

SUMMARY
Cotranslational protein folding requires assistance from elaborate ribosome-associated chaperone networks. It remains unclear how the changing information in a growing nascent polypeptide dictates the recruitment of functionally distinct chaperones. Here, we used ribosome profiling to define the principles governing the cotranslational action of the chaperones TRiC/CCT and Hsp70/Ssb. We show that these chaperones are sequentially recruited to specific sites within domain-encoding regions of select nascent polypeptides. Hsp70 associates first, binding select sites throughout domains, whereas TRiC associates later, upon emergence of nearly complete domains that expose an unprotected hydrophobic surface. This suggests that transient topological properties of nascent folding intermediates drive sequential chaperone association. Surprisingly, cotranslational recruitment of both TRiC and Hsp70 correlated with translation elongation slowdowns. We propose that the temporal modulation of the nascent chain structural landscape is coordinated with local elongation rates to regulate the hierarchical action of Hsp70 and TRiC for cotranslational folding.
Graphical Abstract
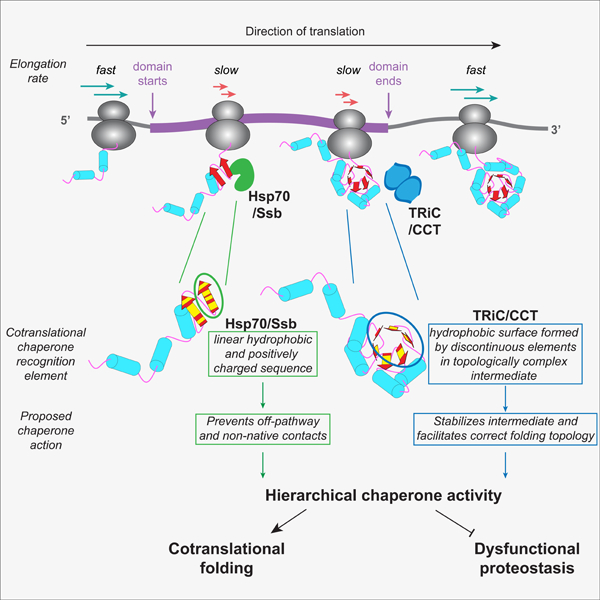
Stein et al. demonstrate a sequential association of chaperones Hsp70/Ssb and TRiC/CCT to nascent polypeptides, essential to maintain proteostasis, and uncover the principles driving this hierarchical recognition. They show Hsp70 and TRiC recognize specific and distinct topological properties in ribosome-bound nascent folding intermediates; acting in coordination with translation elongation slowdown.
INTRODUCTION
Faithfully transforming one-dimensional genetic information into functional three-dimensional proteins depends on efficient folding of newly translated polypeptides (Ellgaard et al., 2016; Kim et al., 2013; Preissler and Deuerling, 2012). This process is critical for cellular health, and many human diseases arise from failures of protein folding (Balch et al., 2008; Chiti and Dobson, 2017). Nascent polypeptides are prone to misfolding and aggregation as they emerge vectorially during synthesis, and thus lack the information necessary to complete folding until translation terminates. Large and multidomain proteins are particularly vulnerable in the crowded cellular environment (Han et al., 2007). To help overcome this challenge, protein biogenesis relies on two complementary strategies. First, folding can begin cotranslationally (Frydman and Hartl, 1996; Frydman et al., 1994; Jansens et al., 2002; Kim et al., 2015); this helps prevent aggregation through sequential domain folding of multidomain proteins ((Frydman and Hartl, 1996; Han et al., 2012; Liu et al., 2019) and cotranslational assembly of oligomeric complexes (Duncan and Mata, 2011; Shiber et al., 2018). Secondly, eukaryotes have an elaborate and conserved ribosome-associated network of chaperones, called CLIPS for chaperones linked to protein synthesis, which bind and process nascent chains as they emerge from the ribosomal exit tunnel (Albanèse et al., 2010; 2006; Preissler and Deuerling, 2012).
Hsp70 and TRiC/CCT are major, structurally-distinct chaperones in the CLIPS network (Kim et al., 2013). Hsp70s recognize linear, hydrophobic sequences that are aggregation-prone and usually buried within folded polypeptides (Clerico et al., 2015). These recognition motifs are extremely common in proteins, predicted to occur every 36 amino acids on average (Rüdiger et al., 1997). Functional analyses of the ribosome-associated yeast Hsp70 isoforms Ssb1/Ssb2 (herein referred to as Ssb) have shown that Ssb is critical for cotranslational folding (Döring et al., 2017; Hanebuth et al., 2016; Koplin et al., 2010; Willmund et al., 2013). Loss of Ssb, or its binding partners in the ribosome-associated chaperone (RAC) complex, leads to widespread aggregation of nascent polypeptides (Koplin et al., 2010; Willmund et al., 2013), highlighting the critical role of Ssb’s cotranslational activity on a large set of proteins.
TRiC/CCT (herein referred to as TRiC) is an essential hetero-oligomer chaperonin that binds co-and post-translationally to a select set of substrates, and is obligately required to fold many essential proteins (Gong et al., 2009; Yam et al., 2008). TRiC is ring-shaped and encapsulates its substrates in a central folding chamber (Cong et al., 2010; Lopez et al., 2015; Spiess et al., 2004). The determinants of TRiC’s substrate selectivity in vivo remain undefined. Biochemical analyses show that, unlike Ssb, TRiC engages its substrates through polyvalent interactions with ill-defined, non-native conformations (Joachimiak et al., 2014; Spiess et al., 2006). Unlike Ssb, the global contribution of TRiC to nascent chain stabilization remains a key unresolved question, as is its cotranslational interplay with Ssb.
Here, we defined the contribution and functional relationship of TRiC and Ssb during cotranslational protein folding. We found that the topological properties of nascent chains dictate their hierarchical recruitment of TRiC and Ssb. Both chaperones are specifically recruited to domain-encoding regions within nascent chains. Whereas Ssb is recruited early to a broad range of substrates, TRiC is recruited later to a subset of domain-specific folding intermediates. Surprisingly, TRiC and Ssb recruitment correlates with elongation slowdowns, suggesting that elongation attenuation promotes chaperone binding. Our work elucidates the rules that coordinate the activity of two major CLIPS during cotranslational folding, establishing a fundamental principle of eukaryotic protein biogenesis.
RESULTS
Cotranslational enrichment of TRiC and Ssb on ribosome-nascent chains
To globally define and compare the principles of cotranslational association of TRiC and Ssb (Figure 1A), we carried out parallel selective ribosome profiling of both chaperones in the yeast S. cerevisiae (Becker et al., 2013; Ingolia et al., 2009). Ribosome-protected mRNA was isolated from the total population of ribosome nascent-chain complexes (RNCs) (i.e. the translatome), as well as TRiC-or Ssb-bound RNCs following immunoprecipitation (IP), and then sequenced to a depth >22 million reads mapping to coding sequences (Figures 1B, S1A, and S1B, and Table S1). This depth enabled us to calculate chaperone enrichment with high statistical power to elucidate the determinants of TRiC and Ssb recruitment.
Figure 1. Cotranslational enrichment of TRiC and Ssb on ribosome-nascent chains.
(A) Investigating the cotranslational dynamics of TRiC and Ssb association with RNCs.
(B) Procedural overview.
(C) Gene-level chaperone enrichment. Few transcripts have significant total enrichment of TRiC (blue) or Ssb (green). Select substrates and IP-targeted subunits of TRiC (red) and Ssb (purple) are demarcated.
(D) Volcano plot of chaperone enrichment with each point representing a codon in the translatome. Colored points show the positions with significant TRiC (blue) or Ssb (green) enrichment classified as binding sites and used to identify substrates, such as actin (residues in red).
(E) Fraction of the identified TRiC (n = 565) and Ssb (n = 1,343) substrates in each of the indicated protein destination categories, as compared to the total translatome. ****P = 1.1e−10, **P = 0.002, hypergeometric test with cytonuclear proteins.
(F) Overlap of TRiC and Ssb substrates. P << 1e−16, hypergeometric test.
(G) Overlap of chaperone enriched positions in substrates shared by TRiC and Ssb.
See also Figures S1 and S2.
We validated our procedure by analyzing the total enrichment of select genes in the chaperone-bound fractions. RNCs translating Ssb or the TRiC subunits to which antibodies were raised, served as internal positive and negative specificity controls of cotranslational enrichment (Figures 1C and S1C). Our TRiC antibodies were raised against three of eight subunits in the hetero-oligomer, i.e. Cct3, Cct5, and Cct8. Only mRNAs encoding these subunits were enriched in the TRiC IPs, whereas neither Ssb nor the other CCT subunits were enriched in the TRiC IP (Figures S1C). In addition, the TRiC IPs were also strongly enriched in mRNAs encoding known cotranslational TRiC substrates, such as tubulin (Figure S1C) (Frydman and Hartl, 1996; Rommelaere et al., 1993). This mRNA enrichment was disrupted by puromycin treatment to release nascent chains prior to chaperone IP (Figure S1D), indicating that the interaction of TRiC or Ssb with RNCs depended on the presence of a nascent chain. However, consistent with previous work (Döring et al., 2017), our gene-level analyses identified relatively few genes as significantly enriched in the chaperone-bound fractions, with the known TRiC substrate of actin being notably unenriched (Figure 1C).
To better resolve the cotranslational substrates of TRiC and Ssb, we developed a statistically robust strategy to determine significant chaperone enrichment at each codon across a transcript (see STAR Methods). We classified chaperone binding sites as regions with five consecutive codons exhibiting statistically significant chaperone enrichment (Table S2), which eliminates positions with spuriously high counts of ribosome-protected reads. We then categorized as substrates the proteins that had at least one of these binding sites, which now included known TRiC substrates such as actin (Figure 1D). Incorporating a statistical test to our pipeline showed improved stringency of identified chaperone binding sites compared to previous analyses of Ssb (Döring et al., 2017) (Figures S1E–I). Demonstrating the robustness of our analysis, we obtained similar results using an alternative statistical approach, DESeq2 (Love et al., 2014), which was previously employed in ribosome profiling analyses of the Signal Recognition Particle (SRP) (Chartron et al., 2016).
We further determined that our addition of a membrane-permeable crosslinker during lysis to stabilize chaperone-bound RNCs enhanced IP reproducibility, but did not affect the overall distribution of positions showing TRiC enrichment (Figures S1J and S1K). We also examined the effect of adding cycloheximide (CHX). Our datasets for TRiC and Ssb were obtained following CHX addition prior to lysis to obtain a more homogenous population of ribosome-protected fragments (Wu et al., 2019), whereas a previous Ssb dataset was obtained without adding CHX (Döring et al., 2017). Applying our analysis pipeline to the Ssb datasets obtained with or without CHX revealed similar Ssb binding sites (Figure S1L), indicating that the use of CHX had negligible effect on our identification of chaperone binding sites. Collectively, these analyses validate the utility of our datasets to define the principles by which TRiC and Ssb are recruited to nascent chains.
