

Abstract
Using enzymes as bioelectrocatalysts is an important step toward the next level of biotechnology for energy production. In such biocatalysts, a sacrificial cofactor as an electron and proton source is needed. This is a great obstacle for upscaling, due to cofactor instability and product separation issues, which increase the costs. Here, we report a cofactor-free electroreduction of CO2 to a high energy density chemical (methanol) catalyzed by enzyme–graphene hybrids. The biocatalyst consists of dehydrogenases covalently bound on a well-defined carboxyl graphene derivative, serving the role of a conductive nanoplatform. This nanobiocatalyst achieves reduction of CO2 to methanol at high current densities, which remain unchanged for at least 20 h of operation, without production of other soluble byproducts. It is thus shown that critical improvements on the stability and rate of methanol production at a high Faradaic efficiency of 12% are possible, due to the effective electrochemical process from the electrode to the enzymes via the graphene platform.
Keywords: bioelectrocatalysis, carbon dioxide reduction, enzyme catalysis, graphene, enzyme immobilization, methanol
1. Introduction
The development of renewable energies is a key prerequisite for a sustainable high-technology level civilization. In the big picture, cleaner and sustainable energy technologies avoiding accumulation of greenhouse gases in the atmosphere, such as CO2, is of utmost interest.1−3 Solar and wind energy are among the most attractive power sources, because they are practically free, inexhaustible, and widely accessible.4 However, such renewable energy sources are “supply driven” energy systems; that is, their potential is currently limited due to lack of large scale energy storage and energy transport technologies from the source to the point of end user. To overcome this challenge, solar energy transformation into chemical energy is a particularly promising solution, for both storage and transportation of the latter.5−10
This idea is not new. Around 3.4 billion years ago, photosynthetic bacteria and natural algae utilized solar energy and later CO2 and H2O to synthesize higher hydrocarbons through photosynthesis. This very effective energy transformation mechanism has inspired scientists to develop catalysts for the reduction of CO2 to hydrocarbons.11−14 Such catalysts include materials such as solid metal surfaces15−17 or molecular systems of synthetic18−21 or biological origin.22,23 The majority of metal-based catalysts, such as Cu-based,17,24−26 Ni–Ga,27 and Pd/SnO2,28 usually are limited from low product selectivity, incomplete reduction, or harsh reaction conditions.
Biocatalysts, such as enzymes and microorganisms, are of special interest because of their large availability from the biosphere and their remarkable high selectivity and activity toward the desired product at particularly mild conditions (ambient pressure, room temperature).11,29,30 Dehydrogenase enzymes (DH) are used for the reduction of CO2 in the presence of electron and proton donors,31,32 and are thus candidates for synthetic fuel. For example, formate dehydrogenase (FateDH) catalyzes CO2 reduction into formate in the presence of nicotinamide adenine dinucleotide (NADH) as electron and proton donor.33,34 In 1999, Obert and Dave first presented CO2 reduction to methanol using three different DHs in three consequent reductions, requiring one NADH molecule in every step.35 This opened the doors for the biocatalytic production of molecules with high energy content and very efficient combustion/energy-dissipation mechanism. Although the NADH cofactor is a very efficient proton and electron donor for CO2 reduction,36 it is irreversibly oxidized to NAD+ (i.e., is sacrificial), which limits its application potential due to its particularly cost-demanding synthesis, separation, and regeneration from the reactions.37,38
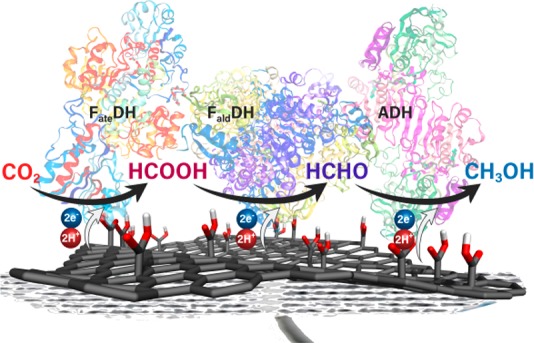
An alternative and very attractive method for avoiding the use of NADH is the electrochemical direct electron injection from appropriately designed electrodes onto the enzymes for a heterogeneous bioelectrocatalysis. This approach not only may bypass the need for NADH but also can minimize the diffusion induced overpotentials and also simplify the product separation at the end.39 The electrochemical addressing of dehydrogenases has been investigated. Amao and Shuto demonstrated the electrochemical conversion of CO2 to formate by using viologen modified FateDH-coated indium tin oxide electrodes.40 This study showed evidence of electron transfer from the electrode directly to the active site of FateDH. Reda et al. reported proposed the electron transfer mechanism of two electrons directly from the electrode to formate dehydrogenase’s active site through iron–sulfur clusters as electron relay units in the electrochemical reduction of CO2 to formate without the addition of NADH (Figure S1).41 This direct electron transfer behavior was reported also in the case of alcohol dehydrogenase (ADH), showing the potential for electrochemical addressing of dehydrogenases.42
A key technology to achieve this direct electron injection is the effective functional integration of the enzyme on electrodes. Biopolymers,43−45 nanowires,46,47 and sol–gel matrices have been proposed in the literature as supports for enzyme-immobilization.48−50 We have recently demonstrated the successful direct electron injection using ADH38 for the conversion of butyraldehyde to butanol, as well as using three enzymes for CO2 to methanol reduction, by noncovalently trapping the enzymes on an alginate-silicate matrix on carbon felt electrodes.37 Any noncovalent immobilization of the enzymes51 raises concerns on possible leakage of the expensive enzymes from the electrodes. Also the use of matrices which are hindering the diffusion of ions, adducts, and products is a limiting factor, because electrochemical reactions are slowed down, current densities are low and higher potentials are required. Therefore, we are on the search of bioelectrocatalytic systems which have higher electrical as well as ionic conductivity.
Recently, graphene, a two-dimensional carbon-based nanomaterial, is proposed for an enzyme immobilization platform due to its large specific surface area, high electronic conductivity, biocompatibility, and high chemical stability.52,53 It has shown great promise in many bioelectrochemical applications, especially biosensors54−56 and biofuel cells.57,58 Several immobilization approaches have been developed for the integration of enzymes on graphene-based electrodes. Electrostatic immobilization of the enzyme itself on graphene oxide (GO),52 or chemically cross-linked protein/enzyme networks on the surface of graphene electrodes have been reported.58 Such noncovalent associates often suffer from low long-term stability, due to leakage and disintegration.59 Enhancing the stability of the enzyme/graphene hybrid electrodes is hence crucial,60 and therefore covalent binding between the enzyme and the electrode mainly by using appropriate molecular linkers would provide more stable materials.61 However, linkers may hinder the effective electron transfer between the electrode and the distant bioreaction centers.54 A significant obstacle for achieving this through conventional graphene chemistry is the dense but uncontrolled functionalization of GO,62−64 which makes it an insulator and limits the yield of the coupling reaction and electron injection, respectively. On the other hand, pristine graphene lacks functional groups and displays very low reactivity.65,66
In the present work, these challenges were tackled by exploiting graphene carboxylic acid (G-COOH), a densely (∼15% degree of functionalization) and selectively functionalized graphene derivative, which was recently prepared by a two-step process: transformation of fluorographene (FG) into cyanographene (G-CN)67 and subsequent hydrolysis into G-COOH (Scheme 1).67 Significant conductivity67−69 combined with selective functionalization with carboxyl groups renders G-COOH an ideal substrate for conjugation reactions, electron propagation, and injection. To prove our hypothesis, G-COOH was covalently modified by direct conjugation with the three dehydrogenases, that is, FateDH, formaldehyde dehydrogenase (FaldDH), and ADH, yielding three graphene-based biocatalysts namely G-FateDH, G-FaldDH, and G-ADH, respectively, which were evaluated for the conversion of CO2 to methanol in two approaches: chemical reduction using NADH as cofactor (Figure 1a) and the NADH-free cascade electroreduction (Figure 1b).
Scheme 1. Schematic Synthesis of G-COOH and Immobilization of Dehydrogenase (DH) Enzyme onto Graphene.
