

Abstract
Immunotherapy for melanoma has undergone significant change since the first attempts to treat patients with high dose IL-2. Herein, strategies to boost patient antitumor immunity through vaccination, treatment with agents that augment host immunity, and adoptive cell transfer will be discussed. The first two strategies have yielded only limited clinical success, but adoptive cell transfer therapy, particularly following a lymphodepleting, preconditioning regimen has resulted in objective response rates approaching 50%. For a number of reasons, lymphodepletion appears to be critical for maintenance of circulating antitumor T cells following adoptive cell transfer. Balancing antitumor efficacy, autoimmunity, and reconstitution of a functioning immune system remain challenging and potentially life-threatening issues.
Introduction
Melanoma is among the most immunogenic of all solid cancers, as supported by the phenomenon of spontaneous regression of primary tumors, which is seen in 3–15% of melanomas with unknown primaries (Morton et al., 1991). Moreover, the presence of tumor antigen-specific antibodies and tumor-specific cytotoxic T cells in the peripheral blood of melanoma patients has been well established (Lee et al., 1999). The ability of T lymphocytes, especially CD8 T cells, to prevent tumor formation has been shown in mice and humans (Shankaran et al., 2001; Chiao and Krown, 2003), and recent evidence indicates that the presence of infiltrating CD8 T cells within tumors is positively correlated with better prognosis in cutaneous melanoma (Ladanyi et al., 2007) as well as in several other types of cancers (Zhang et al., 2003). Thus, melanoma has been of interest as an intensively studied target for immunotherapy for over two decades.
In general, an effective antitumor CD8 T-cell response must fulfill a number of criteria. First, sufficient numbers of antitumor specific CD8 cells need to be generated in vivo or expanded ex vivo. Second, the CD8 T cells that are generated should be able to traffic and infiltrate into tumors. Finally, the CD8 T cells must be sufficiently activated within tumors such that they kill tumor cells, leading to tumor necrosis and/or tumor regression. The development of murine models of melanoma, particularly those deploying transgenic T cells specific for melanoma antigens, has made it possible to better understand mechanisms underlying effective regression of established melanoma tumors and to make adjustments to improve immunotherapy prior to testing new strategies in the clinic (Finkelstein et al., 2004).
The most successful murine studies, however, do not always translate to safe, effective melanoma therapy for patients. Thus, we have attempted to list some of the most important clinical trials involving treatment of advanced melanoma that have been performed in the past few years (Table 1). The definition of clinical responses used in this review generally refers to “objective responses” as defined by World Health Organization (WHO) or WHO-RECIST criteria (James et al., 1999). The RECIST criteria include unidimensional measures of specific tumors and/or metastases with complete response (CR), partial response (PR), and progressive disease responses defined by a complete disappearance of all known lesions (CR), at least 30% decrease in size (PR), or 20% increase in the size of tumors (progressive disease). Patients who do not meet the criteria for PR or progressive disease are considered to have stable disease. Although RECIST criteria allow better comparison of clinical benefit between various trials, some authors have argued that “stable disease” rather than PR and CR may be a more realistic goal for newer biological agents and perhaps immunotherapy (Michaelis and Ratain, 2006).
Table 1.
Selected immunotherapy trials for advanced melanoma
| Clinical objective response | References | |
|---|---|---|
| CTLA-4 blockade | ||
| MDX-010 monotherapy (single dose, 3 mg kg−1) | OR 0/7 | Hodi et al.(2003) |
| MDX-010+gp100 peptide vaccination (3 mg kg−1 every 3 weeks) | OR 3/14 (CR 2/14; PR 1/14) | Phan et al.(2003)1 |
| MDX-010+gp100 peptide vaccination (3 mg kg−1 every 3 weeks) | OR 4/29 (CR 2/29; PR 2/29) | Attia et al.(2005)1 |
| CP-675,206 monotherapy (0.01–15 mg kg−1, single dose) | OR 4/34 (CR 2/34; PR 2/34) | Ribas et al. (2005) |
| Dendritic cell-based | ||
| Peptide or tumor lysate | OR 8/32 (CR 2/32; PR 6/32) | Nestle et al. (1998) |
| Peptide2 vs peptide+GM-CSF2 | OR 1/13 (CR 0/13; PR 1/13; SD 1/13) OR 2/13 (CR 0/13; PR 2/13; SD 2/13) |
Slingluff et al. (2003)1 |
| Peptide/lysate vs DTIC | OR 3/53 (CR 0/53; PR 2/53; SD 8/53) OR 2/55 (CR 0/55; PR 3/55; SD 10/55) |
Schadendorf et al. (2006)1 |
| Synthetic peptide vaccine | ||
| g209-2M (gp100-associated antigen)+IL-2, i.v. | OR 13/31 (CR 1/31; PR 12/31) | Rosenberg et al. (1998a) |
| ACT | ||
| Lymphodepletion | OR 6/13 (CR 0/13; PR 6/13) | Dudley et al. (2002b) |
| Lymphodepletion | OR 18/35 (CR 3/35; PR 15/35) | Dudley et al. (2005) |
| Whole-cell vaccine | ||
| Canvaxin® (n=150) vs no adjuvant treatment (n=113) (resected AJCC stage IV) | Five-year OS 39% Five-year OS 19% |
Hsueh et al. (2002) |
| Melacine® (n=89) vs no adjuvant treatment (resected intermediate-thickness AJCC stage II) | No significant difference in OS and RFS | Sondak et al. (2002) |
| Melacine+low-dose IFN-α2b vs high-dose IFN-α2b (n=604, resected AJCC stage III) | No significant difference in OS and RFS | Mitchell et al. (2007) |
CR, complete response; CTLA, cytotoxic T lymphocyte antigen; i.v., intravenous; OR, objective response; OS, overall survival; PR, partial response; RFS, recurrence-free survival; SD, stable disease.
Clinical Response Evaluation according to RECIST.
All patients were also administered low-dose IL-2.
Currently, there are three major approaches to boost antitumor CD8 T-cell responses in patients with melanoma as summarized in Figure 1: (1) non-specific stimulation of antitumor immune responses by stimulating endogenous effector cells with cytokines or removing inhibitory signals for T-cell activation, (2) active immunization (that is, vaccines) to enhance endogenous antitumor responses in vivo, and (3) adoptive cell-transfer (ACT) therapy as exemplified by ex vivo selection and expansion of autologous antitumor-specific CD8 T cells that are subsequently transferred back to the patient. This review will discuss each of these therapeutic strategies in terms of the preclinical studies that underlie their mechanisms, their reported efficacy in melanoma clinical trials, as well as their distinct toxicity profiles.
Figure 1. Strategy for immunotherapy of melanoma.
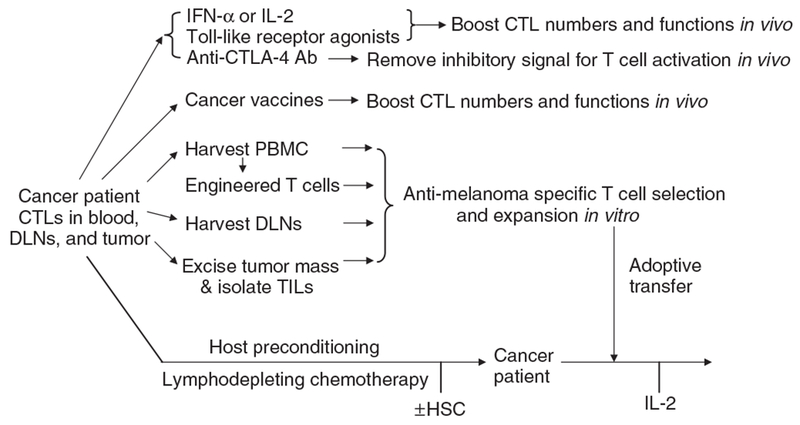
CTL, cytotoxic T lymphocyte; DLN, draining lymph node; PBMC, peripheral blood mononuclear cells; CTLA, cytotoxic T lymphocyte antigen; TIL, tumor-infiltrating lymphocyte; HSC, hematopoietic stem cell.
Nonspecific stimulation of antitumor immune responses
IFN-α and IL-2 therapy.