Properties of TRiC and Ssb nascent chain substrates
To identify features within the nascent polypeptide modulating chaperone recruitment, we first analyzed the general characteristics of each set of chaperone substrates. We found that TRiC and Ssb cotranslational substrates were enriched for cytonuclear proteins, with fewer substrates encoding ER-and mitochondria-bound proteins (Figure 1E). This indicates TRiC and Ssb primarily function in the cytosol, but may also facilitate the proper targeting of some secretory or mitochondrial proteins. Overall, Ssb associated with a broader range of substrates compared to TRiC, consistent with previous analyses (Döring et al., 2017; Koplin et al., 2010; Thulasiraman et al., 1999; Willmund et al., 2013; Yam et al., 2008), but remarkably, most TRiC substrates also associated with Ssb (Figure 1F). However, we found that Ssb and TRiC did not compete for the same binding sites on these shared substrates, and instead were recruited at largely distinct points along substrate transcripts (Figure 1G).
We then observed that both TRiC and Ssb substrates were enriched in biochemical features known to challenge the cotranslational folding process (Chiti and Dobson, 2017; Döring et al., 2017; Hartl et al., 2011; Willmund et al., 2013). This included being generally longer, with higher β-sheet and lower α-helical content, and containing longer domains and more protein-protein interactions (Figure S2). TRiC substrates were further distinguished from Ssb substrates by having increased intrinsic disorder, protein length, and domain length (Figure S2C–E), suggesting that TRiC might be required for particularly recalcitrant substrates. Collectively, these data indicate that TRiC and Ssb might cooperate to cotranslationally fold many of TRiC’s substrates by recognizing different determinants within this shared set of substrates.
Distinct temporal dynamics define TRiC and Ssb recruitment
To identify the determinants of chaperone recruitment, we next examined the timing of TRiC and Ssb recruitment. Although potential Hsp70 recognition motifs across a protein sequence are predicted to occur frequently (Rüdiger et al., 1997), both Ssb and TRiC cotranslational binding events were sparse, with a few discrete binding events per transcript (Figures 2A and 2B). Most substrates had one or two significantly enriched binding events for either TRiC or Ssb (Figure 2B). These findings suggest that chaperones do not coat the nascent polypeptide by binding stably to every available chaperone recognition motif, but instead, TRiC and Ssb are recruited to specific, and mostly distinct, locations within the nascent chain.
Figure 2. Distinct temporal dynamics define TRiC and Ssb recruitment.

(A) TRiC and Ssb enrichment profiles of RPS20 and DPS1. Domains shown in purple.
(B) Fraction of cytonuclear TRiC or Ssb substrates with the indicated number of binding sites.
(C – D) Metagene analysis of (C) TRiC and (D) Ssb enrichment in their respective substrates. Lines represent median enrichment and the shaded region is the interquartile range.
(E) TRiC enrichment as a function of normalized protein length. Points represent residues in TRiC substrates (n = 565) with color showing significant enrichment.
(F) As in (E) for Ssb substrates (n = 1,343).
(G – H) Positional enrichment of (G) TRiC and (H) Ssb according to type of substrate, with n representing the number of substrates. ****P < 1e−4, ***P < 1e−3, **P < 0.01, *P < 0.05, Wilcoxon rank-sum test; pairwise comparisons not shown are not significant.
(I) Heat map of TRiC and Ssb positional enrichment in shared substrates (n = 473 ORFs with 788 TRiC binding sites and 1,138 Ssb binding sites) showing the position of peak enrichment for all binding sites. ORFs are sorted by protein length and colored in gray.
(J) Density distributions of TRiC and Ssb binding sites in (I), P = 3.3e−13, Wilcoxon rank-sum test.
See also Figures S3 and S4.
We then globally compared the positional distribution of TRiC and Ssb recruitment. We found that enrichment of both chaperones occurred after the nascent chain emerges from the ribosome (Figures 2C and 2D), unlike SRP, which is often pre-recruited to RNCs (Chartron et al., 2016). Upon nascent chain emergence, however, TRiC and Ssb exhibited distinct enrichment patterns during translation. Ssb binding events were evenly distributed throughout the length of transcripts, whereas TRiC tended to bind later in translation (Figures 2E, 2F, and S3A). Moreover, whereas Ssb was most highly enriched early in translation of its substrates, enrichment of TRiC on its substrates increased as translation proceeded (Figures S3B and S3C). We also examined the positional enrichment of TRiC and Ssb on polypeptides destined to organelles. As compared to cytonuclear substrates, Ssb binding to mitochondrial transcripts was skewed towards the N-terminus, whereas TRiC recruitment still generally occurred later in translation (Figures 2G, 2H, and S4A–C). Ssb binding was also skewed towards the N-terminus of secretory proteins containing a signal sequence (SS) or a transmembrane domain (TMD), particularly SS proteins (Figures 2H and S4D–E). The distribution of TRiC binding events was also earlier in translation for SS proteins, but not significantly earlier for TMD proteins (Figures 2G and S4D–E). However, for both types of secretory proteins, the distribution of TRiC binding events occurred later than Ssb, and many of the binding sites occurred in cytoplasmic domains of TMD proteins (Figures 2G, 2H, and S4E). These analyses suggest a temporal dynamic of Ssb and TRiC binding, with Ssb associating early and throughout translation and TRiC preferentially binding after a significant portion of the polypeptide has emerged.
We next examined the patterns of temporal association of TRiC and Ssb to the nascent chains of shared substrates. Ssb was generally recruited before TRiC, regardless of the Ssb dataset we used in the analysis (ours or from (Döring et al., 2017)) (Figures 2I, 2J, and S3D–E). When examining the highest enriched binding event in each shared transcript, ~74% of TRiC binding events occurred after or at Ssb binding. These findings support a model of ordered recruitment of these chaperones to translating polypeptides. To examine if Ssb binding was required for TRiC recruitment, we pulse-labelled nascent chains followed by a TRiC IP. Surprisingly, deletion of Ssb tended to enhance cotranslational TRiC association with nascent chains (Figure S3F). This indicates that while Ssb is dispensable for nascent chain interaction with TRiC, its action early in translation reduces TRiC association with nascent chains. Interestingly, despite generally binding later in translation than Ssb, some TRiC binding events occurred long before the full length protein had emerged (Figure 2E), and thus were not determined by chain length or by termination events. In fact, we found that larger proteins tended to recruit TRiC earlier in translation than smaller proteins (Figure S3G). Overall, these data reveal a hierarchical chaperone pathway where nascent polypeptides generally first interact with Ssb, and upon further translation, are then recognized by TRiC.
Structural and sequence elements dictate cotranslational chaperone recruitment
Formation of correctly folded domains is a challenge for cotranslational folding (Kaiser and Liu, 2018). We thus examined whether recruitment of TRiC or Ssb correlated with the translation of protein domains (Figure 3A). Aligning transcripts to the start of predicted protein domains revealed that TRiC and Ssb binding sites concentrated within domain-encoding regions (Figures 3B–D). While Ssb binding sites were distributed throughout the domain, TRiC tended to associate after nearly the entire domain had emerged from the ribosome (Figures 3B–D and S5A–D), even for domains that emerge early in translation (Figures S5E and S5F). These distinct modes of chaperone interaction were very robust, and held true using either of two major domain annotation databases; when analyzing all chaperone binding sites or just the most enriched site within transcripts (Figure S5C); or using our Ssb dataset or a previously published one (Döring et al., 2017) (Figure S5D). We conclude that Ssb associates with specific elements distributed throughout domains, whereas TRiC recognizes nascent chain determinants generated in nearly-complete domains.
Figure 3. Structural and sequence elements dictate cotranslational chaperone recruitment.

(A) Investigating when TRiC and Ssb bind as protein domains are translated.
(B – D) Density distribution and heat maps showing the position of peak enrichment for all binding sites in reference to the start of the nearest emerged protein domain (purple). Gray bars in heat maps denote the remaining protein length after the end of the domain. n = 1,585 Ssb binding sites assigned to 1,125 domains in 904 ORFs, 683 TRiC binding sites assigned to 500 domains in 418 ORFs; P = 7.4e−9, Wilcoxon rank-sum test.
(E – F) Propensity of β sheet secondary structure in the nascent chain at the start of chaperone recruitment for (E) Ssb binding sites (n = 2,263) or (F) TRiC binding sites (n = 902), as compared to an equivalent number of randomly sampled positions in the respective set of substrates. The line represents the mean β sheet propensity and the shaded region is the 95% bootstrapped confidence interval.
(G) Net charge of amino acids in the nascent chain at the start of chaperone recruitment at TRiC binding sites (blue; n = 806) or Ssb binding sites (green; n = 2,220), as compared to 10,000 randomly sampled peptide sequences (gray). The line represents the mean charge and the shaded region is the 95% bootstrapped confidence interval.
(H) Enriched domains in TRiC and Ssb substrates. Dashed lavender line indicates P = 0.05, hypergeometric test.
See also Figures S5 and S6.
We next investigated the determinants driving the distinct TRiC or Ssb recruitment to different sites in protein domains. We first examined secondary structure and sequence properties. Ssb was recruited when the nascent chain segment exiting the ribosome had high β-sheet propensity, was enriched for positively charged and hydrophobic residues, and depleted for negatively charged residues (Figures 3E, 3G, and S5G–H), as previously observed (Döring et al., 2017). By contrast, we did not identify any obvious characteristics in the nascent chain region emerging from the ribosome when TRiC was recruited, with the exception of a slight increase in Ala and Gly residues (Figures 3F, 3G, and S5H). However, we did identify a repeating pattern of higher β sheet propensity in distal segments of the nascent chain upon TRiC recruitment, with these segments having emerged from the ribosome well before TRiC was recruited (Figure 3F). These data suggest that Ssb recognizes properties of linear polypeptides just emerged from the ribosome, whereas TRiC recognition depends on global domain properties, such as formation of specific intermediates linked to β-sheet content.
The topology of protein domains determines their folding kinetics and pathway (Dokholyan et al., 2002; Kaiser and Liu, 2018; Shank et al., 2010). We thus examined whether specific domain topologies were enriched in TRiC and Ssb substrates. Indeed, TRiC and Ssb were preferentially recruited to nascent chains encoding different types of domains. Smaller domains, such as the two-layer α-β plait sandwich preferentially associated with Ssb, but not TRiC (Figures 3H and S6A). Both Ssb and TRiC substrates were enriched in larger, more complex α-β domains, including TIM barrel domains and the three-layer α-β sandwich Rossmann fold. TRiC substrates were also particularly enriched for the mostly β-sheet WD40 propeller domain. These findings led us to examine the positional distribution of TRiC and Ssb binding sites in shared substrates containing either α-β Rossmann folds or β-propeller/WD40 domains. For these distinct domains, TRiC generally associated after Ssb (Figures S6B–G). Of note, TRiC bound earlier in translation for proteins encoding Rossmann folds as compared to WD40 domains (Figures S6B–E), but TRiC was nonetheless recruited near the end of domain synthesis for both domain types (Figures S6F and S6G). Collectively, these data suggest that chaperones are recruited to domain-specific intermediates generated during cotranslational folding.