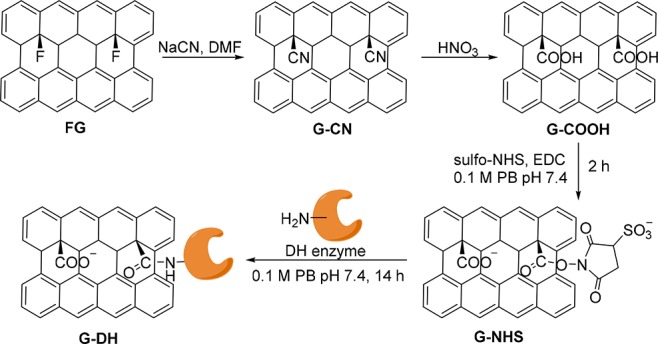
Figure 1.
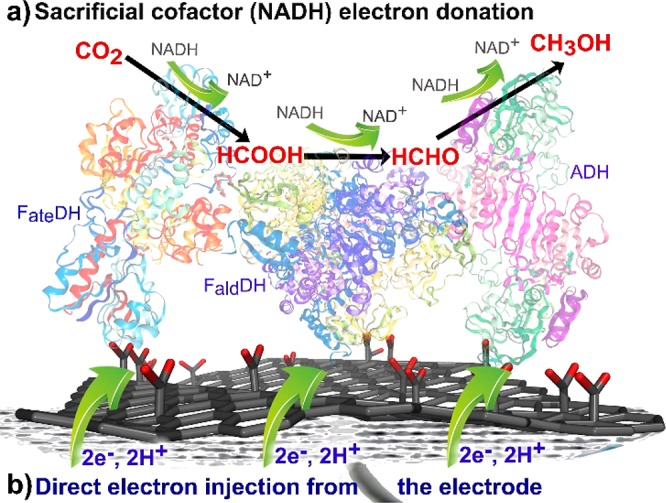
Schematic depiction of the reduction of CO2 to methanol catalyzed by FateDH, FaldDH, and ADH (a) using NADH as a sacrificial cofactor and (b) via a direct electron transfer through a functionalized graphene support and without any cofactors.
2. Experimental Section
2.1. Preparation of Enzyme-Modified Graphene
With a slight modification of the reported procedure,54 a solution of EDC (7.7 mg, 0.015 M) and G-COOH dispersion (0.2 mL of 10 mg·mL–1 solution) in 0.1 M phosphate buffer solution pH 7.4 (2.3 mL) was treated with sulfo-NHS (18 mg, 0.03 M) at room temperature for 2 h. The modified graphene carboxylic acid was purified by centrifugal washing with 0.1 M phosphate buffer (PB) pH 7.4 for four times, and the solid was collected, obtaining carboxylate graphene modified with sulfo-NHS group (G-NHS). For enzyme immobilization, the resulting G-NHS (2 mg) was redispersed in 0.1 M phosphate buffer pH 7.4 (1 mL). The amounts of added enzymes were different in each enzyme due to protein contents. The enzyme solutions were prepared separately by dissolving 2.5 mg of ADH or 10 mg of FateDH or 2.5 mg of FaldDH in 0.1 M phosphate buffer pH 7.4 (0.5 mL) and added into G-NHS suspension. The coupling reaction was performed by incubation at room temperature for 14 h. The biofunctionalized graphene was collected by centrifugation, and the product was purified by centrifugal washing with the phosphate buffer pH 7.4 for four times, resulting in ∼2 mg of G-ADH, G-FateDH, or G-FaldDH.
2.2. Electrode Preparation
Following the reported procedure,38 the G-ADH was immobilized on a sponge-like carbon felt using alginate hydrogel matrix. The alginate solution was prepared by dissolving alginic acid sodium salt (0.05 g) in 1.75 mL of 0.1 M TRIS-HCl buffer solution (pH 7.4). Subsequently, the prepared G-ADH in 0.1 M TRIS-HCl buffer solution pH 7.4 (∼2 mg·mL–1) was added. A carbon felt was soaked in the mixture and transferred to 0.2 M CaCl2 solution for 20 min for gelation. The resulting alginate containing G-ADH covered the carbon felt electrode. In the case of the three-dehydrogenase system, the electrode was prepared by soaking a carbon felt in the mixture of alginic acid sodium salt and G-FateDH (0.5 mL, ∼4 mg·mL–1), G-FaldDH (0.25 mL, ∼4 mg·mL–1), and G-ADH (0.25 mL, ∼4 mg·mL–1) solutions. Then, the soaked carbon felt was transferred to 0.2 M CaCl2 solution, and the electrode was left in the solution for 20 min, yielding the carbon felt modified with G-DHs-containing alginate. The modified electrodes were kept in TRIS-HCl buffer solution.
2.3. Electrochemical Studies
All electrochemical experiments were carried out using an IVIUM CompactStat (The Netherlands) instrument, and the potential values reported in this work referred to Ag/AgCl (3 M KCl). The experiments were performed in a two-compartment electrochemical cell with three-electrode system. Anode and cathode compartments were separated by a glass frit. The carbon felt and modified carbon felt (0.8 × 0.5 × 0.2 cm3) connected with a Pt wire, a Ag/AgCl (3 M KCl) electrode, and a Pt plate (1.4 × 4.1 cm2) were used as a working electrode, a reference electrode, and a counter electrode, respectively. A 0.1 M TRIS-HCl buffer solution served as electrolyte solution. Cyclic voltammograms were recorded at potentials between 0 and −1.20 V with the scan rate of 10 mV·s–1 in the presence of 1 M acetaldehyde under N2-saturated conditions in order to study the reduction of acetaldehyde. The results were recorded under CO2-saturated condition to investigate CO2 conversion. The constant-potential electrolysis was performed at ambient temperature using the above-mentioned electrochemical setup at an applied potential of −1.00 V for the reduction of acetaldehyde and at a potential of −1.20 V for CO2 conversion. After that, the liquid samples were taken for product analysis using a gas chromatograph.
The Faradaic efficiency toward reduction can be calculated by using the following equation:
where moles of products are calculated from the amount of ethanol/methanol/H2 formed during electrolysis, n is the number of electrons needed for the reduction process (n = 2 for the reduction of acetaldehyde to ethanol and the formation of H2, and n = 6 for the conversion of CO2 to methanol) and moles of electrons are calculated by dividing the number of consumed charges during electrolysis by the Faradaic constant of 96485.33 C·mol–1.
3. Results and Discussion
3.1. Characterization of Modified Graphene
The G-ADH biocatalyst was initially evaluated for the reduction of acetaldehyde to ethanol using both technologies: with NADH as a sacrificial cofactor and by electrochemical direct electron injection exploiting the hybrid nanobiocatalyst itself at the electrode. Importantly, this reduction of aldehydes with the ADH metalloenzyme is the final step of alcohol production from CO2,35 and therefore it makes a robust benchmark for preliminary evaluation. It was also recently identified that acetaldehyde reduction is a crucial intermediate in the electrochemical reduction of carbon monoxide to ethanol by oxide-derived copper electrode surfaces.70 Covalent attachment of ADH on G-COOH was achieved by the conjugation of the primary amine units (−NH2) of ADH and carboxylic groups of G-COOH through an amide bond. First, the carboxylic groups of G-COOH were activated through the coupling reaction in the presence of sulfo-NHS and EDC, yielding the G-NHS ester which was considerably stable at physiological conditions. The electrophilic ester groups then reacted with the −NH2 nucleophilic sites on the enzyme by incubation of G-NHS and ADH solution at room temperature overnight, yielding G-ADH, as shown in Scheme 1.