In the 1980s, IFN-α was reported to inhibit the growth of B16 murine melanoma cells in vitro and in vivo (Bart et al., 1980). Kirkwood et al. (1996) demonstrated for the first time that high-dose IFN-α2b not only prolongs disease-free survival, but also overall survival in high-risk patients with melanomas thicker than 4 mm or with lymph node metastasis.
To date, IFN-α has been one of the most intensely investigated immunotherapeutic agents for melanoma, and it is the only currently approved adjuvant therapy for the treatment of high-risk melanoma patients. The effect of IFN-α2b on disease-free survival was confirmed in a subsequent trial, but the study failed to prove significant effect on overall survival (Kirkwood et al., 2000). In an extensive meta-analysis, investigators concluded that adjuvant treatment with IFN-α reduces the incidence of melanoma recurrence by ~26% and provides benefit in overall survival (15% reduction in risk of death) that is not quite statistically significant (P = 0.06) (Wheatley et al., 2003). The use of polyethylene glycol-conjugated IFN, which has a substantially longer half-life than the unconjugated molecule, resulted in significant clinical responses of ~33% when used in combination with temozolomide in patients with advanced disease (Hwu et al., 2006). Polyethylene glycol-conjugated IFN may also have utility in the adjuvant setting in melanoma patients with a high risk of recurrence (Eggermont et al. j Clin Oncol, 2007 ASCO Annual Meeting Proceedings Part I. vol. 25, no. 18S (June 20 Supplement); 2007: 8504). Considering its efficacy and ease of administration, polyethylene glycol-conjugated IFN is worth further investigation to define its place in the treatment of patients with advanced stage melanoma.
IL-2 is known for its potent ability to activate CD8 T cells and natural killer cells, resulting in development of the so-called lymphokine-activated killer cells. Because systemic administration of high dose of IL-2 resulted in regression of established pulmonary metastases in B16 melanoma-bearing mice (Rosenberg et al., 1985b), Rosenberg et al. performed the first clinical trial using autologous lymphokine-activated killer cells and systemic IL-2 in patients with metastatic melanoma. Although initial clinical results were promising (Rosenberg et al., 1985a), subsequent trials demonstrated that high-dose IL-2 provided consistent, but low, overall response rate of ~ 13–17% (7–9% PR and 6–8% CR) (Rosenberg et al., 1994a; Atkins et al., 1999). Currently, IL-2 is the only FDA-approved immunotherapeutic agent for treatment of patients with metastatic melanoma. Notably, within the 15 years of follow-up in these studies, >80% of the complete responders to high-dose IL-2 treatment have not had recurrences and may be considered cured (personal communication, SA Rosenberg, NCI).
The modest benefits of IL-2-based treatment regimens, however, must be weighed against the serious adverse side effects and the high cost of therapy. Moreover, recent studies have shown that IL-2 expands CD4+ CD25+ Foxp3+ -regulatory T cells (Tregs) in vivo, thereby possibly inducing immune tolerance (Malek and Bayer, 2004). Furthermore, administration of high-dose IL-2 resulted in a nearly fourfold increase in the frequency of CD4+ Foxp3+ Tregs in the peripheral blood when compared with pretreatment levels (Ahmadzadeh and Rosenberg, 2006), suggesting that Tregs may play important downstream roles in determining responses to IL-2.
Several studies have been conducted to study combinations of IL-2 and chemotherapy in advanced melanoma in an effort to improve response rates and survival. Unfortunately, recent phase-III trials of chemotherapy with high-dose IL-2 administration resulted in increased serious toxicity to patients and failed to confirm significantly better durable response rates (or overall survival) compared with chemotherapy alone (Rosenberg et al., 1999; Ridolfi et al., 2002; Keilholz et al., 2005).
Anti-CTLA-4 therapy.
Optimal T-cell activation requires signaling through both the T-cell receptor (TCR) and the costimulatory receptor CD28, which is constitutively expressed on T cells. Cytotoxic T lymphocyte-associated antigen-4 (CTLA-4), a member of CD28 immunoglobulin family, is an inhibitory receptor expressed mainly by activated T cells and Tregs. CTLA-4 binds CD80 or CD86 with higher affinity and avidity than does CD28, thereby inhibiting CD28-dependent T-cell activation and decreasing IL-2 production (Teft et al., 2006). The critical negative regulatory function of CTLA-4 was established upon characterization of CTLA-4-deficient mice, which exhibited profound lymphopro-liferation and autoimmunity.
mAbs targeting CTLA-4 have been shown to increase host antitumor immunity in a number of tumor models (Leach et al., 1996). However, anti-CTLA4 mAb alone failed to produce similar antitumor efficacy in a B16 melanoma model, even when the treatment was initiated at the time of tumor inoculation (van Elsas et al., 1999). By contrast, a short course of anti-CTLA-4 blocking antibody in combination with a GM-CSF-expressing irradiated tumor cell vaccine eradicated established B16 melanoma in 80% of wild-type mice and protected those mice from subsequent tumor challenge (van Elsas et al., 1999). When tumors were successfully rejected, depigmentation was observed in 50% of the treated mice. Since depigmentation, a marker of autoimmunity, is an uncommon event following vaccination alone and has not been observed with anti-CTLA-4 monotherapy, these findings suggested that CTLA-4 played a significant role in breaking peripheral tolerance.
Fully humanized antibodies against human CTLA-4, MDX-010 (ipilimumab), and CP-675,206 (ticilimumab) have been evaluated in several published phase-I and phase-II trials (Table 1). Anti-CTLA-4 monotherapy has yielded response rates of 7–15% in heavily pretreated patients with metastatic melanoma (Korman et al., 2005; Ribas et al., 2005). Administration of MDX-010 in conjunction with peptide vaccination to patients with stage-IV melanoma resulted in objective cancer regression in 3 of 14 patients (two CRs and one PR) and grade-III/IV autoimmune manifestations in 6 of 14 patients (43%) (Phan et al., 2003). Combination therapy with anti-CTLA-4 and IL-2 in patients with metastatic melanoma yielded a similar 22% response rate coupled with a 14% rate of grade-II/IV autoimmune toxicities (described below). Given an anticipated 15% response rate to IL-2 alone in the treatment of metastatic melanoma, there does not seem to be a synergistic effect (Maker et al., 2005).
Treatment with anti-CTLA-4 mAb is frequently associated with adverse immune events, most commonly involving the skin and the gastrointestinal tract. Grade-III and grade-IV adverse immune events, especially enterocolitis (21%;Beck et al., 2006) and autoimmune hypophysitis (5%; Blansfield et al., 2005) have been reported. A dermatitis characterized by pruritic macules and papules occurred in 14% of stage-IV melanoma patients treated with ipilimumab as monotherapy (Jaber et al., 2006). The erythermatous macules and papules present in anti-CTLA-4 dermatitis were photodistributed in some patients, and a Koebner-like phenomenon could be elicited in others (Figure 2). Peripheral eosinophil counts were increased by fourfold in patients who had experienced the eruption compared with in those who had not (Jaber et al., 2006). Of note, this pruritic skin eruption was similar in clinical and histological appearance (superficial perivascular lymphocytic infiltrate with frequent eosinophils) to the macular-papular rash most commonly associated with drugs such as penicillins (Jaber et al., 2006). Autoimmune side effects have been correlated with clinical antitumor responses and relapse-free survival (Attia et al., 2005), suggesting that antitumor immunity is achieved by blockade of CTLA-4 at the increased risk of autoimmunity in other tissues.
Figure 2. Skin dermatitis following treatment with anti-CTLA-4 mAb as a single agent as described in Jaber et al. (2006).
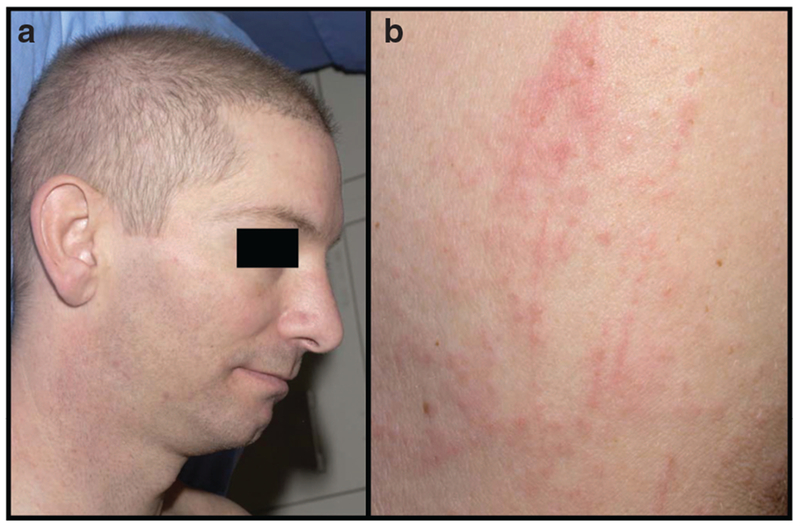
(a) Photodistributed macules and papules on face and neck, and (b) erythematous papules with Koebner-like phenomenon elicited by the trauma of scratching.
