

Abstract
Biglycan, a ubiquitous proteoglycan, acts as a danger signal when released from the extracellular matrix. As such, biglycan triggers the synthesis and maturation of interleukin-1β (IL-1β) in a Toll-like receptor (TLR) 2-, TLR4-, and reactive oxygen species (ROS)-dependent manner. Here, we discovered that biglycan autonomously regulates the balance in IL-1β production in vitro and in vivo by modulating expression, activity and stability of NADPH oxidase (NOX) 1, 2 and 4 enzymes via different TLR pathways. In primary murine macrophages, biglycan triggered NOX1/4-mediated ROS generation, thereby enhancing IL-1β expression. Surprisingly, biglycan inhibited IL-1β due to enhancement of NOX2 synthesis and activation, by selectively interacting with TLR4. Synthesis of NOX2 was mediated by adaptor molecule Toll/IL-1R domain-containing adaptor inducing IFN-β (TRIF). Via myeloid differentiation primary response protein (MyD88) as well as Rac1 activation and Erk phosphorylation, biglycan triggered translocation of the cytosolic NOX2 subunit p47phox to the plasma membrane, an obligatory step for NOX2 activation. In contrast, by engaging TLR2, soluble biglycan stimulated the expression of heat shock protein (HSP) 70, which bound to NOX2, and consequently impaired the inhibitory function of NOX2 on IL-1β expression. Notably, a genetic background lacking biglycan reduced HSP70 expression, rescued the enhanced renal IL-1β production and improved kidney function of Nox2−/y mice in a model of renal ischemia reperfusion injury. Here, we provide a novel mechanism where the danger molecule biglycan influences NOX2 synthesis and activation via different TLR pathways, thereby regulating inflammation severity. Thus, selective inhibition of biglycan-TLR2 or biglycan-TLR4 signaling could be a novel therapeutic approach in ROS-mediated inflammatory diseases.
Keywords: Extracellular matrix, Inflammation, Reactive oxygen species (ROS), Small leucine-rich proteoglycan, Toll-like receptor
Introduction
Inflammation can be triggered by a broad spectrum of stimuli, ranging from external pathogenic factors to endogenous molecules such as damage-associated molecular patterns (DAMPs) [1]. One such molecule is the small leucine-rich proteoglycan (SLRP) biglycan, which carries one or two covalently linked glycosaminoglycan chains at its N-terminus [2]. Biglycan, together with its closest SLRP decorin, is involved in a variety of biological functions [3–10]. Biglycan is ubiquitously distributed and can be proteolytically released from the extracellular matrix upon tissue injury [11–13]. When soluble, biglycan can bind to TLR2/4 [14], thereby inducing the phosphorylation of extracellular signal-regulated kinases (Erk), p38 mitogen-activated protein kinase (MAPK) as well as nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) in a MyD88- and TRIF-dependent manner [15,16]. This process induces transcription and secretion of tumor necrosis factor α (TNF-α), chemokine (C–C motif) ligand (CCL) 2, CCL5, chemokine (C-X-C motif) ligand (CXCL) 1, CXCL2 or CXCL13 [11,14,15]. Various studies have clearly demonstrated, that overexpression of biglycan is associated with a number of pathological conditions, such as acute kidney failure, lupus nephritis, and diabetic nephropathy, suggesting a central role of biglycan in renal inflammation [11,15–18]. However, to date the exact molecular mechanisms of how the pro-inflammatory signaling of soluble biglycan might be regulated and inhibited in these pathologies remains elusive.
Previously, we demonstrated that soluble biglycan regulates the activation of pro-caspase-1 through the NOD-like receptor protein 3 (NLRP3) inflammasome and consequently the maturation of IL-1β [15,19]. Notably, this process is associated with enhanced ROS generation, implicating ROS in biglycan-mediated IL-1β production [19]. However, the source of ROS in this pro-inflammatory signaling event triggered by biglycan, remained unrevealed.
NOX enzymes are the major producers of ROS within immune cell phagosomes in response to inflammation [20], as they transfer electrons to reduce oxygen and thus produce superoxide [21]. The NOX family comprises seven members, NOX1 to NOX5, as well as the dual oxidases DUOX1 and DUOX2. NOX2, the prototype of these transmembrane proteins, requires the assembly of its subunits to gain catalytic activity. This process involves the association of p22phox with the catalytic subunit at the cell surface forming the heterodimer flavocytochromeb558 complex. Subsequently, the cytosolic p47phox is phosphorylated, leading to its translocation to the plasma membrane and interaction with p22phox. This results in the activation of NOX2 and consequently in ROS generation [22–26]. The expression of p22phox is crucial for NOX1–4 activity, but its association with the individual regulatory subunits is relevant only in the activation of NOX1 and NOX2 [27]. In contrast, NOX4 is constitutively active and generates hydrogen peroxide [28,29].
The production of ROS by NOX enzymes relies on tightly-regulated molecular mechanisms and even small imbalances often lead to pathological consequences. The regulation of ROS/NOX axis occurs at various levels, including transcription and translation as well as the formation of alternatively-spliced variants and protein degradation. Proper expression of NOX1 and NOX4 is critical for the development of inflammatory diseases such as liver and lung fibrosis or diabetic nephropathy, and genetic targeting or pharmacologic inhibition of these enzymes is beneficial for the clinical outcome [30–33]. On the other hand, deletion of NOX2 as well as mutations in its regulatory subunits cause chronic granulomatous disease, characterized by an excessive inflammatory response [34]. In the same line, loss of NOX2 in mice leads to increased levels of IL-1β in the early onset of arthritis leading to a more severe inflammatory phenotype, indicating a protective role of NOX2-generated ROS [35]. The stability of NOX2 is essential for ROS production and therefore for its function in inflammatory response. HSP70 has been suggested to bind and to regulate the stability of NOX2 by facilitating ubiquitination and proteasomal degradation [36].
Here we show a novel molecular mechanism where biglycan regulates the balance in IL-1β synthesis and maturation in macrophages by modulating expression, activity, and stability of NOX enzymes via different TLR-pathways. Biglycan triggers the expression and the maturation of IL-1β by inducing the activation of NOX1/4 in macrophages. Moreover, biglycan directly induces synthesis of Nox2 mRNA via TLR4/TRIF and its activation in a TLR4/MyD88-dependent manner, thereby attenuating expression of the pro-inflammatory cytokine IL-1β. We further show that, by engaging TLR2 and triggering Hsp70 mRNA expression, biglycan impairs the function of NOX2 and thus regulates the balance in IL-1β expression. Finally, under pathological conditions, such as in renal ischemia reperfusion injury (IRI), lack of biglycan in Nox2−/y mice, protects the mutant mice from hyper-inflammation and massive macrophage infiltration and consequently improves renal function. Thus, inhibition of biglycan-mediated signaling might be a potential therapeutic strategy in renal inflammation evoked by impaired NOX2 function.
Results
Biglycan-induced IL-1β production in macrophages is regulated by NOX-derived ROS
We have previously linked biglycan-induced IL-1β production in macrophages to ROS-dependent NLRP3 inflammasome activation [19], and thus postulated that NOX-derived ROS might be involved in this process. Therefore, murine peritoneal macrophages isolated from wild-type (WT) C57BL/6 mice were stimulated with soluble biglycan in the presence of the ROS-inhibitor diphenyleneiodonium (DPI) [37] and the NOX inhibitor VAS2870 [38] (Fig. 1A). After 16 h of continuous stimulation with biglycan, we found markedly increased levels of secreted mature IL-1β in the conditioned media (Fig. 1A). Addition of DPI significantly attenuated secretion of mature IL-1β, and this was totally abolished by VAS2870, indicating a critical role for NADPH oxidases in biglycan-induced IL-1β production (Fig. 1A). Western blot analysis revealed increased levels of both pro- and mature IL-1β after stimulation with biglycan. Of note, application of VAS2870 was able to inhibit about 50% of pro-IL-1β gene and protein expression while the maturation of the cytokine was fully abolished by the NOX inhibitor (Fig. 1B to E). Thus, NOX enzymes are required for biglycan-mediated IL-1β expression and maturation.
Fig. 1.

Biglycan-induced IL-1β expression and maturation in macrophages is regulated by NAPDH oxidase-derived ROS. (A) ELISA for mature IL-1β in the cell culture media from wild-type macrophages pre-treated for 1 h with DPI (0.5 μM) or VAS2870 (5 μM), followed by 16 h biglycan (4 μg/ml) stimulation; n = 5. (B) Western blot analysis of pro- and mature IL-1β in macrophages after 1 h pre-incubation with VAS2870 and stimulation with biglycan for 6 h for pro-IL-1β and 16 h for mature IL-1β. β-actin served as loading control. (C) Quantification of pro-IL-1β normalized to β-actin and given as fold induction to untreated control from 3 independent experiments. (D) Quantification of mature IL-1β normalized to β-actin and given as fold induction to untreated control from 3 independent experiments. (E) Quantitative RT-PCR for II1β mRNA expression in primary murine macrophages after 6 h stimulation with soluble biglycan and 1 h pre-incubation with VAS2870, normalized to Gapdh and given as fold induction over untreated controls; n = 5. (A, C–E) Data are given as means ± S.D.; *P < 0.05.
Biglycan-mediated IL-1β production in macrophages is differentially regulated by NOX1/4 and NOX2
Next, we identified the members of the NOX family specifically involved in the regulation of biglycan-induced IL-1β expression and maturation. As NOX1, NOX2 and NOX4 are the major sources of ROS, we isolated peritoneal macrophages from Nox1−/y, No2−/y and Nox4−/− mice and stimulated the primary cultures with soluble biglycan. Both, Nox1−/y and Nox4−/− macrophages displayed a 2-fold decrease in II1β synthesis as compared to WT, while it was about 4-fold upregulated in Nox2−/y macrophages after stimulation with biglycan (Fig. 2A). The same pattern was observed for mature IL-1β as measured by ELISA (Fig. 2B). Interestingly, biglycan failed to induce the release of mature IL-1β in NOX1-deficient macrophages, indicating a crucial role for NOX1 in the regulation of biglycan-mediated synthesis and maturation of IL-1β. This effect was further confirmed using specific inhibitors for NOX1 and NOX1/4, ML-171 [39] and GKT137831 [40], respectively. The application of ML-171 as well as GKT137831 on biglycan-treated macrophages led to an impaired IL-1β release (Fig. 2C and D). On the other hand, administration of the peptidic NOX2 inhibitor Nox2ds-tat [41] on primary WT macrophages resulted in significantly increased IL-1β production after stimulation with biglycan (Fig. 2C and D). These results demonstrate that NOX1/4 and NOX2 have opposite effects on biglycan-induced IL-1β expression and secretion in macrophages.
Fig. 2.

Biglycan-induced IL-1β production in macrophages is differentially regulated by NOX1/4 and NOX2. (A) Quantitative RT-PCR for II1β mRNA expression in macrophages isolated from wild-type (WT), Nox1−/y, Nox2−/y and Nox4−/− mice before and after stimulation with biglycan for 6 h (4 μg/ml). (B) ELISA for mature IL-1β in cell culture media from macrophages isolated from WT, Nox1−/y, Nox2−/y and Nox4−/− mice before and after stimulation with biglycan for 16 h. (C) Quantitative RT-PCR for II1β mRNA expression in WT macrophages after 1 h pre-incubation with NOX1 inhibitor ML-171 (10 nM), NOX2 inhibitor Nox2ds-tat (40 μM) or NOX1/4 inhibitor GKT137831 (200 μM), with or without biglycan treatment for 6 h; scrambled Nox2ds-tat peptide (40 μM) was used as respective control. (A, C) mRNA expression was normalized to Gapdh and is given as fold induction compared to untreated or scrambled Nox2ds-tat WT controls. (D) ELISA for mature IL-1β in cell culture media of WT macrophages pre-treated with the inhibitors ML-171 (10 nM), Nox2ds-tat (40 μM) or GKT137831 (200 μM), with or without biglycan treatment for 16 h. (A–D) n = 5; data are given as means ± S.D.; *P < 0.05.