The successful conjugation of the G-COOH with ADH was verified with IR spectroscopy (Figure 2a,b) The conjugation reaction was further supported by the dramatic drop in the intensity of the carboxyl band in the G-ADH hybrid, as compared with the respective band in the starting G-COOH (band 1). Although the identification of the amide bond in G-ADH could be complicated with the peptide/amide bonds of the pristine enzyme (band 3), Figure 2b shows that the new amide bonds (band 2) appeared at different frequency. Although the IR bands of pristine ADH were not visible in the spectrum of the G-ADH, potentially posing doubts about the successful immobilization of the enzyme, high resolution X-ray photoelectron spectroscopy (HR-XPS) analysis, lifted any ambiguities, showing very clearly the characteristic fingerprint of pure ADH imprinted on the C 1s XPS envelope of G-ADH (Figure 2c). IR vibrations in the C-H bond region in Figure 2a, appearing in the G-ADH spectrum, could possibly arise from the hydrocarbon chains of the enzyme. Bands 4 and 5 (at 1580 cm–1 and at 1210 cm–1, respectively) originate from aromatic ring stretchings of the graphene’s backbone.71,72 Furthermore, the thermogravimetric analysis of the G-COOH before and after immobilization of the enzyme (Figure S3) showed increased mass loss by 9 wt % due to the decomposition of the peptide chains, corroborating the successful conjugation reaction. The morphology of G-COOH before and after covalent modification was evaluated using a transmission electron microscope, showing the preservation of graphene’s morphological features (Figure S4). The amount of ADH attached on graphene was determined by a bicinchoninic acid (BCA) assay (see Supporting Information). Compared with the known standard protein (bovine serum albumin, BSA), the amount of ADH bound on graphene was found to be 0.03 mg·mL–1 in G-ADH solution with the concentration of ∼1 mg·mL–1. Since ADH was covalently bound to graphene carboxylic acid, the three-dimensional structure might differ, and ADH activity would be reduced. Therefore, the enzyme assay of G-ADH was performed in the presence of ethanol as a substrate and NAD+ as cofactor. According to the ADH content and its activity in G-ADH, the enzymatic activity was found to be 14.6 units per milligram of protein. Meanwhile, free ADH showed the activity of 76.2 units per milligram of protein reflecting the effects of the immobilization, possibly due to steric hindrance from the presence of G-COOH and mass transfer reduction of the substrate to the active center, or due to a change in enzyme stereochemistry affecting the binding and/or catalytic sites.73−75
Figure 2.
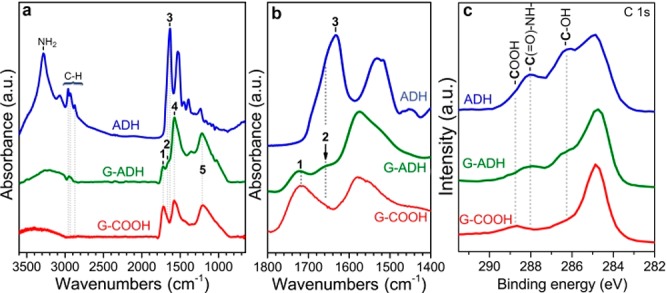
FT-IR (a,b) and C 1s HR-XPS (c) spectra of the starting G-COOH (red solid line), the hybrid after conjugation of G-COOH with ADH (G-ADH, green solid line), and the pristine enzyme (ADH, blue solid line).
3.2. Conversion of Acetaldehyde to Ethanol
The bioactive graphene conjugate was initially applied for acetaldehyde reduction using NADH as a sacrificial cofactor as an electron and proton source by simply mixing the substrate and cofactor, without applying any voltage. The reaction was initiated by NADH addition (1 × 10–6 mol) and after 2 h, liquid samples were analyzed for ethanol with gas chromatography. A 64% conversion efficiency (CE, see Supporting Information, chemical conversion of acetaldehyde to ethanol) was achieved for G-ADH (Table 1, entry 1) while the control samples without added ADH or graphene (Table 1, entry 2) and of G-COOH alone (Table 1, entry 3) displayed a CE of 2% and 6%, respectively. To evaluate any possible catalytic contribution from physically adsorbed ADH units on G-COOH, another control sample was prepared by mixing G-COOH with ADH and thoroughly washing this mixture. The obtained CE of 20% of this control sample (Table 1, entry 4) further verified that G-ADH activity was indeed the outcome of the covalent immobilization of the enzyme on G-COOH, and that physical adsorption interactions are too weak to keep the enzyme fixed on the graphene surface. G-ADH was further tested by immobilization in the alginate matrix, aiming to protect the enzyme from thermal and chemical denaturation/deactivation, as reported previously.74,76−78 The alginate beads containing G-ADH showed higher CE (Table 1, entry 6), as compared to G-ADH in the solution. Although the efficiency of free G-ADH and G-ADH immobilized in alginate beads was lower than that of the free ADH (Table 1, entry 5), the covalent conjugation and hydrogel immobilization offer the advantages of stability and facile product separation.
Table 1. Chemical Conversion Efficiencies toward the Reduction of Acetaldehyde to Ethanol Using NADH as Cofactor during 2 h Reaction.
| entry | sample | ethanol/×10–7 mol | conversion efficiency (%) |
|---|---|---|---|
| 1 | G-ADH (homogeneous) | 6.4 | 64 |
| 2 | blank | 0.2 | 2 |
| 3 | G-COOH (homogeneous) | 0.6 | 6 |
| 4 | G-COOH/ADH (homogeneous) | 2.0 | 20 |
| 5 | free ADH (homogeneous) | 9.2 | 92 |
| 6 | G-ADH immobilized in alginate beads (heterogeneous) | 7.8 | 78 |
The G-ADH biocatalyst in the following was challenged in the same reaction for the electrochemical direct electron transfer to the catalytic sites of ADH, in the absence of the costly NADH. The electrode was prepared by immobilizing G-ADH in an alginate hydrogel matrix and then deposited on carbon felt. The catalyst displayed high activity reflected by the remarkable increase in the reductive current starting at c.a. −0.80 V, shown in the cyclic voltammogram of Figure 3a (red circles). The respective curves using bare carbon felt and alginate matrix-coated carbon felt displayed much lower currents evidencing that the recorded currents are indeed ascribed to the presence of G-ADH.
Figure 3.

Cyclic voltammograms of a bare carbon felt (black square line), an alginate matrix coated on carbon felt electrode (green triangle line), and a carbon felt modified with alginate hydrogel (red circle line) containing (a) G-ADH and (c) G-DHs were recorded at the potentials between 0 to −1.20 V vs Ag/AgCl (3 M KCl) with a scan rate of 10 mV·s–1 in 0.1 M TRIS-HCl solution pH 7.4 containing 1 M acetaldehyde under N2-saturated condition and in 0.1 M TRIS-HCl solution pH 7.0 under CO2-saturated condition, respectively. Gas chromatograms for (b) ethanol and (d) methanol analysis of samples collected before electrolysis, after 5 h electrolysis, and after 20 h electrolysis. The inset shows a transient curve of constant-potential electrolysis at −1.20 V vs Ag/AgCl (3 M KCl) from 5 to 20 h of the modified carbon felt electrode containing G-DHs.
To further substantiate the mechanism of direct electron injection to the active site of ADH, a constant potential of −1.00 V was applied continuously for 5 h. Liquid samples before and immediately after electrolysis were taken for ethanol quantification using liquid-injection gas chromatography. Figure 3b presents the chromatograms of the sample before and after electrolysis. In the case of the sample collected after electrolysis, a peak at the retention time of 5.1 min was detected corresponding to ethanol standard solution peak (Figure S6) indicating ethanol production. The 5 h chromatographic results presented the formation of 5.5 × 10–6 mol of ethanol corresponding to a Faradaic efficiency for the conversion of acetaldehyde to ethanol of 21%. These results support the direct electron injection mechanism using the electrochemical approach of ADH and subsequent reduction of acetaldehyde to ethanol. Control electrolysis experiments of the bare carbon felt and alginate-coated on carbon felt did not show formation of ethanol. For the stability test, the reaction was further performed for an additional 15 h. Ethanol production was observed with a final amount of 19.9 × 10–6 mol, corresponding to a Faradaic efficiency of 22%, showing continuous ethanol production at the same rate. While the reductive current was found to be marginally decreasing slowly from around −0.24 to −0.20 mA over 15 h (Figure S8). This observation highlights the stability of the G-ADH biocatalyst for at least 20 h electrolysis. The further stability investigation was carried out by applying a constant potential at −1.0 V for 132 h. The reductive current (Figure S9) showed a change at around 24 h of reaction, and started declining after 50 h. This observation might be due to the long-term electrochemical reaction. These results from our experiments (at an applied constant potential of −0.36 V vs reversible hydrogen electrode (RHE)) show comparable efficiency to a reported nonenzymatic system, an oxide-derived copper electrode (Faradaic efficiency of about 30% at −0.33 V vs RHE).70
3.3. Conversion of CO2 to Methanol
Encouraged by these results, we expanded the concept on the three-enzyme cascade reaction for the conversion of CO2 to high energy-density chemicals in a quest for “artificial photosynthesis”. The reduction of CO2 to methanol was pursued using the FateDH, FaldDH, and ADH, as described in Figure 1. The three enzymes were covalently bound to G-COOH via amide bond as previously described for ADH. The reactions were performed separately, yielding G-FateDH, G-FaldDH, and G-ADH.