Structural logic underlying TRiC and Ssb recruitment to β-propeller domains
To better understand how structural topology determines the cotranslational recruitment of TRiC and Ssb, we next examined their interaction with β-propeller WD40 domains. While β-propellers are common in eukaryotes due to their stability and functional versatility (Han et al., 2007; Stirnimann et al., 2010), their folding is challenged by intra-domain-swaps between the blades in the propeller, which stabilize off-pathway folding intermediates (Borgia et al., 2015; 2011).. In fact, several β-propeller proteins obligately require TRiC for folding (Camasses et al., 2003; Freund et al., 2014; Miyata et al., 2014). We hypothesized that the precise TRiC and Ssb recruitment sites for nascent β-propellers may reflect how chaperones alleviate these biophysical challenges to the folding process.
To test this hypothesis, we first examined Enp2, which has an N-terminal WD40 domain and was one of the highest enriched substrates in the TRiC-interactome. Positional analysis of chaperone enrichment revealed that Ssb did not bind to every blade or even every hydrophobic β strand, but instead, Ssb bound periodically at discrete sites in the central WD40 blades of the β-propeller (Figures 4A, 4B, and Movie S1). This specificity in the cotranslational recruitment of Ssb was also confirmed using the previous Ssb dataset (Döring et al., 2017) (Figure S6H). By contrast, TRiC was recruited only upon synthesis of the last blade, once the ribosome exposed nearly the entire domain (Figures 4A, 4B, and Movie S1). We next carried out a metagene analysis aligning the subdomain topology of thirty β-propeller WD40 proteins and examined Ssb and TRiC binding sites. Remarkably, the sequential pattern of Ssb and TRiC recruitment was characteristic of nascent WD40 domains: Ssb binding occurred during synthesis of the early repeats, whereas TRiC binding occurred as the last blades emerged from the ribosome (Figure 4C). This pattern rationalizes the distinct roles of these chaperones in addressing the challenges of β-propeller domain folding: Ssb binding to the central blades presumably prevents inappropriate inter-blade packing interactions, whereas TRiC binding the entire domain likely facilitates acquisition of the correct overall topology (Figure 4D). These findings illustrate how the hierarchical activity of Ssb and then TRiC responds to specific folding challenges of the nascent protein to promote on-pathway cotranslational folding.
Figure 4. Structural logic underlying Ssb and TRiC recruitment to β-propeller domains.

(A) Positional enrichment in ENP2. Dashed gray lines indicate where the WD40 repeats emerge.
(B) Structure of Enp2 (PDB ID: 5WLC (Barandun et al., 2017)) denoting the recently emerged nascent chain at the time of TRiC (blue) or Ssb (green) binding.
(C) Metagene analysis of TRiC and Ssb binding in WD40 proteins identified as TRiC substrates (n = 30), aligned where the first WD40 repeat emerges, with dashed gray lines noting the average point at which WD40 repeats emerge. Blue and green lines represent the median enrichment of TRiC and Ssb, respectively, and the shaded region is the interquartile range.
(D) Model of how Ssb and TRiC are involved in the formation of folded WD40 domains.
Linking chaperone recruitment to the structural landscape of domain-specific folding intermediates
To further define the determinants of TRiC recruitment, we next analyzed its binding sites in the context of the final domain structure. Intriguingly, unlike what we observed for Rossmann folds and WD40 domains, TRiC binding to TIM barrels occurred much before translation of the domain was completed, but still after Ssb binding (Figures 5A and 5B). We hypothesized that this difference may arise from the folding intermediates generated by each type of domain during translation. The native domain structure was used to examine possible substructures available to the nascent chain upon TRiC binding and after dissociation. TIM barrels consist of a barrel of β sheets surrounded by α helices. We found that TRiC was recruited after most of the β barrel had emerged from the ribosome, at a point when the β strands collectively form a folding intermediate with a large exposed hydrophobic surface that is presumably recognized by TRiC (Figures 5C and S6I). Dissociation of TRiC occurred upon translation of the C-terminal α-helices that complete folding of the domain and conceal the β sheet surface. This timing of TRiC recruitment and release was observed for several nascent TIM barrel domains (e.g. the cytonuclear Hem2 and the secretory Exg1) (Figures 5C and S6I), indicating a general mechanism of chaperonin-assisted recognition and folding of nascent TIM barrels.
Figure 5. Linking chaperone recruitment to the structural landscape of domain-specific folding intermediates.

(A) Density distribution of TRiC binding sites in reference to TIM barrels (gray) compared to WD40 domains (orange, P = 5.7e−6) and Rossmann domains (purple, P = 0.002), using the position of peak enrichment for all binding sites.
(B) Heat map of peak enriched position of all TRiC binding sites aligned at the start of TIM barrels (dark gray). Light gray lines indicate the length of the protein beyond the domain end. Density plot shows Ssb sites. n = 34 TRiC sites assigned to 25 domains in 25 ORFs, and 67 Ssb sites assigned to 42 domains in 40 ORFs; P = 0.001, Wilcoxon rank-sum test.
(C – D) Structure of (C) Hem2’s TIM barrel (PDB ID: 1H7N (Erskine et al., 2001)) and (D) Gsp1’s Rossmann fold (PDB ID: 3M1I (Koyama and Matsuura, 2010)) exposed from the ribosome at the time TRiC binds (left) as well as the complete domain after TRiC is released (right). Cartoon colored according to secondary structure, with hydrophobic residues in yellow, and surface representation of just the region exposed before TRiC binds. Box denotes the nascent chain that emerges just before TRiC binds.
(E) Schematic of TRiC binding after Ssb as the end of a domain is translated.
(F) Growth of WT and mutant atp2-P353,355A yeast on YPD or media containing the non-fermentable carbon source of glycerol. Representative image of 5 biological replicates is shown.
(G) TRiC enrichment of ATP2 and atp2-P353,355A. Total and TRiC-bound mRNA was isolated from RNCs followed by qPCR. n = 3 biological replicates with mean ± SEM. P = 0.01, Welch’s t-test.
(H) TRiC enrichment ribosome occupancy profile of ATP2 or atp2-P353,355A.
See also Figure S6.
Remarkably, analysis of TRiC binding to nascent chains encoding the three-layer (α-β-α) Rossmann fold revealed a similar folding principle to that of TIM barrels (Figures 5D and S6J). In Rossmann folds, the folding intermediate containing a β sheet exposing a hydrophobic surface emerges only after almost the entire domain was translated. As shown for Gsp1, TRiC was recruited after the first two layers (α-β) had emerged from the ribosome, which creates a hydrophobic surface composed of contributions from previously translated β strands in the sheet (Figures 5D and S6J). TRiC then dissociated upon translation of the second α-helical layer, which buries the hydrophobic surface in the native structure. These analyses rationalize the distinct timing of TRiC recruitment to TIM barrels versus Rossmann folds, whereby ribosome exposure of an intermediate containing a hydrophobic surface comprised of discontinuous β strands occurs earlier in TIM barrels than in Rossmann folds. Yet, both types of domains seem to recruit TRiC to a similar nascent folding intermediate that contains a large hydrophobic patch, which will be subsequently buried within the folded structure upon translation of additional structural elements.
We next asked whether the timing of TRiC-nascent chain association was linked to domain folding (Figure 5E). We utilized the Rossmann domain TRiC substrate Atp2, which is a subunit of the mitochondrial ATP synthase complex. TRiC associated just before the emergence of the last two of seven β strands comprising the β-sheet layer in Atp2’s Rossmann domain. Similar to the examples above, this nascent intermediate exposes a discontinuous, hydrophobic surface created by the β strands (Figure S6K). At the start of TRiC association, residues P353 and P355 occupy the ribosome active site, and upon emergence, these residues help complete domain folding (Figure S6L). Therefore, we hypothesized that the double P353A-P355A mutation should disrupt Atp2 folding. Indeed, this mutant impaired growth on non-fermentable carbon sources (Figure 5F), as well as incorporation of Atp2 into mitochondria (Figure S6M). We then compared the cotranslational TRiC association with this mutant and wild-type using qPCR and ribosome profiling. qPCR indicated that the ATP2 mutations enhanced TRiC-RNC association (Figure 5G). Ribosome profiling further revealed that TRiC binding was enhanced at the point of the Atp2 mutation (Figure 5H). Interestingly, additional TRiC binding events occurred later in translation of the mutant, which mapped to a fully helical C-terminal domain that TRiC does not normally bind stably (Figure 5H). This altered pattern of TRiC association likely reflects disrupted domain folding which generates aberrant nascent folding intermediates. Collectively, these experiments define how domain topology and properties determine the nascent folding intermediates that are recognized co-translationally by TRiC.
Chaperone association is coordinated with changes in elongation rate
Since chaperone recruitment and cotranslational folding both occur on a dynamically changing growing polypeptide, we next considered whether these are coordinated with local elongation rate. Translation kinetics has emerged as an important, albeit poorly understood, determinant of nascent chain fate (Rodnina and Wintermeyer, 2016; Stein and Frydman, 2019). Changes in local elongation rates were determined by analyzing the abundance of ribosome-protected reads as translation proceeds across a transcript, since increased ribosome occupancy relative to adjacent codons indicates an elongation slowdown (Ingolia et al., 2018; Schuller et al., 2017). To assess the relationship between elongation rates and TRiC or Ssb recruitment, we generated total ribosome profiling libraries without CHX treatment, then performed a metagene analysis aligned at the start of either TRiC or Ssb binding. We found an increase in ribosome-protected reads coincided with TRiC binding, and surprisingly, also with Ssb binding, unlike previously suggested (Döring et al., 2017) (Figures 6A and 6B). We also observed a subtle but consistent acceleration in translation just before binding, but maximal chaperone enrichment corresponded with maximal slowdown in the translatome (Figures 6A and 6B). This suggests that chaperone binding occurs in coordination with local ribosome pausing.
Figure 6. Chaperone association is coordinated with changes in elongation rate.

(A – B) Metagene analysis of translation kinetics. Dark lines represent the mean ribosome occupancy of the translatome, and lightly shaded lines represent the mean chaperone enrichment (odds ratio), centered around the peptidyl transferase center (PTC) at the start of (A) TRiC (n = 902 sites) or (B) Ssb (n = 2,263 sites) recruitment. Gray lines represent the mean ribosome occupancy at an equivalent number of randomly sampled positions in the respective set of substrates. Shaded regions are the 95% bootstrapped confidence interval. Arrows demarcate slight acceleration before binding.
(C – D) Metagene analysis of codon optimality at (C) TRiC or (D) Ssb binding sites, as in (A – B).
(E – F) Top left, TRiC and Ssb enrichment profiles of TUB1 and TUB2, domains in purple. Top right, structures of Tub1 and Tub2 (PDB ID: 4FFB (Ayaz et al., 2012)) colored by the region of the protein that is exposed when major chaperone binding occurs. Bottom, ribosome occupancy of the translatome (red) and TRiC enrichment (blue) showing elongation slowdown around positions of TRiC recruitment as compared to N-terminal region in TUB1 when TRiC is not recruited.