Biglycan specifically induces NOX2 expression via TLR4/TRIF-signaling
Next, we investigated whether biglycan induces the expression of the NOX catalytic subunits Nox1-4, and Duox1–2 by quantitative RT-PCR. While we detected a 2-fold increase of Nox2 mRNA upon stimulation with biglycan, the expression of the other NOX subunits was not affected (Fig. 3A). Western blot analysis confirmed that biglycan can induce NOX2 expression not only at the mRNA, but also at the protein level (Fig. 3B).
Fig. 3.

Biglycan specifically induces NOX2 expression in a TLR4/TRIF-dependent manner. (A) Quantitative RT-PCR for Nox1, Nox2, Nox3, Nox4, Duox1 and Duox2 mRNA expression in WT macrophages after stimulation with biglycan for 6 h (4 μg/ml), normalized to Gapdh and given as fold induction to untreated control; n = 5. (B) Western blot analysis for NOX2 expression in WT and Nox2−/y macrophages after stimulation with biglycan for 16 h, NOX2-deficient macrophages were used as control for the specificity of the NOX2 antibody, and β-actin served as loading control. (C, D) Quantitative RT-PCR for Nox2 mRNA expression in (C) WT, Tlr2−/−, Tlr4−/− and Tlr2−/−/Tlr4-m macrophages and (D) WT and Trif−/− macrophages pre-incubated for 2 h with a MyD88 inhibitor (50 μM). Macrophages were stimulated with biglycan for 6 h. (C, D) mRNA expression normalized to Gapdh and given as fold induction to untreated WT control. (A, C, D) n = 5; data are given as means ± S.D.; *P < 0.05.
As soluble biglycan signals via TLR2 and TLR4 [14], we investigated the receptor involved in biglycan-mediated NOX2 expression. Interestingly, quantitative RT-PCR revealed that Nox2 mRNA expression was only induced in WT and Tlr2−/− but not in Tlr4−/− and Tlr2−/−/Tlr4-m macrophages after stimulation with biglycan (Fig. 3C). Hence, biglycan regulates the expression of Nox2 only through TLR4.
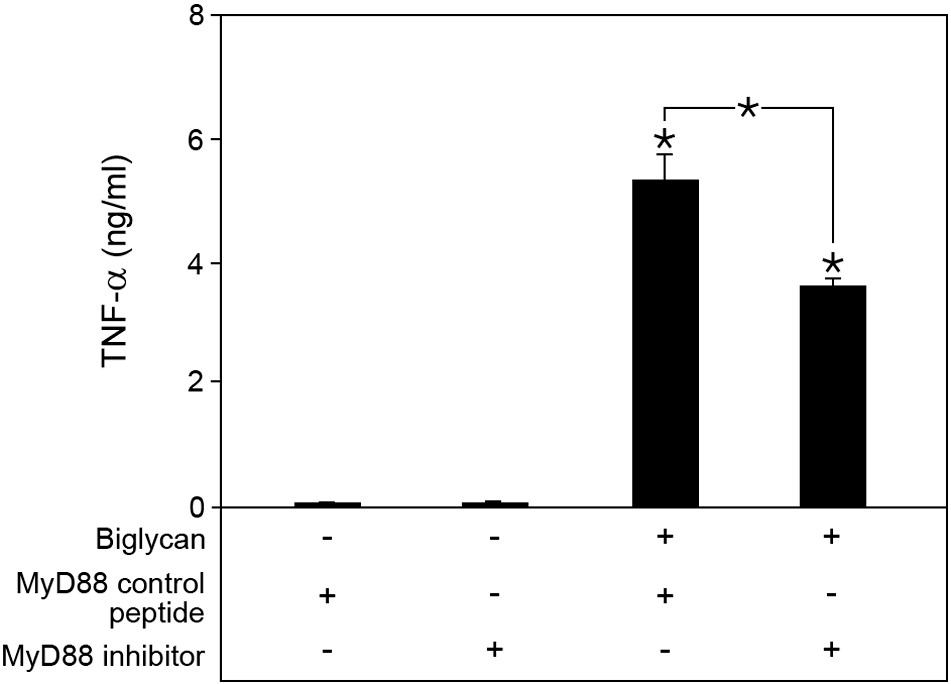
To investigate the downstream mechanisms, we next examined the involvement of the TLR4 adapter molecules MyD88 and TRIF [42]. While the inhibition of MyD88 had no effect on the biglycan-induced Nox2 mRNA expression, no induction was detected in Trif−/− macrophages (Fig. 3D), demonstrating that biglycan-mediated Nox2 expression is TLR4/TRIF-dependent. The successful MyD88 inhibition was confirmed by reduced TNF-α release upon stimulation with biglycan (Suppl. Fig. S1).
Biglycan promotes p47phox translocation to the NOX2 subunit p22phox via a TLR4/MyD88-dependent mechanism
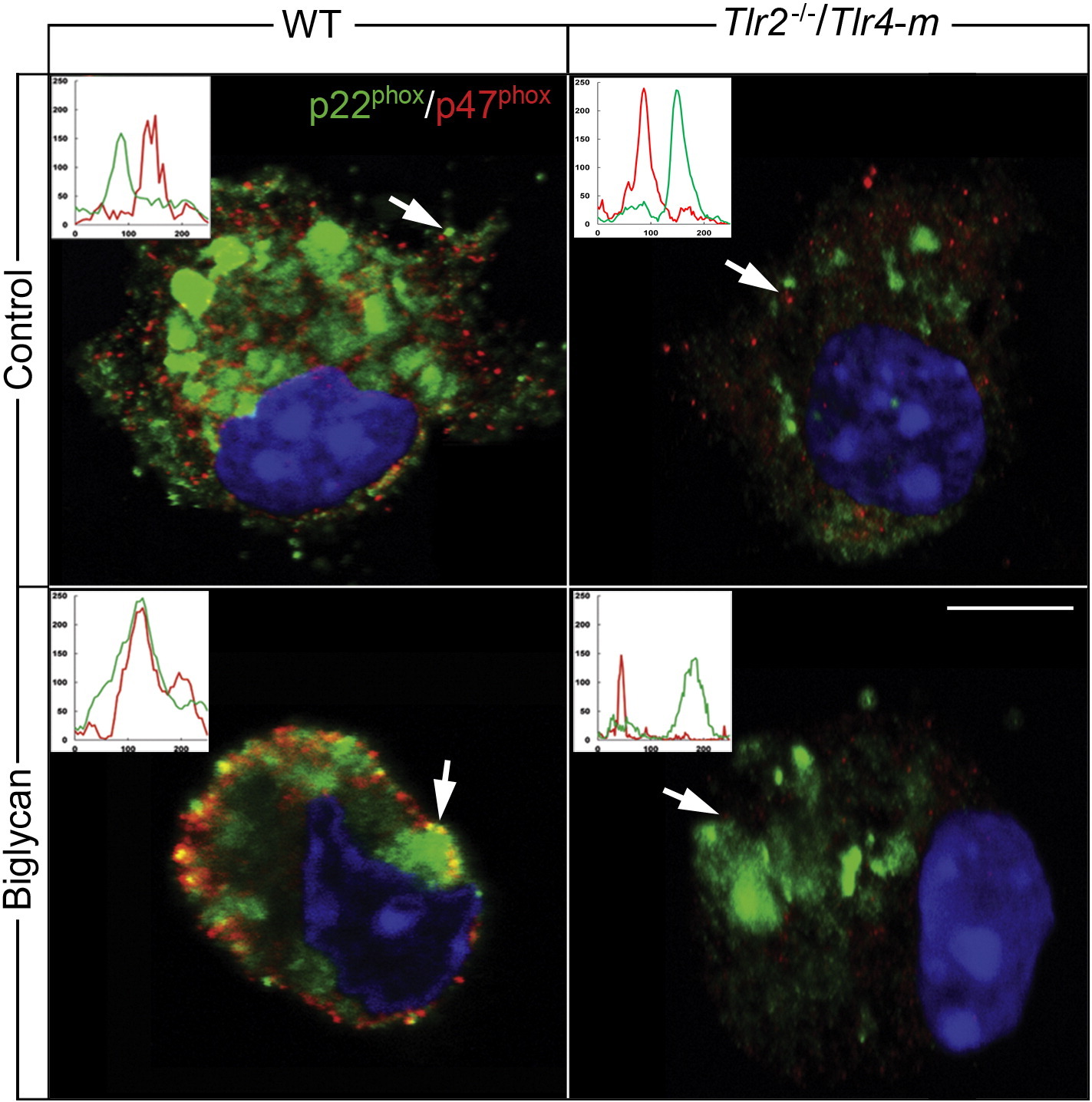
The formation of the NOX2 complex is a crucial step for its activation, and consequently for ROS production. Confocal analysis of WT macrophages revealed that biglycan promoted a time-dependent redistribution of the intracellular regulatory subunit p47phox from the cytosolic compartment to the plasma membrane (Fig. 4A, upper panel). Efficient p47phox translocation was evident as early as 20 min and this further increased at 40 min. The specificity of our anti-p47phox was validated by utilization of p47phoxB/ macrophages, which showed a total lack of immunoreactive p47phox (Fig. 4A, lower panel). Additionally, membrane translocation of p47phox was corroborated by co-localization of p47phox with the macrophage-specific surface marker F4/80 (Fig. 4B). Quantification of the confocal images showed a significant co-localization index evoked by biglycan (Fig. 4C). Consistently, biglycan did not induce translocation of p47phox in Tlr4−/− and Tlr2−/−/Tlr4-m macrophages (Fig. 4B and C), emphasizing the TLR4 dependency of biglycan-evoked NOX2 activation. Importantly, biglycan was not only able to mediate p47phox membrane shift but also its association with the NOX2 subunit p22phox after a 40 min treatment (Suppl. Fig. S2). A similar redistribution was also observed after incubation of WT macrophages with phorbol 12-myristate 13-acetate (PMA), which induces ROS generation (data not shown). Absence of p47phox membrane occupancy was found when NOX2 activity was depleted in Nox2−/y macrophages followed by stimulation with biglycan for 40 min (data not shown).
Fig. 4.

Biglycan triggers p47phox translocation in a TLR4-dependent manner. (A) Confocal images of p47phox (red) in macrophages isolated from WT mice either untreated or stimulated with biglycan (4 μg/ml) for 10, 20 and 40 min. White arrows indicate localization of p47phox (red) to the plasma membrane. Nuclei were stained with DAPI (blue). (B) Confocal images of F4/80 (green) and p47phox (red) in macrophages isolated from WT, Tlr2−/−, Tlr4−/− and Tlr2−/−/Tlr4-m mice either untreated or after 40 min treatment with biglycan (4 μg/ml). Inserts in the confocal images represent line-scanned profiles, while the white arrows indicate the distance within which the scans were generated. Bar = 10 μm; *P < 0.05. (C) Quantification of p47phox co-localization with F4/80 from (B) given as dot plot for the mean correlation index (Icorr) ± S.D. of at least 10 cells per condition.