The protein contents in G-FateDH and G-FaldDH were determined using the mentioned BCA assay, showing the amount of FateDH and FaldDH bound on graphene of 0.3 and 0.1 mg·mL–1 of G-FateDH and G-FaldDH solution (with the concentration of graphene ∼1 mg·mL–1), respectively. While their enzymatic activities were observed as 0.1 and 1.0 enzyme unit per milligram of proteins, respectively (see details in Supporting Information).
In these cascade reductions, the conversion of CO2 to formate occurs slowly. However, the bottleneck is the second step due to the quick reaction forming hydrated formaldehyde (methanediol) from formaldehyde.36,79,80 To further perform the last step, formaldehyde is needed, resulting in a dehydration rate-limit. To facilitate the cascade reaction toward methanol formation, coimmobilization was suggested as the products will be consumed in situ by other enzymes.81 Further, the suspension of each G-DH was coimmobilized in an alginate matrix. Preliminary testing of an effective function of the cascade reaction revealed the catalytic activity toward the conversion of CO2 all the way to methanol in the presence of NADH (2.26 × 10–6 mol of methanol in 14 h, 50% conversion efficiency).
To characterize the electrochemical activities of each modified electrode, cyclic voltammograms were recorded comparing inert and substrate-containing conditions (Figures S10–S12). The graphs revealed the enhancement of reductive currents in the presence of their suitable substrates indicating their catalytic activities toward the substrate conversion. Moreover, cyclic voltammograms of G-DHs modified electrodes were recorded under N2- and CO2-saturated conditions (Figure S13). A slight increase in reductive current at around −1.2 V was observed under CO2-saturated conditions, showing catalytic activities toward CO2 reduction. The direct electron injection bioelectro-catalysis was then investigated using the three G-DHs. The cyclic voltammograms in Figure 3c show high capacitive current and increased reductive current starting from −1.00 V, indicating the effective direct electron transfer mechanism. On the contrary, the bare carbon felt and nonenzymatic alginate matrix presented marginal activity (Figure 3c). By applying constant potential at −1.20 V, the transient graph revealed that the reductive current increased after 5 h up to a stable value at around -0.9 mA (Figure 3d, inset, and Figure S14), without any signs of dropping, even up to 20 h of reaction. Liquid samples were taken after 5 and 20 h of the electrolysis. In Figure 3d, the gas chromatograms showed 12.4 × 10–6 mol (around 20 ppm) of methanol production (a methanol standard solution showed a peak at retention time of around 4.00–4.50 min, Figure S6), which corresponds to a Faradaic efficiency of 12%. As a control, electrolysis of the modified electrode was performed under N2-saturated conditions (Figure S15). This experiment reveals no methanol formation after 20 h electrolysis (Figure S16) confirming the conversion of CO2 to methanol. Another control experiment was performed using unmodified G-COOH immobilized alginate matrix coated on carbon felt (Figures S17 and S18). The experiment revealed high reductive current delivered whereas only a trace of methanol was formed (Figure S19). The three-step cascade enzymatic reaction, involves the formation of formate and formaldehyde as intermediate products. The analysis of formate, formaldehyde, or other related gas products (such as CO, CH4) using ion and high-performance liquid chromatography, and gas chromatography, respectively, showed a nondetectable amount. Since the electrolysis was performed in aqueous solution, H2 is often a side-product.
Headspace analysis revealed H2 formation corresponding to 40%FE which was in the same range as the experiment performed under N2-saturated conditions (41%FE). Moreover, scanning electron microscope images of electrodes before and after 20 h electrolysis were taken (Figure S5). As compared to the bare carbon felt electrode, alginate films containing G-DHs-covered carbon felt were preserved, suggesting no loss of immobilized modified graphene.
The direct electron injection mechanism for CO2 reduction to methanol without covalent immobilization of the enzymes was previously studied by Schlager et al.37 Methanol production was reported with a concentration of 0.1 ppm (corresponding to around 10% Faradaic efficiency) and the recorded reductive current was around −0.08 mA, using the same three dehydrogenases entrapped in the alginate matrix coated on a carbon felt (2 × 0.6 × 0.6 cm3). In the present case, the three-dehydrogenases-modified graphenes showed 1 order of magnitude higher absolute currents delivered to the reaction sites suggesting a far more efficient electron transport from the electrode to the enzymes’ active sites, via the conductive graphene support57,88,89 as well as higher production rate (around 0.6 μmol·h–1). Furthermore, while the reductive current profile in the presented case of the covalent immobilization was preserved for at least 20 h (Figure S14). These observations reflected the advantage of using the conductive graphene carboxylic acid as a platform for covalent enzyme immobilization. The novel nanobiohybrid electrocatalyst has thus been herewith proven to operate even under the three-enzyme electrocatalytic reaction, outperforming the previous studies.
4. Conclusion
In this work we report the use of a nanobiohybrid catalyst, consisting of immobilized enzymes on a densely and selectively functionalized conductive graphene derivative (graphene carboxylic acid). Using this biofunctionalized graphene electrocatalyst, we show the electrocatalytic production of ethanol, fully avoiding the use of sacrificial cofactor (NADH), owing to the effective electron transfer from the electrode onto the nanobiohybrid catalyst. This cofactor- and mediator-free bioelectrocatalytic process simplifies the product separation, stabilizes the catalytic process, and boosts the current density (i.e., production rate). Electrochemical Faradaic efficiencies of around 20% were achieved for aldehyde to ethanol conversion. The same approach employed the more challenging three-dehydrogenase enzymatic system for the conversion of CO2 all the way to methanol, achieving a Faradaic efficiency of 12%. The catalyst’s selectivity toward high energy content products, operation under neutral aqueous solution, and low overpotential of 0.61 V for the six-electron electrochemical reduction of CO2 to methanol are further important advantages, as compared to state-of-the-art metal-based systems, as summarized in Table 2.24,26,82−87 Because of the bio-origin, the dehydrogenases’ system offers high material availability over rare metal composite materials. Exploitation of such enzymatic nanobioelectrocatalysis may be of importance for many other biotechnological conversions which today require costly electron donors but may be completely revolutionized by using this electrochemical method.
Table 2. State-of-the-Art Electrochemical Reduction of CO2 to Methanol.
| catalysts | electrolyte | electrode/substrate | applied potential | overpotentiala/V | j/mA cm–2 | %FEMeOH | other CO2RR product(s) | ref |
|---|---|---|---|---|---|---|---|---|
| Cu2O/Zn2O | 0.5 M KHCO3 | GDEb | –1.16 V vs Ag/AgCl | 0.54 | 10 | 27.5 (17.7c) | C2H6O | Albo et al.25 |
| Pd/SnO2 nanosheet | 0.1 M NaHCO3 | carbon paper | –0.24 V vs RHE | 0.27 | 1.45 | 54.8 | formate | Zhang et al.82 |
| copper selenide (Cu1.63Se) nanoparticle | [Bmim]PF6d (30 wt %) /CH3CN/H2O (5 wt %) | carbon paper | –2.1 V vs Ag/Ag+ (−1.175 V vs RHE) | 1.2 | 41.5 | 77.6 | formate, CO | Yang et al.26 |
| PYD@Cu–Pt alloye | 0.5 M KCl | free standing electrode | –0.6 V vs SCE | 0.07 | 22 | 37 | formate | Yang et al.83 |
| Cu2O-MWCNTsg | 0.5 M KHCO3 | Cu foil | –0.8 V vs Ag/AgCl | f | 6 | 38 | not reported | Irfan Malik et al.84 |
| Cu nanocluster/ZnO | 0.1 M KHCO3 | single crystal ZnO | –1.4 V vs Ag/AgCl | 0.83 | 12 | 2.8 | CO, CH4, C2H4, C2H6O, methyl formate | Andrews et al.85 |
| Ni | 0.1 M KHCO3 | Ni foil | –1.0 V vs RHE | 1.03 | 5 | 2.3 | CH4, C2H4, formate | Kuhl et al.86 |
| BDDh | 1 M NH3 | Si wafer | –1.3 V vs Ag/AgCl | 0.67 | f | 24.3 | CO, CH4 | Jiwanti et al.87 |
| dehydrogenases | 0.05 M phosphate buffer pH 7.6 | carbon felt | –1.2 V vs Ag/AgCl | 0.58 | 0.08 | 10 | not observed | Schlager et al.37 |
| dehydrogenases modified graphene | 0.1 M TRIS-HCl buffer pH 7.0 | carbon felt | –1.2 V vs Ag/AgCl (−0.58 V vs RHE) | 0.61 | 1 | 12 | not observed | This work |
Overpotential is compared with the thermodynamic potential for the conversion of CO2 to methanol of 0.03 V vs RHE86 where E (V vs RHE) is calculated from E(V vs Ag/AgCl) + 0.205 V + 0.0591*pH or E(V vs SCE) + 0.244 V + 0.0591*pH.