See also Figure S7.
To validate our conclusions, we performed a number of additional analyses: (1) we used only the maximally enriched chaperone binding site in each transcript (Figure S7A), (2) we used the Ssb binding sites identified through our statistical pipeline with the previously published Ssb dataset prepared without CHX (Döring et al., 2017) (Figure S7B), and (3) we used previously published total translatome datasets (Figures S7C and S7D). These analyses yielded the same conclusions as our original analyses: chaperone binding coincided with a local elongation slowdown. However, we noted that CHX addition introduced slight variation in where the ribosome pausing event occurred in relation to chaperone binding (Figures S7C and S7D), likely due to subtle offsets in the exact codons classified as chaperone-bound. These effects are minor and did not impact any of our conclusions, as we observed the same relationship between chaperone binding and elongation attenuation when analyzing the previous Ssb dataset prepared without CHX (Döring et al., 2017).
As a comparison to TRiC and Ssb, we also used our analysis strategy to examine elongation rates at previously characterized SRP binding sites (Chartron et al., 2016). In agreement with previous findings (Chartron et al., 2016; Pechmann et al., 2014), we found increased ribosome occupancy around these sites (Figure S7E). The duration of this increase was shorter than that for TRiC and Ssb binding sites, but interestingly, the slight acceleration in translation observed before TRiC and Ssb binding was absent for SRP. These analyses indicate that local slowdowns in elongation are prevalent at sites of cotranslational chaperone or SRP recruitment, with slightly distinct dynamics.
Given that short clusters of optimal or nonoptimal codons help dictate the rate of elongation (Dana and Tuller, 2014; Hussmann et al., 2015; Weinberg et al., 2016), we next examined whether the translation slowdowns during chaperone binding correlated with changes in codon optimality. Indeed, the slowdown coincided with clusters of non-optimal codons in the PTC just after the start of either TRiC or Ssb recruitment (Figures 6C, 6D, and S7F). Notably, clusters of optimal codons also correlated with the subtle elongation acceleration observed just before chaperone binding, as seen for the Ssb binding sites we identified in the Ssb dataset prepared without CHX (Döring et al., 2017) (Figure S7F). These data raise the possibility that codon optimality has evolved to modulate cotranslational chaperone recruitment.
We further found that the link between elongation rate and chaperone recruitment explained our intriguing observation that the two tubulin isoforms, which share a very similar double α-β sandwich structure, had distinct cotranslational association of TRiC. All tubulin isoforms shared a late TRiC binding event at the end of the second α-β sandwich domain (Figures 6E and 6F). However, TUB2, but not TUB1, also had an earlier TRiC binding event around position 170, as the first α-β sandwich domain emerged from the ribosome (Figures 6E and 6F). Analysis of the local elongation rates offered a rationale for the distinct mode of TRiC recruitment. While both TUB1 and TUB2 had a local elongation slowdown at the end of the second α-β sandwich, only the TUB2 transcript encoded an elongation slowdown at the end of the N-terminal α-β sandwich, as no slowdown occurred at the corresponding position in the TUB1 mRNA (Figures 6E and 6F, bottom). This illustrates that TRiC recruitment is coordinated by the integration of translation kinetics with the specific properties of cotranslationally-generated folding intermediates.
DISCUSSION
We here define the determinants and temporal dynamics that drive the cotranslational recruitment of TRiC and Ssb to nascent chains in vivo. Our data explain early in vitro translation experiments showing that Hsp70 and TRiC sequentially bind model nascent polypeptides to achieve folding (Frydman and Hartl, 1996; Frydman et al., 1994). We now define the nascent polypeptide properties driving cotranslational TRiC binding and its hierarchical relationship with Ssb action (Figure 7A). While Ssb recognized linear sequence elements emerging throughout translation of domain-encoding regions (Döring et al., 2017; Willmund et al., 2013), TRiC generally associated once nearly complete domains emerged due to the formation of specific types of topologically complex folding intermediates. Of note, association with either TRiC or Ssb was coordinated with a slowdown in translation, suggesting that local elongation rates are optimized to balance the kinetics of chaperone binding with the cotranslational formation of folding intermediates.
Figure 7. Cotranslational proteostasis balances chaperone association, nascent chain folding, and elongation rate.

(A) Model for how the kinetics of nascent chain folding drive cotranslational chaperone recruitment in coordination with translation elongation. As elongation proceeds, some structural elements fold quickly before chaperones can be recruited (bottom). The longer time required to fold other structural elements allows time for chaperone association based on nascent chain properties.
(B) Cellular model of cotranslational chaperone recruitment. Distinct topological features in the nascent chain recruit Ssb or TRiC to RNCs to form structural folding intermediates during local translation slowdowns. Subcellular localization of substrates also helps dictate chaperone recruitment, with potential roles in: 1) membrane targeting, 2) folding cytoplasmic regions, or 3) generating import-competent structural intermediates.
Our findings help elucidate the global principles by which the cell synergizes the activity of distinct chaperones to efficiently fold nascent polypeptides. Despite Hsp70 recognition motifs being extremely common (Rüdiger et al., 1997), TRiC and Ssb bound sparsely to specific positions in the polypeptide. Although it is possible, in principle, that additional lower affinity sites do not pass our statistical threshold, our data indicate that chaperones do not bind stably to the many available chaperone-binding sites in the emerging nascent chain. Instead, the binding sites identified in our dataset, presumably of higher affinity, appear to correspond with transient folding intermediates that require chaperone action for productive folding. The preferential association of Ssb to linear sequence elements that form β strands, may serve to prevent off-pathway folding events and promote on-pathway intermediates that can either fold or be transferred to TRiC, or possibly other CLIPS (Gestaut et al., 2019). By contrast, our data suggest that TRiC associates with domain-specific folding intermediates exposing a hydrophobic patch composed of discontinuously translated hydrophobic β strands. Binding of TRiC towards the end of such domains likely stabilizes these intermediates within the protected environment of TRiC’s chamber to facilitate folding of the complete domain. TRiC then dissociates upon translation of the elements that complete folding and bury the hydrophobic surface within the core of the folded domain.
The chaperone interactions with β-propeller WD40 domains illuminates the functional cooperation of TRiC and Ssb in cotranslational folding of complex multidomain topologies. Interestingly, with repeating structures such as those in β-propellers, the forces stabilizing the native state also stabilize misfolded trapped intermediates produced by non-native contacts between repeats (Borgia et al., 2011). During the evolution of β-propellers, these non-native contacts have driven β-propeller domains to decrease overall native stability in order to optimize the likelihood of folding correctly (Smock et al., 2016). Our data show how cotranslational Hsp70 and TRiC action might have allowed evolution to resolve this trade-off between stability and foldability. Ssb recruitment earlier in translation to bind central WD40 repeats presumably prevents non-native contacts between β-propeller repeats, whereas TRiC association near the end of the domain likely facilitates the correct topology (Figure 4). The evolution of this chaperone hierarchy may have enabled the expansion and versatility of β-propeller domains.
Although mostly interacting with cytonuclear proteins, we found that Ssb and TRiC also interacted with a subset of organelle-destined proteins. These interactions likely facilitate cotranslational stabilization of translocation precursors, as we demonstrated for Atp2, as well as the maturation of cytoplasmic domains in transmembrane proteins. Presumably, Ssb and TRiC function in concert with additional specialized targeting factors, such as the SRP, the mitochondrial import machinery, or the EMC complex (Chartron et al., 2016; Costa et al., 2018; Plath and Rapoport, 2000; Shurtleff et al., 2018; Tripathi et al., 2017). Interestingly, the timing of TRiC recruitment was similar for Hem2 and Exg1 – occurring shortly before the end of the TIM barrel emerges from the ribosome – despite these proteins being cytonuclear or secretory, respectively. Such remarkable congruence suggests that domains of a given topology share folding landscapes populated by similar kinetically-trapped folding intermediates (Neudecker et al., 2012), and that these folding landscapes may have co-evolved with chaperone machinery.
The finding that local slowdowns in elongation are linked to both TRiC and Ssb binding resonates with the emerging concept that elongation kinetics in vivo are optimized for productive protein folding (Stein and Frydman, 2019). Such local slowdowns may help recruit TRiC and Ssb, as seen for SRP (Chartron et al., 2016; Pechmann et al., 2014), or may balance the kinetics of chaperone recruitment and activity with formation of folding intermediates. Disrupting this balance might impair chaperone-mediated folding, and thereby provide a mechanistic explanation for why increasing codon optimality enhances the likelihood of nascent chain misfolding (Kim et al., 2015). However, our results conflict with previous analyses of cotranslational Ssb binding (Döring et al., 2017). Two primary reasons explain this discrepancy. First, our incorporation of a statistical test to identify chaperone binding sites helped reduce the noise when analyzing elongation rate (not shown). Second, our analysis of local translation rates relied on the standard comparison of ribosome-protected reads in the upstream and downstream regions adjacent to a position of interest (Ingolia et al., 2018; Schuller et al., 2017). By contrast, the previous study grouped codon positions based on their Ssb enrichment or depletion (Döring et al., 2017), which resulted in comparing ribosome-protected reads across distal sites rather than contiguous codons.
Interestingly, in analyzing the elongation kinetics around TRiC and Ssb binding sites, we also found a slight acceleration just before chaperone binding, which was absent for SRP binding sites. The significance of this acceleration remains to be determined, but we posit that it might be associated with either folding or chaperone-mediated pulling on the nascent chain. Folding nascent chains can generate force to overcome ribosome stalling (Goldman et al., 2015), and chaperone binding or domain folding may also alter the rate of translation (Leininger et al., 2019). The slight acceleration that was present at the initial point of TRiC and Ssb binding sites might indicate that initial chaperone recognition may help pull the nascent chain and accelerate translation.
Defining the distinct and hierarchical roles for TRiC and Ssb provides insight into how the cell optimally distributes their cotranslational load for effective proteostasis maintenance. The more abundant Ssb acts first and perhaps is sufficient to resolve less challenging folding problems, whereas the less abundant TRiC acts later on a more restricted set of topologically complex domains. Disrupting this balance, such as by deletion of Ssb, would impair proteostasis by creating a higher load for other chaperones like TRiC, as observed here (Figure S3F), and other Hsp70s as observed previously (Yam et al., 2005). Cotranslational misfolding events might also disrupt this balance through accumulation of intermediates with enhanced chaperone binding (as we found with Atp2), which may deplete the pool of available folding capacity. These considerations might prove to be critical for understanding proteostasis dysfunction in the context of aging or disease.