The underlying mechanism was further investigated in WT and Trif−/− macrophages and by addition of a MyD88 inhibitor. Confirming the solely TLR4/MyD88-dependency of biglycan-mediated p47phox translocation, the MyD88 inhibitor blocked redistribution of p47phox to the cell membrane, while in Trif−/− macrophages p47phox translocation was comparable to WT (Fig. 5A and B). These data emphasize that activation of NOX2 is mediated by biglycan in a TLR4/MyD88-dependent manner by inducing the translocation of p47phox and consequently the association with the membrane-bound p22phox subunit.
Fig. 5.

Biglycan-dependent translocation of p47phox is mediated by MyD88. (A) Confocal images of F4/80 (green) and p47phox (red) in macrophages isolated from WT and Trif−/− mice upon 2 h pre-incubation with MyD88 inhibitor (50 μM) or the scrambled MyD88 inhibitor control, and treatment with biglycan for 40 min (4 μg/ml). Inserts in the confocal images represent line-scanned profiles, while the white arrows indicate the distance within which the scans were generated. Bar = 10 μm. (B) Quantification of p47phox co-localization with F4/80 from (A) given as dot plot for the mean correlation index (Icorr) ± S.D. of at least 10 cells per condition; *P < 0.05.
Biglycan-evoked p47phox redistribution is Erk and Rac1 dependent
The translocation of p47phox to the membrane and the formation of active NOX2 is induced by phosphorylation of this subunit [43,44]. To identify the intracellular kinase responsible for phosphorylating p47phox in response to biglycan, we screened a range of protein kinase and GTP-binding protein inhibitors (Fig. 6). In WT macrophages, biglycan failed to induce translocation of the p47phox subunit after application of either Rac1 or MEK1/2 inhibitors (Fig. 6A and B). In contrast, other kinase inhibitors, p38 MAPK as well as protein kinase C (PKC) inhibitors had no effect on biglycan-mediated p47phox redistribution, and thus NOX2 activation (Fig. 6A and B). Moreover, Akt and phosphatidylinositol 3-kinase (PI3K) inhibitors were also ineffective (data not shown). Thus, biglycan-induced redistribution of p47phox is mediated by Rac1 and Erk activation.
Fig. 6.

Biglycan-mediated NOX2 activation is dependent on Erk and Rac1. (A) Confocal images of F4/80 (green) and p47phox (red) in WT macrophages with or without biglycan treatment for 40 min, after 1 h treatment with Rac1 inhibitor (50 μM), MEK1/2 inhibitor (10 μM), p38 MAPK inhibitor (10 μM), or PKC inhibitor (20 nM). Inserts in the confocal images represent line-scanned profiles, while the white arrows indicate the distance within which the scans were generated. Bar = 10 μm. (B) Quantification of p47phox co-localization with F4/80 from (A) given as dot plot for the mean correlation index (Icorr) ± S.D. of at least 10 cells per condition; *P < 0.05.
Biglycan accentuates the inflammatory phenotype of NOX2-deficient macrophages
Impaired NOX2 activity as a result of reduced p47phox translocation leads to severe consequences, such as hyper-inflammation observed in patients with chronic granulomatous disease [34,45]. Consistently, inhibition of p47phox translocation upon pathogen infection is associated with elevated expression of pro-inflammatory cytokines [46]. Therefore, activation of NOX2 and consequently NOX2-mediated ROS production is crucial for the balance in the pro-inflammatory response. To uncover the role of NOX2 in biglycan-mediated IL-1β maturation, we investigated the role of the upstream mediators caspase-1 and NLRP3 in NOX2-deficient macrophages. In these mutant cells, Casp1 and Nlrp3 mRNA levels were constitutively elevated vis-a-vis WT controls and further enhanced by biglycan (Fig. 7A and B). Thus, NOX2 inhibits the maturation of biglycan-induced IL-1β by regulating the expression of NLRP3 and caspase-1 in physiological and in pathological conditions when soluble biglycan is overexpressed.
Fig. 7.

Biglycan accentuates the inflammatory phenotype of NOX2-deficient macrophages. Quantitative RT-PCR for (A) Caspase-1, (B) Nlrp3, and (C) Ccl2 mRNA expression in WT and Nox2−/y macrophages before and after 6 h stimulation with biglycan (4 μg/ml). (D) Ccl2 mRNA expression in WT macrophages pre-incubated with a NOX2 inhibitor (40 μM) or scrambled Nox2ds-tat control for 1 h, and subsequently stimulated for 6 h with biglycan (4 μg/ml). (A–D) mRNA expression was normalized to Gapdh and given as fold induction to untreated WT control; n = 5. (E, G, I) Western blot analysis for (E) Rac1 activation, (G) Erk phosphorylation, and (I) p38 phosphorylation in WT and Nox2−/y macrophages before and after stimulation with biglycan (4 μg/ml) for 5, 10 and 30 min. (F, H, J) Quantification of (E, G, I) respectively, normalized to (E) total Rac1, (G) total Erk and (I) total p38; n = 3; data are given as means ± S.D.; *P < 0.05.
The hyper-inflammatory phenotype of NOX2-deficient macrophages was confirmed by the increased constitutive Ccl2 mRNA expression in these cells, which was similarly enhanced by biglycan (Fig 7C). Of note, Ccl2 mRNA expression was also up-regulated by biglycan in the presence of the specific NOX2 inhibitor in WT macrophages, confirming that the hyper-inflammatory phenotype in Nox2−/y macrophages is due to the anti-inflammatory role of biglycan-induced NOX2 (Fig. 7D).
Collectively, these data suggest a negative regulatory role for NOX2 in biglycan-mediated NLRP3/caspase-1 inflammasome induction and pro-inflammatory cytokine expression.
To unravel upstream mechanisms involved in the hyper-inflammation of Nox2−/y macrophages, we analyzed the activation and phosphorylation of Rac1, Erk and p38. By pull-down assays, we observed Rac1 activation in WT macrophages already at 5, and 30 min after stimulation with biglycan (Fig. 7E and F). Additionally, Western blot analysis revealed phosphorylation of Erk (Fig. 7G and H) and p38 MAPK (Fig. 7I and J) after 15 and 30 min stimulation with biglycan. Interestingly, in NOX2-deficient macrophages Rac1 activation and Erk phosphorylation were already constitutively increased under resting conditions. Furthermore, upon biglycan stimulation Erk phosphorylation was more enhanced in NOX2-deficient macrophages compared to WT controls, yet no difference was detected in p38 phosphorylation in these cells. This indicates that biglycan-mediated NOX2 activation can selectively signal through the MEK/Erk pathway, thereby attenuating the expression of pro-inflammatory cytokines.
Biglycan evokes HSP70 expression, alters NOX2 activity and promotes IL-1β production
A recent study has linked NOX2 stabilization to the HSP70 co-chaperone system [36]. Enhanced HSP70 expression leads to its binding to NOX2, followed by ubiquitination and proteasomal degradation of the oxidase. Thus, we hypothesized that biglycan could bypass the inhibitory effect of NOX2 in IL-1β production by inducing the expression of HSP70. Quantitative RT-PCR revealed that biglycan induced Hsp70 mRNA expression in a time-dependent manner that peaked between 1 and 2 h, then declined (Fig. 8A). Interestingly, Hsp70 mRNA expression was solely triggered through the TLR2 pathway, as the time-dependent induction was only present in Tlr4−/−, but not in Tlr2−/− and Tlr2−/−/Tlr4-m macrophages (Fig. 8A). Consequently, biglycan-evoked Hsp70 expression correlated with increased II1β mRNA expression at early time points (Fig. 8B), indicating this process is an early pro-inflammatory response.
Fig. 8.

Biglycan induces the expression of HSP70 which subsequently binds to NOX2 and potentiates biglycan-mediated IL-1β production. (A) Quantitative RT-PCR for Hsp70 mRNA expression in WT, Tlr2−/−, Tlr4−/− and Tlr2−/−/Tlr4-m macrophages after biglycan (4 μg/ml) stimulation for 1, 2, 4, and 6 h, normalized to Gapdh and given as fold induction to untreated control; n = 5. (B) Quantitative RT-PCR for II1β mRNA expression in WT macrophages after stimulation with biglycan (4 μg/ml) for 1, 2, 4, and 6 h, normalized to Gapdh and given as fold induction to untreated control; n = 5. (C) Co-immunoprecipitation of NOX2 with HSP70 from lysates of WT macrophages after stimulation with biglycan for 1 h and treatment with HSP70 inhibitor PES (200 μM); n = 3. (D) ELISA for mature IL-1β in cell culture media from WT macrophages before and after 16 h biglycan and PES stimulation; n = 5. All data are given as means ± S.D.; *P < 0.05.
Co-immunoprecipitation studies of NOX2 and HSP70 confirmed that biglycan induced an enhanced binding of these two proteins (Fig. 8C). Addition of the HSP70 inhibitor 2-phenylethyne-sulfonamide (PES) reduced the binding of HSP70 to NOX2 after stimulation with biglycan, thereby stabilizing NOX2 activity and inhibiting the biglycan-mediated pro-inflammatory response. Consequently, secretion of mature IL-1β after 16 h of biglycan-stimulation was decreased by PES (Fig. 8D). These data demonstrate that biglycan regulates the balance of IL-1β production, and thus pro-inflammatory signaling in macrophages, and that this biological process requires specific Toll-like receptors to activate and simultaneously inhibit NOX2.
Lack of biglycan in NOX2-deficient mice rescues the hyper-inflammatory phenotype in renal ischemia reperfusion
We have previously shown that biglycan exacerbates the pathophysiology of renal ischemia reperfusion injury in a TLR2/4-dependent manner [11]. Given the fact that impaired NOX2 activation has pathologic consequences [34,45], and that biglycan triggers activation of NOX2, we postulated that genetic ablation of biglycan in NOX2 knockout mice might be beneficial for the pathophysiology. To this end we generated Nox2−/y/Bgn−/y mice (Fig. 9A). Nox2−/y/Bgn−/y mice have unchanged body weight, fertility, and life span compared to the parental genotypes. To investigate the regulatory role of NOX2 on biglycan-mediated renal inflammation in IRI, we determined mRNA expression of II1β and Ccl2, protein expression of HSP70 as well as renal macrophage infiltration in WT, Bgn−/y, Nox2−/y and Nox2−/y/Bgn−/y mice. In reperfused kidneys of NOX2-deficient mice II1β mRNA expression was significantly higher than in WT kidneys, while biglycan-deficient mice had lower II1β levels after IRI (Fig. 9B). Interestingly, ablation of biglycan in NOX2-deficient mice rescued the hyper-inflammatory phenotype by impeding the expression of II1β (Fig. 9B). Similarly to II1β, markedly elevated Ccl2 mRNA expression was observed in NOX2-deficient mice after IRI, while biglycan-deficient mice expressed significantly lower levels compared to WT mice (Fig. 9C). Of note, the knockout of biglycan in NOX2-deficient mice resulted in reduced Ccl2 expression after IRI, comparable to that in Bgn−/y mice (Fig. 9C), and was therefore protective in renal inflammation.
Fig. 9.

