).
We considered two studies to be at unclear risk of bias because although acute flaccid paralysis (AFP) and virological testing are objective outcomes, VAPP requires interpretation and the method by which cases were identified was not clearly stated (Ivanova 2018; Kapusinszky 2010).
Reliable primary outcome measure(s)
We rated all five studies at low risk of bias on this domain; three because the outcomes were obtained using an automated system (Alexander 2004; Davis 2001; Von Magnus 1984), and two because the surveillance systems had data quality controls (Ivanova 2018; Kapusinszky 2010).
Intervention unlikely to affect data collection
We rated two studies at low risk of bias because data collection methods were the same before and after the intervention (Alexander 2004; Davis 2001), and one study at unclear risk of bias because this information was not reported (Von Magnus 1984).
Completeness of data set
We rated one study at low risk of bias (Alexander 2004). We judged two studies to be at unclear risk of bias because they used national databases (Davis 2001; Von Magnus 1984).
Protection against exclusion bias
Follow‐up of professionals
We rated three studies at low risk of bias on this domain since national AFP surveillance implies a complete nationwide follow‐up; the follow‐up period was long enough; and no changes to protocols were reported (Alexander 2004; Ivanova 2018; Kapusinszky 2010).
Follow‐up of patients
We rated three studies at low risk of bias on this domain, since national AFP surveillance implies a complete nationwide follow‐up; the follow‐up period was long enough; and no changes to protocols were reported (Alexander 2004; Ivanova 2018; Kapusinszky 2010).
Conflict of interest
No study received industry funding, therefore, we rated all five studies at low risk of bias on this domain.
Effects of interventions
See: Table 1; Table 2; Table 3
Summary of findings for the main comparison. IPV‐OPV compared to OPV for preventing poliomyelitis.
| IPV‐OPV compared to OPV for preventing poliomyelitis | ||||||
| Patient or population: infants Setting: USA, UK, China, Thailand, Israel, Oman, Guatemala, Bangladesh Intervention: IPV‐OPV Comparison: OPV | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with OPV | Risk with IPV‐OPV | |||||
|
Paralytic wild polio cases (Change in slope at 3 years) |
−0.2 (−1.3 to 0.9) |
−21% (−137% to 95%) |
(1 ITS) | ⊕⊝⊝⊝ Very lowa | Re‐analysis considering the year of schedule change as transition period | |
|
Vaccine‐associated paralytic polio (VAPP) cases (Range of follow‐up from 2 to 15 years) |
6.6 cases per year | 3.0 cases per year | −54.3% | (1 USA nationwide UBA study) | ⊕⊕⊝⊝ Lowb |
OPV (1990‐6): 46 cases (0.34 cases per million of OPV doses) IPV‐OPV (1997‐9): 13 cases, none with IPV‐OPV or all‐IPV schedules |
| 7.64 cases per million newborns | 1.56 cases per million newborns | −80% | (1 Russian Federation nationwide UBA study) |
OPV (1998‐2007): 1 VAPP case per 1.59 million OPV doses IPV‐OPV (2008‐14): 1 case per 4.18 million doses |
||
| 90 cases | 0 case | −100% | (1 Hungary nationwide UBA study) |
OPV (1959‐92): 90 cases IPV‐OPV (1992‐2006): 0 cases |
||
|
Persons with protective humoral response (Range of follow‐up from 4.4 to 18 months) |
P1: 973 per 1000 | 973 per 1000 (963 to 982) | RR 1.00 (0.99 to 1.01) | 3189 (12 RCTs) |
⊕⊕⊕⊝ Moderatea | No important differences with other vaccination schemes |
| P2: 989 per 1000 #subgroup with 2 IPV doses | 989 per 1000 (979 to 999) | RR 1.00 (0.99 to 1.01) | 2361 (11 RCTs) | ⊕⊕⊝⊝ Lowa,d |
IIbO: 0 (−70 to + 70) persons IbObO vs tOPV: −210 (−38 to −344) persons IbObO vs bOPV: + 672 (± 428 to + 1018) persons |
|
| P3: 962 per 1000 | 953 per 1000 (933 to 972) | RR 0.99 (0.97 to 1.00) | 3184 (12 RCTs) |
IbObO: −300 (−180 to −400) persons IOO / IOOO: −19 (−39 to 0) persons IIO/IIOO/IIIO: −10 (−39 to 10) persons IOI: −9 (−47 to + 28) persons |
||
|
Neutralising antibodies with 2 IPV doses Geometric mean titres (GMT) (Range of follow‐up from 5 to 16 months) |
P1 (IIO) | 244 lower (−827 to + 339)SE | ‐ | 795 (3 RCTs) | ⊕⊕⊝⊝ Lowa,c |
IbObO: + 362 (−330 to + 1054) IOO: −181 (−594 to 232) IIIOO / IIIO: + 439 (−355 to + 1233) |
|
P2 (IIO) P2 (IIbO) |
267 higher (−84 to + 619)SE 218 lower (−305 to −130)LE |
‐ ‐ |
667 (3 RCTs) 125 (1 RCT) |
⊕⊕⊝⊝ Lowa,c ⊕⊕⊕⊝ Moderatea |
IbObO: −260 (−347 to −174) IOO: + 29 (−22 to + 79) IIIOO/IIIO: + 486 (−698 to + 1670) |
|
|
P3 (IIO) P3 (IIbO) |
90 higher (+ 9 to + 171)SE 592 higher (+ 185 to + 998)SE |
‐ ‐ |
667 (3 RCTs) 125 (1 RCT) |
⊕⊕⊕⊝ Moderatea |
IbObO: + 221 (+ 10 to + 432) IOO: + 44 (−1.47 to + 90) IIIOO/IIIO: + 248 (−181 to + 677) |
|
|
Persons with polio faecal excretion after OPV challenge (Range of follow‐up from 4.4 to 18 months) |
P1: 86 per 1000 | 193 per 1000 (60 to 613) | RR 2.24 (0.70 to 7.12) | 916 (2 studies) | ⊕⊕⊝⊝ Lowc,d | ‐ |
| P2: 279 per 1000 | 497 per 1000 (416 to 598) | RR 1.78 (1.49 to 2.14) | 916 (2 RCTs) | ⊕⊕⊕⊝ Moderatec | ||
| P3: 74 per 1000 | 173 per 1000 (108 to 276) | RR 2.35 (1.47 to 3.76) | 916 (2 RCTs) | |||
|
Vaccination coverage (Follow‐up 24 months) |
91.9% | 92.4% |
RR 1.01 (0.96 to 1.06) |
(1 ITS) | ⊕⊕⊝⊝ Low | ‐ |
|
Serious adverse events (classified by MedDRA) (Range of follow‐up from 5 to 16 months) |
48 per 1000 | 42 per 1000 (26 to 75) | RR 0.88 (0.46 to 1.70) | 1948 (4 RCTs) | ⊕⊕⊝⊝ Lowa,c | ‐ |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI).
# 2 IPV doses (IIO) was selected as the main subgroup for this outcome since it is the most studied scheme CI: Confidence interval; RR: Risk ratio; P1, P2, P3: Poliovirus Serotypes 1, 2, 3 respectively; SE: Small effect; ME; Moderate effect.LE: Large effect | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect | ||||||
I = IPV, O = OPV, bO = bOPV (see detailed acronyms in Appendix 1).
aSerious study limitations: most studies have unclear risk of bias regarding random sequence generation and allocation concealment. bThese are data from quasi‐experimental studies and therefore evidence was graded as low, we have not downgraded or upgraded the evidence. cSerious imprecision: confidence interval limits include clinically important increase or reduction of the effect. dSerious inconsistency: considerable heterogeneity but in the same direction.
Summary of findings 2. IPV‐OPV compared to IPV for preventing poliomyelitis.
| IPV‐OPV compared to IPV for preventing poliomyelitis | ||||||
| Patient or population: infants Setting: USA, UK, China, Guatemala, Chile, Bangladesh Intervention: IPV‐OPV Comparison: IPV | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with IPV | Risk with IPV‐OPV | |||||
| Paralytic wild polio cases at 4 years (Change level from IPV‐OPV to IPV) | −1.2 (−6.2 to 3.8) | −100% (−517% to 317%) |
(1 ITS) | ⊕⊝⊝⊝ Very lowa | Re‐analysis considering the year of schedule change as transition period | |
| Vaccine‐associated paralytic polio cases | ‐ | ‐ | ‐ | (0 studies) | ‐ | No data available |
|
Persons with protective humoral response (Range of follow‐up from 4.4 to 18 months) |
P1: 970 per 1000 | 972 per 1000 (961 to 980) | RR 1.00 (0.99 to 1.01) | 2858 (10 RCTs) | ⊕⊕⊕⊝ Moderateb |
IOO: −0 (−20 to + 20) persons IIO(O): 0 (−10 to + 10) persons IbOI: + 46 (0 to + 91) persons |
| P2: 960 per 1000 | 931 per 1000 (985 to 1000) | RR 0.97 (0.95 to 1.00) | 2907 (10 RCTs) | ⊕⊕⊝⊝ Lowb,c |
IbObO: −236 (−89 to −354) persons IOO: 0 (−39 to + 39) persons IbOI: −43 (−103 to + 17) persons |
|
| P3: 972 per 1000 | 962 per 1000 (953 to 982) | RR 0.99 (0.97 to 1.01) | 2620 (9 RCTs) | ⊕⊕⊕⊝ Moderateb |
IOO: −10 (−30 to + 20) persons IIO(O): −10 (−40 to + 20) persons IbOI: + 9 (−37 to + 47) persons |
|
| Comment: A nationwide ITS (Denmark) reported a median proportion of persons with protective humoral response for PV1, 2 and 3 as 82.06%, 91.94% and 76.67%, respectively, during IPV scheme; the proportions were higher during IPV‐OPV scheme: 98.44%; 97.67%; and 97.57%, respectively. | ||||||
|
Neutralising antibodies with 2 IPV doses Geometric mean titres (Follow‐up 5 months) |
P1 (IIO) P1 (IIbO) |
768 higher (+ 338 to + 1198)ME 867 higher (+ 479 to + 1254)ME |
‐ ‐ |
363 (2 RCTs) 127 (1 RCT) |
⊕⊕⊝⊝ Lowb,c ⊕⊕⊕⊝ Moderated |
IbObO: + 1521 (+ 1085 to + 1956) IOO: + 799 (+ 531 to + 1068) |
|
P2 (IIO) P2 (IIbO) |
2224 higher (−1146 to + 5594)LE 83 lower (−133 to −34)LE |
‐ ‐ |
362 (2 RCTs) 127 (1 RCT) |
⊕⊕⊝⊝ Very lowa,b,e ⊕⊕⊕⊝ Moderated |
IbObO: −126 (−175 to −77) IOO: + 142 (+ 58 to + 227) |
|
|
P3 (IIO) P3 (IIbO) |
185 higher (−212 to + 581)SE 698 higher (+ 301 to + 1096)SE |
‐ | 360 (2 RCTs) 127 (1 RCT) |
⊕⊕⊝⊝ Very lowa,b,c ⊕⊕⊕⊝ Moderated |
IbObO: + 328 (+ 135 to + 520) IOO: + 110 (−78 to + 299) |
|
|
Persons with faecal polio excretion after OPV challenge (Range of follow‐up from 4.4 to 18 months) |
P1: 427 per 1000 | 222 per 1000 (60 to 841) | RR 0.52 (0.14 to 1.97) | 822 (2 RCTs) | ⊕⊝⊝⊝ Very lowa,b,c |
ibOI: −356 (−297 to −394) persons IIO/IIOO: + 7 (−72 to + 148) persons |
| P2: 572 per 1000 | 309 per 1000 (177 to 537) | RR 0.55 (0.31 to 0.94) | 1351 (3 RCTs) | ⊕⊕⊝⊝ Lowb,c |
ibOI/IIbO: + 7 (−72 to + 148) persons IIO/IIOO: −329 (−282 to −357) persons |
|
| P3: 450 per 1000 | 176 per 1000 (144 to 212) | RR 0.39 (0.32 to 0.47) | 822 (2 RCTs) | ⊕⊕⊕⊝ Moderateb |
ibOI: −233 (−170 to −274) persons IIO/IIOO: −470 (−392 to −533) persons |
|
| Vaccination coverage | ‐ | ‐ | ‐ | (0 studies) | ‐ | No data available |
|
Serious adverse events (≥ 1 symptom related to study drug or not) (Range of follow‐up from 5 to 7 months) |
94 per 1000 | 87 per 1000 (57 to 135) | RR 0.92 (0.60 to 1.43) | 1063 (2 RCTs) | ⊕⊕⊝⊝ Lowb,c | ‐ |
| *The risk in the intervention group (and its 95% CI) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). # 2 IPV doses (IIO) was selected as the main result for this outcome since it is the most frequent scheme. CI: Confidence interval; RR: Risk ratio; P1, P2, P3: Poliovirus Serotypes 1, 2, 3 respectively. | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect | ||||||
I = IPV, O = OPV, bO = bOPV (see detailed acronyms in Appendix 1).
aSerious imprecision: confidence interval limits include clinically important increase or reduction of the effect. bSerious study limitations: most studies have unclear risk of bias regarding random sequence generation and allocation concealment. cConsiderable heterogeneity but in the same direction. dSerious imprecision: only one study with low number of participants. eVery serious imprecision: confidence interval limits include a marked effect increase or reduction.
Summary of findings 3. IPV(3)‐OPV compared to IPV(2)‐OPV for preventing poliomyelitis.
| 3 IPV‐OPV compared to 2 IPV‐OPV for preventing poliomyelitis | ||||||
| Patient or population: preventing poliomyelitis Setting: India Intervention: 3 IPV‐OPV Comparison: 2 IPV‐OPV | ||||||
| Outcomes | Anticipated absolute effects* (95% CI) | Relative effect (95% CI) | № of participants (studies) | Certainty of the evidence (GRADE) | Comments | |
| Risk with IPV(2)‐OPV | Risk with IPV(3)‐OPV | |||||
| Paralytic polio | ‐ | ‐ | ‐ | (0 studies) | ‐ | No data available |
| VAPP cases | ‐ | ‐ | ‐ | (0 studies) | ‐ | No data available |
|
Persons with protective humoral response (Follow‐up 7 months) |
P1: 1000 per 1000 | 980 per 1000 (930 to 1000) | RR 0.98 (0.93 to 1.03) | 137 (1 RCT) | ⊕⊕⊕⊝ Moderatea | ‐ |
| P2: 1000 per 1000 | 1000 per 1000 (970 to 1000) | RR 1.00 (0.97 to 1.03) | 137 (1 RCT) | |||
| P3: 989 per 1000 | 998 per 1000 (959 to 1000) | RR 1.01 (0.97 to 1.05) | 137 (1 RCT) | |||
| Neutralising antibodies | ‐ | ‐ | ‐ | (0 studies) | ‐ | No data available |
| Persons with faecal excretion after OPV challenge | ‐ | ‐ | ‐ | (0 studies) | ‐ | No data available |
| Vaccination coverage | ‐ | ‐ | ‐ | (0 studies) | ‐ | No data available |
| Serious adverse events | ‐ | ‐ | ‐ | (0 studies) | ‐ | No data available |
| *The risk in the intervention group (and its 95% Cl) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; RR: Risk ratio | ||||||
| GRADE Working Group grades of evidence High certainty: we are very confident that the true effect lies close to that of the estimate of the effect Moderate certainty: we are moderately confident in the effect estimate; the true effect is likely to be close to the estimate of the effect, but there is a possibility that it is substantially different Low certainty: our confidence in the effect estimate is limited; the true effect may be substantially different from the estimate of the effect Very low certainty: we have very little confidence in the effect estimate; the true effect is likely to be substantially different from the estimate of effect | ||||||
aSerious imprecision: confidence interval limits include clinically important increase or reduction of the effect.
See Appendix 1 Glossary to facilitate the reading of this section particularly for long‐combined schedules. In general, 'I' represents Inactivated Polio Vaccine (IPV) and 'O' represents Oral Polio Vaccine (OPV). Where O or OPV is used below we refer to trivalent OPV. Where bO or bOPV is used we refer to Bivalent OPV. P1, P2, and P3 refer respectively to Poliovirus Serotype 1, 2 and 3.
Comparison 1: IPV‐OPV versus OPV
See Table 1.
This comparison includes 12 RCTs with 4813 participants (Anand 2015; Asturias 2007; Faden 1990; Jain 1997; Li 2016a; Modlin 1997; Qiu 2017; Ramsay 1994; Simasathien 1994; Sutter 1997; West 2001; Yeh 2001).
We also included one interrupted time series (ITS) study (Davis 2001, n = 28,330), and three nationwide studies: two uncontrolled before‐and‐after (UBA) studies (Ivanova 2018; Kapusinszky 2010), and one study that used a mixed design (ITS + UBA analysis for vaccine‐associated paralytic polio (VAPP) cases) (Alexander 2004).
Paralytic polio
One nationwide ITS study assessed this outcome (Alexander 2004). It is uncertain if IPV‐OPV compared to OPV reduces the number of cases of wild poliovirus (WPV) since this study provided very low‐certainty evidence of the effect of this outcome.
In Alexander 2004, the national vaccination policy in the USA changed in 1997 from a reliance on OPV to options for a sequential IPV‐OPV schedule. In 2000, an exclusive IPV schedule was adopted. We re‐analysed the data considering the year 1997 as transition period, our re‐analysis of the OPV to IPV‐OPV schedule on reported cases of paralytic poliomyelitis showed a change in level of −0.3 (95% confidence interval (CI) −2.2 to 1.5; −36.0%), a change in slope of 0.1 (95% CI −1.0 to 1.2), and an estimated effect at three years of −0.2 (95% CI −1.3 to 0.9; −21.0%; very low‐certainty evidence).
Not considering a transition period, the ITS showed a change in level of 0.4 (95% CI −0.6 to 1.3; 59.0%), change in slope of −0.3 (95% CI −0.9 to 0.3), and an estimated effect at three years of −0.3 (95% CI −1.3 to 0.8; −32.0%).
Vaccine‐associated paralytic polio (VAPP)
Three nationwide, UBA studies showed that the sequential IPV‐OPV scheme compared with the previous OPV schemes may substantially reduce cases of VAPP (Alexander 2004; Ivanova 2018; Kapusinszky 2010). Although there is no international standard, we judged the preventive effect size of the intervention as large (low certainty‐evidence).
Alexander 2004, in the USA, analysed national acute flaccid paralysis (AFP) surveillance data conducted by the Centers for Disease Control and Prevention from 1990 to 1999.
Before. Exclusive use of OPV scheme OOOO (OPV at aged two, four, and 12 to 18 months and again at four to six years) between 1990 and 1996: 46 VAPP cases occurred, 1 VAPP case per 2.9 million doses or 0.34 VAPP cases per million of OPV doses.
After. Sequential scheme IIOO (IPV at two and four months of age followed by OP\/ at 12 to 18 months, and again at four to six years of age) between 1997 and 1999: 13 VAPP cases occurred, none of them occurred in persons who had followed the sequential IPV‐OPV or IPV‐only schedules.
Before‐After. Comparing 1990 to 1996 with 1998 to 1999, the average number of VAPP cases per year decreased from 6.6 to 3.0, which equates to a reduction of 54.3%. The average number of sporadic contact cases decreased by 68%; community‐acquired cases and immunologically abnormal cases both decreased by 100%.
Ivanova 2018, in the Russian Federation, analysed national acute flaccid paralysis (AFP) surveillance data from 1998 to 2014. Out of the 6643 cases of AFP, 127 cases were VAPP: 82 cases were observed in OPV recipients and 45 cases in non‐vaccinated contacts. Poliovirus type 2 (23.7%) and type 3 (39.5%) were isolated most often. Half of the children had a burdened premorbid status and 67% had various immunological disorders before presenting with VAPP.
Before. Exclusive use of OPV scheme OOOO (OPV at ages six, 18, and 20 months and again at 14 years) between 1998 and 2007: 1 VAPP case per 1.59 million OPV doses, 7.64 VAPP cases per million newborns (5.64 and 1.99 VAPP cases per million newborns in OPV recipients (rVAPP) and non‐vaccinated contacts (cVAPP), respectively: rVAPP/cVAPP ratio = 2.83). The VAPP cases yielded mostly type 3 (38.5%): rVAPP isolates were type 3 (69.2%) or mixtures of types 2 and 3 (56.7%); and cVAPP cases yielded types 2 (44%) and 3 (40%).
After. Sequential scheme IIIOOOO ( IPV at ages three and 4.5 months; OPV at ages six, 18 and 20 months and again at 14 years) between 2008 and 2014: 1 case per 4.18 million doses, 1.56 cases per million newborns (0.23 and 1.33 VAPP cases per million newborns in OPV recipients and contacts, respectively: rVAPP/cVAPP ratio = 0.17). VAPP cases occurred mainly in unvaccinated children as a result of contact with an OPV recipient and also in recipients, who, for various reasons, received OPV in violation of the vaccination schedule. Individual serotypes were mostly isolated: 85% from cases of VAPP in general and 100% from cVAPP. During the same time period, no case was associated with poliovirus type 1.
Before‐After. Observed reduction of 79.58% of cases per million newborns with the sequential scheme (95.92% and 33.17% reduction in VAPP cases per million newborns in OPV recipients and contacts, respectively).
Kapusinszky 2010, in Hungary, analysed national AFP surveillance data from 1992 to 2006. During that period, 90 VAPP cases were reported.
Before. Exclusive use of OPV scheme (mOPV1, mOPV2, mOPV3, separated by six weeks, at two to 38 months of age) between 1959 and 1992: 90 cases of VAPP.
After. Sequential scheme IOOOOO (IPV at two to 38 months of age, tOPV six weeks later‐OPV) between 1992 and 2006; zero cases of VAPP.
Before‐After. Observed reduction of 100% of cases.
Out of the 90 VAPP cases, 52 were associated with Sabin 3‐related virus (76% of VAPP cases with virologic data). No evidence was found for prolonged monovalent OPV type 3 (mOPV3) replication in the VAPP patients or for spread of Sabin 3‐related viruses beyond close vaccinee contacts. We could not judge the clinical meaningfulness of this level of change, due to the lack of international standards.
Protective immune responses
Humoral and intestinal immunity
Twelve RCTs provided data on this outcome (Anand 2015; Asturias 2007; Faden 1990; Jain 1997; Li 2016a; Modlin 1997; Qiu 2017; Ramsay 1994; Rennels 2000; Simasathien 1994; West 2001; Yeh 2001).
We grouped, analysed and presented the results according to P1, P2 and P3 protective humoral and intestinal immunity. We also present the respective results from subgroup analyses by serotype. The efficacy of IPV‐OPV by serotype P1, P2 and P3 is presented in additional Table 7.
4. Efficacy of IPV‐OPV by serotype P1, P2 and P3.
| Serotype | Outcome | Measure | Effect (95% CI) | Favours |
| IPV‐OPV vs OPV | ||||
| P1 | 1.1 Persons with protective humoral response | RR | 1.00 (0.99 to 1.01) | = |
| 1.1.1 Sequential IOO/IOOO | RR | 1.00 (0.98 to 1.02) | = | |
| 1.1.2 Sequential IIO/IIOO/IIIO | RR | 1.00 (0.99 to 1.01) | = | |
| 1.1.3 Sequential IOI | RR | 1.03 (0.93 to 1.14) | = | |
| 1.13 Mean titres of neutralising antibody | ||||
| 1.13.1 Sequential IbObO | MD | 362.12 (−329.70 to 1053.94) | IPV‐OPV (NS) | |
| 1.13.2 Sequential IOO | MD | −181.13 (−594.25 to 231.99) | OPV (NS) | |
| 1.13.3 Sequential IIO | MD | −244.37 (−827.31 to 338.57) | OPV (NS) | |
| 1.13.4 Sequential IIIOO/IIIO | MD | 439.07 (−354.63 to 1232.77) | IPV‐OPV (NS) | |
| 1.16 Long‐term mean titres of neutralising antibody | ||||
| 1.16.1 Sequential IOO | MD | 0.00 (−0.48 to 0.48) | = | |
| 1.16.2 Sequential IOO + O | MD | 0.20 (−0.33 to 0.73) | = | |
| 1.16.3 Sequential IIO | MD | 0.60 (0.22 to 0.98) | IPV‐OPV | |
| 1.16.4 Sequential IIO + O | MD | 0.50 (0.01 to 0.99) | IPV‐OPV | |
| 1.19.1 Persons with polio faecal excretion after OPV challenge | RR | 1.86 (1.21 to 2.86) | OPV (NS) | |
| P2 | 1.7 Persons with protective humoral response | RR | 1.00 (0.97 to 1.04) | = |
| 1.7.1 Sequential IbObO (bOPV) | RR | 0.85 (0.78 to 0.91) | OPV | |
| 1.7.2 Sequential IOO/IOOO | RR | 1.00 [0.98 to 1.02) | = | |
| 1.7.3 Sequential IIO/IIOO/IIIO | RR | 1.00 (0.99 to 1.01) | = | |
| 1.7.4 Sequential IbOI (vs bObObO) | RR | 5.85 (4.10 to 8.34) | IPV‐OPV | |
| 1.7.5 Sequential IbOI | RR | 0.82 (0.75 to 0.90) | OPV | |
| 1.7.6 Sequential IOI | RR | 1.08 (0.99 to 1.17) | IPV‐OPV (NS) | |
| 1.14 Mean titres of neutralising antibody | ||||
| 1.14.1 Sequential IbObO | MD | −260.38 (−347.21 to −173.55) | OPV | |
| 1.14.2 Sequential IOO | MD | 28.64 (−22.16 to 79.43) | = | |
| 1.14.3 Sequential IIbO | MD | −217.90 (−305.36 to −130.44) | OPV | |
| 1.14.4 Sequential IIO | MD | 267.40 (−83.95 to 618.76) | IPV‐OPV (NS) | |
| 1.14.5 Sequential IIIOO/IIIO | MD | 486.17 (−698.02 to 1670.37) | IPV‐OPV (NS) | |
| 1.17 Long‐term mean titres of neutralising antibody | ||||
| 1.17.1 Sequential IOO | MD | 0.10 (−0.26 to 0.46) | = | |
| 1.17.2 Sequential IOO + O | MD | 0.10 (−0.30 to 0.50) | = | |
| 1.17.3 Sequential IIO | MD | 0.00 (−0.38 to 0.38) | = | |
| 1.17.4 Sequential IIO + O | MD | 0.30 (−0.11 to 0.71) | = | |
| 1.10.2 Persons with polio faecal excretion after OPV challenge | RR | 1.79 (1.49 to 2.15) | OPV | |
| P3 | 1.11 Persons with protective humoral response | RR | 0.98 (0.96 to 1.00) | = |
| 1.11.1 Sequential IbObO | RR | 0.70 (0.60 to 0.82) | OPV | |
| 1.11.2 Sequential IOO/IOOO | RR | 0.98 ([0.96 to 1.00) | = | |
| 1.11.3 Sequential IIO/IIOO/IIIO | RR | 0.99 (0.96 to 1.01) | = | |
| 1.11.4 Sequential IOI | RR | 0.99 (0.95 to 1.03) | = | |
| 1.15 Mean titres of neutralising antibody | ||||
| 1.15.1 Sequential IbObO | MD | 221.03 (9.66 to 432.40) | IPV‐OPV | |
| 1.15.2 Sequential IOO | MD | 44.07 (−1.47 to 89.61) | IPV‐OPV (NS) | |
| 1.15.3 Sequential IIbO | MD | 591.78 (185.14 to 998.42) | IPV‐OPV (NS) | |
| 1.15.4 Sequential IIO | MD | 89.97 (8.98 to 170.97) | IPV‐OPV | |
| 1.15.5 Sequential IIIOO/IIIO | MD | 248.39 (−180.58 to 677.37) | IPV‐OPV (NS) | |
| 1.18 Long‐term mean titres of neutralising antibody | ||||
| 1.18.1 Sequential IOO | MD | −0.50 (−1.14 to 0.14) | OPV (NS) | |
| 1.18.2 Sequential IOO + O | MD | 0.00 (−0.61 to 0.61) | = | |
| 1.18.3 Sequential IIO | MD | 0.40 (0.02 to 0.78) | IPV‐OPV | |
| 1.18.4 Sequential IIO + O | MD | 0.20 (−0.34 to 0.74) | = | |
| 1.19.3 Persons with polio faecal excretion after OPV challenge | RR | 2.42 (1.60 to 3.67) | OPV | |
| IPV‐OPV vs IPV | ||||
| P1 | 2.1 Persons with P1 protective humoral response | RR | 1.00 (0.99 to 1.01) | = |
| 2.1.1 Sequential IOO | RR | 0.99 (0.98 to 1.01) | = | |
| 2.1.2 Sequential IIO(O) | RR | 1.00 (0.99 to 1.01) | ||
| 2.1.3 Sequential IbOI | RR | 1.05 (1.00 to 1.10) | IPV‐OPV | |
| 2.4 Mean titres of P1 neutralising antibody | Subtotals only | |||
| 2.4.1 Sequential IbObO | MD | 1520.59 (1084.80 to 1956.38) | IPV‐OPV | |
| 2.4.2 Sequential IOO | MD | 799.47 (530.82 to 1068.12) | IPV‐OPV | |
| 2.4.3 Sequential IIbO | MD | 866.53 (478.83 to 1254.23) | IPV‐OPV | |
| 2.4.4 Sequential IIO | MD | 767.90 (337.75 to 1198.06) | IPV‐OPV | |
| 2.7 Long‐term mean titres of P1 neutralising antibody | Subtotals only | |||
| 2.7.1 Sequential IOO | MD | −0.20 (−0.73 to 0.33) | = | |
| 2.7.2 Sequential IOO + O | MD | −0.30 (−0.83 to 0.23) | = | |
| 2.7.3 Sequential IIO | MD | 0.40 (−0.04 to 0.84) | IPV‐OPV (NS) | |
| 2.7.4 Sequential IIO + O | MD | 0.90 (0.40 to 1.40) | IPV‐OPV | |
| 2.10.1 Persons with faecal excretion after OPV challenge (ibOI) | RR | 0.27 (0.19 to 0.39) | IPV‐OPV | |
| 2.10.2 Persons with faecal excretion after OPV challenge (IIO/IIOO) | RR | 1.04 (0.59 to 1.84) | = | |
| P2 | 2.2 Persons with protective humoral response | RR | 0.97 (0.94 to 1.00) | = |
| 2.2.1 Sequential IbObO | RR | 0.76 (0.64 to 0.91) | IPV | |
| 2.2.2 Sequential IOO | RR | 1.00 (0.96 to 1.04) | = | |
| 2.2.3 Sequential IIO(O) | RR | 1.00 (0.99 to 1.01) | = | |
| 2.2.4 Sequential IbOI | RR | 0.95 (0.88 to 1.02) | = | |
| 2.5 Mean titres of neutralising antibody | Subtotals only | |||
| 2.5.1 Sequential IbObO | MD | −125.93 (−174.77 to −77.09) | IPV | |
| 2.5.2 Sequential IOO | MD | 142.25 (57.65 to 226.85) | IPV‐OPV | |
| 2.5.3 Sequential IIbO | MD | −83.45 (−133.40 to −33.50) | IPV | |
| 2.5.4 Sequential IIO | MD | 787.97 (573.84 to 1002.09) | IPV‐OPV | |
| 2.8 Long‐term mean titres of neutralising antibody | Subtotals only | |||
| 2.8.1 Sequential IOO | MD | 0.20 (−0.06 to 0.46) | IPV‐OPV (NS) | |
| 2.8.2 Sequential IOO + O | MD | −0.60 (−0.96 to −0.24) | IPV | |
| 2.8.3 Sequential IIO | MD | 0.10 (−0.19 to 0.39) | = | |
| 2.8.4 Sequential IIO + O | MD | −0.40 (−0.78 to −0.02) | IPV | |
| 2.10.3 Persons with faecal excretion after OPV challenge (ibOI/IIbO) | RR | 0.85 (0.68 to 1.06) | IPV‐OPV (NS) | |
| 2.10.4 Persons with faecal excretion after OPV challenge (IIO/IIOO) | RR | 0.16 (0.09 to 0.28) | IPV‐OPV | |
| P3 | 2.3 Persons with protective humoral response | RR | 0.99 (0.98 to 1.01) | = |
| 2.3.1 Sequential IOO | RR | 0.99 (0.97 to 1.02) | = | |
| 2.3.2 Sequential IIO(O) | RR | 0.99 (0.96 to 1.01) | = | |
| 2.3.3 Sequential IbOI | RR | 1.01 (0.96 to 1.05) | = | |
| 2.6 Mean titres of neutralising antibody | Subtotals only | |||
| 2.6.1 Sequential IbObO | MD | 327.62 (134.82 to 520.42) | IPV‐OPV | |
| 2.6.2 Sequential IOO | MD | 110.39 (−77.98 to 298.76) | IPV‐OPV (NS) | |
| 2.6.3 Sequential IIbO | MD | 698.37 (301.06 to 1095.68) | IPV‐OPV | |
| 2.6.4 Sequential IIO | MD | 184.52 (−211.93 to 580.97) | IPV‐OPV (NS) | |
| 2.9 Long‐term mean titres of neutralising antibody | Subtotals only | |||
| 2.9.1 Sequential IOO | MD | −0.60 (−1.22 to 0.02) | IPV | |
| 2.9.2 Sequential IOO + O | MD | −0.30 (−0.76 to 0.16) | IPV (NS) | |
| 2.9.3 Sequential IIO | MD | 0.30 (−0.04, 0.64) | IPV‐OPV (NS) | |
| 2.9.4 Sequential IIO + O | MD | −0.10 (−0.46 to 0.26) | = | |
| 2.10.5 Persons with faecal excretion after OPV challenge(ibOI) | RR | 0.37 (0.26 to 0.54) | IPV‐OPV | |
| 2.10.6 Persons with faecal excretion after OPV challenge (IIO/IIOO) | RR | 0.40 (0.32 to 0.50) | IPV‐OPV | |
| CI: confidence interval; IPV: inactivated poliovirus vaccine; MD: mean difference; NS: non‐statistically significant; OPV: oral poliovirus vaccine; P: poliovirus; RR: risk ratio; =: non‐statistically and non‐clinically significant | ||||
For definition of each sequence (e.g. sequential IOO), see Glossary in Appendix 1.
P1 protective humoral immunity
There is no difference between the two treatments for number of persons with P1 protective humoral immunity (risk ratio (RR) 1.00, 95% CI 0.99 to 1.01; P = 0.60; 12 studies, 3189 participants; Analysis 1.1; moderate‐certainty evidence). There is no heterogeneity (Tau = 0.00; Chi2 = 7.80, df = 14 (P = 0.90); I2 = 0%).
1.1. Analysis.
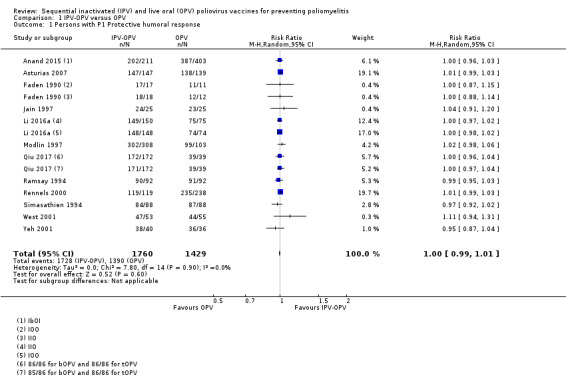
Comparison 1 IPV‐OPV versus OPV, Outcome 1 Persons with P1 Protective humoral response.
Subgroup analyses
Timing of the first dose: the test for subgroup differences indicates that there is no statistically significant subgroup effect (Test for subgroup differences: Chi2 = 0.33, df = 1 (P = 0.57), I2 = 0%; Analysis 1.2). First dose given at birth (RR 1.04 CI 0.91 to 1.20; 1 study, 50 participants); First dose given at two months (RR 1.00 CI 0.99 to 1.01; 12 studies, 3139 participants); there is no heterogeneity (Tau2 = 0.00; Chi = 7.61, df = 10 (P = 0.67), I2 = 0%).
Type of dose sequence: the test for subgroup differences indicates that there is no statistically significant subgroup effect (Chi2 = 0.51, df = 2 (P = 0.78), I2 = 0%; Analysis 1.3). IOO/IOOO dose sequence: RR 1.00, 95% CI 0.98 to 1.01; 5 studies, 695 participants; moderate‐certainty evidence. There is no heterogeneity (Tau2 = 0.00; Chi2 = 0.69, df = 4 (P = 0.95), I2 = 0%). IIO/IIOO/IIIO dose sequence: RR 1.00, 95% CI 0.99 to 1.01; 8 studies, 1772 participants; moderate‐certainty evidence. There is no heterogeneity (Tau2 = 0.00; Chi2 = 5.51, df = 7 (P = 0.60), I2 = 0%). IOI dose sequence: RR 1.03 CI 0.93 to 1.14; 2 studies, 722 participants; moderate‐certainty evidence. Heterogeneity is moderately high (Tau2 = 0.00; Chi2 = 1.95; df = 1 (P = 0.16); I2 = 49%).
Country or location: the test for subgroup differences indicates that there is no statistically significant subgroup effect (Test for subgroup differences: Chi2 = 0.41, df = 1 (P = 0.52), I2 = 0%; Analysis 1.4). Low and middle‐income countries: RR 1.00, 95% CI 0.99 to 1.01; 4 studies, 1331 participants. Heterogeneity is low (Tau2 = 0.00; Chi2 = 3.13, df = 3 (P = 0.37); I2 = 4%). High‐income countries: RR 1.01, 95% CI 0.99 to 1.02; 8 studies, 1858 participants. There is no heterogeneity (Tau2 = 0.00; Chi2 = 5.08, df = 7 (P = 0.65); I2 = 0%).
1.2. Analysis.