GDE: gas diffusion electrode.
The experiment was performed without supplying CO2 to GDE.
[Bmim]PF6:1-butyl-3-methylimidazolium hexafluorophosphate.
PYD: 4-(3-phenoxy-2,2-bis(phenoxymethyl)propoxy)pyridine.
The information was not given.
Multiwall carbon nanotubes (MWCNTs) impregnated with Cu2O.
Boron-doped diamond electrode.
Acknowledgments
Financial support by the Austrian Climate and Energy Fund within the MELOS (861392) project is gratefully acknowledged. Further support of the Austrian Science Foundation (FWF) within the Wittgenstein Prize for Prof. Sariciftci is gratefully acknowledged (Z222-N19). The authors gratefully acknowledge support from the Operational Programme Research, Development and Education—European Regional Development Fund, Project No. CZ.02.1.01/0.0/0.0/16_019/0000754 of the Ministry of Education, Youth, and Sports of the Czech Republic from the Czech Science Foundation (project GA CR-EXPRO, 19-27454X) and from an ERC Consolidator Grant (H2020) No. 683024. Dr. Juri Ugolotti and Jana Havláková (TGA), Jana Stráska and Dr. Klara Cepe (TEM), and Martin Petr (XPS) are acknowledged for the measurements.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsami.9b17777.
Experimental procedures, additional results, references (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Baede A. P. M.; van der Linden P.; Verbruggen A.. Annex II to IPCC Fourth Assessment Report; Cambridge University Press, 2007.
- Lüthi D.; Le Floch M.; Bereiter B.; Blunier T.; Barnola J.-M.; Siegenthaler U.; Raynaud D.; Jouzel J.; Fischer H.; Kawamura K.; et al. High-Resolution Carbon Dioxide Concentration Record 650,000–800,000 Years before Present. Nature 2008, 453 (7193), 379–382. 10.1038/nature06949. [DOI] [PubMed] [Google Scholar]
- Arrhenius S. XXXI. On the Influence of Carbonic Acid in the Air upon the Temperature of the Ground. Philos. Mag. S. 5 1896, 41 (251), 237–276. 10.1080/14786449608620846. [DOI] [Google Scholar]
- Lewis N. S.; Nocera D. G. Powering the Planet: Chemical Challenges in Solar Energy Utilization. Proc. Natl. Acad. Sci. U. S. A. 2006, 103 (43), 15729–15735. 10.1073/pnas.0603395103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aresta M.; Dibenedetto A. Utilisation of CO2 as a Chemical Feedstock: Opportunities and Challenges. Dalt. Trans. 2007, (28), 2975–2992. 10.1039/b700658f. [DOI] [PubMed] [Google Scholar]
- Kondratenko E. V.; Mul G.; Baltrusaitis J.; Larrazábal G. O.; Pérez-Ramírez J. Status and Perspectives of CO2 Conversion into Fuels and Chemicals by Catalytic, Photocatalytic and Electrocatalytic Processes. Energy Environ. Sci. 2013, 6 (11), 3112–3135. 10.1039/c3ee41272e. [DOI] [Google Scholar]
- Li H.; Opgenorth P. H.; Wernick D. G.; Rogers S.; Wu T.-Y.; Higashide W.; Malati P.; Huo Y.-X.; Cho K. M.; Liao J. C. Integrated Electromicrobial Conversion of CO2 to Higher Alcohols. Science 2012, 335 (6076), 1596. 10.1126/science.1217643. [DOI] [PubMed] [Google Scholar]
- Aresta M.; Dibenedetto A.. Key Issues in Carbon Dioxide Utilization as a Building Block for Molecular Organic Compounds in the Chemical Industry. CO2 Conversion and Utilization; American Chemical Society, 2002; Chapter 4. [Google Scholar]
- Olah G. A. Beyond Oil and Gas: The Methanol Economy. Angew. Chem., Int. Ed. 2005, 44 (18), 2636–2639. 10.1002/anie.200462121. [DOI] [PubMed] [Google Scholar]
- Olah G. A.; Goeppert A.; Prakash G. K. S. Chemical Recycling of Carbon Dioxide to Methanol and Dimethyl Ether: From Greenhouse Gas to Renewable, Environmentally Carbon Neutral Fuels and Synthetic Hydrocarbons. J. Org. Chem. 2009, 74 (2), 487–498. 10.1021/jo801260f. [DOI] [PubMed] [Google Scholar]
- Schlager S.; Dibenedetto A.; Aresta M.; Apaydin D. H.; Dumitru L. M.; Neugebauer H.; Sariciftci N. S. Biocatalytic and Bioelectrocatalytic Approaches for the Reduction of Carbon Dioxide Using Enzymes. Energy Technol. 2017, 5 (6), 812–821. 10.1002/ente.201600610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Apaydin D. H.; Schlager S.; Portenkirchner E.; Sariciftci N. S. Organic, Organometallic and Bioorganic Catalysts for Electrochemical Reduction of CO2. ChemPhysChem 2017, 18 (22), 3094–3116. 10.1002/cphc.201700148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh R. K.; Singh R.; Sivakumar D.; Kondaveeti S.; Kim T.; Li J.; Sung B. H.; Cho B.-K.; Kim D. R.; Kim S. C.; et al. Insights into Cell-Free Conversion of CO2 to Chemicals by a Multienzyme Cascade Reaction. ACS Catal. 2018, 8 (12), 11085–11093. 10.1021/acscatal.8b02646. [DOI] [Google Scholar]
- Ji X.; Su Z.; Wang P.; Ma G.; Zhang S. Tethering of Nicotinamide Adenine Dinucleotide Inside Hollow Nanofibers for High-Yield Synthesis of Methanol from Carbon Dioxide Catalyzed by Coencapsulated Multienzymes. ACS Nano 2015, 9 (4), 4600–4610. 10.1021/acsnano.5b01278. [DOI] [PubMed] [Google Scholar]
- Peterson A. A.; Nørskov J. K. Activity Descriptors for CO2 Electroreduction to Methane on Transition-Metal Catalysts. J. Phys. Chem. Lett. 2012, 3 (2), 251–258. 10.1021/jz201461p. [DOI] [Google Scholar]
- Hori Y.Electrochemical CO2 Reduction on Metal Electrodes. In Modern Aspects of Electrochemistry; Springer New York: New York, 2008; pp 89–189. [Google Scholar]
- Kuhl K. P.; Cave E. R.; Abram D. N.; Jaramillo T. F. New Insights into the Electrochemical Reduction of Carbon Dioxide on Metallic Copper Surfaces. Energy Environ. Sci. 2012, 5 (5), 7050. 10.1039/c2ee21234j. [DOI] [Google Scholar]
- Kumar B.; Llorente M.; Froehlich J.; Dang T.; Sathrum A.; Kubiak C. P. Photochemical and Photoelectrochemical Reduction of CO2. Annu. Rev. Phys. Chem. 2012, 63 (1), 541–569. 10.1146/annurev-physchem-032511-143759. [DOI] [PubMed] [Google Scholar]
- Hawecker J.; Lehn J.-M.; Ziessel R. Photochemical and Electrochemical Reduction of Carbon Dioxide to Carbon Monoxide Mediated by (2,2’-Bipyridine)Tricarbonylchlororhenium(I) and Related Complexes as Homogeneous Catalysts. Helv. Chim. Acta 1986, 69 (8), 1990–2012. 10.1002/hlca.19860690824. [DOI] [Google Scholar]
