

Abstract
Inflammasomes are molecular platforms that assemble upon sensing various intracellular stimuli. Inflammasome assembly leads to activation of caspase-1, thereby promoting the secretion of bioactive interleukin-1β and interleukin-18 and inducing an inflammatory cell death called pyroptosis. Effectors of the inflammasome efficiently drive an immune response, primarily providing protection against microbial infections and mediating control over sterile insults. However, aberrant inflammasome signaling is associated with pathogenesis of inflammatory and metabolic diseases, neurodegeneration, and malignancies. Chronic inflammation perpetuated by inflammasome activation plays a central role in all stages of tumorigenesis, including immunosuppression, proliferation, angiogenesis, and metastasis. Conversely, inflammasome signaling also contributes to tumor suppression by maintaining intestinal barrier integrity, which portrays the diverse roles of inflammasomes in tumorigenesis. Studies have underscored the significance of environmental factors, such as diet and gut microbiota [G] in inflammasome signaling, which in turn influences tumorigenesis. In this review, we deliver an overview of the interplay between inflammasomes and tumorigenesis and discuss their potential as a therapeutic target.
ToC blurb
Inflammasome signaling in myeloid cells largely protects against microbial infections. Aberrant inflammasome signaling promotes chronic inflammation, which contributes to tumorigenesis. This Review presents an overview of the diverging roles of inflammasomes in cancer and discuss its targeting potential in anti-cancer therapy.
Introduction
Inflammasomes are macromolecular complexes that trigger central and rapid inflammatory responses to cytosolic insults. Inflammasome sensors are grouped based on their structural features into nucleotide-binding oligomerization domain and leucine-rich repeat receptors (NLRs), absent in melanoma 2 (AIM2)–like receptors (ALRs), and the recently identified Pyrin. The NLR family is subdivided into NLRP or NLRC based on whether the N-terminus contains a pyrin domain (PYD) or caspase activation and recruitment domain (CARD), respectively1. NLRP1 (mouse NLRP1b), NLRP3, and NLR family apoptosis inhibitory protein (NAIP)-NLRC4 are well-established NLRs for their ability to assemble inflammasome2,3. Several other NLR sensors, including NLRP6, NLRP7, NLRP9, NLRP12, NLRC3, NLRC5, and non-NLR sensors, such as interferon gamma-inducible protein 16, and retinoic acid-inducible gene I may form inflammasome complexes in context-dependent manners4–6. Inflammasomes are largely described in immune cells, such as macrophages and dendritic cells, but are also expressed and assembled in non-hematopoietic cells4.
Previous studies have indicated that the inflammasome-dependent biological responses– both inflammation and cell death– regulate the pathogenesis of a broad range of diseases including obesity, diabetes, atherosclerosis, gout, and ulcerative colitis7. Additionally, the complex roles of inflammasomes in tumorigenesis and antitumor immunity have been revealed over the past decade. The hallmarks of tumor are determined by the central biological characteristics that tumors acquire during the multistep process of tumorigenesis8. Remarkably, inflammasome signaling is implicated in virtually every aspect of tumor development, performing either tumor-suppressive or pro-tumorigenic functions9(TABLE 1). In this review, we describe the dynamic and divergent roles of inflammasome signaling in multiple tissues and organs, highlighting its major functions in shaping inflammatory responses, cellular proliferation and survival, immunosuppression, angiogenesis, and metastasis and gut microbiota, all of which are critical for modulating tumorigenesis. We also discuss recent advances made in translational research that motivate potential pharmacological approaches aiming the inflammasome crossroads in tumorigenesis.
Table 1.
Diverse roles of inflammasomes across different cancers
| Cancer Type | Animal Model | In vitro systems |
|---|---|---|
| Lung cancer (implanted tumors) | ■ Rapid growth of LLC cells in mice transduced with human IL-1β–expressing vector54. ■ Decreased lung metastases of B16F10 melanoma, RM-1 prostate, and E0771 mammary adenocarcinoma in Nlrp3−/− mice66. ■ Decreased lung metastases, and tumor growth of B16 melanoma in Il1b−/− mice59 or Il18r−/− mice42. ■ Increased lung metastases of LLC in Casp1−/− Casp11−/− mice63. ■ Reduced tumor growth in nude mice injected with Gsdmd-silenced PC9 cells40. |
■ Decreased proliferation and migration of Nlrp3-silenced, CASP1-inhibited, and IL-18BP–added A549 cells174. ■ Augmentation of IL-1α release from AIM2-activated lung tumor–associated pDCs50. ■ Decreased viability and increased apoptosis in Gsdmd-silenced PC9, H1703, and H1975 cells40. |
| Breast cancer (implanted tumors) | ■ Reduced tumor growth of Py8119 in Il1b−/− mice130. ■ Reduced tumor growth of EO771 or PyT8 or MDA-MB-231 in Casp1−/− and Nlrp3−/− mice65. ■ Reduced tumor growth of MDA-MB-435 in mice treated with AIM2/SN2 DNA108. ■ Reduced tumor progression in 4T1 tumor-bearing mice treated with IL-1R antibody49. ■ Retarded tumor growth in E0771 cells–bearing mice treated with anakinra49. ■ Reduced tumor growth of Py8119 or EO771 cells in diet-induced obese Casp1−/− Casp11−/− or Nlrc4−/− mice130. |
■ Increased migration and invasion of IL-10-treated BT474 cells73. ■ Increased migration of IL-18–treated and decreased migration of Il18-silenced MCF-7 cells 74. ■ Decreased viability and colony-forming ability of Aim2-overexpressed MCF-7, MDA-MB-231, MDA-MB-435, and MDA-MB-453 cells109,108. |
| Fibrosarcoma | ■ Reduced MCA-induced tumor incidence in Nlrp3−/−, Caspl−/− and Il1r−/− mice66. ■ Reduced tumor growth in IL-18-injected mice after implantation of T241 cells175. |
N/A |
| Gastric Cancer | ■ Increased gastric cancer in IL-1B transgenic mice infected with Helicobacter felis33. ■ Reduced gastric preneoplasia in Il1r−/− mice infected with Helicobacter felis176. ■ Reduced tumor volume of Il18-silenced gastric cancer cells in nude mice43. ■ Reduced tumor incidence in gp130F/F Asc−/− or gp130F/F Il18−/− mice32. ■ Decreased tumor growth of Gsdmd-expressing BGC-823 cells in nude mice177. |
■ Suppressed colony formation of AGS and MKN1 cells treated with IL18 neutralizing antibody32. ■ Suppressed colony formation in Asc−/− and Casp1−/− AGS cells32. ■ Decreased proliferation and colony formation in Gsdmd-overexpressed BGC-823 cells and increased proliferation in Gsdmd-silenced BGC-823 cells177. |
| Hepatic Cancer (Implanted tumors) | ■ Regression of CT26 tumors in the liver of WT mice after IL-18 gene transfer178. ■ Decreased hepatic metastases of B16M cells in mice injected with IL-18BP62. ■ Reduced tumor incidence in DEN-treated Casp1−/− and Aim2−/− mice179. ■ Decreased hepatic metastasis of B16 melanoma in Il1b−/− and Casp1−/− Casp11−/− mice58. ■ Increased metastasis in mice injected with Aim2-silenced HCC cells180. ■ Decreased MC38 metastatic tumors in the liver of Nlrp3−/− mice64. ■ Increased MC38 metastatic tumors in the livers of Casp1−/− Casp11−/−, Nlrp3−/−, Il18−/− and Il18r−/− mice63. |
■ Increased migration and invasion of Aim2-silenced HCC cells 180. |
| Colon Cancer (AOM/DSS) | ■ Increased tumors in the colons of Nlrp3−/−78,79, Aim2−/−80,81, Nlrc4−/−82,83, Naip1–6−/−102, Nlrp1b−/−77, Nlrp6−/−83,181,182, Nlrp12−/−115,116, Pyrin−/−84, Asc−/−77–80,83, Casp1−/−78,82, Il18−/−
79,88 and Il18r−/−88 mice. ■ Decreased tumors in colons of IL-18 injected Casp1−/− 79, Pyrin−/−84 or II18−/−84 mice. ■ Decreased tumor in the colons of Casp1−/−183 and Nlrp3R258W mice97. ■ Decreased tumors in the colons of IL-1Ra–injected WT mice184. ■ Increased tumors in the colons of IL-1β–treated complement deficient mice185. ■ More tumors in the colons of Ptpn2r−/− mice treated with a vaccine against IL-1β or in Ptpn2−/− Asc−/− mice98. |
■ Increased proliferation and invasion of HT29, Caco-2, HCA7 and HCT116 cells cocultured with IL-1β–treated normal or cancer-associated fibroblasts186. ■ Decreased survival of AIM2-expressing HCT116 cells104. ■ Decreased migration and invasion of Nlrp3-silenced HCT116, HT29 and SW620 cells76. |
| Prostate Cancer | ■ Decreased metastatic lesions in bones of WT mice injected with Il1b-silenced PC-3 cells and increased metastatic lesions in bones of WT mice injected with IL-1β–overexpressed DU-145 cells187. | ■ Increased migration of IL-1β–stimulated PC-3 cells188. |
| Glioblastoma | ■ Reduced growth of 9L cells in WT mice or WT rats inoculated with BMSCs expressing IL-18189,190 ■ Increased survival and decreased tumor size in WT rats following injection of IL-18-transduced C6 glioma cells191. ■ Decreased tumor size in SR-B10-injected mice followed by administration of recombinant IL-18192. ■ Reduced tumor growth of GL261 and U87 cells in Nlrp3-silenced mice following IR-treatment193. |
■ Increased proliferation, migration and invasion of IL-1β-treated U87 and U251 cells194. ■ Increased apoptosis of IL-1β-treated hypoxic U87 cells195. ■ Decreased viability and increased apoptosis of 9L glioma cells co-cultured with BMSCs expressing IL-18189,190. |
| Skin Cancer Melanoma | ■ Delayed tumor onset in 3-MCA–treated Il1b
−/− mice196. ■ Reduced tumor growth in mice injected with Nlrp1b-silenced 1205Lu cells197. ■ Reduced tumor incidence in DMBA/TPA-treated Il1r−/− and Casp1−/− mice100. ■ Reduced tumor incidence in LysM-Asc−/− mice and increased tumor incidence in K14-Asc−/− mice after DMBA/TPA treatment100. ■ Reduced papilloma lesions in Nlrp3−/− mice198. ■ Increased tumor size in Nlrc4−/− mice challenged with B16F10 cells101. ■ Reduced B16 tumor growth in chimeric WT mice adoptively transferred with Casp1−/− BMCs199. |
■ Increased proliferation of IL-18–treated B16M, A375, HMB2, VUP, SK23, MJM melanoma cells62. ■ Decreased viability and increased apoptosis of Nlrp1b-silenced WM115, 1205Lu and Hs294T cells197. |
| Squamous Cell Carcinoma | ■ Reduced tumor burden in the head and neck of NLRP3 inhibitor (MCC950)-treated Tgfbr1-−/−
Pten−/− mice200. ■ Delayed onset and smaller tumor area in the tongue of 4-NQO–administered and 5-FU–treated Nlrp3−/− and Casp1−/− mice159. ■ Reduced tumor growth in SCID mice injected with Aim2-silenced UT-SCC7 cells39. ■ Reduced tumor growth in nude mice injected with Nlrp3-silenced WSU-HN6 cells69. |
■ More colony formation in LPS+ATP–treated CAL27 cells200. ■ Reduced viability and increased apoptosis of Nlrp3-silenced WSU-HN6 and CAL27 cells treated with 5-FU159. ■ Reduced viability and increased apoptosis of Aim2-silenced UT-SCC7 cells39. ■ Increased proliferation and migration of IL-1β–treated DOK and TW2.6 cells201. |
| Pancreatic Cancer | ■ Reduced pancreatic neoplasia and improved survival of Aim2-deficient KCPink1−/− and KCPark2−/− mice51. ■ Delayed malignant progression and extended survival in Nlrp3−/− KC mice48. ■ Reduced PDA tumor growth in the pancreata of Nlrp3−/−, Asc−/− and Casp1−/− mice48. ■ Reduced Panc02 tumor growth in chimeric WT mice adoptively transferred with Casp1−/− BMCs199. |
N/A |
| Hematopoietic neoplasms | ■ Delayed multiple myeloma progression and prolonged survival in Nlrp1−/− and Il18−/− mice challenged with myeloma cell lines52. | ■ Enhanced proliferation of IL-18–treated Pfeiffer cells38. |
| Malignant Mesothelioma | ■ Delayed onset and reduced tumor incidence in Asc−/− mice202. | N/A |
Abbreviations: AOM, azoxymethane; BMCs, bone marrow cells; BMSCs, bone marrow stem cells; DEN, N,N-diethylnitrosamine; DMBA, dimethylbenz[a]anthracene; DSS, dextran sodium sulfate; HCC, hepatocellular carcinoma; KC, Pdx1creKrasG12D/+; LLC, Lewis lung carcinoma; MCA, methylcholanthrene; N/A, information not available;
NSCLC, non–small cell lung cancer; PDA, pancreatic ductal adenocarcinoma; pDCs, plasmacytoid dendritic cells; SCID, severe combined immunodeficiency; TPA, 12-O-tetradecanoylphorbol-13-acetate; 4-NQO; 4-nitroquinoline 1-oxide; 5-FU, 5-Fluorouracil.
Cell lines: A375, B16F10, HMB2, VUP, SK23, MJM, WM115, 1205Lu, Hs294T: melanoma; RM-1, PC3-ML, DU145: prostate cancer; E0771, Py8119, PyT8, MDA-MB-231, MDA-MB-435, MDA-MB-453, 4T1, BT474, MCF-7: breast cancer; PC, A549, H1703, H1975: lung cancer; T241: fibrosarcoma; BGC-823, AGS, MKN1: gastric cancer; CT26, MC38, HCT116, HPCC, HT29, Caco-2, HCA7, SW620: colon cancer; Hepa1–6: liver cancer; 9L, C6, SR-B10, GL-261, U87, T98G, U251: glioma; UT-SCC7, WSU-HN6, CAL27, DOK, TW2.6: squamous cell carcinoma; Panc02: Pancreatic cancer
Inflammasome activation
Upon sensing stimuli, inflammasome sensors generally recruit the common adaptor molecule apoptosis-associated speck-like protein containing a CARD (ASC), which forms a platform for the activation of caspase-1. Recruitment and oligomerization of these components by homotypic CARD-CARD or PYD-PYD interactions are the basis of inflammasome assembly, which is followed by proximity-induced autoprocessing of caspase-1. ASC is a bipartite protein containing both PYD and CARD. PYD-containing AIM2, NLRP3, and Pyrin interact with CARD of caspase-1 via ASC, whereas, CARD-containing NLRP1b and NLRC4 do not necessarily require ASC for their interaction with caspase-14. Active caspase-1 proteolytically cleaves the pro-inflammatory cytokines pro-IL-1β and pro-IL-18 into bioactive IL-1β and IL-182. Similarly, active caspase-1 cleaves gasdermin D (GSDMD), which subsequently forms pores in the cell membrane, thereby allowing the secretion of mature IL-1β and IL-18 as well as certain damage-associated molecular patterns (DAMPs). IL-1β can pass through the GSDMD [G] pore in the absence of membrane rupture suggesting that pyroptosis is not prerequisite for the release of this cytokine10. Moreover, vesicular release has been described as an alternative mechanism for slow release of mature IL-1β, however, effective and robust release of IL-1β depends on GSDMD11,12. Unlike in myeloid cells where caspase-1 is required for processing of IL-18 and IL-1β, it appears that caspase-1–independent IL-18 processing by caspase-4 occurs in human intestinal epithelial cells13. These distinctive features suggest mechanistic differences in the processing and release of cytokines between cell types.