Comparison 1 IPV‐OPV versus OPV, Outcome 2 Persons with P1 Protective humoral response by time of first dose.
1.3. Analysis.

Comparison 1 IPV‐OPV versus OPV, Outcome 3 Persons with P1 Protective humoral response by type of dose sequence.
1.4. Analysis.

Comparison 1 IPV‐OPV versus OPV, Outcome 4 Persons with P1 Protective humoral response by countries' income.
P2 protective humoral immunity
The test for subgroup differences indicates that there is a high probability of real differences between the different types of doses (Chi2 = 99.08, df = 3 (P < 0.001), I2 = 97.0%; Analysis 1.5); therefore, we did not present potentially misleading overall effect. There is probably no difference between the treatments in terms of the number of persons with P2 protective humoral immunity for the IPV‐tOPV and IIbO dosing schemes. IbObO may be worse than tOPV and IbObO is probably better than bOPV.
1.5. Analysis.

Comparison 1 IPV‐OPV versus OPV, Outcome 5 Persons with P2 Protective humoral response.
We present below a post hoc subgroup analysis of the bOPV component of the sequential schedule IPV‐tOPV versus IPV‐bOPV.
IPV‐tOPV (none using bOPV)
There is probably no difference between the treatments in terms of the number of persons with P2 protective humoral immunity for the IPV‐tOPV and IIbO dosing schemes.
IIO/IIOO/IIIO/IOO/IOOO: RR 1.00, 95% CI 0.99 to 1.01; 11 studies, 2361 participants; I2 = 0%; low‐certainty evidence.
IPV‐bOPV
There is probably no difference between the treatments in terms of the number of persons with P2 protective humoral immunity for IIbO dosing schemes, IbObO may be worse than tOPV and IbObO is probably better than bOPV.
IIbO: RR 1.00, 95% CI 0.93 to 1.07; 1 study, 105 participants; I2 = 0%; moderate‐certainty evidence).
IbObO vs tOPV: RR 0.78, 95% CI 0.64 to 0.96; 2 studies, 411 participants; I2 = 80%; low‐certainty evidence).
IbObO vs bOPV; RR 5.80, 95% CI 4.06 to 8.27; 1 study, 306 participants; I2 = 0%; moderate‐certainty evidence).
Subgroup analyses
Timing of the first dose: the test for subgroup differences indicates that there is no statistically significant subgroup effect (Test for subgroup differences: Chi2 = 0.06, df = 1 (P = 0.80), I2 = 0%; Analysis 1.6.)
Type of dose sequence: the test for subgroup differences indicates that there is a statistically significant subgroup effect (Test for subgroup differences: Chi2 = 134.11, df = 5 (P < 0.001), I2 = 96.3%; Analysis 1.7). Vaccination with OPV using the IbObO (bOPV) (RR 0.85, 95% CI 0.78 to 0.91; 1 study, 211 participants; moderate‐certainty evidence) and IbOI (RR 0.82, 95% CI 0.75 to 0.90; 1 study, 309 participants; moderate‐certainty evidence) dose sequences, or vaccination with IPV‐OPV using the IbOI (vs bObObO) dose sequence (RR 5.85, 95% CI 4.10 to 8.34; 1 study, 305 participants; moderate‐certainty evidence) may increase the number of persons with P2 protective humoral immunity. There are no differences between the two treatments with the other dose sequences.
Country or location: the test for subgroup differences indicates that there is no statistically significant subgroup effect (Test for subgroup differences: Chi2 = 2.05, df = 1 (P = 0.15), I2 = 51.2%; Analysis 1.8).
1.6. Analysis.

Comparison 1 IPV‐OPV versus OPV, Outcome 6 Persons with P2 Protective humoral response by time of first dose.
1.7. Analysis.

Comparison 1 IPV‐OPV versus OPV, Outcome 7 Persons with P2 Protective humoral response by type of dose sequence.
1.8. Analysis.
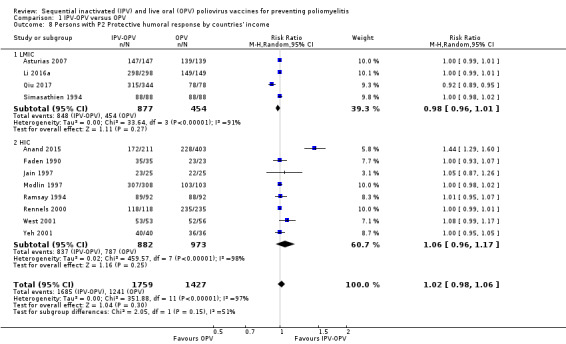
Comparison 1 IPV‐OPV versus OPV, Outcome 8 Persons with P2 Protective humoral response by countries' income.
P3 humoral immunity
Vaccination with IPV‐OPV may make little or no difference to the number of people with P3 protective humoral immunity (RR 0.99, 95% CI 0.97 to 1.00; 12 studies, 3184 participants; Analysis 1.9; low‐certainty evidence). There is moderate heterogeneity (Tau2 = 0.00; Chi2 = 28.21, df = 16 (P = 0.03); I2 = 43%).
1.9. Analysis.

Comparison 1 IPV‐OPV versus OPV, Outcome 9 Persons with P3 Protective humoral response.
Subgroup analyses
Timing of the first dose: the test for subgroup differences indicates that there is no statistically significant subgroup effect (Test for subgroup differences: Chi2 = 1.20, df = 1 (P = 0.27), I2 = 16.7%; Analysis 1.10). First dose at birth: RR 1.17 CI 0.87 to 1.57; 1 study, 50 participants. First dose at two months: RR 0.99 CI 0.97 to 1.01; 11 studies, 2963 participants; however, heterogeneity is high (Tau2 = 0.00; Chi2 = 29.12, df = 11 (P = 0.002); I2 = 62%).
Type of dose sequence: the test for subgroup differences indicates that there is a statistically significant subgroup effect (Test for subgroup differences: Chi2 = 17.48, df = 3 (P < 0.001), I2 = 82.8%; Analysis 1.11). Vaccination with OPV using the IbObO dose sequence may increase the number of persons with P3 protective humoral immunity (RR 0.70, 95% CI 0.60 to 0.82; 1 study, 105 participants; moderate‐certainty evidence). There are no differences between the two treatments with the other dose sequences.
Country or location: the test for subgroup differences indicates that there is no statistically significant subgroup effect (Test for subgroup differences: Chi2 = 0.71, df = 1 (P = 0.40), I2 = 0%; Analysis 1.12).
1.10. Analysis.
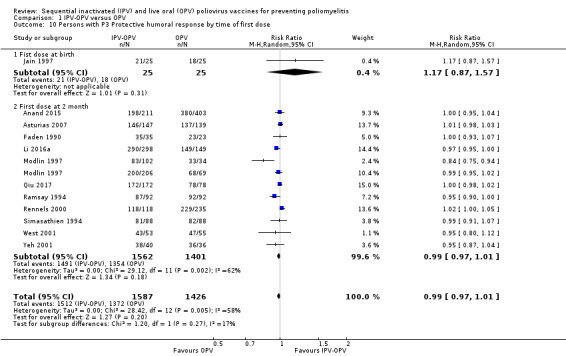
Comparison 1 IPV‐OPV versus OPV, Outcome 10 Persons with P3 Protective humoral response by time of first dose.
1.11. Analysis.

Comparison 1 IPV‐OPV versus OPV, Outcome 11 Persons with P3 Protective humoral response by type of dose sequence.
1.12. Analysis.

Comparison 1 IPV‐OPV versus OPV, Outcome 12 Persons with P3 Protective humoral response by countries' income.
Mean titres of neutralising antibodies
Six studies contributed data on this outcome (Li 2016a; Qiu 2017; Ramsay 1994; Rennels 2000; Simasathien 1994; Sutter 1997). We excluded the oldest RCT, Faden 1990, from these analyses because it used a very different method to measure antibody titres than the more recent studies. In that study, sequential schemes showed higher neutralising antibodies titre than OPV, for all serotypes except for P3 when used only one dose of IPV:
sequential IIO: P1 mean titre = 3044; P2 mean titre = 10693; and P3 mean titre = 2347;
sequential IOO: P1 mean titre = 2174: P2 mean titre = 11110; and P3 mean titre = 857; and
sequential OOO: P1 mean titre = 1470; P2 mean titre = 3378; and P3 mean titre = 1522.
For this outcome, we only present the results of the subgroup analyses because there was considerable statistical heterogeneity, even in each subgroup (I2 range from 83% to 97%).
P1 neutralising antibody
The test for subgroup differences indicates that there is no statistically significant subgroup effect by type of dose sequence (Test for subgroup differences: Chi2 = 3.61, df = 3 (P = 0.31), I2 = 16.9%). None of the individual results were statistically significant and thus we are uncertain of the effects of vaccination with IPV‐OPV, compared with OPV, on mean titres of P1 neutralising antibodies (Analysis 1.13).
1.13. Analysis.
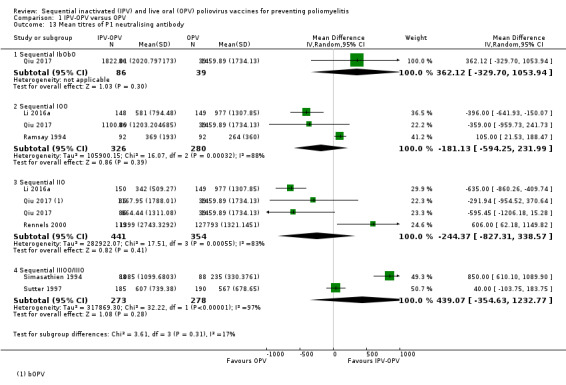
Comparison 1 IPV‐OPV versus OPV, Outcome 13 Mean titres of P1 neutralising antibody.
IbObO dose sequence: MD 362.12, 95% CI −329.70 to 1053.94; 1 study, 125 participants; low‐certainty evidence.
IOO dose sequence: MD −181.13, 95% CI −594.25 to 231.99; 3 studies, 606 participants; low‐certainty evidence. However, heterogeneity is high (Tau2 = 105900.15, Chi2 = 16.07, df = 2 (P < 0.001; I2 = 88%).
IIO dose sequence: MD −244.37, 95% CI −827.31 to 338.57; 3 studies/substudies, 795 participants; low‐certainty evidence. However, heterogeneity is high (Tau2 = 282922.07, Chi2 = 17.51, df = 3 (P < 0.001); I2 = 83%).
IIIOO/IIIO dose sequence: MD 439.07, 95% CI −354.63 to 1232.77; 2 studies, 551 participants; low‐certainty evidence. However, heterogeneity is high (Tau2 = 317869.30, Chi2 = 32.22, df = 1 (P < 0.001); I2 = 97%).
P2 neutralising antibody
The test for subgroup differences indicates that there is a statistically significant subgroup effect by type of dose sequence (Test for subgroup differences: Chi2 = 48.27, df = 4 (P < 0.001), I2 = 91.7%). Vaccination with OPV using the IbObO and IIbO dose sequences may reduce mean titres of P2 neutralising antibodies compared to IPV‐OPV, whereas vaccination with IPV‐OPV using the IOO, IIO, and IIIOO/IIIO dose sequences may make little or no difference on the mean titres of P2 neutralising antibodies compared with OPV. See Analysis 1.14.
1.14. Analysis.

Comparison 1 IPV‐OPV versus OPV, Outcome 14 Mean titres of P2 neutralising antibody.
IbObO dose sequence: MD −260.38, 95% CI −347.21 to −173.55; 1 study, 125 participants; moderate‐certainty evidence.
IOO dose sequence: MD 28.64, 95% CI −22.16 to 79.43; 3 studies, 606 participants; low‐certainty evidence. There is no heterogeneity (Tau2 = 0.00; Chi2 = 0.20, df = 2 (P = 0.91); I2= 0%).
IIbO dose sequence: MD −217.90, 95% CI −305.36 to −130.44; 1 study, 125 participants; moderate‐certainty of evidence of large effect.
IIO dose sequence: MD 267.40, 95% CI −83.95 to 618.76; 3 studies, 667 participants; low‐certainty of evidence. There is considerable heterogeneity (Tau2 = 57792.38; Chi2 = 9.40, df = 2 (P = 0.009); I2 = 79%).
IIIOO/IIIO dose sequence: MD 486.17, 95% CI −698.02 to 1670.37; 2 studies, 551 participants; low‐certainty evidence. There is high heterogeneity (Tau2 = 702371.09; Chi2 = 25.67, df = 1 (P < 0.001); I2 = 96%).
P3 neutralising antibody
The test for subgroup differences indicates that there is a statistically significant subgroup effect by type of dose sequence (Test for subgroup differences: Chi2 = 10.41, df = 4 (P = 0.03), I2 = 61.6%). Vaccination with IPV‐OPV using the IbObO, IIbO, and IIO dose sequences may increase mean titres of P3 neutralising antibodies compared to OPV, whereas vaccination with IPV‐OPV using the IOO and IIIOO/IIIO dose sequences may make little or no difference on the mean titres of P3 neutralising antibodies compared with OPV. See Analysis 1.15.
1.15. Analysis.

Comparison 1 IPV‐OPV versus OPV, Outcome 15 Mean titres of P3 neutralising antibody.
IbObO dose sequence: MD 221.03, 95% CI 9.66 to 432.40; 1 study, 125 participants; moderate‐certainty evidence.
IOO dose sequence: MD 44.07, 95% CI −1.47 to 89.61; 3 studies, 606 participants; low‐certainty evidence. There is no heterogeneity (Tau = 0.00; Chi = 1.12, df = 2 (P = 0.57); I2 = 0%).
IIbO dose sequence: MD 591.78, 95% CI 185.14 to 998.42; 1 study, 125 participants; moderate‐certainty of evidence of moderate effect.
IIO dose sequence: MD 89.97, 95% CI 8.98 to 170.97; 3 studies, 667 participants; moderate‐certainty of evidence of large effect. Heterogeneity is moderate (Tau2 = 1653.80; Chi2 = 2.91, df = 2 (P = 0.23); I2 = 31%).
IIIOO/IIIO dose sequence: MD 248.39, 95% CI −180.58 to 677.37; 2 studies, 551 participants; very low‐certainty evidence. Heterogeneity is high (Tau2 = 91317.08; Chi2 = 20.83, df = 1 (P < 0.001); I2 = 95%).
Long‐term mean titres of neutralising antibody
One study contributed data on this outcome (Faden 1990).
We grouped, analysed and presented the results according to P1, P2 and P3 protective humoral immunity. We also presented the respective results from subgroup analyses by serotype and by scheme.
P1 neutralising antibody
Vaccination with IPV‐OPV may increase the long‐term mean titres of P1 neutralising antibodies, compared to OPV (MD 0.35, 95% CI 0.07 to 0.63; 1 study, 86 participants; Analysis 1.16; low‐certainty evidence; heterogeneity: Tau2 = 0.02; Chi2 = 4.33, df = 3 (P = 0.23); I2 = 31%).
1.16. Analysis.

Comparison 1 IPV‐OPV versus OPV, Outcome 16 Long term mean titres of P1 neutralising antibody.
The test for subgroup differences indicates that there is no statistically significant subgroup effect by type of dose sequence (Test for subgroup differences: Chi2 = 4.33, df = 3 (P = 0.23), I2 = 30.7%). Vaccination with IPV‐OPV may increase the long‐term mean titres of P1 neutralising antibodies, compared to OPV, but only with the IIO and IIO + O dose sequences (Analysis 1.16; low‐certainty evidence).
IOO dose sequence: MD 0.00, 95% CI −0.48 to 0.48; 1 study, 20 participants; low‐certainty evidence.
IOO + O dose sequence: MD 0.20, 95% CI −0.33 to 0.73; 1 study, 20 participants; low‐certainty evidence.
IIO dose sequence: MD 0.60, 95% CI 0.22 to 0.98; 1 study, 23 participants; low‐certainty evidence.
IIO + O dose sequence: MD 0.50, 95% CI 0.01 to 0.99; 1 study, 23 participants; low‐certainty evidence.
P2 neutralising antibody
Vaccination with IPV‐OPV may produce minimal or no difference on the long‐term mean titres of P2 neutralising antibodies, compared to OPV (MD 0.12, 95% CI −0.07 to 0.31; 1 study, 86 participants; Analysis 1.17; low‐certainty evidence; heterogeneity: Tau2 = 0.00; Chi2 = 1.15, df = 3 (P = 0.77); I2 = 0%).
1.17. Analysis.

Comparison 1 IPV‐OPV versus OPV, Outcome 17 Long term mean titres of P2 neutralising antibody.
The test for subgroup differences indicates that there is no statistically significant subgroup effect by type of dose sequence (Test for subgroup differences: Chi2 = 1.15, df = 3 (P = 0.77), I2 = 0%). Vaccination with IPV‐OPV may make little or no difference on the long‐term mean titres of P2 neutralising antibodies compared with OPV (Analysis 1.17; low‐certainty evidence).
IOO dose sequence: MD 0.10, 95% CI −0.26 to 0.46; 1 study, 20 participants; low‐certainty evidence.
IOO + O dose sequence: MD 0.10, 95% CI −0.30 to 0.50; 1 study, 20 participants; low‐certainty of evidence.
IIO dose sequence: MD 0.00, 95% CI −0.38 to 0.38; 1 study, 23 participants; low‐certainty evidence.
IIO + O dose sequence: MD 0.30, 95% CI −0.11 to 0.71; 1 study, 23 participants; low‐certainty evidence.
P3 neutralising antibody
Vaccination with IPV‐OPV may produce minimal or no difference on the long‐term mean titres of P3 neutralising antibodies, compared to OPV (MD 0.08, 95% CI −0.29 to 0.45; 1 study, 86 participants; Analysis 1.18; low‐certainty evidence; heterogeneity: Tau2 = 0.07; Chi2 = 5.92, df = 3 (P = 0.12); I2 = 49%).
1.18. Analysis.

Comparison 1 IPV‐OPV versus OPV, Outcome 18 Long term mean titres of P3 neutralising antibody.
The test for subgroup differences indicates that there is no statistically significant subgroup effect by type of dose sequence (Test for subgroup differences: Chi2 = 5.92, df = 3 (P = 0.12), I2 = 49.3%). Vaccination with IPV‐OPV may increase the long‐term mean titres of P3 neutralising antibodies, compared to OPV, but only with the IIO dose sequence (Analysis 1.18; low‐certainty of evidence).
IOO dose sequence: MD −0.50, 95% CI −1.14 to 0.14; 1 study, 20 participants; low‐certainty evidence.
IOO + O dose sequence: MD 0.00, 95% CI −0.61 to 0.61; 1 study, 20 participants; low‐certainty evidence.
IIO dose sequence: MD 0.40, 95% CI 0.02 to 0.78; 1 study, 23 participants; low‐certainty evidence.
IIO + O dose sequence: MD 0.20, 95% CI −0.34 to 0.74; 1 study, 23 participants; low‐certainty evidence.
Intestinal immunity
Two studies with 916 participants contributed data on this outcome (Anand 2015; Modlin 1997).
The test for subgroup differences indicates that there is no statistically significant subgroup effect by serotype (Test for subgroup differences: Chi2 = 1.25, df = 2 (P = 0.54), I2 = 0%). IPV‐OPV probably increases P2 and P3 poliovirus faecal excretion after OPV challenge, compared to OPV. It may also increase or decrease P1 faecal excretion after OPV challenge.
P1 faecal excretion after OPV challenge: RR 2.24, 95% CI 0.70 to 7.12; 2 studies, 916 participants; low‐certainty evidence. There is moderate heterogeneity (Tau2 = 0.53; Chi2 = 3.68, df = 1 (P = 0.06); I2 = 73%.
P2 faecal excretion after OPV challenge: RR 1.78, 95% CI 1.49 to 2.14; 2 studies, 916 participants; moderate‐certainty evidence. There is no heterogeneity (Tau2 = 0.00; Chi2 = 0.09, df = 1 ( P = 0.76); I2 = 0).
P3 faecal extraction after OPV challenge: RR 2.35, 95% CI 1.47 to 3.76; 2 studies, 916 participants; moderate‐certainty evidence. There is low heterogeneity (Tau2 = 0.03; Chi2 = 1.25, df = 1 (P = 0.26); I2 = 20%).
Thus, compared to OPV, vaccination with IPV‐OPV probably increases the number of people with polio faecal excretion after OPV challenge; Analysis 1.19; moderate‐certainty evidence).
1.19. Analysis.

Comparison 1 IPV‐OPV versus OPV, Outcome 19 Persons with polio faecal excretion after OPV challenge.
Vaccination coverage in children
One ITS study that involved 28,330 participants (Group Health Cooperative Puget Sound (GHC) = 2721 participants and Kaiser Permanente Northern California (KPNC) = 25,609 enrollees) assessed this outcome (Davis 2001). The changeover from an OPV‐only schedule to one containing IPV had little (if any) negative impact on vaccine coverage (average vaccine coverage: 91.9% with OPV and 92.4% with IPV‐OPV; low‐certainty evidence). There was no difference between the two treatments (RR 1.01, 95% CI 0.96 to 1.06; Analysis 1.20; low‐certainty evidence); the RR for KPNC was 1.04 (95% CI 1.00 to 1.08) and for GHC was 0.99 (95% CI 0.98 to 1.00). This outcome had high levels of heterogeneity (Tau2 = 0.00; Chi2 = 5.69, df = 1 (P = 0.02); I2 = 82%).
1.20. Analysis.

Comparison 1 IPV‐OPV versus OPV, Outcome 20 Vaccination coverage.
At GHC, children who received IPV were less likely to have a missed opportunity visit by 12 months of age (odds ratio (OR) 0.46, 95% CI 0.31 to 0.70), but this finding did not persist at 24 months of age. At Kaiser Permanente, children who received IPV were more likely to have a missed opportunity by 12 months of age (OR 2.06, 95% CI 1.84 to 2.30) and 24 months of age (OR 1.50, 95% CI 1.36 to 1.67). Use of IPV was associated with a small increase in the likelihood of being up to date in polio vaccinations at two years of age at one of the HMOs and, conversely, was associated with a small increase in the likelihood of having a missed opportunity visit in the other HMO.
Safety
Four studies involving 1948 participants assessed serious adverse events (SAE) as classified by the Medical Dictionary for Regulatory Activities Terminology (MedDRA): Anand 2015; Li 2016a; O'Ryan 2015; and Qiu 2017. The evidence suggests that vaccination with IPV‐OPV may make little or no difference to the number of persons with SAEs compared with OPV (RR 0.88, 95% CI 0.46 to 1.70; Analysis 1.21; low‐certainty evidence); heterogeneity was moderate (Tau2 = 0.17, Chi2 = 4.93, df = 3 (P = 0.18); I2 = 39%).
1.21. Analysis.

Comparison 1 IPV‐OPV versus OPV, Outcome 21 Serious adverse events classified by MedDRA.
Sensitivity analysis
To test the robustness of the results, we performed a sensitivity analysis by method of meta‐analysis, comparing the effects of using a random‐effects model (default analysis) to a fixed‐effect model (analyses not shown). We found changes in statistical significance only, not in the direction of the effect. The results of the following analyses changed from non‐statistically significant to statistically significant with a random‐effects model compared to a fixed‐effect model respectively.
-
Analysis 1.13. Mean titres of P1 neutralising antibody:
Sequential IIO: MD −244.37, 95% CI −827.31 to 338.57, P = 0.41 versus MD −453.75, 95% CI −642.57 to −264.93, P < 0.001; and
Sequential IIIOO/IIIO: MD 439.07, 95% CI −354.63 to 1232.77, P = 0.28 versus MD 253.99, 95% CI 130.68 to 377.29, P < 0.001.
Analysis 1.14. Mean titre of P2 neutralising antibody > Sequential IIO: MD 267.40, 95% CI −83.95 to 618.76, P = 0.14 versus MD 127.27, 95% CI 56.53 to 198.00, P < 0.001.
Analysis 1.15. Mean titres of P3 neutralising antibody > Sequential IIIOO/IIIO: MD 248.39, 95% CI −180.58 to 677.37, P = 0.26 versus MD 97.56, 95% CI 32.63 to 162.48, P = 0.003.
Analysis 1.19. Persons with polio faecal extraction after OPV challenge > Persons with P1 faecal excretion: RR 2.24, 95% CI 0.70 to 7.12, P = 0.17 versus RR 1.86, 95% CI 1.21 to 2.86, P = 0.005).
A post hoc sensitivity analysis including studies at low risk of bias for allocation concealment showed an identical effect: RR 1.00, 95% CI 0.99 to 1.02; 4 studies, 1303 participants; I2 = 0% (analysis not shown).
Comparison 2: IPV‐OPV versus IPV
See Table 2.
This comparison includes 10 RCTs with 4675 participants (Anand 2015; Asturias 2007; Faden 1990; Halsey 1997; Jain 1997; Modlin 1997; O'Ryan 2015; Qiu 2017; Rennels 2000; Yeh 2001). We also included one ITS study, which included 2.7 million people (Von Magnus 1984) and one study that used a mixed design (ITS + UBA analysis for VAPP cases) (Alexander 2004).
Paralytic polio
Two nationwide ITS studies assessed this outcome (Alexander 2004; Von Magnus 1984). It is uncertain if IPV‐OPV compared to IPV reduces the number of wild polio cases since both studies provided very low‐certainty evidence of the effect of this outcome. We were unable to combine the data in a meta‐analysis, since the studies reported the data in very different ways. Therefore, we provide a narrative summary of their results.
We re‐analysed the ITS data of the change from an IPV‐OPV to IPV schedule in Alexander 2004 and provided absolute change in level and relative change in level if it was estimable. Considering the year 1997 as transition period, we obtained a change in level or 'step' of −1.2 (95% CI −2.9 to 0.6; −100%), a change in slope trend of 0.0 (95% CI −1.2 to 1.2), and an estimated effect at three years of −1.2 (95% CI −5.0 to 2.7, % not estimable) and at four years of −1.2 (95% CI −6.2 to 3.8; −100%, representing a potential complete elimination of cases; very low‐certainty evidence).
Not considering a transition period, the ITS data showed a change in level of −0.7 (95% CI −2.1 to 0.7; −100%), change in slope of 0.4 (95% CI −1.2 to 0.4), and estimated effect at three years of 0.1 (95% CI −2.5 to 2.7, % not estimable) and at four years of 0.5 (95% CI −2.9 to 3.8, % not estimable).
The polio vaccination program in the Von Magnus 1984 study in Denmark reported 35 cases of AFP due to WPV with an IPV schedule and 1 case with a sequential IPV‐OPV schedule.
Vaccine‐associated paralytic polio (VAPP) cases
No study reported on this outcome.
Vaccine‐derived poliovirus
No study reported on this outcome.
Protective immune responses
Humoral and intestinal immunity
The nationwide ITS study in Denmark, Von Magnus 1984, assessed the use of an IPV‐only schedule before 1961 and the use of the sequential IPV‐OPV schedule during the period 1973 to 1980. The median proportion of people with protective humoral response for poliovirus serotypes 1, 2 and 3 was 82.06%, 91.94% and 76.67% with IPV, and it was higher with IPV‐OPV: 98.44%; 97.67%; and 97.57% respectively (low‐certainty evidence).
We were able to combine data from 10 studies (4204 participants) that provided data on this outcome (Anand 2015; Asturias 2007; Faden 1990; Halsey 1997; Jain 1997; Modlin 1997; O'Ryan 2015; Qiu 2017; Rennels 2000; Yeh 2001.
We grouped, analysed and presented the results according P1, P2 and P3 protective humoral and intestinal immunity.
P1 protective humoral and intestinal immunity
There is no difference between the two treatments for number of people with P1 protective humoral and intestinal immunity (RR 1.00, 95% CI 0.99 to 1.01; P = 0.96; 10 studies, 2858 participants; Analysis 2.1; moderate‐certainty evidence). There is no heterogeneity (Tau2 = 0.00; Chi2 = 13.27, df = 12 (P = 0.35); I2 = 10%).
2.1. Analysis.
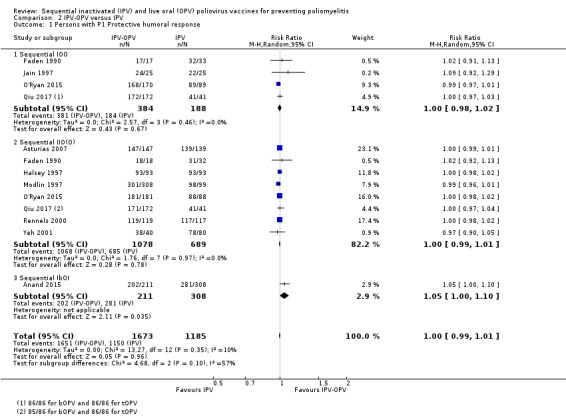
Comparison 2 IPV‐OPV versus IPV, Outcome 1 Persons with P1 Protective humoral response.
Subgroup analyses
The test for subgroup differences indicates that there is no statistically significant subgroup effect by type of dosing sequence although there is moderate heterogeneity (Test for subgroup differences: Chi2 = 4.68, df = 2 (P = 0.10), I2 = 57.3%). Compared with IPV, vaccination with IPV‐OPV using the IbOI dose sequence may increase slightly the number of persons with P1 protective humoral and intestinal immunity (RR 1.05, 95% CI 1.00 to 1.10; 1 study, 519 participants; moderate‐certainty evidence. There are no differences between the two treatment with the other dose sequences:
sequential IOO: RR 1.00, 95% CI 0.98 to 1.02; 4 studies, 572 participants; moderate‐certainty evidence; heterogeneity: Tau2 = 0.00; Chi2 = 2.57, df = 3 (P = 0.46); I2 = 0%; and
sequential IIO(O): RR 1.00, 95% CI 0.99 to 1.01; 8 studies, 1767 participants; moderate‐certainty evidence; heterogeneity: Tau2 = 0.00, Chi2 = 1.76, df = 7 (P = 0.97); I = 0%.
P2 protective humoral and intestinal immunity
There is no difference between the two treatments for the number of people with P2 protective humoral and intestinal immunity (RR 0.97, 95% CI 0.95 to 1.00; P = 0.08; 10 studies, 2907; Analysis 2.2; low‐certainty evidence). However, heterogeneity is high (Tau2 = 0.00; Chi2 = 135.19, df = 13 (P < 0.001); I2 = 90%).
2.2. Analysis.
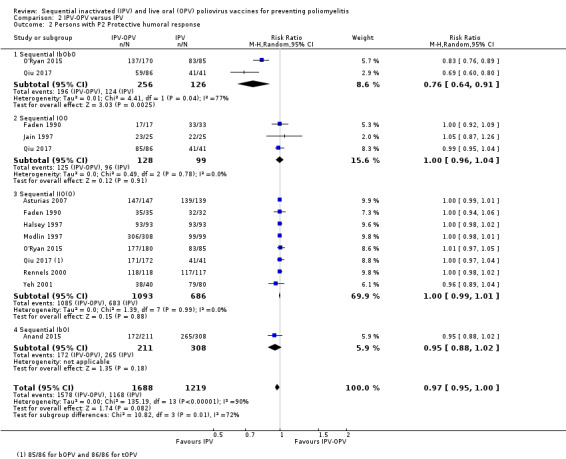
Comparison 2 IPV‐OPV versus IPV, Outcome 2 Persons with P2 Protective humoral response.
Subgroup analyses
The test for subgroup differences indicates that there is a statistically significant subgroup effect by type of dose sequence (Test for subgroup differences: Chi2 = 10.82, df = 3 (P = 0.01), I2 = 72.3%). Vaccination with OPV using the IbObO dose sequence may decrease the number of persons with P2 protective humoral and intestinal immunity (RR 0.76, 95% CI 0.64 to 0.91; 2 studies, 382 participants; low‐certainty evidence); however, there is considerable heterogeneity for this outcome (Tau2 = 0.01; Chi2 = 4.41, df = 1 (P = 0.04); I2 = 77%). There are no differences between the two treatments for:
sequential IOO: RR 1.00, 95% CI 0.96 to 1.04; 3 studies, 227 participants; moderate‐certainty evidence; heterogeneity: Tau2 = 0.00; Chi2 = 0.49, df = 2 (P = 0.78); I2 = 0%;
sequential IIO(O): RR 1.00, 95% CI 0.99 to 1.01; 8 studies, 1779 participants; moderate‐certainty evidence; heterogeneity: Tau2 = 0.00; Chi2 = 1.39, df = 7 (P = 0.99); I2 = 0%); and
sequential IbOI: RR 1.01, 95% CI 0.88 to 1.02; 1 study, 519 participants; moderate‐certainty evidence.
P3 protective humoral and intestinal immunity
There is no difference between the two treatments for the number of people with P3 protective humoral and intestinal immunity (RR 0.99, 95% CI 0.97 to 1.01; P = 0.27; 9 studies, 2620 participants; Analysis 2.3; moderate‐certainty evidence). There is moderate heterogeneity (Tau2 = 0.00; Chi2 = 23.72, df = 11 (P = 0.01); I2 = 54%).
2.3. Analysis.