- Lehn J. M.; Ziessel R. Photochemical Generation of Carbon Monoxide and Hydrogen by Reduction of Carbon Dioxide and Water under Visible Light Irradiation. Proc. Natl. Acad. Sci. U. S. A. 1982, 79 (2), 701–704. 10.1073/pnas.79.2.701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rakowski Dubois M.; Dubois D. L. Development of Molecular Electrocatalysts for CO2 Reduction and H2 Production/Oxidation. Acc. Chem. Res. 2009, 42 (12), 1974–1982. 10.1021/ar900110c. [DOI] [PubMed] [Google Scholar]
- Schuchmann K.; Müller V. Direct and Reversible Hydrogenation of CO2 to Formate by a Bacterial Carbon Dioxide Reductase. Science 2013, 342 (6164), 1382–1385. 10.1126/science.1244758. [DOI] [PubMed] [Google Scholar]
- Chiu S.-Y.; Kao C.-Y.; Chen C.-H.; Kuan T.-C.; Ong S.-C.; Lin C.-S. Reduction of CO2 by a High-Density Culture of Chlorella Sp. in a Semicontinuous Photobioreactor. Bioresour. Technol. 2008, 99 (9), 3389–3396. 10.1016/j.biortech.2007.08.013. [DOI] [PubMed] [Google Scholar]
- Albo J.; Sáez A.; Solla-Gullón J.; Montiel V.; Irabien A. Production of Methanol from CO2 Electroreduction at Cu2O and Cu2O/ZnO-Based Electrodes in Aqueous Solution. Appl. Catal., B 2015, 176–177, 709–717. 10.1016/j.apcatb.2015.04.055. [DOI] [Google Scholar]
- Albo J.; Irabien A. Cu2O-Loaded Gas Diffusion Electrodes for the Continuous Electrochemical Reduction of CO2 to Methanol. J. Catal. 2016, 343, 232–239. 10.1016/j.jcat.2015.11.014. [DOI] [Google Scholar]
- Yang D.; Zhu Q.; Chen C.; Liu H.; Liu Z.; Zhao Z.; Zhang X.; Liu S.; Han B. Selective Electroreduction of Carbon Dioxide to Methanol on Copper Selenide Nanocatalysts. Nat. Commun. 2019, 10 (1), 677. 10.1038/s41467-019-08653-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Studt F.; Sharafutdinov I.; Abild-Pedersen F.; Elkjær C. F.; Hummelshøj J. S.; Dahl S.; Chorkendorff I.; Nørskov J. K. Discovery of a Ni-Ga Catalyst for Carbon Dioxide Reduction to Methanol. Nat. Chem. 2014, 6 (4), 320–324. 10.1038/nchem.1873. [DOI] [PubMed] [Google Scholar]
- Qiao J.; Liu Y.; Hong F.; Zhang J. A Review of Catalysts for the Electroreduction of Carbon Dioxide to Produce Low-Carbon Fuels. Chem. Soc. Rev. 2014, 43, 631–675. 10.1039/C3CS60323G. [DOI] [PubMed] [Google Scholar]
- Appel A. M.; Bercaw J. E.; Bocarsly A. B.; Dobbek H.; DuBois D. L.; Dupuis M.; Ferry J. G.; Fujita E.; Hille R.; Kenis P. J. A.; et al. Frontiers, Opportunities, and Challenges in Biochemical and Chemical Catalysis of CO2 Fixation. Chem. Rev. 2013, 113 (8), 6621–6658. 10.1021/cr300463y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aresta M.; Dibenedetto A.; Pastore C. Biotechnology to Develop Innovative Syntheses Using CO2. Environ. Chem. Lett. 2005, 3 (3), 113–117. 10.1007/s10311-005-0009-y. [DOI] [Google Scholar]
- Mandler D.; Willner I. Photochemical Fixation of Carbon Dioxide: Enzymic Photosynthesis of Malic, Aspartic, Isocitric, and Formic Acids in Artificial Media. J. Chem. Soc., Perkin Trans. 2 1988, (6), 997. 10.1039/p29880000997. [DOI] [Google Scholar]
- Parkinson B. A.; Weaver P. F. Photoelectrochemical Pumping of Enzymatic 2 Reduction. Nature 1984, 309 (5964), 148–149. 10.1038/309148a0. [DOI] [Google Scholar]
- Andreesen J. R.; Ljungdahl L. G. Formate Dehydrogenase of Clostridium Thermoaceticum: Incorporation of Selenium-75, and the Effects of Selenite, Molybdate, and Tungstate on the Enzyme. J. Bacteriol. 1973, 116 (2), 867–873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seelbach K.; Riebel B.; Hummel W.; Kula M.-R.; Tishkov V. I.; Egorov A. M.; Wandrey C.; Kragl U. A Novel, Efficient Regenerating Method of NADPH Using a New Formate Dehydrogenase. Tetrahedron Lett. 1996, 37 (9), 1377–1380. 10.1016/0040-4039(96)00010-X. [DOI] [Google Scholar]
- Obert R.; Dave B. C. Enzymatic Conversion of Carbon Dioxide to Methanol: Enhanced Methanol Production in Silica Sol-Gel Matrices. J. Am. Chem. Soc. 1999, 121, 12192–12193. 10.1021/ja991899r. [DOI] [Google Scholar]
- Singh R. K. R.; Singh R. K. R.; Sivakumar D.; Kondaveeti S.; Kim T.; Li J.; Sung B. H.; Cho B.-K.; Kim D. R.; Kim S. C.; et al. Insights into Cell-Free Conversion of CO2 to Chemicals by a Multienzyme Cascade Reaction. ACS Catal. 2018, 8 (12), 11085–11093. 10.1021/acscatal.8b02646. [DOI] [Google Scholar]
- Schlager S.; Dumitru L. M.; Haberbauer M.; Fuchsbauer A.; Neugebauer H.; Hiemetsberger D.; Wagner A.; Portenkirchner E.; Sariciftci N. S. Electrochemical Reduction of Carbon Dioxide to Methanol by Direct Injection of Electrons into Immobilized Enzymes on a Modified Electrode. ChemSusChem 2016, 9 (6), 631–635. 10.1002/cssc.201501496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schlager S.; Neugebauer H.; Haberbauer M.; Hinterberger G.; Sariciftci N. S. Direct Electrochemical Addressing of Immobilized Alcohol Dehydrogenase for the Heterogeneous Bioelectrocatalytic Reduction of Butyraldehyde to Butanol. ChemCatChem 2015, 7 (6), 967–971. 10.1002/cctc.201402932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabaey K.; Rozendal R. A. Microbial Electrosynthesis — Revisiting the Electrical Route for Microbial Production. Nat. Rev. Microbiol. 2010, 8 (10), 706–716. 10.1038/nrmicro2422. [DOI] [PubMed] [Google Scholar]
- Amao Y.; Shuto N. Formate Dehydrogenase-Viologen-Immobilized Electrode for CO2 Conversion, for Development of an Artificial Photosynthesis System. Res. Chem. Intermed. 2014, 40 (9), 3267–3276. 10.1007/s11164-014-1832-1. [DOI] [Google Scholar]
- Reda T.; Plugge C. M.; Abram N. J.; Hirst J. Reversible Interconversion of Carbon Dioxide and Formate by an Electroactive Enzyme. Proc. Natl. Acad. Sci. U. S. A. 2008, 105 (31), 10654–10658. 10.1073/pnas.0801290105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lima F.; Maia G. Direct Electron Transfer from Alcohol Dehydrogenase. RSC Adv. 2014, 4 (43), 22575–22588. 10.1039/c4ra02946a. [DOI] [Google Scholar]
- Zhao X.; Mai Z.; Kang X.; Zou X. Direct Electrochemistry and Electrocatalysis of Horseradish Peroxidase Based on Clay-Chitosan-Gold Nanoparticle Nanocomposite. Biosens. Bioelectron. 2008, 23, 1032–1038. 10.1016/j.bios.2007.10.012. [DOI] [PubMed] [Google Scholar]