Toll-like receptor agonists.
Synthetic deoxycytidyl-deoxyguanosine oligodeoxynucleotides contain unmethylated CG motifs similar to those observed in bacterial DNA. By triggering Toll-like receptor-9 (TLR9), these oligodeoxynucleotides trigger an immunostimulatory pathway that includes dendritic cell (DC) activation and production of proin-flammatory cytokines such as type-I IFN, thus potentially favoring antitumor immunity. In humans, only plasma-cytoid DCs and B cells are known to express TLR9 (Krieg, 2004).
Because TLR9 agonists have shown efficacy in animal tumor models (Lonsdorf et al., 2003), they are being evaluated for their therapeutic potential in clinical trials for melanoma patients. The first human trial of a synthetic deoxycytidyl-deoxyguanosine oligodeoxynucleotides combined with a melanoma peptide showed that use of CpG 7909 as an adjuvant to Melan-A/Mart-1 peptide vaccination resulted in 10-fold increase in circulating Melan-A-specific CD8 T cells compared with use of peptide alone. Clinical response rates were not obtained in this phase-I study due to the short follow-up period (Speiser et al., 2005).
Imiquimod, a TLR7 agonist that is currently FDA-approved for the topical treatment of actinic keratosis, external genital warts, and superficial basal cell carcinoma, also appears to hold potential for the treatment of melanoma. Several cases of successful treatment with topical 5% imiquimod of otherwise untreatable cutaneous metastases of malignant melanoma (Steinmann et al., 2000) and melanoma in situ (Ray et al., 2005) have been reported. Therapeutic approaches based on TLR agonists may be of particular relevance in cases where standard therapies have been refused or are contraindicated.
Cancer vaccines
Peptide-based vaccination strategies.
The identification of defined tumor antigens that can be the targets of T cells has led to development of new immunotherapeutic strategies (Boon et al., 1997). Tumor antigens isolated from melanomas include MART-1/Melan-A, gp100, and tyrosinase, all of which are melanocytic differentiation antigens that are also expressed by normal melanocytes. Anti-melanoma-specific CTLs have been found in peripheral blood, draining lymph nodes, and within tumors in melanoma patients (Pittet et al., 1999; Zippelius et al., 2004). It has been suggested that these cells may be unable to eradicate invasive tumors due to low frequency and/or an immunosuppressive host microenvironment. Therefore, administration of cancer vaccines may boost endogenous antitumor immune responses in vivo.
Vaccines are classified as either univalent, stimulating the immune system against a particular antigen, or polyvalent, eliciting immune responses against multiple antigens. The most extensively studied melanoma vaccines are polyvalent whole cells or cell lysates derived from allogenic or autologous tumor cells. One advantage of this approach is that the vaccine immunizes the patient against a variety of tumor antigens, without attempting to predict which antigen will elicit the most effective antitumor responses. Since these vaccines are expected to be immunogenic in patients with various HLA types, they are potentially beneficial for a larger patient population.
Early trials of polyvalent whole-melanoma-cell vaccines (Mitchell et al., 1988; Morton et al., 1992; Hsueh et al., 2002) suggested vaccines could elicit immunological responses in melanoma patients and provide clinical benefit to some patients. However, later trials (Table 1) involving larger numbers of patients failed to show a clinical benefit (Sondak et al., 2002; Faries and Morton, 2005). One polyvalent whole-cell vaccine (Melacine®) combined with low-dose cyclophosphamide was shown to provide superior quality of life during therapy, although there was no demonstrable difference in response rates and survival compared with patients treated with a four-drug chemotherapy regimen (Mitchell, 1998).
Immunization with melanoma-associated peptide antigens is a strategy that has been vigorously pursued. In one of the first clinical trials using melanoma cell-derived peptides, Rosenberg et al. (1998a) vaccinated stage-IV melanoma patients with g209-2M, a modified immunodominant peptide of the gp100 antigen. On the basis of immunological assays, 91% of patients could be successfully immunized with this synthetic peptide, and 13 of 31 patients (42%) receiving the peptide vaccine plus IL-2 had objective cancer responses. All patients, however, eventually developed progressive disease. Nevertheless, this study provided essential proof-of-principle of the concept that immune responses against self-antigens can be elicited in patients with advanced melanoma. Although encouraging, this and subsequent clinical studies also highlighted the fact that tumor progression can occur despite induction of an antigen-specific immune response (Powell et al., 2006). Strategies using expression of melanoma peptides via recombinant adenoviral vectors (Rosenberg et al., 1998b) or with plasmid vectors encoding tumor antigen (Rosenberg et al., 2003) were initially attractive due to their low cost and ease of administration, but have not resulted in clear elicitation of immune or clinical responses in patients. Cancer-testis antigens, expressed in different tumors and normal testis, are also potential targets for melanoma immunotherapy. While an initial study of the cancer-testis antigen known as MAGE-3 resulted in tumor regression in some patients (Marchand et al., 1999), later studies with this cancer-testis antigen showed poor clinical and immunological responses (Kruit et al., 2005).
DC-based vaccination strategies.
Effective presentation of the defined tumor antigens to the immune system of a cancer patient remains to be one of the most important challenges in the field. The presentation of tumor antigen by DCs or other antigen-presenting cells is a central step in the induction of an antigen-specific T-cell response, and DCs, therefore, have been proposed to be an ideal tool for the induction and augmentation of an immune response in a vaccination setting (Grabbe et al., 1995). In recent years, the clinical use of DCs has been facilitated by development of techniques to generate large numbers of these cells in vitro from blood monocytes or CD34+ progenitor cells. The first human trial using monocyte-derived peptide- or tumor lysate-pulsed DCs for antigen delivery enrolled 16 patients with advanced melanoma and yielded five objective responses (ORs) (2/16 CR, 3/16 PR) (Nestle et al., 1998). Using a similar vaccination strategy with larger numbers of patients, however, Schadendorf et al. (2006) demonstrated only a 3.8% OR rate that was similar to the OR rate for dacarbazine. Other DC vaccination trials have not resulted in significant objective clinical responses, but have shown delayed-type hypersensitivity to the peptide antigen (Thurner et al., 1999) or a delay in tumor progression when patients were able to mount immune responses to multiple antigens used in the multivalent DC-based vaccine (Banchereau et al., 2001). Thus, although immunological responses to selected vaccination strategies have often been detected, their clinical utility as single agents is not promising. In summarizing the results from 35 representative published vaccine studies, Rosenberg et al. (2004) calculated an OR rate of 3.8% (7.1% for DCs, 4.2% for modified tumor cells, 4.0% for peptide-based vaccines, and 0% for pox viruses) for cancer vaccines. Currently there are no vaccination strategies that consistently induce melanoma regression.
ACT therapy
CTL therapy.
In CTL therapy, peripheral blood mononuclear cells are isolated from melanoma patients and stimulated with autologous antigen-presenting cells pulsed with HLA-restricted peptide epitope of MART-1 or gp100. After several rounds of stimulation, single clones are selected in vitro for their abilities to specifically kill antigen-positive tumor targets. CTL clones are further expanded in vitro before being adoptively transferred back to patients (Ho et al., 2002). Early clinical trials with CTL therapy met with limited success as antigen-specific CD8 T cells persisted in vivo only for a short period of time. In addition, recurrent tumors were found to selectively lose the targeted antigen(s) (Yee et al., 2002).
Tumor-infiltrating lymphocyte (TIL) therapy.
In contrast to cancer vaccines, which activate the immune system in situ, TIL therapy relies upon (1) isolation and propagation of autologous T cells present in patient tumors (usually metastases) in the presence of high levels of IL-2, (2) selection of highly avid clones that produce high levels of IFN-γ against multiple melanoma cell lines, and (3) expansion of those cells in vitro with anti-CD3 and IL-2 to large numbers that are then transferred back to the patient (Dudley and Rosenberg, 2003).