Comparison 2 IPV‐OPV versus IPV, Outcome 3 Persons with P3 Protective humoral response.
Subgroup analyses
The test for subgroup differences indicates that there is no statistically significant subgroup effect by any of the three types of dose sequences (Test for subgroup differences: Chi2 = 0.52, df = 2 (P = 0.77), I2 = 0%):
sequential IOO: RR 0.99, 95% CI 0.96 to 1.02; 4 studies, 570 participants; I2 = 0%; moderate‐certainty evidence; heterogeneity: Tau2 = 0.00; Chi2 = 4.47, df = 3 (P = 0.21); I2 = 33%;
sequential IIO(O): RR 0.99 CI 0.96 to 1.01; 7 studies, 1531 participants; I2 = 72%; low‐certainty evidence; heterogeneity: Tau2 = 0.00; Chi2 = 21.11, df = 6 (P = 0.002); I2 = 72%; and
sequential IbOI: RR 1.01, 95% CI 0.96 to 1.05; 1 study, 519 participants; moderate‐certainty evidence.
Mean titres of neutralising antibodies
We identified three studies for this outcome (Faden 1990; Qiu 2017; Rennels 2000). Only two of the studies were included in the analysis, Qiu 2017 and Rennels 2000. We excluded the oldest RCT, Faden 1990, from these analyses because it used a very different method to measure antibody titres than the more recent studies. In that study, sequential schemes showed higher neutralising antibodies titre for all serotypes except for P3 for OPV:
sequential IIO: P1 mean titres = 3044; P2 mean titre = 10693; and P3 mean titre = 2347;
sequential IOO: P1 mean titres = 2174; P2 mean titre = 11110; and P3 mean titre = 857; and
sequential III: P1 mean titre = 1954; P2 mean titre = 5835; and P3 mean titre = 5187.
P1 neutralising antibody
The test for subgroup differences indicates that there is a statistically significant subgroup effect by type of dose sequence (Test for subgroup differences: Chi2 = 8.61, df = 3 (P = 0.03), I2 = 65.2%; Analysis 2.4). Vaccination with IPV‐OPV, compared to IPV, may increase the mean titres of P1 neutralising antibodies across all dose sequences shown below.
2.4. Analysis.

Comparison 2 IPV‐OPV versus IPV, Outcome 4 Mean titres of P1 neutralising antibody.
Sequential IbObO: MD 1520.59, 95% CI 1084.80 to 1956.38; 1 study, 127 participants; moderate‐certainty evidence.
Sequential IOO: MD 799.47, 95% CI 530.82 to 1068.12; 1 study, 127 participants; moderate‐certainty evidence.
Sequential IIbO: MD 866.53, 95% CI 478.83 to 1254.23; 1 study, 127 participants; moderate‐certainty evidence.
Sequential IIO: MD 767.90, 95% CI 337.75 to 1198.06; 2 studies, 363 participants; low‐certainty evidence; heterogeneity: Tau2 = 68203.59; Chi2 = 3.39, df = 1 (P = 0.07); I2 = 70%).
P2 neutralising antibody
The test for subgroup differences indicates that there is a statistically significant subgroup effect by type of dose sequence (Test for subgroup differences: Chi2 = 31.27, df = 3 (P < 0.001), I2 = 90.4%; Analysis 2.5). Vaccination with IPV using the IbObO and IIbO sequences may reduce mean titres of P2 neutralising antibodies compared to IPV‐OPV, whereas vaccination with IPV‐OPV using the IOO and IIO sequences may increase mean titres of P2 neutralising antibodies compared with IPV.
2.5. Analysis.

Comparison 2 IPV‐OPV versus IPV, Outcome 5 Mean titres of P2 neutralising antibody.
Sequential IbObO: MD −125.93, 95% CI −174.77 to −77.09; 1 study, 127 participants; moderate‐certainty evidence.
Sequential IOO: MD 142.25, 95% CI 57.65 to 226.85; 1 study, 127 participants; moderate‐certainty evidence.
Sequential IIbO: MD −83.45, 95% CI −133.40 to −33.50; 1 study, 127 participants; moderate‐certainty evidence.
Sequential IIO: MD 2224.48, 95% CI −1145.70 to 5594.67; 2 studies, 362 participants; very low‐certainty evidence of large effect; heterogeneity: Tau = 5829599.24; Chi2 = 69.81, df = 1 (P < 0.001); I2 = 99%.
P3 neutralising antibody
The test for subgroup differences indicates that there is a statistically significant subgroup effect by type of dose sequence (Test for subgroup differences: Chi2 = 7.71, df = 3 (P = 0.05), I2 = 61.1%; Analysis 2.6). Compared to IPV, vaccination with IPV‐OPV using the IbObO and IIbO sequences increases mean titres of P3 neutralising antibodies, but makes little or no difference when using the IOO and IIO sequences.
2.6. Analysis.

Comparison 2 IPV‐OPV versus IPV, Outcome 6 Mean titres of P3 neutralising antibody.
Sequential IbObO: MD 327.62, 95% CI 134.82 to 520.42; 1 study, 127 participants; moderate‐certainty evidence.
Sequential IOO: MD 110.39, 95% CI −77.98 to 298.76; 1 study, 127 participants; low‐certainty evidence.
Sequential IIbO: MD 698.37, 95% CI 301.06 to 1095.68; 1 study, 127 participants; moderate‐certainty evidence.
Sequential IIO: MD 184.52, 95% CI −211.93 to 580.97; 2 studies, 360 participants; I2 = 87%; very low‐certainty evidence; heterogeneity: Tau2 = 71965.06; Chi2 = 7.76, df = 1 (P = 0.005); I2 = 87%.
Long‐term mean titres of neutralising antibody
A single study contributed data on this outcome (Faden 1990).
P1 neutralising antibody
The test for subgroup differences indicates that there is a statistically significant subgroup effect by type of dose sequence (Test for subgroup differences: Chi2 = 13.89, df = 3 (P = 0.003), I2 = 78.4%; Analysis 2.7). Of the four dose sequences, only one was statistically significant, and suggests that vaccination with IPV‐OPV using a IIO + O dose sequence may increase long‐term mean titres of P1 neutralising antibody compared with IPV.
2.7. Analysis.

Comparison 2 IPV‐OPV versus IPV, Outcome 7 Long term mean titres of P1 neutralising antibody.
Sequential IOO: MD −0.20, 95% CI −0.73 to 0.33; 1 study, 37 participants; very low‐certainty evidence.
Sequential IOO + O: MD −0.30, 95% CI −0.83 to 0.23; 1 study, 37 participants; very low‐certainty evidence.
Sequential IIO: MD 0.40, 95% CI −0.04 to 0.84; 1 study, 40 participants; very low‐certainty evidence.
Sequential IIO + O: MD 0.90, 95% CI 0.40 to 1.40; 1 study, 40 participants; low‐certainty evidence.
P2 neutralising antibody
The test for subgroup differences indicates that there is a statistically significant subgroup effect by type of dose sequence (Test for subgroup differences: Chi2 = 16.46, df = 3 (P < 0.001), I2 = 81.8%; Analysis 2.8. Of the four dose sequences, only two were statistically significant; both suggest that IPV with either a IOO + O or a IIO + O dose sequence may reduce long‐term mean titres of P2 neutralising antibody.
2.8. Analysis.

Comparison 2 IPV‐OPV versus IPV, Outcome 8 Long term mean titres of P2 neutralising antibody.
Sequential IOO: MD 0.20, 95% CI −0.06 to 0.46; 1 study, 37 participants; very low‐certainty evidence.
Sequential IOO + O: MD −0.60, 95% CI −0.96 to −0.24; 1 study, 37 participants; low‐certainty evidence.
Sequential IIO: MD 0.10, 95% CI −0.19 to 0.39; 1 study, 40 participants; very low‐certainty evidence.
Sequential IIO + O: MD −0.40, 95% CI −0.78 to −0.02; 1 study, 40 participants low‐certainty evidence.
P3 neutralising antibody
The test for subgroup differences indicates that there is a statistically significant subgroup effect by type of dose sequence (Test for subgroup differences: Chi2 = 8.29, df = 3 (P = 0.04), I2 = 63.8%; Analysis 2.9). None of the individual results were statistically significant and thus we are uncertain of the effects of vaccination with IPV‐OPV, compared with IPV, on long‐term mean titres of P3 neutralising antibody.
2.9. Analysis.

Comparison 2 IPV‐OPV versus IPV, Outcome 9 Long term mean titres of P3 neutralising antibody.
Sequential IOO: MD −0.60, 95% CI −1.22 to 0.02; 1 study, 37 participants; very low‐certainty evidence.
Sequential IOO + O: MD −0.30, 95% CI −0.76 to 0.16; 1 study, 37 participants; low‐certainty evidence.
Sequential IIO: MD 0.30, 95% CI −0.04 to 0.64; 1 study, 40 participants; very low‐certainty evidence.
Sequential IIO + O: MD −0.10, 95% CI −0.46 to 0.26; 1 study, 40 participants; very low‐certainty evidence.
Intestinal immunity
Three relevant studies with 2995 participants provided data on intestinal immunity assessed as polio faecal excretion after OPV challenge (Anand 2015; Modlin 1997; O'Ryan 2015).
People with P1 faecal excretion
There is no difference between the two treatments for the number of people with P1 faecal excretion after OPV challenge (RR 0.52, 95% CI 0.14 to 1.97; 2 studies, 822 participants I2 = 94%; data not shown in analysis; very low‐certainty evidence).
People with P2 faecal excretion
There is evidence that IPV‐OPV may reduce P2 faecal excretion after OPV challenge (RR 0.55, 95% CI 0.31 to 0.94; 3 studies, 1351 participants; I2 = 94%; data not shown in analysis; low‐certainty evidence).
People with P3 faecal excretion
There is evidence that IPV‐OPV probably reduces P3 poliovirus faecal excretion after OPV challenge compared to IPV (RR 0.39, 95% CI 0.32 to 0.47; 2 studies, 822 participants; I2 = 0%; data not shown in analysis; moderate‐certainty evidence).
Subroup analysis: type of dose sequence
The test for subgroup differences indicates that there is a statistically significant subgroup effect by type of dose sequence (Test for subgroup differences: Chi2 = 61.83, df = 5 (P < 0.001), I2 = 91.9%; Analysis 2.10).
2.10. Analysis.
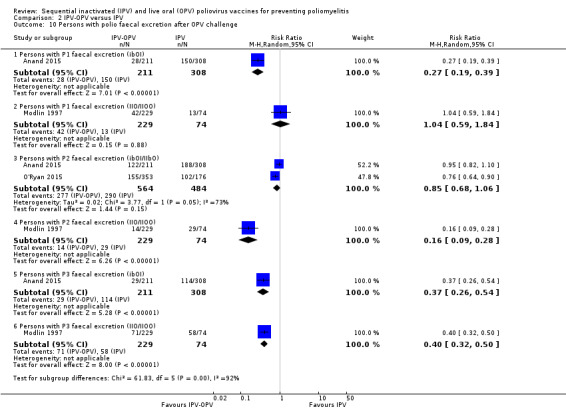
Comparison 2 IPV‐OPV versus IPV, Outcome 10 Persons with polio faecal excretion after OPV challenge.
There is evidence that, compared to IPV, vaccination with IPV‐OPV probably reduces:
P1 faecal excretion after OPV challenge with sequential ibOI (RR 0.27, 95% CI 0.19 to 0.39; 1 study, 519 participants; moderate‐certainty evidence but not with IIO/IIOO (RR 1.04, 95% CI 0.59 to 1.84; 1 study, 303 participants; low‐certainty evidence);
P2 faecal excretion after OPV challenge with sequential IIO/IIOO (RR 0.16, 95% CI 0.09 to 0.28; 1 study, 303 participants; moderate‐certainty evidence but not with ibOI/IIbO (RR 0.85, 95% CI 0.68 to 1.06; 2 studies, 1048 participants; low‐certainty evidence; heterogeneity: Tau2 = 0.02; Chi2 = 3.77, df = 1 (P = 0.05); I2 = 73%); and
P3 faecal excretion after OPV challenge with sequential ibOI (RR 0.37, 95% CI 0.26 to 0.54; 1 study, 519 participants; moderate‐certainty evidence of large effect) and with IIO/IIOO RR 0.40, 95% CI 0.32 to 0.50; 1 study, 303 participants; moderate‐certainty evidence.
Vaccine coverage in children
No study reported on this outcome.
Safety
Two studies reported on this outcome. O'Ryan 2015 reported 81 serious adverse events (SAE), of which one was thought to possibly be vaccine‐related (intestinal intussusception four days after receiving the mOPV2 challenge). In Qiu 2017, no vaccine‐related SAEs were reported. Infectious pneumonia was the main SAE (10 participants, 1.67%), followed by bronchitis (four participants, 0.67%) and hand‐foot‐and‐mouth disease (four participants, 0.67%).
There was little or no difference between the schedules on number of people experiencing one or more serious adverse event (vaccine related or not) (RR 0.92, 95% CI 0.60 to 1.43; 2 studies, 1063 participants; Analysis 2.11; low‐certainty evidence; heterogeneity: Tau2 = 0.00; Chi2 = 0.82, df = 1 (P = 0.37); I2 = 0%).
2.11. Analysis.
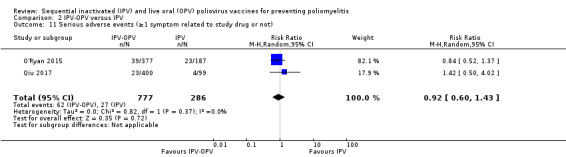
Comparison 2 IPV‐OPV versus IPV, Outcome 11 Serious adverse events (≥1 symptom related to study drug or not).
Sensitivity analysis
To test the robustness of the results, we performed a sensitivity analysis comparing using a random‐effects model (default analysis) to a fixed‐effect model (analyses not shown). We found changes in statistical significance only, not in the direction of the effect. The results of the following analyses changed from non‐statistically significant to statistically significant with a random‐effects model compared to a fixed‐effect model, respectively.
Analysis 2.2. Persons with P2 protective humoral response: RR 0.97, 95% CI 0.95 to 1.00, P = 0.07 versus RR 0.96, 95% CI 0.94 to 0.98, P < 0.001.
Analysis 2.3.2. Persons with P3 protective humoral response > Sequential IIO(O): RR 0.99, 95% CI 0.96 to 1.01, P = 0.39 versus RR 0.98, 95% CI 0.97 to 1.00, P = 0.03.
Analysis 2.5.4. Mean titres of P2 neutralising antibody > Sequential IIO: MD 2224.48, 95% CI −1145.70 to 5594.67, P = 0.20 versus MD 787.97, 95% CI 573.84 to 1002.09, P < 0.001.
Analysis 2.10.3. Persons with polio faecal extraction after OPV challenge > persons with P2 faecal excretion after OPV challenge (IbOI/IIbO): RR 0.85, 95% CI 0.68 to 1.06, P = 0.15 versus RR 0.86, 95% CI 0.77 to 0.96, P = 0.007.
Comparison 3: IPV(3)‐OPV versus IPV(2)‐OPV
In this comparison, a single study with 137 participants reported on one outcome only: protective humoral response (Linder 2000).
Protective immune responses: humoral and intestinal immunity
There is no difference between the two treatments for P1 (RR 0.98, 95% CI 0.93 to 1.03); P2 (RR 1.00, 95% CI 0.97 to 1.03); or P3 (RR 1.01, 95% CI 0.97 to 1.05) protective humoral and intestinal response (see Analysis 3.1; all moderate‐certainty evidence, Table 3).
3.1. Analysis.