There are two distinct molecular mechanisms governing the ability of different inflammasome sensors to initiate inflammasome assembly. NLRP1, NLRP3, and Pyrin assemble inflammasomes without directly binding ligands, but caspase-11 (caspase-4 and caspase-5 in humans; sensor for non-canonical NLRP3), AIM2, and NAIPs directly bind with their ligands for inflammasome activation7.
Non-ligand binding sensors
NLRP1
Human NLRP1 has 3 paralogs NLRPI(a–c) in mouse. Unlike human NLRP1, mouse NLRP1 lacks PYD. NLRP1 b responds to lethal toxin of Bacillus anthracis. Lethal factor component of the anthrax lethal toxin or any protease that induces N-terminal proteolytic cleavage at the amino terminus or function to find domain of NLRP1 can activate this inflammasome14,15 (FIG. 1 a). It has been proposed that lethal factor-mediated cleavage relieves an intramolecular autoinhibition or induces conformational changes that subsequently leads to oligomerization of the receptor.
Fig. 1 |. Diverse mechanisms of inflammasome activation.

a| Bacillus anthracis toxin, which contains protective antigen (PA) and lethal factor (LF), activates NLRP1 inflammasome by inducing cleavage at the N-terminal linker region (red dotted line). PA forms pores in the host cell membrane which is used by LF to enter the cell. Auto-proteolytic cleavage at the FIIND domain (black dotted line) has also been observed. Caspase-1 is recruited into the complex via ASC- or by direct association with NLRP1 through CARD–CARD interactions. b| NLRP3 inflammasome is activated by various pathogen associated molecular patterns (PAMPs) and damage associated molecular patterns (DAMPs). NEK7 is an upstream activator of NLRP3 inflammasome assembly. Noncanonical NLRP3 inflammsome activation induced by cytosolic LPS is caspase-4/5/11 dependent. Caspase 11 cleaves the N-terminal domain of gasdermin-D to induce pyroptosis. c| Bacterial type 3 secretion system (T3SS) components (needle and inner rod) and flagellin are sensed by NAIPs to assemble and activate NLRC4 inflammsome. The basal transcription of NAIPs is regulated by IRF8. NLRC4 enables ASC-dependent or ASC-independent CARD-CARD interaction for casapse-1 activation. d| AIM2 is activated by host or pathogen-derived dsDNA. During Francisella tularensis infection, interferon regulatory factor 1 (IRF1) transcriptionally regulate the expression of GBPs and IRGs to liberate bacterial dsDNA which is recognized by AIM2. e| Pyrin inflammasome is activated when Pyrin senses the modification of Rho induced by Rho-inactivating toxins. Inflammasome activation results in activated caspase 1 inducing pyroptosis via cleavage of the N-terminal domain of gasdermin-D that generates pores on the host plasma membrane, which leads to the release of bioactive IL-1β and IL-18.
Canonical NLRP3
Although no direct ligands of NLRP3 have been identified yet, it is known to respond to cellular aberrations characterized by lysosomal rupture, potassium efflux, mitochondrial DNA or disruption, calcium influx or decrease in cellular cAMP levels, all caused by various PAMPs and DAMPs, including toxins, pathogens, crystalline substances, and metabolites7. The serine/threonine-protein kinase NEK7 and MAP kinase TGF-β activated kinase-1 (TAK1) are shown to regulate NLRP3 activation16–18(FIG. 1b). In addition, Z-DNA-binding protein regulates NLRP3 inflammasome activation during influenza A virus infection19. Due to the broad nature of NLRP3 activation, it is unlikely that direct structural recognition or ligand binding is involved for its activation. NLRP3 may act as a sensor for homeostasis-altering molecular processes (HAMPs)20.
Pyrin
Activation of the most recently identified inflammasome, Pyrin, depends on the inactivation of host small GTPases of the Rho family via Rho-modifying toxins, which inhibits the action of serine-threonine kinases PKN1 and PKN2 to relieve the phosphorylation of Pyrin and promote its activation21,22 (FIG. 1e). RhoA inactivation occurs in different residues which can be sensed by Pyrin, indicating that Pyrin responds to perturbation of cytoplasmic homeostasis rather than directly recognizing a conserved PAMP/DAMP20.
Ligand- binding sensors
Caspase-11
Mouse caspase-11 or the human analogues, caspase-4 and caspase-5 directly bind to cytoplasmic lipopolysaccharide that subsequently leads to non-canonical NLRP3 inflammasome activation23 (FIG. 1b). The molecular events by which caspase-11 acquires protease function have been obscure. A recent report has shown that caspase-11 dimerization is sufficient for autoprocessing that generates the fully active caspase-11 responsible for cleaving GSDMD24. Although it has been demonstrated that GSDMD is required for non-canonical NLRP3 inflammasome activation, the proximal events leading to inflammasome activation are not fully understood.
AIM2
The HIN domain of AIM2 binds to cytoplasmic double-stranded DNA (dsDNA), irrespective of the sequence of the dsDNA25,26 (FIG. 1d). Binding of the sugar-phosphate backbone of dsDNA with the positively charged HIN-200 domain relieves PYD for self-oligomerization and its interaction with ASC for inflammasome activation.
NAIP-NLRC4
Recognition of bacterial ligands by NAIPs is an initial event in NLRC4 inflammasome activation. Human NAIP recognizes flagellin and components of the type 3 secretion system; mouse NAIP5 and NAIP6 bind directly to flagellin, and mouse NAIP1 and NAIP2 bind the needle and inner rod of the type 3 secretion system, respectively27,28. This ligand-binding event is followed by NLRC4 recruitment and subsequent oligomerization for the assembly of a functional NAIP–NLRC4 inflammasome complex29 (FIG. 1c). The production of NAIPs is under the control of IRF8, which therefore is required for optimal NLRC4 inflammasome activation30.
Promoting tumorigenesis
Although the primary role of inflammasome activation is to restrict pathogenic insults, persistent activation of inflammasomes propagates undesirable inflammatory responses7. It is well established that chronic inflammation contributes to most stages of tumorigenesis31. The pro-tumorigenic roles of inflammasome components have been mostly studied in tumor cell transplantation models, and are centered on proliferation and survival, immunosuppression, angiogenesis, and metastasis (FIG. 2).
Fig. 2 |. Protumorigenic roles of inflammasome components.
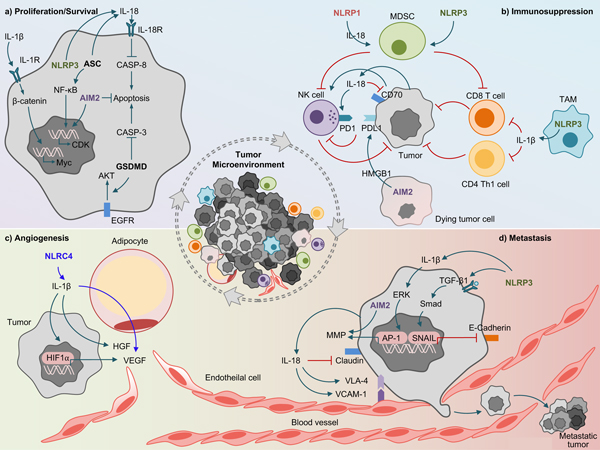
a| Proliferation. NLRP3-mediated release of IL-18 or IL-1β activates β-catenin which induces the oncogene c-Myc, that enhances proliferation of lymphoma cells. ASC in gastric epithelial cancer cells induces IL-18 production and NF-κB activation that leads to cellular proliferation. Gasdermin-D (GSDMD) promotes epidermal growth factor receptor-mediated protein kinase B (AKT) signaling in non-small cell lung cancer. b| Immunosuppression. NLRP1-mediated IL-18 production in multiple myeloma enhances the generation of myeloid-derived suppressor cells (MDSCs) in the immune niche that leads to tumorigenesis via inhibiting CD8+ T-cells and natural killer (NK)-cells. NLRP3-mediated IL-1β in tumor associated macrophages (TAMs) inhibits anti-tumor immunity of CD4+ Th1 cells and CD8+ T-cells. High mobility group box 1 (HMGB1) released from the disrupted mitochondrial iron metabolism in an AIM2-dependent manner promotes the upregulation of the immune checkpoint programmed death-ligand 1 (PDL1) in pancreatic ductal adenocarcinoma. IL-18 facilitates immune escape of gastric cancer cells by upregulating programmed cell death protein 1 (PD1) on NK-cells and downregulating CD70 on tumor cells. CD70 increases the cytotoxicity of NK-cells and induces tumor-specific T-cell memory. c| Angiogenesis. NLRC4 inflammasome mediated release of IL-1β acts on adipocytes to induce the production of vascular endothelial growth factor (VEGF). In tumor cells, IL-1β stimulates the secretion of hepatocyte growth factor (HGF) and induces hypoxia-inducible factor-1 (HIF1α) in tumor cells to transcriptionally regulate VEGF production. d| Metastasis. NLRP3 promotes epithelial-mesenchymal transition (EMT) by enhancing transforming growth factor beta 1 (TGF-β1) mediated Smad signaling that induces Snail expression which downregulates E-cadherin in squamous cell carcinoma and gastric carcinoma. AIM2 enhances the production of matrix-metalloproteinases (MMPs) that leads to the invasion of cutaneous squamous cell carcinoma. IL-1β increases the transcriptional activity of activator protein (AP-1) that induces MMPs increasing the invasiveness of breast ductal cancer cells. Tumor-derived IL-18 induces vascular cell adhesion molecule-1 (VCAM-1) expression in hepatic sinusoidal endothelial cells (HSECs) and very late antigen 4 (VLA-4) in melanoma cells that facilitates VCAM-1 dependent melanoma cell adhesion to HSECs.
Proliferation and Survival.
A common feature of all cancers is that they possess the ability to undergo increased proliferation and reduced cell death, both of which are stimulated by inflammation-driven mechanisms31. The inflammasome effector cytokines IL-1β and IL-18 released during acute and chronic inflammation may function in both a paracrine and an autocrine manner to induce cellular proliferation (FIG. 2a)
Notably, IL-18 but not IL-1β, produced from gastric epithelium promotes the development of gastric tumor in gp130F/F mice [G] as a downstream effect of inflammasome activation. IL-18 inhibits caspase 8–mediated apoptosis in gastric cancer cells thereby promoting cell survival in a cell-autonomous manner32. Furthermore, IL-18 production from immune cells in the particular study is not sufficient to drive gastric tumorigenesis, suggesting that IL-18 production from epithelial tumor cells rather than immune cells is detrimental. Perhaps IL-18 is produced in higher amounts from gastric epithelium, as the study has showed higher gene expression of IL-18 in these cells compared to immune cells. Although IL-1β does not have such a role in survival, it also accelerates gastric tumorigenesis by a different mechanism, discussed in detail in the immunosuppressive section33. Therefore, the functional dichotomy between IL-18 and IL-1β in gastric cancer could be explained, at least in part, by IL-18 acting on epithelial (tumor) cells expressing higher levels of IL-18R to promote cell survival32, whereas IL-1β-responsive immune cells provide a tumor-supporting microenvironment33. However, IL-1R signaling is important for proliferation of tumor cells in a spontaneous model of breast cancer34. IL-1β acts on breast cancer cells to enhance nuclear translocation of β-catenin that results in induction of multiple oncogenes responsible for cell proliferation35. IL-18 production from gastric epithelial cancer cells is dependent on ASC, and ablation of ASC correlates with reduced NF-κB activation32 (FIG. 2a). NF-κB being a mediator of ASC in the gastric epithelium is consistent with NF-κB promoting cecal carcinogenesis in which ASC has been assigned a pro-tumorigenic role36. ASC suppresses IL-1R–mediated NF-κB signaling in primary melanoma cells to inhibit proliferation, whereas such a suppressive function is inert in metastatic melanoma cells and instead ASC contributes to an inflammasome-mediated positive feedback loop of IL-1R signaling in metastatic melanoma to promote proliferation37.
Furthermore, studies have highlighted the role of individual inflammasome sensors in tumor proliferation and survival. NLRP3 activation in lymphoma cells produce IL-18 which attenuates dexamethasone-induced apoptosis38 thereby promoting their survival (FIG. 2a). The decreased tumor growth of cutaneous squamous cell carcinoma (SCC) in the absence of AIM2 is associated with its ability to suppress cell death and promote cellular proliferation by inducing the expression of several genes involved in cell cycle regulation39. Although the precise regulatory mechanisms by which AIM2 induces these genes has not been reported, this might be due to the indirect effect of cytokines released by SCC cells in response to AIM2 inflammasome activation.
Another consequence of inflammasome activation, besides cytokine maturation, is GSDMD cleavage, which generates cytokine-releasing pores and culminates in pyroptosis3. The precise relevance and function of the pyroptosis executioner GSDMD in tumorigenesis is largely unknown. Gsdmd-silenced non-small cell lung cancer cells exhibit reduced EGFR–AKT signaling and enhanced caspase-3 cleavage and apoptosis, leading to suppression of tumor growth in transplanted mice40. Together, these studies show how inflammasome signaling provides proliferative and survival cues by activating NF-κB or β-catenin and inhibiting apoptosis during tumorigenesis. But how these diverse signals are distributed among, and finally integrated in different cell types in distinct tumor settings requires further investigation.
Immunosuppression.
In response to invading tumor cells, our immune system launches a potent anti-tumor response with an influx of inflammatory cells into the tumor microenvironment [G] (TME)41. However, cancer cells can employ multiple mechanisms to evade immune surveillance. The release of inflammasome-dependent cytokines IL-18 and IL-1β, and other costimulatory molecules (either directly from cancer cells or from neighboring cells) is a well-known process for shaping an immunosuppressive TME during the progression of various cancers33,42,43.
Myeloid-derived suppressor cells (MDSCs) are one of the key components of the TME and are characterized by their ability to produce inhibitory cytokines and inducible nitric oxide synthase, express arginase and induce regulatory T-cells, all of which collectively exert potent immunosuppressive activity44. IL-1β is instrumental in suppressing the tumour immune response by recruiting MDSCs. Xenograft tumors that overexpress IL-1β show a greater accumulation of immunosuppressive MDSCs and more rapid tumor progression45, and Il1r−/− mice implanted with mammary carcinomas exhibit delayed accumulation of MDSCs and tumor growth46. The key role of IL-1β in MDSCs recruitment and activation is also shown in gastric cancer. Here, overexpression of IL-1β leads to early recruitment of MDSCs during the spontaneous development of gastric adenocarcinoma, and IL-1β directly activates these MDSCs33. IL-18 suppresses the immune response through a distinct mechanism, which involves activation of T-cell immune checkpoints. In particular, IL-18 promotes melanoma or colon carcinoma metastasis likely by inhibiting natural killer (NK)-cell tumoricidal function through surface induction of the immunosuppressive co-stimulatory molecule PD-142 (FIG. 2b). IL-18 also facilitates the immune escape of gastric cancer cells by downregulating CD70, a costimulatory molecule that increases the cytotoxicity of NK-cells and induces tumor-specific T-cell memory43. Although these studies illustrate the direct immunosuppressive functions of IL-1β and IL-18, the specific inflammasome complexes to regulate these effectors are still being explored.