Early attempts with TILs and high-dose IL-2 in melanoma patients achieved limited clinical success (Rosenberg et al., 1994b) and were generally characterized by poor persistence of TIL clones in vivo following adoptive transfer. Although cultured TIL clones often showed high avidity toward tumor-associated melanoma peptides, only rarely were clinical remissions achieved in excess of that expected for IL-2 therapy alone. Thus, high-avidity T-cell clones alone were not sufficient for treatment efficacy.
Remarkable results in this field were first reported by Rosenberg et al., who used non-myeloablative, lymphodepleting preconditioning followed by infusion of autologous TILs and IL-2 (Dudley et al., 2002a). Subsequent studies at the NCI confirmed response rates of ~50% in advanced melanoma patients with metastatic disease (Dudley et al., 2005). Several patients receiving treatment regimen achieved impressive clinical regression of large bulky tumors (figure 3). The rationale for this lymphodepleting preconditioning regimen was based on early animal studies demonstrating that depletion of endogenous lymphocytes by chemotherapy (prior to adoptive transfer of T cells) resulted in improved antitumor responses (Cheever et al., 1980; North, 1982; Dummer et al., 2002). The mechanisms by which lymphodepleting regimens improve outcome of ACT therapy have been shown to involve elimination of Tregs, increasing the availability of T-cell growth-promoting cytokines, and improvement of the function and/ or availability of antigen-presenting cells as discussed below (Gattinoni et al., 2006).
Figure 3. Response of a melanoma tumor to a lymphodepleting chemotherapy regimen combined with adoptive transfer of tumor-infiltrating T cells (Dudley et al., 2002a).
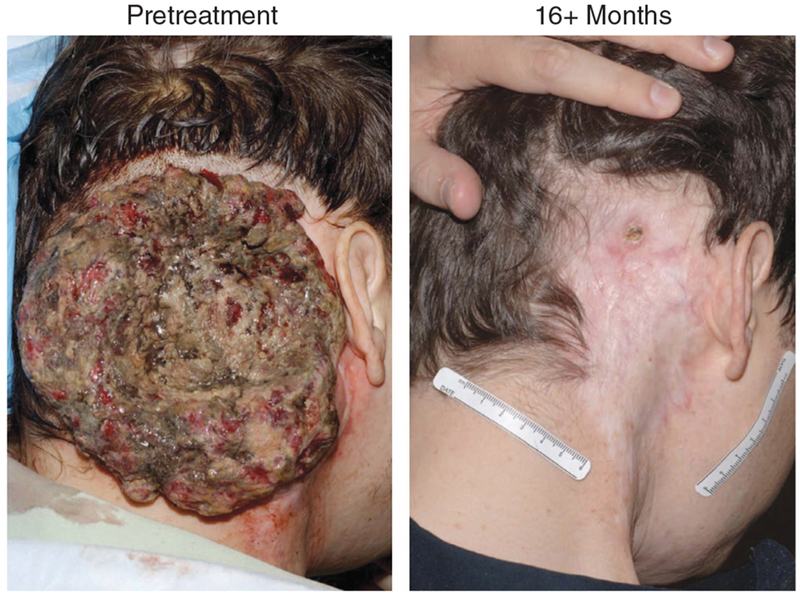
(photo courtesy of Dr Steven A. Rosenberg, Surgery Branch, NCI).
Naturally occurring Tregs possess the ability to suppress or antagonize the function of other T cells. Tregs are crucial for maintenance of peripheral self-tolerance and suppression of antitumor immunity (Sakaguchi, 2005). The Tregs described in this review are characterized by expression of the forkhead box P3 (FoxP3) transcription factor, CD4, and activated T-cell markers, including CD25 (IL-2 receptor-a), glucocorticoid-induced tumor-necrosis factor-receptor family-regulated gene, and CTLA-4.
CD4+ CD25hiFoxp3+ cells are relevant to immunotherapy for melanoma for a number of reasons. Co-transfer of CD4+ CD25+ Tregs reduced the ability of transgenic, antigen-specific CD8 T cells to induce B16 murine melanoma tumor regression in vivo (Antony et al., 2005). Clinically, Foxp3+ Tregs were overrepresented in metastatic lymph nodes from patients with melanoma. Following isolation, these Tregs inhibited proliferation and cytokine production of tumor-infiltrating CD4 and CD8 cells in vitro (Viguier et al., 2004). In ovarian cancer patients, reduced survival was associated with increased tumor-infiltrating Treg levels (Curiel et al., 2004), whereas increased survival was associated with a high ratio of intratumoral CD8 T cells to Tregs (Sato et al., 2005). Even though the presence of Treg cells in tumor lesions has not been conclusively connected with progression of cancers, the suppressive function of Treg cells may contribute to poor clinical response in non-lymphodepleting patients receiving ACT therapy.
Lymphodepleting preconditioning regimens may also be effective because they reduce the size of the host endogenous lymphocyte pool, allowing newly transferred T cells access to growth promoting cytokines (Goldrath et al., 2000). Because the size of the T-cell pool in humans and mice is tightly maintained at a nearly constant level, adoptive transfer of cells into a lymphopenic host will result in rapid expansion of the newly transferred antigen-specific T cells in an IL-7- and IL-15-dependent manner (Tan et al., 2002). Dummer et al. (2002) first reported that the antitumor benefits of lymphodepletion were dependent on homeostatic expansion of a polyclonal T-cell population within lymph nodes, indicating that T cells can be induced to mount an effective autoimmune response against self-antigens when homeostatic expansion occurs at the time of antigen encounter. In the pmel-1 mouse model of ACT therapy for melanoma, lymphodepletion also enhanced antitumor efficacy of adoptively transferred T cells in an IL-7- and IL-15 dependent manner (Gattinoni et al., 2005a).
Preconditioning of the host with systemic chemotherapy or total-body irradiation before ACT therapy was initially thought to result in apoptosis or necrosis of tumor cells, allowing more efficient presentation of tumor antigens by host DCs to the adoptively transferred CD8 cells. Recent results, however, showed that total-body irradiation caused mucosal injury, resulting in microbial translocation from gastrointestinal tract and systemic release of lipopolysaccharide. Signaling through TLR4, lipopolysaccharide increased the absolute numbers of activated DCs, which subsequently secreted high levels of T-cell-activating cytokines (Paulos et al., 2007).
While lymphodepletion remarkably improves the outcome of ACT therapy, it is not the only factor that determines the clinical response. Preclinical and clinical studies have implied that the differentiation state of transferred CD8 T cells may be crucial for the success of ACT therapy. Upon encoun-ter with antigen, naïve CD8 T cells proliferate and differentiate through early, intermediate, and late effector stages depending on signal strength (Lanzavecchia and Sallusto, 2002). Differentiation from early stage to late stage of effector CD8 T cells is characterized by progressive downregulation of CD62L, CCR7, β7-integrin, and CD27 and concurrent upregulation of CD44, CD69, CD25, granzyme B, and perforin. After multiple rounds of in vitro stimulation with antigen and IL-2, activated pmel-1-transgenic CD8 T cells acquired terminally differen-tiated effector properties, including increased cytolytic activity and higher levels of IFN-γ production. How-ever, when adoptively transferred into lymphodepleted B16 tumor-bearing wild-type mice, terminally differentiated pmel-1 CD8 T cells were at least 100-fold less effective than early effector CD8 cells in antitumor efficacy (Gattinoni et al., 2005b). In addition, terminally differentiated pmel-1 CD8 T cells proliferated poorly in vivo, suggesting they might have already exhausted their abilities to proliferate and to persist in vivo once adoptively transferred. On the other hand, early effector CD8 T cells possessing essential adhesion molecules for trafficking to lymph nodes showed superior antitumor efficacy compared with T cells that could not traffic to lymph nodes (Gattinoni et al., 2005b). These findings suggest that trafficking of less differentiated CD8 T cells to lymph nodes (where they can be effectively stimulated by DCs) may be more effective in ACT for cancer.