Comparison 3 IPV(3)‐OPV versus IPV(2)‐OPV, Outcome 1 Persons with polio protective humoral response.
Subgroup analyses
The test for subgroup differences indicates that there is no statistically significant subgroup effect by type of protective humoral response (test for subgroup differences: Chi2 = 0.98, df = 2 (P = 0.61), I2 = 0%).
Discussion
Summary of main results
The review includes 21 studies involving 6407 healthy infants in 16 randomised controlled trials (RCTs), 28,330 infants in one interrupted time series (ITS) and also four nationwide studies (from USA Denmark and Hungary). Below we summarise the main findings and outstanding uncertainties by comparison.
Comparison 1: IPV‐OPV versus OPV
See Table 1.
It is uncertain if IPV‐OPV compared to OPV reduces the number of wild polio cases (very low‐certainty evidence); however, it may substantially reduce cases of vaccine‐associated paralytic polio (VAPP) (low‐certainty‐evidence).
We found no important difference between the two treatments for the number of persons with P1, P2 and P3 protective humoral response (moderate‐ and low‐certainty evidence); however; for P2; the sequential scheme IbObO may be worse than tOPV‐only scheme.
The subgroup analysis showed variability in the results for all three poliovirus serotypes depending on the schedule used. The most recommended schedule, two doses of IPV followed by one or more doses of OPV (IIO/OO), probably makes little or no difference to the number of people with protective humoral response (moderate‐certainty evidence for P1 and low‐certainty evidence for P2 and P3). There were no differences with other schedules with only one IPV dose for P1, but the response for poliovirus serotype P2 may be lower with IbOI than tOPV only scheme (low‐certainty evidence) (subgroup differences I2 = 96.3%). The response for poliovirus serotype P3 is probably lower with IbObO (moderate‐certainty evidence) (subgroup differences I2 = 82.8%).
Two doses of IPV followed by one or more doses of OPV (IIO/OO), may make little or no difference to P1 mean titres of neutralising antibodies across all types of dose sequences (low‐certainty evidence). There are differences for poliovirus serotypes P2 and P3. Specifically, for P2, the IIbO and IbObO dose sequences may reduce mean titres of neutralising antibodies (moderate‐certainty evidence), whereas the IOO, IIO and IIIOO/IIIO (all low‐certainty evidence) dose sequences may make little to no difference (subgroup differences: I2 = 91.7%). Regarding P3, vaccination with IPV‐OPV using the IbObO, IIbO, and IIO dose sequences may increase mean titres of P3 neutralising antibodies compared to OPV, whereas using the IOO and IIIOO/IIIO dose sequences may make little or no difference on the mean titres of P3 neutralising antibodies compared with OPV (low‐certainty and very low‐certainty evidence respectively) (subgroup differences: I2 = 61.6%).
IPV‐OPV seems to generate less mucosal immunity, since it probably increases polio virus serotype P2 and P3 faecal excretion after OPV challenge (both moderate‐certainty evidence), and it may increase or decrease the number of persons with P1 polio faecal excretion after OPV challenge (low‐certainty evidence).
There were no significant subgroup effects by time of first dose or country or location.
Compared to OPV, vaccination with IPV‐OPV may make little or no difference to vaccination coverage or the incidence of serious adverse events (both low certainty‐evidence).
Changing the model used for analysis, from a random‐effects to a fixed‐effect model, only changed the level of statistical significance for a few outcomes (mean titres of neutralising antibodies for IIO (P1 and P2) and IIIOO/IIIO (P1 and P3) dose sequences, and the number of people with P1 polio faecal excretion after OPV challenge). It did not change the direction or the interpretation of the results.
Comparison 2: IPV‐OPV versus IPV
See Table 2.
It is uncertain if IPV‐OPV compared to IPV reduces the number of wild polio cases (very low‐certainty evidence). There were no data on the number of VAPP cases.
Compared to IPV, IPV‐OPV probably makes little or no difference to the number of people with protective humoral response for poliovirus serotypes P1 and P3 (moderate‐certainty evidence) and makes little or no difference for P2 (low‐certainty evidence). The subgroup analysis showed no statistically significant subgroup effect for P1 and P3, but did for P2: the number of people with a protective humoral response is probably lower for P2 only, with an IbObO sequence.
A subgroup analysis indicated differences in mean titres of neutralising antibodies for poliovirus serotypes 1, 2 and 3 by type of dose sequence. Vaccination with IPV‐OPV, compared to IPV, may increase the mean titres of P1 neutralising antibodies across all dose sequences (low‐ and moderate‐certainty evidence). Using the IbObO and IIbO sequences may reduce mean titres of P2 neutralising antibodies compared to IPV‐OPV, whereas using the IOO and IIO sequences may increase mean titres of P2 neutralising antibodies compared with IPV (moderate‐ and very low‐certainty evidence). The IbObO and IIbO sequences increases mean titres of P3 neutralising antibodies, but makes little or no difference when using the IOO and IIO sequences (very low, low, and moderate‐certainty evidence).
IPV‐OPV probably reduces the number of people with P3 poliovirus faecal excretion after OPV challenge (moderate‐certainty evidence), and may reduce the number of people with P2 poliovirus faecal excretion after OPV challenge (low‐certainty evidence). However, there was no difference between the two treatments for P1 (very low‐certainty evidence). A subgroup analysis by dose sequence showed that the following sequences probably reduce the number of people with poliovirus faecal excretion after OPV challenge: ibOI for P1 and P3 (moderate‐certainty evidence); and IIO/IIOO for P2 and P3 (both moderate‐certainty evidence). There was no difference between the two treatments for the number of people with poliovirus faecal excretion after OPV challenge with the following dose sequences: IIO/IIOO for P1; and ibOI/IIbO for P2 (both low‐certainty evidence).
IPV‐OPV compared to IPV may make little or no difference to the incidence of serious adverse events.
Changing the model used for analysis, from a random‐effects to a fixed‐effect model, only changed the level of statistical significance for a few outcomes (number of people with P2 protective humoral response and P3 protective humoral response with the IIO(O) dose sequence, P2 mean titres of neutralising antibodies with the IIO dose sequence, and number of people with P2 polio faecal excretion after OPV challenge with the IbIO/IIbO dose sequence). It did not change the direction or the interpretation of the results.
Comparison 3: IPV(3)‐OPV versus IPV(2)‐OPV
See Table 3.
The only outcome assessed by this study was protective humoral response. Three doses of IPV followed by two doses of OPV, compared to two doses of IPV followed by two doses of OPV probably makes little or no difference to the number of persons with protective humoral response (moderate‐certainty evidence).
Overall completeness and applicability of evidence
The relevance of the evidence identified in this review is completely applicable to the review question regarding participants and interventions but not completely applicable to the reported outcomes. Our broad systematic review, in terms of eligible study designs, allowed us to deal, in part, with this limitation. The included RCTs provided a reasonable body of evidence about immunogenicity and short‐term safety concerns. Quasi‐experimental designs (most nationwide) two ITS studies, two uncontrolled before‐after (UBA) studies, and one study that used a mixed ITS and UBA design provided uncertain evidence about wild polio cases, but probably constitute the best available (albeit low‐certainty) evidence regarding the reduction of VAPP cases associated with IPV‐OPV compared to OPV. Based on these considerations we are confident in the external validity of our review.
Considering the most recent Wordl Health Organization (WHO) recommendations, our findings align with current practice. In 2012, SAGE recommended the replacement of trivalent OPV (tOPV; containing serotypes 1, 2, and 3) with bivalent OPV (bOPV), containing only serotypes 1 and 3, in all countries by 2016, preceded by the introduction of at least one dose of IPV in routine immunisation programmes (WHO 2012). The Polio Eradication and Endgame Strategic Plan 2013 to 2018 recommended completion of IPV introduction and globally synchronised withdrawal of OPV serotype 2 in 2016 (WHO 2015a). These recommendations are based on the fact that wild polio virus (WPV) serotype 2 has not been detected since 1999 and serotype 3 since 2012. All cases detected from 2013 onwards are due to serotype 1 (WHO 2015d, WHO 2016). However, approximately 400 to 500 cases per year of VAPP are caused by OPV (Platt 2014), which means that the cases of paralytic poliomyelitis caused by vaccination already exceed those caused by WPV, and are becoming the main source of polio paralysis in the world. This means that polio‐free is not polio risk‐free because live‐attenuated Sabin viruses from OPV could revert to virulence causing VAPP or cVDPV (Minor 2009). Serotype 2 vaccine‐related viruses continue to induce paralysis and cause 26% of cases of VAPP in vaccinees, 31% in contacts and more than 90% of all cVDPVs (Platt 2014, WHO 2018). After SAGE's recommendation, 155 countries using OPV in their immunisation programs changed from tOPV to bOPV (WHO 2012). This globally synchronised switch in vaccination policy, during a two‐week period, represents an historical milestone reached in the polio eradication effort (Garon 2016). Even if the two most recent cases of WPV, in February 2018 in Afghanistan, were the last in the world, bOPV will not be retired until 2022 to 2023, after more than three years have passed since the last case (Peng 2018). For this reason, WHO also decided that at least one IPV, containing the three serotypes of inactivated poliovirus, should be included in the immunisation programs of countries using only OPV schemes to protect against possible future outbreaks of poliovirus serotype 2. The main challenge to this plan is the availability of IPV; the insufficient production of these vaccines, which is unlikely to be remedied in the short term (Anand 2017). In an unpublished, ongoing study, Peng and colleagues observed five new cases of VAPP in 2016 after administration of the first dose of bOPV, due to a shortage of IPV (Peng 2018).
Given that bOPV will continue to be used widely for several more years, and that new cases of VAPP will inevitably occur, it is essential to discuss key issues such as sequence, timing, pre‐vaccination contraindication screening, relative risk of VAPP with different doses of OPV, and accessibility to IPV. A more rigorous screening of contraindications for bOPV is truly important, since the WHO suggests that immunocompromised patients should avoid vaccination with bivalent bOPV. The evidence suggests that, although bOPV administration is much safer after IPV administration, VAPP can still occur (Desai 2014). This finding reminds us that the detection of contraindications cannot be avoided while using OPV. The WHO recommends one or two doses of IPV followed by at least two doses of bOPV in countries with high vaccination coverage (greater than 90%) and low risk of imported WPV (population movement with similar vaccination coverage). Conversely, in countries with endemic polio, lower vaccination coverage or high importation risk (polio importation history or countries bordering with areas of endemic polio or with recurrent outbreaks), the WHO recommends one or more doses of bOPV followed by IPV (WHO 2015d). The reason for this difference is based on how WHO weights the relative risks of VAPP versus WPV in these two classifications of countries. There is a long‐standing perception that the relative risk of VAPP with the first dose of OPV is lower in low‐income countries than in high‐income countries. This is clearly reflected in a response from Zhang 2016 to the hypothesis posited by Asturias 2016: that IPV‐bOPV sequential administration could be safer than bOPV‐IPV. Asturias and colleagues state that, in high‐ and middle‐income countries, the risk of VAPP is 6.6 times higher with the first dose of OPV than with later doses, while in low‐income countries, the majority of VAPP cases appear after receiving multiple, previous doses of OPV (Asturias 2016). However, as highlighted by Peng 2018, this conclusion comes from a misinterpretation of a single study conducted in India, which showed that the risk of VAPP with the first dose of OPV was one case per 2.8 million of doses and the risk with subsequent doses was one case per 13.9 million doses (Kohler 2002). Therefore, the risk of VAPP with the first dose of OPV was almost five times greater than the risk associated with subsequent doses. Other studies in countries of different socioeconomic status, such as the USA (Alexander 2004), Brazil (de Oliveira 2000), China (Wu 2018) and Latin America and the Caribbean (Landaverde 2014), showed that the risk of VAPP with the first dose of OPV was between 4 and 13 times greater than the risk with subsequent doses. Considering the history of OPV vaccination, available for 49% of the total VAPP cases, 74% of cases occurred after the first dose of OPV, and 8%, 7%, and 11% occurred after the second, third, or fourth or more doses, respectively (Platt 2014). Among contacts, 52% of cases had never been vaccinated with OPV and the risk of VAPP with the first dose of OPV seems more balanced, with 11%, 8%, and 28% after receiving 1, 2, or 3 or more previous OPV doses, respectively. A low number of recipient and contact VAPP cases reported a history of previous vaccination with IPV, almost any of these cases with enhanced‐potency IPV, that was introduced in the late 1960s (Platt 2014). Although the studies were from tOPV, there is no reason to think that with the bOPV it would be very different.
The evidence also suggests that the first dose of IPV can induce seroconversion and 'priming' (rapid seroconversion a week after challenge with OPV) in more than 90% of immunised children (Resik 2013). A systematic review and meta‐analysis of studies documenting seroconversion following one or two full or fractional doses of enhanced‐potency IPV showed that routine immunisation with two full or fractional doses of IPV given after 10 weeks of age is likely to protect more than 80% of recipients against poliomyelitis (Grassly 2014). More importantly, we found that VAPP was substantially prevented after the use of the sequential IPV‐OPV scheme in the immunisation programs in countries regardless of their income classification, suggesting that IPV makes subsequent doses of OPV safer. Additionally, multiple intramuscular injection during the 30‐day period after immunisation with OPV was found to be a risk factor for VAPP (Strebel 1995).
The burden caused by VDPV is another problem that is becoming increasingly important to manage. WHO databases reported 798 cases of cVDPV in 25 countries around the world between 2000 and 2016; none caused by IPV, two by IPV‐OPV and the remainder of cases (99.7%) caused by OPV or OPV‐IPV. A proportion meta‐analysis using a random‐effects model estimated an annual incidence of 14 cases of cVDPV per million. Although the cVDPV incidence is low, there is no evidence that cVDPV tends to disappear on its own (Ciapponi 2017). Long‐term risks include the reintroduction of poliovirus serotype 2 from a laboratory or manufacturing facility breach, as occurred in 2002 to 2003 in India (Deshpande 2003). For these reasons, immunity levels against polioviruses should be kept as high as possible in the population by the use of IPV, and both clinical and environmental surveillance should be maintained as high as possible. Moreover, if the use of mOPV serotype 2 is required to control an outbreak, it will be easier to reach the levels of immunity necessary to stop transmission in a population previously vaccinated with IPV. Therefore, the introduction of IPV could facilitate the control of outbreaks in the future (Gentile 2016).
Many countries (n = 49, see Table 4) use IPV as the first dose in a sequential vaccination scheme. However, according to the WHO database of national vaccination schedules, in 2018, 89 countries used bOPV as the first dose of a sequential or combined scheme with IPV, and five used bOPV exclusively (WHO 2019). The decision to use bOPV as the first dose is usually taken because bOPV can confer early protection, since it can be administered from birth (WHO 2015d). In contrast, IPV is generally used at an older age (around two months old), to avoid interference from maternal antibodies and maximise immunogenicity. The initial use of bOPV is a good policy when the risk of infection with WPV is high. However, given that the global incidence of WPV has decreased to an unprecedented level and the relative risk of VAPP and cVDPV in low‐ and middle‐income countries (LMICs) is not as low as previously thought, it is perhaps time to review this recommendation.
Availability and affordability of IPV is another key issue to consider. In countries with a shortage of IPV, two doses of intradermal fIPV prior to bOPV could be considered (Anand 2017; WHO 2015d). In this review, we identified a study, Anand 2015, which showed no differences between the sequential IbOI scheme (two fIPV) and two complete doses of IPV, and even found a slight, statistically significant effect in favour of IbOI in the P1 protective humoral response. In addition, a review found that two doses of flPV were more immunogenic than a single full dose (Anand 2017). Two doses of flPV represent only two fifths of a full IPV dose and would attenuate the current shortage of IPV. In fact, the scheme of two flPVs at six and 14 weeks of age has been supported by technical supervision committees and has been introduced in some affected countries (WHO 2016). However it should be considered that implementation of intradermal vaccines for fIPV could be more difficult because require trained personnel. Currently, 21 out of 49 countries using IPV‐OPV (43%) and 82 out of 86 countries using OPV‐IPV (95%) apply a single dose of IPV. Replacing this full IPV dose with two doses of fIPV, could improve immunogenicity, reduce costs and difficult coverage in a context of shortage of IPV (Lewis 2017).
Several economic evaluations from different settings have studied the costs and cost‐effectiveness of introducing IPV into immunisation programs (Biffi 2003; Duintjer Tebbens 2006; Duintjer Tebbens 2015; Mascareñas 2005; Miller 1996; Sartori 2015). Although inconsistent, their results (summarised in Table 8), provide useful evidence for the design of future immunisation programs, taking into consideration their different frameworks, time periods, and discount over time (discounting tell us how much future benefits and costs are worth today). One way to make IPV more affordable is to reduce the dose by adding adjuvants (compounds that augment the immune response to the vaccine). A systematic review summarised the evidence from studies evaluating the potential efficacy and safety of adjuvants used with IPV (Hawken 2012).
5. Main results of economic evaluations identified.
| Study ID Country | Study period | Monetary unit | Schedule | Total program cost $ millions | Total program saving $ millions | Total benefits $ millions | Net incremental cost $ millions per year | Cost per case of VAPP prevented $ millions per year |
| Miller 1996 USA | 1980‐1991 | USD 1995 | 4 OPV (Ref) | 375 | ‐ | ‐ | ‐ | ‐ |
| 4 IPV | 414.5 | ‐ | 11.4 | 28.1 | 3 | |||
| 2 OPV 2 IPV | 395.4 | ‐ | 5.7 | 14.7 | 3.1 | |||
| Summary: cost‐benefit and cost‐effectiveness models were formulated to compare the USA national 4‐OPV dose with a 4‐dose IPV schedule or a sequential schedule of 2 doses of IPV followed by 2 doses of OPV. Changing to an IPV‐only or a sequential schedule would cost $28.1 million and $14.7 million, respectively. The costs per case of VAPP prevented were estimated as $3.0 million and $3.1 million for each option, respectively. It concluded that the introduction of IPV into the routine vaccination schedule would not be cost‐beneficial at 1995 vaccine prices and with the current compensation awards paid to VAPP cases since, the costs are higher than other public health prevention programs. | ||||||||
| Biffi 2003 Italy | 2000 (life expectancy of 75 years) | Euros 2000 | 4 OPV (Ref) | ‐ | ‐ | ‐ | ‐ | ‐ |
| 2 OPV 2 IPV | ‐ | ‐ | ‐ | 2.8 | 2.9 | |||
| Summary: in Italy, a sequential schedule based on two IVP doses followed by two OPV doses replaced in 1999 the OPV‐only schedule to reduce the incidence of VAPP, the most dangerous adverse event of OPV. Assuming an hypothetical VAPP reduction, an economic evaluation estimated that a sequential schedule would avoid 0.768 cases/year; however, the costs of the sequential schedule outweigh the expected economic benefits associated with a decreased incidence of VAPP. | ||||||||
| Mascareñas 2005 Mexico | 2002 | USD 2002 | 4 OPV (Ref) | ‐ | ‐ | ‐ | ‐ | ‐ |
| NIW | 100,454 to 156,614 | ‐ | ‐ | ‐ | ‐ | |||
| 4 IPV | ‐ | 28.8 | ‐ | ‐ | ‐ | |||
| 2 OPV 2 IPV | ‐ | 18.6 | ‐ | ‐ | ‐ | |||
| Summary: a prospective Mexican, micro‐costing study estimated that changing from the current OPV‐based intensive and routine schedule to a sequential IPV‐OPV routine schedule would save US $14.52 per vaccinated child, and changing to a full IPV routine schedule would save US $9.41 per vaccinated child. It also estimated a national immunisation week (NIW) cost. | ||||||||
| Sartori 2015 Brazil | 2011 | USD 2011 | 5 OPV + 2 NIDs (Ref) | 19,873,170 | ‐ | ‐ | ‐ | ‐ |
| 1 IPV 4 OPV | 14,608,419 | −26.50% | ‐ | ‐ | ‐ | |||
| 2 IPV 3 OPV | 22,852,799 | 15.00% | ‐ | ‐ | ‐ | |||
| 3 IPV 2 OPV | 31,283,072 | 57.40% | ‐ | ‐ | ‐ | |||
| 4 IPV 4 | 38,936,547 | 95.90% | ‐ | ‐ | ‐ | |||
| 5 IPV | 41,681,259 | 109.70% | ‐ | ‐ | ‐ | |||
| 1 IPV 4 OPV + 1 NID | 21,409,159 | 7.70% | ‐ | ‐ | ‐ | |||
| 2 IPV 3 OPV + 1 NID | 29,653,539 | 49.20% | ‐ | ‐ | ‐ | |||
| 3 IPV 2 OPV + 1 NID | 38,083,812 | 91.60% | ‐ | ‐ | ‐ | |||
| 4 IPV 4 + 1 NID | 45,737,287 | 130.20% | ‐ | ‐ | ‐ | |||
| 5 IPV + 1 NID | 48,481,999 | 143.90% | ‐ | ‐ | ‐ | |||
| 1 IPV 4 OPV + 2 NID | 28,209,899 | 41.90% | ‐ | ‐ | ‐ | |||
| 2 IPV 3 OPV + 2 NID | 36,454,279 | 83.40% | ‐ | ‐ | ‐ | |||
| 3 IPV 2 OPV + 2 NID | 44,884,552 | 125.90% | ‐ | ‐ | ‐ | |||
| 4 IPV 4 + 2 NID | 52,538,027 | 164.40% | ‐ | ‐ | ‐ | |||
| 5 IPV + 2 NID | 55,282,740 | 178.20% | ‐ | ‐ | ‐ | |||
| Summary: the introduction of IPV in Brazil increased the annual costs of the polio vaccines by 49.2% compared with the oral vaccine‐only regimen. This increase represented 1.13% of the expenditure of the national immunisation program on the purchase of vaccines in 2011. | ||||||||
| Duintjer Tebbens 2006 Wordwide | ‐ | ‐ | HIC: IPV | 6100 | ‐ | ‐ | ‐ | ‐ |
| HIC: AFP | 800 | ‐ | ‐ | ‐ | ‐ | |||
| Total | 6900 | ‐ | ‐ | ‐ | ‐ | |||
| UMIC: IPV | 1300 | ‐ | ‐ | ‐ | ‐ | |||
| UMIC: SIAs | 1700 | ‐ | ‐ | ‐ | ‐ | |||
| UMIC: OPV | 700 | ‐ | ‐ | ‐ | ‐ | |||
| UMIC: AFP | 400 | ‐ | ‐ | ‐ | ‐ | |||
| Total | 4100 | ‐ | ‐ | ‐ | ‐ | |||
| LMIC: IPV | 3100 | ‐ | ‐ | ‐ | ‐ | |||
| LMIC: SIAs | 2100 | ‐ | ‐ | ‐ | ‐ | |||
| LMIC: OPV | 1100 | ‐ | ‐ | ‐ | ‐ | |||
| LMIC: AFP | 700 | ‐ | ‐ | ‐ | ‐ | |||
| Total | 7000 | ‐ | ‐ | ‐ | ‐ | |||
| LIC: IPV | 3900 | ‐ | ‐ | ‐ | ‐ | |||
| LIC: SIAs | 1700 | ‐ | ‐ | ‐ | ‐ | |||
| LIC: OPV | 1900 | ‐ | ‐ | ‐ | ‐ | |||
| LIC: AFP | 1100 | ‐ | ‐ | ‐ | ‐ | |||
| LIC: Total | 8600 | ‐ | ‐ | ‐ | ‐ | |||
| Summary: a model for the expected future costs of different polio strategies estimated that a global transition from routine immunisation with OPV to IPV would increase the costs of managing polio globally, although routine IPV use remains less costly than routine OPV use with supplemental immunisation activities. The uncertainty in the aggregated costs, the discount rate and price and administration cost of IPV drives the expected incremental cost of routine IPV vs OPV immunisation. | ||||||||
| Duintjer Tebbens 2015 200 countries | 2013–2052 | USD 2013 | tOPV (Ref) | ‐ | ‐ | ‐ | ‐ | ‐ |
| LIC: ≥ 1 IPV no SIAs | ‐ | ‐ | 4700 | ‐ | ‐ | |||
| LMIC: ≥ 1 IPV no SIAs | ‐ | ‐ | 15,000 | ‐ | ‐ | |||
| UMIC: ≥ 1 IPV no SIAs | ‐ | ‐ | −400 | ‐ | ‐ | |||
| HIC: ≥ 1 IPV no SIAs | ‐ | ‐ | −3500 | ‐ | ‐ | |||
| World: ≥ 1 IPV no SIAs | ‐ | ‐ | 16,000 | ‐ | ‐ | |||
| Summary: an integrated dynamic poliovirus transmission and stochastic risk model simulated possible futures and estimate the health and economic outcomes of maintaining the 2013 status quo of continued OPV use in most developing countries compared with OPV cessation policies with various assumptions about global IPV adoption. The authors estimated a global incremental net benefits during 2013‐2052 of approximately 16 US $2013 billion (almost 20 billion in LMICs) with at least one IPV routine immunisation dose in all countries until 2024 compared to continued OPV use, although significant uncertainty remains associated with the frequency of exportations between populations and the implementation of long‐term risk‐management policies. | ||||||||
| AFP: acute flaccid paralysis; HIC: high‐income country; IPV: inactivated poliovirus vaccine; LIC: low‐income country;LMIC: lower‐middle income country; NID: national immunisation days;NIW: national immunisation week; OPV: oral poliovirus vaccine; SIA: supplemental immunisation days; UM: upper‐middle‐income country; USD: US dollars; VAPP: vaccine‐associated paralytic poliomyelitis. | ||||||||
For definition of schedules (e.g. sequential IOO), see Glossary in Appendix 1.
The Sabin IPV (sIPV) could also play an important role in the final phase of global polio eradication. sIPV has high production safety and low production cost, compared with the conventional wild‐virus‐derived IPV (Dong 2016). Additionally, an RCT showed that sequential schedules of sIPV‐bOPV have good safety and immunogenicity, with the two‐dose sIPV group showing slight superiority to the one‐dose sIPV group (Ye 2018). It is likely that most new IPVs will be made with Sabin strain viruses, which reduce the risks to the population from a breach of containment. sIPV combined with tetanus and diphtheria toxoids and acellular pertussis vaccine was introduced in Japan in 2012, and two stand‐alone sIPVs have been licensed for distribution in China (Modlin 2019). Additional to availability and affordability issues, good immunisation strategy can lead to high coverage with a new vaccine without negative impact attached to other vaccines (Domingues 2014).
The main potential benefit of the sequential IPV‐OPV scheme, compared to OPV alone, is that it may reduce or eliminate VAPP cases while limiting the risks of VDPVs and maintaining good mucosal immunity. The success in controlling outbreaks of polio during the final stage of eradication will depend, to a large extent, on the degree of immunity of the intestinal mucosa. A systematic review found that, compared with unvaccinated individuals, IPV did not induce intestinal mucosal immunity to reduce faecal vaccine virus shedding after challenge with OPV (OR 0.81, 95% CI 0.59 to 1.11) (Hird 2012). An analytical cohort comprising 152 infants (Brickley 2018), derived from O'Ryan 2015, supports this concept; 37% of infants, in the IPV‐bOPV groups and 26% in the IPV‐only arm had detectable serotype 2–specific stool neutralisation after the primary vaccine series and almost half of all study participants continued to shed the virus four weeks after the challenge. However, a challenge dose of mOPV2 induced intestinal immune responses in all groups, reflected by statistically significant (P < 0.001) rises in serotype 2–specific stool neutralisation titres and immunoglobulin A concentrations in both arms, but Infants who received IPV alone presented with inferior serotype 1 intestinal immunity than participants who had received at least one dose of bOPV.
IPV‐only shedding studies have shown that the odds of excreting poliovirus serotype 2 after OPV challenge are similar between unvaccinated children and children vaccinated with two or three doses of IPV (Cuba 2007; Laassri 2005). These findings reinforce concerns that immunisation by inactivated, rather than live, vaccines have the potential to maintain transmission if OPV or WPV strains (or both) remain in circulation.
Sequential schedules that currently use bOPV (IPV–bOPV) have the potential to achieve immunity to poliovirus serotype 2 by giving one or more IPV doses before bOPV, which could also prevent serotype 2 VAPP and VDPV. The overall protection rate against serotype 2 after an IPV–bOPV–bOPV schedule would be up to 92%, compared with the 98% to 100% noted after schedules with two or three doses of IPV. Additionally, some benefits could arise from enhanced mucosal immunity due to bOPV use, following the global introduction of IPV and switch to bOPV. The induction of cross‐protective intestinal immunity from bOPV to serotype 2 could provide enhanced individual immunity, and could also decrease transmission of WPV serotype 2 or vaccine‐related virus after withdrawal of tOPV from vaccination schedules (O'Ryan 2015),
Results from a randomised trial by Lund 2015 suggest that OPV might have beneficial, non‐specific effects that reduce all‐cause mortality by 17% and possibly to a greater extent in boys than in girls, whereas previous evidence suggests that IPV increases all‐cause mortality by 10% (Aaby 2007). Although controversial, these results support the idea that a sequential IPV‐OPV schedule might decrease VAPP without increasing mortality (Fish 2016).
Additionally, poliovirus importations into polio‐free countries around the world represent a major concern during the final phases of global eradication of WPV. Extended dynamic transmission models demonstrate that as population immunity declines below the threshold required for preventing transmission, countries become at risk for re‐established transmission. All countries should invest in active management of population immunity to avoid the potential circulation of imported live polio viruses (Thompson 2015).
Although the WHO strategy is to stop OPV vaccination completely and replace it with IPV vaccination, this goal is not close yet, and the sequential IPV‐OPV vaccination schedule could have an important role during the transition period. Our findings reinforce the applicability of a sequential IPV‐OPV scheme during this transition period, which could reduce inequities between high‐ and low‐income countries due to limited access to IPV.
Quality of the evidence
This review included 21 studies, 16 of which were RCTs involving 6407 healthy infants; 28,330 infants in one ITS and four nationwide studies (two of which were nationwide ITSs, and two of which were nationwide UBAs).
Below, we describe the key risk of bias of the studies (see also Figure 2; Figure 3 and Table 1; Table 2; Table 3).
RCTs (n = 16)
The number of studies at low risk of bias varied across the seven domains: six studies for allocation concealment; 16 studies for blinding of participants and personnel and blinding of outcome assessment; four studies for incomplete outcome data; 16 studies for selective reporting; seven studies for potential bias related to vested interests and 16 studies seems to be free of other sources of bias. Incomplete outcome data was the only domain with studies rated at high risk of bias (n = 4).
ITS studies (n = 3)
Regarding protection against secular changes we considered all three ITS studies to be at low risk of bias for the domain independent of other changes and the number of points. Only one ITS specified the shape of the intervention effect but we considered it to be at unclear risk of bias because we reanalysed the data as an ITS. We rated all three ITS studies at low risk of bias for the four domains related to detection bias also, but two ITSs considered as unclear risk of bias because they used national databases No study received industry funding and therefore we considered all studies at low risk of bias for vested interests.
UBA studies (n = 3)
We considered two of the three UBA studies to be at unclear risk of bias for blinded assessment of primary outcomes. Although acute flaccid paralysis (AFP) and virological testing are objective outcomes, interpretation of VAPP is partially subjective, and we classified this as unclear risk of bias because the identification method was not clearly stated in the report. We rated the other, remaining study at low risk of bias on this domain.
We rated all three UBA studies at low risk of bias for primary outcome reliability since the surveillance systems are of good quality. We also rated all three studies at low risk of bias for follow‐up issues, since national AFP surveillance suggests a complete, nationwide follow‐up; the follow‐up period was long enough; and no changes to the protocols were reported. No study received industry funding and therefore we considered all three studies to be at low risk of bias for vested interests.
For the comparisons IPV‐OPV versus OPV (Table 1) and IPV‐OPV versus IPV (Table 2), we downgraded the certainty of the body of evidence for each outcome for the following reasons: study limitations (most studies were at unclear risk of bias regarding random sequence generation and allocation concealment), imprecision (the confidence intervals (CIs) were consistent with both a clinically important increase or reduction of the effect), and inconsistency (there was considerable heterogeneity; however, it was in the same direction of the effect). For the comparison IPV(3)‐OPV versus IPV(2)‐OPV (Table 3), we downgraded for imprecision only. We present our reasons for downgrading the certainty of the evidence for each outcome in the footnotes of each ‘Summary of findings’ table.
We rated the certainty of the evidence for most outcomes as low or moderate. Overall, there is insufficient evidence to draw conclusions about the effectiveness of IPV‐OPV for preventing paralytic polio, but the evidence is quite solid for VAPP, protective immune response (humoral and intestinal) and serious adverse events.
Potential biases in the review process
Our review methods, which followed Cochrane guidelines, were unlikely to have introduced bias. We conducted a comprehensive search without restriction on date or language and we undertook independent screening of eligible studies. Although we are confident we were able to obtain most of the relevant data, our review may have omitted important unpublished data that were not reported to WHO from countries' health databases. This could be important, particularly for infrequent events like polio, VAPP or cVDPV cases. Another potential source of bias is that we were unable to obtain further data from many authors of included studies to clarify certain aspects of methodology that would have enabled a more thorough assessment of the risk of bias.
Agreements and disagreements with other studies or reviews
Our finding that IPV‐OPV may reduce cases of VAPP without affecting vaccination coverage or safety, compared to OPV, is consistent with several UBA sub‐nationwide studies which did not meet our nationwide inclusion criteria. Zhao 2017, in a province‐wide study in China, assessed the introduction of one dose of IPV into Beijing's Expanded Program on Immunization on December 2014, changing the schedule from OPV only to sequential IPV‐OPV. Coverage with the first dose of polio vaccine was maintained from 96.2% to 96.9%, similar to coverage with the first doses of diphtheria and tetanus toxoids and pertussis vaccine (DTP; 96.5% in 2014, and 97.2% in 2015); the percentage of children who received the first dose of polio vaccine but failed to complete the series was 1.0% in 2015 and 0.4% in 2016, which again, was similar to that for DTP. No cases of VAPP were identified between 2014 and 2016. Liu 2017 analysed 566,894 Chinese children, born between 2010 and 2014 and registered in Hangzhou's Immunization Information System, who were exposed to OPV‐only, IPV‐only and sequential IPV‐OPV, and found consistent results with our findings. VAPP cases were detected through the acute flaccid paralysis surveillance system. The incidence of VAPP in the 2010 to 2014 birth cohorts was 3.76 per 1 million doses of OPV. Five VAPP cases were identified during the study years; all cases occurred following the first OPV.
In the early 1970s, the WHO Consultative Group conducted an extensive epidemiological study of VAPP cases in 13 countries (Cockburn 1988). After 15 years of continual surveillance, they found evidence that P3 strains caused most cases of post‐vaccination paralysis; the P1 strain was almost never implicated, suggesting that it is safe and effective, as is the P2 strain, although occasionally, it can cause paralysis in contacts of the vaccine. The only country where children were primarily immunised with IPV and given reinforcing doses of tOPV ("hidden name of the country #2 with a population of 5.1 million", quote from Cockburn 1988), reported zero confirmed cases of VAPP (Cockburn 1988).
Platt 2014 conducted a systematic review summarising the epidemiology of VAPP, and estimated the global VAPP burden applying a bootstrap method. Since many high‐income countries have replaced OPV with IPV, over 90% of the VAPP burden is concentrated in low‐ and middle‐income countries in Southeast Asia, Africa, the Western Pacific, and Eastern Mediterranean regions. Platt 2014 estimated that the planned universal introduction of IPV is likely to substantially decrease the global VAPP burden by 80% to 90%.
Two reviews addressed our review question. Bandyopadhyay 2018 conducted a narrative review of studies that assessed humoral and intestinal immunogenicity induced by the newly recommended IPV‐bOPV schedules. They included five sequential and three non‐sequential IPV‐bOPV RCTs and reported that differences in seroconversion rates were closely associated with both timing of first IPV administration and number of doses administered. Consistent with our larger body of evidence, they found that all studies demonstrated high levels of immunity for poliovirus serotypes 1 and 3 regardless of immunisation schedule, and that IPV doses and administration schedules showed limited impact on poliovirus serotype 2 excretion following challenge. They also reported that a second dose of IPV narrows the humoral immunity gap for poliovirus serotype 2, largely irrespective of the primary immunisation schedule.
Tang 2018 performed a systematic review and meta‐analysis to compare the immunogenicity of sequential IPV‐OPV versus IPV‐alone in healthy infants. They included four case‐series studies and two RCTs. They concluded that seroconversion rates against all three poliovirus serotypes were non‐inferior and geometric mean antibody titres were superior in sequential schedules, compared with an IPV‐only schedule. We included five additional RCTs for this comparison in our review, and we obtained similar findings for seroconversion rates for all three poliovirus serotypes but different results for mean antibody titres depending the scheme used.
Authors' conclusions
Implications for practice.
Compared to OPV, sequential IPV‐OPV may reduce cases of vaccine‐associated paralytic polio (VAPP) without affecting vaccination coverage, safety, or humoral response, except P2 with sequential schemes without P2 components, but reduce mucosal immunity as revealed by increased poliovirus faecal excretion after OPV challenge for some poliovirus serotypes.