NLRP3 is crucial for accumulating MDSCs in tumors and inhibiting antitumor T-cell immunity after dendritic cell vaccination47. NLRP3 signaling in tumor-associated macrophages (TAMs) [G] drives immunosuppressive CD4+ T-cell polarization in the TME of pancreatic ductal adenocarcinoma (PDA) via IL-1β 48. Also, NLRP3-dependent release of IL-1β affects immune cells, primarily CD4+ T-cells, to express and release IL-22, which has been associated with aggressive growth of multiple cancers including lung, breast, gastric and skin cancer49. In contrast to NLRP3 which mediates its effects by secreting inflammasome effector cytokines, studies point to AIM2 having a role in inflammasome-dependent release of alarmins [G] that promotes tumorigenesis. Immunosuppressive tumor-associated plasmacytoid dendritic cells [G] in lung cancer are associated with a heightened ability to secrete IL-1α that depends on AIM2 and subsequent calpain activation50. Mitophagy proteins PINK1 and PARK2 suppress oncogenic Kras-driven pancreatic tumorigenesis by keeping mitochondrial iron levels in check. Mechanistically, disrupted mitochondrial iron metabolism in the absence of these proteins induces AIM2-dependent HMGB1 release in PDA cells, which promotes upregulation of the immune checkpoint protein PD-L1. Blocking of HMGB1, but not IL-1β or IL-18, reduces neoplastic lesions and prolongs survival of the mice51. These two studies highlight the significance of understudied inflammasome-mediated alarmins in tumorigenesis.
Lastly, NLRP1 inflammasome-mediated IL-18 production in multiple myeloma augments the generation of MDSCs in the immune niche, leading to accelerated disease progression52 (FIG. 2b). While the NLRP1 inflammasome is known to be activated by anthrax lethal toxin, Toxoplasma infection, and hematopoietic stress15, the mechanism of its activation in the TME is largely unknown. NLRP1 may sense hematopoietic stress or the disturbance of intracellular homeostasis in the TME of multiple myeloma. Overall, inflammasomes and their effector cytokines lead to the accumulation of MDSCs and expression of immune checkpoint molecules, thereby inhibiting tumoricidal function of T-cells and NK-cells.
Angiogenesis.
Angiogenesis, the process by which new capillaries and vessels emerge from pre-existing vasculature, is vital for the progression from a small localized tumor to an enlarging tumor with the ability to metastasize. It is tightly regulated by numerous proangiogenic and antiangiogenic factors53. Inflammasome effector cytokines have been shown to regulate angiogenesis in several settings. Rapid tumor growth in mice implanted with IL-1β–expressing lung cancer cells is associated with hyperneovascularization induced by proangiogenic factors secreted into the TME54. IL-1β produced from tumor cells induces the production of proangiogenic factors, such as vascular endothelial growth factor (VEGF) and hepatocyte growth factor54, and chemokines such as CXCL2. Moreover, IL-1β–mediated hypoxia-inducible factor-1α upregulation stimulates VEGF production in lung cancer cells55 (FIG. 2c). Although these studies exemplify the angiogenic functions of IL-1β, how distinct inflammasome complexes regulate this cytokine to modulate angiogenesis need further investigations.
Metastasis.
Tumor metastasis is a complex process that involves all sequential events from tumor cell dissemination from the primary foci to the formation of metastatic nodules in distant organs56. The environment of the distant metastatic target organs undergoes re-programming, mostly by recruiting immune cells, to favor the growth of the tumor56,57. Tumor cell invasion and migration are key events in the metastatic cascade57. Several mechanisms are shown for IL-1β or IL-18–mediated invasion and migration of cancer cells (FIG. 2d). These cytokines facilitate the invasion of malignant cells into the circulatory system and enhance the expression of adhesion molecules on endothelial and malignant cells, allowing dissemination and implantation into remote tissues58. Il1b−/− mice have reduced lung metastasis and increased survival following inoculation of B16 melanoma cells59. Similarly, systemic administration of IL-1 receptor antagonist (IL-1RA) reduces the size and numbers of hepatic metastases of melanoma and improves survival of the mice60. Il1b−/− mice are comparatively less resistant to hepatic metastasis than Casp1−/−Casp11−/− mice, which fail to produce both mature IL-1β and IL-18, suggesting that both cytokines contribute to metastasis. In this particular model, IL-18 acts downstream of IL-1β to facilitate inflammation-augmented hepatic metastasis by increasing vascular cell adhesion molecule-1 (VCAM-1) expression in hepatic-sinusoidal endothelial cells (HSECs) [G], which allows cancer cell adhesion58. IL-18 also induces very late antigen-4 in melanoma cells61, which facilitates VCAM-1–dependent melanoma cell adhesion to HSECs62. Depletion of IL-18 in transplanted melanoma cells or systemic neutralization of IL-18 in recipient mice reduces lung metastasis42 indicating that IL-18 from cancer cells also contributes to metastasis. The outcome of metastasis appears to be greatly affected by the cancer cell type and source of IL-18 production. For instance, IL-18 promotes melanoma58, but suppresses colon cancer63 metastasis to the liver. Colon carcinoma–induced IL-18 production from liver Kupfer cells promotes Fas-Fas ligand (FasL)-driven cell death63 (FIG. 2d). It is interesting to note that despite using the same intrasplenic injection model, these two studies presented with contrasting outcomes of hepatic metastasis indicating that IL-18 production induced by different cancer cell types dictates metastasis.
Similarly, the effect of NLRP3 activation may differ according to the cancer type, route of inoculation, and metastatic site. Hepatic metastasis following splenic injection of colon cancer cells is enhanced in Casp1−/−Casp11−/− and Nlrp3−/− mice, which is dependent on IL-18 but not IL-1β63. In contrast, a similar study has suggested that NLRP3-dependent IL-1β secretion from macrophages may shift tumor cells to a more migratory phenotype to drive hepatic dissemination and metastasis of colon cancer 64. However, a critical flaw in the latter study is that different strains of WT and KO mice have been used, which generates difficulty in assessing the contribution of the gene of interest. On the other hand, the reduced lung metastasis in Casp1−/− and Nlrp3−/− mice after orthotopic implantation or intravenous injection of EO771 breast cancer cells supports the idea that inflammasome activation regulated by NLRP3 encourages metastasis in this setting65. Furthermore, there is evidence that NLRP3 also promotes metastasis independently of its inflammasome activity. Nlrp3−/− mice, but not Casp1−/− mice, have lower numbers of lung metastases after intravenous inoculation of melanoma or prostate carcinoma cells. Resistance to metastasis is attributed to enhanced NK-cell activity in the Nlrp3−/− mice66. Although tumor growth and lung metastasis in Casp1−/−Casp11−/− mice are comparable to that in WT mice in a model of breast cancer with spontaneous metastasis (PyMT-MMTV transgenic mice)34, lung metastasis is reduced in Nlrp3−/− mice implanted with tumors derived from PyMT-MMTV transgenic mice65. Further study is required to explain whether this discrepancy results from inflammasome-independent functions of NLRP3 or from the differential contribution of inflammasomes in tumor cells versus host cells.
Tumor cells lose epithelial markers and gain mesenchymal traits during primary tumor invasion and metastasis67. IL-1β and IL-18 have been reported to induce epithelial–mesenchymal transition [G] (EMT) in multiple cell types68. IL-1β downregulates E-cadherin expression and upregulates Snail expression in SCC69,70 and gastric cancer cells71. Tumor invasion also involves degradation of the extracellular matrix by matrix metalloproteinases72[G]. IL-1β synergistically acts with growth factors to upregulate MMP-9 by increasing transcriptional activity of AP-1, thereby enhancing the invasiveness of breast ductal cancer cells73. Furthermore, interruption of cell–cell adhesion is a crucial step in invasion and metastasis. IL-18 enhances breast cancer cell migration via downregulation of claudins, which are junctional adhesion molecules74. Of interest, NLRP3 can promote EMT by enhancing TGF-β1 signaling and Smad activation independently of caspase-1 or inflammasome-regulated cytokines75,76 and reduced invasion of cutaneous SCC in the absence of AIM2 is associated with decreased production of MMP-13 and MMP-139.
Together, these studies show that the role of inflammasomes in metastasis is bidirectional and the outcomes vary depending on factors such as the type of cancer and route of inoculation. Extending our knowledge gained from transplantable tumors, it would be exciting to explore how inflammasomes are involved in metastasis from primary tumor settings.
Suppressing tumorigenesis
The tumor-suppressive function of the inflammasome has been mostly demonstrated in the colorectal cancer (CRC) model. Mice lacking the inflammasome-initiating sensors NLRP1b77, NLRP378,79, AIM280,81, NLRC482,83, and Pyrin84 are hypersusceptible to colitis-associated cancer induced by the DNA-damaging agent azoxymethane [G] (AOM) and chemical colitogen dextran sulfate sodium (DSS). AOM is a precursor of methylazoxymethanol, which can damage DNA by methylation of guanine. Tumors induced by AOM frequently carry mutations in K-Ras and β-catenin mimicking human CRC. AOM combined with DSS accelerates tumor development and is commonly used to study inflammation-mediated CRC85. Inflammasome sensors assemble a fully functional inflammasome complex by recruiting ASC and caspase-1, which are also important in mediating protection against CRC77–80,82,83 (TABLE 1).
Protection in colorectal cancer
The ability of NLRP1b, NLRP3, and Pyrin to protect against CRC is attributed to the effector function of caspase-1 to mediate secretion of IL-18, a key cytokine that promotes epithelial barrier regeneration during the early stages of colitis78,79,86–89 (FIG. 3b). In addition, the ability of IL-18 to mount NK- or T-cell–mediated anti-tumor immune responses may contribute to this protection90. Injection of recombinant IL-18 into Casp1−/− mice reduces the prevalence of tumors in response to AOM and DSS79. In contrast, another study found that mice with a conditional deletion of IL-18 in either epithelial cells or hematopoietic cells are more resistant to DSS-induced colitis91. Furthermore, amelioration of intestinal inflammation by neutralization of IL-18 suggests a detrimental role of IL-18 in colitis92,93. Notably, IL-18 can inhibit the expression of soluble IL-22 binding protein largely in hematopoietic cells, which modulates the bioavailability of IL-22, a cytokine that suppresses early intestinal damage but also promotes tumorigenesis over time94 (FIG. 3b). Therefore, early local production of IL-18 by enterocytes or cells residing in the lamina propria may contribute to epithelial repair after injury, whereas its excessive production during chronic inflammation potentially promotes tumorigenesis.
Fig. 3 |. Protective roles of inflammasomes in cancer.

a| IL-18 released from Kupffer cells in an NLRP3 inflammasome dependent manner controls liver metastasis of colon carcinoma by enhancing Fas ligand (FasL) expression on natural killer (NK) cells, promoting their tumoricidal function. b| NLRP3, NLRP1b, Pyrin, and NLRP6 inflammasomes mediate the production of IL-18 that enhances the barrier function and regenerates epithelial cells to protect against colorectal cancer (CRC). IL-18 modulates the bioavailability of IL-22 which has dual effects in CRC. Pyrin-mediated release of IL-18 promotes CD8+ T-cells to inhibit CRC. c| NLRP6 and NLRP12 function in both the myeloid cells and enterocytes to negatively regulate the expression of NF-κB-mediated inflammatory cytokines and chemokines. These inflammatory mediators induce the chemotaxis of immune cells into the lamina propria during colitis. AIM2 inhibits DNA-dependent protein kinase (DNA-PK)–mediated phosphorylation of AKT and cMyc induction to inhibit overt proliferation of intestinal stem cells. NAIPs inhibit signal transducer and activator of transcription 3 (STAT3) hyperactivation and cellular proliferation to prevent CRC, whereas NLRC4 restricts proliferation and drives apoptosis of epithelial cells. d| NLRC4 in myeloid cells releases chemokines which potentiate the production of IFN-γ from CD4+ and CD8+ T-cells to suppress melanoma growth.
Signaling through the NLRP3 inflammasome in the hematopoietic78 and non-hematopoietic compartment86 are essential for mediating protection against colonic tumorigenesis. One study has suggested that Nlrp3−/− mice are more resistant to DSS-induced colitis than are WT mice95, whereas another study has found similar tumor prevalence between WT mice and Nlrp3−/− mice82. However, colorectal tumorigenesis induced by AOM in the presence of a high-cholesterol diet is mediated by NLRP3 inflammasome activation96. Could these contrasting roles be explained to a certain extent by the differences in the gut microbiota between different animal facilities or the use of littermate versus non-littermate controls? It is important to note that genetically modified mice carrying the gain-of-function mutation Nlrp3R258W [G], which is homologous to the human NLRP3R260W mutation, are strongly resistant to experimental colitis and CRC. In these mice, enhanced IL-1β production in colon reshapes intestinal microbiota which in turn supports the development of Treg-cells to restrict gut inflammation97. Furthermore, increased production of IL-1β by ASC-dependent inflammasome activation in the absence of PTPN2 in myeloid cells protects against CRC. In this particular model, more colitis in the mice lacking PTPN2 is associated with decreased tumorigenesis98. Injection of recombinant IL-1β into Nlrp1b−/− mice mediates protection against colitis77. This suggests that IL-1β plays an important role in inflammasome-mediated protection against colitis. In contrast, a previous study has shown that Il1r−/− mice suffer similar susceptibility as WT to CRC88, which might be due to a nullified outcome of differential IL-1R signaling in different tissues. Indeed, a recent study has elegantly demonstrated opposing roles of cell-type specific IL-1R signaling in CRC. The pro-tumorigenic roles of IL-1R in epithelial and T-cells are counteracted by its effects on myeloid cells, particularly neutrophils, where IL-1R signaling contributes to deter tumor-infiltrating bacteria and dampen CRC promoting inflammation99. A similar approach has been studied for the role of ASC in skin tumorigenesis, where inflammasome activation driven by ASC in myeloid cells favors epithelial skin tumorigenesis, whereas ASC in keratinocytes serves to limit proliferation, possibly through p53 activation in an inflammasome-independent manner100. Thus, inflammasome signaling elicits cell-type specific responses, which altogether, determine the propensity for tumorigenesis.
Inflammasome-independent protection in colorectal cancer
The ability of inflammasome sensors to provide protection against cancer does not always rely on the effector cytokines. The protective role of NLRC4 in the development of CRC is associated with its intrinsic ability to restrict the proliferation and drive apoptosis of epithelial cells in both steady-state and the early phase of tumorigenesis82 (FIG. 3c). Apart from CRC, NLRC4 amplifies inflammatory signaling pathways in macrophages independently of inflammasome assembly and potentiates the production of IFN-γ in CD4+ and CD8+ T-cells to suppress melanoma growth in mice101 (FIG. 3d). Although mouse NAIP1–6 are components of the NLRC4 inflammasome, NAIP-mediated protection against CRC is related to their ability to inhibit hyperactivation of the transcription factor STAT3 and the expression of genes encoding anti-apoptotic and proliferation-related molecules, an NLRC4 inflammasome–independent and epithelium-intrinsic function of NAIPs102 (FIG. 3c). Furthermore, simultaneous recognition of flagellin in tumor cell lines by NAIPs and TLR5 induces tumor cell clearance by innate immune cells and activation of tumor-specific T-cell responses in mice103.
An inflammasome-autonomous function of AIM2 in inhibiting inflammation-induced and spontaneous colorectal tumorigenesis has been suggested by two independent studies80,81. AIM2 interacts with the DNA-dependent protein kinase to limit PI3K/AKT activation80,104, thereby suppressing overt proliferation of colonic stem cells and inducing cell death81,104 (FIG. 3c). A similar negative regulatory effect of the putative inflammasome sensor NLRC3 on PI3K-AKT signaling in limiting mTOR activation has been described to inhibit cellular proliferation105. Although the specific ligand that AIM2 senses in the colon has yet to be determined, host-DNA released after intestinal injury or DNA derived from the gut microbiota might activate AIM2106. Indeed, AIM2 localizes to DNA in the nucleus of intestinal epithelial cells and bone marrow cells in response to dsDNA breaks caused by ionizing radiation or chemotherapeutic agents107. Whether the DNA-sensing property of AIM2 is required for its tumor suppressive function is not known. Another mechanism by which AIM2 restricts tumorigenesis is through antagonizing NF-κB activity to induce apoptosis, as shown in breast cancer cells108,109.