STAR METHODS
LEAD CONTACT AND MATERIALS AVAILABILITY
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Judith Frydman (jfrydman@stanford.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Yeast strains
All experiments were performed using derivatives of BY4741 (MATa his3Δ1 leu2Δ0 met15Δ0 ura3Δ0). Point mutations in ATP2 of BY4741 were generated using the CRISPR-Cas9 method following established techniques (Ryan et al., 2016). A plasmid expressing Cas9 and the single guide RNA expression cassette was a gift from Jamie Cate (Addgene plasmid #60847) (Ryan et al., 2014). A guide sequence specific to ATP2 was cloned using oligonucleotides 5’CGGGTGGCGAATGGGACTTTGCAAAAGTAGTGGCAGGAGCGTTTTAGAGCTAGAAATAGC and 5’GCTATTTCTAGCTCTAAAACGCTCCTGCCACTACTTTTGCAAAGTCCCA TTCGCCACCCG. The DNA repair template containing the atp2-P353A and atp2-P355A mutations was generated by annealing oligonucleotides 5’GAAGGGTTCTGTCACTTCTGTGC AAGCCGTTTATGTTCCAGCCGATGATTTAACAGATGCTGCTGCTGCCACTAC and 5’CCTAATTCTGAAATACCTCTTGACAAGACGGTAGTAGCGTCCAAATGGGCAAAAGTA GTGGCAGCAGCAGCATC. Correct strains were confirmed by amplification of genomic DNA and sequencing.
METHOD DETAILS
Ribosome profiling
To isolate chaperone-bound ribosome nascent chain complexes (RNCs) for selective ribosome profiling, overnight yeast cultures were diluted in 500 mL YPD to OD600 0.05 and grown at 30°C to OD600 ~0.5. Cells were harvested by vacuum filtration in the presence of 100 µg mL−1 cycloheximide (CHX), which was added immediately prior to collection and was in contact with cells for less than 2 min during collection, as described elsewhere (Chartron et al., 2016). No CHX was added to cells used for puromycin-treated samples. Cells were lysed in lysis buffer (50 mM HEPES-KOH pH 7.5, 140 mM KCl, 5 mM MgCl2, 0.1% NP-40, 100 µg mL−1 CHX, 1X cOmplete EDTA-free Protease Inhibitor Cocktail (Roche), 0.5 mM DTT, 1 mM PMSF) supplemented with 20 U mL−1 apyrase (Sigma-Aldrich) and 1 mg mL−1 DSP (Thermo Fisher Scientific) for crosslinking, as described elsewhere (Becker et al., 2013). (Preliminary libraries were also prepared without DSP crosslinker. We found these decreased the reproducibility of immunoprecipitation efficiency, but did not affect the positional enrichment of chaperones during translation (Figures S1J and S1K).) For puromycin treatment, 5 mM puromycin was added to the lysis buffer and CHX was omitted. Cells were lysed with glass beads by vortexing for 20 min at 4°C, followed by centrifugation at 8,000 g for 5 min at 4°C. Crosslinking reactions were quenched with 25 mM glycine. Ribosomal pellets were isolated using a 25% (w/v) sucrose cushion prepared in lysis buffer without NP-40 and centrifuged at 72,000 rpm in a TLA 100.2 rotor (Beckman) for 20 min at 4°C. Pellets were resuspended in lysis buffer and digestion was performed with 0.25 µl RNase I (Ambion) per 100 µg RNA for 45 minutes at room temperature, followed by adding 2 µl SUPERase·In (Ambion) per 100 µl to stop digestion. Samples of total RNA from the ribosomal pellets were removed for ribosome profiling of the total translatome. Immunoprecipitation of chaperone-bound RNCs was performed using antibodies generated in rabbits against the apical domains of Cct3, Cct5, and Cct8, and against Ssb1 and Ssb2. Antibodies were crosslinked to Dynabeads™ Protein G (Invitrogen), then added to samples that were rotated at 4°C for 2 hr. Samples were washed twice in lysis buffer, and then five times with lysis buffer supplemented with 0.2% NP-40 and 10% glycerol. RNA extraction was performed using a miRNeasy kit (Qiagen). Ribosomal RNA was removed using the Ribo-Zero Gold kit (Illumina) after isolating 24–35 nucleotide footprints. Library preparation continued as described previously (Ingolia et al., 2012).
Ribosome profiling libraries to monitor translation kinetics were prepared with minor differences. Briefly, yeast were not treated with CHX prior to harvesting. Lysis was performed by combining 2 mL of lysis buffer (20 mM Tris-HCl pH 7.5, 140 mM KCl, 1.5 mM MgCl2, 0.5 mM DTT, 100 µg mL−1, 1% Triton X-100) frozen in liquid nitrogen with cell pellets, and pulverizing using a MM-301 mixer mill at 20 Hz for 1 min. Lysate was thawed in a water bath at room temperature and immediately centrifuged at 15,000 g at 4°C for 10 min. After quantifying RNA concentration, RNase I (Ambion) digestion followed by sucrose gradient centrifugation and fractionation were performed as described previously (Chartron et al., 2016). Total RNA was extracted using the hot SDS-phenol-chloroform method, 24–35 nucleotide RNA footprints were isolated, and libraries were prepared as described elsewhere (Ingolia et al., 2012). For all samples, libraries of two independent biological replicates were quantified by qPCR (Kapa Biosystems) and sequenced using a HiSeq 4000 (Illumina).
Data processing and computational analysis
Data processing.
After demultiplexing, sequencing reads were trimmed of adaptor sequences using Cutadapt v1.4.2, followed by removal of the 5’ nucleotide using FASTX-Trimmer. Reads that mapped to ribosomal RNAs using Bowtie v1.0.0 (http://bowtie-bio.sourceforge.net/index.shtml) (Langmead et al., 2009) were removed. Remaining reads were aligned to a library of protein coding sequences consisting of 5,793 ORFs that excluded ORFs characterized as dubious in the Saccharomyces Genome Database (SGD), or that overlapped with other genes. Sequences of these ORFs contained 21 nucleotides flanking upstream of the start codon and downstream of the stop codon. Bowtie v1.0.0 alignment of sequencing reads to this library used the following parameters to allow for two mismatches and keep only uniquely mapped reads for further analysis:-y -a -m 1 -v 2 --norc --best --strata. For each footprint length, customized python scripts were used to sum reads at each nucleotide. Metagene analysis was performed separately on each fragment length, and lengths that did not exhibit the characteristic 3-nucleotide periodicity were removed. Remaining reads had a nucleotide offset empirically determined from the 5’ end of each fragment length, using the characteristic large ribosome density at the start codon, so that each read was assigned to the first A-site nucleotide. Nucleotide reads at each codon were then summed and used for all additional analysis.
Gene level enrichment.
Gene level counts and tpm were calculated for both total and chaperone pulldown libraries after excluding the first five and last five sense codons to avoid known ribosome profiling biases. Raw counts were used with DESeq2 (Love et al., 2014) to determine significantly enriched genes. Enrichment was then analyzed by only including genes that had greater than 100 reads between the two biological replicates of both the total and pulldown fractions.
Positional chaperone enrichment identification.
To identify positions of cotranslational interaction of chaperones with RNCs, we first applied two thresholds to restrict our analysis to genes with adequate coverage and reproducibility between replicates. First, we only kept genes that had an average sequencing coverage of ≥ 0.5 reads per codon in both replicates of the pulldown and total fractions when excluding the first five and last five sense codons. Secondly, for this same internal region of each gene, we calculated Pearson’s correlation coefficients using the reads per million (rpm) calculated at each position between replicates. Only genes with high reproducibility between replicates of both the total and pulldown libraries (Pearson’s r ≥ 0.5) were kept for analysis. With these two thresholds, we continued with 2,766 genes from our Ssb dataset, 1,568 genes from our TRiC dataset, and 937 genes from the Ssb dataset previously published (Döring et al., 2017). Next, we developed a statistically robust strategy to analyze positional chaperone enrichment. We first summed the reads at each position between replicates in order to increase coverage. We then created 2 × 2 contingency tables at each position of a transcript and performed two-tailed Fisher’s exact tests to compare the ratio of reads in the total and pulldown fractions at a given position to the ratio at all other positions in that transcript (i.e. the summed reads in each fraction for the entire transcript minus the position of interest). In other words, this compares the observed ratio of ribosome reads from our pulldown and total fractions at a given position to the expected ratio based on the total number of reads that map to this transcript. At each position, this allows us to calculate the odds ratio as a measure of enrichment, along with an adjusted p-value to test significance, using the Benjamini-Hochberg correction for multiple hypothesis testing for each gene. The odds ratio is:
where PD and TT are the ribosomal reads of the pulldown and total translatome fractions, respectively, at position i of gene j that has length k codons. Finally, we established thresholds at the codon level to identify positions of chaperone enrichment. We required five or more consecutive codons to meet these criteria: (1) adjusted p-value < 0.05, (2) 1 < odds ratio < ∞, (3) to control for background, the pulldown reads must be greater than the average number of reads across the transcript (i.e. each position in the pulldown fraction had to have higher ribosome occupancy than the background coverage for that transcript), and (4) position > 30 to avoid anomalous positions that result from known ribosome profiling technicalities and because we found chaperone binding depended on the presence of the nascent chain (Figures 2C, 2D, and S1D). The peak positions of chaperone enrichment were determined by identifying the position within each consecutive set of residues having the maximum odds ratio. Peaks within 5 residues of each other were counted as one peak by retaining the position with the higher odds ratio. As indicated, analyses included either all identified peaks, or just the one site within a gene that had the highest chaperone enrichment (i.e. highest odds ratio) so that each gene was given equal weight to confirm that our conclusions were not biased by genes with multiple sites of chaperone enrichment. As one exception, for clarity in showing the Ssb binding events in Enp2 in Figure 4B and Movie S1, we lowered our threshold for identifying Ssb binding events to requiring 3 consecutive codons having an odds ratio > 1 and adjusted p-value < 0.05 since unlike other WD40 proteins, Enp2 did not pass our stringent threshold for identifying Ssb binding events that we used for our global analysis. Finally, we also adapted an alternative statistical approach that we have used previously (Chartron et al., 2016) to identify positions with significant chaperone enrichment. This approach used DESeq2 (Love et al., 2014) with a negative binomial distribution to fit a model to the sequencing count distribution at all codons for each gene. This approach generated a similar distribution of binding sites and we were able to reach all of the same conclusions presented in this study.
Ribosome occupancy analysis.
For ribosome occupancy plots of chaperone enrichment for individual substrates (e.g. Figure 2A), odds ratios were calculated after adding 1 to each position to eliminate division by 0. Where indicated (e.g. Figures 2E and 2F), protein length was normalized such that the start codon was at 0 and the stop codon was at 1. Metagene plots in Figures 2C, 2D, and S4 exclude substrates with length < 300 amino acids.
Domain analysis.
Chaperone binding sites were assigned to annotated domains by calculating the distance from the position of peak chaperone enrichment to the start of each domain in a gene. Binding sites were then assigned to the nearest domain, i.e. the domain for which this distance was minimized. Moreover, due to the dependence of chaperone enrichment on a nascent chain (Figure S1D), the peak of chaperone enrichment had to be greater than 30 residues after the start of the domain such that the domain has started emerging from the ribosome exit tunnel when the chaperone is recruited, and before any downstream domain has started to emerge. Significance was tested by random sampling of the stretch of residues that fit this same criteria and included the position of peak chaperone enrichment. Peaks that could not be unambiguously assigned to a domain, such as for the substrates with poor domain annotation, were excluded from the analysis. Where indicated (e.g. Figures 3B and S5), domain length was normalized such that the start of the domain was at 0 and the end was at 1.