These findings pose new challenges for ACT-based immunotherapy. Currently, the only criteria clinically used to screen for TIL clones is their ability to produce high levels of IFN-γ and to kill antigen-specific target cells in vitro. Selected TIL clones undergo several rounds of expansion with anti-CD3, IL-2, and allogeneic antigen-presenting cells, which inevitably led to selection of late-stage or terminal differentiated TIL clones for adoptive transfer. Two ACT clinical studies using tumor-reactive CD8 T cells generated and expanded ex vivo through multiple stimulations did not show substantial ORs, although transferred CD8 cells showed potent antitumor activity in vitro (Dudley et al., 2002b; Yee et al., 2002). Thus, balancing the quality and quantity of the transferred cells poses one of the greatest challenges for ACT therapy.
While lymphodepletion combined with ACT shows great promise, lymphodepletion appears to increase the risk of viral infections and virus-associated cancers, perhaps because of the long period of time required for lymphocyte recovery following chemotherapy (Dudley et al., 2002b). Furthermore, use of high-dose IL-2 following transfer of T cells exposes patients to many of the same risks incurred by IL-2 therapy in the past. The use of genetically engineered T cells that endogenously express IL-2 may remove the need to give patients high systemic doses of IL-2 (see below). Alternatively, modifications of IL-2 that result in more T-cell (vs natural killer cell) selectivity may reduce systemic toxicity (Shanafelt et al., 2000). Clearly, advances in reducing the use of systemic IL-2 (or replacing it with less toxic alternatives) would make ACT a more attractive (and safer) therapy for melanoma.
Additional strategies for improving immunotherapy for melanoma
Engineered T-cell therapy.
Although ACT therapy has achieved objective tumor regression in patients with metastatic melanoma, not all the biopsy specimens yield high-avidity TIL clones for adoptive transfer (Dudley et al., 2003). One approach to overcome this limitation is to generate and adoptively transfer engineered autologous T cells that express cloned high-affinity TCRs for melanoma-specific antigens. TILs recognize tumor-associated antigens through major histocompatibility complex-restricted TCRs that are composed of TCR-α and -β chains. Rosenberg et al. have identified individual patient TIL clones showing high affinity in vitro to specific melanoma antigens (for example, gp100); they cloned the TCR-α and -β chains from these TILs and subsequently expressed these TCRs using retroviruses in peripheral blood T cells from other patients (Morgan et al., 2003). The genetically engineered T cells secreted high levels of IFN-γ and were cytolytic against both melanoma cell lines and autologous melanoma cells. Importantly, following adoptive transfer into melanoma patients lymphodepleted by chemotherapy, transduced T cells expressing the cloned TCR chains persisted at high levels in the peripheral blood for at least 2 months (and up to 1 year) in patients (Morgan et al., 2006).
Although clinical response rate in this study was lower than that achieved using autologous TILs (Dudley et al., 2005), several approaches to increase efficiency of TCR expression may potentially enhance clinical efficacy (Cohen et al., 2007; Zhao et al., 2007). Furthermore, transducing tumor-reactive T cells with IL-2 (Liu and Rosenberg, 2001) or IL-15 (Klebanoff et al., 2004) may promote survival of TILs without subjecting patients to the toxicities of systemically administered IL-2.
Potential synergy of immunotherapy and blockade of chemokine receptor-mediated survival pathways
Clinical studies showed that selected chemokine receptors, particularly CXCR4, are often upregulated in a large number of common human cancers, including melanoma, and that chemokine receptors (in concert with their chemokine ligands) facilitate cancer survival and metastasis through a number of mechanisms (Muller et al., 2001; Murakami et al., 2002; Kakinuma and Hwang, 2006). The CXCR4 ligand, CXCL12, protected B16 cells from killing by activated pmel-1 CD8 T cells in vitro (Lee et al., 2006), presumably through activation of the phosphoinositide-3-kinase and its downstream effector, Akt (Murakami et al., 2003). Inhibition of CXCR4 by a peptide antagonist, T22, in combination with cyclophosphamide or anti-CTLA-4 antibody significantly reduced metastatic tumor burden in the lungs compared with treatment with cyclophosphamide or anti-CTLA-4 alone (Lee et al., 2006). This study suggests that pretreatment of patients with a chemo-kine-receptor antagonist prior to immunotherapy may result in better clinical responses.
Summary
Until recently, immunotherapy for melanoma has made only small incremental improvements since the first attempts to treat patients with high-dose IL-2. Vaccination strategies alone have shown little efficacy, whereas attempts to boost the endogenous host antitumor response with agents such as anti-CTLA4 have met with marginal success. In the case of the latter agent, clinical efficacy in shrinking tumors has often come at a high price in terms of autoimmune toxicities that have been potentially life threatening. By contrast, ACT therapy, particularly following lymphodepleting, preconditioning regimen has resulted in response rates approaching 50%. Lymphodepletion appears to be particularly critical for the success of these therapies because (1) it provides access to cytokines that promote growth and survival of newly transferred T cells and (2) it removes suppressive or regulatory T-cell populations.
Developing safe lymphodepleting regimens will be a challenge, since toxicities of high-dose IL-2 and delays in immune reconstitution remain major impediments to ACT. Future enhancements to ACT include using total-body irradiation to activate antigen-presenting cells via TLRs (in addition to reducing endogenous T-cell populations through bone marrow suppression). These experiments suggest that the growing library of clinical-grade TLR agonists may prove to be valuable adjuvants for ACT. Lastly, genetic manipulation of either allogeneic or autologous T cells with highly avid cloned TCRs selected for clinical efficacy or with cloned cytokines such as IL-2 and IL-15 may allow production of highly tumor-reactive T cells that can be delivered far more quickly and with less cost than the current methods of culturing tumor-infiltrating T cells from resected tumors. While prognosis for the majority of patients with advanced metastatic melanoma remains relatively poor, almost half of the patients with advanced melanoma can obtain substantial ORs using current ACT regimens. Improvements to current treatment strategies that are now being refined in mouse models of melanoma suggest that future enhancements in clinical efficacy are forthcoming.
ACKNOWLEDGMENTS
We thank Drs Mark C. Udey (Dermatology Branch, NCI) and Steven A. Rosenberg (Surgery Branch, NCI) for helpful comments and suggestions. ASL is a recipient of a German Research Foundation (DFG)-NIH Career Transition Award. This work was supported by the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research.
Abbreviations:
- ACT
adoptive cell transfer
- CR
complete response
- CTLA
cytotoxic T lymphocyte-associated antigen
- DC
dendritic cell
- OR
objective cyte
- TLR
Toll-like receptor
- Tregs
regulatory T cells
Footnotes
CONFLICT OF INTEREST
The authors state no conflict of interest.