NLRP6110–112 and NLRP12113 are potential inflammasome sensors. NLRP6-dependent secretion of mucin 2 [G] (MUC2) in the intestinal epithelium (FIG. 4) is required to clear colitogenic bacteria. However, MUC2 secretion can be either dependent or independent of the inflammasome111,114. Likewise, despite of its ability to form inflammasomes, the protective function of NLRP12 against colitis and associated tumorigenesis is attributed to the negative regulation of inflammation via suppression of NF-κB pathways115,116 (FIG. 3c).
Fig. 4 |. Inflammasome–microbiota axis in intestinal homeostasis.

Several immune mechanisms work in concert with the intestinal microbiota to maintain intestinal homeostasis and provide protection against colorectal cancer (CRC). Dysbiosis inhibits NLRP6 inflammasome activation in enterocytes. Conversely, NLRP6 in enterocytes inhibits intestinal dysbiosis and CCL5-mediated recruitment of immune cells into the lamina propria. Increased IL-6 production from myeloid cells in the absence of NLRP6 acts on neighboring enterocytes, to activate the oncogenic transcription factor signal transducer and activator of transcription 3 (STAT3). The microbial metabolite taurine activates, whereas histamine and spermine suppress NLRP6– mediated IL-18 secretion. NLRP6 in sentinel goblet cells induces the secretion of mucin 2 (MUC2) which expels intruding bacteria found in the inner mucous layer providing protection against CRC.
Inflammasome-microbiota–diet axis.
The composition of symbiotic microorganisms that live in our gut, the so-called gut microbiome, is one of the principal environmental factors that contributes to immune homeostasis in the intestine117. Disturbance in the microbial ecology results in the development and progression of CRC, which can be modulated by the use of broad-spectrum antibiotics118. In addition to their conventional role as a guardian of cellular integrity, inflammasomes can serve as a surveillance system, regulating the host/microbiota crosstalk in health and disease. The contrasting phenotypic outcomes in mice with identical genetic deficiencies are intriguing and can largely be attributed to different microbial composition across mice. The notion of inflammasomes regulating the gut microbial landscape emerged from a study that found that a ‘colitogenic’ gut microbial community, defined by an increased or decreased relative abundance of Prevotellaceae or Lactobacillus, respectively, predisposes Nlrp6−/− mice to exacerbated DSS-induced colitis and tumorigenesis or low-grade intestinal inflammation110. Dysbiosis has also been reported in mice that are deficient in inflammasome components AIM2, NLRP3, ASC, caspase-1, and IL-18110,119,120. The distinct microbiota profile characterized by an increased relative abundance of Lactobacillus murinus, and decreased abundance of colitogenic bacteria, Akkermensia muciniphila, is associated with decreased gut inflammation and CRC in Nlrp3R258W mice97. The colitogenic gut microbiota can be transmissible, as suggested by altered susceptibility of mice lacking NLRP6, AIM2, or NLRP12 following reciprocal exchange of gut microbiota with WT mice106,110,121. Dysbiosis driven by NLRP6 deficiency elevates the production of CCL5, a chemokine that recruits immune cells in the intestinal lamina propria and mediates the secretion of IL-6, which in turn acts on epithelial cells to drive a pro-tumorigenic response83 (FIG. 4). Consistently, CCL5 deficiency prevents dysbiosis-induced colitis and tumorigenesis, suggesting that CCL5 mediates immune dysregulation downstream of the microbial community83,110. Furthermore, metabolites produced by gut microbiota may influence inflammasome-mediated disease outcome. The microbiota-derived metabolite taurine promotes the production of IL-18 in an NLRP6-dependent manner, whereas histamine and spermine inhibit NLRP6122 (FIG. 4). An additional mechanism that NLRP6 employs to protect against tumorigenesis is to induce the secretion of MUC2 in sentinel goblet cells that expels intruding bacteria from the inner mucous layer111,114 (FIG. 4). This phenotype is unaffected by microbiota transfer, highlighting that certain biological functions of NLRP6 are not influenced by gut microbiota.
The effects of the inflammasome–gut microbiota axis have been extended from the gut to systemic metabolic and inflammatory processes. Microbiota composition influences the susceptibility of mice lacking NLRP3 to non-alcoholic steatohepatitis, which can progress to hepatocellular carcinoma123. However, more recent studies argue that dysbiosis observed in inflammasome-deficient mice is dependent on the facilities used to house the animals124,125. Diet and the aseptic techniques used by different facilities may affect the ecology of gut microbiota, which in turn affects the activation status of inflammasomes. Indeed, mice fed with high-fat diet (HFD) have different microbial communities than those fed a normal diet or low-fat diet126. Dietary cholesterol and dietary-derived deoxycholic acid [G] serve as endogenous danger signals that activate the NLRP3 inflammasome and contribute to HFD-related colonic inflammation and CRC96,127. On the other hand, NLRP3 activation by short-chain fatty acids or inhibition by omega-3 fatty acids prevent inflammation128,129. Lastly, obesity is a risk factor in numerous cancers, and recent evidence presents inflammasome involvement as one link between this form of metabolic dysregulation and tumor-sustaining angiogenesis. Tumorigenesis in HFD-fed mice following orthotopic implantation of breast cancer cells is mediated by NLRC4 inflammasome activation. There is increased gene expression of NLRC4 in the obese TME of humans and mice. Immune cells with pronounced NLRC4 inflammasome activation are recruited in the TME of obese mice, leading to IL-1β release, which acts on adipocytes to promote VEGF production130. Metabolites of HFD or the alteration of gut microbiota in HFD-fed mice may activate the NLRC4 inflammasome122,126. These studies highlight a dynamic and emerging association among diet, microbiota, and inflammasome activation, indicating that dysbiosis influenced by inflammasomes should be revisited and interpreted with care.
Inflammasomes in human cancer
Evidence presented above from studies of murine models highlights the multifaceted roles of inflammasomes in cancer. Inflammasome signaling in human cancer is controlled by a combination of genetic factors, such as inherited genomic variations and acquired somatic mutations, and environmental factors that can affect epigenetics, gene expression, and ultimately provide stimulation for activation. Our understanding of the significance of inflammasomes in human cancer susceptibility has been enhanced by investigations of the genotype–phenotype correlations and gene expression profiling in cancers.
Gene polymorphisms and mutations
Polymorphisms in the genes encoding inflammasome components are associated with increased predisposition to multiple cancers. Several single-nucleotide polymorphisms (SNPs) in the NLRP3 region are associated with susceptibility to Crohn’s disease [G] 131,132, which is a strong risk factor for CRC. Among those SNPs, the gain-of-function NLRP3 (Q705K) is associated with poorer survival in advanced-stage CRC133. The same NLRP3 variant is a risk allele for melanoma in Swedish males134. Individuals with polymorphisms in NLRP3, NLRP12, and CASP1 have a greater risk of gastric cancer when they are infected with Helicobacter pylori, displaying the interplay between genetic and environmental factors in tumorigenesis135. A hyperactive NLRP1 mutation causes spontaneous skin inflammation and a predisposition to skin cancer136. However, other reports on the association of NLRP1 polymorphisms with asbestos-associated mesothelioma137,138 are controversial. The AIM2 gene contains a site for microsatellite instability that results in frequent gene mutation in CRC and small bowel cancers139,140. In addition, AIM2 is a potential oncogenic driver in endometrial cancer, though most microsatellite-containing genes are bystander genes141. Altogether, SNP associations provide clues for determining which inflammasome components are important for the pathogenesis of specific cancer types. Going further, it would be helpful to examine the functional outcome of individual SNPs, such as alterations in inflammasome function or changes in expression levels.
Although inflammation is associated with a higher risk of cancer, it might be protective in some cases. Patients with familial Mediterranean fever [G] suffer from autoinflammation due to hyperactive Pyrin inflammasome, however, have a lower combined incidence of cancer than the general population142. It could be that pyrin inflammasome activation is beneficial in inhibiting tumorigenesis, but these results should be interpreted with caution, as the drugs used to treat FMF patients have anti-tumor effects. Further studies are needed to define the relation between genetic polymorphisms in inflammasomes and general cancer incidence or susceptibility to certain cancers.
Differential expression
Gene-expression profiling in many cancers has revealed differential gene expression of inflammasomes, which imply that inflammasome signaling is altered in cancer. In certain types of cancer, expression of inflammasome components are upregulated. Components of NLRP3 and AIM2 inflammasomes are highly expressed in nasopharyngeal and lung cancer tissues, compared with normal tissue, and this molecular signature is correlated with improved probability of survival of nasopharyngeal cancer143,144. On the other hand, the NLRP3 inflammasome is overexpressed in oral SCC tissue and is associated with unfavorable pathology. In oral SCC tissue, enhanced expression of ASC is an independent predictor of poor prognosis145. The discrepancy in the role of NLRP3 in cancer of anatomically adjacent oral and nasopharyngeal epithelia could be due to the distinct etiology of cancers in these tissues. While oral SCC is more attributed to chronic exposure of carcinogens such as tobacco, nasopharyngeal carcinoma is strongly linked with Epstein-Barr Virus infection. Therefore, it would be interesting to check whether inflammasome may have a protective role in other virus-associated cancers. The NLRP3 inflammasome is associated with the development of lymphoproliferative malignancy secondary to Sjogren syndrome [G]. Patients who later develop non-Hodgkin lymphoma express higher levels of NLRP3 inflammasome at the time of diagnosis146. Inflammasome activity also has a crucial role in drug resistance in acute lymphoblastic leukemia. Due to hypomethylation, CASP1 and NLRP3 expression is higher in glucocorticoid-resistant cells and cells at relapse. In vitro inhibition of caspase-1 blocks glucocorticoid receptor cleavage, thereby restoring sensitivity to treatment147. NLRP3 and NAIP expression is increased in urine sediments of patients diagnosed with bladder cancer. Combination of NAIP along with the established marker CK20 results in greater sensitivity for predicting the histological outcome of bladder cancer, suggesting that inflammasome genes could be useful as non-invasive diagnostic tools for bladder cancer148. Also, NLRP12 is highly expressed in prostate cancer epithelium compared to that of adjacent benign tissues. Despite high expression of pro-IL-1β and pro-IL-18, mature IL-1β or IL-18 are not detected in malignant prostate cancer cells, implying that NLRP12 may promote progression of prostate cancer independently of inflammasome activity149.
Reduced expression of inflammasome components in certain tumors may be indicative of their tumor suppressor function. For instance, histone methyltransferase G9A mediates invasion and migration of lung cancer cells through repressing CASP1 expression. CASP1 is downregulated in lung adenocarcinoma compared to normal tissue, and reduced CASP1 expression is associated with poor survival, indicating that CASP1 may serve as a prognostic marker in lung cancer150. The level of AIM2 expression is lower in CRC tumors than in adjacent normal tissue. The lack of AIM2 expression is correlated with higher tumor grade and is associated with poorer survival151. This finding is supported by gene expression analysis from the TCGA database showing lower levels of NLRP1, NLRP3, NLRC4 and AIM2 expression in CRC than in healthy controls, but results vary when different databases are used for the analysis152. Evaluation of inflammasome expression in hepatocellular carcinoma reveals a dynamic pattern during the progression of the disease: NLRP3 and AIM2 inflammasomes are upregulated in cirrhotic tissue but then are lost in cancer. Moreover, lower expression of these inflammasome components correlates with advanced clinicopathological features of HCC. These results are not straightforward but may provide insight into how chronic inflammation fosters malignant transformation. Cells proliferating in the presence of persistent inflammation and inflammasome activation may accumulate mutations, lose expression of tumor-suppressive NLRP3, ultimately leading to cancer progression 153. Although not well-studied as an inflammasome sensor, NLRC5 is a key transcriptional coactivator of MHC class I genes. Reduced NLRC5 expression is associated with impaired CD8+ T-cell activation and immune evasion in cancers. Lower NLRC5 expression corelates with reduced survival in melanoma, bladder and cervical cancers, suggesting that NLRC5 may be used as a prognostic marker for multiple cancers154.
Therapeutic approaches
Therapeutics targeting inflammasome activity have been shown effective for autoinflammatory periodic fever syndromes and used experimentally for treating inflammation-driven neurodegenerative diseases and metabolic disorders. Although there is ample evidence that inappropriate inflammasome regulation contributes to the pathogenesis of human cancers, where and how inflammasome therapy is applicable in cancer should be evaluated critically. One emerging approach could be modulating inflammasome activation while administering chemotherapeutics or cell therapeutics to maximize their efficacy. Prospectively, cytokine blockade may prove useful in various clinical settings, ranging from preventive regimens for individuals predisposed to chronic inflammation, to therapeutic intervention to restrict tumor progression in precancerous settings, and to palliative or adjuvant treatment for advanced cancers.
Inflammasome signaling during therapy
Recent findings address the interactions between chemotherapeutic agents and inflammasome activation. Cancer chemotherapeutics, such as anthracyclines and platinum-based drugs, elicit cytotoxicity against malignant cells. This is achieved in part, by inducing immunogenic cell death which can be modulated by inflammasome signaling155. The observation that caspase activity augments antitumor immune responses induced by anthracyclin156 led to the discovery that NLRP3 inflammasome activation is required for effective cancer chemotherapy157. The release of various DAMPs, including ATP, HMGB1, and IL-1α from dying tumor cells activates the NLRP3 inflammasome in dendritic cells (DC) to release IL-1β, which is crucial for priming IFN-γ producing CD8+ T-cells and activating NK-cells157 (FIG. 5). Furthermore, a loss-of-function polymorphism in the purinergic receptor P2RX7 in patients with breast cancer who were treated with anthracyclines, has been correlated with more metastasis157. On the other hand, gemcitabine and 5-fluorouracil (5-FU) activate NLRP3 inflammasome via cathepsin B release in MDSCs, thereby producing IL-1β levels insufficient for CD8+ T-cell priming but sufficient to drive IL-17-producing CD4+ T cells158 (FIG. 5). Likewise, NLRP3 inflammasome mediates resistance to 5-FU treatment for oral SCC159, and reduces the efficacy of DC-derived anti-tumor vaccines against grafted tumor cells47.
Fig. 5 |. Inflammasome signaling during chemotherapy.

Gemcitabine and 5-fluorouracil (5-FU) activate NLRP3 inflammasome in myeloid-derived suppressor cells (MDSCs) to produce IL-1β, which primes CD4+ T-cells to release IL-17. High mobility group box 1 (HMGB1), IL-1α, and adenosine triphosphate (ATP) released from dying tumor cells activate NLRP3 inflammasome in dendritic cells (DCs) to release IL-1β which is crucial for activating natural-killer (NK) cells and priming CD8+ T-cells to produce IFN-γ. Antibody-dependent cellular phagocytosis of tumor cells by macrophages leads to AIM2 inflammasome activation. IL-1β upregulates the expression of programmed death-ligand 1 (PD-L1) and indoleamine 2,3-dioxygenase (IDO) in macrophages that inhibit NK cell and cytotoxic T lymphocyte (CTL)-mediated cytotoxicity.
A recent study showed that AIM2 inflammasome activation by antibody-dependent cellular phagocytosis of tumor DNA into macrophages following monoclonal antibody-based cancer treatment suppresses anti-tumor immunity by upregulating PD-L1 and indoleamine 2,3-dioxygenase160 This finding suggests that the use of immune checkpoint blockade in combination with antibody treatment may be beneficial to boost treatment efficacy.