Genetic ablation of biglycan in Nox2−/y mice rescues the hyper-inflammatory phenotype in renal ischemia reperfusion injury. (A) Schematic drawing of the generation of Nox2−/y/Bgn−/y mice and the control PCR. (B, C) Quantitative RT-PCR of (B) II1β and (C) Ccl2 mRNA expression in sham-operated (Control) kidneys or kidneys 20 h after ischemia reperfusion injury (IRI) in WT, Bgn−/y, Nox2−/y and Nox2−/y/Bgn−/y mice, normalized to Gapdh and given as fold induction to WT control mice; n = 7. (D) Western blot analysis for HSP70 expression in sham-operated control or 20 h reperfused IRI kidneys of WT, Bgn−/y, Nox2−/y and Nox2−/y/Bgn−/y mice, β-actin served as loading control; n = 3. (E) Immunohistochemical staining for F4/80 (brown) in sham-operated control or 20 h reperfused IRI kidneys of WT, Bgn−/y, Nox2−/y and Nox2−/y/Bgn−/y mice. Bar = 50 μm. (F) Quantification of F4/80+ cells in (E) given as cell count per high-power field (HPF); n = 7. (G) Serum creatinine levels from sham-operated or 20 h post IRI in WT, Bgn−/y, Nox2−/y and Nox2−/y/Bgn−/y mice. Data are given as means ± S.D.; *P < 0.05.
As biglycan regulates the in vitro expression of IL-1β through the induction of HSP70, we tested whether this mechanism would be also operational in vivo. HSP70 protein expression was markedly decreased in both Bgn−/y and Nox2−/y/Bgn−/y mice; however, in NOX2-deficient mice, HSP70 expression was comparable to that in WT mice (Fig. 9D), confirming that biglycan triggers HSP70 expression in vivo. In agreement with previous studies [47], 20 h of IRI did not result in enhanced HSP70 expression (Fig. 9D).
Infiltration of immune cells is an important contributor to ischemia reperfusion injury [48]. Since Ccl2 mRNA expression was increased in Nox2−/y mice after IRI, we analyzed macrophage infiltration into the kidney 20 h after ischemia reperfusion by immunohistochemistry. As expected, all analyzed genotypes displayed an increased macrophage infiltration 20 h after IRI compared to control (Fig. 9E and F). Interestingly, kidneys of Nox2−/y mice displayed a significantly higher constitutive macrophage infiltration compared to WT mice, which was absent in Nox2−/y/Bgn−/y kidneys (Fig. 9E and F). Consistent with the renal expression of the chemoattractant Ccl2 in Bgn−/y mice after IRI (Fig. 9C), macrophage infiltration was significantly lower in these kidneys compared to those of WT (Fig. 9E and F). On the other hand, deletion of Nox2 resulted in a massive macrophage infiltration after IRI, which could be rescued in Nox2−/y/Bgn−/y kidneys. Additionally, serum creatinine levels reflected the hyper-inflammatory phenotype of NOX2-deficient mice (Fig. 9G). While Nox2−/y mice had significantly higher serum creatinine level after IRI compared to WT mice, biglycan-deficient mice were protected from this increase (Fig. 9G). Additionally, biglycan-knockout in Nox2−/y mice significantly reduced the serum creatinine level after IRI, indicating that inhibition of biglycan might be beneficial to the clinical outcome in sterile inflammatory kidney disease.
Discussion
In this study, we describe a novel concept whereby the danger signal biglycan can either promote or self-limit inflammation by specifically targeting a single TLR and diverse downstream signaling pathways involving various NOX family members. A major biological effect of this complex interplay is the biglycan-triggered synthesis and maturation of IL-1β. A working model of our findings is provided in Fig. 10.
Fig. 10.

Schematic drawing of biglycan-mediated balance in IL-1β production in macrophages through regulation of NOX enzymes via different TLR-pathways. Soluble biglycan triggers the expression and maturation of IL-1β in macrophages through TLR2/4 in a NOX1/4-dependent manner. Moreover, biglycan directly induces the expression of Nox2 mRNA via TLR4/TRIF and the activation of NOX2 in a TLR4/MyD88-dependent manner, thereby attenuating the expression of the pro-inflammatory cytokine IL-1β. By binding to TLR4/MyD88 soluble biglycan activates Rac1 and induces Erk phosphorylation, which promote the translocation of the cytosolic NOX2 subunit p47phox to the membrane bound p22phox, thus activating NOX2. In turn, active NOX2 inhibits biglycan-mediated expression of IL-1β. In contrast, by engaging TLR2, soluble biglycan triggers the expression of HSP70, which binds to NOX2, and consequently impairs the inhibitory function of NOX2 on biglycan-mediated IL-1β expression and maturation.
We show for the first time how soluble biglycan autonomously regulates the balance in IL-1β production in vitro and in vivo by modulating expression, activity and stability of NOX enzymes via different TLR-pathways. We further demonstrate that in primary murine macrophages soluble biglycan not only induces a pro-inflammatory response via NOX1/4, but it can also inhibit the synthesis and maturation of IL-1β by triggering NOX2 activation (Fig. 10). Additionally, biglycan directly induces Nox2 mRNA expression via TLR4/TRIF, while the activation of this oxidase is TLR4/MyD88-dependent. Notably, by binding to TLR4/MyD88, soluble biglycan activates Rac1 and induces Erk phospho-rylation, which promote the translocation of the cytosolic NOX2 subunit p47phox to the plasma membrane. The latter binds p22phox and thus activates NOX2 [49]. In turn, active NOX2 inhibits biglycan-mediated expression of IL-1β (Fig. 10). In contrast, by engaging TLR2, soluble biglycan triggers the expression of HSP70, which binds to NOX2, and consequently impairs the inhibitory function of NOX2 on IL-1β expression (Fig. 10). These in vitro studies were corroborated by robust evidence utilizing a genetic background lacking endogenous biglycan. Lack of biglycan in Nox2−/y mice protects from hyper-inflammation as well as from massive macrophage infiltration after renal ischemic-reperfusion injury and consequently improves renal function.
It is well established that ROS production is involved in the control of IL-1β synthesis and secretion [50]. However, the exact stimuli and mechanisms in this redox-regulated IL-1β expression remain elusive [50,51]. In macrophages, biglycan-evoked IL-1β production is associated with ROS-dependent NLRP3 inflammasome activation [15,19]. In this study we discover that NOX enzymes are the major source of ROS in biglycan-mediated IL-1β expression and maturation. When ROS is inhibited in primary murine macrophages, biglycan-mediated IL-1β expression and maturation are concurrently suppressed. VAS2870, which inhibits ROS generation by NOX enzymes [38], fully abolishes this process, indicating that NOX enzymes are involved in biglycan-mediated IL-1β expression and maturation. Moreover, primary macrophages isolated from Nox1−/y, Nox2−/y, and Nox4−/− mice and application of specific NOX1/4 and NOX2 inhibitors reveal opposing effects for NOX1/4 and NOX2 in biglycan-mediated pro-inflammatory signaling. NOX1- and NOX4-derived ROS is implicated in the development of inflammatory diseases and mediates inflammation as well as cell death in macrophages [52–55]. Therefore, it is not surprising that genetic ablation of NOX1 and NOX4 or inhibition of these enzymes in primary murine macrophages results in a decreased biglycan-mediated IL-1β production. Of note, Nox4 expression in murine macrophages is not detectible by quantitative RT-PCR, implying biglycan-mediated IL-1β production is mainly dependent on NOX1. In contrast to NOX1 and NOX4, loss of NOX2 significantly increases the release of the pro-inflammatory cytokine IL-1β after stimulation with biglycan. This is in agreement with the previous findings that NOX2 deficiency in mice leads to increased levels of IL-1β in the early onset of arthritis and therefore to a more severe inflammatory phenotype [35]. Moreover, NOX2-derived ROS is essential for the phagosome function and it has been considered as a barrier of sustained inflammation [56–58]. Hence, we hypothesize that this protective role of NOX2-generated ROS in inflammation and IL-1β production could be regulated by biglycan.
Interestingly, Nox2 mRNA expression is induced by biglycan in a TLR4/TRIF-dependent manner, while NOX2 activation [49] is triggered by biglycan through TLR4/MyD88, by promoting the translocation of the cytosolic subunit p47phox to the plasma membrane (Fig. 10). To date, various pathogenic agonists of TLR2 [59] and TLR4 [60,61] can induce NOX2 activation by triggering phosphorylation of p47phox. In addition, activation of TLR adapter molecules, such as MyD88 and TRIF, is crucial to this process [62,63]. Of note, ROS stimuli, such as PMA, induce p47phox phosphorylation [64–69], and facilitate p47phox translocation from the cytosol to the plasma membrane in a TLR4/MyD88-dependent manner. Our present findings posit soluble biglycan as the endogenous danger signal that differentially induces and regulates NOX2-dependent pro-inflammatory signaling. We clearly demonstrate that binding of soluble biglycan to TLR4/MyD88 leads to activation of Rac1 and Erk phosphorylation, which both promote the redistribution of p47phox to the plasma membrane. Upon translocation, p47phox binds p22phox and thus activates NOX2 [49]. Our data suggest that ROS produced by active NOX2 inhibits biglycan-mediated expression of IL-1β. Therefore, biglycan not only triggers the pro-inflammatory response, but it also autonomously controls the fine balance of pro-inflammatory cytokine production by modulating the expression and activity of NOX2, albeit through different pathways.
The activity of the NADPH oxidase NOX2 is tightly-regulated [49]. Impaired NOX2 activity due to reduced p47phox translocation can cause pathological consequences such as the hyper-inflammatory phenotype of patients with chronic granulomatous disease [34,45,46]. Interestingly, we could show that biglycan accentuates the hyper-inflammatory phenotype in Nox2−/y macrophages or after application of the specific NOX2 inhibitor. This is associated with increased mRNA expression of Nlrp3 as well as the IL-1β converting enzyme caspase-1, hyper-phosphorylation of Erk, and Rac1 activation. Additionally, inhibition of NOX2-generated ROS increases biglycan-mediated IL-1β secretion. Thus, biglycan acts as a crucial regulator of IL-1β synthesis and maturation promoting inflammation via NOX1/4-mediated ROS production and self-limiting IL-1β synthesis by inducing NOX2. It is likely that the imbalance of this biglycan-mediated process, i.e., loss of NOX2 activity, could lead to severe pathological consequences.
There is growing clinical evidence that alterations in NOX2 activity lead to enhanced inflammation [34,35,45]. As a consequence, the lack of functional NOX2 leads to chronic granulomatous disease and patients with this condition are not only prone to serious infections, but they also exhibit chronic inflammatory conditions [34,45]. However, the role of biglycan as a potential trigger of these disorders has not been addressed yet. It is conceivable that in pathological conditions associated with enhanced levels of soluble biglycan [11,14–16,19], biglycan-mediated NOX2 expression and activation could provide a protective mechanism to limit IL-1β synthesis and maturation.
HSP70 can bind and regulate the stability of NOX2 by facilitating ubiquitination and proteasomal degradation [36]. With respect to stress responses, we identified a rapid induction of HSP70 upon stimulation with biglycan as an early pro-inflammatory event, along with increased II1β expression. Interestingly, soluble biglycan can trigger HSP70 expression in a TLR2-dependent manner, and can induce the binding of this chaperone protein to NOX2. We hypothesize that this could lead to NOX2 degradation and subsequent overexpression of IL-1β. Thus, by inducing NOX2 activation via TLR4 and by inhibiting NOX2 activity through HSP70 induction via TLR2, biglycan can autonomously orchestrate the balance of IL-1β expression and maturation in macrophages. Collectively, our results provide a novel negative mechanism through which biglycan regulates the severity of tissue inflammation by activating and inhibiting ROS production and pro-inflammatory signaling via different TLRs. Moreover, our new finding of the dual role of biglycan expands the knowledge regarding the biological role of this SLRP solely considered, up to now, as a pro-inflammatory danger molecule [11,14–16,19].