REFERENCES
- Ahmadzadeh M, Rosenberg SA (2006) IL-2 administration increases CD4+ CD25(hi) Foxp3+ regulatory T cells in cancer patients. Blood 107:2409–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antony PA, Piccirillo CA, Akpinarli A, Finkelstein SE, Speiss PJ, Surman DR et al. (2005) CD8+ T cell immunity against a tumor/self-antigen is augmented by CD4+ T helper cells and hindered by naturally occurring T regulatory cells. J Immunol 174:2591–601 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins MB, Lotze MT, Dutcher JP, Fisher RI, Weiss G, Margolin K et al. (1999) High-dose recombinant interleukin 2 therapy for patients with metastatic melanoma: analysis of 270 patients treated between 1985 and 1993. J Clin Oncol 17:2105–16 [DOI] [PubMed] [Google Scholar]
- Attia P, Phan GQ, Maker AV, Robinson MR, Quezado MM, Yang JC et al. (2005) Autoimmunity correlates with tumor regression in patients with metastatic melanoma treated with anti-cytotoxic T-lymphocyte antigen-4. J Clin Oncol 23:6043–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banchereau J, Palucka AK, Dhodapkar M, Burkeholder S, Taquet N, Rolland A et al. (2001) Immune and clinical responses in patients with metastatic melanoma to CD34(+) progenitor-derived dendritic cell vaccine. Cancer Res 61:6451–8 [PubMed] [Google Scholar]
- Bart RS, Porzio NR, Kopf AW, Vilcek JT, Cheng EH, Farcet Y (1980) Inhibition of growth of B16 murine malignant melanoma by exogenous interferon. Cancer Res 40:614–9 [PubMed] [Google Scholar]
- Beck KE, Blansfield JA, Tran KQ, Feldman AL, Hughes MS, Royal RE et al. (2006) Enterocolitis in patients with cancer after antibody blockade of cytotoxic T-lymphocyte-associated antigen 4. J Clin Oncol 24:2283–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blansfield JA, Beck KE, Tran K, Yang JC, Hughes MS, Kammula US et al. (2005) Cytotoxic T-lymphocyte-associated antigen-4 blockage can induce autoimmune hypophysitis in patients with metastatic melanoma and renal cancer. J Immunother 28:593–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boon T, Coulie PG, Van den Eynde B (1997) Tumor antigens recognized by T cells. Immunol Today 18:267–8 [DOI] [PubMed] [Google Scholar]
- Cheever MA, Greenberg PD, Fefer A (1980) Specificity of adoptive chemoimmuno-therapy of established syngeneic tumors. J Immunol 125:711–4. [PubMed] [Google Scholar]
- Chiao EY, Krown SE (2003) Update on non-acquired immunodeficiency syndrome-defining malignancies. Curr Opin Oncol 15:389–97 [DOI] [PubMed] [Google Scholar]
- Cohen CJ, Li YF, El-Gamil M, Robbins PF, Rosenberg SA, Morgan RA (2007) Enhanced antitumor activity of T cells engineered to express T-cell receptors with a second disulfide bond. Cancer Res 67:3898–903 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Curiel TJ, Coukos G, Zou L, Alvarez X, Cheng P, Mottram P et al. (2004) Specific recruitment of regulatory T cells in ovarian carcinoma fosters immune privilege and predicts reduced survival. Nat Med 10:942–9 [DOI] [PubMed] [Google Scholar]
- Dudley ME, Rosenberg SA (2003) Adoptive-cell-transfer therapy for the treatment of patients with cancer. Nat Rev 3:666–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley ME, Wunderlich JR, Robbins PF, Yang JC, Hwu P, Schwartzentruber DJ et al. (2002a) Cancer regression and autoimmunity in patients after clonal repopulation with antitumor lymphocytes. Science 298:850–4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley ME, Wunderlich JR, Shelton TE, Even J, Rosenberg SA (2003) Generation of tumor-infiltrating lymphocyte cultures for use in adoptive transfer therapy for melanoma patients. J Immunother 26:332–42 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley ME, Wunderlich JR, Yang JC, Hwu P, Schwartzentruber DJ, Topalian SL et al. (2002b) A phase I study of nonmyeloablative chemotherapy and adoptive transfer of autologous tumor antigen-specific T lymphocytes in patients with metastatic melanoma. J Immunother 25:243–51 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dudley ME, Wunderlich JR, Yang JC, Sherry RM, Topalian SL, Restifo NP et al. (2005) Adoptive cell transfer therapy following non-myeloablative but lymphodepleting chemotherapy for the treatment of patients with refractory metastatic melanoma. J Clin Oncol 23:2346–57 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dummer W, Niethammer AG, Baccala R, Lawson BR, Wagner N, Reisfeld RA et al. (2002) T cell homeostatic proliferation elicits effective antitumor autoimmunity. J Clin Invest 110:185–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faries MB, Morton DL (2005) Therapeutic vaccines for melanoma: current status. BioDrugs 19:247–60 [DOI] [PubMed] [Google Scholar]
- Finkelstein SE, Heimann DM, Klebanoff CA, Antony PA, Gattinoni L, Hinrichs CS et al. (2004) Bedside to bench and back again: how animal models are guiding the development of new immunotherapies for cancer. J Leukoc Biol 76:333–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Finkelstein SE, Klebanoff CA, Antony PA, Palmer DC, Spiess PJ et al. (2005a) Removal of homeostatic cytokine sinks by lymphodepletion enhances the efficacy of adoptively transferred tumor-specific CD8+ T cells. J Exp Med 202:907–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Klebanoff CA, Palmer DC, Wrzesinski C, Kerstann K, Yu Z et al. (2005b) Acquisition of full effector function in vitro paradoxically impairs the in vivo antitumor efficacy of adoptively transferred CD8+ T cells. J Clin Invest 115:1616–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gattinoni L, Powell DJ Jr, Rosenberg SA, Restifo NP (2006) Adoptive immunotherapy for cancer: building on success. Nat Rev Immunol 6:383–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldrath AW, Bogatzki LY, Bevan MJ (2000) Naive T cells transiently acquire a memory like phenotype during homeostasis-driven proliferation. J Exp Med 192:557–64 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grabbe S, Beissert S, Schwarz T, Granstein RD (1995) Dendritic cells as initiators of tumor immune responses: a possible strategy for tumor immunotherapy? Immunol Today 16: 117–21 [DOI] [PubMed] [Google Scholar]
- Ho WY, Yee C, Greenberg PD (2002) Adoptive therapy with CD8(+) T cells: it may get by with a little help from its friends. J Clin Invest 110:1415–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hodi FS, Mihm MC, Soiffer RJ, Haluska FG, Butler M, Seiden MV et al. (2003) Biologic activity of cytotoxic T lymphocyte-associated antigen 4 antibody blockade in previously vaccinated metastatic melanoma and ovarian carcinoma patients. Proc Natl Acad Sci USA 100:4712–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsueh EC, Essner R, Foshag LJ, Ollila DW, Gammon G, O’Day SJ et al. (2002) Prolonged survival after complete resection of disseminated melanoma and active immunotherapy with a therapeutic cancer vaccine. J Clin Oncol 20:4549–54 [DOI] [PubMed] [Google Scholar]
- Hwu WJ, Panageas KS, Menell JH, Lamb LA, Aird S, Krown SE et al. (2006) Phase II study of temozolomide plus pegylated interferon-alpha-2b for metastatic melanoma. Cancer 106:2445–51 [DOI] [PubMed] [Google Scholar]
- Jaber SH, Cowen EW, Haworth LR, Booher SL, Berman DM, Rosenberg SA et al. (2006) Skin reactions in a subset of patients with stage IV melanoma treated with anti-cytotoxic T-lymphocyte antigen 4 monoclonal antibody as a single agent. Arch Dermatol 142:166–72 [DOI] [PubMed] [Google Scholar]
- James K, Eisenhauer E, Christian M, Terenziani M, Vena D, Muldal A et al. (1999) Measuring response in solid tumors: unidimensional versus bidimensional measurement. J Natl Cancer Inst 91:523–8 [DOI] [PubMed] [Google Scholar]
- Kakinuma T, Hwang ST (2006) Chemokines, chemokine receptors, and cancer metastasis. J Leukoc Biol 79:639–51 [DOI] [PubMed] [Google Scholar]
- Keilholz U, Punt CJ, Gore M, Kruit W, Patel P, Lienard D et al. (2005) Dacarbazine, cisplatin, and interferon-alfa-2b with or without interleukin-2 in metastatic melanoma: a randomized phase III trial (18951) of the European Organisation for Research and Treatment of Cancer Melanoma Group. J Clin Oncol 23:6747–55 [DOI] [PubMed] [Google Scholar]