Although IL-1β signaling enhances the efficacy of doxorubicin against breast adenocarcinomas and fibrosarcomas161, the efficacy of cisplatin against malignant mesothelioma tumor cells is increased by concomitant use of IL-1Ra162. Also, impaired doxorubicin efficacy towards breast cancer cells in the presence of IL-18 suggests a role of IL-18 in the development of chemotherapy resistance163. Eradicating large solid tumors at advanced stages remains challenging, and new attempts have been made to harness inflammasome signaling for better treatments. Adoptive therapy with chimeric antigen receptor–redirected T-cells engineered with inducible IL-18 release mounts acute inflammation and reduces the number of repressor cells, resulting in an augmented immune attack against large established pancreatic tumors164.
In general, GSDMD has been viewed as an executioner of pyroptosis downstream of caspase-1 and caspase-11. However, a recent study has demonstrated that apoptosis-inducing chemotherapies are capable of inducing pyroptosis by activating gasdermin E (GSDME) in a caspase-3 dependent manner165. Based on this finding, it would be expected that chemotherapy would be more effective in GSDME–expressing cancers. It is worth to investigate whether GSDME mediated pyroptosis is a preferred mode of cell death that subsequently activates inflammasome in neighboring cancer cells.
Inflammasome-targeting therapy
Multiple approaches can be used to target inflammasome activity, including abrogation of upstream signaling pathways, inhibition of inflammasome components, and antagonism of end-products of inflammasome activation (TABLE 2). Pharmacological inhibition of the NLRP3 inflammasome has been widely implemented in laboratory studies, which have been reviewed elsewhere166,167. In clinical studies, cytokine blockade is the most successful approach, and evidence of the effectiveness of IL-1 inhibition in anticancer treatment is just emerging. A Phase II clinical trial investigating the effect of IL-1Ra anakinra in patients with early-stage multiple myeloma indicates that IL-1Ra suppresses markers of disease progression168,169. IL-1Ra blocks signaling induced by both IL-1α and IL-1β. While the production of the latter cytokine is virtually inflammasome-dependent, studies have shown IL-1α release can be either dependent or independent of inflammasome activation170. A Phase III trial of MABp1 (a monoclonal antibody targeting IL-1α) treatment in refractory CRC has shown clinical benefit, and an increase in median survival171. This suggests that the efficacy of anakinra in cancer treatment may in part come from the inhibition of IL-1α.
Table 2.
Therapeutic targets of inflammasomes in cancer
| Therapy | Inflammasome Target |
Cancer type | Clinical trial Phase |
|---|---|---|---|
| Anakinra | IL-1β | Metastatic breast cancer | Phase I/NCT01802970 203 |
| Anakinra | IL-1β | Metastatic CRC | PhaseII/NCT02090101173 |
| Anakinra | IL-1β | Multiple myeloma Plasma cell neoplasm | PhaseII/NCT00635154168,169 |
| Canakinumab/Ilaris | IL-1β | CRC, breast cancer, NSCLC, adenocarcinoma | Phase I/NCT02900664 |
| Thalidomide | Caspase-1 | Multiple myeloma204,205 | N/A |
| MCC950/CRID3 | NLRP3 | Head and neck SCC206 | N/A |
| Glyburide | NLRP3207 | N/A | N/A |
| BOT-4-one | NLRP3 | Lymphoma167 | N/A |
| IL-18BP | IL-1890 | N/A | |
| Methylene blue | NLRP3, AIM2, NLRC4208 | N/A | N/A |
| CY-09 | NLRP3209 | N/A | N/A |
Abbreviations: CRC, colorectal cancer; IL-18BP, IL-18–binding protein, N/A, information not available; NSCLC, non–small cell lung cancer; SCC, squamous cell carcinoma.
The Canakinumab Anti-inflammatory Thrombosis Outcomes Study [G] (CANTOS) was a clinical trial to determine whether IL-1β inhibition prevents cardiovascular events in patients who had high markers of inflammation after a myocardial infarction. In a secondary analysis, it was found that canakinumab reduced lung cancer incidence and mortality but did not affect other types of cancer172. The results are encouraging for cancers in which inflammation is highly involved in cancer development and progression. In a Phase II trial for metastatic CRC, co-administration of anakinra with 5-FU and bevacizumab has shown a favorable median survival similar to that of other palliative therapies173. Therefore, targeted therapy against inflammasome activation could be useful in adjuvant or palliative therapies for cancer.
Conclusion
Understanding how inflammasome signaling affects tumor initiation and progression has been one of the most intriguing questions in the field. Inflammasomes exhibit distinct and sometimes conflicting roles in multiple facets of tumorigenesis. The influence of the inflammasome in tumor promotion ranges from suppressing antitumor immunity and cell death to fostering proliferation, angiogenesis, and metastasis. The relative expression of inflammasome components differs in various cell types4, which suggests that inflammasomes perform distinct functions in different cellular compartments. The tumor-suppressive function of the inflammasome is largely reflected in its preventive role in colitis and colon cancer, which is achieved by tumor immunosurveillance, maintaining epithelial integrity, mucus production, and suppressing the proliferation of intestinal epithelial cells. Regardless of whether the dysregulated inflammasome signaling is a result of dysbiosis or vice versa, which has not been conclusively investigated yet, both affect intestinal inflammation and cancer development. In addition to genetic factors, environmental factors (e.g., diet) and experimental conditions associated with animal models influence the ecology of gut microbiota and, in turn, inflammasome activation.
Given that the outcomes of inflammasome signaling are diverse across tumor types, gaining insight about how to manage this diversity will be an important area for future investigations. Clinical trials that specifically inhibit inflammasome components in human cancers are yet to be performed (Table 2). Although a major clinical trial on inhibiting the downstream effector molecule IL-1β has recently yielded promising results172, blocking IL-1β or IL-18 individually is not the same as caspase-1 inhibition. Caspase-1 blockade can inhibit global inflammatory responses regulated by both inflammasome-dependent cytokines and pyroptosis. In addition, inhibition of a specific inflammasome like NLRP3 by MCC950 can block pathological effects of NLRP3 without compromising beneficial effects from other inflammasomes. Indeed, the development of highly specific NLRP3 inhibitors, such as MCC950, and caspase-1 inhibitors have opened exciting new avenues for translational research with the potential for therapeutic targeting of tumorigenesis. The studies highlighted in this Review outline a roadmap to developing specific anticancer therapy by modulating inflammasome signaling, which could set the stage for the concept of “tumor immunoediting” by inflammasome signaling.
Acknowledgements
Research studies from our laboratory are supported by the U.S. National Institutes of Health (AR056296, CA163507, AI124346, and AI101935 to T.-D.K.), the American Lebanese Syrian Associated Charities (to T.-D.K.). We apologize to our colleagues whose work was not cited due to space limitation.
Glossary
- Gut microbiota
Ecological community of microorganisms that harbors in gastrointestinal tract
- Pyroptosis
An inflammatory and lytic form of cell death mediated by inflammatory caspases
- gp130F/F mice
Mice are generated using a phenylalanine knock-in substitution at tyrosine 757 in the cytoplasmic domain of the IL-6 receptor β-chain Gp130. They rapidly develop tumors in the epithelium of the glandular stomach and highlight a key role for STAT3 signaling in gastric tumorigenesis
- Tumor microenvironment
Cellular and molecular environment where tumor cells interact with infiltrating immune cells, fibroblasts, blood vessels and extracellular matrix
- Tumor associated macrophages
A class of mostly abundant immune cells present in the tumor microenvironment. They have tumor-promoting phenotype via modulating tumor cell proliferation, tumor angiogenesis, invasion and metastasis
- Alarmins
Damage-associated molecular patterns such as HMGB1 or IL-1α released by damaged or necrotic cells
- Plasmacytoid dendritic cells
Type I interferon–producing subset of dendritic cells with antigen presenting potential
- Hepatic sinusoidal endothelial cells
A special type of endothelial cells which represent the interface between blood cells on the one side and hepatocytes and hepatic stellate cells on the other side
- Epithelial to mesenchymal transition
A process by which epithelial cells lose their motility and cell-cell adhesion properties and acquire mesenchymal fibroblast like properties
- Matrix metalloproteinases
A family of endopeptidases capable of degrading extracellular matrix components, influencing multiple cellular processes such as migration and adhesion
- Azoxymethane
A potent carcinogen which is used to induce colon carcinoma in mice and rats. It is metabolized to methyl-azoxymethanol in the liver and then reaches to the colon via the blood stream rather than the bile
- Nlrp3R258W mice
Genetically modified mice carrying R258W mutation in the Nlrp3 gene, which corresponds to R260W mutation in the NLRP3 gene in human. These mice develop severe cutaneous lesions, including erythema, scaling and the thickening of both epidermis and dermis, symptoms that recapitulate urticaria-like skin lesions reported in Muckle-Wells syndrome patients
- Mucin 2
A glycoprotein which is particularly prominent in the gut and is secreted from goblet cells in the epithelial lining into the lumen of the large intestine
- Deoxycholic acid
A bile-acid which emulsifies and solubilizes dietary fats in the intestine
- Crohn’s disease
An inflammatory bowel disease that involves chronic inflammation of digestive tract
- Familial Mediterranean fever (FMF)
Mutations in gene encoding pyrin (MEFV) are associated with FMF, which is an autosomal-recessive, autoinflammatory disorder characterized by episodic fever and neutrophil-mediated inflammation of serosal tissues
- Sjogren syndrome
An autoimmune disease that mostly affects the salivary and lacrimal glands, resulting in dry mouth and dry eyes
- Canakinumab Anti-inflammatory Thrombosis Outcomes Study
A randomized, double blinded, placebo-controlled trial to evaluate the effect of canakinumab, a human monoclonal antibody that selectively neutralizes IL-1β, in the prevention of recurrent vascular events in patients with previous myocardial infarction
Footnotes
Conflicts of Interest
The authors have no conflicts of interest to disclose.
References
- 1.Sharma D. & Kanneganti TD The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J Cell Biol 213, 617–629, doi: 10.1083/jcb.201602089 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Schroder K. & Tschopp J. The inflammasomes. Cell 140, 821–832, doi: 10.1016/j.cell.2010.01.040 (2010). [DOI] [PubMed] [Google Scholar]
- 3.Kesavardhana S. & Kanneganti TD Mechanisms governing inflammasome activation, assembly and pyroptosis induction. Int Immunol 29, 201–210, doi: 10.1093/intimm/dxx018 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Man SM Inflammasomes in the gastrointestinal tract: infection, cancer and gut microbiota homeostasis. Nat Rev Gastroenterol Hepatol, doi: 10.1038/s41575-018-0054-1 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Gultekin Y, Eren E. & Ozoren N. Overexpressed NLRC3 acts as an anti-inflammatory cytosolic protein. J Innate Immun 7, 25–36, doi: 10.1159/000363602 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Davis BK et al. Cutting edge: NLRC5-dependent activation of the inflammasome. J Immunol 186, 1333–1337, doi: 10.4049/jimmunol.1003111 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Man SM, Karki R. & Kanneganti TD Molecular mechanisms and functions of pyroptosis, inflammatory caspases and inflammasomes in infectious diseases. Immunol Rev 277, 61–75, doi: 10.1111/imr.12534 (2017).This review provide an overview on the functions and mechanisms of inflammatory caspases and pyroptosis.
- 8.Hanahan D. & Weinberg RA Hallmarks of cancer: the next generation. Cell 144, 646–674, doi: 10.1016/j.cell.2011.02.013 (2011). [DOI] [PubMed] [Google Scholar]
- 9.Karki R, Man SM & Kanneganti TD Inflammasomes and Cancer. Cancer Immunol Res 5, 94–99, doi: 10.1158/2326-6066.CIR-16-0269 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Evavold CL et al. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity 48, 35–44 e36, doi: 10.1016/j.immuni.2017.11.013 (2018).This study demonstrates that GSDMD facilitates active secretion of IL-1 family cytokines from live cells independently of its role as the effector of pyroptosis.
- 11.Monteleone M. et al. Interleukin-1beta Maturation Triggers Its Relocation to the Plasma Membrane for Gasdermin-D-Dependent and -Independent Secretion. Cell Rep 24, 1425–1433, doi: 10.1016/j.celrep.2018.07.027 (2018). [DOI] [PubMed] [Google Scholar]
- 12.Semino C, Carta S, Gattorno M, Sitia R. & Rubartelli A. Progressive waves of IL-1beta release by primary human monocytes via sequential activation of vesicular and gasdermin D-mediated secretory pathways. Cell Death Dis 9, 1088, doi: 10.1038/s41419-018-1121-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Knodler LA et al. Noncanonical inflammasome activation of caspase-4/caspase-11 mediates epithelial defenses against enteric bacterial pathogens. Cell Host Microbe 16, 249–256, doi: 10.1016/j.chom.2014.07.002 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Levinsohn JL et al. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PLoS Pathog 8, e1002638, doi: 10.1371/journal.ppat.1002638 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Chavarria-Smith J. & Vance RE The NLRP1 inflammasomes. Immunol Rev 265, 22–34, doi: 10.1111/imr.12283 (2015). [DOI] [PubMed] [Google Scholar]
- 16.He Y, Zeng MY, Yang D, Motro B. & Nunez G. NEK7 is an essential mediator of NLRP3 activation downstream of potassium efflux. Nature 530, 354–357, doi: 10.1038/nature16959 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shi H. et al. NLRP3 activation and mitosis are mutually exclusive events coordinated by NEK7, a new inflammasome component. Nat Immunol 17, 250–258, doi: 10.1038/ni.3333 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Malireddi RKS et al. TAK1 restricts spontaneous NLRP3 activation and cell death to control myeloid proliferation. J Exp Med 215, 1023–1034, doi: 10.1084/jem.20171922 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuriakose T. et al. ZBP1/DAI is an innate sensor of influenza virus triggering the NLRP3 inflammasome and programmed cell death pathways. Sci Immunol 1, doi: 10.1126/sciimmunol.aag2045 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Liston A. & Masters SL Homeostasis-altering molecular processes as mechanisms of inflammasome activation. Nat Rev Immunol 17, 208–214, doi: 10.1038/nri.2016.151 (2017).This article provides a concept that how inflammasomes sense changes in cellular homeostasis.