Secondary structure prediction and net charge analysis.
The secondary structure propensity at each position of all TRiC and Ssb substrates was calculated using PSIPRED v4.01 (http://bioinf.cs.ucl.ac.uk/psipred/) (McGuffin et al., 2000). This was also done at an equivalent number of randomly sampled positions from the substrate set. The net charge of the nascent chain was calculated with the Peptides package in R using a six residue moving window and the Lehninger pKa scale, and compared to 10,000 random peptide sequences generated from the 5,793 ORFs included in our ribosome profiling analysis. Metagene analysis was performed using the mean at each position and bootstrapped 95% confidence interval.
Amino acid composition analysis.
We defined the recently emerged nascent chain as 31–60 amino acids upstream of the start of chaperone recruitment, and used this sequence to calculate amino acid composition. As background, 10,000 random peptide sequences of equivalent length as our input (30 residues) were generated from the 5,793 ORFs included in our ribosome profiling analysis to examine the average amino acid composition within the proteome and test significance using Wilcoxon rank-sum tests.
Translation kinetics and codon optimality.
In order to examine the kinetics of translation elongation around the sites of chaperone enrichment, we analyzed wild-type translatome datasets (Table S1) of cells that were not treated with CHX, as CHX can produce artifacts of increased ribosome occupancy. These datasets were all processed as described above. For metagene analysis of translation kinetics, genes were aligned to the ribosome A-site at the start of TRiC or Ssb chaperone enrichment, or at sites of pronounced SRP binding identified previously (Chartron et al., 2016). Differences in gene expression and coverage were normalized by dividing the reads at each codon by the mean number of reads per codon across the analysis window. Low coverage genes were excluded by including only the genes that had an average of ≥0.5 reads per codon across the analysis window, and chaperone binding sites before position 75 were also excluded in case differences in initiation impacted normalization. The mean and bootstrapped 95% confidence intervals were calculated at each position and compared to an equivalent number of randomly sampled positions from the substrate set (i.e. for each binding event of a given substrate, a random position was chosen from this same substrate). Metagene analysis of codon optimality was similarly performed using the species-specific tRNA adaptation index, which is not dependent on gene expression data (Sabi and Tuller, 2014), and smoothed with a 3 residue rolling average. Equivalent results were observed using other metrics, such as the classical tRNA adaptation index (Reis et al., 2004) and codon usage frequency.
Gene and domain categorization
Protein localization and classification of secretory proteins as transmembrane domain or signal sequence proteins were taken from previous work (Chartron et al., 2016). Proteins classified as “other” in Figure 1E included tail-anchored proteins and those classified as “exception” by (Chartron et al., 2016) in which there was ambiguity in identifying the targeting signal. Protein properties and annotations of domains and their boundaries, as identified by the CATH/Gene3D (Sillitoe et al., 2015) and SCOP/Superfamily databases (Gough et al., 2001), were downloaded from SGD. Domains were grouped using the topology level of the hierarchical CATH/Gene3D nomenclature with names generalized from the CATH, SCOP, and Interpro databases: WD40 repeat b-propeller (2.130.10), Rossmann fold (3.40.50), TIM barrel (3.20.20), and Nucleotide-binding a-b plait (3.30.70). Overall intrinsic disorder and secondary structure prediction of proteins were taken from previous work (Willmund et al., 2013). Annotation of WD40 repeats and cytoplasmic domains were obtained from UniProt (www.uniprot.org) (The UniProt Consortium, 2017). Boundaries of transmembrane domains and signal sequences were determined using UniProt and previous work (Chartron et al., 2016). Protein structures were obtained from the Protein Data Bank (www.rcsb.org) (Berman et al., 2000).
Nascent chain labeling
Wild-type and ssb1,2∆ cells were diluted in 50 mL of YPD to an OD600 of 0.05 and grown at 30°C to OD600 ~0.5. Cells were then starved for 30 min in synthetic medium that lacks methionine, followed by pulsing with 100 µCi mL−1 35S-Methionine for 1.5 min. Upon adding sodium azide to 250 mM and CHX to 0.5 mg mL−1, cells were harvested on ice, followed by lysing cell pellets and isolating total and TRiC-bound ribosomes as described above. Pre-immune beads not crosslinked to CCT antibodies served as a negative control. Samples were run on a 15% SDS-PAGE gel, which was then dried and quantified by autoradiography.
Growth assay
Cells from WT or mutant atp2 strains were grown at 30°C in YPD to an OD600 of ~0.7. Cell number was normalized to an OD600 of 0.4 followed by 5-fold serial dilutions spotted on plates containing YPD or YP + 3% glycerol media.
Subcellular fractionation and immunoblotting
Cells from WT or mutant atp2 strains were grown at 30°C in YPD and harvested at an OD600 of ~0.7. Frozen cell pellets were resuspended in 150 µl of native lysis buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1 mM EDTA, 5% glycerol, 0.5 mM DTT, 1 mM PMSF, Roche cOmplete EDTA-free Protease Inhibitor Cocktail), and lysed by vortexing with glass beads at 3000 rpm at 4°C for 2 min, incubating on ice for 3 min, and vortexing again at 3000 rpm at 4°C for 2 min. Protein concentration was normalized followed by centrifugation of cell lysates at 12,000 rpm at 4°C for 10 min. The supernatant was removed as the total soluble fraction. Pellets were resuspended using 150 µl RIPA buffer (50 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% SDS, 0.5 mM DTT, 1 mM PMSF, Roche cOmplete EDTA-free Protease Inhibitor Cocktail), vortexed at 3000 rpm at 4°C for 2 min, and centrifuged as before. The detergent-extracted supernatant was retained as the total membrane fraction. Sample buffer was added to each fraction, boiled for 5 min, run on a 12% SDS-PAGE gel, and transferred to nitrocellulose membrane. Blots were then subjected to immunblotting using rabbit anti-ATPB antibody (Abcam ab128743, 1:1000 dilution) and rat anti-tubulin antibody (Abcam ab6161, 1:1000 dilution), and visualized using the LI-COR system with IRDye® 800CW Goat anti-Rabbit IgG (LI-COR 926–32211, 1:10,000 dilution) and IRDye® 680RD Goat anti-Rat IgG (LI-COR 926–68076, 1:10,000 dilution).
qPCR
Overnight yeast cultures were diluted in 100 mL of YPD media to an OD600 of 0.05, grown at 30°C to OD600 ~0.7, treated with CHX to 100 µg mL−1, and immediately harvested by fast filtration and flash freezing in liquid nitrogen. Cells were lysed and fractionated as described above for preparing selective ribosome profiling libraries. RNA concentrations were normalized to 100 ng for cDNA synthesis using iScript (Bio-Rad). qPCR was performed using a CFX96 thermocycler (Bio-Rad) and iTaq Universal SYBR Green Supermix (Bio-Rad) with oligonucleotides to ATP2 (5’TTTGCCCATTTGGACGCTAC and 5’GGCGGCATCCAATAACCTTG) and RPL3 (5’CTGGGCTCGTGAACATTTCG and 5’ACACCTTCGAAACCGTGACC). Using the non-TRiC substrate RPL3 as the reference gene for each sample, TRiC enrichment was calculated as the fold change of the TRiC-bound mRNA over the total mRNA. Enrichment was then averaged between three technical replicates followed by calculating the mean and standard error of three biological replicates.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical Analysis
All analysis was performed in R (https://www.r-project.org). Distributions for categorical variables shown in box plots, violin plots, or density plots used two-sided Wilcoxon rank-sum tests to assess statistical significance. For box plots, the center line represents the median, box limits indicate the upper and lower quartiles, whiskers indicate the 1.5x interquartile range, and points are outliers. Significance of enrichment of protein domains (Figures 3H and S6E), cytonuclear proteins (Figure 1E), and the overlap of TRiC and Ssb substrates (Figure 1F), was assessed using hypergeometric tests. Two-sided Welch’s t-tests were used to assess significance of TRiC enrichment from qPCR (Figure 5G) and TRiC-bound nascent chain labeling (Figure S3F). Significance of codon optimality (Figure S7F) was determined using Wilcoxon rank-sum tests with the 3 residue window before and after the start of chaperone binding, as compared to the randomly sampled distribution. Additional statistical details are mentioned in the Method Details section of the STAR Methods, as well as in the figures or figure legends, including the values of n and P. None of the experiments involved blinding or randomization, and sample size was not predetermined.
Supplementary Material
This file consists of a data and annotation table that includes: positional enrichment data at TRiC and Ssb binding sites, annotation of domains assigned to binding sites, TRiC and Ssb cotranslational substrate annotation. Peaks of chaperone enrichment require at least 5 consecutive codons with an odds ratio > 1, adjusted p-value < 0.05, and a codon position > 30.