Excessive ROS production is one of the hallmarks of ischemia reperfusion injury particularly during the early phase of reperfusion, leading to oxidative stress, which further contributes to tissue damage [70]. Among potential sources of ROS, NOX1, NOX2 and NOX4 are the major ROS producing oxidases in the kidney [71–74]. However, the mechanism of how these enzymes are regulated upon tissue injury and inflammation and how they might contribute to inflammatory signaling is not completely understood. In inflammatory response, macrophages are recruited to the place of tissue damage where they secrete pro-inflammatory cytokines and produce ROS [75–77]. Accordingly, we observed a massive macrophage infiltration in WT kidneys 20 h post IRI, and this process was more pronounced in NOX2-deficient mice, along with increased levels of II1β and Ccl2. It is well established that in the course of renal IRI, the endogenous danger signal biglycan is released to induce pro-inflammatory signaling in a TLR2/4 dependent manner [11]. Consequently, we show here that biglycan-deficient mice are protected from excessive II1β and Ccl2 induction and massive macrophage infiltration in IRI. Furthermore, we provide an important genetic proof for a crucial role of biglycan in the hyper-inflammatory phenotype of Nox2−/y mice.
Several studies have addressed the role of NOX enzymes in IRI, but often with contradictory results [78,79]. Especially unresolved is the role of NOX2 in different organs and cell types [80]. In the present model of renal IRI, impaired NOX2 function results in severe inflammation and macrophage infiltration in a biglycan-dependent manner, especially in the early stages of acute kidney failure. Here we confirm that genetic ablation of biglycan protects against the severe Ccl2 expression and macrophage infiltration as well as elevated II1β in the reperfused kidney of NOX2-deficient mice and consequently improves renal outcome. This correlated with decreased levels of HSP70 in kidneys of Nox2−/y mice when biglycan was depleted. Interestingly, in all analyzed genotypes no induction of HSP70 was observed in the reperfused kidneys compared to sham-operated controls. Of note, this observation is in line with a previous report, which demonstrated that the protein expression of HSP70 was not significantly increased in IRI compared to sham-operated kidneys on days 1 to 5 [47]. Nevertheless, we show that loss of biglycan leads to reduction of HSP70 expression, which is associated with a decreased pro-inflammatory signaling and a less severe inflammation. Several studies have shown that pharmacological HSP70 induction protects renal tissue from IRI damage [81–83]. Our data suggest that in ROS-mediated renal inflammation targeting biglycan and consequently inhibiting HSP70 expression might limit renal pathology.
In conclusion, we provide new insights on how different NADPH oxidases are involved in biglycan-mediated pro-inflammatory signaling in health and disease. We show for the first time how the endogenous danger molecule biglycan autonomously regulates the physiological balance in pro-inflammatory signaling by triggering NOX enzymes via different TLR pathways. As loss of biglycan in ROS-mediated pathological conditions such as renal IRI is beneficial to the clinical outcome, a selective inhibition of biglycan-TLR2 or biglycan-TLR4 signaling could represent a novel therapeutic approach in inflammatory diseases.
Experimental procedures
Animal experiments
8-week old male wild-type C57BL/6 mice were purchased from Charles River Laboratories (Sulzfeld, Germany). Nox1−/y, Nox2−/y and Nox4−/− were provided by Prof. R. P. Brandes (University of Frankfurt, Frankfurt, Germany). Tlr2−/− and Tlr4−/− mice were generously provided by Prof. M. Freudenberg (Max Planck Institute for Immunology, Freiburg, Germany). Tlr2−/−/Tlr4-m mice (Tlr2−/− mice carrying a TLR4 mutation) were generously provided by Prof. C. Kirschning (Technical University of Munich, Munich, Germany). Trif −/− mice were a generous gift from Prof. H. H. Radeke (University of Frankfurt, Frankfurt, Germany). Nox2−/y/Bgn−/y mice were generated by intercrossing Nox2−/y mice with Bgn−/y mice. Biglycan-deficient mice were described previously [84]. Total genomic DNA was isolated from tail clippings by KAPA Mouse Genotyping Hot Start Kit (PEQLAB, Germany). DNA was subjected to PCR amplification of exon 2 of the Bgn gene and the following primers were used: forward primer 5′-CAG GAA CAT TGA CCA TG-3′ and a reverse primer 5′-GAA AGG ACA CAT GGC ACT GAA G-3′, yielding a PCR product of 238 base pairs (bp) [85]. Another primer complementary to the PGKneo cassette 5′-TGG ATG TGG AAT GTG TGC GAG G-3′ was added to amplify the inserted PGKneo cassette within the exon 2 of Bgn gene resulting in a PCR product of 161 bp. For amplification of exon 3 of the Nox2 gene as well as the inserted PGKneo cassette [86] the following primers were used: forward primer 5′-AAG AGA AAC TCC TCT GCT GTG AA-3′, 5′-GTT CTA ATT CCA TCA GAA GCT TAT CG-3′, and reverse primer 5′-CGC ACT GGA ACC CCT GAG AAA GG-3′, yielding bands of 240 bp for Nox2 gene, and 195 bp when the PGKneo cassette was inserted [87]. All animal work was conducted in accordance with the German Animal Protection Law and was approved by the Ethics Review Committee for laboratory animals of the District Government of Darmstadt, Germany.
Induction of renal ischemia reperfusion injury
For induction of kidney ischemia reperfusion injury, mice were anesthetized with Ketamin/Xylazin (100 mg/kg and 10 mg/kg, respectively; Ketavet from Pfizer, Germany and Rompun from Bayer, Germany). A midline incision was made, the left renal pedicle was tied off with a suture, then distally sectioned and the kidney was removed. Next, renal artery of the right kidney was clamped for 25 min with an atraumatic microaneurysm clamp. After clamp removal kidneys were inspected for 1 min for restoration of blood flow and returning to their original color, then the incision was closed. Sham-operated mice (control, n = 7 per group) received identical surgical procedures except that neither nephrectomy nor application of the microaneurysm clamps was performed. Mice were sacrificed 20 h after reperfusion. Serum was taken for ELISA. Kidney tissues were divided to snap frozen for subsequent RNA extraction and Western blot analysis, and fixation in 4% paraformaldehyde for histological and immunohisto-chemical analysis.
Determination of renal function and histopathology
Serum creatinine was determined by colorimetric Microplate Assay (Cayman Chemical, Germany). Sections (4 μm) of paraffin-embedded kidneys from mice were blocked with 5% milk in TBS with 0.05% Tween 20 for 1 h at room temperature and incubated with the primary antibody rat anti-mouse F4/80 (MCA497, AbDSerotec, Germany) for 2 h at room temperature. The staining was developed with 3,3′-diaminobenzidine (Vector Laboratories, UK). Counterstaining was performed with Mayer’s Hematoxylin (Sigma Aldrich, Germany). Specificity controls included omitting or replacement of primary antiserum with rat IgG or goat serum. The number of macrophages was estimated per high-power field (HPF 400×, with a minimum of 7 fields counted) (Soft Imaging System, Olympus, Germany). Histological examinations were performed by two observers blinded to the conditions.
Purification of human biglycan
Expression of human biglycan in 293 HEK cells has been described previously [88]. For purification of the native proteoglycan with attached glycosaminoglycan chains, the conditioned medium was supplemented with 0.1% Triton X-100, 20 mM Tris/HCl pH 7.4 and proteinase inhibitors (0.1 M amino-n-caproic acid, 10 mM EDTA, 5 mM benzamidine, 10 mM N-ethylmaleimide, and 1 mM phenyl-methylsulfonyl fluoride); further passed over a DEAE-Trisacryl-M packed column (Tosoh Bioscience, Japan). Elution was performed with 1 M NaCl in the same isotonic condition. The eluent was dialyzed overnight against 20 mM Tris/HCl, pH 7.4, containing 150 mM NaCl and separated by high performance liquid chromatography (Prominence LC; Shimadzu) on a TSK-GELDEAE-5PW, 7.5 mm inner diameter × 7.5 cm, 10-μm column (Tosoh Bioscience, Japan) by a discontinuous binary NaCl gradient. The protein purity was confirmed by silver staining on SDS gel electrophoresis after dialysis against phosphate-buffered saline (PBS).
Cell culture and stimulation
Murine thioglycolate-elicited macrophages were isolated from peritoneal lavage and grown in RPMI 1640 (Life Technologies, Germany) supplemented with 1% penicillin and streptomycin and 2% fetal bovine serum (Biochrom, Germany). Cells were stimulated with 4 μg/ml (80 nM) human biglycan in serum-free medium for indicated time periods. Phorbol 12-myristate 13-acetate (PMA) (100 nM; Sigma, Germany) served as positive control for p47phox translocation studies. For ROS inhibition diphenyleneiodonium chloride (DPI, 0.5 μM; Sigma, Germany), VAS2870 (5 μM; Sigma, Germany), ML-171 (10 nM; Merck, Germany), Nox2ds-tat (40 μM; AnaSpec, USA) and GKT137831 (200 μM) were applied to the macrophages 1 h prior to stimulation with biglycan. For immunofluorescence studies Akt inhibitor 124008 (1 μM; Calbiochem, Germany), p38 MAPK inhibitor SB203580 (10 μM; Calbiochem, Germany), MEK1/2 inhibitor UO126 (10 μM; Cell Signaling, Germany), PI3K inhibitor LY294002 (10 μM; Cell Signaling, Germany), Rac1 inhibitor (50 μM; Calbiochem, Germany), PKC inhibitor GO6976 (20 nM; Sigma, Germany) and MyD88 inhibitor peptide NBP2–29328 (50 μM; Novus Biologicals, Germany) were applied 1 h prior to stimulation with biglycan. For HSP70 inhibition phenylsulfonamide (PES, pifithrin-μ) (200 μM; Sigma, Germany) was applied to the cells in addition to biglycan.
Pull-down assay
Macrophages were stimulated with 4 μg/ml soluble biglycan for indicated time points and subsequently lysed in the buffer supplied by either active Rac1 pull-down and detection kit (Thermo Fisher Scientific, Germany) or Pierce Co-immunoprecipitation kit (Thermo Fisher Scientific, Germany) according to manufacturer’s instructions. 500 μg of cell lysate was incubated with 20 μg purified GST-Pak1-protein binding domain and Glutathione Resin overnight at 4 °C. For NOX2 co-immunoprecipitation studies, NOX2 antibody (ab80508, Abcam, Germany) was coupled with AminoLink Plus Coupling Resin before use. 500 μg lysate was applied to antibody-coupled resin overnight at 4 °C. All steps were performed according to the manufacturer’s protocol.