- 21.Xu H. et al. Innate immune sensing of bacterial modifications of Rho GTPases by the Pyrin inflammasome. Nature 513, 237–241, doi: 10.1038/nature13449 (2014). [DOI] [PubMed] [Google Scholar]
- 22.Park YH, Wood G, Kastner DL & Chae JJ Pyrin inflammasome activation and RhoA signaling in the autoinflammatory diseases FMF and HIDS. Nat Immunol 17, 914–921, doi: 10.1038/ni.3457 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Shi J. et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature 514, 187–192, doi: 10.1038/nature13683 (2014). [DOI] [PubMed] [Google Scholar]
- 24.Ross C, Chan AH, Von Pein J, Boucher D. & Schroder K. Dimerization and autoprocessing induce caspase-11 protease activation within the non-canonical inflammasome. Life Sci Alliance 1, e201800237, doi: 10.26508/lsa.201800237 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fernandes-Alnemri T, Yu JW, Datta P, Wu J. & Alnemri ES AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 458, 509–513, doi: 10.1038/nature07710 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hornung V. et al. AIM2 recognizes cytosolic dsDNA and forms a caspase-1-activating inflammasome with ASC. Nature 458, 514–518, doi: 10.1038/nature07725 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kortmann J, Brubaker SW & Monack DM Cutting Edge: Inflammasome Activation in Primary Human Macrophages Is Dependent on Flagellin. J Immunol 195, 815–819, doi: 10.4049/jimmunol.1403100 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Reyes Ruiz VM et al. Broad detection of bacterial type III secretion system and flagellin proteins by the human NAIP/NLRC4 inflammasome. Proc Natl Acad Sci U S A 114, 13242–13247, doi: 10.1073/pnas.1710433114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Zhao Y. et al. The NLRC4 inflammasome receptors for bacterial flagellin and type III secretion apparatus. Nature 477, 596–600, doi: 10.1038/nature10510 (2011). [DOI] [PubMed] [Google Scholar]
- 30.Karki R. et al. IRF8 Regulates Transcription of Naips for NLRC4 Inflammasome Activation. Cell 173, 920–933 e913, doi: 10.1016/j.cell.2018.02.055 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Grivennikov SI, Greten FR & Karin M. Immunity, inflammation, and cancer. Cell 140, 883–899, doi: 10.1016/j.cell.2010.01.025 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Deswaerte V. et al. Inflammasome Adaptor ASC Suppresses Apoptosis of Gastric Cancer Cells by an IL18-Mediated Inflammation-Independent Mechanism. Cancer Res 78, 1293–1307, doi: 10.1158/0008-5472.CAN-17-1887 (2018). [DOI] [PubMed] [Google Scholar]
- 33.Tu S. et al. Overexpression of interleukin-1beta induces gastric inflammation and cancer and mobilizes myeloid-derived suppressor cells in mice. Cancer Cell 14, 408–419, doi: 10.1016/j.ccr.2008.10.011 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Dagenais M. et al. The Interleukin (IL)-1R1 pathway is a critical negative regulator of PyMT-mediated mammary tumorigenesis and pulmonary metastasis. Oncoimmunology 6, e1287247, doi: 10.1080/2162402X.2017.1287247 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Perez-Yepez EA, Ayala-Sumuano JT, Lezama R. & Meza I. A novel beta-catenin signaling pathway activated by IL-1beta leads to the onset of epithelial-mesenchymal transition in breast cancer cells. Cancer Lett 354, 164–171, doi: 10.1016/j.canlet.2014.08.015 (2014). [DOI] [PubMed] [Google Scholar]
- 36.Ikuta T. et al. ASC-associated inflammation promotes cecal tumorigenesis in aryl hydrocarbon receptor-deficient mice. Carcinogenesis 34, 1620–1627, doi: 10.1093/carcin/bgt083 (2013). [DOI] [PubMed] [Google Scholar]
- 37.Liu W. et al. Dual role of apoptosis-associated speck-like protein containing a CARD (ASC) in tumorigenesis of human melanoma. J Invest Dermatol 133, 518–527, doi: 10.1038/jid.2012.317 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Zhao X. et al. NLRP3 inflammasome activation plays a carcinogenic role through effector cytokine IL-18 in lymphoma. Oncotarget 8, 108571–108583, doi: 10.18632/oncotarget.21010 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Farshchian M. et al. Tumor cell-specific AIM2 regulates growth and invasion of cutaneous squamous cell carcinoma. Oncotarget 8, 45825–45836, doi: 10.18632/oncotarget.17573 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gao J. et al. Downregulation of GSDMD attenuates tumor proliferation via the intrinsic mitochondrial apoptotic pathway and inhibition of EGFR/Akt signaling and predicts a good prognosis in nonsmall cell lung cancer. Oncol Rep 40, 1971–1984, doi: 10.3892/or.2018.6634 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Gajewski TF, Schreiber H. & Fu YX Innate and adaptive immune cells in the tumor microenvironment. Nat Immunol 14, 1014–1022, doi: 10.1038/ni.2703 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Terme M. et al. IL-18 induces PD-1-dependent immunosuppression in cancer. Cancer Res 71, 5393–5399, doi: 10.1158/0008-5472.CAN-11-0993 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Kang JS et al. Interleukin-18 increases metastasis and immune escape of stomach cancer via the downregulation of CD70 and maintenance of CD44. Carcinogenesis 30, 1987–1996, doi: 10.1093/carcin/bgp158 (2009). [DOI] [PubMed] [Google Scholar]
- 44.Marvel D. & Gabrilovich DI Myeloid-derived suppressor cells in the tumor microenvironment: expect the unexpected. J Clin Invest 125, 3356–3364, doi: 10.1172/JCI80005 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Song X. et al. CD11b+/Gr-1+ immature myeloid cells mediate suppression of T cells in mice bearing tumors of IL-1beta-secreting cells. J Immunol 175, 8200–8208 (2005). [DOI] [PubMed] [Google Scholar]
- 46.Bunt SK et al. Reduced inflammation in the tumor microenvironment delays the accumulation of myeloid-derived suppressor cells and limits tumor progression. Cancer Res 67, 10019–10026, doi: 10.1158/0008-5472.CAN-07-2354 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.van Deventer HW et al. The inflammasome component NLRP3 impairs antitumor vaccine by enhancing the accumulation of tumor-associated myeloid-derived suppressor cells. Cancer Res 70, 10161–10169, doi: 10.1158/0008-5472.CAN-10-1921 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Daley D. et al. NLRP3 signaling drives macrophage-induced adaptive immune suppression in pancreatic carcinoma. J Exp Med 214, 1711–1724, doi: 10.1084/jem.20161707 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Voigt C. et al. Cancer cells induce interleukin-22 production from memory CD4(+) T cells via interleukin-1 to promote tumor growth. Proc Natl Acad Sci U S A 114, 12994–12999, doi: 10.1073/pnas.1705165114 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Sorrentino R. et al. Human lung cancer-derived immunosuppressive plasmacytoid dendritic cells release IL-1alpha in an AIM2 inflammasome-dependent manner. Am J Pathol 185, 3115–3124, doi: 10.1016/j.ajpath.2015.07.009 (2015). [DOI] [PubMed] [Google Scholar]
- 51.Li C. et al. PINK1 and PARK2 Suppress Pancreatic Tumorigenesis through Control of Mitochondrial Iron-Mediated Immunometabolism. Dev Cell 46, 441–455 e448, doi: 10.1016/j.devcel.2018.07.012 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Nakamura K. et al. Dysregulated IL-18 Is a Key Driver of Immunosuppression and a Possible Therapeutic Target in the Multiple Myeloma Microenvironment. Cancer Cell 33, 634–648 e635, doi: 10.1016/j.ccell.2018.02.007 (2018). [DOI] [PubMed] [Google Scholar]
- 53.McMahon G. VEGF receptor signaling in tumor angiogenesis. Oncologist 5 Suppl 1, 3–10 (2000). [DOI] [PubMed] [Google Scholar]
- 54.Saijo Y. et al. Proinflammatory cytokine IL-1 beta promotes tumor growth of Lewis lung carcinoma by induction of angiogenic factors: in vivo analysis of tumor-stromal interaction. J Immunol 169, 469–475 (2002). [DOI] [PubMed] [Google Scholar]
- 55.Jung YJ, Isaacs JS, Lee S, Trepel J. & Neckers L. IL-1beta-mediated up-regulation of HIF-1alpha via an NFkappaB/COX-2 pathway identifies HIF-1 as a critical link between inflammation and oncogenesis. FASEB J 17, 2115–2117, doi: 10.1096/fj.03-0329fje (2003). [DOI] [PubMed] [Google Scholar]
- 56.Valastyan S. & Weinberg RA Tumor metastasis: molecular insights and evolving paradigms. Cell 147, 275–292, doi: 10.1016/j.cell.2011.09.024 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lambert AW, Pattabiraman DR & Weinberg RA Emerging Biological Principles of Metastasis. Cell 168, 670–691, doi: 10.1016/j.cell.2016.11.037 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vidal-Vanaclocha F. et al. IL-18 regulates IL-1beta-dependent hepatic melanoma metastasis via vascular cell adhesion molecule-1. Proc Natl Acad Sci U S A 97, 734–739 (2000).This article shows that IL-18 contributes to hepatic metastasis by increasing VCAM-1 expression in hepatic-sinusoidal enothelial cells.
- 59.Voronov E. et al. IL-1 is required for tumor invasiveness and angiogenesis. Proc Natl Acad Sci U S A 100, 2645–2650, doi: 10.1073/pnas.0437939100 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Vidal-Vanaclocha F, Amezaga C, Asumendi A, Kaplanski G. & Dinarello CA Interleukin-1 receptor blockade reduces the number and size of murine B16 melanoma hepatic metastases. Cancer Res 54, 2667–2672 (1994). [PubMed] [Google Scholar]
- 61.Valcarcel M, Carrascal T, Crende O. & Vidal-Vanaclocha F. IL-18 regulates melanoma VLA-4 integrin activation through a Hierarchized sequence of inflammatory factors. J Invest Dermatol 134, 470–480, doi: 10.1038/jid.2013.342 (2014). [DOI] [PubMed] [Google Scholar]
- 62.Carrascal MT et al. Interleukin-18 binding protein reduces b16 melanoma hepatic metastasis by neutralizing adhesiveness and growth factors of sinusoidal endothelium. Cancer Res 63, 491–497 (2003). [PubMed] [Google Scholar]
- 63.Dupaul-Chicoine J. et al. The Nlrp3 Inflammasome Suppresses Colorectal Cancer Metastatic Growth in the Liver by Promoting Natural Killer Cell Tumoricidal Activity. Immunity 43, 751–763, doi: 10.1016/j.immuni.2015.08.013 (2015).This study has elegantly demonstrated that IL-18 production as a result of NLRP3 activation in the immune cells contributes to maturation of hepatic NK cells, surface expression of the death ligand FasL, and capacity to kill FasL-sensitive tumors.
- 64.Deng Q. et al. NLRP3 inflammasomes in macrophages drive colorectal cancer metastasis to the liver. Cancer Lett, doi: 10.1016/j.canlet.2018.10.030 (2018). [DOI] [PubMed] [Google Scholar]
- 65.Guo B, Fu S, Zhang J, Liu B. & Li Z. Targeting inflammasome/IL-1 pathways for cancer immunotherapy. Sci Rep 6, 36107, doi: 10.1038/srep36107 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chow MT et al. NLRP3 suppresses NK cell-mediated responses to carcinogen-induced tumors and metastases. Cancer Res 72, 5721–5732, doi: 10.1158/0008-5472.CAN-12-0509 (2012). [DOI] [PubMed] [Google Scholar]
- 67.Heerboth S. et al. EMT and tumor metastasis. Clin Transl Med 4, 6, doi: 10.1186/s40169-015-0048-3 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Fu XT et al. Macrophage-secreted IL-8 induces epithelial-mesenchymal transition in hepatocellular carcinoma cells by activating the JAK2/STAT3/Snail pathway. Int J Oncol 46, 587–596, doi: 10.3892/ijo.2014.2761 (2015). [DOI] [PubMed] [Google Scholar]
- 69.Wang H. et al. NLRP3 promotes tumor growth and metastasis in human oral squamous cell carcinoma. BMC Cancer 18, 500, doi: 10.1186/s12885-018-4403-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.St John MA et al. Proinflammatory mediators upregulate snail in head and neck squamous cell carcinoma. Clin Cancer Res 15, 6018–6027, doi: 10.1158/1078-0432.CCR-09-0011 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jee YS, Jang TJ & Jung KH Prostaglandin E(2) and interleukin-1beta reduce E-cadherin expression by enhancing snail expression in gastric cancer cells. J Korean Med Sci 27, 987–992, doi: 10.3346/jkms.2012.27.9.987 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Duffy MJ, Maguire TM, Hill A, McDermott E. & O’Higgins N. Metalloproteinases: role in breast carcinogenesis, invasion and metastasis. Breast Cancer Res 2, 252–257 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ma L. et al. Epidermal growth factor (EGF) and interleukin (IL)-1beta synergistically promote ERK1/2-mediated invasive breast ductal cancer cell migration and invasion. Mol Cancer 11, 79, doi: 10.1186/1476-4598-11-79 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang Y. et al. Interleukin-18 enhances breast cancer cell migration via down-regulation of claudin-12 and induction of the p38 MAPK pathway. Biochem Biophys Res Commun 459, 379–386, doi: 10.1016/j.bbrc.2015.02.108 (2015). [DOI] [PubMed] [Google Scholar]
- 75.Wang W. et al. Inflammasome-independent NLRP3 augments TGF-beta signaling in kidney epithelium. J Immunol 190, 1239–1249, doi: 10.4049/jimmunol.1201959 (2013). [DOI] [PubMed] [Google Scholar]
- 76.Wang H. et al. Inflammasome-independent NLRP3 is required for epithelial-mesenchymal transition in colon cancer cells. Exp Cell Res 342, 184–192, doi: 10.1016/j.yexcr.2016.03.009 (2016). [DOI] [PubMed] [Google Scholar]
- 77.Williams TM et al. The NLRP1 inflammasome attenuates colitis and colitis-associated tumorigenesis. J Immunol 194, 3369–3380, doi: 10.4049/jimmunol.1402098 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Allen IC et al. The NLRP3 inflammasome functions as a negative regulator of tumorigenesis during colitis-associated cancer. J Exp Med 207, 1045–1056, doi: 10.1084/jem.20100050 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zaki MH, Vogel P, Body-Malapel M, Lamkanfi M. & Kanneganti TD IL-18 production downstream of the Nlrp3 inflammasome confers protection against colorectal tumor formation. J Immunol 185, 4912–4920, doi: 10.4049/jimmunol.1002046 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wilson JE et al. Inflammasome-independent role of AIM2 in suppressing colon tumorigenesis via DNA-PK and Akt. Nat Med 21, 906–913, doi: 10.1038/nm.3908 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Man SM et al. Critical Role for the DNA Sensor AIM2 in Stem Cell Proliferation and Cancer. Cell 162, 45–58, doi: 10.1016/j.cell.2015.06.001 (2015).Irrespective of its role in inflammasome activation, this study has identified a role of AIM2 in suppressing AKT activation, thereby preventing excessive proliferation of stem cells in the colon.