Compared to IPV, sequential IPV‐OPV may make little or no difference to serious adverse events, probably make little or no difference to the number of people with protective humoral response, may increase neutralising antibodies and probably improve mucosal immunity for some polio serotypes, depending on the schedule used.
Sequential schedules of two full doses of IPV, compared to three, may provide adequate protective humoral response and may not affect mucosal immunity against poliovirus. Both short‐ and long‐term antibody titres and protective response of the poliovirus serotype 2 are probably lower using the most recent bOPV schedules without this component.
Sequential schedules during the transition from an OPV to IPV‐only immunisation schedule seem a reasonable option aligned with current World Health Organization (WHO) recommendations. Our findings could help decision‐makers to optimise polio vaccination policies, and might contribute to the expansion of IPV use in OPV‐using countries, thereby reducing inequities between countries.
Implications for research.
It is mandatory to improve registers of infrequent events beyond cases of wild polio virus (WPV), to include, for example, VAPP or vaccine‐derived polioviruses, and to assess the impact of the recent implementation of the WHO's recommendation to introduce of at least one dose of IPV in routine OPV immunisation programmes worldwide.
It is imperative to find the most cost‐effective and far‐reaching schemes in a context of scarcity of IPV in vast regions of the world.
Acknowledgements
The Cochrane Developmental, Psychosocial and Learning Problems Group for all the support provided during the whole process of this review.
Daniel Comande, from IECS, contributed to the search strategy developed by Margaret Anderson from Cochrane Developmental, Psychosocial and Learning Problems. Marina Romano contributed to the first round of study selection for the present review.
Jan Odgaard‐Jensen of Norwegian Knowledge Centre for the Health Services for the re‐analysis of the ITSs
We would also like to acknowledge the following reviewers, who commented on an earlier version of this review: Nishant Premnath Jaiswal, Department of Pediatrics, PGIMER Chandigarh; Mike Famulare, Institute for Disease Modeling; and Yinghui Wei, University of Plymouth.
Appendices
Appendix 1. Glossary
| Advisory Committee on Immunization Practices | ACIP |
| Auto regressive integrated moving average | ARIMA |
| Ambiguous vaccine‐derived poliovirus | aVDPV |
| Bivalent oral polio vaccine | bOPV |
| Two doses of IPV followed by one dose of bOPV | IIbO |
| Two doses of IPV followed by two doses of bOPV | IIbObO |
| One dose of IPV followed by two doses of bOPV | IbObO |
| Certainty of the Evidence | CoE |
| Circulating vaccine‐derived poliovirus | cVDPV |
| contact VAPP | cVAPP |
| Controlled before and after | CBA |
| Confidence intervals | CI |
| Controlled interrupted time series | CITS |
| Expanded program for immunization | EPI |
| Effective Practice and Organisation of Care | EPOC |
| Early review organizing software | EROS |
| Fractional Inactivated poliovirus vaccine | fIPV |
| Geometric Mean Titre | GMT |
| High‐income countries | HICs |
| Human immunodeficiency virus | HIV |
| Health maintenance organisations | HMOs |
| Schedule of IPV sequence | I |
| Intracluster correlation | ICC |
| Two doses of IPV followed by one dose of tOPV | IIO |
| Two doses of IPV followed by two doses of tOPV | IIOO |
| One dose of IPV followed by two doses of tOPV | IOO |
| One dose of IPV followed by three doses of tOPV | IOOO |
| IPV and tOPV given simultaneously | (I+O) |
| Inactivated poliovirus vaccine | IPV |
| Sequential inactivated poliovirus vaccine‐oral poliovirus vaccine | IPV‐OPV |
| Interrupted time series studies | ITS |
| Intention‐to‐treat | ITT |
| Immunodeficiency‐related vaccine‐derived poliovirus | iVDPV |
| Low‐ and middle‐income countries | LMICs |
| Mean difference | MD |
| Medical Dictionary for Regulatory Activities Terminology | MedDRA |
| Monovalent oral polio vaccine type 1 | mOPV1 |
| Monovalent oral polio vaccine type 2 | mOPV2 |
| Monovalent oral polio vaccine type 3 | mOPV3 |
| Schedule of OPV sequence (trivalent) | O |
| Schedule of OPV sequence (bivalent) | bO |
| Oral poliovirus vaccine | OPV |
| Odds ratio | OR |
| Poliovirus Serotype 1 | P1 |
| Poliovirus Serotype 2 | P2 |
| Poliovirus Serotype 3 | P3 |
| Quasi‐randomised controlled trial | Quasi‐RCT |
| Randomised controlled trial | RCT |
| recipient VAPP | rVAPP |
| Risk ratio | RR |
| Risk difference | RD |
| Sabin IPV | sIPV |
| Serious Adverse Event | SAE |
| Standardised mean difference | SMD |
| Trivalent oral polio vaccine | tOPV |
| Uncontrolled before and after | UBA |
| United States | US |
| Vaccine‐associated paralytic poliomyelitis | VAPP |
| Vaccine‐derived polioviruses | VDPVs |
| World Health Organization | WHO |
| Wild poliovirus | WPV |
Appendix 2. EPOC Data Collection Checklist: Study design
Data Collection Checklist: Study design
Randomised controlled trial (RCT) i.e. a trial in which the participants (or other units) were definitely assigned prospectively to one or two (or more) alternative forms of health care using a process of random allocation (e.g. random number generation, coin flips).
Controlled clinical trial (CCT)may be a trial in which participants (or other units) were:
a) definitely assigned prospectively to one or two (or more) alternative forms of health care using a quasi‐random allocation method (e.g. alternation, date of birth, patient identifier) or;
b) possibly assigned prospectively to one or two (or more) alternative forms of health care using a process of random or quasi‐random allocation.
Controlled before and after study (CBA) i.e. involvement of intervention and control groups other than by random process, and inclusion of baseline period of assessment of main outcomes. There are two minimum criteria for inclusion of CBAs in EPOC reviews:
a) Contemporaneous data collection
Score DONE pre‐ and post‐intervention periods for study and control sites are the same.
Score NOT CLEAR if it is not clear in the paper, e.g. dates of collection are not mentioned in the text. (N.B. the paper should be discussed with the contact editor for the review before data extraction is undertaken).
Score NOT DONE if data collection was not conducted contemporaneously during pre‐ and post‐intervention periods for study and control sites.
b) Appropriate choice of control site
Studies using second site as controls:
Score DONE if study and control sites are comparable with respect to dominant reimbursement system, level of care, setting of care, and academic status.
Score NOT CLEAR if not clear from paper whether study and control sites are comparable. (N.B. the paper should be discussed with the contact editor for the review before data extraction is undertaken).
Score NOT DONE if study and control sites are not comparable.
Interrupted time series (ITS) i.e. a change in trend attributable to the intervention. There are two minimum criteria for inclusion of ITS designs in EPOC reviews:
a) Clearly defined point in time when the intervention occurred
Score DONE if reported that intervention occurred at a clearly defined point in time.
Score NOT CLEAR if not reported in the paper (will be treated as NOT DONE if information cannot be obtained from the authors).
Score NOT DONE if reported that intervention did not occur at a clearly defined point in time.
b) At least three data points before and three after the intervention
Score DONE if three or more data points before and three or more data points recorded after the intervention.
Score NOT CLEAR if not specified in paper e.g. number of discrete data points not mentioned in text or tables (will be treated as NOT DONE if information cannot be obtained from the authors).
Score NOT DONE if less than three data points recorded before and three data points recorded after intervention.
Uncontrolled before and after study (UBA), also called before and after study. There is only one criteria, which is: nationwide studies evaluating the impact of changing the vaccination policy to sequential IPV‐OPV vaccination schemes.
Appendix 3. Record of searches
| Database | Search date | Date range/issue | Number of records |
| CENTRAL (Cochrane Library) | 15 August 2014 | 2014 Issue 7 (July) | 99 |
| 11 August 2015 | 2015 Issue 7 (July ) | 7 | |
| 22 July 2016 | 2016 Issue 7 (July) | 2 | |
| 11 May 2018 | 2018 Issue 5 (May) | 0 | |
| 14 May 2019 | 2019 Issue 5 (May) | 29 | |
| MEDLINE (OVID) | 13 August 2014 | 1946 to July Week 5 2014 | 1075 |
| 11 August 2015 | 1946 to July Week 5 2015 | 48 | |
| 22 July 2016 | 1946 to July Week 5 2016 | 119 | |
| 11 May 2018 | 1946 to July Week 3 2018 | 145 | |
| 14 May 2019 | 1946 to April Week 3 2019 | 71 | |
| Embase (OVID) | 13 August 2014 | 1980 to 2014 Week 32 | 1811 |
| 11 August 2015 | 1980 to 2015 Week 32 | 126 | |
| 22 July 2016 | 1980 to 2016 Week 29 | 124 | |
| 11 May 2018 | 1980 to 2018 Week 18 | 54 | |
| 14 May 2019 | 1980 to 2019 Week 18 | 54 | |
| Science Citation Index (Web of Science) | 13 August 2014 | 1970 to 8 August 2014 | 435 |
| 11 August 2015 | 1970 to 11 August 2015 | 71 | |
| 22 July 2016 | 1970 to 22 July 2015 | 117 | |
| 11 May 2018 | 1970 to 11 May 2015 | 109 | |
| 14 May 2019 | all available years | 93 | |
| CCPI ‐S | 13 August 2014 | 1990 to 8 August 2014 | 38 |
| 11 August 2015 | 1990 to 11 August 2015 | 0 | |
| 21 May 2018 | 1990 to 11 May 2018 | 3 | |
| 23 July 2019 | 1990 to 23 July 2019 | 1 | |
| SCOPUS | 13 August 2014 | current issue | 1600 |
| 11 August 2015 | current issue | 80 | |
| 21 May 2018 | current issue | 223 | |
| 23 July 2019 | current issue | 82 | |
| LILACS | 13 August 2014 | current issue | 12 |
| 11 August 2015 | current issue | 0 | |
| 22 July 2016 | current issue | 1 | |
| 11 May 2018 | current issue | 1 | |
| 14 May 2019 | current issue | 1 | |
| Cochrane Database of Systematic Reviews | 15 August 2014 | 2014 Issue 8 (August) | 1 |
| 11 August 2015 | 2015 Issue 8 (August) | 0 | |
| 22 July 2016 | 2016 Issue 7 (July) | 0 | |
| 11 May 2018 | 2018 Issue 5 (May) | 2 | |
| 31 July 2019 | 2019 Issue 7 (July) | 0 | |
| Database of Abstracts of Reviews of Effects | 15 August 2014 | 2014 Issue 3 (July) | 2 |
| 11 August 2015 | 2015 Issue 2 (April) | 0 | |
| IndMEd (indmed.nic.in) |
15 August 2014 | current issue | 7 |
| 11 August 2015 | current issue | 1 | |
| 30 August 2016 | current issue | 0 | |
| 11 May 2018 | current issue | 0 | |
| 31 July 2019 | Not accessible | 0 | |
| IBECS regional.bvsaud.org/php/index.php?lang=en |
13 August 2014 | current issue | 8 |
| 11 August 2015 | current issue | 0 | |
| 11 May 2018 | current issue | 0 | |
| 14 May 2019 | current issue | 0 | |
| PAHO‐ PAHO HQ Library Catalog regional.bvsalud.org/php/index.php?lang=en |
13 August 2014 | current issue | 12 |
| 11 August 2015 | current issue | 0 | |
| 22 July 2016 | current issue | 0 | |
| 11 May 2018 | current issue | 0 | |
| 2 June 2019 | current issue | 0 | |
| WHOLIS regional.bvsalud.org/php/index.php?lang=en |
13 August 2014 | current issue | 11 |
| 11 August 2015 | current issue | 0 | |
| 11 May 2018 | current issue | 0 | |
| 23 July 2019 | current issue | 0 | |
| IMSEAR (imsear.li.mahidol.ac.th/) | 15 August 2014 | current issue | 28 |
| 11 August 2015 | current issue | 1 | |
| 30 August 2016 | current issue | 0 | |
| 11 May 2018 | current issue | ‐ | |
| IMSEAR (www.globalhealthlibrary.net/php/index.php) | 31 July 2019 | current issue | 1 |
| SciELO | 15 August 2014 | current issue | 5 |
| 11 August 2015 | current issue | 16 | |
| 22 July 2016 | current issue | 0 | |
| 11 May 2018 | current issue | 3 | |
| 1 June 2019 | current issue | 4 | |
| African Index Medicus (indexmedicus.afro.who.int) | 15 August 2014 | current issue | 1 |
| 11 August 2015 | current issue | 0 | |
| 11 May 2018 | current issue | 0 | |
| African Index Medicus (www.globalhealthlibrary.net/php/index.php) | 31 July 2019 | current issue | 0 |
| IMEMR (www.emro.who.int/information‐resources/imemr‐database/) |
15 August 2014 | current issue | 5 |
| 11 August 2015 | current issue | 0 | |
| 22 July 2016 | current issue | 0 | |
| 11 May 2018 | current issue | 0 | |
| IMEMR (www.globalhealthlibrary.net/php/index.php) |
31 July 2019 | current issue | 1 |
| ICTRP (apps.who.int/trialsearch/AdvSearch.aspx) |
11 May 2018 | current issue | 11 |
| 11 July 2019 | current issue | 0 | |
| ClinicalTrials.gov (www.clinicaltrials.gov/ct2/home) |
11 May 2018 | current issue | 33 |
| 2 June 2019 | current issue | 11 | |
| ISRCTN (www.controlled‐trials.com) |
11 May 2018 | current issue | 3 |
| ISRCTN (www.isrctn.com) |
2 June 2019 | current issue | 0 |
| All databases | Deduplicated | ||
| Subtotal 2014 | 5292 | 2997 | |
| Subtotal 2015 | 350 | 231 | |
| Subtotal 2016 | 363 | 80 | |
| Subtotal 2018 | 585 | 458 | |
| Subtotal 2019 | 368 | 136 | |
| Total | 6958 | 3902 |
Appendix 4. Search strategies
Cochrane Central Register of Controlled Trials (CENTRAL), in the Cochrane Library
#1 [mh "Poliovirus Vaccine,oral"] #2 (polio* near/5 (activ* or oral* or live or attenuated)) #3 (OPV* or mOPV* or bOPV* or tOPV* or Sabin) #4 #1 or #2 or #3 #5 [mh "Poliovirus Vaccine, Inactivated"] #6 (polio* near/5 (inactiv* or in‐activ* or injectable or injection* or killed)) #7 (Salk or IPV* or eIPV*) #8 #5 or #6 or #7 #9 #4 and #8 in Trials
MEDLINE Ovid
1 Poliovirus Vaccine,oral/ 2 (polio$ adj5 (activ$ or oral$ or live or attenuated)).tw. 3 (OPV$ or mOPV$ or bOPV$ or tOPV$ or Sabin).tw. 4 or/1‐3 5 Poliovirus Vaccine, Inactivated/ 6 (polio$ adj5 (inactiv$ or in‐activ$ or injectable or injection$ or killed)).tw. 7 (Salk or IPV$ or eIPV$).tw. 8 or/5‐7 9 4 and 8 10 exp animals/ not humans/ 11 9 not 10
Embase Ovid
1 oral poliomyelitis vaccine/ 2 (polio$ adj5 (activ$ or oral$ or live or attenuated)).tw. 3 (OPV$ or mOPV$ or bOPV$ or tOPV$ or Sabin).tw. 4 or/1‐3 5 poliomyelitis vaccine/ 6 (polio$ adj5 (inactiv$ or in‐activ$ or injection$ or injectable or killed)).tw. 7 (Salk or IPV$ or eIPV$).tw. 8 5 or 6 or 7 9 4 and 8 10 exp animals/ or exp invertebrate/ or animal experiment/ or animal model/ or animal tissue/ or animal cell/ or nonhuman/ 11 human/ or normal human/ or human cell/ 12 10 and 11 13 10 not 12 14 9 not 13 15 remove duplicates from 14
Science Citation Index Web of Science
#7 #6 AND #3 DocType=All document types; Language=All languages; #6 #5 OR #4 DocType=All document types; Language=All languages; #5TS=(Salk or IPV* or eIPV*) DocType=All document types; Language=All languages; #4TS=(polio* NEAR/5 (inactiv* or in‐activ* or injectable or injection* or killed)) DocType=All document types; Language=All languages; #3 #2 OR #1 DocType=All document types; Language=All languages; #2TS=(OPV* or mOPV* or bOPV* or tOPV* or Sabin) DocType=All document types; Language=All languages; #1TS=(polio* NEAR/5 (activ* OR oral* OR live OR attenuated)) DocType=All document types; Language=All languages;
Conference Proceedings Citation Index ‐ Science Web of Science
#7 #6 AND #3 DocType=All document types; Language=All languages; #6 #5 OR #4 DocType=All document types; Language=All languages; #5TS=(Salk or IPV* or eIPV*) DocType=All document types; Language=All languages; #4TS=(polio* NEAR/5 (inactiv* or in‐activ* or injectable or injection* or killed)) DocType=All document types; Language=All languages; #3 #2 OR #1 DocType=All document types; Language=All languages; #2TS=(OPV* or mOPV* or bOPV* or tOPV* or Sabin) DocType=All document types; Language=All languages; #1TS=(polio* NEAR/5 (activ* OR oral* OR live OR attenuated)) DocType=All document types; Language=All languages;
SCOPUS Elsevier
((TITLE‐ABS‐KEY((polio* W/5 (activ* OR oral*)))) OR ((TITLE‐ABS‐KEY(opv OR mopv OR bopv OR topv OR sabin)) AND (TITLE‐ABS‐KEY(polio*)))) AND (((TITLE‐ABS‐KEY(polio*)) AND (TITLE‐ABS‐KEY((salk OR ipv OR eipv)))) OR (TITLE‐ABS‐KEY(polio* W/5 (inactiv* OR in‐activ*))
LILACS regional.bvsalud.org/php/index.php?lang=en
(tw:((tw:((polio* AND oral) OR sabin OR opv )) OR (mh:("poliovirus vaccine, oral")) AND (tw:((polio* AND inactiv*) OR (polio* AND in‐activ*) OR salk OR ipv )) OR (mh:(("poliovirus vaccine, inactivated")))) AND (instance:"regional") AND ( db:("LILACS"))
Cochrane Database of Systematic Reviews (CDSR), part of the Cochrane Library
#1[mh "Poliovirus Vaccine,oral"] #2(polio* near/5 (activ* or oral* or live or attenuated)):ti,ab #3(OPV* or mOPV* or bOPV* or tOPV* or Sabin) #4#1 or #2 or #3 #5[mh "Poliovirus Vaccine, Inactivated"] #6(polio* near/5 (inactiv* or in‐activ* or injectable or injection* or killed)):ti,ab #7(Salk or IPV* or eIPV*):ti,ab #8#5 or #6 or #7 in CDSR
Database of Abstracts of Reviews of Effects (DARE), part of the Cochrane Library
#1[mh "Poliovirus Vaccine,oral"] #2(polio* near/5 (activ* or oral* or live or attenuated)):ti,ab #3(OPV* or mOPV* or bOPV* or tOPV* or Sabin) #4#1 or #2 or #3 #5[mh "Poliovirus Vaccine, Inactivated"] #6(polio* near/5 (inactiv* or in‐activ* or injectable or injection* or killed)):ti,ab #7(Salk or IPV* or eIPV*):ti,ab #8#5 or #6 or #7 in Other Reviews
IndMED (indmed.nic.in/)
((polio AND inactiv) OR (polio AND killed) OR (polio AND inject) OR IPV OR Salk) AND ((polio AND oral) OR OPV OR (polio AND live) OR sabin)
IBECS (regional.bvsalud.org/php/index.php?lang=en)
(tw:((polio* AND oral))) OR (tw:(sabin OR opv)) OR (tw:("poliovirus vaccine oral")) AND (tw:((polio* AND inactiv*))) OR (tw:(salk)) OR (tw:(ipv )) OR (mh:("poliovirus vaccine, inactivated")) AND (instance:"regional")) AND (instance:"regional") with IBECS filter selected
PAHO‐ PAHO HQ Library Catalog (regional.bvsalud.org/php/index.php?lang=en)
(tw:((polio* AND oral))) OR (tw:(sabin OR opv)) OR (tw:("poliovirus vaccine oral")) AND (tw:((polio* AND inactiv*))) OR (tw:(salk)) OR (tw:(ipv )) OR (mh:("poliovirus vaccine, inactivated")) AND (instance:"regional")) with PAHO filter selected
WHOLIS (regional.bvsalud.org/php/index.php?lang=en)
(tw:((polio* AND oral))) OR (tw:(sabin OR opv)) OR (tw:("poliovirus vaccine oral")) AND (tw:((polio* AND inactiv*))) OR (tw:(salk)) OR (tw:(ipv )) OR (mh:("poliovirus vaccine, inactivated")) ) AND (instance:"regional") with WHOLIS filter selected
IMSEAR (Index Medicus for South East Asia Region)
Between 2014 and 2016, IMSEAR was searched viaimsear.li.mahidol.ac.th/
Advanced search (("inactivated poliovirus" OR "inactivated polio vaccine" OR IPV OR "killed polio vaccine" OR "killed polio virus" OR "injectable polio vacccine") AND ("oral poliovirus" OR "oral polio vaccine" OR "live polio vaccine" OR OPV OR bOPV OR tOPV))
In 2019 IMSEAR was searched via Global Index Medicus (www.globalhealthlibrary.net/php/index.php)
(tw:(polio*)) AND (tw:(salk OR ipv OR eipv OR inactiv* OR in‐activ* )) AND (tw:(opv* OR mopv* OR bopv* OR topv* OR sabin OR activ* OR oral)) AND (instance:"ghl") AND ( db:("IMSEAR"))
SciELO ( scielo.org/php/index.php?lang=en)
#3Expression: #1 and #3
#2Expression: (oral AND polio$) OR (live AND polio$) OR OPV OR bOPV OR tOP
#1Expression: ( polio$ AND inactiv$) OR (polio$ AND inject$) OR (polio$ AND killed) OR IPV
African Index Medicus (indexmedicus.afro.who.int/)
(polio$ AND inactiv$ ) OR IPV OR (polio$ AND killed) OR (polio$ AND inject$) [Key Word] and (polio$ AND oral) OR OPV OR bOPB OR tOPV OR (live AND polio$) [Key Word]
IMEMR (Index Medicus for the Eastern Mediterranean Region)
Between 2014 and 2016, IMEMR was searched via www.emro.who.int/information‐resources/imemr‐database/
(inactiv$ AND polio$) OR (inject$ AND polio$) OR (IPV AND polio$) OR (killed AND polio$) [KeyWords] and (oral AND polio$) OR (live AND polio$) OR OPV [KeyWords]
In 2019 IMEMR was searched via Global Index Medicus (www.globalhealthlibrary.net/php/index.php)
(tw:(polio*)) AND (tw:(salk OR ipv OR eipv OR inactiv* OR in‐activ* )) AND (tw:(opv* OR mopv* OR bopv* OR topv* OR sabin OR activ* OR oral)) AND (instance:"ghl") AND ( db:("IMEMR"))
ICTRP (International Clinical Trials Registry Platform apps.who.int/trialsearch/)
Polio AND Vaccine
ClinicalTrials.gov (clinicaltrials.gov/)
IPV OR OPV
ISRCTN registry (www.isrctn.com/)
Polio AND Vaccine
Appendix 5. Criteria for judging the risk of bias in RCTs and quasi‐RCTs
RANDOM SEQUENCE GENERATION
Selection bias (biased allocation to interventions) due to inadequate generation of a randomised sequence.
| Criteria for a judgement of 'low' risk of bias. | The investigators describe a random component in the sequence generation process such as:
*Minimization may be implemented without a random element, and this is considered to be equivalent to being random. |
| Criteria for the judgement of 'high' risk of bias. | The investigators describe a non‐random component in the sequence generation process. Usually, the description would involve some systematic, non‐random approach, for example:
Other non‐random approaches happen much less frequently than the systematic approaches mentioned above and tend to be obvious. They usually involve judgement or some method of non‐random categorization of participants, for example:
|
| Criteria for the judgement of 'unclear' risk of bias. | Insufficient information about the sequence generation process to permit judgement of ‘low' or ‘high’ risk of bias. |
|
ALLOCATION CONCEALMENT Selection bias (biased allocation to interventions) due to inadequate concealment of allocations prior to assignment. | |
| Criteria for a judgement of 'low' risk of bias. | Participants and investigators enrolling participants could not foresee assignment because one of the following, or an equivalent method, was used to conceal allocation:
|
| Criteria for the judgement of 'high' risk of bias. | Participants or investigators enrolling participants could possibly foresee assignments and thus introduce selection bias, such as allocation based on:
|
| Criteria for the judgement of 'unclear' risk of bias. | Insufficient information to permit judgement of 'low' or 'high' risk. This is usually the case if the method of concealment is not described or not described in sufficient detail to allow a definite judgement – for example, if the use of assignment envelopes is described but it remains unclear whether envelopes were sequentially numbered, opaque, and sealed. |
|
BLINDING OF PARTICIPANTS AND PERSONNEL Performance bias due to knowledge of the allocated interventions by participants and personnel during the study. | |
| Criteria for a judgement of 'low' risk of bias. | Any one of the following:
|
| Criteria for the judgement of 'high' risk of bias. | Any one of the following:
|
| Criteria for the judgement of 'unclear' risk of bias. | Any one of the following:
|
|
BLINDING OF OUTCOME ASSESSMENT Detection bias due to knowledge of the allocated interventions by outcome assessors. | |
| Criteria for a judgement of 'low' risk of bias. | Any one of the following:
|
| Criteria for the judgement of 'high' risk of bias. | Any one of the following:
|
| Criteria for the judgement of 'unclear' risk of bias. | Any one of the following:
|
|
INCOMPLETE OUTCOME DATA Attrition bias due to amount, nature or handling of incomplete outcome data. | |
| Criteria for a judgement of 'low' risk of bias. | Any one of the following:
|
| Criteria for the judgement of 'high' risk of bias. | Any one of the following:
|
| Criteria for the judgement of 'unclear' risk of bias. | Any one of the following:
|
|
SELECTIVE REPORTING Reporting bias due to selective outcome reporting. | |
| Criteria for a judgement of ‘low' risk of bias. | Any of the following:
|
| Criteria for the judgement of ‘high' risk of bias. | Any one of the following:
|
| Criteria for the judgement of ‘unclear' risk of bias. | Insufficient information to permit judgement of ‘low’ or ‘high' risk of bias. It is likely that the majority of studies will fall into this category. |
|
OTHER BIAS Bias due to problems not covered elsewhere in the table. | |
| Criteria for a judgement of 'low' risk of bias. | The study appears to be free of other sources of bias. |
| Criteria for the judgement of 'high' risk of bias. | There is at least one important risk of bias. For example, the study:
|
| Criteria for the judgement of 'unclear' risk of bias. | There may be a risk of bias, but there is either:
|
Appendix 6. Criteria for judging the risk of bias in controlled and uncontrolled before‐and‐after studies, and in (controlled) interrupted time series studies
QUALITY CRITERIA FOR CONTROLLED BEFORE AND AFTER (CBA) DESIGNS
Seven standard criteria are used for CBAs included in EPOC reviews:
a) Baseline measurement:
LOW RISK if performance or patient outcomes were measured prior to the intervention, and no substantial differences were present across study groups (e.g. where multiple pre‐intervention measures describe similar trends in intervention and control groups);
UNCLEAR RISK if baseline measures are not reported, or if it is unclear whether baseline measures are substantially different across study groups;
HIGH RISK if there are differences at baseline in main outcome measures likely to undermine the post‐intervention differences (e.g. are differences between the groups before the intervention similar to those found post‐intervention).
b) Characteristics for studies using second site as control:
LOW RISK if characteristics of study and control providers are reported and similar;
UNCLEAR RISK if it is not clear in the paper e.g. characteristics are mentioned in the text but no data are presented;
HIGH RISK if there is no report of characteristics either in the text or a table OR if baseline characteristics are reported and there are differences between study and control providers.
c) Blinded assessment of primary outcome(s)* (protection against detection bias):
LOW RISK if the authors state explicitly that the primary outcome variables were assessed blindly OR the outcome variables are objective e.g. length of hospital stay, drug levels as assessed by a standardised test;
UNCLEAR RISK if not specified in the paper;
HIGH RISK if the outcomes were not assessed blindly.
* Primary outcome(s) are those variables that correspond to the primary hypothesis or question as defined by the authors. In the event that some of the primary outcome variables were assessed in a blind fashion and others were not, score each separately and label each outcome variable clearly.
d) Protection against contamination:
Studies using second site as control:
LOW RISK if allocation was by community, institution, or practice and is unlikely that the control group received the intervention;
UNCLEAR RISK if providers were allocated within a clinic or practice and communication between experimental and group providers was likely to occur;
HIGH RISK if it is likely that the control group received the intervention (e.g. cross‐over studies or if patients rather than providers were randomised).
e) Reliable primary outcome measure(s):
LOW RISK if two or more raters with at least 90% agreement or kappa greater than or equal to 0.8 OR the outcome is obtained from some automated system e.g. length of hospital stay, drug levels as assessed by a standardised test;
UNCLEAR RISK if reliability is not reported for outcome measures that are obtained by chart extraction or collected by an individual;
HIGH RISK if agreement is less than 90% or kappa is less than 0.8.
* In the event that some outcome variables were assessed in a reliable fashion and others were not, score each separately and label each outcome variable clearly.
f) Follow‐up of professionals (protection against exclusion bias):
LOW RISK if outcome measures obtained 80‐100% subjects allocated to groups. (Do not assume 100% follow‐up unless stated explicitly.);
UNCLEAR RISK if not specified in the paper;
HIGH RISK if outcome measures obtained for less than 80% of patients allocated to groups.
g) Follow‐up of patients:
LOW RISK if outcome measures obtained 80‐100% of patients allocated to groups or for patients who entered the study. (Do not assume 100% follow‐up unless stated explicitly.);
UNCLEAR RISK if not specified in the paper;
HIGH RISK if outcome measures obtained for less than 80% of patients allocated to groups or for less than 80% of patients who entered the study.
QUALITY CRITERIA FOR INTERRUPTED TIME SERIES (ITS)
The following seven standard criteria should be used to assess the methodology quality of ITS designs included in EPOC reviews. Each criterion is scored DONE, NOT CLEAR or NOT DONE but here we use 'low risk', 'unclear risk', and 'high risk' respectively to be consistent with the 'Risk of bias' assessment tool for RCTs (Appendix 5).
Protection against secular changes:
a) The intervention is independent of other changes.
LOW RISK if the intervention occurred independently of other changes over time;
UNCLEAR RISK if not specified (will be treated as HIGH RISK if information cannot be obtained from the authors);
HIGH RISK if reported that intervention was not independent of other changes in time.
b) Data were analysed appropriately:
LOW RISK if ARIMA models were used OR time series regression models were used to analyse the data and serial correlation was adjusted or tested for;
UNCLEAR RISK if not specified (will be treated as HIGH RISK if information cannot be obtained from the authors);
HIGH RISK if it is clear that neither of the conditions above not met.
c) Reason for the number of points pre‐ and post‐intervention given:
LOW RISK if rationale for the number of points stated (e.g. monthly data for 12 months post‐intervention was used because the anticipated effect was expected to decay) OR sample size calculation performed;
UNCLEAR RISK if not specified (will be treated as HIGH RISK if information cannot be obtained from the authors);
HIGH RISK if it is clear that neither of the conditions above met.
d) Shape of the intervention effect was specified:
LOW RISK if a rational explanation for the shape of intervention effect was given by the author(s);
UNCLEAR RISK if not specified (will be treated as HIGH RISK if information cannot be obtained from the authors);
HIGH RISK if it is clear that the condition above is not met.
Protection against detection bias:
e) Intervention unlikely to affect data collection:
LOW RISK if reported that intervention itself was unlikely to affect data collection (for example, sources and methods of data collection were the same before and after the intervention);
UNCLEAR RISK if not reported (will be treated as HIGH RISK if information cannot be obtained from the authors);
HIGH RISK if the intervention itself was likely to affect data collection (for example, any change in source or method of data collection reported).
f) Blinded assessment of primary outcome(s)*:
LOW RISK if the authors state explicitly that the primary outcome variables were assessed blindly OR the outcome variables are objective e.g. length of hospital stay, drug levels as assessed by a standardised test;
UNCLEAR RISK if not specified (will be treated as HIGH RISK if information cannot be obtained from the authors);
HIGH RISK if the outcomes were not assessed blindly.
* Primary outcome(s) are those variables that correspond to the primary hypothesis or question as defined by the authors. In the event that some of the primary outcome variables were assessed in a blind fashion and others were not, score each separately and label each outcome variable clearly.
g) Completeness of data set:
LOW RISK if data set covers 80‐100% of total number of participants or episodes of care in the study;
UNCLEAR RISK if not specified (will be treated as HIGH RISK if information cannot be obtained from the authors);
HIGH RISK if data set covers less than 80% of the total number of participants or episodes of care in the study.
h) Reliable primary outcome measure(s)*:
LOW RISK if two or more raters with at least 90% agreement or kappa greater than or equal to 0.8 OR the outcome is obtained from some automated system e.g. length of hospital stay, drug levels as assessed by a standardised test;
UNCLEAR RISK if reliability is not reported for outcome measures that are obtained by chart extraction or collected by an individual (will be treated as HIGH RISK if information cannot be obtained from the authors);
HIGH RISK if agreement is less than 90% or kappa is less than 0.8.
* In the event that some outcome variables were assessed in a reliable fashion and others were not, score each separately.
QUALITY CRITERIA FOR CONTROLLED INTERRUPTED TIME SERIES (CITS)
a) Protection against secular changes:
The intervention is independent of other changes.
LOW RISK if the intervention occurred independently of other changes over time;
UNCLEAR RISK if not specified (will be treated as HIGH RISK if information cannot be obtained from the authors);
HIGH RISK if reported that intervention was not independent of other changes in time.
b) Data were analysed appropriately:
LOW RISK if ARIMA models were used OR time series regression models were used to analyse the data and serial correlation was adjusted or tested for;
UNCLEAR RISK if not specified (will be treated as HIGH RISK if information cannot be obtained from the authors);
HIGH RISK if it is clear that neither of the conditions above not met.
c) Reason for the number of points pre‐ and post‐intervention given:
LOW RISK if rationale for the number of points stated (e.g. monthly data for 12 months post‐intervention was used because the anticipated effect was expected to decay) OR sample size calculation performed;
UNCLEAR RISK if not specified (will be treated as HIGH RISK if information cannot be obtained from the authors);
HIGH RISK if it is clear that neither of the conditions above met.
d) Shape of the intervention effect was specified:
LOW RISK if a rational explanation for the shape of intervention effect was given by the author(s);
UNCLEAR RISK if not specified (will be treated as HIGH RISK if information cannot be obtained from the authors);
HIGH RISK if it is clear that the condition above is not met.
e) Protection against detection bias:
Intervention unlikely to affect data collection:
LOW RISK if reported that intervention itself was unlikely to affect data collection (for example, sources and methods of data collection were the same before and after the intervention);
UNCLEAR RISK if not reported (will be treated as HIGH RISK if information cannot be obtained from the authors);
HIGH RISK if the intervention itself was likely to affect data collection (for example, any change in source or method of data collection reported).
Blinded assessment of primary outcome(s)*:
LOW RISK if the authors state explicitly that the primary outcome variables were assessed blindly OR the outcome variables are objective e.g. length of hospital stay, drug levels as assessed by a standardised test;
UNCLEAR RISK if not specified (will be treated as HIGH RISK if information cannot be obtained from the authors);
HIGH RISK if the outcomes were not assessed blindly.
* Primary outcome(s) are those variables that correspond to the primary hypothesis or question as defined by the authors. In the event that some of the primary outcome variables were assessed in a blind fashion and others were not, score each separately and label each outcome variable clearly.
f) Completeness of data set:
LOW RISK if data set covers 80‐100% of total number of participants or episodes of care in the study;
UNCLEAR RISK if not specified (will be treated as HIGH RISK if information cannot be obtained from the authors);
HIGH RISK if data set covers less than 80% of the total number of participants or episodes of care in the study.
g) Reliable primary outcome measure(s)*:
LOW RISK if two or more raters with at least 90% agreement or kappa greater than or equal to 0.8 OR the outcome is obtained from some automated system e.g. length of hospital stay, drug levels as assessed by a standardised test;
UNCLEAR RISK if reliability is not reported for outcome measures that are obtained by chart extraction or collected by an individual (will be treated as HIGH RISK if information cannot be obtained from the authors);
HIGH RISK if agreement is less than 90% or kappa is less than 0.8.
* In the event that some outcome variables were assessed in a reliable fashion and others were not, score each separately.
For CITSs, as for CBAs, we will include three additional domains that assess design‐specific threats to validity covered by the Cochrane EPOC group: imbalance of outcome measures at baseline; comparability of intervention and control group characteristics at baseline; and protection against contamination.
h) Baseline measurement:
LOW RISK if performance or patient outcomes were measured prior to the intervention, and no substantial differences were present across study groups (e.g. where multiple pre‐intervention measures describe similar trends in intervention and control groups);
UNCLEAR RISK if baseline measures are not reported, or if it is unclear whether baseline measures are substantially different across study groups;
HIGH RISK if there are differences at baseline in main outcome measures likely to undermine the post‐intervention differences (e.g. are differences between the groups before the intervention similar to those found post‐intervention).
m) Characteristics for studies using second site as control:
LOW RISK if characteristics of study and control providers are reported and similar;
UNCLEAR RISK if it is not clear in the paper e.g. characteristics are mentioned in the text but no data are presented;
HIGH RISK if there is no report of characteristics either in the text or a table OR if baseline characteristics are reported and there are differences between study and control providers.
i) Protection against contamination:
Studies using second site as control:
LOW RISK if allocation was by community, institution, or practice and is unlikely that the control group received the intervention;
UNCLEAR RISK if providers were allocated within a clinic or practice and communication between experimental and group providers was likely to occur;
HIGH RISK if it is likely that the control group received the intervention (e.g. cross‐over studies or if patients rather than providers were randomised).
QUALITY CRITERIA FOR UNCONTROLLED BEFORE AND AFTER (UBA) DESIGNS
Four standard criteria are used for UBAs (Derived from CBAs EPOC criteria):
a) Blinded assessment of primary outcome(s)* (protection against detection bias):
LOW RISK if the authors state explicitly that the primary outcome variables were assessed blindly OR the outcome variables are objective e.g. length of hospital stay, drug levels as assessed by a standardised test;
UNCLEAR RISK if not specified in the paper;
HIGH RISK if the outcomes were not assessed blindly.
* Primary outcome(s) are those variables that correspond to the primary hypothesis or question as defined by the authors. In the event that some of the primary outcome variables were assessed in a blind fashion and others were not, score each separately and label each outcome variable clearly.
b) Reliable primary outcome measure(s):
LOW RISK if two or more raters with at least 90% agreement or kappa greater than or equal to 0.8 OR the outcome is obtained from some automated system e.g. length of hospital stay, drug levels as assessed by a standardised test;
UNCLEAR RISK if reliability is not reported for outcome measures that are obtained by chart extraction or collected by an individual;
HIGH RISK if agreement is less than 90% or kappa is less than 0.8.
* In the event that some outcome variables were assessed in a reliable fashion and others were not, score each separately and label each outcome variable clearly.
c) Follow‐up of professionals (protection against exclusion bias):