- Zhou Y.; Yang H.; Chen H.-Y. Direct Electrochemistry and Reagentless Biosensing of Glucose Oxidase Immobilized on Chitosan Wrapped Single-Walled Carbon Nanotubes. Talanta 2008, 76, 419–423. 10.1016/j.talanta.2008.03.028. [DOI] [PubMed] [Google Scholar]
- Shan D.; Wang S.; Xue H.; Cosnier S. Direct Electrochemistry and Electrocatalysis of Hemoglobin Entrapped in Composite Matrix Based on Chitosan and CaCO3 Nanoparticles. Electrochem. Commun. 2007, 9 (4), 529–534. 10.1016/j.elecom.2006.10.032. [DOI] [Google Scholar]
- Liu J.; Guo C.; Li C. M.; Li Y.; Chi Q.; Huang X.; Liao L.; Yu T. Carbon-Decorated ZnO Nanowire Array: A Novel Platform for Direct Electrochemistry of Enzymes and Biosensing Applications. Electrochem. Commun. 2009, 11, 202–205. 10.1016/j.elecom.2008.11.009. [DOI] [Google Scholar]
- Zang J.; Li C. M.; Cui X.; Wang J.; Sun X.; Dong H.; Sun C. Q. Tailoring Zinc Oxide Nanowires for High Performance Amperometric Glucose Sensor. Electroanalysis 2007, 19 (9), 1008–1014. 10.1002/elan.200603808. [DOI] [Google Scholar]
- Jia J.; Wang B.; Wu A.; Cheng G.; Li Z.; Dong S. A Method to Construct a Third-Generation Horseradish Peroxidase Biosensor: Self-Assembling Gold Nanoparticles to Three-Dimensional Sol–Gel Network. Anal. Chem. 2002, 74 (9), 2217–2223. 10.1021/ac011116w. [DOI] [PubMed] [Google Scholar]
- Zhang Q.; Zhang L.; Liu B.; Lu X.; Li J. Assembly of Quantum Dots-Mesoporous Silicate Hybrid Material for Protein Immobilization and Direct Electrochemistry. Biosens. Bioelectron. 2007, 23, 695–700. 10.1016/j.bios.2007.08.008. [DOI] [PubMed] [Google Scholar]
- Nadzhafova O.; Etienne M.; Walcarius A. Direct Electrochemistry of Hemoglobin and Glucose Oxidase in Electrodeposited Sol-Gel Silica Thin Films on Glassy Carbon. Electrochem. Commun. 2007, 9 (5), 1189–1195. 10.1016/j.elecom.2007.01.010. [DOI] [Google Scholar]
- Blankenship R. E.; Tiede D. M.; Barber J.; Brudvig G. W.; Fleming G.; Ghirardi M.; Gunner M. R.; Junge W.; Kramer D. M.; Melis A.; et al. Comparing Photosynthetic and Photovoltaic Efficiencies and Recognizing the Potential for Improvement. Science 2011, 332 (6031), 805–809. 10.1126/science.1200165. [DOI] [PubMed] [Google Scholar]
- Zhang J. J.; Zhang F.; Yang H.; Huang X.; Liu H.; Zhang J. J.; Guo S. Graphene Oxide as a Matrix for Enzyme Immobilization. Langmuir 2010, 26 (9), 6083–6085. 10.1021/la904014z. [DOI] [PubMed] [Google Scholar]
- Zuo X.; He S.; Li D.; Peng C.; Huang Q.; Song S.; Fan C. Graphene Oxide-Facilitated Electron Transfer of Metalloproteins at Electrode Surfaces. Langmuir 2010, 26 (3), 1936–1939. 10.1021/la902496u. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Yu D.; Zeng C.; Miao Z.; Dai L. Biocompatible Graphene Oxide-Based Glucose Biosensors. Langmuir 2010, 26 (9), 6158–6160. 10.1021/la100886x. [DOI] [PubMed] [Google Scholar]
- Shao Y.; Wang J.; Wu H.; Liu J.; Aksay I. A.; Lin Y. Graphene Based Electrochemical Sensors and Biosensors: A Review. Electroanalysis 2010, 22 (10), 1027–1036. 10.1002/elan.200900571. [DOI] [Google Scholar]
- Urbanová V.; Holá K.; Bourlinos A. B.; Čépe K.; Ambrosi A.; Loo A. H.; Pumera M.; Karlický F.; Otyepka M.; Zbořil R. Thiofluorographene-Hydrophilic Graphene Derivative with Semiconducting and Genosensing Properties. Adv. Mater. 2015, 27 (14), 2305–2310. 10.1002/adma.201500094. [DOI] [PubMed] [Google Scholar]
- Liu C.; Alwarappan S.; Chen Z.; Kong X.; Li C.-Z. Membraneless Enzymatic Biofuel Cells Based on Graphene Nanosheets. Biosens. Bioelectron. 2010, 25 (7), 1829–1833. 10.1016/j.bios.2009.12.012. [DOI] [PubMed] [Google Scholar]
- Prasad K. P.; Chen Y.; Chen P. Three-Dimensional Graphene-Carbon Nanotube Hybrid for High-Performance Enzymatic Biofuel Cells. ACS Appl. Mater. Interfaces 2014, 6 (5), 3387–3393. 10.1021/am405432b. [DOI] [PubMed] [Google Scholar]
- Walcarius A.; Minteer S. D.; Wang J.; Lin Y.; Merkoçi A. Materials for Biology and Medicine Nanomaterials for Bio-Functionalized Electrodes: Recent Trends. J. Mater. Chem. B 2013, 1, 4878–4908. 10.1039/c3tb20881h. [DOI] [PubMed] [Google Scholar]
- Guo K.; Qian K.; Zhang S.; Kong J.; Yu C.; Liu B. Bio-Electrocatalysis of NADH and Ethanol Based on Graphene Sheets Modified Electrodes. Talanta 2011, 85 (2), 1174–1179. 10.1016/j.talanta.2011.05.038. [DOI] [PubMed] [Google Scholar]
- Besharati Vineh M.; Saboury A. A.; Poostchi A. A.; Rashidi A. M.; Parivar K. Stability and Activity Improvement of Horseradish Peroxidase by Covalent Immobilization on Functionalized Reduced Graphene Oxide and Biodegradation of High Phenol Concentration. Int. J. Biol. Macromol. 2018, 106, 1314–1322. 10.1016/j.ijbiomac.2017.08.133. [DOI] [PubMed] [Google Scholar]
- Eng A. Y. S.; Chua C. K.; Pumera M. Refinements to the Structure of Graphite Oxide: Absolute Quantification of Functional Groups via Selective Labelling †. Nanoscale 2015, 7, 20256–20266. 10.1039/C5NR05891K. [DOI] [PubMed] [Google Scholar]
- Marcano D. C.; Kosynkin D. V.; Berlin J. M.; Sinitskii A.; Sun Z.; Slesarev A.; Alemany L. B.; Lu W.; Tour J. M. Improved Synthesis of Graphene Oxide. ACS Nano 2010, 4 (8), 4806–4814. 10.1021/nn1006368. [DOI] [PubMed] [Google Scholar]
- Lerf A.; He H.; Forster M.; Klinowski J. Structure of Graphite Oxide Revisited∥. J. Phys. Chem. B 1998, 102 (23), 4477–4482. 10.1021/jp9731821. [DOI] [Google Scholar]
- Park J.; Yan M. Covalent Functionalization of Graphene with Reactive Intermediates. Acc. Chem. Res. 2013, 46 (1), 181–189. 10.1021/ar300172h. [DOI] [PubMed] [Google Scholar]
- Liao L.; Peng H.; Liu Z. Chemistry Makes Graphene beyond Graphene. J. Am. Chem. Soc. 2014, 136, 12194–12200. 10.1021/ja5048297. [DOI] [PubMed] [Google Scholar]