- 82.Hu B. et al. Inflammation-induced tumorigenesis in the colon is regulated by caspase-1 and NLRC4. Proc Natl Acad Sci U S A 107, 21635–21640, doi: 10.1073/pnas.1016814108 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Hu B. et al. Microbiota-induced activation of epithelial IL-6 signaling links inflammasome-driven inflammation with transmissible cancer. Proc Natl Acad Sci U S A 110, 9862–9867, doi: 10.1073/pnas.1307575110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sharma D. et al. Pyrin Inflammasome Regulates Tight Junction Integrity to Restrict Colitis and Tumorigenesis. Gastroenterology 154, 948–964 e948, doi: 10.1053/j.gastro.2017.11.276 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.De Robertis M. et al. The AOM/DSS murine model for the study of colon carcinogenesis: From pathways to diagnosis and therapy studies. J Carcinog 10, 9, doi: 10.4103/1477-3163.78279 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zaki MH et al. The NLRP3 inflammasome protects against loss of epithelial integrity and mortality during experimental colitis. Immunity 32, 379–391, doi: 10.1016/j.immuni.2010.03.003 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dupaul-Chicoine J. et al. Control of intestinal homeostasis, colitis, and colitis-associated colorectal cancer by the inflammatory caspases. Immunity 32, 367–378, doi: 10.1016/j.immuni.2010.02.012 (2010). [DOI] [PubMed] [Google Scholar]
- 88.Salcedo R. et al. MyD88-mediated signaling prevents development of adenocarcinomas of the colon: role of interleukin 18. J Exp Med 207, 1625–1636, doi: 10.1084/jem.20100199 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Takagi H. et al. Contrasting action of IL-12 and IL-18 in the development of dextran sodium sulphate colitis in mice. Scand J Gastroenterol 38, 837–844 (2003). [DOI] [PubMed] [Google Scholar]
- 90.Fabbi M, Carbotti G. & Ferrini S. Context-dependent role of IL-18 in cancer biology and counter-regulation by IL-18BP. J Leukoc Biol 97, 665–675, doi: 10.1189/jlb.5Ru0714-360RR (2015). [DOI] [PubMed] [Google Scholar]
- 91.Nowarski R. et al. Epithelial IL-18 Equilibrium Controls Barrier Function in Colitis. Cell 163, 1444–1456, doi: 10.1016/j.cell.2015.10.072 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sivakumar PV et al. Interleukin 18 is a primary mediator of the inflammation associated with dextran sulphate sodium induced colitis: blocking interleukin 18 attenuates intestinal damage. Gut 50, 812–820 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Siegmund B. et al. Neutralization of interleukin-18 reduces severity in murine colitis and intestinal IFN-gamma and TNF-alpha production. Am J Physiol Regul Integr Comp Physiol 281, R1264–1273, doi: 10.1152/ajpregu.2001.281.4.R1264 (2001). [DOI] [PubMed] [Google Scholar]
- 94.Huber S. et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature 491, 259–263, doi: 10.1038/nature11535 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Bauer C. et al. Colitis induced in mice with dextran sulfate sodium (DSS) is mediated by the NLRP3 inflammasome. Gut 59, 1192–1199, doi: 10.1136/gut.2009.197822 (2010). [DOI] [PubMed] [Google Scholar]
- 96.Du Q. et al. Dietary cholesterol promotes AOM-induced colorectal cancer through activating the NLRP3 inflammasome. Biochem Pharmacol 105, 42–54, doi: 10.1016/j.bcp.2016.02.017 (2016). [DOI] [PubMed] [Google Scholar]
- 97.Yao X. et al. Remodelling of the gut microbiota by hyperactive NLRP3 induces regulatory T cells to maintain homeostasis. Nat Commun 8, 1896, doi: 10.1038/s41467-017-01917-2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Spalinger MR et al. PTPN2 Regulates Inflammasome Activation and Controls Onset of Intestinal Inflammation and Colon Cancer. Cell Rep 22, 1835–1848, doi: 10.1016/j.celrep.2018.01.052 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dmitrieva-Posocco O. et al. Cell-Type-Specific Responses to Interleukin-1 Control Microbial Invasion and Tumor-Elicited Inflammation in Colorectal Cancer. Immunity 50, 166–180 e167, doi: 10.1016/j.immuni.2018.11.015 (2019).Using conditional gene deletion approaches in a model of colorectal cancer, this study has demonstrated opposing roles of IL-1R signaling on different cell types.
- 100.Drexler SK et al. Tissue-specific opposing functions of the inflammasome adaptor ASC in the regulation of epithelial skin carcinogenesis. Proc Natl Acad Sci U S A 109, 18384–18389, doi: 10.1073/pnas.1209171109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Janowski AM et al. NLRC4 suppresses melanoma tumor progression independently of inflammasome activation. J Clin Invest 126, 3917–3928, doi: 10.1172/JCI86953 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Allam R. et al. Epithelial NAIPs protect against colonic tumorigenesis. J Exp Med 212, 369–383, doi: 10.1084/jem.20140474 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Garaude J, Kent A, van Rooijen N. & Blander JM Simultaneous targeting of toll- and nod-like receptors induces effective tumor-specific immune responses. Sci Transl Med 4, 120ra116, doi: 10.1126/scitranslmed.3002868 (2012). [DOI] [PubMed] [Google Scholar]
- 104.Chen J, Wang Z. & Yu S. AIM2 regulates viability and apoptosis in human colorectal cancer cells via the PI3K/Akt pathway. Onco Targets Ther 10, 811–817, doi: 10.2147/OTT.S125039 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Karki R. et al. NLRC3 is an inhibitory sensor of PI3K-mTOR pathways in cancer. Nature, doi: 10.1038/nature20597 (2016).The tumor suppressive effect of NLRC3 is associated with its inhibitory effect in mTOR signaling pathway to suppress cellular proliferation and stem-cell-derived organoid formation.
- 106.Man SM, Karki R. & Kanneganti TD AIM2 inflammasome in infection, cancer, and autoimmunity: Role in DNA sensing, inflammation, and innate immunity. Eur J Immunol 46, 269–280, doi: 10.1002/eji.201545839 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hu B. et al. The DNA-sensing AIM2 inflammasome controls radiation-induced cell death and tissue injury. Science 354, 765–768, doi: 10.1126/science.aaf7532 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Chen IF et al. AIM2 suppresses human breast cancer cell proliferation in vitro and mammary tumor growth in a mouse model. Mol Cancer Ther 5, 1–7, doi: 10.1158/1535-7163.MCT-05-0310 (2006). [DOI] [PubMed] [Google Scholar]
- 109.Liu ZY, Yi J. & Liu FE The molecular mechanism of breast cancer cell apoptosis induction by absent in melanoma (AIM2). Int J Clin Exp Med 8, 14750–14758 (2015). [PMC free article] [PubMed] [Google Scholar]
- 110.Elinav E. et al. NLRP6 inflammasome regulates colonic microbial ecology and risk for colitis. Cell 145, 745–757, doi: 10.1016/j.cell.2011.04.022 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wlodarska M. et al. NLRP6 inflammasome orchestrates the colonic host-microbial interface by regulating goblet cell mucus secretion. Cell 156, 1045–1059, doi: 10.1016/j.cell.2014.01.026 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Hara H. et al. The NLRP6 Inflammasome Recognizes Lipoteichoic Acid and Regulates Gram-Positive Pathogen Infection. Cell, doi: 10.1016/j.cell.2018.09.047 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Vladimer GI et al. The NLRP12 inflammasome recognizes Yersinia pestis. Immunity 37, 96–107, doi: 10.1016/j.immuni.2012.07.006 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Birchenough GM, Nystrom EE, Johansson ME & Hansson GC A sentinel goblet cell guards the colonic crypt by triggering Nlrp6-dependent Muc2 secretion. Science 352, 1535–1542, doi: 10.1126/science.aaf7419 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Allen IC et al. NLRP12 suppresses colon inflammation and tumorigenesis through the negative regulation of noncanonical NF-kappaB signaling. Immunity 36, 742–754, doi: 10.1016/j.immuni.2012.03.012 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zaki MH et al. The NOD-like receptor NLRP12 attenuates colon inflammation and tumorigenesis. Cancer Cell 20, 649–660, doi: 10.1016/j.ccr.2011.10.022 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Brennan CA & Garrett WS Gut Microbiota, Inflammation, and Colorectal Cancer. Annu Rev Microbiol 70, 395–411, doi: 10.1146/annurev-micro-102215-095513 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zackular JP et al. The gut microbiome modulates colon tumorigenesis. MBio 4, e00692–00613, doi: 10.1128/mBio.00692-13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Hirota SA et al. NLRP3 inflammasome plays a key role in the regulation of intestinal homeostasis. Inflamm Bowel Dis 17, 1359–1372, doi: 10.1002/ibd.21478 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Ratsimandresy RA, Indramohan M, Dorfleutner A. & Stehlik C. The AIM2 inflammasome is a central regulator of intestinal homeostasis through the IL-18/IL-22/STAT3 pathway. Cell Mol Immunol 14, 127–142, doi: 10.1038/cmi.2016.35 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chen L. et al. NLRP12 attenuates colon inflammation by maintaining colonic microbial diversity and promoting protective commensal bacterial growth. Nat Immunol 18, 541–551, doi: 10.1038/ni.3690 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Levy M. et al. Microbiota-Modulated Metabolites Shape the Intestinal Microenvironment by Regulating NLRP6 Inflammasome Signaling. Cell 163, 1428–1443, doi: 10.1016/j.cell.2015.10.048 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Pierantonelli I. et al. Lack of NLRP3-inflammasome leads to gut-liver axis derangement, gut dysbiosis and a worsened phenotype in a mouse model of NAFLD. Sci Rep 7, 12200, doi: 10.1038/s41598-017-11744-6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Mamantopoulos M. et al. Nlrp6- and ASC-Dependent Inflammasomes Do Not Shape the Commensal Gut Microbiota Composition. Immunity 47, 339–348 e334, doi: 10.1016/j.immuni.2017.07.011 (2017).This study concludes that NLRP6 and ASC-dependent inflammasomes don’t contribute in shaping commensal gut microbiota and hightlight the necessity of littermate-controlled experiments to study the gene functions in the gut.
- 125.Lemire P. et al. The NLR Protein NLRP6 Does Not Impact Gut Microbiota Composition. Cell Rep 21, 3653–3661, doi: 10.1016/j.celrep.2017.12.026 (2017). [DOI] [PubMed] [Google Scholar]
- 126.Lukens JR et al. Dietary modulation of the microbiome affects autoinflammatory disease. Nature 516, 246–249, doi: 10.1038/nature13788 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zhao S. et al. Deoxycholic Acid Triggers NLRP3 Inflammasome Activation and Aggravates DSS-Induced Colitis in Mice. Front Immunol 7, 536, doi: 10.3389/fimmu.2016.00536 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Macia L. et al. Metabolite-sensing receptors GPR43 and GPR109A facilitate dietary fibre-induced gut homeostasis through regulation of the inflammasome. Nat Commun 6, 6734, doi: 10.1038/ncomms7734 (2015). [DOI] [PubMed] [Google Scholar]
- 129.Yan Y. et al. Omega-3 fatty acids prevent inflammation and metabolic disorder through inhibition of NLRP3 inflammasome activation. Immunity 38, 1154–1163, doi: 10.1016/j.immuni.2013.05.015 (2013). [DOI] [PubMed] [Google Scholar]
- 130.Kolb R. et al. Obesity-associated NLRC4 inflammasome activation drives breast cancer progression. Nat Commun 7, 13007, doi: 10.1038/ncomms13007 (2016).This study demonstrates that IL-1b production as a result of NLRC4 inflammasome activation in immune cells of the tumor microenvironment of obese mice acts on adipocytes to promote VEGF production and angiogenesis.
- 131.Villani AC et al. Common variants in the NLRP3 region contribute to Crohn’s disease susceptibility. Nat Genet 41, 71–76, doi: 10.1038/ng.285 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Schoultz I. et al. Combined polymorphisms in genes encoding the inflammasome components NALP3 and CARD8 confer susceptibility to Crohn’s disease in Swedish men. Am J Gastroenterol 104, 1180–1188, doi: 10.1038/ajg.2009.29 (2009). [DOI] [PubMed] [Google Scholar]
- 133.Ungerback J. et al. Genetic variation and alterations of genes involved in NFkappaB/TNFAIP3- and NLRP3-inflammasome signaling affect susceptibility and outcome of colorectal cancer. Carcinogenesis 33, 2126–2134, doi: 10.1093/carcin/bgs256 (2012). [DOI] [PubMed] [Google Scholar]
- 134.Verma D. et al. Inflammasome polymorphisms confer susceptibility to sporadic malignant melanoma. Pigment Cell Melanoma Res 25, 506–513, doi: 10.1111/j.1755-148X.2012.01008.x (2012). [DOI] [PubMed] [Google Scholar]
- 135.Castano-Rodriguez N, Kaakoush NO, Goh KL, Fock KM & Mitchell HM The NOD-like receptor signalling pathway in Helicobacter pylori infection and related gastric cancer: a case-control study and gene expression analyses. PLoS One 9, e98899, doi: 10.1371/journal.pone.0098899 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zhong FL et al. Germline NLRP1 Mutations Cause Skin Inflammatory and Cancer Susceptibility Syndromes via Inflammasome Activation. Cell 167, 187–202 e117, doi: 10.1016/j.cell.2016.09.001 (2016). [DOI] [PubMed] [Google Scholar]
- 137.Borelli V, Moura RR, Trevisan E. & Crovella S. NLRP1 and NLRP3 polymorphisms in mesothelioma patients and asbestos exposed individuals a population-based autopsy study from North East Italy. Infect Agent Cancer 10, 26, doi: 10.1186/s13027-015-0022-0 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Girardelli M. et al. NLRP1 polymorphisms in patients with asbestos-associated mesothelioma. Infect Agent Cancer 7, 25, doi: 10.1186/1750-9378-7-25 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Mori Y. et al. Instabilotyping: comprehensive identification of frameshift mutations caused by coding region microsatellite instability. Cancer Res 61, 6046–6049 (2001). [PubMed] [Google Scholar]
- 140.Schulmann K. et al. HNPCC-associated small bowel cancer: clinical and molecular characteristics. Gastroenterology 128, 590–599 (2005). [DOI] [PubMed] [Google Scholar]
- 141.Woerner SM. et al. Pathogenesis of DNA repair-deficient cancers: a statistical meta-analysis of putative Real Common Target genes. Oncogene 22, 2226–2235, doi: 10.1038/sj.onc.1206421 (2003). [DOI] [PubMed] [Google Scholar]
- 142.Brenner R. et al. Familial Mediterranean Fever and Incidence of Cancer: An Analysis of 8,534 Israeli Patients With 258,803 Person-Years. Arthritis Rheumatol 70, 127–133, doi: 10.1002/art.40344 (2018). [DOI] [PubMed] [Google Scholar]
- 143.Chen LC. et al. Tumour inflammasome-derived IL-1beta recruits neutrophils and improves local recurrence-free survival in EBV-induced nasopharyngeal carcinoma. EMBO Mol Med 4, 1276–1293, doi: 10.1002/emmm.201201569 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kong H. et al. Differential expression of inflammasomes in lung cancer cell lines and tissues. Tumour Biol 36, 7501–7513, doi: 10.1007/s13277-015-3473-4 (2015). [DOI] [PubMed] [Google Scholar]
- 145.Wu CS et al. ASC contributes to metastasis of oral cavity squamous cell carcinoma. Oncotarget 7, 50074–50085, doi: 10.18632/oncotarget.10317 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Baldini C, Santini E, Rossi C, Donati V. & Solini A. The P2X7 receptor-NLRP3 inflammasome complex predicts the development of non-Hodgkin’s lymphoma in Sjogren’s syndrome: a prospective, observational, single-centre study. J Intern Med 282, 175–186, doi: 10.1111/joim.12631 (2017). [DOI] [PubMed] [Google Scholar]
- 147.Paugh SW et al. NALP3 inflammasome upregulation and CASP1 cleavage of the glucocorticoid receptor cause glucocorticoid resistance in leukemia cells. Nat Genet 47, 607–614, doi: 10.1038/ng.3283 (2015).The findings from this study establish a regulatory mechanism by which the NLRP3–mediated caspase-1 activation modulates cellular levels of the glucocorticoid receptor and diminishes cell sensitivity to glucocorticoids.