RNA isolation and quantitative real-time PCR
Total RNA was isolated using the TRI Reagent (Sigma Aldrich, Germany). cDNA was reverse transcribed using the High Capacity cDNA Reverse Transcription Kit (Applied Biosystems, Germany). Real-time quantitative PCR was performed using AbiPrism 7500 Sequence Detection System. Quantitative RT-PCR was performed using TaqMan Fast Universal PCR Master Mix and the following primers: Caspase1 (Mm00438023_m1), Ccl2 (Mm00441212_m1), Gapdh (Mm99999915_g1), II1β (Mm00434228_m1), Nlrp3 (Mm00840904_m1), and Nox2 (Mm01287743_m1) (Applied Biosystems, Germany). Alternatively, Luminaris High Green Low ROX qPCR Mastermix (Thermo Fisher Scientific, Germany) with the following primer pairs was used: mouse Gapdh, 5′-CAT GGC CTT CCG TGT TCC TA-3′ and 5′-CCT GCT TCA CCA CCT TCT TGA T-3′; mouse Nox1, 5′-AGG TCG TGA TTA CCA AGG TTG TC-3′ and 5′-AAG CCT CGC TTC CTC ATC TG-3′; mouse Nox3, 5′- GCT GGC TGC ACT TTC CAA AC-3′ and 5′-AAG GTG CGG ACT GGA TTG AG-3′; mouse Nox4, 5′-CCA GAA TGA GGA TCC CAG AA-3′ and 5′-TGG AAC TTG GGT TCT TCC AG-3′; mouse Duox1, 5′-CCA CCA TGC TGT ACA TCT GTG A-3′, and 5′-AGG GAG GGC GAC CAA AGT-3′; mouse Duox2, 5′-TCC AGA AGG CGC TGA ACA G-3′ and 5′-GCG ACC AAA GTG GGT GAT G-3′; mouse Hsp70, 5′-GCA AGG CCA ACA AGA TCA CCA T-3′ and 5′-GGC GCT CTT CAT GTT GAA GC-3′. Relative changes in gene expression compared to control and normalized to Gapdh were quantified by the 2−ΔΔCt method.
SDS-PAGE and Western blot
Total kidneys as well as macrophage were lysed in buffer containing 50 mM Tris/HCl (pH 8), 150 mM NaCl, 0.02% NaN3, 0.1% SDS, 1 μg/ml aprotinin, 1% Nonidet P-40, 0.5% sodium deoxycholate, 100 μg/ml phenyl methyl sulphonyl fluoride and protease inhibitors consisting of 0.1 M ε-amino-ncaproic acid, 13 μM EDTA, 5 mM benzammoniumchloride monohydrate and 10 mM N-ethyl maleimide. Alternatively, conditioned media collected from biglycan-stimulated macrophages were precipitated with 100% trichloroacetic acid and further washed twice with cold acetone. For SDS-PAGE and Western blotting, 100 μg of total protein from each sample were mixed with loading buffer (250 mM Tris/HCl pH 6.8, 8% sodium dodecyl sulfate, 40% Glycerol, 8% β-mercaptoethanol, 0.02% Bromo phenol blue) and boiled at 95 °C for 5 min.
Primary antibodies used were: mouse anti-β-actin (A5441, Sigma Aldrich, Germany), mouse anti-HSP70/HSP72 (C92F3A-5, Enzo Life Sciences, Germany), rabbit anti-mouse IL-1β (12507S, Cell Signaling, Germany), rabbit anti-NOX2 (ab80508, abcam, Germany), rabbit anti-p38 MAPK (9212, Cell Signaling, Germany), rabbit anti-P-p38 MAPK (T180/Y182) (9215, Cell Signaling, Germany), rabbit anti-p44/42 MAPK (9102, Cell Signaling, Germany), rabbit anti-P-p44/42 MAPK (Thr202/Tyr204) (9101, Cell Signaling, Germany). Secondary antibodies were HRP-coupled donkey anti-rabbit (NA934V, GE Healthcare, UK) and HRP-coupled sheep anti-mouse (NA931V, GE Healthcare, UK).
ELISA
Cell culture media were collected from biglycan-stimulated macrophages. Mouse IL-1β/IL-1F2 DuoSet (R&D Systems, Germany) ELISA kit was employed and followed according to the manufacturer’s protocol.
Immunofluorescence
Macrophages (4 × 105 cells/ml) were seeded on tissue culture treated eight-chamber glass slides (BD Falcon, USA). Following stimulation, cells were washed with PBS and fixed for 30 min with 4% paraformaldehyde at 4 °C. After permeabilization with 0.2% Triton X-100 for 30 sec and blocking in 5% albumin fraction V containing PBS for 1 h, cells were incubated overnight with primary antibodies rat anti--mouse F4/80 (MCA497, AbDSerotec, Germany) or goat anti-mouse p22phox and rabbit anti-mouse p47phox (sc-11,712 and sc-14,015, Santa Cruz Biotechnology, Germany). After washing with PBS, cells were stained with donkey anti-rat or anti-goat Alexa Fluor 488 and donkey anti-rabbit Alexa Fluor 594 secondary antibodies (Life Technologies, Germany) for 1 h in the dark. Nuclei were visualized with DAPI (Vector Laboratories, UK). A Zeiss LSM-510 confocal laser-scanning microscope was utilized for the image acquisition with a ×63, 1.3 oil-immersion objective. Merged images represent single optical sections (<0.8 μm), collected with the pinhole set to 1 airy unit for the red channel, and adjusted to give the same optical slice thickness in the green and blue channels. Images were acquired in single confocal planes to determine association of the proteins using Zeiss LSM-510 software, with filters set to 488/594 nm for dual-channel imaging. All images were analyzed using ImageJ and Adobe Photoshop CS6. Line scanning was used to further quantify co-localization of p47phox with F4/80 or p22phox [89]. This allows the measurements of pixels on a single defined axis along the specimen in order to define localization of two differentially labeled fluorophores. A qualitative assessment of a proximity-dependent localization between the two potentially interacting molecules is obtained from measuring the extent of overlap, defined as two different fluorescent labels displaying independent emission wavelengths that occupy the same pixel. In addition, the ImageJ co-localization color map plugin was utilized to corroborate our analysis. The index of co-localization (ICorr value) is represented on the y-axis. Co-localization analysis was performed on single, segmented cells.
Statistics
All data are expressed as means ± S.D. Two-sided Student’s t-test was used to evaluate significance of differences between groups. Differences were considered significant at P < 0.05.
Supplementary Material
{kind=link}
{kind=link}
Acknowledgments
The original research in our laboratories was supported by the German Research Council (SFB 815, project A5 for LS and A1 for KS and RB, SFB 1039, project B2, SFB 1177, project C2, and SCHA 1082/6-1 to LS), and LOEWE program Ub-Net (LS) and the NIH grants CA39481 and NIH CA47282 (RVI). We thank Riad Haceni for performing the immunohistochemistry analysis and purifying the recombinant human biglycan. We thank Susanne Schütz for the Nox1−/y, Nox2−/y mice genotyping.
Abbreviations used:
- CCL
chemokine (C–C motif) ligand
- CXCL
chemokine (C-X-C motif) ligand
- DAMPs
damage-associated molecular patterns
- DPI
diphenyliodonium
- DUOX
dual oxidase
- ECM
extracellular matrix
- Erk
extracellular signal-regulated kinases
- HSP
heat shock protein
- IL-1β
interleukin-1β
- IRI
ischemia reperfusion injury
- MAPK
mitogen-activated protein kinase
- NOX
NADPH oxidase
- NLRP3
NOD-like receptor protein 3
- PES
2-phenylethynesulfonamide
- Phox
phagocytic oxidase
- ROS
reactive oxygen species
- SLRP
small leucine-rich proteoglycan
- TLR
Toll-like receptor
- TNF-α
tumor necrosis factor α
- MyD88
myeloid differentiation primary response protein
- TRIF
Toll/IL-1R domain-containing adaptor inducing IFN-β
- WT
wild-type
Footnotes
Supplementary data to this article can be found online at http://dx.doi.org/10.1016/j.matbio.2015.12.005.
Competing financial interests
The authors declare no competing financial interests.
References
- [1].Iozzo RV, Schaefer L, Proteoglycan form and function: a comprehensive nomenclature of proteoglycans, Matrix Biol. 42 (2015) 11–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bianco P, Fisher LW, Young MF, Termine JD, Robey PG, Expression and localization of the two small proteoglycans biglycan and decorin in developing human skeletal and non-skeletal tissues, J. Histochem. Cytochem 38 (1990) 1549–1563. [DOI] [PubMed] [Google Scholar]
- [3].Morcavallo A, Buraschi S, Xu SQ, Belfiore A, Schaefer L, Iozzo RV, Morrione A, Decorin differentially modulates the activity of insulin receptor isoform A ligands, Matrix Biol. 35 (2014) 82–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Nikolovska K, Renke JK, Jungmann O, Grobe K, Iozzo RV, Zamfir AD, Seidler DG, A decorin-deficient matrix affects skin chondroitin/dermatan sulfate levels and keratinocyte function, Matrix Biol. 35 (2014) 91–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Chen S, Young MF, Chakravarti S, Birk DE, Interclass small leucine-rich repeat proteoglycan interactions regulate collagen fibrillogenesis and corneal stromal assembly, Matrix Biol. 35 (2014) 103–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Skandalis SS, Afratis N, Smirlaki G, Nikitovic D, Theocharis AD, Tzanakakis GN, Karamanos NK, Cross-talk between estradiol receptor and EGFR/IGF-IR signaling pathways in estrogen-responsive breast cancers: focus on the role and impact of proteoglycans, Matrix Biol. 35 (2013) 182–193. [DOI] [PubMed] [Google Scholar]
- [7].Berendsen AD, Pinnow EL, Maeda A, Brown AC, McCartney-Francis N, Kram V, Owens RT, Robey PG, Holmbeck K, de Castro LF, Kilts TM, Young MF, Biglycan modulates angiogenesis and bone formation during fracture healing, Matrix Biol. 35 (2014) 223–231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Dunkman AA, Buckley MR, Mienaltowski MJ, Adams SM, Thomas SJ, Kumar A, Beason DP, Iozzo RV, Birk DE, Soslowsky LJ, The injury response of aged tendons in the absence of biglycan and decorin, Matrix Biol. 35 (2013) 232–238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Gubbiotti MA, Neill T, Frey H, Schaefer L, Iozzo RV, Decorin is an autophagy-inducible proteoglycan and is required for proper in vivo autophagy, Matrix Biol. 48 (2015) 14–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Gubbiotti MA, Iozzo RV, Proteoglycans regulate autophagy via outside-in signaling: an emerging new concept, Matrix Biol. 48 (2015) 6–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Moreth K, Frey H, Hubo M, Zeng-Brouwers J, Nastase MV, Hsieh LT, Haceni R, Pfeilschifter J, Iozzo RV, Schaefer L, Biglycan-triggered TLR-2- and TLR-4-signaling exacerbates the pathophysiology of ischemic acute kidney injury, Matrix Biol. 35 (2014) 143–151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Frey H, Schroeder N, Manon-Jensen T, Iozzo RV, Schaefer L, Biological interplay between proteoglycans and their innate immune receptors in inflammation, FEBS J. 280 (2013) 2165–2179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Nastase MV, Young MF, Schaefer L, Biglycan: a multivalent proteoglycan providing structure and signals, J. Histochem. Cytochem 60 (2012) 963–975. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Schaefer L, Babelova A, Kiss E, Hausser HJ, Baliova M, Krzyzankova M, Marsche G, Young MF, Mihalik D, Gotte M, Malle E, Schaefer RM, Grone HJ, The matrix component biglycan is proinflammatory and signals through Toll-like receptors 4 and 2 in macrophages, J. Clin. Invest 115 (2005) 2223–2233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Moreth K, Brodbeck R, Babelova A, Gretz N, Spieker T, Zeng-Brouwers J, Pfeilschifter J, Young MF, Schaefer RM, Schaefer L, The proteoglycan biglycan regulates expression of the B cell chemoattractant CXCL13 and aggravates murine lupus nephritis, J. Clin. Invest 120 (2010) 4251–4272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Zeng-Brouwers J, Beckmann J, Nastase MV, Iozzo RV, Schaefer L, De novo expression of circulating biglycan evokes an innate inflammatory tissue response via MyD88/TRIF pathways, Matrix Biol. 35 (2013) 132–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Thompson J, Wilson P, Brandewie K, Taneja D, Schaefer L, Mitchell B, Tannock LR, Renal accumulation of biglycan and lipid retention accelerates diabetic nephropathy, Am. J. Pathol 179 (2011) 1179–1187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Schaefer L, Raslik I, Grone HJ, Schonherr E, Macakova K, Ugorcakova J, Budny S, Schaefer RM, Kresse H, Small proteoglycans in human diabetic nephropathy: discrepancy between glomerular expression and protein accumulation of decorin, biglycan, lumican, and fibromodulin, FASEB J. 15 (2001) 559–561. [DOI] [PubMed] [Google Scholar]
- [19].Babelova A, Moreth K, Tsalastra-Greul W, Zeng-Brouwers J, Eickelberg O, Young MF, Bruckner P, Pfeilschifter J, Schaefer RM, Grone HJ, Schaefer L, Biglycan, a danger signal that activates the NLRP3 inflammasome via toll-like and P2X receptors, J. Biol. Chem 284 (2009) 24035–24048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Nunes P, Demaurex N, Dinauer MC, Regulation of the NADPH oxidase and associated ion fluxes during phagocytosis, Traffic 14 (2013) 1118–1131. [DOI] [PubMed] [Google Scholar]
- [21].Lambeth JD, NOX enzymes and the biology of reactive oxygen, Nat. Rev. Immunol 4 (2004) 181–189. [DOI] [PubMed] [Google Scholar]
- [22].Groemping Y, Lapouge K, Smerdon SJ, Rittinger K, Molecular basis of phosphorylation-induced activation of the NADPH oxidase, Cell 113 (2003) 343–355. [DOI] [PubMed] [Google Scholar]
- [23].Yu L, Quinn MT, Cross AR, Dinauer MC, Gp91(phox) is the heme binding subunit of the superoxide-generating NADPH oxidase, Proc. Natl. Acad. Sci. U. S. A 95 (1998) 7993–7998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Bedard K, Krause KH, The NOX family of ROS-generating NADPH oxidases: physiology and pathophysiology, Physiol. Rev 87 (2007) 245–313. [DOI] [PubMed] [Google Scholar]
- [25].Parkos CA, Allen RA, Cochrane CG, Jesaitis AJ, Purified cytochrome b from human granulocyte plasma membrane is comprised of two polypeptides with relative molecular weights of 91,000 and 22,000, J. Clin. Invest 80 (1987) 732–742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Ambasta RK, Kumar P, Griendling KK, Schmidt HH, Busse R, Brandes RP, Direct interaction of the novel Nox proteins with p22phox is required for the formation of a functionally active NADPH oxidase, J. Biol. Chem 279 (2004) 45935–45941. [DOI] [PubMed] [Google Scholar]
- [27].Kawahara T, Ritsick D, Cheng G, Lambeth JD, Point mutations in the proline-rich region of p22phox are dominant inhibitors of Nox1- and Nox2-dependent reactive oxygen generation, J. Biol. Chem 280 (2005) 31859–31869. [DOI] [PubMed] [Google Scholar]
- [28].Martyn KD, Frederick LM, von Loehneysen K, Dinauer MC, Knaus UG, Functional analysis of Nox4 reveals unique characteristics compared to other NADPH oxidases, Cell. Signal 18 (2006) 69–82. [DOI] [PubMed] [Google Scholar]
- [29].Takac I, Schroder K, Zhang L, Lardy B, Anilkumar N, Lambeth JD, Shah AM, Morel F, Brandes RP, The E-loop is involved in hydrogen peroxide formation by the NADPH oxidase Nox4, J. Biol. Chem 286 (2011) 13304–13313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Aoyama T, Paik YH, Watanabe S, Laleu B, Gaggini F, Fioraso-Cartier L, Molango S, Heitz F, Merlot C, Szyndralewiez C, Page P, Brenner DA, Nicotinamide adenine dinucleotide phosphate oxidase in experimental liver fibrosis: GKT137831 as a novel potential therapeutic agent, Hepatology 56 (2012) 2316–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Carnesecchi S, Deffert C, Donati Y, Basset O, Hinz B, Preynat-Seauve O, Guichard C, Arbiser JL, Banfi B, Pache JC, Barazzone-Argiroffo C, Krause KH, A key role for NOX4 in epithelial cell death during development of lung fibrosis, Antioxid. Redox Signal 15 (2011) 607–619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Sedeek M, Gutsol A, Montezano AC, Burger D, Nguyen Dinh Cat A, Kennedy CR, Burns KD, Cooper ME, Jandeleit-Dahm K, Page P, Szyndralewiez C, Heitz F, Hebert RL, Touyz RM, Renoprotective effects of a novel Nox1/4 inhibitor in a mouse model of type 2 diabetes, Clin. Sci. (Lond.) 124 (2012) 191–202. [DOI] [PubMed] [Google Scholar]
- [33].Gorin Y, Cavaglieri RC, Khazim K, Lee DY, Bruno F, Thakur S, Fanti P, Szyndralewiez C, Barnes JL, Block K, Abboud HE, Targeting NADPH oxidase with a novel dual Nox1/Nox4 inhibitor attenuates renal pathology in type 1 diabetes, Am. J. Physiol. Renal Physiol 308 (2015) F1276–F1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Deffert C, Carnesecchi S, Yuan H, Rougemont AL, Kelkka T, Holmdahl R, Krause KH, Schappi MG, Hyperinflammation of chronic granulomatous disease is abolished by NOX2 reconstitution in macrophages and dendritic cells, J. Pathol 228 (2012) 341–350. [DOI] [PubMed] [Google Scholar]
- [35].Huang YF, Lo PC, Yen CL, Nigrovic PA, Chao WC, Wang WZ, Hsu GC, Tsai YS, Shieh CC, Redox regulation of pro-IL-1beta processing may contribute to the increased severity of serum-induced arthritis in NOX2-deficient mice, Antioxid. Redox Signal 23 (2015) 973–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Chen F, Yu Y, Qian J, Wang Y, Cheng B, Dimitropoulou C, Patel V, Chadli A, Rudic RD, Stepp DW, Catravas JD, Fulton DJ, Opposing actions of heat shock protein 90 and 70 regulate nicotinamide adenine dinucleotide phosphate oxidase stability and reactive oxygen species production, Arterioscler. Thromb. Vasc. Biol 32 (2012) 2989–2999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Ratz JD, McGuire JJ, Anderson DJ, Bennett BM, Effects of the flavoprotein inhibitor, diphenyleneiodonium sulfate, on ex vivo organic nitrate tolerance in the rat, J. Pharmacol. Exp. Ther 293 (2000) 569–577. [PubMed] [Google Scholar]
- [38].Ten Freyhaus H, Huntgeburth M, Wingler K, Schnitker J, Baumer AT, Vantler M, Bekhite MM, Wartenberg M, Sauer H, Rosenkranz S, Novel Nox inhibitor VAS2870 attenuates PDGF-dependent smooth muscle cell chemotaxis, but not proliferation, Cardiovasc. Res 71 (2006) 331–341. [DOI] [PubMed] [Google Scholar]
- [39].Gianni D, Taulet N, Zhang H, DerMardirossian C, Kister J, Martinez L, Roush WR, Brown SJ, Bokoch GM, Rosen H, A novel and specific NADPH oxidase-1 (Nox1) smallmolecule inhibitor blocks the formation of functional invadopodia in human colon cancer cells, ACS Chem. Biol 5 (2010) 981–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Laleu B, Gaggini F, Orchard M, Fioraso-Cartier L, Cagnon L, Houngninou-Molango S, Gradia A, Duboux G, Merlot C, Heitz F, Szyndralewiez C, Page P, First in class, potent, and orally bioavailable NADPH oxidase isoform 4 (Nox4) inhibitors for the treatment of idiopathic pulmonary fibrosis, J. Med. Chem 53 (2010) 7715–7730. [DOI] [PubMed] [Google Scholar]
- [41].Csanyi G, Cifuentes-Pagano E, Al Ghouleh I, Ranayhossaini DJ, Egana L, Lopes LR, Jackson HM, Kelley EE, Pagano PJ, Nox2 B-loop peptide, Nox2ds, specifically inhibits the NADPH oxidase Nox2, Free Radic. Biol. Med 51 (2011) 1116–1125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Yamamoto M, Sato S, Hemmi H, Hoshino K, Kaisho T, Sanjo H, Takeuchi O, Sugiyama M, Okabe M, Takeda K, Akira S, Role of adaptor TRIF in the MyD88-independent toll-like receptor signaling pathway, Science 301 (2003) 640–643. [DOI] [PubMed] [Google Scholar]
- [43].Bolscher BG, van Zwieten R, Kramer IM, Weening RS, Verhoeven AJ, Roos D, A phosphoprotein of Mr 47,000, defective in autosomal chronic granulomatous disease, copurifies with one of two soluble components required for NADPH:O2 oxidoreductase activity in human neutrophils, J. Clin. Invest 83 (1989) 757–763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Yuzawa S, Suzuki NN, Fujioka Y, Ogura K, Sumimoto H, Inagaki F, A molecular mechanism for autoinhibition of the tandem SH3 domains of p47phox, the regulatory subunit of the phagocyte NADPH oxidase, Genes Cells 9 (2004) 443–456. [DOI] [PubMed] [Google Scholar]
- [45].Kuhns DB, Alvord WG, Heller T, Feld JJ, Pike KM, Marciano BE, Uzel G, DeRavin SS, Priel DA, Soule BP, Zarember KA, Malech HL, Holland SM, Gallin JI, Residual NADPH oxidase and survival in chronic granulomatous disease, N. Engl. J. Med 363 (2010) 2600–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Griffith B, Pendyala S, Hecker L, Lee PJ, Natarajan V, Thannickal VJ, NOX enzymes and pulmonary disease, Antioxid. Redox Signal 11 (2009) 2505–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Wu H, Chen G, Wyburn KR, Yin J, Bertolino P, Eris JM, Alexander SI, Sharland AF, Chadban SJ, TLR4 activation mediates kidney ischemia/reperfusion injury, J. Clin. Invest 117 (2007) 2847–2859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Ascon DB, Lopez-Briones S, Liu M, Ascon M, Savransky V, Colvin RB, Soloski MJ, Rabb H, Phenotypic and functional characterization of kidney-infiltrating lymphocytes in renal ischemia reperfusion injury, J. Immunol 177 (2006) 3380–3387. [DOI] [PubMed] [Google Scholar]
- [49].Brandes RP, Kreuzer J, Vascular NADPH oxidases: molecular mechanisms of activation, Cardiovasc. Res 65 (2005) 16–27. [DOI] [PubMed] [Google Scholar]