This file contains a video that models the cotranslational recruitment of TRiC and Ssb to the WD40 domain of Enp2. Cartoon of Enp2 (PDB ID: 5WLC (Barandun et al., 2017)) colored according to secondary structure, with the indicated nascent chain sites that just emerged from the ribosome colored in green or blue when Ssb or TRiC bind, respectively.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti-Cct3, anti-Cct5, anti-Cct8 | This paper | N/A |
| Rabbit polyclonal anti-Ssb | This paper | N/A |
| Rabbit polyclonal anti-ATPB | Abcam | Cat#ab128743 |
| Rat monoclonal anti-tubulin | Abcam | Cat#ab6161 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| DSP (dithiobis(succinimidyl propionate)), Lomant’s Reagent | Thermo Fisher Scientific | Cat#22585 |
| Apyrase | Sigma-Aldrich | Cat#A6410 |
| Dynabeads™ Protein G | Invitrogen | Cat#10003D |
| Ribo-Zero Gold rRNA Removal Kit (Yeast) | Illumina | Cat#MRZY1324 |
| Deposited Data | ||
| Raw and analyzed data | This paper | GEO: GSE114882 |
| Protein properties and domain annotation | Saccharomyces Genome Database | https://downloads.yeastgenome.org/curation/calculated_protein_info/ |
| WD40 domain annotation | UniProt | www.uniprot.org |
| Membrane fractionation, Figure S6M | This paper; Mendeley Data | http://dx.doi.org/10.17632/54zrpyzrt8.1 |
| Experimental Models: Organisms/Strains | ||
| S. cerevisiae strain background: BY4741 | Dharmacon | Cat#YSC1048 |
| BY4741 atp2-P353A,P355A | This paper | yKS208 |
| Oligonucleotides | ||
| CGGGTGGCGAATGGGACTTTGCAAAAGTAGTGGCAGGAGCGTTTTAGAGCTAGAAATAGC | This paper | 5’ ATP2-gRNA |
| GCTATTTCTAGCTCTAAAACGCTCCTGCCACTACTTTTGCAAAGTCCCATTCGCCACCCG | This paper | 3’ ATP2-gRNA |
| GAAGGGTTCTGTCACTTCTGTGCAAGCCGTTTATGTTCCAGCCGATGATTTAACAGATGCTGCTGCTGCCACTAC | This paper | 5’ ATP2-P353,355A |
| CCTAATTCTGAAATACCTCTTGACAAGACGGTAGTAGCGTCCAAATGGGCAAAAGTAGTGGCAGCAGCAGCATC | This paper | 3’ ATP2-P353,355A |
| TTTGCCCATTTGGACGCTAC | This paper | 5’ ATP2 qPCR1 |
| GGCGGCATCCAATAACCTTG | This paper | 3’ ATP2 qPCR1 |
| CTGGGCTCGTGAACATTTCG | This paper | 5’ RPL3 qPCR2 |
| ACACCTTCGAAACCGTGACC | This paper | 3’ RPL3 qPCR2 |
| Universal miRNA cloning linker: rAppCTGTAGGCACCATCAAT-NH2 | NEB | Cat# S1315S |
| /5Phos/AGATCGGAAGAGCGTCGTGTAGGGAAAGAGTGTAGATCTCGGTGGTCGC/Sp-C18/CACTCA/Sp-C18/TTCAGACGTGTGCTCTTCCGATCTATTGATGGTGCCTACAG | Ingolia et al., 2012 | RT primer |
| Recombinant DNA | ||
| pCAS | Ryan et al., 2014 | Addgene Cat#60847 |
| Software and Algorithms | ||
| Bowtie v1.0.0 | Langmead et al., 2009 | http://bowtie-bio.sourceforge.net/index.shtml |
| PSIPRED v4.01 | McGuffin et al., 2000 | http://bioinf.cs.ucl.ac.uk/psipred/ |
Highlights.
Uncovers principles of cotranslational action of chaperones TRiC/CCT and Hsp70/Ssb
Topological features of protein domains dictate sequential binding of Ssb then TRiC
TRiC recognizes nearly complete domains exposing an unprotected hydrophobic surface
Local slowdown in translation elongation rates correlate with chaperone recruitment
ACKNOWLEDGMENTS
We thank T. Lopez and R. Andino for comments on this manuscript, P. Dolan, M. Aguilar, and J. Chartron for advice on data analysis. We thank J. Weissman and members of the Frydman lab for advice and discussions. Sequencing was performed at the UCSF Center for Advanced Technology. K.C.S was supported by NIH NRSA grant AG047126. A.K. was supported by NIH NRSA GM108244. This work was supported by NIH grant GM056433 to J.F.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
The authors declare no competing interests.
DATA AND CODE AVAILABILITY
The datasets generated for this study are deposited in NCBI’s Gene Expression Omnibus (GEO) (Edgar et al., 2002) under GEO Series accession number GEO: GSE114882. Additional datasets used in this study are also available in GEO with accession numbers provided in Table S1. Unprocessed data associated with the membrane fractionation shown in Figure S6M, with relevant proteins demarcated and superfluous lanes cropped, is available at http://dx.doi.org/10.17632/54zrpyzrt8.1. All customized python or R scripts used for data processing and analysis are available upon request.
REFERENCES
- Albanèse V, Reissmann S, and Frydman J. (2010). A ribosome-anchored chaperone network that facilitates eukaryotic ribosome biogenesis. J Cell Biol 189, 69–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albanèse V, Yam AY-W, Baughman J, Parnot C, and Frydman J. (2006). Systems analyses reveal two chaperone networks with distinct functions in eukaryotic cells. Cell 124, 75–88. [DOI] [PubMed] [Google Scholar]
- Ayaz P, Ye X, Huddleston P, Brautigam CA, and Rice LM. (2012). A TOG:αβ-tubulin complex structure reveals conformation-based mechanisms for a microtubule polymerase. Science 337, 857–860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balch WE, Morimoto RI, Dillin A, and Kelly JW. (2008). Adapting proteostasis for disease intervention. Science 319, 916–919. [DOI] [PubMed] [Google Scholar]
- Barandun J, Chaker-Margot M, Hunziker M, Molloy KR, Chait BT, and Klinge S. (2017). The complete structure of the small-subunit processome. Nat Struct Mol Biol 24, 944–953. [DOI] [PubMed] [Google Scholar]
- Becker AH, Oh E, Weissman JS, Kramer G, and Bukau B. (2013). Selective ribosome profiling as a tool for studying the interaction of chaperones and targeting factors with nascent polypeptide chains and ribosomes. Nat Protoc 8, 2212–2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berman HM, Westbrook J, Feng Z, Gilliland G, Bhat TN, Weissig H, Shindyalov IN, and Bourne PE. (2000). The Protein Data Bank. Nucleic Acids Res 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgia A, Kemplen KR, Borgia MB, Soranno A, Shammas S, Wunderlich B, Nettels D, Best RB, Clarke J, and Schuler B. (2015). Transient misfolding dominates multidomain protein folding. Nature Communications 6, 8861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borgia MB, Borgia A, Best RB, Steward A, Nettels D, Wunderlich B, Schuler B, and Clarke J. (2011). Single-molecule fluorescence reveals sequence-specific misfolding in multidomain proteins. Nature 474, 662–665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Camasses A, Bogdanova A, Shevchenko A, and Zachariae W. (2003). The CCT chaperonin promotes activation of the anaphase-promoting complex through the generation of functional Cdc20. Mol Cell 12, 87–100. [DOI] [PubMed] [Google Scholar]
- Chartron JW, Hunt KCL, and Frydman J. (2016). Cotranslational signal-independent SRP preloading during membrane targeting. Nature 536, 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiti F, and Dobson CM. (2017). Protein Misfolding, Amyloid Formation, and Human Disease: A Summary of Progress Over the Last Decade. Annu Rev Biochem 86, 27–68. [DOI] [PubMed] [Google Scholar]
- Clerico EM, Tilitsky JM, Meng W, and Gierasch LM. (2015). How Hsp70 Molecular Machines Interact with Their Substrates to Mediate Diverse Physiological Functions. J Mol Biol 427, 1575–1588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cong Y, Baker ML, Jakana J, Woolford D, Miller EJ, Reissmann S, Kumar RN, Redding-Johanson AM, Batth TS, Mukhopadhyay A, et al. (2010). 4.0-A resolution cryo-EM structure of the mammalian chaperonin TRiC/CCT reveals its unique subunit arrangement. Proc Natl Acad Sci USA 107, 4967–4972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa EA, Subramanian K, Nunnari J, and Weissman JS. (2018). Defining the physiological role of SRP in protein-targeting efficiency and specificity. Science 359, 689–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dana A, and Tuller T. (2014). The effect of tRNA levels on decoding times of mRNA codons. Nucleic Acids Res 42, 9171–9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dokholyan NV, Li L, Ding F, and Shakhnovich EI. (2002). Topological determinants of protein folding. Proc Natl Acad Sci USA 99, 8637–8641. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Döring K, Ahmed N, Riemer T, Suresh HG, Vainshtein Y, Habich M, Riemer J, Mayer MP, O’Brien EP, Kramer G, et al. (2017). Profiling Ssb-Nascent Chain Interactions Reveals Principles of Hsp70-Assisted Folding. Cell 170, 298–311.e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan CDS, and Mata J. (2011). Widespread cotranslational formation of protein complexes. PLoS Genet 7, e1002398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edgar R, Domrachev M, and Lash AE. (2002). Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res 30, 207–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellgaard L, McCaul N, Chatsisvili A, and Braakman I. (2016). Co-and Post-Translational Protein Folding in the ER. Traffic 17, 615–638. [DOI] [PubMed] [Google Scholar]
- Erskine PT, Newbold R, Brindley AA, Wood SP, Shoolingin-Jordan PM, Warren MJ, and Cooper JB. (2001). The x-ray structure of yeast 5-aminolaevulinic acid dehydratase complexed with substrate and three inhibitors. J Mol Biol 312, 133–141. [DOI] [PubMed] [Google Scholar]
- Freund A, Zhong FL, Venteicher AS, Meng Z, Veenstra TD, Frydman J, and Artandi SE. (2014). Proteostatic control of telomerase function through TRiC-mediated folding of TCAB1. Cell 159, 1389–1403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frydman J, and Hartl FU. (1996). Principles of chaperone-assisted protein folding: differences between in vitro and in vivo mechanisms. Science 272, 1497–1502. [DOI] [PubMed] [Google Scholar]
- Frydman J, Nimmesgern E, Ohtsuka K, and Hartl FU. (1994). Folding of nascent polypeptide chains in a high molecular mass assembly with molecular chaperones. Nature 370, 111–117. [DOI] [PubMed] [Google Scholar]
- Garreau de Loubresse N, Prokhorova I, Holtkamp W, Rodnina MV, Yusupova G, and Yusupov M. (2014). Structural basis for the inhibition of the eukaryotic ribosome. Nature 513, 517–522. [DOI] [PubMed] [Google Scholar]
- Gestaut D, Roh S-H, Ma B, Pintilie G, Joachimiak LA, Leitner A, Walzthoeni T, Aebersold R, Chiu W, and Frydman J. (2019). The Chaperonin TRiC/CCT Associates with Prefoldin through a Conserved Electrostatic Interface Essential for Cellular Proteostasis. Cell 177, 751–765.e15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman DH, Kaiser CM, Milin A, Righini M, Tinoco I, and Bustamante C. (2015). Ribosome. Mechanical force releases nascent chain-mediated ribosome arrest in vitro and in vivo. Science 348, 457–460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y, Kakihara Y, Krogan N, Greenblatt J, Emili A, Zhang Z, and Houry WA. (2009). An atlas of chaperone-protein interactions in Saccharomyces cerevisiae: implications to protein folding pathways in the cell. Mol Syst Biol 5, 275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gough J, Karplus K, Hughey R, and Chothia C. (2001). Assignment of homology to genome sequences using a library of hidden Markov models that represent all proteins of known structure. J Mol Biol 313, 903–919. [DOI] [PubMed] [Google Scholar]
- Han J-H, Batey S, Nickson AA, Teichmann SA, and Clarke J. (2007). The folding and evolution of multidomain proteins. Nat Rev Mol Cell Biol 8, 319–330. [DOI] [PubMed] [Google Scholar]
- Han Y, David A, Liu B, Magadán JG, Bennink JR, Yewdell JW, and Qian S-B. (2012). Monitoring cotranslational protein folding in mammalian cells at codon resolution. Proc Natl Acad Sci USA 109, 12467–12472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanebuth MA, Kityk R, Fries SJ, Jain A, Kriel A, Albanèse V, Frickey T, Peter C, Mayer MP, Frydman J, et al. (2016). Multivalent contacts of the Hsp70 Ssb contribute to its architecture on ribosomes and nascent chain interaction. Nature Communications 7, 13695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hartl FU, Bracher A, and Hayer-Hartl M. (2011). Molecular chaperones in protein folding and proteostasis. Nature 475, 324–332. [DOI] [PubMed] [Google Scholar]