- 148.Poli G. et al. Expression of inflammasome-related genes in bladder cancer and their association with cytokeratin 20 messenger RNA. Urol Oncol 33, 505 e501–507, doi: 10.1016/j.urolonc.2015.07.012 (2015). [DOI] [PubMed] [Google Scholar]
- 149.Karan D, Tawfik O. & Dubey S. Expression analysis of inflammasome sensors and implication of NLRP12 inflammasome in prostate cancer. Sci Rep 7, 4378, doi: 10.1038/s41598-017-04286-4 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Huang T. et al. G9A promotes tumor cell growth and invasion by silencing CASP1 in non-small-cell lung cancer cells. Cell Death Dis 8, e2726, doi: 10.1038/cddis.2017.65 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dihlmann S. et al. Lack of Absent in Melanoma 2 (AIM2) expression in tumor cells is closely associated with poor survival in colorectal cancer patients. Int J Cancer 135, 2387–2396, doi: 10.1002/ijc.28891 (2014). [DOI] [PubMed] [Google Scholar]
- 152.Liu R. et al. Expression profile of innate immune receptors, NLRs and AIM2, in human colorectal cancer: correlation with cancer stages and inflammasome components. Oncotarget 6, 33456–33469, doi: 10.18632/oncotarget.5587 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Wei Q. et al. Deregulation of the NLRP3 inflammasome in hepatic parenchymal cells during liver cancer progression. Lab Invest 94, 52–62, doi: 10.1038/labinvest.2013.126 (2014). [DOI] [PubMed] [Google Scholar]
- 154.Yoshihama S. et al. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc Natl Acad Sci U S A 113, 5999–6004, doi: 10.1073/pnas.1602069113 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Locher C. et al. Desirable cell death during anticancer chemotherapy. Ann N Y Acad Sci 1209, 99–108, doi: 10.1111/j.1749-6632.2010.05763.x (2010). [DOI] [PubMed] [Google Scholar]
- 156.Casares N. et al. Caspase-dependent immunogenicity of doxorubicin-induced tumor cell death. J Exp Med 202, 1691–1701, doi: 10.1084/jem.20050915 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Ghiringhelli F. et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med 15, 1170–1178, doi: 10.1038/nm.2028 (2009).Using tumor transplantation models, this study has demonstrated that chemotherapy mediated DAMPs release from dying tumor cells activate NLRP3 inflammasome in dendritic cells to regulate adaptive immune cell repertoire, resulting in enhanced T cell-mediated tumor-cell death.
- 158.Bruchard M. et al. Chemotherapy-triggered cathepsin B release in myeloid-derived suppressor cells activates the Nlrp3 inflammasome and promotes tumor growth. Nat Med 19, 57–64, doi: 10.1038/nm.2999 (2013). [DOI] [PubMed] [Google Scholar]
- 159.Feng X. et al. The role of NLRP3 inflammasome in 5-fluorouracil resistance of oral squamous cell carcinoma. J Exp Clin Cancer Res 36, 81, doi: 10.1186/s13046-017-0553-x (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Su S. et al. Immune Checkpoint Inhibition Overcomes ADCP-Induced Immunosuppression by Macrophages. Cell 175, 442–457 e423, doi: 10.1016/j.cell.2018.09.007 (2018). [DOI] [PubMed] [Google Scholar]
- 161.Mattarollo SR et al. Pivotal role of innate and adaptive immunity in anthracycline chemotherapy of established tumors. Cancer Res 71, 4809–4820, doi: 10.1158/0008-5472.CAN-11-0753 (2011). [DOI] [PubMed] [Google Scholar]
- 162.Westbom C. et al. Inflammasome Modulation by Chemotherapeutics in Malignant Mesothelioma. PLoS One 10, e0145404, doi: 10.1371/journal.pone.0145404 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Yao L, Zhang Y, Chen K, Hu X. & Xu LX Discovery of IL-18 as a novel secreted protein contributing to doxorubicin resistance by comparative secretome analysis of MCF-7 and MCF-7/Dox. PLoS One 6, e24684, doi: 10.1371/journal.pone.0024684 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Chmielewski M. & Abken H. CAR T Cells Releasing IL-18 Convert to T-Bet(high) FoxO1(low) Effectors that Exhibit Augmented Activity against Advanced Solid Tumors. Cell Rep 21, 3205–3219, doi: 10.1016/j.celrep.2017.11.063 (2017). [DOI] [PubMed] [Google Scholar]
- 165.Wang Y. et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature 547, 99–103, doi: 10.1038/nature22393 (2017). [DOI] [PubMed] [Google Scholar]
- 166.Baldwin AG, Brough D. & Freeman S. Inhibiting the Inflammasome: A Chemical Perspective. J Med Chem 59, 1691–1710, doi: 10.1021/acs.jmedchem.5b01091 (2016). [DOI] [PubMed] [Google Scholar]
- 167.Mangan MSJ et al. Targeting the NLRP3 inflammasome in inflammatory diseases. Nat Rev Drug Discov 17, 588–606, doi: 10.1038/nrd.2018.97 (2018).This review highlights the evolving landscape of NLRP3 modulators and discuss opportunities for pharmacologically targeting NLRP3 with novel small molecules.
- 168.Lust JA et al. Induction of a chronic disease state in patients with smoldering or indolent multiple myeloma by targeting interleukin 1{beta}-induced interleukin 6 production and the myeloma proliferative component. Mayo Clin Proc 84, 114–122, doi: 10.4065/84.2.114 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Lust JA et al. Reduction in C-reactive protein indicates successful targeting of the IL-1/IL-6 axis resulting in improved survival in early stage multiple myeloma. Am J Hematol 91, 571–574, doi: 10.1002/ajh.24352 (2016). [DOI] [PubMed] [Google Scholar]
- 170.Gross O. et al. Inflammasome activators induce interleukin-1alpha secretion via distinct pathways with differential requirement for the protease function of caspase-1. Immunity 36, 388–400, doi: 10.1016/j.immuni.2012.01.018 (2012). [DOI] [PubMed] [Google Scholar]
- 171.Hickish T. et al. MABp1 as a novel antibody treatment for advanced colorectal cancer: a randomised, double-blind, placebo-controlled, phase 3 study. Lancet Oncol 18, 192–201, doi: 10.1016/S 1470–2045(17)30006–2 (2017). [DOI] [PubMed] [Google Scholar]
- 172.Ridker PM et al. Effect of interleukin-1beta inhibition with canakinumab on incident lung cancer in patients with atherosclerosis: exploratory results from a randomised, double-blind, placebo-controlled trial. Lancet 390, 1833–1842, doi: 10.1016/S0140-6736(17)32247-X (2017).An additional finding of the CANTOS, where administration of Canakinumab reduced lung cancer incidence and mortality, shed light on IL-1b as a potential therapeutic target in lung cancer.
- 173.Isambert N. et al. Fluorouracil and bevacizumab plus anakinra for patients with metastatic colorectal cancer refractory to standard therapies (IRAFU): a single-arm phase 2 study. Oncoimmunology 7, e1474319, doi: 10.1080/2162402X.2018.1474319 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Wang Y. et al. Activation of NLRP3 inflammasome enhances the proliferation and migration of A549 lung cancer cells. Oncol Rep 35, 2053–2064, doi: 10.3892/or.2016.4569 (2016). [DOI] [PubMed] [Google Scholar]
- 175.Cao R, Farnebo J, Kurimoto M. & Cao Y. Interleukin-18 acts as an angiogenesis and tumor suppressor. FASEB J 13, 2195–2202 (1999). [DOI] [PubMed] [Google Scholar]
- 176.Hitzler I. et al. Caspase-1 has both proinflammatory and regulatory properties in Helicobacter infections, which are differentially mediated by its substrates IL-1beta and IL-18. J Immunol 188, 3594–3602, doi: 10.4049/jimmunol.1103212 (2012). [DOI] [PubMed] [Google Scholar]
- 177.Wang WJ et al. Downregulation of gasdermin D promotes gastric cancer proliferation by regulating cell cycle-related proteins. J Dig Dis 19, 74–83, doi: 10.1111/1751-2980.12576 (2018). [DOI] [PubMed] [Google Scholar]
- 178.Chang CY et al. Intratumoral delivery of IL-18 naked DNA induces T-cell activation and Th1 response in a mouse hepatic cancer model. BMC Cancer 7, 87, doi: 10.1186/1471-2407-7-87 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Martinez-Cardona C. et al. AIM2 deficiency reduces the development of hepatocellular carcinoma in mice. Int J Cancer, doi: 10.1002/ijc.31827 (2018). [DOI] [PubMed] [Google Scholar]
- 180.Chen SL et al. HBx-mediated decrease of AIM2 contributes to hepatocellular carcinoma metastasis. Mol Oncol 11, 1225–1240, doi: 10.1002/1878-0261.12090 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Normand S. et al. Nod-like receptor pyrin domain-containing protein 6 (NLRP6) controls epithelial self-renewal and colorectal carcinogenesis upon injury. Proc Natl Acad Sci U S A 108, 9601–9606, doi: 10.1073/pnas.1100981108 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Chen GY, Liu M, Wang F, Bertin J. & Nunez G. A functional role for Nlrp6 in intestinal inflammation and tumorigenesis. J Immunol 186, 7187–7194, doi: 10.4049/jimmunol.1100412 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Blazejewski AJ et al. Microbiota Normalization Reveals that Canonical Caspase-1 Activation Exacerbates Chemically Induced Intestinal Inflammation. Cell Rep 19, 2319–2330, doi: 10.1016/j.celrep.2017.05.058 (2017). [DOI] [PubMed] [Google Scholar]
- 184.Wang Y. et al. Neutrophil infiltration favors colitis-associated tumorigenesis by activating the interleukin-1 (IL-1)/IL-6 axis. Mucosal Immunol 7, 1106–1115, doi: 10.1038/mi.2013.126 (2014). [DOI] [PubMed] [Google Scholar]
- 185.Ning C. et al. Complement activation promotes colitis-associated carcinogenesis through activating intestinal IL-1beta/IL-17A axis. Mucosal Immunol 8, 1275–1284, doi: 10.1038/mi.2015.18 (2015). [DOI] [PubMed] [Google Scholar]
- 186.Zhu Y, Zhu M. & Lance P. IL1beta-mediated Stromal COX-2 signaling mediates proliferation and invasiveness of colonic epithelial cancer cells. Exp Cell Res 318, 2520–2530, doi: 10.1016/j.yexcr.2012.07.021 (2012). [DOI] [PubMed] [Google Scholar]
- 187.Liu Q. et al. Interleukin-1beta promotes skeletal colonization and progression of metastatic prostate cancer cells with neuroendocrine features. Cancer Res 73, 3297–3305, doi: 10.1158/0008-5472.CAN-12-3970 (2013). [DOI] [PubMed] [Google Scholar]
- 188.Tsai CY, Lee TS, Kou YR & Wu YL Glucosamine inhibits IL-1beta-mediated IL-8 production in prostate cancer cells by MAPK attenuation. J Cell Biochem 108, 489–498, doi: 10.1002/jcb.22278 (2009). [DOI] [PubMed] [Google Scholar]
- 189.Xu G, Guo Y, Seng Z, Cui G. & Qu J. Bone marrow-derived mesenchymal stem cells co-expressing interleukin-18 and interferon-beta exhibit potent antitumor effect against intracranial glioma in rats. Oncol Rep 34, 1915–1922, doi: 10.3892/or.2015.4174 (2015). [DOI] [PubMed] [Google Scholar]
- 190.Xu G. et al. Adenoviral-mediated interleukin-18 expression in mesenchymal stem cells effectively suppresses the growth of glioma in rats. Cell Biol Int 33, 466–474, doi: 10.1016/j.cellbi.2008.07.023 (2009). [DOI] [PubMed] [Google Scholar]
- 191.Zhang Y, Wang C, Zhang Y. & Sun M. C6 glioma cells retrovirally engineered to express IL-18 and Fas exert FasL-dependent cytotoxicity against glioma formation. Biochem Biophys Res Commun 325, 1240–1245, doi: 10.1016/j.bbrc.2004.10.165 (2004). [DOI] [PubMed] [Google Scholar]
- 192.Kikuchi T. et al. Antitumor activity of interleukin-18 on mouse glioma cells. J Immunother 23, 184–189 (2000). [DOI] [PubMed] [Google Scholar]
- 193.Li L. & Liu Y. Aging-related gene signature regulated by Nlrp3 predicts glioma progression. Am J Cancer Res 5, 442–449 (2015). [PMC free article] [PubMed] [Google Scholar]
- 194.Fathima Hurmath K, Ramaswamy P. & Nandakumar DN IL-1beta microenvironment promotes proliferation, migration, and invasion of human glioma cells. Cell Biol Int 38, 1415–1422, doi: 10.1002/cbin.10353 (2014). [DOI] [PubMed] [Google Scholar]
- 195.Sun W, Depping R. & Jelkmann W. Interleukin-1beta promotes hypoxia-induced apoptosis of glioblastoma cells by inhibiting hypoxia-inducible factor-1 mediated adrenomedullin production. Cell Death Dis 5, e1020, doi: 10.1038/cddis.2013.562 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Krelin Y. et al. Interleukin-1beta-driven inflammation promotes the development and invasiveness of chemical carcinogen-induced tumors. Cancer Res 67, 1062–1071, doi: 10.1158/0008-5472.CAN-06-2956 (2007). [DOI] [PubMed] [Google Scholar]
- 197.Zhai Z. et al. NLRP1 promotes tumor growth by enhancing inflammasome activation and suppressing apoptosis in metastatic melanoma. Oncogene 36, 3820–3830, doi: 10.1038/onc.2017.26 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Chow MT, Tschopp J, Moller A. & Smyth MJ NLRP3 promotes inflammation-induced skin cancer but is dispensable for asbestos-induced mesothelioma. Immunol Cell Biol 90, 983–986, doi: 10.1038/icb.2012.46 (2012). [DOI] [PubMed] [Google Scholar]
- 199.Zeng Q. et al. Caspase-1 from Human Myeloid-Derived Suppressor Cells Can Promote T Cell-Independent Tumor Proliferation. Cancer Immunol Res 6, 566–577, doi: 10.1158/2326-6066.CIR-17-0543 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Huang CF et al. NLRP3 inflammasome activation promotes inflammation-induced carcinogenesis in head and neck squamous cell carcinoma. J Exp Clin Cancer Res 36, 116, doi: 10.1186/s13046-017-0589-y (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Lee CH et al. IL-1beta promotes malignant transformation and tumor aggressiveness in oral cancer. J Cell Physiol 230, 875–884, doi: 10.1002/jcp.24816 (2015). [DOI] [PubMed] [Google Scholar]
- 202.Kadariya Y. et al. Inflammation-Related IL1beta/IL1R Signaling Promotes the Development of Asbestos-Induced Malignant Mesothelioma. Cancer Prev Res (Phila) 9, 406–414, doi: 10.1158/1940-6207.CAPR-15-0347 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Wu TC et al. IL1 Receptor Antagonist Controls Transcriptional Signature of Inflammation in Patients with Metastatic Breast Cancer. Cancer Res 78, 5243–5258, doi: 10.1158/0008-5472.CAN-18-0413 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Palumbo A. et al. Thalidomide for treatment of multiple myeloma: 10 years later. Blood 111, 3968–3977, doi: 10.1182/blood-2007-10-117457 (2008). [DOI] [PubMed] [Google Scholar]
- 205.Zitvogel L, Kepp O, Galluzzi L. & Kroemer G. Inflammasomes in carcinogenesis and anticancer immune responses. Nat Immunol 13, 343–351, doi: 10.1038/ni.2224 (2012). [DOI] [PubMed] [Google Scholar]
- 206.Chen L. et al. Blockage of the NLRP3 inflammasome by MCC950 improves anti-tumor immune responses in head and neck squamous cell carcinoma. Cell Mol Life Sci 75, 2045–2058, doi: 10.1007/s00018-017-2720-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Lamkanfi M. et al. Glyburide inhibits the Cryopyrin/Nalp3 inflammasome. J Cell Biol 18T, 61–70, doi: 10.1083/jcb.200903124 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Ahn H. et al. Methylene blue inhibits NLRP3, NLRC4, AIM2, and non-canonical inflammasome activation. Sci Rep 7, 12409, doi: 10.1038/s41598-017-12635-6 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Jiang H. et al. Identification of a selective and direct NLRP3 inhibitor to treat inflammatory disorders. J Exp Med 214, 3219–3238, doi: 10.1084/jem.20171419 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]