- [50].Tassi S, Carta S, Vene R, Delfino L, Ciriolo MR, Rubartelli A, Pathogen-induced interleukin-1beta processing and secretion is regulated by a biphasic redox response, J. Immunol 183 (2009) 1456–1462. [DOI] [PubMed] [Google Scholar]
- [51].Davis BK, Ting JP, NLRP3 has a sweet tooth, Nat. Immunol 11 (2010) 105–106. [DOI] [PubMed] [Google Scholar]
- [52].Maitra U, Singh N, Gan L, Ringwood L, Li L, IRAK-1 contributes to lipopolysaccharide-induced reactive oxygen species generation in macrophages by inducing NOX-1 transcription and Rac1 activation and suppressing the expression of antioxidative enzymes, J. Biol. Chem 284 (2009) 35403–35411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Lee CF, Qiao M, Schroder K, Zhao Q, Asmis R, Nox4 is a novel inducible source of reactive oxygen species in monocytes and macrophages and mediates oxidized low density lipoprotein-induced macrophage death, Circ. Res 106 (2010) 1489–1497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Lan T, Kisseleva T, Brenner DA, Deficiency of NOX1 or NOX4 prevents liver inflammation and fibrosis in mice through inhibition of hepatic stellate cell activation, PLoS One 10 (2015), e0129743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Deliyanti D, Wilkinson-Berka JL, Inhibition of NOX1/4 with GKT137831: a potential novel treatment to attenuate neuroglial cell inflammation in the retina, J. Neuroinflammation 12 (2015) 136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Diebold BA, Smith SM, Li Y, Lambeth JD, NOX2 as a target for drug development: indications, possible complications, and progress, Antioxid. Redox Signal 23 (2015) 375–405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Sareila O, Kelkka T, Pizzolla A, Hultqvist M, Holmdahl R, NOX2 complex-derived ROS as immune regulators, Antioxid. Redox Signal 15 (2011) 2197–2208. [DOI] [PubMed] [Google Scholar]
- [58].Whitmore LC, Hilkin BM, Goss KL, Wahle EM, Colaizy TT, Boggiatto PM, Varga SM, Miller FJ, Moreland JG, NOX2 protects against prolonged inflammation, lung injury, and mortality following systemic insults, J. Innate Immun 5 (2013) 565–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Huang J, Canadien V, Lam GY, Steinberg BE, Dinauer MC, Magalhaes MA, Glogauer M, Grinstein S, Brumell JH, Activation of antibacterial autophagy by NADPH oxidases, Proc. Natl. Acad. Sci. U. S. A 106 (2009) 6226–6231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Bae YS, Lee JH, Choi SH, Kim S, Almazan F, Witztum JL, Miller YI, Macrophages generate reactive oxygen species in response to minimally oxidized low-density lipoprotein: toll-like receptor 4- and spleen tyrosine kinase-dependent activation of NADPH oxidase 2. Circ. Res 104 (2009) 210–218 (21 pp. following 218). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Kampfrath T, Maiseyeu A, Ying Z, Shah Z, Deiuliis JA, Xu X, Kherada N, Brook RD, Reddy KM, Padture NP, Parthasarathy S, Chen LC, Moffatt-Bruce S, Sun Q, Morawietz H, Rajagopalan S, Chronic fine particulate matter exposure induces systemic vascular dysfunction via NADPH oxidase and TLR4 pathways, Circ. Res 108 (2011) 716–726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Laroux FS, Romero X, Wetzler L, Engel P, Terhorst C, Cutting edge: MyD88 controls phagocyte NADPH oxidase function and killing of Gram-negative bacteria, J. Immunol 175 (2005) 5596–5600. [DOI] [PubMed] [Google Scholar]
- [63].Park HS, Jung HY, Park EY, Kim J, Lee WJ, Bae YS, Cutting edge: direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B, J. Immunol 173 (2004) 3589–3593. [DOI] [PubMed] [Google Scholar]
- [64].Dusi S, Donini M, Rossi F, Mechanisms of NADPH oxidase activation: translocation of p40phox, Rac1 and Rac2 from the cytosol to the membranes in human neutrophils lacking p47phox or p67phox, Biochem. J 314 (Pt 2) (1996) 409–412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Koshkin V, Lotan O, Pick E, The cytosolic component p47(phox) is not a sine qua non participant in the activation of NADPH oxidase but is required for optimal superoxide production, J. Biol. Chem 271 (1996) 30326–30329. [DOI] [PubMed] [Google Scholar]
- [66].Usatyuk PV, Gorshkova IA, He D, Zhao Y, Kalari SK, Garcia JG, Natarajan V, Phospholipase D-mediated activation of IQGAP1 through Rac1 regulates hyperoxiainduced p47phox translocation and reactive oxygen species generation in lung endothelial cells, J. Biol. Chem 284 (2009) 15339–15352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Hoyal CR, Gutierrez A, Young BM, Catz SD, Lin JH, Tsichlis PN, Babior BM, Modulation of p47PHOX activity by site-specific phosphorylation: Akt-dependent activation of the NADPH oxidase, Proc. Natl. Acad. Sci. U. S. A 100 (2003) 5130–5135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Olavarria VH, Valdivia S, Salas B, Villalba M, Sandoval R, Oliva H, Valdebenito S, Yanez A, ISA virus regulates the generation of reactive oxygen species and p47phox expression in a p38 MAPK-dependent manner in Salmo salar, Mol. Immunol 63 (2015) 227–234. [DOI] [PubMed] [Google Scholar]
- [69].Belambri SA, Hurtado-Nedelec M, Senator A, Makni-Maalej K, Fay M, Gougerot-Pocidalo MA, Marie JC, Dang PM, El-Benna J, Phosphorylation of p47phox is required for receptor-mediated NADPH oxidase/NOX2 activation in Epstein–Barr virus-transformed human B lymphocytes, Am. J. Blood Res 2 (2012) 187–193. [PMC free article] [PubMed] [Google Scholar]
- [70].Ashraf MI, Ebner M, Wallner C, Haller M, Khalid S, Schwelberger H, Koziel K, Enthammer M, Hermann M, Sickinger S, Soleiman A, Steger C, Vallant S, Sucher R, Brandacher G, Santer P, Dragun D, Troppmair J, A p38MAPK/MK2 signaling pathway leading to redox stress, cell death and ischemia/reperfusion injury, Cell Commun. Signal 12 (2014) 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Holterman CE, Read NC, Kennedy CR, Nox and renal disease, Clin. Sci. (Lond.) 128 (2015) 465–481. [DOI] [PubMed] [Google Scholar]
- [72].Li JM, Shah AM, ROS generation by nonphagocytic NADPH oxidase: potential relevance in diabetic nephropathy, J. Am. Soc. Nephrol 14 (2003) S221–S226. [DOI] [PubMed] [Google Scholar]
- [73].Jones SA, Hancock JT, Jones OT, Neubauer A, Topley N, The expression of NADPH oxidase components in human glomerular mesangial cells: detection of protein and mRNA for p47phox, p67phox, and p22phox, J. Am. Soc. Nephrol 5 (1995) 1483–1491. [DOI] [PubMed] [Google Scholar]
- [74].Lee HB, Yu MR, Yang Y, Jiang Z, Ha H, Reactive oxygen species-regulated signaling pathways in diabetic nephropathy, J. Am. Soc. Nephrol 14 (2003) S241–S245. [DOI] [PubMed] [Google Scholar]
- [75].Torres M, Forman HJ, Redox signaling and the MAP kinase pathways, Biofactors 17 (2003) 287–296. [DOI] [PubMed] [Google Scholar]
- [76].Yang CS, Lee DS, Song CH, An SJ, Li S, Kim JM, Kim CS, Yoo DG, Jeon BH, Yang HY, Lee TH, Lee ZW, El-Benna J, Yu DY, Jo EK, Roles of peroxiredoxin II in the regulation of proinflammatory responses to LPS and protection against endotoxin-induced lethal shock, J. Exp. Med 204 (2007) 583–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Forman HJ, Torres M, Signaling by the respiratory burst in macrophages, IUBMB Life 51 (2001) 365–371. [DOI] [PubMed] [Google Scholar]
- [78].Kahles T, Brandes RP, Which NADPH oxidase isoform is relevant for ischemic stroke? The case for Nox 2, Antioxid. Redox Signal 18 (2012) 1400–1417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Radermacher KA, Wingler K, Langhauser F, Altenhofer S, Kleikers P, Hermans JJ, Hrabe de Angelis M, Kleinschnitz C, Schmidt HH, Neuroprotection after stroke by targeting NOX4 as a source of oxidative stress, Antioxid. Redox Signal 18 (2012) 1418–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Anilkumar N, Weber R, Zhang M, Brewer A, Shah AM, Nox4 and Nox2 NADPH oxidases mediate distinct cellular redox signaling responses to agonist stimulation, Arterioscler. Thromb. Vasc. Biol 28 (2008) 1347–1354. [DOI] [PubMed] [Google Scholar]
- [81].Bidmon B, Endemann M, Muller T, Arbeiter K, Herkner K, Aufricht C, Heat shock protein-70 repairs proximal tubule structure after renal ischemia, Kidney Int. 58 (2000) 2400–2407. [DOI] [PubMed] [Google Scholar]
- [82].Bidmon B, Kratochwill K, Rusai K, Kuster L, Herzog R, Eickelberg O, Aufricht C, Increased immunogenicity is an integral part of the heat shock response following renal ischemia, Cell Stress Chaperones 17 (2012) 385–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Kim MG, Jung Cho E, Won Lee J, Sook Ko Y, Young Lee H, Jo SK, Cho WY, Kim HK, The heat-shock protein-70-induced renoprotective effect is partially mediated by CD4+ CD25+ Foxp3+ regulatory T cells in ischemia/reperfusion-induced acute kidney injury, Kidney Int. 85 (2014) 62–71. [DOI] [PubMed] [Google Scholar]
- [84].Xu T, Bianco P, Fisher LW, Longenecker G, Smith E, Goldstein S, Bonadio J, Boskey A, Heegaard AM, Sommer B, Satomura K, Dominguez P, Zhao C, Kulkarni AB, Robey PG, Young MF, Targeted disruption of the biglycan gene leads to an osteoporosis-like phenotype in mice, Nat. Genet 20 (1998) 78–82. [DOI] [PubMed] [Google Scholar]
- [85].Chen XD, Shi S, Xu T, Robey PG, Young MF, Agerelated osteoporosis in biglycan-deficient mice is related to defects in bone marrow stromal cells, J. Bone Miner. Res 17 (2002) 331–340. [DOI] [PubMed] [Google Scholar]
- [86].Pollock JD, Williams DA, Gifford MA, Li LL, Du X, Fisherman J, Orkin SH, Doerschuk CM, Dinauer MC, Mouse model of X-linked chronic granulomatous disease, an inherited defect in phagocyte superoxide production, Nat. Genet 9 (1995) 202–209. [DOI] [PubMed] [Google Scholar]
- [87].Chandra R, Federici S, Hasko G, Deitch EA, Spolarics Z, Female X-chromosome mosaicism for gp91phox expression diversifies leukocyte responses during endotoxemia, Crit. Care Med 38 (2010) 2003–2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Kresse H, Seidler DG, Muller M, Breuer E, Hausser H, Roughley PJ, Schonherr E, Different usage of the glycosaminoglycan attachment sites of biglycan, J. Biol. Chem 276 (2001) 13411–13416. [DOI] [PubMed] [Google Scholar]
- [89].Jaskolski F, Mulle C, Manzoni OJ, An automated method to quantify and visualize colocalized fluorescent signals, J. Neurosci. Methods 146 (2005) 42–49. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.