- Bakandritsos A.; Pykal M.; Błonski P.; Jakubec P.; Chronopoulos D. D.; Polakova K.; Georgakilas V.; Cepe K.; Tomanec O.; Ranc V.; Bourlinos A. B.; Zboril R.; Otyepka M. Cyanographene and Graphene Acid: Emerging Derivatives Enabling High-Yield and Selective Functionalization of Graphene. ACS Nano 2017, 11, 2982–2991. 10.1021/acsnano.6b08449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heng Cheong Y.; Nasir M. Z. M.; Bakandritsos A.; Pykal M.; Jakubec P.; Zbořil R.; Otyepka M.; Pumera M. Cyanographene and Graphene Acid: The Functional Group of Graphene Derivative Determines the Application in Electrochemical Sensing and Capacitors. ChemElectroChem 2019, 6, 229–234. 10.1002/celc.201800675. [DOI] [Google Scholar]
- Mosconi D.; Blanco M.; Gatti T.; Calvillo L.; Otyepka M.; Bakandritsos A.; Menna E.; Agnoli S.; Granozzi G. Arene CH Insertion Catalyzed by Ferrocene Covalently Heterogenized on Graphene Acid. Carbon 2019, 143, 318–328. 10.1016/j.carbon.2018.11.010. [DOI] [Google Scholar]
- Bertheussen E.; Verdaguer-Casadevall A.; Ravasio D.; Montoya J. H.; Trimarco D. B.; Roy C.; Meier S.; Wendland J.; Nørskov J. K.; Stephens I. E. L.; et al. Acetaldehyde as an Intermediate in the Electroreduction of Carbon Monoxide to Ethanol on Oxide-Derived Copper. Angew. Chem., Int. Ed. 2016, 55, 1450–1454. 10.1002/anie.201508851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Szabó T.; Berkesi O.; Forgó P.; Josepovits K.; Sanakis Y.; Petridis D.; Dékány I. Evolution of Surface Functional Groups in a Series of Progressively Oxidized Graphite Oxides. Chem. Mater. 2006, 18 (11), 2740–2749. 10.1021/cm060258+. [DOI] [Google Scholar]
- Mayo D. W.Characteristic Frequencies of Aromatic Compounds (Group Frequencies of Arenes). In Course Notes on the Interpretation of Infrared and Raman Spectra; Dana W. M., Foil A. M., Robert W. H., Eds.; John Wiley & Sons, Inc., 2004; pp 101–140. [Google Scholar]
- Hanefeld U.; Gardossi L.; Magner E. Understanding Enzyme Immobilisation. Chem. Soc. Rev. 2009, 38 (2), 453–468. 10.1039/B711564B. [DOI] [PubMed] [Google Scholar]
- Mohamad N. R.; Marzuki N. H. C.; Buang N. A.; Huyop F.; Wahab R. A. An Overview of Technologies for Immobilization of Enzymes and Surface Analysis Techniques for Immobilized Enzymes. Biotechnol. Biotechnol. Equip. 2015, 29 (2), 205–220. 10.1080/13102818.2015.1008192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevan M. D.Enzyme Immobilization by Covalent Bonding. In New Protein Techniques; Humana Press: NJ, 1988; pp 495–510. [DOI] [PubMed] [Google Scholar]
- Zhao F.; Li H.; Wang X.; Wu L.; Hou T.; Guan J.; Jiang Y.; Xu H.; Mu X. CRGO/Alginate Microbeads: An Enzyme Immobilization System and Its Potential Application for a Continuous Enzymatic Reaction. J. Mater. Chem. B 2015, 3 (48), 9315–9322. 10.1039/C5TB01508A. [DOI] [PubMed] [Google Scholar]
- Won K.; Kim S.; Kim K. J.; Park H. W.; Moon S. J. Optimization of Lipase Entrapment in Ca-Alginate Gel Beads. Process Biochem. 2005, 40 (6), 2149–2154. 10.1016/j.procbio.2004.08.014. [DOI] [Google Scholar]
- Dibenedetto A.; Stufano P.; Macyk W.; Baran T.; Fragale C.; Costa M.; Aresta M. Hybrid Technologies for an Enhanced Carbon Recycling Based on the Enzymatic Reduction of CO2 to Methanol in Water: Chemical and Photochemical NADH Regeneration. ChemSusChem 2012, 5 (2), 373–378. 10.1002/cssc.201100484. [DOI] [PubMed] [Google Scholar]
- Winkelman J. G. M.; Voorwinde O. K.; Ottens M.; Beenackers A. A. C. M.; Janssen L. P. B. M. Kinetics and Chemical Equilibrium of the Hydration of Formaldehyde. Chem. Eng. Sci. 2002, 57 (19), 4067–4076. 10.1016/S0009-2509(02)00358-5. [DOI] [Google Scholar]
- Ma K.; Yehezkeli O.; Park E.; Cha J. N. Enzyme Mediated Increase in Methanol Production from Photoelectrochemical Cells and CO2. ACS Catal. 2016, 6 (10), 6982–6986. 10.1021/acscatal.6b02524. [DOI] [Google Scholar]
- Luo J.; Meyer A. S.; Mateiu R. V.; Pinelo M. Cascade Catalysis in Membranes with Enzyme Immobilization for Multi-Enzymatic Conversion of CO2 to Methanol. New Biotechnol. 2015, 32 (3), 319–327. 10.1016/j.nbt.2015.02.006. [DOI] [PubMed] [Google Scholar]
- Zhang W.; Qin Q.; Dai L.; Qin R.; Zhao X.; Chen X.; Ou D.; Chen J.; Chuong T. T.; Wu B.; et al. Electrochemical Reduction of Carbon Dioxide to Methanol on Hierarchical Pd/SnO 2 Nanosheets with Abundant Pd-O-Sn Interfaces. Angew. Chem., Int. Ed. 2018, 57 (30), 9475–9479. 10.1002/anie.201804142. [DOI] [PubMed] [Google Scholar]
- Yang H. P.; Yue Y. N.; Qin S.; Wang H.; Lu J. X. Selective Electrochemical Reduction of CO2 to Different Alcohol Products by an Organically Doped Alloy Catalyst. Green Chem. 2016, 18 (11), 3216–3220. 10.1039/C6GC00091F. [DOI] [Google Scholar]
- Irfan Malik M.; Malaibari Z. O.; Atieh M.; Abussaud B. Electrochemical Reduction of CO2 to Methanol over MWCNTs Impregnated with Cu2O. Chem. Eng. Sci. 2016, 152, 468–477. 10.1016/j.ces.2016.06.035. [DOI] [Google Scholar]
- Andrews E.; Ren M.; Wang F.; Zhang Z.; Sprunger P.; Kurtz R.; Flake J. Electrochemical Reduction of CO2 at Cu Nanocluster/(1010) ZnO Electrodes. J. Electrochem. Soc. 2013, 160 (11), H841. 10.1149/2.105311jes. [DOI] [Google Scholar]
- Kuhl K. P.; Hatsukade T.; Cave E. R.; Abram D. N.; Kibsgaard J.; Jaramillo T. F. Electrocatalytic Conversion of Carbon Dioxide to Methane and Methanol on Transition Metal Surfaces. J. Am. Chem. Soc. 2014, 136 (40), 14107–14113. 10.1021/ja505791r. [DOI] [PubMed] [Google Scholar]
- Jiwanti P. K.; Natsui K.; Nakata K.; Einaga Y. Selective Production of Methanol by the Electrochemical Reduction of CO2 on Boron-Doped Diamond Electrodes in Aqueous Ammonia Solution. RSC Adv. 2016, 6 (104), 102214–102217. 10.1039/C6RA20466J. [DOI] [Google Scholar]
- Campbell A. S.; Jeong Y. J.; Geier S. M.; Koepsel R. R.; Russell A. J.; Islam M. F. Membrane/Mediator-Free Rechargeable Enzymatic Biofuel Cell Utilizing Graphene/Single-Wall Carbon Nanotube Cogel Electrodes. ACS Appl. Mater. Interfaces 2015, 7 (7), 4056–4065. 10.1021/am507801x. [DOI] [PubMed] [Google Scholar]
- Shen F.; Cao X.; Pankratov D.; Zhang J.; Chi Q. Nanoengineering of Graphene-Supported Functional Composites for Performance-Enhanced Enzymatic Biofuel Cells. Graphene Bioelectron. 2018, 219–240. 10.1016/B978-0-12-813349-1.00010-X. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.