LOW RISK if outcome measures obtained 80‐100% subjects at baseline. (Do not assume 100% follow‐up unless stated explicitly.);
UNCLEAR RISK if not specified in the paper;
HIGH RISK if outcome measures obtained for less than 80% of patients at baseline.
d) Follow‐up of patients:
LOW RISK if outcome measures obtained 80‐100% of patients who entered the study. (Do not assume 100% follow‐up unless stated explicitly.);
UNCLEAR RISK if not specified in the paper;
HIGH RISK if outcome measures obtained for less than 80% of patients who entered the study.
Data and analyses
Comparison 1. IPV‐OPV versus OPV.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Persons with P1 Protective humoral response | 12 | 3189 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.99, 1.01] |
| 2 Persons with P1 Protective humoral response by time of first dose | 12 | 3189 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.99, 1.01] |
| 2.1 First dose at birth | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.91, 1.20] |
| 2.2 First dose at 2 month | 11 | 3139 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.99, 1.01] |
| 3 Persons with P1 Protective humoral response by type of dose sequence | 12 | 3189 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.99, 1.01] |
| 3.1 Sequential IOO/IOOO | 5 | 695 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.98, 1.01] |
| 3.2 Sequential IIO/IIOO/IIIO | 8 | 1772 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.99, 1.01] |
| 3.3 Sequential IOI | 2 | 722 | Risk Ratio (M‐H, Random, 95% CI) | 1.03 [0.93, 1.14] |
| 4 Persons with P1 Protective humoral response by countries' income | 12 | 3189 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.99, 1.01] |
| 4.1 LMIC | 4 | 1331 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.99, 1.01] |
| 4.2 HIC | 8 | 1858 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.99, 1.02] |
| 5 Persons with P2 Protective humoral response | 12 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 5.1 IPV‐tOPV | 11 | 2361 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.99, 1.01] |
| 5.2 IIbO | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.93, 1.07] |
| 5.3 IbObO vs tOPV | 2 | 411 | Risk Ratio (M‐H, Random, 95% CI) | 0.78 [0.64, 0.96] |
| 5.4 IbObO vs bOPV | 1 | 306 | Risk Ratio (M‐H, Random, 95% CI) | 5.80 [4.06, 8.27] |
| 6 Persons with P2 Protective humoral response by time of first dose | 12 | 3186 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.98, 1.06] |
| 6.1 First dose at birth | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [0.87, 1.26] |
| 6.2 First dose at 2 month | 11 | 3136 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.98, 1.06] |
| 7 Persons with P2 Protective humoral response by type of dose sequence | 12 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 7.1 Sequential IbObO (bOPV) | 1 | 211 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.78, 0.91] |
| 7.2 Sequential IOO/IOOO | 4 | 484 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.98, 1.02] |
| 7.3 Sequential IIO/IIOO/IIIO | 8 | 1768 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.99, 1.01] |
| 7.4 Sequential IbOI (vs bObObO) | 1 | 305 | Risk Ratio (M‐H, Random, 95% CI) | 5.85 [4.10, 8.34] |
| 7.5 Sequential IbOI | 1 | 309 | Risk Ratio (M‐H, Random, 95% CI) | 0.82 [0.75, 0.90] |
| 7.6 Sequential IOI | 1 | 109 | Risk Ratio (M‐H, Random, 95% CI) | 1.08 [0.99, 1.17] |
| 8 Persons with P2 Protective humoral response by countries' income | 12 | 3186 | Risk Ratio (M‐H, Random, 95% CI) | 1.02 [0.98, 1.06] |
| 8.1 LMIC | 4 | 1331 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.96, 1.01] |
| 8.2 HIC | 8 | 1855 | Risk Ratio (M‐H, Random, 95% CI) | 1.06 [0.96, 1.17] |
| 9 Persons with P3 Protective humoral response | 12 | 3184 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.97, 1.00] |
| 10 Persons with P3 Protective humoral response by time of first dose | 12 | 3013 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.97, 1.01] |
| 10.1 Fist dose at birth | 1 | 50 | Risk Ratio (M‐H, Random, 95% CI) | 1.17 [0.87, 1.57] |
| 10.2 First dose at 2 month | 11 | 2963 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.97, 1.01] |
| 11 Persons with P3 Protective humoral response by type of dose sequence | 12 | 3184 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.96, 1.00] |
| 11.1 Sequential IbObO | 1 | 105 | Risk Ratio (M‐H, Random, 95% CI) | 0.70 [0.60, 0.82] |
| 11.2 Sequential IOO/IOOO | 5 | 590 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.96, 1.00] |
| 11.3 Sequential IIO/IIOO/IIIO | 8 | 1767 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.96, 1.01] |
| 11.4 Sequential IOI | 2 | 722 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.95, 1.03] |
| 12 Persons with P3 Protective humoral response by countries' income | 12 | 3013 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.97, 1.01] |
| 12.1 LMIC | 4 | 1159 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.98, 1.01] |
| 12.2 HIC | 8 | 1854 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.94, 1.01] |
| 13 Mean titres of P1 neutralising antibody | 6 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 13.1 Sequential IbObO | 1 | 125 | Mean Difference (IV, Random, 95% CI) | 362.12 [‐329.70, 1053.94] |
| 13.2 Sequential IOO | 3 | 606 | Mean Difference (IV, Random, 95% CI) | ‐181.13 [‐594.25, 231.99] |
| 13.3 Sequential IIO | 3 | 795 | Mean Difference (IV, Random, 95% CI) | ‐244.37 [‐827.31, 338.57] |
| 13.4 Sequential IIIOO/IIIO | 2 | 551 | Mean Difference (IV, Random, 95% CI) | 439.07 [‐354.63, 1232.77] |
| 14 Mean titres of P2 neutralising antibody | 6 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 14.1 Sequential IbObO | 1 | 125 | Mean Difference (IV, Random, 95% CI) | ‐260.38 [‐347.21, ‐173.55] |
| 14.2 Sequential IOO | 3 | 606 | Mean Difference (IV, Random, 95% CI) | 28.64 [‐22.16, 79.43] |
| 14.3 Sequential IIbO | 1 | 125 | Mean Difference (IV, Random, 95% CI) | ‐217.90 [‐305.36, ‐130.44] |
| 14.4 Sequential IIO | 3 | 667 | Mean Difference (IV, Random, 95% CI) | 267.40 [‐83.95, 618.76] |
| 14.5 Sequential IIIOO/IIIO | 2 | 551 | Mean Difference (IV, Random, 95% CI) | 486.17 [‐698.02, 1670.37] |
| 15 Mean titres of P3 neutralising antibody | 6 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 15.1 Sequential IbObO | 1 | 125 | Mean Difference (IV, Random, 95% CI) | 221.03 [9.66, 432.40] |
| 15.2 Sequential IOO | 3 | 606 | Mean Difference (IV, Random, 95% CI) | 44.07 [‐1.47, 89.61] |
| 15.3 Sequential IIbO | 1 | 125 | Mean Difference (IV, Random, 95% CI) | 591.78 [185.14, 998.42] |
| 15.4 Sequential IIO | 3 | 667 | Mean Difference (IV, Random, 95% CI) | 89.97 [8.98, 170.97] |
| 15.5 Sequential IIIOO/IIIO | 2 | 551 | Mean Difference (IV, Random, 95% CI) | 248.39 [‐180.58, 677.37] |
| 16 Long term mean titres of P1 neutralising antibody | 1 | 86 | Mean Difference (IV, Random, 95% CI) | 0.35 [0.07, 0.63] |
| 16.1 Sequential IOO | 1 | 20 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐0.48, 0.48] |
| 16.2 Sequential IOO+O | 1 | 20 | Mean Difference (IV, Random, 95% CI) | 0.20 [‐0.33, 0.73] |
| 16.3 Sequential IIO | 1 | 23 | Mean Difference (IV, Random, 95% CI) | 0.60 [0.22, 0.98] |
| 16.4 Sequential IIO+O | 1 | 23 | Mean Difference (IV, Random, 95% CI) | 0.5 [0.01, 0.99] |
| 17 Long term mean titres of P2 neutralising antibody | 1 | 86 | Mean Difference (IV, Random, 95% CI) | 0.12 [‐0.07, 0.31] |
| 17.1 Sequential IOO | 1 | 20 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐0.26, 0.46] |
| 17.2 Sequential IOO+O | 1 | 20 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐0.30, 0.50] |
| 17.3 Sequential IIO | 1 | 23 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐0.38, 0.38] |
| 17.4 Sequential IIO+O | 1 | 23 | Mean Difference (IV, Random, 95% CI) | 0.30 [‐0.11, 0.71] |
| 18 Long term mean titres of P3 neutralising antibody | 1 | 86 | Mean Difference (IV, Random, 95% CI) | 0.08 [‐0.29, 0.45] |
| 18.1 Sequential IOO | 1 | 20 | Mean Difference (IV, Random, 95% CI) | ‐0.50 [‐1.14, 0.14] |
| 18.2 Sequential IOO+O | 1 | 20 | Mean Difference (IV, Random, 95% CI) | 0.0 [‐0.61, 0.61] |
| 18.3 Sequential IIO | 1 | 23 | Mean Difference (IV, Random, 95% CI) | 0.40 [0.02, 0.78] |
| 18.4 Sequential IIO+O | 1 | 23 | Mean Difference (IV, Random, 95% CI) | 0.20 [‐0.34, 0.74] |
| 19 Persons with polio faecal excretion after OPV challenge | 2 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 19.1 Persons with P1 faecal excretion | 2 | 916 | Risk Ratio (M‐H, Random, 95% CI) | 2.24 [0.70, 7.12] |
| 19.2 Persons with P2 faecal excretion | 2 | 916 | Risk Ratio (M‐H, Random, 95% CI) | 1.78 [1.49, 2.14] |
| 19.3 Persons with P3 faecal excretion | 2 | 916 | Risk Ratio (M‐H, Random, 95% CI) | 2.35 [1.47, 3.76] |
| 20 Vaccination coverage | 1 | Risk Ratio (Random, 95% CI) | 1.01 [0.96, 1.06] | |
| 21 Serious adverse events classified by MedDRA | 4 | 1948 | Risk Ratio (M‐H, Random, 95% CI) | 0.88 [0.46, 1.70] |
Comparison 2. IPV‐OPV versus IPV.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Persons with P1 Protective humoral response | 10 | 2858 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.99, 1.01] |
| 1.1 Sequential IOO | 4 | 572 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.98, 1.02] |
| 1.2 Sequential IIO(O) | 8 | 1767 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.99, 1.01] |
| 1.3 Sequential IbOI | 1 | 519 | Risk Ratio (M‐H, Random, 95% CI) | 1.05 [1.00, 1.10] |
| 2 Persons with P2 Protective humoral response | 10 | 2907 | Risk Ratio (M‐H, Random, 95% CI) | 0.97 [0.95, 1.00] |
| 2.1 Sequential IbObO | 2 | 382 | Risk Ratio (M‐H, Random, 95% CI) | 0.76 [0.64, 0.91] |
| 2.2 Sequential IOO | 3 | 227 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.96, 1.04] |
| 2.3 Sequential IIO(O) | 8 | 1779 | Risk Ratio (M‐H, Random, 95% CI) | 1.00 [0.99, 1.01] |
| 2.4 Sequential IbOI | 1 | 519 | Risk Ratio (M‐H, Random, 95% CI) | 0.95 [0.88, 1.02] |
| 3 Persons with P3 Protective humoral response | 9 | 2620 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.97, 1.01] |
| 3.1 Sequential IOO | 4 | 570 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.96, 1.02] |
| 3.2 Sequential IIO(O) | 7 | 1531 | Risk Ratio (M‐H, Random, 95% CI) | 0.99 [0.96, 1.01] |
| 3.3 Sequential IbOI | 1 | 519 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.96, 1.05] |
| 4 Mean titres of P1 neutralising antibody | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 4.1 Sequential IbObO | 1 | 127 | Mean Difference (IV, Random, 95% CI) | 1520.59 [1084.80, 1956.38] |
| 4.2 Sequential IOO | 1 | 127 | Mean Difference (IV, Random, 95% CI) | 799.47 [530.82, 1068.12] |
| 4.3 Sequential IIbO | 1 | 127 | Mean Difference (IV, Random, 95% CI) | 866.53 [478.83, 1254.23] |
| 4.4 Sequential IIO | 2 | 363 | Mean Difference (IV, Random, 95% CI) | 767.90 [337.75, 1198.06] |
| 5 Mean titres of P2 neutralising antibody | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 5.1 Sequential IbObO | 1 | 127 | Mean Difference (IV, Random, 95% CI) | ‐125.93 [‐174.77, ‐77.09] |
| 5.2 Sequential IOO | 1 | 127 | Mean Difference (IV, Random, 95% CI) | 142.25 [57.65, 226.85] |
| 5.3 Sequential IIbO | 1 | 127 | Mean Difference (IV, Random, 95% CI) | ‐83.45 [‐133.40, ‐33.50] |
| 5.4 Sequential IIO | 2 | 362 | Mean Difference (IV, Random, 95% CI) | 2224.48 [‐1145.70, 5594.67] |
| 6 Mean titres of P3 neutralising antibody | 2 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 6.1 Sequential IbObO | 1 | 127 | Mean Difference (IV, Random, 95% CI) | 327.62 [134.82, 520.42] |
| 6.2 Sequential IOO | 1 | 127 | Mean Difference (IV, Random, 95% CI) | 110.39 [‐77.98, 298.76] |
| 6.3 Sequential IIbO | 1 | 127 | Mean Difference (IV, Random, 95% CI) | 698.37 [301.06, 1095.68] |
| 6.4 Sequential IIO | 2 | 360 | Mean Difference (IV, Random, 95% CI) | 184.52 [‐211.93, 580.97] |
| 7 Long term mean titres of P1 neutralising antibody | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 7.1 Sequential IOO | 1 | 37 | Mean Difference (IV, Random, 95% CI) | ‐0.20 [‐0.73, 0.33] |
| 7.2 Sequential IOO+O | 1 | 37 | Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.83, 0.23] |
| 7.3 Sequential IIO | 1 | 40 | Mean Difference (IV, Random, 95% CI) | 0.40 [‐0.04, 0.84] |
| 7.4 Sequential IIO+O | 1 | 40 | Mean Difference (IV, Random, 95% CI) | 0.90 [0.40, 1.40] |
| 8 Long term mean titres of P2 neutralising antibody | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 8.1 Sequential IOO | 1 | 37 | Mean Difference (IV, Random, 95% CI) | 0.20 [‐0.06, 0.46] |
| 8.2 Sequential IOO+O | 1 | 37 | Mean Difference (IV, Random, 95% CI) | ‐0.60 [‐0.96, ‐0.24] |
| 8.3 Sequential IIO | 1 | 40 | Mean Difference (IV, Random, 95% CI) | 0.10 [‐0.19, 0.39] |
| 8.4 Sequential IIO+O | 1 | 40 | Mean Difference (IV, Random, 95% CI) | ‐0.40 [‐0.78, ‐0.02] |
| 9 Long term mean titres of P3 neutralising antibody | 1 | Mean Difference (IV, Random, 95% CI) | Subtotals only | |
| 9.1 Sequential IOO | 1 | 37 | Mean Difference (IV, Random, 95% CI) | ‐0.60 [‐1.22, 0.02] |
| 9.2 Sequential IOO+O | 1 | 37 | Mean Difference (IV, Random, 95% CI) | ‐0.30 [‐0.76, 0.16] |
| 9.3 Sequential IIO | 1 | 40 | Mean Difference (IV, Random, 95% CI) | 0.30 [‐0.04, 0.64] |
| 9.4 Sequential IIO+O | 1 | 40 | Mean Difference (IV, Random, 95% CI) | ‐0.10 [‐0.46, 0.26] |
| 10 Persons with polio faecal excretion after OPV challenge | 3 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 10.1 Persons with P1 faecal excretion (ibOI) | 1 | 519 | Risk Ratio (M‐H, Random, 95% CI) | 0.27 [0.19, 0.39] |
| 10.2 Persons with P1 faecal excretion (IIO/IIOO) | 1 | 303 | Risk Ratio (M‐H, Random, 95% CI) | 1.04 [0.59, 1.84] |
| 10.3 Persons with P2 faecal excretion (ibOI/IIbO) | 2 | 1048 | Risk Ratio (M‐H, Random, 95% CI) | 0.85 [0.68, 1.06] |
| 10.4 Persons with P2 faecal excretion (IIO/IIOO) | 1 | 303 | Risk Ratio (M‐H, Random, 95% CI) | 0.16 [0.09, 0.28] |
| 10.5 Persons with P3 faecal excretion (ibOI) | 1 | 519 | Risk Ratio (M‐H, Random, 95% CI) | 0.37 [0.26, 0.54] |
| 10.6 Persons with P3 faecal excretion (IIO/IIOO) | 1 | 303 | Risk Ratio (M‐H, Random, 95% CI) | 0.40 [0.32, 0.50] |
| 11 Serious adverse events (≥1 symptom related to study drug or not) | 2 | 1063 | Risk Ratio (M‐H, Random, 95% CI) | 0.92 [0.60, 1.43] |
Comparison 3. IPV(3)‐OPV versus IPV(2)‐OPV.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Persons with polio protective humoral response | 1 | Risk Ratio (M‐H, Random, 95% CI) | Subtotals only | |
| 1.1 P1 protective humoral response | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 0.98 [0.93, 1.03] |
| 1.2 P2 protective humoral response | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 1.0 [0.97, 1.03] |
| 1.3 P3 protective humoral response | 1 | 137 | Risk Ratio (M‐H, Random, 95% CI) | 1.01 [0.97, 1.05] |
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Alexander 2004.
| Methods |
Study design: interrupted time series, uncontrolled before‐and‐after. National surveillance data from 1990 to 2003 for cases of confirmed paralytic poliomyelitis. The Centers for Disease Control and Prevention (CDC) has maintained national poliomyelitis surveillance since 1955. This system relies on voluntary reporting of suspected cases from healthcare providers and laboratories through local and state health departments to the CDC, the Vaccine Adverse Events Reporting System, and the National Vaccine Injury Compensation Program. Although laboratory technology evolved during the study period, polioviruses were isolated and identified using conventional procedures of inoculation of processed specimens onto susceptible cell cultures. Isolates were then determined to be vaccine‐related by 1 of several standard molecular methods Setting: USA Study dates: 1990 to 2003 |
|
| Participants | Age: range = 19 to 35 months | |
| Interventions | To reduce the VAPP burden, national vaccination policy changed in 1997 from reliance on OPV to options for a sequential schedule of inactivated poliovirus vaccine (IPV) followed by OPV. In 2000, an exclusive IPV schedule was adopted. Before: exclusive use of OPV scheme OOOO (OPV at 2, 4, 12 to 18 months and again at 4 to 6 years) between 1990 and 1996 After: sequential scheme IIOO (IPV at 2 and 4 months of age followed by OP\/ at 12 to 18 months and again at 4 to 6 years) between 1997 and 1999. 13 cases of VAPP occurred, none of them in persons who had followed the sequential IPV‐OPV or all‐IPV schedules. |
|
| Outcomes |
*The sources were national information systems. Timing of outcome assessment: before, during, and after implementation of policy changes Follow‐up: 3 years |
|
| Notes | The last case of poliomyelitis in the USA due to indigenously acquired wild poliovirus occurred in 1979; however, as a consequence of oral poliovirus vaccine (OPV) use that began in 1961, an average of 9 cases of vaccine‐associated paralytic poliomyelitis (VAPP) were confirmed each year between 1961 and 1989. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| ITS: protection against secular changes | Low risk | Comment: the intervention appears to occur independently of other changes over time for VAPP cases. |
| ITS: data were analysed appropriately | Unclear risk | Comment: data were presented but not analysed as ITS, therefore, we re‐analysed the data. |
| ITS: reason given for the number of points pre‐ and post‐intervention | Low risk | Comment: the rationale for the yearly points is justified in the context of national poliomyelitis surveillance. |
| ITS: shape of the intervention effect was specified | Low risk | Comment: data were analysed as an ITS but were re‐analysed as time series regression models were used to analyse the data |
| ITS/UBA: blinded assessment of primary outcome(s) | Low risk | Comment: outcome variable is objective (case of paralytic poliomyelitis). |
| ITS/UBA: reliable primary outcome measure(s) | Low risk | Comment: Centers for Disease Control and Prevention has maintained national poliomyelitis surveillance since 1955. |
| ITS: intervention unlikely to affect data collection | Low risk | Comment: sources and methods of data collection were the same before and after the intervention. The outcome variable is objective (case of paralytic poliomyelitis). |
| ITS: completeness of data set | Low risk | Comment: data set probably covers 80% to 100% of total number of episodes in study |
| UBA: follow‐up of professionals | Low risk | Comment: low risk of bias in the context of a proper national poliomyelitis surveillance |
| UBA: follow‐up of patients | Low risk | Comment: low risk of bias in the context of a proper national poliomyelitis surveillance |
| Conflict of interest | Low risk | Comment: not clearly stated. Probably internal support from study authors' affiliation institution: National Immunization Program and National Center for Infectious Diseases, Centers for Disease Control and Prevention, Atlanta, Georgia |
Anand 2015.
| Methods |
Study design: open‐label, randomised trial Setting: Mirpur, an urban neighbourhood in Dhaka, Bangladesh Study dates: 2012 to 2013 |
|
| Participants |
Sample size: 975 infants recruited from 27 November 2012 to 30 November 2013 Age: median age = 44 days Sex: male = 462, female = 513 Dropouts/withdrawals: not reported Inclusion criteria: not reported Exclusion criteria: "(1) receipt of any polio vaccine before enrolment; (2) diagnosis or suspicion of immunodeficiency or a bleeding disorder; (3) known allergy to polio vaccines or constituents; (4) any acute illness such as vomiting, diarrhoea or infection immediately before enrolment; and (5) an infant who was part of a multiple birth" (quote) |
|
| Interventions |
|
|
| Outcomes |
Timing of outcome assessment: 6, 14, and 18 weeks |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Comment: randomisation mentioned but not described |
| Allocation concealment (selection bias) | Unclear risk | Comment: randomisation mentioned but not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: no blinding but performance is not likely to be influenced by lack of blinding in terms of intervention or co‐interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: no blinding but objective outcome (antibody titres) is not likely to be influenced by lack of blinding; adverse events may be influenced |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: 5.4% lost to follow‐up |
| Selective reporting (reporting bias) | Low risk | Comment: reported outcomes are the same as those reported in the trial register (NCT01813604) |
| Other bias | Low risk | Comment: study seems to be free of other bias |
| Conflict of interest | Low risk | Comment: funded by the Centers for Disease Control and Prevention. All study authors declare that they have no conflict of interest. |
Asturias 2007.
| Methods |
Study design: open‐label, randomised trial Setting: 3 well‐child public clinics, Guatemala city, Guatemala Study dates: April to November 2004 |
|
| Participants |
Sample size: 500 healthy, full‐term infants naive to polio vaccination were recruited from April to November 2004 Age: mean = 59.5 days Sex: male = 253, female = 247 Inclusion criteria: "Healthy, full‐term, 6–11‐week old infants attending 3 well‐child public clinics in Guatemala City were eligible" (quote) Exclusion criteria: "(1) received polio, hepatitis B (HB), Haemophilus influenzae type b (Hib), or diphtheria‐tetanus toxoids–pertussis (DTP) vaccines; (2) a history of any disease preventable by these vaccines; (3) a confirmed immunosuppressive condition; (4) received immunosuppressive drugs or blood‐derived products; (5) major congenital defects or serious chronic illness; (6) a history of any neurological disorders or seizures; or (7) allergies to any component of the vaccines." (quote) |
|
| Interventions |
Group A (n = 166): IIII at 2, 4, 6 and 12 months Group B (n = 168): IIOO at 2, 4,6 and 12 months Group C (n = 166): OOOO at 2, 4, 6 and 12 months |
|
| Outcomes |
Timing of outcome assessment: 2, 6, 7, 12, and 13 months |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk |
Quote: "Infants were randomized using a permuted block design of 6–12 at a 1:1:1 ratio within each study" Comment: probably done properly, since the table of baseline characteristics of included participants is balanced |
| Allocation concealment (selection bias) | Low risk | Quote: "Allocation to study group was done by opening sealed, sequentially numbered envelopes" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: no blinding but performance is not likely to be influenced by lack of blinding in terms of intervention or co‐interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: no blinding. Objective outcome (antibody titres) is not likely to be influenced by lack of blinding. Diary cards and digital thermometers were used by parents to record adverse events. Diary cards were collected at the next scheduled visit, and the parent was interviewed to ensure completeness of the information. These outcomes could have an unclear risk of bias but they were not meta‐analysed. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: Out of 500 infants enrolled, 444 (88.8%) were available for primary end‐point analysis, and 439 (87.8%) completed the last study visit. |
| Selective reporting (reporting bias) | Low risk | Comment: no evidence of selecting reporting |
| Other bias | Low risk | Comment: no evidence of other bias |
| Conflict of interest | Unclear risk | Financial support: financially supported by Sanofi Pasteur. Independent funds were used to support statistical analyses, and support for one of the authors was provided by a Fogarty International Research Scientist Development Award (grant KO1 TW006659). |
Davis 2001.
| Methods |
Study design: interrupted time series Setting: 2 large health maintenance organisations (HMOs), USA; Group Health Co‐operative Puget Sound (GHC) in Seattle (530,000 enrollees) and Kaiser Permanente (KPNC) in Northern California (2.8 million enrollees) Study dates: 1996 and 1997 |
|
| Participants |
Participants: children who were born between October 1, 1996, and December 31, 1997, resided in the greater metropolitan areas of Seattle and the Northern California region, were continuously enrolled in the HMO through the first year of life, and received at least 1 polio vaccination Sample size: ˜ 2721 GHC and 25,609 KPNC enrollees. Age: 12 months |
|
| Interventions |
Before (quarter 4, 1995 to quarter 3, 1997): 4 doses of OPV (OOOO) After (quarter 4, 1996 to quarter 1, 1997): 2 doses of IPV followed by 2 OPV doses (IIOO). Fewer than 5%, of GHC children and 3% of KPNC children received a mixed schedule. |
|
| Outcomes |
Timing of outcome assessment: 12 and 24 months |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| ITS: protection against secular changes | Low risk | Comment: The intervention appears to occur independently of other changes over time |
| ITS: data were analysed appropriately | Unclear risk | Quote: "As the follow‐up time was the same for each. child in the study, we estimated the relative risk of being up to date in tile IPV vs the OPV groups using Mante‐Haenszel estimation on stratified cumulative incidence data. We modelled cumulative Up‐to‐date time by comparing its mean value in the IPV group with the OPV group using analysis of variance. Finally, to examine the effect of IPV on missed opportunity visits, we modelled the likelihood of having a missed opportunity visit using logistic regression." |
| ITS: reason given for the number of points pre‐ and post‐intervention | Low risk | Comment: the rationale for the quarterly points is justified in the context of automated immunisation tracking systems. |
| ITS: shape of the intervention effect was specified | Unclear risk | Comment: data were analysed as an ITS but we re‐analysed the data |
| ITS/UBA: blinded assessment of primary outcome(s) | Low risk | Comment: the outcome (Imunisation status) is an objective measure, data is registered by information systems. |
| ITS/UBA: reliable primary outcome measure(s) | Low risk | Comment: both sites have automated immunisation tracking systems that allow for assessment of vaccination coverage by region, clinic, and individual patient. |
| ITS: intervention unlikely to affect data collection | Low risk | Comment: sources and methods of data collection were the same before and after the intervention. The outcome variable is objective ('up‐to‐date status' is an objective measure). |
| ITS: completeness of data set | Unclear risk | Comment: data set probably covers 80% to 100% of total number of episodes in the study |
| Conflict of interest | Low risk | Comment: this work was supported by grant R95‐074 from the Centers for Disease Control and Prevention's Comprehensive Linked Data Collection of Medical Events and Immunization. |
Faden 1990.
| Methods |
Study design: open‐label, randomised trial Setting: children's hospital, Buffalo, USA Study dates: not reported |
|
| Participants |
Sample size: 123 children naive to polio vaccination Dropouts/withdrawals: 35 Age: mean ages of children in each group ranged between 8.9 and 9.6 weeks at enrolment Sex: not reported Inclusion criteria: "Male and female infants 6‐10 weeks old were enrolled in the study from physicians' practices and the hospital's well child clinic. The children were free from apparent illness at the time of immunization." (quote) Exclusion criteria: "Children with major medical problems, in particular immune deficiency disorders, were excluded." (quote); "Patients were excluded from the efficacy analysis if they had fewer than three visits, were >13.5 weeks old at visit 1, were given the wrong vaccine, or had an unacceptable amount of time between visits[*]" (quote) |
|
| Interventions |
Group A (n = 23): OOO at 2, 4 and 12 months Group B (n = 65): III at 2 4 and 12 months Group C (n = 17): IOO at 2, 4 and 12 months Group D (n = 18): IIO at 2, 4 and 12 months Oral vaccine (Orimune; Lederle, Wayne, NJ) prepared in monkey kidney cells contained antigen doses of: poliovirus type 1, 2, and 3. Inactivated vaccine (Imovax Polio; Merieux Institute, Lyon, France) prepared in Vero cells contained antigen doses of: poliovirus type 1.40 D antigen units (DAU); type 2, 8 DAU; and type 3.32 DAU. |
|
| Outcomes |
Timing of outcome assessment: 2, 4, 5, 12, 13 and 60 months |
|
| Notes | *"The acceptable time between visits was visit 1 (2 months) to visit 2 (4 months), 6‐10 weeks; visit 2 to visit 3 (5 months), 3‐8 weeks; visit 3 to visit 4 (12 months), 6‐10 months; visit 4 to visit 5 (13 months), 3‐8 weeks. A patient excluded at one visit was excluded at all subsequent visits." (quote) | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote: "Infants were randomized to one of four treatment groups" Quote: "Differences in mean age at visit 1 were not significant (P = .49, one‐way analysis of variance)." |
| Allocation concealment (selection bias) | Unclear risk | Comment: not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: no blinding but performance is not likely to be influenced by lack of blinding in terms of intervention or co‐interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: no blinding but the objective outcomes (antibody titres and poliovirus shedding) are not likely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: patients were excluded from the efficacy analysis if they had fewer than three visits, were > 13.5 weeks old at visit 1, were given the wrong vaccine, or had an unacceptable amount of time between visits. Of the 158 children enrolled in the study, 35 were excluded; thus, 123 were analysed and 86 of them were assessed for their long‐term immunity. |
| Selective reporting (reporting bias) | Low risk | Comment: no evidence of selecting reporting |
| Other bias | Low risk | Comment: no evidence of other bias |
| Conflict of interest | Unclear risk | Comment: supported by grant from Merieux Institute, Miami, and National Institutes of Health (AI‐15939), and for long‐term immunity assessment, from Pasteur Merieux, Swiftwater, Pennsylvania |
Halsey 1997.
| Methods |
Study design: open‐label, randomised trial Setting: children's hospital, Buffalo, USA Assessment of immunogenicity. Venous blood samples were obtained at 2, 6 and 7 months of age and from participants at two of the study sites at 15 and 16 months of age. Polio antibody responses were evaluated at 2, 6, 7, 15 and 16 months of age. Poliovirus‐neutralising antibody titres were measured by a micrometabolic inhibition assay in Vero cells, a modification of a method previously described. Study dates: not reported |
|
| Participants |
Sample size: 295 children aged 6‐10 weeks old, recruited from 1991 Dropouts/withdrawals: 22 (12 = voluntarily withdrawn by their parents, 7 = noncompliant with study visits, 2 = lost to follow‐up, 1 = due to unusually high‐pitched cry after dose 1) Age: mean = 8.7 weeks Sex: male = 49.8%, female = 50.2% Inclusion criteria: healthy infants 6 to 12 weeks of age Exclusion criteria: not reported |
|
| Interventions |
Group A (n = not reported): IIIO at birth, 2, 4, 6 and 15 months; Group B (n = not reported): II(I+O)O at birth, 2, 4, 6 and 15 months; Group C (n = not reported): IIIOO at birth, 2, 4, 6 and 15 months. (Measurement at month 7 was used as III group) Each 0.5‐ml dose of IPV (IPOL™; Pasteur Merieux Connaught, Swiftwater, PA) contains 40 D antigen units of type I (Mahoney strain), 8 D antigen units of type 2 (MEFI strain) and 32 D antigen units of type 3 (Saukett strain) poliovirus grown in Vero cell cultures. Orimune® OPV (Lederle Laboratories, Pearl River, NY). |
|
| Outcomes |
Timing of outcome assessment: 2, 6, 7, 15 and 16 months Follow‐up: 4.6 months |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote: "Infants were randomly assigned to one of three groups at the time of enrolment" In favour of a appropriate randomisation process Quote: "Baseline characteristics were similar by study group for the 295 children enrolled in the trial." |
| Allocation concealment (selection bias) | Unclear risk | Comment: not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: no blinding but performance is not likely to be influenced by lack of blinding in terms of intervention or co‐interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "All serologic testing was performed by technicians blinded to the study group" |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: 12 infants were voluntarily withdrawn from the study by their parents. 7 infants were noncompliant with study visits, 2 were lost to follow‐up and 1 infant was withdrawn because of an unusual high‐pitched cry after dose 1. The distribution of withdrawals was similar by study group. Information was collected on 280 (94.9%) enrolled children at 6 months of age and 134 children at 15 and 16 months of age. |
| Selective reporting (reporting bias) | Low risk | Comment: no evidence of selecting reporting |
| Other bias | Low risk | Comment: no evidence of other bias |
| Conflict of interest | Unclear risk | Comment: grant from Pasteur Merieux Connaught, Inc |
Ivanova 2018.
| Methods |
Study design: nationwide uncontrolled before‐and‐after study. National AFP surveillance data from 2006 to 2013. Virological testing in WHO‐accredited laboratories of faecal specimens adequately taken and handled. VAPP cases were identified through the acute flaccid paralysis surveillance system, classified by the National Expert Classification Committee Setting: Russian Federation Study dates: 1998 to 2014 |
|
| Participants |
Participants: out of 6643 cases of AFP during the period 1998–2014, 127 cases were VAPP. 82 cases were observed in OPV recipients (rVAPP), whereas 45 cases were observed in non‐vaccinated contacts and classified as ‘contact VAPP’ (cVAPP). Age: the age of the patients varied from 1 month to 5.4 years (8.4 ± 8.1 months old). Children younger than 1 year constituted 74% of the group Sex: boys were dominant (80%) Ninety (70.9%) of the 127 VAPP patients were vaccinated against poliomyelitis. The majority of them (85.6%) had received one dose of OPV, while 10% had received two doses, 3.3% three doses, and 1.1% four doses. The time between OPV administration and the onset of paralysis was 20.9 8.7 days (ranging from 2 to 35 days). |
|
| Interventions |
Before: exclusive use of OPV scheme OOOO (OPV at 6, 18, 20 months; 14 years) during the period 1998‐2007 After: Sequential scheme IIIOOOO ( IPV at 3, 4.5 months; OPV at 6, 18, 20 months; 14 years) during the period 2008‐2014. |
|
| Outcomes |
Timing of outcome assessment: 10‐year period of tOPV use and a 7‐year period of the use of the sequential IPV–tOPV. Follow‐up: 10‐year period of tOPV use and a 7‐year period of the use of the sequential IPV–tOPV. |
|
| Notes | The research was carried out with support from the Federal Budget of the Russian Federation allocated for the implementation of the Polio Eradication Programme in the Russian Federation, the WHO Polio Eradication Programme, the WHO Regional Office for Europe, and a Russian Science Foundation grant (project No.15‐15‐00147). The work of OEI, KLI, and APG were partially supported by Russian academic excellence project “5–100”. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| ITS/UBA: blinded assessment of primary outcome(s) | Unclear risk | Comment: AFP and virological testing are objective outcomes; however, VAPP requires interpretation. |
| ITS/UBA: reliable primary outcome measure(s) | Low risk | Comment: national AFP surveillance and virological testing of faecal specimens in WHO‐accredited laboratories adequately taken and handled |
| UBA: follow‐up of professionals | Low risk | Comment: national AFP surveillance suggests a complete nationwide follow‐up. The follow‐up period was long enough. The AFP surveillance was unchanged from 2006 through to 2014. |
| UBA: follow‐up of patients | Low risk | Comment: national AFP surveillance suggests a complete nationwide follow‐up. The follow‐up period was long enough. The AFP surveillance was unchanged from 2006 through to 2014. |
| Conflict of interest | Low risk | Comment: the study was financed from the Russian Federation budget within the framework of the Program for eradication of poliomyelitis in the Russian Federation, WHO Polio eradication initiative, WHO's European Regional Bureau, Russian Foundation for Basic Research (project number 15‐15‐00147). |
Jain 1997.
| Methods |
Study design: open‐label, randomised trial Setting: Tel Aviv, Israel Study dates: not reported |
|
| Participants |
Sample size: 150 neonates Dropouts/withdrawals: 45 neonates; 25 from group II and 20 from group III Age: neonates Sex: not reported Inclusion criteria: healthy term, appropriate for date, newborns Exclusion criteria: newborns with significant problems during the first week of life |
|
| Interventions |
Group IA (n = 25): III, at birth, 1.5 and 2.5 months Group IB (n = 25): IOOO, at birth, 1.5, 2.5 and 3.5 months Group II (n = 50): OOOO, at birth, 1.5, 2.5 and 3.5 months Group III (n = 50): OOO at 1.5, 2.5 and 3.5 months tOPV was obtained from Central Government Hospital supply. Each batch of the vaccine was tested for potency at NICD. The IPV used was prepared at Merieux Institute, France. Each dose provides 40, 8 and 32 D antigen units against the three poliovirus types. |
|
| Outcomes |
Timing of assessment: 6 weeks, 10 weeks and 20 weeks Follow‐up: 4.6 months |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote: "..all newborns who were randomly divided into three groups" Quote: "The babies in the three groups after drop outs were comparable as regards their age and sex distribution, feeding pattern socioeconomic status and pre immunization antibody titers." Comment: this description is insufficient to classify as high or low risk of bias |
| Allocation concealment (selection bias) | Unclear risk | Comment: not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: no blinding but performance is not likely to be influenced by lack of blinding in terms of intervention or co‐interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: no blinding but objective outcome (antibody titres) is not likely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "A total of 45 neonates who dropped out from the study belonged to Group II (25) and Group III (20)." |
| Selective reporting (reporting bias) | Low risk | Comment: no evidence of selecting reporting |
| Other bias | Low risk | Comment: no evidence of other bias |
| Conflict of interest | Unclear risk | Comment: not described |
Kapusinszky 2010.
| Methods |
Study design: nationwide uncontrolled before‐and‐after study Setting: Hungary National AFP surveillance data from 1992 to 2006. Type 3 polioviruses isolated from the stools of patients with onset of AFP were recovered from archived specimens at the National Institute of Public Health, Budapest, Hungary. Study dates: 1959 to 1992 |
|
| Participants |
Participants: Out of all vaccinated Hungarian children, 90 cases of vaccine‐associated paralytic poliomyelitis (VAPP) were reported, 52 of which were associated with Sabin 3‐related virus (76% of VAPP cases with virologic data). Age: range = 1 to 54 months (mean = 15.6 months, median = 9 months) |
|
| Interventions |
Before (from 1959 to 1992, duration 396 months) O: (mOPV1, mOPV2, mOPV3) separated by 6 weeks at age 2‐38 months After (from 1992 to 2006, duration 168 months) IOOOOO: IPV at 3 months, 5 tOPV |
|
| Outcomes |
Follow‐up: 15 years |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | |
| ITS/UBA: blinded assessment of primary outcome(s) | Unclear risk | Comment: AFP and virological testing are objective outcomes; however, VAPP requires interpretation. |
| ITS/UBA: reliable primary outcome measure(s) | Low risk | Comment: In Hungary, the surveillance of AFP continued permanently with high standards. Additianalyy, there was a molecular characterization of poliovirus isolates from children who contracted VAPP. |