- Kirkwood JM, Ibrahim JG, Sondak VK, Richards J, Flaherty LE, Ernstoff MS et al. (2000) High- and low-dose interferon alfa-2b in high-risk melanoma: first analysis of intergroup trial E1690/S9111/C9190. J Clin Oncol 18: 2444–58 [DOI] [PubMed] [Google Scholar]
- Kirkwood JM, Strawderman MH, Ernstoff MS, Smith TJ, Borden EC, Blum RH (1996) Interferon alfa-2b adjuvant therapy of high-risk resected cutaneous melanoma: the Eastern Cooperative Oncology Group Trial EST 1684. J Clin Oncol 14:7–17 [DOI] [PubMed] [Google Scholar]
- Klebanoff CA, Finkelstein SE, Surman DR, Lichtman MK, Gattinoni L, Theoret MR et al. (2004) IL-15 enhances the in vivo antitumor activity of tumor-reactive CD8+ T cells. Proc Natl Acad Sci USA 101:1969–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korman A, Yellin M, Keler T (2005) Tumor immunotherapy: preclinical and clinical activity of anti-CTLA4 antibodies. Curr Opin Investig Drugs 6:582–91 [PubMed] [Google Scholar]
- Krieg AM (2004) Antitumor applications of stimulating toll-like receptor 9 with CpG oligodeoxynucleotides. Curr Oncol Rep 6: 88–95 [DOI] [PubMed] [Google Scholar]
- Kruit WH, van Ojik HH, Brichard VG, Escudier B, Dorval T, Dreno B et al. (2005) Phase 1/2 study of subcutaneous and intradermal immunization with a recombinant MAGE-3 protein in patients with detectable metastatic melanoma. Int J Cancer 117:596–604 [DOI] [PubMed] [Google Scholar]
- Ladanyi A, Kiss J, Somlai B, Gilde K, Fejos Z, Mohos A et al. (2007) Density of DC-LAMP(+) mature dendritic cells in combination with activated T lymphocytes infiltrating primary cutaneous melanoma is a strong independent prognostic factor. Cancer Immunol Immunother 56:1459–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lanzavecchia A, Sallusto F (2002) Progressive differentiation and selection of the fittest in the immune response. Nat Rev Immunol 2: 982–7 [DOI] [PubMed] [Google Scholar]
- Leach DR, Krummel MF, Allison JP (1996) Enhancement of antitumor immunity by CTLA-4 blockade. Science 271:1734–6 [DOI] [PubMed] [Google Scholar]
- Lee CH, Kakinuma T, Wang J, Zhang H, Palmer DC, Restifo NP et al. (2006) Sensitization of B16 tumor cells with a CXCR4 antagonist increases the efficacy of immunotherapy for established lung metastases. Mol Cancer Ther 5:2592–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee PP, Yee C, Savage PA, Fong L, Brockstedt D, Weber JS et al. (1999) Characterization of circulating T cells specific for tumor-associated antigens in melanoma patients. Nat Med 5:677–85 [DOI] [PubMed] [Google Scholar]
- Liu K, Rosenberg SA (2001) Transduction of an IL-2 gene into human melanoma-reactive lymphocytes results in their continued growth in the absence of exogenous IL-2 and maintenance of specific antitumor activity. J Immunol 167:6356–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdorf AS, Kuekrek H, Stern BV, Boehm BO, Lehmann PV, Tary-Lehmann M (2003) Intratumor CpG-oligodeoxynucleotide injection induces protective antitumor T cell immunity. J Immunol 171:3941–6 [DOI] [PubMed] [Google Scholar]
- Maker AV, Phan GQ, Attia P, Yang JC, Sherry RM, Topalian SL et al. (2005) Tumor regression and autoimmunity in patients treated with cytotoxic T lymphocyte-associated antigen 4 blockade and interleukin 2: a phase I/II study. Ann Surg Oncol 12:1005–16 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malek TR, Bayer AL (2004) Tolerance, not immunity, crucially depends on IL-2. Nat Rev Immunol 4:665–74 [DOI] [PubMed] [Google Scholar]
- Marchand M, van Baren N, Weynants P, Brichard V, Dreno B, Tessier MH et al. (1999) Tumor regressions observed in patients with metastatic melanoma treated with an antigenic peptide encoded by gene MAGE-3 and presented by HLA-A1. Int J Cancer 80: 219–30 [DOI] [PubMed] [Google Scholar]
- Michaelis LC, Ratain MJ (2006) Measuring response in a post-RECIST world: from black and white to shades of grey. Nat Rev 6: 409–14 [DOI] [PubMed] [Google Scholar]
- Mitchell MS, Abrams J, Thompson JA, Kashani-Sabet M, DeConti RC, Hwu WJ et al. (2007) Randomized trial of an allogeneic melanoma lysate vaccine with low-dose interferon Alfa-2b compared with high-dose interferon Alfa-2b for resected stage III cutaneous melanoma. J Clin Oncol 25:2078–85 [DOI] [PubMed] [Google Scholar]
- Mitchell MS, Kan-Mitchell J, Kempf RA, Harel W, Shau HY, Lind S (1988) Active specific immunotherapy for melanoma: phase I trial of allogeneic lysates and a novel adjuvant. Cancer Res 48:5883–93 [PubMed] [Google Scholar]
- Mitchell MS (1998) Perspective on allogeneic melanoma lysates in active specific immunotherapy. Semin Oncol 25:623–35 [PubMed] [Google Scholar]
- Morgan RA, Dudley ME, Wunderlich JR, Hughes MS, Yang JC, Sherry RM et al. (2006) Cancer regression in patients after transfer of genetically engineered lymphocytes. Science 314: 126–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morgan RA, Dudley ME, Yu YY, Zheng Z, Robbins PF, Theoret MR et al. (2003) High efficiency TCR gene transfer into primary human lymphocytes affords avid recognition of melanoma tumor antigen glycoprotein 100 and does not alter the recognition of autologous melanoma antigens. J Immunol 171:3287–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton DL, Foshag LJ, Hoon DS, Nizze JA, Famatiga E, Wanek LA et al. (1992) Prolongation of survival in metastatic melanoma after active specific immunotherapy with a new polyvalent melanoma vaccine. Ann Surg 216:463–82 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morton DL, Wanek L, Nizze JA, Elashoff RM, Wong JH (1991) Improved long-term survival after lymphadenectomy of melanoma metastatic to regional nodes. Analysis of prognostic factors in 1134 patients from the John Wayne Cancer Clinic. Ann Surg 214:491–9, discussion 499–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muller A, Homey B, Soto H, Ge N, Catron D, Buchanan ME et al. (2001) Involvement of chemokine receptors in breast cancer metastasis. Nature 410:50–6 [DOI] [PubMed] [Google Scholar]
- Murakami T, Cardones AR, Finkelstein SE, Restifo NP, Klaunberg BA, Nestle FO et al. (2003) Immune evasion by murine melanoma mediated through CC chemokine receptor-10. J Exp Med 198:1337–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murakami T, Maki W, Cardones AR, Fang H, Tun Kyi A, Nestle FO et al. (2002) Expression of CXC chemokine receptor-4 enhances the pulmonary metastatic potential of murine B16 melanoma cells. Cancer Res 62: 7328–34 [PubMed] [Google Scholar]
- Nestle FO, Alijagic S, Gilliet M, Sun Y, Grabbe S, Dummer R et al. (1998) Vaccination of melanoma patients with peptide- or tumor lysate-pulsed dendritic cells. Nat Med 4: 328–32 [DOI] [PubMed] [Google Scholar]
- North RJ (1982) Cyclophosphamide-facilitated adoptive immunotherapy of an established tumor depends on elimination of tumor-induced suppressor T cells. J Exp Med 155: 1063–74 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulos CM, Wrzesinski C, Kaiser A, Hinrichs CS, Chieppa M, Cassard L et al. (2007) Microbial translocation augments the function of adoptively transferred self/tumor-specific CD8 T cells via TLR4 signaling. J Clin Invest 117: 2197–204 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phan GQ, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ et al. (2003) Cancer regression and autoimmunity induced by cytotoxic T lymphocyte-associated antigen 4 blockade in patients with metastatic melanoma. Proc Natl Acad Sci USA 100:8372–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pittet MJ, Valmori D, Dunbar PR, Speiser DE, Lienard D, Lejeune F et al. (1999) High frequencies of naive Melan-A/MART-1-specific CD8(+) T cells in a large proportion of human histocompatibility leukocyte antigen (HLA)-A2 individuals. J Exp Med 190: 705–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell DJ Jr, Dudley ME, Hogan KA, Wunderlich JR, Rosenberg SA (2006) Adoptive transfer of vaccine-induced peripheral blood mononuclear cells to patients with metastatic melanoma following lymphodepletion. J Immunol 177:6527–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ray CM, Kluk M, Grin CM, Grant-Kels JM (2005) Successful treatment of malignant melanoma in situ with topical 5% imiquimod cream. Int J Dermatol 44:428–34 [DOI] [PubMed] [Google Scholar]