- Hussmann JA, Patchett S, Johnson A, Sawyer S, and Press WH. (2015). Understanding Biases in Ribosome Profiling Experiments Reveals Signatures of Translation Dynamics in Yeast. PLoS Genet 11, e1005732–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, and Weissman JS. (2012). The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc 7, 1534–1550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JRS, and Weissman JS. (2009). Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324, 218–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Hussmann JA, and Weissman JS. (2018). Ribosome Profiling: Global Views of Translation. Cold Spring Harbor Perspectives in Biology a032698–20. [DOI] [PMC free article] [PubMed]
- Jansens A, van Duijn E, and Braakman I. (2002). Coordinated nonvectorial folding in a newly synthesized multidomain protein. Science 298, 2401–2403. [DOI] [PubMed] [Google Scholar]
- Joachimiak LA, Walzthoeni T, Liu CW, Aebersold R, and Frydman J. (2014). The structural basis of substrate recognition by the eukaryotic chaperonin TRiC/CCT. Cell 159, 1042–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser CM, and Liu K. (2018). Folding up and Moving on-Nascent Protein Folding on the Ribosome. J Mol Biol 430, 4580–4591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SJ, Yoon JS, Shishido H, Yang Z, Rooney LA, Barral JM, and Skach WR. (2015). Protein folding. Translational tuning optimizes nascent protein folding in cells. Science 348, 444–448. [DOI] [PubMed] [Google Scholar]
- Kim YE, Hipp MS, Bracher A, Hayer-Hartl M, and Hartl FU. (2013). Molecular chaperone functions in protein folding and proteostasis. Annu Rev Biochem 82, 323–355. [DOI] [PubMed] [Google Scholar]
- Koplin A, Preissler S, Ilina Y, Koch M, Scior A, Erhardt M, and Deuerling E. (2010). A dual function for chaperones SSB-RAC and the NAC nascent polypeptide-associated complex on ribosomes. 189, 57–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koyama M, and Matsuura Y. (2010). An allosteric mechanism to displace nuclear export cargo from CRM1 and RanGTP by RanBP1. Embo J 29, 2002–2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, and Salzberg SL. (2009). Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol 10, R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leininger SE, Trovato F, Nissley DA, and O’Brien EP. (2019). Domain topology, stability, and translation speed determine mechanical force generation on the ribosome. Proc Natl Acad Sci USA 116, 5523–5532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu K, Maciuba K, and Kaiser CM. (2019). The Ribosome Cooperates with a Chaperone to Guide Multi-domain Protein Folding. Mol Cell [DOI] [PMC free article] [PubMed]
- Lopez T, Dalton K, and Frydman J. (2015). The Mechanism and Function of Group II Chaperonins. J Mol Biol 427, 2919–2930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S. (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGuffin LJ, Bryson K, and Jones DT. (2000). The PSIPRED protein structure prediction server. Bioinformatics 16, 404–405. [DOI] [PubMed] [Google Scholar]
- Miyata Y, Shibata T, Aoshima M, Tsubata T, and Nishida E. (2014). The molecular chaperone TRiC/CCT binds to the Trp-Asp 40 (WD40) repeat protein WDR68 and promotes its folding, protein kinase DYRK1A binding, and nuclear accumulation. J Biol Chem 289, 33320–33332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neudecker P, Robustelli P, Cavalli A, Walsh P, Lundström P, Zarrine-Afsar A, Sharpe S, Vendruscolo M, and Kay LE. (2012). Structure of an intermediate state in protein folding and aggregation. Science 336, 362–366. [DOI] [PubMed] [Google Scholar]
- Pechmann S, Chartron JW, and Frydman J. (2014). Local slowdown of translation by nonoptimal codons promotes nascent-chain recognition by SRP in vivo. Nat Struct Mol Biol 21, 1100–1105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plath K, and Rapoport TA. (2000). Spontaneous Release of Cytosolic Proteins from Posttranslational Substrates before Their Transport into the Endoplasmic Reticulum. J Cell Biol 151, 167–178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Preissler S, and Deuerling E. (2012). Ribosome-associated chaperones as key players in proteostasis. Trends Biochem Sci 37, 274–283. [DOI] [PubMed] [Google Scholar]
- Reis, dos M, Savva R, and Wernisch L. (2004). Solving the riddle of codon usage preferences: a test for translational selection. Nucleic Acids Res 32, 5036–5044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodnina MV, and Wintermeyer W. (2016). Protein Elongation, Co-translational Folding and Targeting. J Mol Biol 428, 2165–2185. [DOI] [PubMed] [Google Scholar]
- Rommelaere H, Van Troys M, Gao Y, Melki R, Cowan NJ, Vandekerckhove J, and Ampe C. (1993). Eukaryotic cytosolic chaperonin contains t-complex polypeptide 1 and seven related subunits. Proc Natl Acad Sci USA 90, 11975–11979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rüdiger S, Germeroth L, Schneider-Mergener J, and Bukau B. (1997). Substrate specificity of the DnaK chaperone determined by screening cellulose-bound peptide libraries. Embo J 16, 1501–1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryan OW, Poddar S, and Cate JHD. (2016). CRISPR-Cas9 Genome Engineering in Saccharomyces cerevisiae Cells. Cold Spring Harb Protoc 2016. [DOI] [PubMed]
- Ryan OW, Skerker JM, Maurer MJ, Li X, Tsai JC, Poddar S, Lee ME, DeLoache W, Dueber JE, Arkin AP, et al. (2014). Selection of chromosomal DNA libraries using a multiplex CRISPR system. eLife 3, 345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabi R, and Tuller T. (2014). Modelling the efficiency of codon-tRNA interactions based on codon usage bias. DNA Res. 21, 511–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schuller AP, Wu CC-C, Dever TE, Buskirk AR, and Green R. (2017). eIF5A Functions Globally in Translation Elongation and Termination. Mol Cell 66, 1–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shank EA, Cecconi C, Dill JW, Marqusee S, and Bustamante C. (2010). The folding cooperativity of a protein is controlled by its chain topology. Nature 465, 637–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiber A, Döring K, Friedrich U, Klann K, Merker D, Zedan M, Tippmann F, Kramer G, and Bukau B. (2018). Cotranslational assembly of protein complexes in eukaryotes revealed by ribosome profiling. Nature 561, 268–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shurtleff MJ, Itzhak DN, Hussmann JA, Schirle Oakdale NT, Costa EA, Jonikas M, Weibezahn J, Popova KD, Jan CH, Sinitcyn P, et al. (2018). The ER membrane protein complex interacts cotranslationally to enable biogenesis of multipass membrane proteins. eLife 7, 382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sillitoe I, Lewis TE, Cuff A, Das S, Ashford P, Dawson NL, Furnham N, Laskowski RA, Lee D, Lees JG, et al. (2015). CATH: comprehensive structural and functional annotations for genome sequences. Nucleic Acids Res 43, D376–D381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smock RG, Yadid I, Dym O, Clarke J, and Tawfik DS. (2016). De Novo Evolutionary Emergence of a Symmetrical Protein Is Shaped by Folding Constraints. Cell 164, 476–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiess C, Meyer AS, Reissmann S, and Frydman J. (2004). Mechanism of the eukaryotic chaperonin: protein folding in the chamber of secrets. Trends Cell Biol 14, 598–604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spiess C, Miller EJ, McClellan AJ, and Frydman J. (2006). Identification of the TRiC/CCT Substrate Binding Sites Uncovers the Function of Subunit Diversity in Eukaryotic Chaperonins. Mol Cell 24, 25–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein KC, and Frydman J. (2019). The stop-and-go traffic regulating protein biogenesis: How translation kinetics controls proteostasis. J Biol Chem 294, 2076–2084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stirnimann CU, Petsalaki E, Russell RB, and Müller CW. (2010). WD40 proteins propel cellular networks. Trends Biochem Sci 35, 565–574. [DOI] [PubMed] [Google Scholar]
- Stock D, Leslie AG, and Walker JE. (1999). Molecular architecture of the rotary motor in ATP synthase. Science 286, 1700–1705. [DOI] [PubMed] [Google Scholar]
- Taylor SC, Ferguson AD, Bergeron JJM, and Thomas DY. (2004). The ER protein folding sensor UDP-glucose glycoprotein-glucosyltransferase modifies substrates distant to local changes in glycoprotein conformation. Nat Struct Mol Biol 11, 128–134. [DOI] [PubMed] [Google Scholar]
- The UniProt Consortium (2017). UniProt: the universal protein knowledgebase. Nucleic Acids Res 45, D158–D169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thulasiraman V, Yang CF, and Frydman J. (1999). In vivo newly translated polypeptides are sequestered in a protected folding environment. Embo J 18, 85–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tripathi A, Mandon EC, Gilmore R, and Rapoport TA. (2017). Two alternative binding mechanisms connect the protein translocation Sec71-Sec72 complex with heat shock proteins. J Biol Chem 292, 8007–8018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg DE, Shah P, Eichhorn SW, Hussmann JA, Plotkin JB, and Bartel DP. (2016). Improved Ribosome-Footprint and mRNA Measurements Provide Insights into Dynamics and Regulation of Yeast Translation. CellReports 14, 1787–1799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willmund F, del Alamo M, Pechmann S, Chen T, Albanèse V, Dammer EB, Peng J, and Frydman J. (2013). The cotranslational function of ribosome-associated Hsp70 in eukaryotic protein homeostasis. Cell 152, 196–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CC-C, Zinshteyn B, Wehner KA, and Green R. (2019). High-Resolution Ribosome Profiling Defines Discrete Ribosome Elongation States and Translational Regulation during Cellular Stress. Mol Cell 1–18. [DOI] [PMC free article] [PubMed]
- Yam AY, Xia Y, Lin H-TJ, Burlingame A, Gerstein M, and Frydman J. (2008). Defining the TRiC/CCT interactome links chaperonin function to stabilization of newly made proteins with complex topologies. Nat Struct Mol Biol 15, 1255–1262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yam AY-W, Albanèse V, Lin H-TJ, and Frydman J. (2005). Hsp110 cooperates with different cytosolic HSP70 systems in a pathway for de novo folding. J Biol Chem 280, 41252–41261. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
This file consists of a data and annotation table that includes: positional enrichment data at TRiC and Ssb binding sites, annotation of domains assigned to binding sites, TRiC and Ssb cotranslational substrate annotation. Peaks of chaperone enrichment require at least 5 consecutive codons with an odds ratio > 1, adjusted p-value < 0.05, and a codon position > 30.
This file contains a video that models the cotranslational recruitment of TRiC and Ssb to the WD40 domain of Enp2. Cartoon of Enp2 (PDB ID: 5WLC (Barandun et al., 2017)) colored according to secondary structure, with the indicated nascent chain sites that just emerged from the ribosome colored in green or blue when Ssb or TRiC bind, respectively.