| UBA: follow‐up of professionals | Low risk | Comment: national AFP surveillance suggests a complete nationwide follow‐up. The follow‐up period was long enough. |
| UBA: follow‐up of patients | Low risk | Comment: national AFP surveillance suggests a complete nationwide follow‐up. The follow‐up period was long enough. |
| Conflict of interest | Unclear risk | Comment: supported by the RiViGene Project (Genomic inventory, forensic markers, and assessment of potential therapeutic and vaccine targets for viruses relevant in biological crime and terrorism; Contract number: SSPE‐CT‐2005‐022639) |
Li 2016a.
| Methods |
Study design: phase IV, randomised, open‐label trial Setting: Guangxi, China Study dates: 2011 to 2013 |
|
| Participants |
Sample size: 456 Dropouts/withdrawals: 7 dropped all (voluntarily withdrawn; 4 from Group A, 2 from Group B and 1 from Group C) Age: range = 2–3 months Sex: not reported Inclusion criteria: healthy infants aged 2‐3 months, born at full term (> 37 weeks) and weighing > 2.5 kg, were eligible for enrolment. Exclusion criteria: "participants in a separate clinical study, receipt of a non‐study vaccine before or during the study (excluding DTaP, Hib, BCG, and hepatitis B, which were to be administered 7 days prior or after any of study vaccinations), receipt of any poliomyelitis vaccine or poliomyelitis infection or any blood or blood‐derived products before the study, congenital or acquired immunodeficiency (in either the participant or in his/her close contacts), known hypersensitivity to any vaccine component, and any bleeding disorder contraindicating intramuscular injection." (quote) |
|
| Interventions |
Group A (n = 152): IOO at 2, 3, and 4 months Group B (n = 152): IIO at 2, 3, and 4 months Group C (n = 152): OOO at 2, 3, and 4 months Commercially available OPV and Sanofi Pasteur's IPV IMOVAX Polio. |
|
| Outcomes |
Secondary outcomes:
Timing of outcome assessment: pre‐dose 1, 1 month post dose 3, 14 months post dose 3. Follow‐up: 16 months |
|
| Notes | ClinicalTrials.gov NCT01475539 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote: "The Sponsor’s statistics department created a 1:1:1 randomisation list" Comment: insufficient information about the sequence generation process |
| Allocation concealment (selection bias) | Unclear risk | Comment: not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: no blinding but performance is not likely to be influenced by lack of blinding in terms of intervention or co‐interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: no blinding but objective outcome (antibody titres) is not likely to be influenced by lack of blinding; adverse events may be influenced |
| Incomplete outcome data (attrition bias) All outcomes | Low risk |
Comment:
|
| Selective reporting (reporting bias) | Low risk | Comment: the study register is available (NCT01475539) and all of the study’s pre‐specified (primary and secondary) outcomes have been reported in the pre‐specified way. |
| Other bias | Low risk | Comment: the study appears to be free of other sources of bias. |
| Conflict of interest | High risk | Comment: sponsored by Sanofi Pasteur, a Sanofi Company. The study director was the medical director of Sanofi Pasteur, China. |
Linder 2000.
| Methods |
Study design: open‐label, randomised trial Setting: Neonatal Division, Department of Pediatrics, Kalawati Saran Children's Hospital, Delhi, India Study dates: June to December 1994 |
|
| Participants |
Sample size: 177 infants (127 preterm infants and 50 full‐term infants) Dropouts/withdrawals: 40 children (22 = received blood products, 3 preterm infants = with sepsis, and 15 full‐term infants = withdrawn by their parents after the first blood test) Age: neonates (preterm experimental group median age = 32 weeks, full‐term control group = 40 weeks, preterm control group = 32 weeks) Sex: male = 62, female = 75 Inclusion criteria: preterm infants born between June and December 1994 (gestational age 30–35 weeks, weight > 1000 g); Fifty healthy full‐term infants (gestational age > 37 weeks, weight > 2500 g), born consecutively in the morning hours between 1 June 1994 and 15 June 1994. Exclusion criteria: not reported |
|
| Interventions |
Preterm experimental? group (n = 50): II (I + O) O at birth, 2, 4 and 6 months Full‐term group (n = 50): I (I + O) O at 2, 4 and 6 months Preterm group (n = 52): I (I + O) O at 2, 4 and 6 months Fifty preterm infants received IPV intramuscularly within 24 hours of birth, in addition to routine recommended childhood immunisations. Fifty‐two preterm infants and 35 full‐term infants received routine immunisations only (routine vaccination timing: HBV at birth, 1 and 6 months of age; IPV at 2 and 4 months; oral polio vaccine (OPV) at 4 and 6 months; diphtheria‐tetanus‐ pertussis (DTP) at 2, 4, and 6 months; and Haemophilus influenzae B vaccine at 2 and 4 months). Blood samples were taken at birth, 3 and 7 months of age from all infants, and at 1 month of age from preterm infants only. |
|
| Outcomes |
Timing of outcome assessment: 7 months Follow‐up: 7 months |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk |
Quote: "The preterm infants were divided into a study group (group A; n = 50) and a control group (group B; n = 52) by 1:1 randomisation..." Comment: probably done properly, since the table of baseline characteristics of randomised participants is balanced |
| Allocation concealment (selection bias) | Low risk | Quote: "...using a blinded envelope drawn by the parents. All full term infants were included in control group C." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: no blinding but performance is not likely to be influenced by lack of blinding in terms of intervention or co‐interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: no blinding but the objective outcome (antibody titres) is not likely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Low risk | Comment: no lost to follow‐up. At the bottom of Table 5 "A, group A (n = 50); B, group B (n = 52); C, group C (n = 35)" (quote) |
| Selective reporting (reporting bias) | Low risk | Comment: no evidence of selecting reporting |
| Other bias | Low risk | Comment: no evidence of other bias |
| Conflict of interest | Low risk | Comment: this study was supported by the chief scientist of the Israel Ministry of Health. |
Modlin 1997.
| Methods |
Study design: open‐label, randomised trial Setting: 3 Baltimore area paediatric practices, USA Study dates: 1991 to 1993 |
|
| Participants |
Sample size: 510 infants Dropouts/withdrawals: 101* (42* due to relocation or change of physician, 34* due to noncompliance with study visits, 28* due to parental request) Age: Not reported Sex: male = 266, female = 244 Inclusion criteria: not reported Exclusion criteria: not reported |
|
| Interventions |
Group A (n = 102): IIO at 2, 4, 15 months Group B (n = 105): IIOO at 2, 4, 6, 15 months Group C (n = 101): I (I + O) OO at 2, 4, 6, 15 months Group D (n = 99): III at 2, 4, 15 months Group E (n = 103): OOO at 2, 4, 15 months |
|
| Outcomes |
Timing of outcome assessment: 2, 6, 15, 18 months |
|
| Notes | *Exact figures taken from study report | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk |
Quote: "Infants were assigned by the Moses‐Oakford randomization algorithm" Quote: "There were no meaningful differences among the study groups in sex, race, mean age at each study visit, or withdrawal rate (data not shown)." |
| Allocation concealment (selection bias) | Unclear risk | Comment: not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: no blinding but performance is not likely to be influenced by lack of blinding in terms of intervention or co‐interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: no blinding but objective outcome (antibody titres) is not likely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Quote: "Of the subjects, 101 (20%) withdrew from the study before the 18month visit." |
| Selective reporting (reporting bias) | Low risk | Comment: no evidence of selecting reporting |
| Other bias | Low risk | Comment: no evidence of other bias |
| Conflict of interest | Unclear risk | Comment: financially supported by Connaught Laboratories, Inc, Swiftwater, Pennsylvania (now Pasteur‐Merieux‐Connaught); National Immunization Program, Centers for Disease Control and Prevention, Atlanta |
O'Ryan 2015.
| Methods |
Study design: multi‐centre, open‐label, non‐inferiority RCT Setting: 6 well‐child clinics in community health‐care centres, Santiago, Chile Study dates: 2013 |
|
| Participants |
Participants: 570 infants (51% males). Sample size: 570 infants Dropouts/withdrawals: 33 (20 = withdrawn by parents, 9 withdrawn due to protocol violations, 2 incorrectly enrolled, and 2 safety dropouts) Age: Mean = 57 days Sex: male = 283, female = 275 Inclusion criteria: "healthy, full‐term infants aged 8 weeks (± 7 days) with no obvious medical disorders, who weighed more than 2.5 kg at birth" (quote) Exclusion criteria: "infants were excluded if they had a sibling who had received, or was scheduled to receive, tOPV during the 6 months before or after the study, to avoid passive exposure to vaccine viruses. Other exclusion criteria were typical for vaccine studies—i.e., any disorder or treatment likely to interfere with normal immune responses to vaccination, or known allergy to vaccine components. Participants were excluded from any supplementary polio immunisation activity during the study." (quote) |
|
| Interventions |
Group A (n = 190): IOO at 2 months (IPV), 4.5, 6 months (bOPV) Group B (n = 192): IIO at 2 4.5 months (IPV), 6 months (bOPV) Group C (n = 188): III at 2, 4.5, 6 months Participants were challenged mOPV2 at age 28 weeks (6.4 months), and obtained another blood sample 1 week later at age 29 weeks (6.7 months). |
|
| Outcomes |
Primary outcome:
Secondary outcomes:
Timing of outcome assessment: weeks 8, 16, 24, 28, 29 Follow‐up: 7 months |
|
| Notes | Quote: "All analyses were done in a masked manner". Mail 7/24/2016 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "We did the allocation using randomisation lists supplied by the study sponsor with blocks of 12, stratified for the six study sites" |
| Allocation concealment (selection bias) | Unclear risk | Comment: not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: no blinding but performance is not likely to be influenced by lack of blinding in terms of intervention or co‐interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: no blinding but objective outcome (antibody titres) is not likely to be influenced by lack of blinding; adverse events may be influenced |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: 564 (99%) infants were included in the intention‐to‐treat cohort and 537 (94%) were vaccinated according to protocol: group IOO (91.4%), group IIO (95.7%) and group III (94.4%). Across groups, the 33 dropouts were mainly due to withdrawal by parents (n = 20) or protocol deviations (n = 9), with 2 being incorrectly enrolled, and 2 dropouts because of safety (heart defect and febrile seizure). Immunogenicity and shedding analyses were done for the per‐protocol population who received all vaccinations. Safety analyses were done in the intention‐to treat population. |
| Selective reporting (reporting bias) | Low risk | Comment: the trial registration is available (NCT01841671) and all of the study’s pre‐specified (primary and secondary) outcomes have been reported in the pre‐specified way. |
| Other bias | Low risk | Comment: the study appears to be free of other sources of bias. |
| Conflict of interest | Low risk | Comment: funded by the Bill & Melinda Gates Foundation. GlaxoSmithKline and Sanofi Pasteur donated the vaccines to the study. |
Qiu 2017.
| Methods |
Study design: multi‐centre, open‐label, non‐inferiority RCT Setting: Hezhou County and Zhongshan County, Guangxi Zhuang Autonomous Region of China Study dates: from April 8 and August 23, 2015 |
|
| Participants |
Participants: 600 healthy full‐term (37 to 42 weeks) infants who weighed more than 2.5 kg at birth with no obvious medical disorders, no polio vaccination, no immunoglobulin vaccination, with no other attenuated vaccine administered in the past 14 days and no other inactivated vaccine administered. Sample size: 600 Dropouts/withdrawals: 48 (35 = withdrawn, 5 = adverse event, 3 = protocol deviation, 3 = move, 2 = other) Age: range = 2‐3 months Sex: male = 231, female = 369 Inclusion criteria: "Eligible participants were healthy full‐term (37–42 weeks) infants aged 60–90 days who weighed more than 2.5 kg at birth with no obvious medical disorders, no polio vaccination, no immunoglobulin vaccination, with no other attenuated vaccine administered in the past 14 days and no other inactivated vaccine administered." (quote) Exclusion criteria: "Participants were excluded if meet one or more of the following criteria: had or were at risk of immunodeficiency, severe allergic reaction, acute fever or infectious diseases, severe chronic diseases, family history of allergies, convulsions, seizures, encephalopathy or psychiatric diseases, oral steroids during at least 14 consecutive days of the preceding month,auxiliary temperature equal or greater than 38.0C during the past 3 days, diarrhoea (defection frequency equal or greater than3 times per day) in the past 7 days, and participated in other drug clinical trials." (quote) |
|
| Interventions |
Administration: 3 doses administered sequentially at 4 to 6 weeks interval after collecting baseline blood sample |
|
| Outcomes |
Primaryoutcome
Secondary outcomes
Timing of outcome assessment: 30 days after last vaccination Follow‐up: 5 months |
|
| Notes | NCT02785705 | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Serial numbers from 1–600 were equally randomized (1:1:1:1:1:1) into 6 sequential vaccination schedules" |
| Allocation concealment (selection bias) | Low risk | Quote: "Sites were provided with sealed envelopes" |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk |
Quote: "Considering that the formulations are different, the vaccines could not be completely masked (oral vs. injectable), however, the bOPV and tOPV vaccines could be masked" Comment: performance, in terms of intervention or co‐interventions, is not likely to be influenced by lack of blinding |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Laboratory investigators were blinded to group assignments. A statistician would analyze data unblinded with the allocation schedule after the database was locked". |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: overall, 84% analysed per protocol for immunogenicity outcomes, and 95% for safety outcomes |
| Selective reporting (reporting bias) | Low risk | Comment: the study register is available (NCT02785705) and all of the study’s pre‐specified (primary and secondary) outcomes have been reported in the pre‐specified way. |
| Other bias | Low risk | Comment: the study appears to be free of other sources of bias. |
| Conflict of interest | Low risk | Comment: sponsored by Guangxi Zhuang Autonomous Region Center for Disease Prevention and Control, Fourth Military Medical University, and Beijing Tiantan Biological Products Co, Ltd |
Ramsay 1994.
| Methods |
Study design: open‐label, randomised trial Setting: North Hertfordshire, UK Study dates: not reported |
|
| Participants |
Sample size: 193 Dropouts/withdrawals: not reported Age: median 9 months at entry Sex: males in the IOO group 55% and in the OOO group 42% Inclusion criteria: infants undergoing routine immunisation in North Hertfordshire Exclusion criteria: no reported |
|
| Interventions |
Administration: 2, 3, 4 months |
|
| Outcomes |
Timing of outcome assessment: 5.5 months Follow‐up: 5.5 months |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | Quote: "Simple randomization of 200 study numbers was performed using a computer program and children were sequentially allocated to study numbers at recruitment." |
| Allocation concealment (selection bias) | Unclear risk | Comment: not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: no blinding but performance is not likely to be influenced by lack of blinding in terms of intervention or co‐interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: no blinding but the objective outcomes (antibody titres and faecal virus excretion) are not likely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Quote: "92 participants per group evaluated" |
| Selective reporting (reporting bias) | Low risk | Comment: no evidence of selecting reporting |
| Other bias | Low risk | Comment: no evidence of other bias |
| Conflict of interest | Low risk | Comment: this study was funded by Action Research. |
Rennels 2000.
| Methods |
Study design: open‐label, randomised trial Setting: North Hertfordshire, UK Study dates: not reported (the study was first presented November 14, 1998) |
|
| Participants |
Sample size: 567 (between 140 and 144 children were randomly allocated to each group) Dropouts/withdrawals: 0 Age: 2 months at entry per protocol Sex: male = 49%, female = 51% Inclusion criteria: healthy infants between 6 and 12 weeks of age Exclusion criteria: not reported |
|
| Interventions | Group A= DTaP + Hib + OPV; Group B, DTaP/Hib + OPV; Treatment Arm C, DTaP/Hib + IPV at 2 and 4 months and OPV at 6 months; or Treatment Arm D, DTaP/Hib + IPV Group A (n = 144): OOO at 2,4,6 months +DTP+Hib Group B (n = 140): OOO at 2,4,6 months + DTP/Hib Group C (n = 142): IIO at 2, 4, 6,15 months Group D: (n = 141): III at 2,4, 6 months Administration: groups A, B, and C doses given at 2, 4, and 6 months. Doses for group C given at 2, 4, 6 and 15 months |
|
| Outcomes |
Timing of outcome assessment: 7 months. Follow‐up: 5 months |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Children were equally randomized to receive at ˜ 2, 4 and 6 months of age (± 4 weeks) one of the vaccine schedules described in Table 1." |
| Allocation concealment (selection bias) | Unclear risk | Comment: not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk |
Quote: "Study nurses were not blinded, but the parents did not know what vaccine was given at which site." Comment: performance is not likely to be influenced by lack of blinding in terms of intervention or co‐interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "Laboratory personnel were blinded as to which was the pre‐ and postvaccination specimen and what vaccines each subject had received." |
| Incomplete outcome data (attrition bias) All outcomes | Low risk |
Quote: "Twenty‐one children were prematurely terminated from the study, and attempts to draw the post‐Dose 3 blood specimen were unsuccessful on an additional 7 infants. Protocol violations occurred with 51 children during the course of the study. Most of these were minor inaccuracies in the timing of vaccination or blood sampling." Quote: "There were no important differences between the results of the intention to treat and per protocol analyses" |
| Selective reporting (reporting bias) | Low risk | Comment: no evidence of selecting reporting |
| Other bias | Low risk | Comment: no evidence of other bias |
| Conflict of interest | Unclear risk | Comment: this work was supported by the National Institute of Allergy and Infectious Diseases, National Institutes of Health Contracts NO1‐AI5096 (Maryland), NO1‐AI72629 (Baylor), NO1‐AI45252 (Cincinnati), NO1‐AI05051 (St. Louis) and NO1‐AI5049 (Rochester). The vaccine was supplied by Aventis Pasteur. |
Simasathien 1994.
| Methods |
Study design: placebo‐controlled (for both Enhanced IPV and OPV), randomised trial Setting: Phramongkutkloa Hospital, Bangkok, Thailand The study was conducted as part of a trial of an oral rhesus‐human reassortant rotavirus tetravalent vaccine (RRV‐TV) given together with OPV. Enhanced potency inactivated poliovirus vaccine, combined with diphtheria‐tetanus‐pertussis (DTP) vaccine, was compared with oral poliovirus vaccine (OPV) regarding immunogenicity in Thai infants, vaccinated at 2, 4 and 6 months of age. Study dates: not reported |
|
| Participants |
Sample size: 330 Dropouts/withdrawals: not reported Age: 2 months at entry Sex: not reported Inclusion criteria: healthy full‐term ( 2 37 weeks of gestation) infants with birth weight of ≥ 2500 g were recruited for the study at birth. Exclusion criteria: not reported |
|
| Interventions |
OPV (or the respective placebo) was given first in 2 drops. Parenteral immunization (DTP plus placebo or DTP plus EIPV) was given after the oral vaccination. A standard lot of oral poliovirus vaccine (OPV), prepared by SmithKline Beecham Biologicals (Rixensart, Belgium) for studies with the WHO Expanded Programme on Immunization (EPI), was donated by the manufacturer for the study. The vaccine was of the 10: 1 : 3 type, and contained the following amounts of vaccine viruses: serotype 1, 106 TCID50 serotype 2, 105 TCID50and serotype 3, 105 TCID50. EIPV lot number 311, prepared by the Dutch National Institute of Public Health and Environmental Protection (RIVM. Bilthoven, The Netherlands) was used. The EIPV contained 40 D‐antigen units (12) of serotype I, 8 DU of serotype 2, and 32 DU of serotype 3. |
|
| Outcomes |
Timing of outcome assessment: 12 months. Follow‐up: 10 months |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk |
Quote: "The randomization was done by World Health Organization in Geneva..." Comment: not described |
| Allocation concealment (selection bias) | Low risk | Quote: "The randomization was done by WHO in Geneva, and the manufacturers were requested to code their vaccines accordingly. The code was held in Geneva and was not made available to the investigators until completion of the study." |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: placebo‐controlled trial |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: probably blinded. Additionally, the objective outcome (antibody titres) is not likely to be influenced by lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: 15% of patients were unavailable for serology at 12 months |
| Selective reporting (reporting bias) | Low risk | Comment: no evidence of selecting reporting |
| Other bias | Low risk | Comment: no evidence of other bias |
| Conflict of interest | Low risk | Comment: the study was financially supported by the Diarrhoea Diseases Control Programme, WHO. |
Sutter 1997.
| Methods |
Study design: placebo‐controlled (for both IPV and OPV), randomised trial Setting: Sohar Hospital, North Batinah, Oman Study dates: not reported |
|
| Participants |
Sample size: 547 Dropouts/withdrawals: not reported Age: mean = 32.6 weeks at 7‐month OPV vaccination Sex: not reported Inclusion criteria: infants born at the Sohar regional hospital in North Batinah were randomised at birth Exclusion criteria: not reported |
|
| Interventions |
Group 1 (n = 185): OOOO (O1) OO at birth, 1.5, 2.5 and 3.5 weeks, and 6, 7, and 9 months Group 2 (n = 172): O (O + I) (O + I) (O + I) (O1) OO at birth, 1.5, 2.5 , and 3.5 weeks, and 6, 7 and 9 months Group 3 (n = 190): III (O1) OO at birth, 1.5, 2.5, and 3.5 weeks, and 6, 7, and 9 months Placebo for IPV was diphtheria‐tetanus toxoid‐pertussis vaccine; placebo for OPV was molar magnesium chloride. IPV was manufactured by Pasteur‐Merieux Serums et Vaccins (Lyon, France) and formulated to contain 40, 8, and 32 D antigen units of poliovirus types 1, 2, and 3, respectively, and was combined with DTP per 0.5 mL dose. OPV was manufactured by SmithKline Beecham Biologicals (Rixensart, Belgium) |
|
| Outcomes |
Timing of outcome assessment: 6 and 10 months. Follow‐up: 10 months |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "...were randomized at birth" |
| Allocation concealment (selection bias) | Unclear risk | Comment: not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: no blinding but performance is not likely to be influenced by lack of blinding in terms of intervention or co‐interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: probably blinded. Additionally, the objective outcome (antibody titres) is not likely to be influenced by lack of blinding. |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: not reported |
| Selective reporting (reporting bias) | Low risk | Comment: no evidence of selecting reporting |
| Other bias | Low risk | Comment: no evidence of other bias |
| Conflict of interest | Low risk | Comment: financial support received from Global Programme for Vaccines and Immunization, WHO, Geneva, using funds donated by the governments of Finland and Sweden, the Rockefeller Foundation, and the United Nations Development Programme |
Von Magnus 1984.
| Methods |
Study design: interrupted time series. National surveillance data from 1935 to 1980 for cases of confirmed paralytic poliomyelitis. Recollection of paired serum samples for type 1 antibodies Setting: Denmark |
|
| Participants |
Sample size: 2.7 million people. In connection with the vaccination, paired serum samples from 300 individuals were collected and examined for type 1 antibodies Age: younger than 40 years of age |
|
| Interventions |
Before: III from 1961‐1967 After: IIIOOO from 1968‐1973 In the years between 1962 and 1967 the polio vaccination program in Denmark consisted of three injections of inactivated poliovirus vaccine (IPV). From 1968 onwards the polio vaccination program changed to include three injections of IPV when children are five, six, and 15 months of age and three vaccinations with tOPV administered at the age of three, four, and five years. |
|
| Outcomes |
Follow‐up: 13 years |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| ITS: protection against secular changes | Low risk | Comment: intervention appears to occur independently of other changes over time |
| ITS: data were analysed appropriately | High risk | Comment: crude numbers |
| ITS: reason given for the number of points pre‐ and post‐intervention | Low risk | Comment: rationale for the yearly points is justified in the context of national surveillance system |
| ITS: shape of the intervention effect was specified | Unclear risk | Comment: data were not analysed as an ITS but we re‐analysed the data. |
| ITS/UBA: blinded assessment of primary outcome(s) | Low risk | Comment: The outcomes 'paralytic poliomyelitis cases' and 'antibody titres' are objective. The source were national information systems. |
| ITS/UBA: reliable primary outcome measure(s) | Low risk | Number of paralytic poliomyelitis cases. Cases occurred in close relation to the vaccinations were considered as confirmed and the other as doubtful. Antibody titres is a more objective outcome. |
| ITS: intervention unlikely to affect data collection | Unclear risk | Comment: sources and methods of data collection were the same before and after the intervention. The outcome variables are objective measures. |
| ITS: completeness of data set | Unclear risk | Comment: unclear report |
| Conflict of interest | Low risk | Study supported by the Epidemiology Department and the Enterovirus Department,Statens Seruminstitut, Copenhagen, Denmark. |
West 2001.
| Methods |
Study design: open‐label, randomised trial Setting: 3 sites, USA Study dates: not reported |
|
| Participants |
Sample size: 126 healthy infants Dropouts/withdrawals: 5 infants did not receive all 3 vaccines. Age: not reported Sex: male = 72, female = 54 Inclusion criteria: "Healthy infants approximately 2 months of age, who had previously received a dose of monovalent HB vaccine shortly after birth and had a negative history for both Hib disease and HBV infection, were recruited for the study" (quote) Exclusion criteria: not reported |
|
| Interventions | 2 treatment groups one in which infants were given diphtheria‐tetanus‐pertussis whole cell vaccine (DTP) + IPV at two months followed by two OPV doses and the other group received DTP + OPV at 2 months followed by two further OPV doses
|
|
| Outcomes |
Timing of outcome assessment: 6 months Follow‐up: 14 months |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Infants were randomly allocated (1 : 1) to 1 of 2 treatment groups designated DTP/IPV and DTP+OPV" |
| Allocation concealment (selection bias) | Unclear risk | Comment: not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk | Comment: no blinding but performance is not likely to be influenced by lack of blinding in terms of intervention or co‐interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Comment: no blinding but objective outcome (antibody titres) is not likely to be influenced by lack of blinding |
| Incomplete outcome data (attrition bias) All outcomes | Unclear risk | Comment: not reported |
| Selective reporting (reporting bias) | Low risk | Comment: no evidence of selecting reporting |
| Other bias | Low risk | Comment: no evidence of other bias |
| Conflict of interest | Unclear risk | This study was supported financially by the Merck Research Laboratories. |
Yeh 2001.
| Methods |
Study design: open‐label, randomised trial Setting: 3 Southern California Kaiser Permanente Health Care Program clinics, USA Study dates: not reported |
|
| Participants |
Sample size: 400 healthy infants Dropout/withdrawals: 53 (2 due to adverse events that were determined to be unrelated to the vaccine); and "follow‐up venipuncture was refused by the parents of an additional 15 subjects" (quote) Age: mean age of 1.9 months (1.2 to 3.1) for the first dose, 3.9 months (3.0 to 6.2) for the second dose, 5.9 months (4.8 to 7.8) for the third dose and 14.1 months (13.0 to 17.0) for the booster dose. Sex: not reported Inclusion criteria: healthy infants Exclusion criteria: "Subjects were excluded from participation if they: were younger than 6 weeks or older than 12 weeks at entry; had rectal temperatures 100.4°F (immunizations deferred); had received blood products or immunoglobulin, including hepatitis B immunoglobulin; were born to a mother known to be HIV‐positive or HBsAg‐positive; had major congenital defects or other serious illnesses; had a history of neurologic disorder or seizures; had known or suspected immune dysfunction or family history of congenital immune dysfunction; had history of prior receipt of any vaccine; had history of hypersensitivity to yeast or any components of the vaccines; had an immunocompromised household member known or one known to be HBsAg‐positive; or had no telephone access." (quote) |
|
| Interventions |
Group A (n = 100): III at 2, 4, 6 months Group B (n = 100): IIO at 2, 4, 6 months Group C (n = 100): III at 2, 4, 6, 15 months Group D (n = 100): OOO at 2, 4, 6 months |
|
| Outcomes |
Timing of outcome assessment: 7 months, 16 months |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | Quote: "Subjects were prospectively randomized into 4 equal study groups. There were no significant differences among groups by age, gender or race/ethnicity." |
| Allocation concealment (selection bias) | Unclear risk | Comment: not described |
| Blinding of participants and personnel (performance bias) All outcomes | Low risk |
Quote: "Given the different types of oral and injectable vaccines and their different schedules, the blinding of parents and clinical investigators could not be assured" Comment: performance is not likely to be influenced by lack of blinding in terms of intervention or co‐interventions |
| Blinding of outcome assessment (detection bias) All outcomes | Low risk | Quote: "...however, all laboratory evaluations were performed blindly" |
| Incomplete outcome data (attrition bias) All outcomes | High risk | Comment: 9% to 14% lost to follow‐up at 7 months and 38% to 69% lost to follow‐up at 18 months |
| Selective reporting (reporting bias) | Low risk | Comment: no evidence of selecting reporting |
| Other bias | Low risk | Comment: no evidence of other bias |
| Conflict of interest | Unclear risk | Comment: study was supported by a grant from GlaxoSmithKline Biologicals |
Most used acronyms
AFP: acute flaccid paralysis ; IPV: inactivated poliovirus vaccine; ITS: interrupted time series; OPV: oral poliovirus vaccine; RCT: randomised controlled trial; VAPP: vaccine‐associated paralytic polio; UBA: uncontrolled before‐and‐after studies; WHO: World Health Organization. See all acronyms in Appendix 1
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| Li 2016b | Ineligible intervention: any sequential IPV‐OPV among the four trials summarised in this study |
| McCollough 1969 | Ineligible study design: uncontrolled before and after study |
| Moǐseieva 2002 | Ineligible study design: not a randomised controlled trial |
| Swartz 1998 | Ineligible study design: cohort study |
| Wattigney 2001 | Ineligible study design: less than three points after intervention to be considered as a short interrupted time series |
| Ye 2018 | Ineligible intervention: sequential schedules of IPV made from sabin strain (sIPV) and not from WPVs |
IPV‐OPV: sequential inactivated poliovirus vaccine‐oral poliovirus vaccine; sIPV: Sabin IPV; WPV: wild poliovirus.
Characteristics of ongoing studies [ordered by study ID]
NCT02412514.
| Trial name or title |
Public title: Intestinal humoral immunity of sequential polio vaccination schedules Scientific title: Assessing the intestinal and humoral immunity of sequential schedules of inactivated poliovirus vaccine and bivalent oral poliovirus vaccine for routine childhood immunization in Bangladesh |
| Methods | Design: randomised, open‐label trial |
| Participants |
Location: Dhaka, Bangladesh Participants: healthy infants Sample size: 456 Age: 6 weeks (range: 42‐48 days) |
| Interventions |
Arm A (I+O)OO: IPV + bOPV, bOPV and bOPV, administered at 6, 10, and 14 weeks, respectively Arm B (IOO): IPV, bOPV and bOPV, administered at 6, 10, and 14 weeks, respectively |
| Outcomes |
Primary outcomes
Secondary outcome
|
| Starting date |
Start date: April 2015 Completion date: December 2015 |
| Contact information |
Sponsor: Centers for Disease Control and Prevention Collabator: International Centre for Diarrhoeal Disease Research, Bangladesh Principal Investigator: not stated |
| Notes | Recruitment status: completed |
NCT03430349.
| Trial name or title |
Public title: Phase 1 novel live attenuated serotype 2 oral polio vaccine study in IPV primed adults (nOPV2M4a) Scientific title: A phase 1 study to evaluate the safety and immunogenicity of 2 novel live attenuated serotype 2 oral poliovirus vaccines, in healthy adults previously primed with inactivated polio vVaccine (IPV) |
| Methods | Design: RCT, phase 1 study; designed to evaluate, in contained conditions, the safety, immunogenicity, shedding and genetic stability of nOPV2 vaccine candidates in IPV‐primed adults before testing in a larger adult and adolescent (> 15 years of age) population, and then in young children and infants; triple blinding (participant, care provider, investigator) |
| Participants |
Location: Antwerp, Belgium Participants: IPV‐primed adults Sample size: 30 Age: 18 to 50 years |
| Interventions | Novel OPV2 candidate vaccine 1 & 2. The study will be conducted with each candidate vaccine sequentially: Quote: "After randomization of the first subject of group 1 the next 14 subjects will all be enrolled in the same Group and receive the same nOPV2 candidate and the next 15 subjects will be enrolled in the other Group and receive the corresponding nOPV2 candidate. Prior to the start of the study the CRO will provide the site with 2 randomization envelopes for the first subject. By randomly choosing 1 of the envelopes first subject will be dedicated to a certain nOPV2 candidate and this will determine the allocation of the next 14 subjects to the same Group." |
| Outcomes |
Primary outcomes
Secondary outcomes
Other outcome measures
|
| Starting date |
Start date: 22 May 2017 Completion date: 30 July 2018 (estimated) |
| Contact information |
Sponsor: Dr Pierre van Damme (Centre for the Evaluation of Vaccination) Collaborators: Bill and Melinda Gates Foundation; Centers for Disease Control and Prevention; PATH; Celerion Principal investigator: Dr Pierre van Damme (Centre for the Evaluation of Vaccination) |
| Notes | Recruitment status: active, not recruiting |
NCT03614702.
| Trial name or title |
Public title: Clinic trial to evaluate the safety and immunogenicity by different sequential schedules of bOPV and IPV Scientific title: Safety and immunogenicity evaluation of different sequential immunization schedules of type 1 + 2 bivalent oral poliovirus vaccine(bOPV) co‐administered with inactived poliovirus vaccine (IPV) in infants aged 2 months: a randomized, double blind, single centre, parallel trial |
| Methods | Design: randomised, double‐blind, single‐centre, parallel trial |
| Participants |
Location: Guangxi Province, China Participants: infants Age: 2 months Sample size: 1200 |
| Interventions |
|
| Outcomes |
Primary outcomes
Secondaary outcomes
Other outcomes
|
| Starting date |
Start date: 15 September 2015 Completion date: August 2016 |
| Contact information |
Sponsor: Chinese Academy of Medical Sciences Collaborator: Guangxi Province Center for Diseases Control and Prevention Principal Investigator: Zhaojun Mo |
| Notes | Recruitment status: completed |
AE: adverse events; bOPV: bivalent oral polio vaccine; CRO: country responsible officer; IPV: inactivated polio vaccine; cIPV: conventional inactivated polio virus; nOPV2: novel monovalent oral type 2 polio vaccine; RCT: randomised controlled trial; PCR: polymerase chain reaction; sIPV: Sabin strain of inactivated polio vaccine; tOPV: trivalent oral polio vaccine.
Differences between protocol and review
Data collection and analysis. We were unable to use all of our preplanned methods. See Table 6.
Summarising and interpreting results (beneath Data synthesis). We decided to use the current version of GRADE (Hultcrantz 2017), so we used the expression ‘certainty’ instead of ‘quality’ as was stated in our protocol (Ciapponi 2014).
We grouped, analysed and presented the results according to serotypes P1, P2 and P3 protective humoral and intestinal response (outcomes 3, 4, and 5) because the effect is partially independent of each other, and in that sense, many policies, like the replacement of tOPV by bOPV, are serotype specific. If we were not able to pool the data in a meta‐analysis due to considerable heterogeneity, we presented the scheme of two IPV doses (IIO) as the main subgroup for this outcome, since it is the most studied scheme.
Since 2012, the WHO has recommended the replacement of the trivalent OPV (tOPV) by the bivalent OPV (bOPV) (WHO 2012). Many of the recent studies included both tOPV and bOPV arms; we decided to include post‐hoc subgroups analysis by type of OPV to ensure that the findings were relevant to the most recent guidelines.
Contributions of authors
All authors contributed to drafting the protocol, selecting studies for inclusion, extracting data, and contributing to drafting the review. Agustín Ciapponi and Ariel Bardach analysed and interpreted the data.
Agustín Ciapponi, as lead author, is the guarantor for the review.
Sources of support
Internal sources
-
Instituto de Efectividad Clínica y Sanitaria [Institute for Clinical Effectiveness and Health Policy], Argentina.
Technical assistance and protected IECS authors' time to develop the protocol and the review
External sources
-
Pan American Health Organization (PAHO), Other.
Our institution, IECS, received an independent grant from the PAHO, which covered salaries and all expenses related to a preliminary version of the review.
Declarations of interest