- Ribas A, Camacho LH, Lopez-Berestein G, Pavlov D, Bulanhagui CA, Millham R et al. (2005) Antitumor activity in melanoma and anti-self responses in a phase I trial with the anti-cytotoxic T lymphocyte-associated antigen 4 monoclonal antibody CP-675,206. J Clin Oncol 23:8968–77 [DOI] [PubMed] [Google Scholar]
- Ridolfi R, Chiarion-Sileni V, Guida M, Romanini A, Labianca R, Freschi A et al. (2002) Cisplatin, dacarbazine with or without subcutaneous interleukin-2, and interferon alpha-2b in advanced melanoma outpatients: results from an Italian multicenter phase III randomized clinical trial. J Clin Oncol 20: 1600–7 [DOI] [PubMed] [Google Scholar]
- Rosenberg SA, Lotze MT, Muul LM, Leitman S, Chang AE, Ettinghausen SE et al. (1985a) Observations on the systemic administration of autologous lymphokine-activated killer cells and recombinant interleukin-2 to patients with metastatic cancer. N Engl J Med 313:1485–92 [DOI] [PubMed] [Google Scholar]
- Rosenberg SA, Mule JJ, Spiess PJ, Reichert CM, Schwarz SL (1985b) Regression of established pulmonary metastases and subcutaneous tumor mediated by the systemic administration of high-dose recombinant interleukin 2. J Exp Med 161:1169–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Yang JC, Restifo NP (2004) Cancer immunotherapy: moving beyond current vaccines. Nat Med 10:909–15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL et al. (1998a) Immunologic and therapeutic evaluation of a synthetic peptide vaccine for the treatment of patients with metastatic melanoma. Nat Med 4:321–7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM, Topalian SL et al. (1999) Prospective randomized trial of the treatment of patients with metastatic melanoma using chemotherapy with cisplatin, dacarbazine, and tamoxifen alone or in combination with interleukin-2 and interferon alfa-2b. J Clin Oncol 17:968–75 [DOI] [PubMed] [Google Scholar]
- Rosenberg SA, Yang JC, Sherry RM, Hwu P, Topalian SL, Schwartzentruber DJ et al. (2003) Inability to immunize patients with metastatic melanoma using plasmid DNA encoding the gp100 melanoma-melanocyte antigen. Hum Gene Ther 14:709–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenberg SA, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS, Parkinson DR et al. (1994a) Treatment of 283 consecutive patients with metastatic melanoma or renal cell cancer using high-dose bolus interleukin 2. JAMA 271:907–13 [PubMed] [Google Scholar]
- Rosenberg SA, Yannelli JR, Yang JC, Topalian SL, Schwartzentruber DJ, Weber JS et al. (1994b) Treatment of patients with metastatic melanoma with autologous tumor-infiltrating lymphocytes and interleukin 2. J Natl Cancer Inst 86:1159–66 [DOI] [PubMed] [Google Scholar]
- Rosenberg SA, Zhai Y, Yang JC, Schwartzentruber DJ, Hwu P, Marincola FM et al. (1998b) Immunizing patients with metastatic melanoma using recombinant adenoviruses encoding MART-1 or gp100 melanoma antigens. J Natl Cancer Inst 90:1894–900 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaguchi S (2005) Naturally arising Foxp3-expressing CD25+CD4+ regulatory T cells in immunological tolerance to self and nonself. Nat Immunol 6:345–52 [DOI] [PubMed] [Google Scholar]
- Sato E, Olson SH, Ahn J, Bundy B, Nishikawa H, Qian F et al. (2005) Intraepithelial CD8+ tumor-infiltrating lymphocytes and a high CD8+/regulatory T cell ratio are associated with favorable prognosis in ovarian cancer. Proc Natl Acad Sci USA 102:18538–43 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schadendorf D, Ugurel S, Schuler-Thurner B, Nestle FO, Enk A, Brocker EB et al. (2006) Dacarbazine (DTIC) versus vaccination with autologous peptide-pulsed dendritic cells (DC) in first-line treatment of patients with metastatic melanoma: a randomized phase III trial of the DC study group of the DeCOG. Ann Oncol 17:563–70 [DOI] [PubMed] [Google Scholar]
- Shanafelt AB, Lin Y, Shanafelt MC, Forte CP, Dubois-Stringfellow N, Carter C et al. (2000) A T-cell-selective interleukin 2 mutein exhibits potent antitumor activity and is well tolerated in vivo. Nat Biotechnol 18: 1197–202 [DOI] [PubMed] [Google Scholar]
- Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ et al. (2001) IFNgamma and lymphocytes prevent primary tumour development and shape tumour immuno-genicity. Nature 410:1107–11 [DOI] [PubMed] [Google Scholar]
- Slingluff CL Jr, Petroni GR, Yamshchikov GV, Barnd DL, Eastham S, Galavotti H et al. (2003) Clinical and immunologic results of a randomized phase II trial of vaccination using four melanoma peptides either administered in granulocyte-macrophage colony-stimulating factor in adjuvant or pulsed on dendritic cells. J Clin Oncol 21: 4016–26 [DOI] [PubMed] [Google Scholar]
- Sondak VK, Liu PY, Tuthill RJ, Kempf RA, Unger JM, Sosman JA et al. (2002) Adjuvant immunotherapy of resected, intermediate-thickness, node-negative melanoma with an allogeneic tumor vaccine: overall results of a randomized trial of the Southwest Oncology Group. J Clin Oncol 20:2058–66 [DOI] [PubMed] [Google Scholar]
- Speiser DE, Lienard D, Rufer N, Rubio-Godoy V, Rimoldi D, Lejeune F et al. (2005) Rapid and strong human CD8+ T cell responses to vaccination with peptide, IFA, and CpG oligodeoxynucleotide 7909. J Clin Invest 115:739–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinmann A, Funk JO, Schuler G, von den Driesch P (2000) Topical imiquimod treatment of a cutaneous melanoma metastasis. J Am Acad Dermatol 43:555–6 [PubMed] [Google Scholar]
- Tan JT, Ernst B, Kieper WC, LeRoy E, Sprent J, Surh CD (2002) Interleukin (IL)-15 and IL-7 jointly regulate homeostatic proliferation of memory phenotype CD8+ cells but are not required for memory phenotype CD4+ cells. J Exp Med 195:1523–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teft WA, Kirchhof MG, Madrenas J (2006) A molecular perspective of CTLA-4 function. Annu Rev Immunol 24:65–97 [DOI] [PubMed] [Google Scholar]
- Thurner B, Haendle I, Roder C, Dieckmann D, Keikavoussi P, Jonuleit H et al. (1999) Vaccination with mage-3A1 peptide-pulsed mature, monocyte-derived dendritic cells expands specific cytotoxic T cells and induces regression of some metastases in advanced stage IV melanoma. J Exp Med 190:1669–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Elsas A, Hurwitz AA, Allison JP (1999) Combination immunotherapy of B16 melanoma using anti-cytotoxic T lymphocyte-associated antigen 4 (CTLA-4) and granulocyte/macrophage colony-stimulating factor (GM-CSF)-producing vaccines induces rejection of subcutaneous and metastatic tumors accompanied by autoimmune depigmentation. J Exp Med 190:355–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Viguier M, Lemaitre F, Verola O, Cho MS, Gorochov G, Dubertret L et al. (2004) Foxp3 expressing CD4+CD25(high) regulatory T cells are overrepresented in human metastatic melanoma lymph nodes and inhibit the function of infiltrating T cells. J Immunol 173:1444–53 [DOI] [PubMed] [Google Scholar]
- Wheatley K, Ives N, Hancock B, Gore M, Eggermont A, Suciu S (2003) Does adjuvant interferon-alpha for high-risk melanoma provide a worthwhile benefit? A meta-analysis of the randomised trials. Cancer Treat Rev 29:241–52 [DOI] [PubMed] [Google Scholar]
- Yee C, Thompson JA, Byrd D, Riddell SR, Roche P, Celis E et al. (2002) Adoptive T cell therapy using antigen-specific CD8+ T cell clones for the treatment of patients with metastatic melanoma: in vivo persistence, migration, and antitumor effectof transferred T cells. Proc Natl Acad Sci USA 99:16168–73 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Conejo-Garcia JR, Katsaros D, Gimotty PA, Massobrio M, Regnani G et al. (2003) Intra-tumoral T cells, recurrence, and survival in epithelial ovarian cancer. N Engl J Med 348: 203–13 [DOI] [PubMed] [Google Scholar]
- Zhao Y, Parkhurst MR, Zheng Z, Cohen CJ, Riley JP, Gattinoni L et al. (2007) Extrathymic generation of tumor-specific T cells from genetically engineered human hematopoietic stem cells via Notch signaling. Cancer Res 67:2425–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zippelius A, Batard P, Rubio-Godoy V, Bioley G, Lienard D, Lejeune F et al. (2004) Effector function of human tumor-specific CD8 T cells in melanoma lesions: a state of local functional tolerance. Cancer Res 64: 2865–73 [DOI] [PubMed] [Google Scholar]