A preliminary version of this review was supported by a grant from the Pan American Health Organization (PAHO). The PAHO commissioned the review team to analyse the evidence for sequential inactivated (IPV) and live oral (OPV) poliovirus vaccines for the prevention of poliomyelitis, to assist their decision‐making process. Agustín Ciapponi, Ariel Bardach, Lucila Rey‐Ares, Demián Glujovsky, María Luisa Cafferata, and Silvana Cesaroni all received payments from this grant. They have no other conflicts of interest to declare.
Aikant Bhatti ‐ none known.
Disclaimer: the views herein are those of the authors and not necessarily those of the PAHO.
New
References
References to studies included in this review
Alexander 2004 {published data only}
- Alexander LN, Seward JF, Santibanez TA, Pallansch MA, Kew OM, Prevots DR, et al. Vaccine policy changes and epidemiology of poliomyelitis in the United States. JAMA 2004;292(14):1696‐701. [DOI: 10.1001/jama.292.14.1696; PUBMED: 15479934] [DOI] [PubMed] [Google Scholar]
Anand 2015 {published data only}
- Anand A, Zaman K, Estívariz CF, Yunus M, Gary HE, Weldon WC, et al. Early priming with inactivated poliovirus vaccine (IPV) and intradermal fractional dose IPV administered by a microneedle device: a randomized controlled trial. Vaccine 2015;33(48):6816‐22. [DOI: 10.1016/j.vaccine.2015.09.039; PUBMED: 26476367] [DOI] [PMC free article] [PubMed] [Google Scholar]
Asturias 2007 {published data only}
- Asturias EJ, Dueger EL, Omer SB, Melville A, Nates SV, Laassri M, et al. Randomized trial of inactivated and live polio vaccine schedules in Guatemalan infants. Journal of Infectious Diseases 2007;196(5):692‐8. [DOI: 10.1086/520546; PUBMED: 17674310] [DOI] [PubMed] [Google Scholar]
Davis 2001 {published data only}
- Davis RL, Lieu TA, Mell LK, Capra AM, Zavitkovsky A, Quesenberry CP Jr, et al. Impact of the change in polio vaccination schedule on immunization coverage rates: a study in two large health maintenance organizations. Pediatrics 2001;107(4):671‐6. [DOI: 10.1542/peds.107.4.671; PUBMED: 11335742] [DOI] [PubMed] [Google Scholar]
Faden 1990 {published data only}
- Faden H. Results of a clinical study of polio vaccine: the Buffalo experience. Pediatric Infectious Disease Journal 1991;10(12):973‐5. [PUBMED: 1766725] [DOI] [PubMed] [Google Scholar]
- Faden H, Duffy L, Sun M, Shuff C. Long‐term immunity to poliovirus in children immunized with live attenuated and enhanced‐potency inactivated trivalent poliovirus vaccines. Journal of Infectious Diseases 1993;168(2):452‐4. [DOI: 10.1093/infdis/168.2.452; PUBMED: 8335983] [DOI] [PubMed] [Google Scholar]
- Faden H, Modlin JF, Thoms ML, McBean AM, Ferdon MB, Ogra PL. Comparative evaluation of immunization with live attenuated and enhanced‐potency inactivated trivalent poliovirus vaccines in childhood: systemic and local immune responses. Journal of Infectious Diseases 1990;162(6):1291‐7. [DOI: 10.1093/infdis/162.6.1291; PUBMED: 2172403] [DOI] [PubMed] [Google Scholar]
Halsey 1997 {published data only}
- Halsey NA, Blatter M, Bader G, Thoms ML, Willingham FF, O'Donovan JC, et al. Inactivated poliovirus vaccine alone or sequential inactivated and oral poliovirus vaccine in two‐, four‐ and six‐month‐old infants with combination Haemophilus influenzae type b/hepatitis B vaccine. Pediatric Infectious Disease Journal 1997;16(7):675‐9. [DOI: 10.1097/00006454-199707000-00010; PUBMED: 9239772] [DOI] [PubMed] [Google Scholar]
Ivanova 2018 {published data only}
- Ivanova OE, Eremeeva TP, Morozova NS, Shakaryan AK, Gmyl AP, Yakovenko ML, et al. Vaccine‐associated paralytic poliomyelitis in Russian Federation during the period of changes in vaccination schedule (2006‐2013 yy.) [Вакцинный паралитический полиомиелит в Российской Федерации в период изменений (2006‐2013 гг.)]. Voprosy Virusologii 2016;61(1):9‐15. [DOI: 10.18821/0507-4088-2016-61-1-9-15; PUBMED: 27145594] [DOI] [PubMed] [Google Scholar]
- Ivanova OE, Eremeeva TP, Morozova NS, Shakaryan AK, Korotkova EA, Kozlovskaya LI, et al. Vaccine‐associated paralytic poliomyelitis in the Russian Federation in 1998‐2014. International Journal of Infectious Diseases 2018;76:64‐9. [DOI: 10.1016/j.ijid.2018.08.017; PUBMED: 30201507] [DOI] [PubMed] [Google Scholar]
Jain 1997 {published data only}
- Jain PK, Dutta AK, Nangia S, Khare S, Saili A. Seroconversion following killed polio vaccine in neonates. Indian Journal of Pediatrics 1997;64(4):511‐5. [DOI: 10.1007/BF02737758; PUBMED: 10771880] [DOI] [PubMed] [Google Scholar]
Kapusinszky 2010 {published data only}
- Kapusinszky B, Molnár Z, Szomor KN, Berencsi G. Molecular characterization of poliovirus isolates from children who contracted vaccine‐associated paralytic poliomyelitis (VAPP) following administration of monovalent type 3 oral poliovirus vaccine in the 1960s in Hungary. FEMS Immunology and Medical Microbiology 2010;58(2):211‐7. [DOI: 10.1111/j.1574-695X.2009.00621.x; PUBMED: 19863665] [DOI] [PubMed] [Google Scholar]
Li 2016a {published data only}
- Li RC, Li CG, Wang HB, Luo HM, Li YP, Wang JF, et al. Immunogenicity of two different sequential schedules of inactivated polio vaccine followed by oral polio vaccine versus oral polio vaccine alone in healthy infants in China. Journal of the Pediatric Infectious Diseases Society 2016;5(3):287‐96. [DOI: 10.1093/jpids/piv017; PUBMED: 26407255] [DOI] [PubMed] [Google Scholar]
Linder 2000 {published data only}
- Linder N, Handsher R, German B, Sirota L, Bachman M, Zinger S, et al. Controlled trial of immune response of preterm infants to recombinant hepatitis B and inactivated poliovirus vaccines administered simultaneously shortly after birth. Archives of Disease in Childhood. Fetal and Neonatal Edition 2000;83(1):F24‐7. [DOI: 10.1136/fn.83.1.f24; PMC1721105; PUBMED: 10873167] [DOI] [PMC free article] [PubMed] [Google Scholar]
Modlin 1997 {published data only}
- Modlin JF, Halsey NA, Thoms ML, Meschievitz CK, Patriarca PA. Humoral and mucosal immunity in infants induced by three sequential inactivated poliovirus vaccine‐live attenuated oral poliovirus vaccine immunization schedules. Baltimore Area Polio Vaccine Study Group. Journal of Infectious Diseases 1997;175 Suppl 1:S228‐34. [DOI: 10.1093/infdis/175.Supplement_1.S228; PUBMED: 9203721] [DOI] [PubMed] [Google Scholar]
O'Ryan 2015 {published data only}
- Brickley EB, Wieland‐Alter W, Connor RI, Ackerman ME, Boesch AW, Arita M, et al. Intestinal immunity to poliovirus following sequential trivalent inactivated polio vaccine/bivalent oral polio vaccine and trivalent inactivated polio vaccine‐only immunization schedules: analysis of an open‐label, randomized, controlled trial in Chilean infants. Clinical Infectious Diseases 2018;67(Suppl 1):S42‐50. [DOI: 10.1093/cid/ciy603; PMC6206105; PUBMED: 30376086] [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Ryan M, Bandyopadhyay AS, Villena R, Espinoza M, Novoa J, Weldon WC, et al. Inactivated poliovirus vaccine given alone or in a sequential schedule with bivalent oral poliovirus vaccine in Chilean infants: a randomised, controlled, open‐label, phase 4, non‐inferiority study. Lancet. Infectious Diseases 2015;15(11):1273‐82. [DOI: 10.1016/S1473-3099(15)00219-4] [DOI] [PubMed] [Google Scholar]
Qiu 2017 {published data only}
- Qiu J, Yang Y, Huang L, Wang L, Jiang Z, Gong J, et al. Immunogenicity and safety evaluation of bivalent types 1 and 3 oral poliovirus vaccine by comparing different poliomyelitis vaccination schedules in China: a randomized controlled non‐inferiority clinical trial. Human Vaccines & Immunotherapeutics 2017;13(6):1359‐68. [DOI: 10.1080/21645515.2017.1288769; PMC5489289 ; PUBMED: 28362135] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ramsay 1994 {published data only}
- Ramsay ME, Begg NT, Gandhi J, Brown D. Antibody response and viral excretion after live polio vaccine or a combined schedule of live and inactivated polio vaccines. Pediatric Infectious Disease Journal 1994;13(12):1117‐21. [DOI: 10.1097/00006454-199412000-00009; PUBMED: 7892081] [DOI] [PubMed] [Google Scholar]
Rennels 2000 {published data only}
- Laassri M, Lottenbach K, Belshe R, Rennels M, Plotkin S, Chumakov K. Analysis of reversions in the 5′‐untranslated region of attenuated poliovirus after sequential administration of inactivated and oral poliovirus vaccines. Journal of Infectious Diseases 2006;193(10):1344‐9. [DOI: 10.1086/503366; PUBMED: 16619180] [DOI] [PubMed] [Google Scholar]
- Laassri M, Lottenbach K, Belshe R, Wolff M, Rennels M, Plotkin S, et al. Effect of different vaccination schedules on excretion of oral poliovirus vaccine strains. Journal of Infectious Diseases 2005;192(12):2092‐8. [DOI: 10.1086/498172; PUBMED: 16288372] [DOI] [PubMed] [Google Scholar]
- Rennels MB, Englund JA, Bernstein DI, Losonsky GA, Anderson EL, Pichichero ME, et al. Diminution of the anti‐polyribosylribitol phosphate response to a combined diphtheria‐tetanus‐acellular pertussis/Haemophilus influenzae type b vaccine by concurrent inactivated poliovirus vaccination. Pediatric Infectious Disease Journal 2000;19(5):417‐23. [DOI: 10.1097/00006454-200005000-00006; PUBMED: 10819337] [DOI] [PubMed] [Google Scholar]
Simasathien 1994 {published data only}
- Simasathien S, Migasena S, Beuvery C, Steenis G, Samakoses R, Pitisuttitham P, et al. Comparison of enhanced potency inactivated poliovirus vaccine (EIPV) versus standard oral poliovirus vaccine (OPV) in Thai infants. Scandinavian Journal of Infectious Diseases 1994;26(6):731‐8. [DOI: 10.3109/00365549409008643; PUBMED: 7747098] [DOI] [PubMed] [Google Scholar]
Sutter 1997 {published data only}
- Sutter RW, Suleiman AJ, Malankar PG, Mehta FR, Medany MA, Arif MA, et al. Sequential use of inactivated poliovirus vaccine followed by oral poliovirus vaccine in Oman. Journal of Infectious Diseases 1997;175 Suppl 1:S235‐40. [DOI: 10.1093/infdis/175.Supplement_1.S235; PUBMED: 9203722] [DOI] [PubMed] [Google Scholar]
Von Magnus 1984 {published data only}
- Magnus H, Petersen I. Vaccination with inactivated poliovirus vaccine and oral poliovirus vaccine in Denmark. Reviews of Infectious Diseases 1984;6 Suppl 2:S471‐4. [DOI: 10.1093/clinids/6.Supplement_2.S471; PUBMED: 6740095] [DOI] [PubMed] [Google Scholar]
West 2001 {published data only}
- West DJ, Rabalais GP, Watson B, Keyserling HL, Matthews H, Hesley TM. Antibody responses of healthy infants to concurrent administration of a bivalent Haemophilus influenzae type b‐hepatitis B vaccine with diphtheria‐tetanus‐pertussis, polio and measles‐mumps‐rubella vaccines. Biodrugs 2001;15(6):413‐8. [DOI: 10.2165/00063030-200115060-00007; PUBMED: 11520252] [DOI] [PubMed] [Google Scholar]
Yeh 2001 {published data only}
- Yeh SH, Ward JI, Partridge S, Marcy SM, Lee H, Jing J, et al. Safety and immunogenicity of a pentavalent diphtheria, tetanus, pertussis, hepatitis B and polio combination vaccine in infants. Pediatric Infectious Disease Journal 2001;20(10):973‐80. [DOI: 10.1097/00006454-200110000-00011; PUBMED: 11642632] [DOI] [PubMed] [Google Scholar]
References to studies excluded from this review
Li 2016b {published data only}
- Li RC, Li CG, Li YP, Liu YP, Zhao H, Chen XL, et al. Primary and booster vaccination with an inactivated poliovirus vaccine (IPV) is immunogenic and well‐tolerated in infants and toddlers in China. Vaccine 2016;34(12):1436‐43. [DOI: 10.1016/j.vaccine.2016.02.010; PUBMED: 26873055] [DOI] [PubMed] [Google Scholar]
McCollough 1969 {published data only}
- McCollough RH, Glezen WP, Lamb GA, Chin TD. Booster effect of oral poliovaccine. Trials in persons previously immunized with inactivated vaccine. American Journal of Diseases of Children 1969;117(2):161‐8. [PUBMED: 4303394] [PubMed] [Google Scholar]
Moǐseieva 2002 {published data only}
- Moǐseieva HV, Zadorozhna VI. Effectiveness of different strategies of vaccine prophylaxis for poliomyelitis in the Ukraine. Likars'ka sprava 2002;‐(2):85‐8. [PUBMED: 12073271] [PubMed] [Google Scholar]
Swartz 1998 {published data only}
- Swartz TA, Handsher R, Manor Y, Stoeckel P, Barkay A, Mendelson E, et al. Immune response to an intercalated enhanced inactivated polio vaccine/oral polio vaccine programme in Israel: impact on the control of poliomyelitis. Vaccine 1998;16(20):2090‐5. [DOI: 10.1016/S0264-410X(98)00071-1; PUBMED: 9796069] [DOI] [PubMed] [Google Scholar]
Wattigney 2001 {published data only}
- Wattigney WA, Mootrey GT, Braun MM, Chen RT. Surveillance for poliovirus vaccine adverse events, 1991 to 1998: impact of a sequential vaccination schedule of inactivated poliovirus vaccine followed by oral poliovirus vaccine. Pediatrics 2001;107(5):E83. [DOI: 10.1542/peds.107.5.e83; PUBMED: 11331733] [DOI] [PubMed] [Google Scholar]
Ye 2018 {published data only}
- Ye H, Huang T, Ying ZF, Li GL, Che YC, Zhao ZM, et al. [Comparing the immunogenicity and safety of sequential inoculation of sIPV followed by bOPV (I + III) in different dosage forms] [株脊髓灰质炎灭活疫苗与不同剂型Ⅰ+Ⅲ型脊髓灰质炎减毒活疫苗序贯接种的免疫原性和安全性比较]. Zhonghua Yu Fang Yi Xue Za Zhi [Chinese Journal of Preventive Medicine] 2018;52(1):43‐9. [DOI: 10.3760/cma.j.issn.0253-9624.2018.01.009; PUBMED: 29334707] [DOI] [PubMed] [Google Scholar]
References to ongoing studies
NCT02412514 {published data only}
- NCT02412514. Intestinal and humoral immunity of sequential polio vaccination schedules. [Assessing the intestinal and humoral immunity of sequential schedules of inactivated poliovirus vaccine and bivalent oral poliovirus vaccine for routine childhood immunization in Bangladesh]. clinicaltrials.gov/ct2/show/NCT02412514 (first received 1 April 2015).
NCT03430349 {published data only}
- NCT03430349. Phase 1 novel live attenuated serotype 2 oral polio vaccine study in IPV primed adults (nOPV2M4a) [A phase 1 study to evaluate the safety and immunogenicity of 2 novel live attenuated serotype 2 oral poliovirus vaccines, in healthy adults previously primed with inactivated polio vaccine (IPV)]. clinicaltrials.gov/ct2/show/NCT03430349 (first received 21 January 2018).
NCT03614702 {published data only}
- NCT03614702. Clinic trial to evaluate the safety and immunogenicity by different sequential schedules of bOPV and IPV [Safety and immunogenicity evaluation of different sequential immunization schedules of type 1 + 2 bivalent oral poliovirus vaccine (bOPV) co‐administered with inactived poliovirus vaccine (IPV) in infants aged 2 months: a randomized, double blind, single center, parallel trial]. clinicaltrials.gov/show/nct03614702 (first received 3 March 2017).
Additional references
Aaby 2007
- Aaby P, Garly ML, Nielsen J, Ravn H, Martins C, Balé C, et al. Increased female‐male mortality ratio associated with inactivated polio and diphtheria‐tetanus‐pertussis vaccines: observations from vaccination trials in Guinea‐Bissau. Pediatric Infectious Disease Journal 2007;26(3):247‐52. [PUBMED: 17484223] [DOI] [PubMed] [Google Scholar]
Anand 2017
- Anand A, Molodecky NA, Pallansch MA, Sutter RW. Immunogenicity to poliovirus type 2 following two doses of fractional intradermal inactivated poliovirus vaccine: a novel dose sparing immunization schedule. Vaccine 2017;35(22):2993‐8. [DOI: 10.1016/j.vaccine.2017.03.008; PUBMED: 28434691] [DOI] [PMC free article] [PubMed] [Google Scholar]
Asturias 2016
- Asturias EJ, Bandyopadhyay AS, Self S, Rivera L, Saez‐Llorens X, Lopez E, et al. Humoral and intestinal immunity induced by new schedules of bivalent oral poliovirus vaccine and one or two doses of inactivated poliovirus vaccine in Latin American infants: an open‐label randomised controlled trial. Lancet 2016;388(10040):158‐69. [DOI: 10.1016/S0140-6736(16)00703-0; PUBMED: 27212429] [DOI] [PubMed] [Google Scholar]
Bandyopadhyay 2018
- Bandyopadhyay AS, Modlin JF, Wenger J, Gast C. Immunogenicity of new primary immunization schedules with inactivated poliovirus vaccine and bivalent oral polio vaccine for the polio endgame: a review. Clinical Infectious Diseases 2018;67(Suppl 1):S35‐41. [DOI: 10.1093/cid/ciy633; PMC6206125 ; PUBMED: 30376081] [DOI] [PMC free article] [PubMed] [Google Scholar]
Biffi 2003
- Biffi MR. Economic evaluation of the sequential schedule of polio vaccination in Italy [Valutazione economica dell’attivazione in Italia della schedula sequenziale per la vaccinazione antipoliomielitica]. PharmacoEconomics ‐ Italian Research Articles 2003;5(Suppl 1):47‐53. [DOI: 10.1007/BF03320615] [DOI] [Google Scholar]
Brickley 2018
- Brickley EB, Wieland‐Alter W, Connor RI, Ackerman ME, Boesch AW, Arita M, et al. Intestinal immunity to poliovirus following sequential trivalent inactivated polio vaccine/bivalent oral polio vaccine and trivalent inactivated polio vaccine‐only immunization schedules: analysis of an open‐label, randomized, controlled trial in Chilean infants. Clinical Infectious Diseases 2018;67(Suppl 1):S42‐50. [DOI: 10.1093/cid/ciy603; NCT01841671; PMC6206105 ; PUBMED: 30376086] [DOI] [PMC free article] [PubMed] [Google Scholar]
Campbell 2000
- Campbell M, Grimshaw J, Steen N. Sample size calculations for cluster randomised trials. Changing Professional Practice in Europe Group (EU BIOMED II Concerted Action). Journal of Health Services Research and Policy 2000;5(1):12‐6. [DOI: 10.1177/135581960000500105; PUBMED: 10787581] [DOI] [PubMed] [Google Scholar]
CDC 1997
- Centers for Disease Control and Prevention. Poliomyelitis prevention in the United States: introduction of a sequential vaccination schedule of inactivated poliovirus vaccine followed by oral poliovirus vaccine. Recommendations of the Advisory Committee on Immunization Practices (ACIP). Morbidity and Mortality Weekly Report. Recommendations and Reports 1997;46(RR‐3):1‐25. [www.cdc.gov/mmwr/PDF/rr/rr4603.pdf; PUBMED: 9026708] [PubMed] [Google Scholar]
CDC 2015
- Centers for Disease Control and Prevention. Updates on CDC’s polio eradication efforts. www.cdc.gov/polio/updates/2015/2015‐1211.htm (accessed 9 December 2015).
Ciapponi 2011
- Ciapponi A, Glujovsky D, Bardach A, García Martí S, Comande D. EROS: a new software for early stage of systematic reviews. HTA for Health Systems Sustainability 8th Annual Meeting; 2011 Jun 27‐9; Rio de Janeiro (BR). 2011:66. [242; www.gapcongressos.com.br/eventos/z0082/documentos/Program_HTAi.pdf]
Ciapponi 2017
- Ciapponi A, Gibbons L. Global incidence of circulating vaccine‐derived poliovirus (cVDPV) during the period 2000‐2016: meta‐analysis [Incidencia mundial de poliovirus circulantes de origen vacunal (cVDPV) durante el período 2000‐2016: meta‐análisis]. XVI Reunión Anual de la Red Cochrane Iberoamericana: Síntesis y transferencia del conocimiento; 2017 Jun 5‐7; Medellín (CO). 2017.
Cockburn 1988
- Cockburn WC. The work of the WHO Consultative Group on poliomyelitis vaccines. Bulletin of the World Health Organization 1988;66(2):143‐54. [PMC2491049; PUBMED: 2840219] [PMC free article] [PubMed] [Google Scholar]
Cuba 2007
- Cuba IPV Study Collaborative Group. Randomized, placebo‐controlled trial of inactivated poliovirus vaccine in Cuba. New England Journal of Medicine 2007;356(15):1536‐44. [DOI: 10.1056/NEJMoa054960; NCT00260312; PUBMED: 17429085] [DOI] [PubMed] [Google Scholar]
Cáceres 2001
- Cáceres VM, Sutter RW. Sabin monovalent oral polio vaccines: review of past experiences and their potential use after polio eradication. Clinical Infectious Diseases 2001;33(4):531‐41. [DOI: 10.1086/321905; PUBMED: 11462191] [DOI] [PubMed] [Google Scholar]
de Oliveira 2000
- Oliveira LH, Struchiner CJ. Vaccine‐associated paralytic poliomyelitis: a retrospective cohort study of acute flaccid paralyses in Brazil. International Journal of Epidemiology 2000;29(4):757‐63. [DOI: 10.1093/ije/29.4.757; PUBMED: 10922356] [DOI] [PubMed] [Google Scholar]
DerSimonian 1986
Desai 2014
- Desai S, Diener T, Tan BK, Lowry NJ, Talukdar C, Chrusch W, et al. An unusual case of vaccine‐associated paralytic poliomyelitis. Canadian Journal of Infectious Diseases & Medical Microbiology 2014;25(4):227‐8. [DOI: 10.1155/2014/378320; PMC4173946; PUBMED: 25285130] [DOI] [PMC free article] [PubMed] [Google Scholar]
Deshpande 2003
- Deshpande JM, Nadkarni SS, Siddiqui ZA. Detection of MEF‐1 laboratory reference strain of poliovirus type 2 in children with poliomyelitis in India in 2002 & 2003. Indian Journal of Medical Research 2003;118:217‐23. [PUBMED: 14870793] [PubMed] [Google Scholar]
Domingues 2014
- Domingues CM, Fátima Pereira S, Cunha Marreiros AC, Menezes N, Flannery B. Introduction of sequential inactivated polio vaccine‐oral polio vaccine schedule for routine infant immunization in Brazil's National Immunization Program. Journal of Infectious Diseases 2014;210 Suppl 1:S143‐51. [DOI: 10.1093/infdis/jit588; PMC4758462; PUBMED: 25316829] [DOI] [PMC free article] [PubMed] [Google Scholar]
Dong 2016
- Dong SZ, Zhu WB. The role of Sabin inactivated poliovirus vaccine in the final phase of global polio eradication [萨宾灭活脊灰病毒疫苗在全球消灭脊髓灰质炎最后阶段的作用]. Zhonghua Yu Fang Yi Xue Za Zhi [Chinese Journal of Preventive Medicine] 2016;50(12):1032‐5. [DOI: 10.3760/cma.j.issn.0253-9624.2016.12.003; PUBMED: 28057104] [DOI] [PubMed] [Google Scholar]
Duintjer Tebbens 2006
- Duintjer Tebbens RJ, Sangrujee N, Thompson KM. The costs of future polio risk management policies. Risk Analysis 2006;26(6):1507‐31. [DOI: 10.1111/j.1539-6924.2006.00842.x; PUBMED: 17184394] [DOI] [PubMed] [Google Scholar]
Duintjer Tebbens 2015
- Duintjer Tebbens RJ, Pallansch MA, Cochi SL, Wassilak SGF, Thompson KM. An economic analysis of poliovirus risk management policy options for 2013‐2052. BMC Infectious Diseases 2015;15:389. [DOI: 10.1186/s12879-015-1112-8; PMC4582932; PUBMED: 26404632] [DOI] [PMC free article] [PubMed] [Google Scholar]
EPOC 2011a
- Effective Practice, Organisation of Care. Cochrane Effective Practice and Organisation of Care Review Group: data collection checklist. bit.ly/1zXPg9y (accessed 12 May 2014).
EPOC 2011b
- Effective Practice, Organisation of Care. Draft EPOC methods paper: including Interrupted Time Series (ITS) designs in a EPOC review. bit.ly/1Eq6Zbf (accessed 16 March 2012).
EPOC 2011c
- Effective Practice, Organisation of Care. Presentation of data from EPOC studies (RCTs and CBAs). bit.ly/1y5dRDI (accessed 16 March 2012).
EPOC 2017
- Cochrane Effective Practice, Organisation of Care (EPOC). Suggested risk of bias criteria for EPOC reviews. epoc.cochrane.org/sites/epoc.cochrane.org/files/public/uploads/Resources‐for‐authors2017/suggested_risk_of_bias_criteria_for_epoc_reviews.pdf (accessed 7 April 2017).
Faden 1993
- Faden H. Poliovirus vaccination: a trilogy. Journal of Infectious Diseases 1993;168(1):25‐8. [DOI: 10.1093/infdis/168.1.25; PUBMED: 8515124] [DOI] [PubMed] [Google Scholar]
Fish 2016
- Fish EN, Flanagan KL, Furman D, Klein SL, Kollmann TR, Jeppesen DL, et al. Changing oral vaccine to inactivated polio vaccine might increase mortality. Lancet 2016;387(10023):1054‐5. [DOI: 10.1016/S0140-6736(16)00661-9; PMC4922306; PUBMED: 27025182] [DOI] [PMC free article] [PubMed] [Google Scholar]
Garon 2016
- Garon J, Seib K, Orenstein WA, Ramirez Gonzalez A, Chang Blanc D, Zaffran M, et al. Polio endgame: the global switch from tOPV to bOPV. Expert Review of Vaccines 2016;15(6):693‐708. [DOI: 10.1586/14760584.2016.1140041; PUBMED: 26751187] [DOI] [PubMed] [Google Scholar]
Gentile 2016
- Gentile Á, Abate H. A new challenge for the world: the eradication of polio. Archivos Argentinos De Pediatria 2016;114(6):557‐62. [DOI: 10.5546/aap.2016.eng.557; PUBMED: 27869415] [DOI] [PubMed] [Google Scholar]
Glujovsky 2010
- Glujovsky D, Bardach A, García Martí S, Comande D, Ciapponi A. New software for early stage of systematic reviews. XVIII Cochrane Colloquium. The Joint Colloquium of the Cochrane and Campbell Collaborations; 2010 Oct 18‐22. 2010.
Glujovsky 2011
- Glujovsky D, Bardach A, García Martí S, Comande D, Ciapponi A. EROS: a new software for early stage of systematic reviews. Value in Health 2011;14(7):A564. [DOI: 10.1016/j.jval.2011.08.1689; PRM2] [DOI] [Google Scholar]
Gold 1994
- Gold R. Enhanced inactivated poliovirus vaccine. Canadian Journal of Infectious Diseases 1994;5(4):152. [DOI: 10.1155/1994/153783] [DOI] [PMC free article] [PubMed] [Google Scholar]
GPEI 2018
- Global Polio Eradication Initiative. Global wild poliovirus 2012 ‐ 2018. polioeradication.org/wp‐content/uploads/2018/01/global‐wild‐poliovirus‐2012‐2018‐20180110.pdf (accessed 10 June 2018).
GRADEpro GDT [Computer program]
- McMaster University (developed by Evidence Prime). GRADEpro GDT. Hamilton (ON): McMaster University (developed by Evidence Prime), accessed 05 September 2015.
Grassly 2006
- Grassly NC, Fraser C, Wenger J, Deshpande JM, Sutter RW, Heymann DL, et al. New strategies for the elimination of polio from India. Science 2006;314(5802):1150‐3. [DOI: 10.1126/science.1130388; PUBMED: 17110580] [DOI] [PubMed] [Google Scholar]
Grassly 2009
- Grassly NC, Jafari H, Bahl S, Durrani S, Wenger J, Sutter RW, et al. Mucosal immunity after vaccination with monovalent and trivalent oral poliovirus vaccine in India. Journal of Infectious Diseases 2009;200(5):794‐801. [DOI: 10.1086/605330; PUBMED: 19624278] [DOI] [PubMed] [Google Scholar]
Grassly 2014
- Grassly NC. Immunogenicity and effectiveness of routine immunization with 1 or 2 doses of inactivated poliovirus vaccine: systematic review and meta‐analysis. Journal of Infectious Diseases 2014;210 Suppl 1:S439‐46. [DOI: 10.1093/infdis/jit601; PMC4197908; PUBMED: 24634499] [DOI] [PMC free article] [PubMed] [Google Scholar]
Guyatt 2011
Hawken 2012
- Hawken J, Troy SB. Adjuvants and inactivated polio vaccine: a systematic review. Vaccine 2012;30(49):6971‐9. [DOI: 10.1016/j.vaccine.2012.09.059; PMC3529007 ; PUBMED: 23041122] [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2003
- Higgins JP, Thompson SG, Deeks JJ, Altman DG. Measuring inconsistency in meta‐analyses. BMJ 2003; Vol. 327, issue 7414:557‐60. [DOI: 10.1136/bmj.327.7414.557; PMC192859; PUBMED: 12958120] [DOI] [PMC free article] [PubMed]
Higgins 2011a
- Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (Updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011b
- Higgins JP, Altman DG, Sterne JAC, on behalf of the Cochrane Statistical Methods Group and the Cochrane Bias Methods Group. Chapter 8: Assessing risk of bias in included studies. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Higgins 2011c
- Higgins JP, Deeks JJ, Altman DG, on behalf of the Cochrane Statistical Methods Group. Chapter 16: Special topics in statistics. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Hird 2012
- Hird TR, Grassly NC. Systematic review of mucosal immunity induced by oral and inactivated poliovirus vaccines against virus shedding following oral poliovirus challenge. PLOS Pathogens 2012;8(4):e1002599. [DOI: 10.1371/journal.ppat.1002599; PMC3330118; PUBMED: 22532797] [DOI] [PMC free article] [PubMed] [Google Scholar]
Hultcrantz 2017
- Hultcrantz M, Rind D, Akl EA, Treweek S, Mustafa RA, Iorio A, et al. The GRADE Working Group clarifies the construct of certainty of evidence. Journal of Clinical Epidemiology 2017;87:4‐13. [DOI: 10.1016/j.jclinepi.2017.05.006; PMC6542664; PUBMED: 28529184] [DOI] [PMC free article] [PubMed] [Google Scholar]
Jorba 2016
- Jorba J, Diop OM, Iber J, Sutter RW, Wassilak SG, Burns CC. Update on vaccine‐derived polioviruses ‐ worldwide, January 2015 ‐ May 2016. Morbidity and Mortality Weekly Report 2016;65(30):763‐9. [DOI: 10.15585/mmwr.mm6530a3; PUBMED: 27491079] [DOI] [PubMed] [Google Scholar]
Kew 2004
- Kew OM, Wright PF, Agol VI, Delpeyroux F, Shimizu H, Nathanson N, et al. Circulating vaccine‐derived polioviruses: current state of knowledge. Bulletin of the World Health Organization 2004;82(1):16‐23. [PMC2585883; PUBMED: 15106296] [PMC free article] [PubMed] [Google Scholar]
Kohler 2002
- Kohler KA, Banerjee K, Gary Hlady W, Andrus JK, Sutter RW. Vaccine‐associated paralytic poliomyelitis in India during 1999: decreased risk despite massive use of oral polio vaccine. Bulletin of the World Health Organization 2002;80(3):210‐6. [PMC2567745; PUBMED: 11984607] [PMC free article] [PubMed] [Google Scholar]
Krugman 1977
- Krugman RD, Hardy GE Jr, Sellers C, Parkman PD, Witte JJ, Meyer BC, et al. Antibody persistence after primary immunization with trivalent oral poliovirus vaccine. Pediatrics 1977;60(1):80‐2. [PUBMED: 195265] [PubMed] [Google Scholar]
Laassri 2005
- Laassri M, Lottenbach K, Belshe R, Wolff M, Rennels M, Plotkin S, et al. Effect of different vaccination schedules on excretion of oral poliovirus vaccine strains. Journal of Infectious Diseases 2005;192(12):2092‐8. [DOI: 10.1086/498172; PUBMED: 16288372] [DOI] [PubMed] [Google Scholar]
Landaverde 2014
- Landaverde JM, Trumbo SP, Danovaro‐Holliday MC, Cochi SE, Gandhi R, Ruiz‐Matus C. Vaccine‐associated paralytic poliomyelitis in the postelimination era in Latin America and the Caribbean, 1992‐2011. Journal of Infectious Diseases 2014;209(9):1393‐402. [DOI: 10.1093/infdis/jit602; PUBMED: 24520126] [DOI] [PubMed] [Google Scholar]
Lewis 2017
- Lewis I, Ottosen A, Rubin J, Blanc DC, Zipursky S, Wootton E. A supply and demand management perspective on the accelerated global introductions of inactivated poliovirus vaccine in a constrained supply market. Journal of Infectious Diseases 2017;216(Suppl 1):S33‐9. [DOI: 10.1093/infdis/jiw550; PMC5853471; PUBMED: 28838159] [DOI] [PMC free article] [PubMed] [Google Scholar]
Liberati 2009
- Liberati A, Altman DG, Tetzlaff J, Mulrow C, Gøtzsche PC, Ioannidis JPA, et al. The PRISMA statement for reporting systematic reviews and meta‐analyses of studies that evaluate health care interventions: explanation and elaboration. PLOS Medicine 2009; Vol. 6, issue 7:e1000100. [DOI: 10.1371/journal.pmed.1000100] [DOI] [PMC free article] [PubMed]
Liu 2017
- Liu Y, Wang J, Liu S, Du J, Wang L, Gu W, et al. Introduction of inactivated poliovirus vaccine leading into the polio eradication endgame strategic plan; Hangzhou, China, 2010‐2014. Vaccine 2017;35(9):1281‐6. [DOI: 10.1016/j.vaccine.2017.01.034; PUBMED: 28161421] [DOI] [PubMed] [Google Scholar]
Lopalco 2017
- Lopalco PL. Wild and vaccine‐derived poliovirus circulation, and implications for polio eradication. Epidemiology and Infection 2017;145(3):413‐9. [DOI: 10.1017/S0950268816002569; PUBMED: 27866483] [DOI] [PMC free article] [PubMed] [Google Scholar]
Lund 2015
- Lund N, Andersen A, Hansen AS, Jepsen FS, Barbosa A, Biering‐Sørensen S, et al. The effect of oral polio vaccine at birth on infant mortality: a randomized trial. Clinical Infectious Diseases 2015;61(10):1504‐11. [DOI: 10.1093/cid/civ617; NCT00710983; PMC4614411; PUBMED: 26219694] [DOI] [PMC free article] [PubMed] [Google Scholar]
Mascareñas 2005
- Mascareñas A, Salinas J, Tasset‐Tisseau A, Mascareñas C, Khan MM. Polio immunization policy in Mexico: economic assessment of current practice and future alternatives. Public Health 2005;119(6):542‐9. [DOI: 10.1016/j.puhe.2004.08.020; PUBMED: 15826896] [DOI] [PubMed] [Google Scholar]
McBean 1988
- McBean AM, Thoms ML, Albrecht P, Cuthie JC, Bernier R. Serologic response to oral polio vaccine and enhanced‐potency inactivated polio vaccines. American Journal of Epidemiology 1988;128(3):615‐28. [DOI: 10.1093/oxfordjournals.aje.a115009; PUBMED: 2843039] [DOI] [PubMed] [Google Scholar]
McCain 1979
- McCain LJ, McCleary R. The statistical analysis of the simple interrupted time‐series quasi‐experiment. In: Cook TD, Campbell DT editor(s). Quasi‐Experimentation: Design & Analysis Issues for Field Settings. Boston (MA): Houghton Mifflin, 1979:233‐93. [Google Scholar]
Miller 1996
- Miller MA, Sutter RW, Strebel PM, Hadler SC. Cost‐effectiveness of incorporating inactivated poliovirus vaccine into the routine childhood immunization schedule. JAMA 1996;276(12):967‐71. [PUBMED: 8805731] [PubMed] [Google Scholar]
Minor 2009
- Minor P. Vaccine‐derived poliovirus (VDPV): impact on poliomyelitis eradication. Vaccine 2009;27(20):2649‐52. [DOI: 10.1016/j.vaccine.2009.02.071; PUBMED: 19428874] [DOI] [PubMed] [Google Scholar]
Modlin 2019
Moher 2009
- Moher D, Liberati A, Tetzlaff J, Altman DG, PRISMA Group. Preferred reporting items for systematic reviews and meta‐analyses: the PRISMA statement. PLOS Medicine 2009;6(7):e1000097. [DOI: 10.1371/journal.pmed.1000097; PMC2707599 ; PUBMED: 19621072] [DOI] [PMC free article] [PubMed] [Google Scholar]
PAHO 2017
- Pan American Health Organization. Switch from tOPV to bOPV. www.paho.org/hq/index.php?option=com_content&view=article&id=11015:topv‐bopv‐supporting‐technical‐documents&Itemid=1707&lang=en (accessed 6 May 2017).
Patriarca 1991
- Patriarca PA, Wright PF, John TJ. Factors affecting the immunogenicity of oral poliovirus vaccine in developing countries: review. Reviews of Infectious Diseases 1991;13(5):926‐39. [PUBMED: 1660184] [DOI] [PubMed] [Google Scholar]
Peng 2018
- Peng X, Hu X, Salazar MA. On reducing the risk of vaccine‐associated paralytic poliomyelitis in the global transition from oral to inactivated poliovirus vaccine. Lancet 2018;392(10147):610‐2. [DOI: 10.1016/S0140-6736(18)30483-5; PUBMED: 29605427] [DOI] [PubMed] [Google Scholar]
Platt 2014
- Platt LR, Estívariz CF, Sutter RW. Vaccine‐associated paralytic poliomyelitis: a review of the epidemiology and estimation of the global burden. Journal of Infectious Diseases 2014;210 Suppl 1:S380‐9. [DOI: 10.1093/infdis/jiu184; PUBMED: 25316859] [DOI] [PMC free article] [PubMed] [Google Scholar]
Resik 2013
- Resik S, Tejeda A, Sutter RW, Diaz M, Sarmiento L, Alemañi N, et al. Priming after a fractional dose of inactivated poliovirus vaccine. New England Journal of Medicine 2013;368(5):416‐24. [DOI: 10.1056/NEJMoa1202541] [DOI] [PubMed] [Google Scholar]
Review Manager 2014 [Computer program]
- Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager 5 (RevMan 5). Version 5.3. Copenhagen: Nordic Cochrane Centre, The Cochrane Collaboration, 2014.
Sartori 2015
- Sartori AM, Vicentine MP, Gryninger LC, Coelho de Soárez P, Novaes HM. Polio inactivated vaccine costs into routine childhood immunization in Brazil. Revista de Saúde Pública 2015;49(8):1‐10. [DOI: 10.1590/S0034-8910.2015049005492] [DOI] [PMC free article] [PubMed] [Google Scholar]
Schünemann 2011b
- Schünemann HJ, Oxman AD, Vist GE, Higgins JPT, Deeks JJ, Glasziou P, et al. on behalf of the Cochrane Applicability and Recommendations Methods Group. Chapter 12: Interpreting results and drawing conclusion. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Schünemann 2013
- Schünemann H, Brożek J, Guyatt G, Oxman A, editor(s). Handbook for grading the quality of evidence and the strength of recommendations using the GRADE approach (updated October 2013). GRADE Working Group, 2013. Available from gdt.guidelinedevelopment.org/app/handbook/handbook.html.
Sterne 2011
- Sterne JA, Egger M, Moher D, on behalf of the Cochrane Bias Methods Group. Chapter 10: Addressing reporting biases. In: Higgins JP, Green S, editor(s). Cochrane Handbook for Systematic Reviews of Interventions Version 5.1.0 (updated March 2011). The Cochrane Collaboration, 2011. Available from handbook.cochrane.org.
Strebel 1995
- Strebel PM, Ion‐Nedelcu N, Baughman AL, Sutter RW, Cochi SL. Intramuscular injections within 30 days of immunization with oral poliovirus vaccine‐‐a risk factor for vaccine‐associated paralytic poliomyelitis. New England Journal of Medicine 1995;332(8):500‐6. [DOI: 10.1056/NEJM199502233320804; PUBMED: 7830731] [DOI] [PubMed] [Google Scholar]
Sutter 2000
- Sutter RW, Prevots DR, Cochi SL. Poliovirus vaccines. Progress toward global poliomyelitis eradication and changing routine immunization recommendations in the United States. Pediatric Clinics of North America 2000;47(2):287‐308. [PUBMED: 10761505] [DOI] [PubMed] [Google Scholar]
Sutter 2010
- Sutter RW, John TJ, Jain H, Agarkhedkar S, Ramanan PV, Verma H, et al. Immunogenicity of bivalent types 1 and 3 oral poliovirus vaccine: a randomised, double‐blind, controlled trial. Lancet 2010;376(9753):1682‐8. [DOI: 10.1016/S0140-6736(10)61230-5; PUBMED: 20980048] [DOI] [PubMed] [Google Scholar]
Tang 2018
- Tang G, Yin W, Cao Y, Tan L, Wu S, Cao Y, et al. Immunogenicity of sequential inactivated and oral poliovirus vaccines (OPV) versus inactivated poliovirus vaccine (IPV) alone in healthy infants: a systematic review and meta‐analysis. Human Vaccines & Immunotherapeutics 2018;14(11):2636‐43. [DOI: 10.1080/21645515.2018.1489188; PMC6314415 ; PUBMED: PMC6314415 ] [DOI] [PMC free article] [PubMed] [Google Scholar]
Thompson 2015
- Thompson KM, Kalkowska DA, Duintjer Tebbens RJ. Managing population immunity to reduce or eliminate the risks of circulation following the importation of polioviruses. Vaccine 2015;33(13):1568‐77. [DOI: 10.1016/j.vaccine.2015.02.013; PUBMED: 25701673] [DOI] [PMC free article] [PubMed] [Google Scholar]
Ukoumunne 1999
- Ukoumunne OC, Gulliford MC, Chinn S, Sterne JA, Burney PG. Methods for evaluating area‐wide and organisation‐based interventions in health and health care: a systematic review. Health Technology Assessment 1999; Vol. 3, issue 5:iii‐92. [PUBMED: 10982317] [PubMed]
WHO 2012
- WHO. Meeting of the Strategic Advisory Group of Experts on Immunization, November 2012 ‐ conclusions and recommendations. Relevé épidémiologique hebdomadaire 2013;88(1):1‐16. [www.who.int/wer/2013/wer8801.pdf; PUBMED: 23311010] [PubMed] [Google Scholar]
WHO 2015a
- World Health Organization. Global Polio Eradication Initiative. Polio eradication & endgame strategic plan 2013–2018. polioeradication.org/wp‐content/uploads/2016/07/PEESP_EN_A4.pdf (accessed 17 January 2016).
WHO 2015b
- World Health Organization. Poliomyelitis. Key facts. www.who.int/en/news‐room/fact‐sheets/detail/poliomyelitis (accessed 17 January 2016).
WHO 2015c
- World Health Organization. Circulating vaccine‐derived poliovirus cases, 2000‐2015. https://drive.google.com/file/d/1sJqwL3jaZZcBjMWU3TXcMBBB7MKIKQ7H/view?usp=sharing (accessed 17 January 2016).
WHO 2015d
- World Health Organization. Global wild poliovirus 2013‐2018. polioeradication.org/wp‐content/uploads/2018/03/global‐wild‐poliovirus‐2013‐2018‐20180320.pdf (accessed 6 March 2018).
WHO 2016
- World Health Organization. Polio vaccines: WHO position paper ‐ March, 2016. Relevé épidémiologique hebdomadaire 2016;91(12):145‐68. [www.who.int/wer/2016/wer9112.pdf?ua=1] [PubMed] [Google Scholar]
WHO 2017
- World Health Organization. Polio case count. extranet.who.int/polis/public/CaseCount.aspx (accessed 6 May 2017).
WHO 2018
- World Health Organization. Global Polio Eradication Initiative. Global circulating vaccine‐derived poliovirus cases, 2000–17. polioeradication.org/polio‐today/polio‐now/this‐week/circulating‐vaccine‐derived‐poliovirus/ (accessed 6 June 2018).
WHO 2019
- World Health Organization. WHO vaccine‐preventable diseases: monitoring system. 2019 global summary. apps.who.int/immunization_monitoring/globalsummary/schedules (accessed 6 June 2018).
Wu 2018
- Wu W, Wang H, Li K, Pekka Nuorti J, Liu D, Xu D, et al. Recipient vaccine‐associated paralytic poliomyelitis in China, 2010‐2015. Vaccine 2018;36(9):1209‐13. [DOI: 10.1016/j.vaccine.2018.01.019; PUBMED: 29395524] [DOI] [PubMed] [Google Scholar]
Zhang 2016
- Zhang Q, Leppold C, Shao Y, Mura Y, Tanimoto T. New poliovirus vaccine schedules. Lancet 2016;388(10059):2477‐8. [DOI: 10.1016/S0140-6736(16)32177-8] [DOI] [PubMed] [Google Scholar]
Zhao 2017
- Zhao D, Ma R, Zhou T, Yang F, Wu J, Sun H, et al. Introduction of inactivated poliovirus vaccine and impact on vaccine‐associated paralytic poliomyelitis ‐ Beijing, China, 2014‐2016. Morbidity and Mortality Weekly Report 2017;66(49):1357‐61. [DOI: 10.15585/mmwr.mm6649a4; PMC5730215; PUBMED: 29240729] [DOI] [PMC free article] [PubMed] [Google Scholar]
References to other published versions of this review
Ciapponi 2014
- Ciapponi A, Bardach A, Rey Ares L, Glujovsky D, Cafferata ML, Romano M, et al. Sequential inactivated (IPV) and live oral (OPV) poliovirus vaccines for preventing poliomyelitis. Cochrane Database of Systematic Reviews 2014, Issue 8. [DOI: 10.1002/14651858.CD011260] [DOI] [PMC free article] [PubMed] [Google Scholar]