

Abstract
Introduction
Familial frontotemporal lobar degeneration (f‐FTLD) due to autosomal dominant mutations is an important entity for developing treatments for FTLD. The Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) and Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS) longitudinal studies were designed to describe the natural history of f‐FTLD.
Methods
We summarized recent publications from the ARTFL and LEFFTDS studies, along with other recent publications describing the natural history of f‐FTLD.
Results
Published and emerging studies are producing data on all phases of f‐FTLD, including the asymptomatic and symptomatic phases of disease, as well as the transitional phase when symptoms are just beginning to develop. These data indicate that rates of change increase along with disease severity, which is consistent with commonly cited models of neurodegeneration, and that measurement of biomarkers may predict onset of symptoms.
Discussion
Data from large multisite studies are producing important data on the natural history of f‐FTLD that will be critical for planning intervention trials.
Keywords: C9orf72, familial, frontotemporal lobar degeneration, genetic, GRN, MAPT
1. INTRODUCTION
Frontotemporal lobar degeneration (FTLD) is the overarching term for a group of neurodegenerative disorders that are associated with accumulation of protein aggregates in the central nervous system (CNS), most commonly comprising one of two major proteins—microtubule associated protein tau (abbreviated tau; shown in blue in Figure 1) and transactive response DNA‐binding protein molecular weight 43 (abbreviated TDP; shown in orange).1, 2 FTLD is at least as common as Alzheimer's disease (AD) in people younger than 65 years of age.3, 4, 5 In US and UK cohorts, the prevalence of the behavioral and cognitive syndromes is ≈10 to 22 per 100,000, with similar prevalence of motor syndromes.6, 7 All FTLD disorders are uniformly fatal and generally lead to death within 10 years of diagnosis, sometimes much earlier.8 When compared with AD, FTLD is thought to have an even greater effect on the lives of patients and their families because it is associated with an earlier age at onset, often developing when affected individuals are still working and raising children,9 as well as a more rapid rate of decline8 and effects on personality and behavior that cause considerable stress for families.10, 11, 12 These features reinforce the importance of efforts to develop treatments for this devastating disorder. Although treatments are not yet available, there is great enthusiasm for a number of potential disease‐modifying therapies, including anti‐tau antibodies, tau aggregation inhibitors, microtubule stabilizers, progranulin modulators, and antisense oligonucleotides,13, 14, 15, 16, 17, 18, 19, 20, 21, 22 with some agents already being tested in clinical trials (Boxer et al, special collection).
Figure 1.
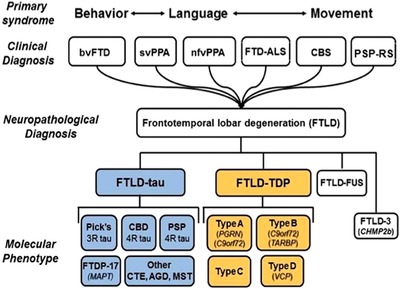
Clinical and neuropathological classification of FTLD. TDP, TAR DNA binding protein 43; FUS, fused in sarcoma; 3R, 3 repeat; 4R, 4 repeat; CBD, corticobasal degeneration; FTDP‐17, frontotemporal dementia Parkinsonism linked to chromosome 17; CTE, chronic traumatic encephalopathy; AGD, argyrophilic grain disease; MST, multisystem tauopathy; see text for additional abbreviations; italicized words are abbreviations for autosomal dominant genes that cause FTLD. (After Seeley et al.)
In preparation for clinical trials in FTLD, it is critical that the medical community develops tools for tracking disease states that are capable of dealing with the unique challenges in FTLD. Some of the most important of these challenges come from clinical and genetic diversity. Clinically, FTLD can present with a variety of syndromes including the behavioral variant frontotemporal dementia (bvFTD), bvFTD plus amyotrophic lateral sclerosis (FTD‐ALS), the semantic variant of primary progressive aphasia (svPPA), the nonfluent/agrammatic variant of PPA (nfPPA), corticobasal syndrome (CBS), and progressive supranuclear palsy (PSP‐RS; the classic PSP syndrome called Richardson syndrome). Each of these syndromes is characterized by its own set of symptoms, signs, and abnormalities on neuropsychological testing. The complexity of FTLD is further increased through its association with genetic mutations, which occur in at least 20% of all FTLD patients and cause a dominantly inherited familial disorder (f‐FTLD). The most common mutations associated with FTLD occur in the microtubule‐associated protein tau (MAPT 23), progranulin (GRN 24), and chromosome 9 open reading frame 72 (C9orf72 or C9ORF72 25, 26) genes. These mutations together account for at least 50% of f‐FTLD.27, 28 Thus, far from being a single entity, FTLD actually represents a variety of clinical and genetic syndromes.
RESEARCH IN CONTEXT
Systematic review: The authors reviewed the literature using traditional sources (eg, PubMed) and meeting abstracts and presentations.
Interpretation: Our results indicate that the published and emerging literature, including papers being published in a special collection of Alzheimer's and Dementia describing recent work from the Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) and Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS) studies indicates that familial frontotemporal dementia (f‐FTLD) is characterized by increasing rates of change as disease severity increases, and that biomarker measurements may predict symptom onset.
Future directions: Ongoing studies are continuing to characterize the natural history of f‐FTLD. These studies will generate important data for planning treatment trials in FTLD.
HIGHLIGHTS
Familial frontotemporal dementia (f‐FTLD) is an important entity for drug development in FTLD.
f‐FTLD appears to show increasing rates of change as disease progresses.
Patterns of change in f‐FTLD will influence planning for clinical trials.
Another major challenge in FTLD is the absence of effective strategies for early diagnosis. In AD, the concept of mild cognitive impairment (MCI) revolutionized diagnosis and treatment by defining a clinical syndrome that often represents the early phase of disease.29 Delineation of MCI, combined with the development of biomarkers for tracking the accumulation of CNS amyloid and tau,30 the proteins that aggregate in AD, have led to clinical trials of disease‐modifying treatments in this early phase of AD.31 Furthermore, these biomarkers have spurred the development of models of disease evolution in non‐AD disorders (Figure 2), which propose that prodromal, asymptomatic stages can be monitored with biomarkers and evolve into symptomatic stages wherein clinical measures can track progression.32 Individuals who are likely in this prodromal stage of AD can now be identified, and are being enrolled in prevention trials.33, 34 Such models should be equally relevant to FTLD. Although there have been attempts to define MCI‐like stages in FTLD,35 their sensitivity, specificity, and prognostic value for future decline have not been established. Furthermore, no biomarkers capable of identifying prodromal stages of FTLD have yet been developed. Here, we review data from recent studies that examine the applicability of the model in Figure 2 to FTLD, in particular highlighting data from two studies called Advancing Research and Treatment for Frontotemporal Lobar Degeneration (ARTFL) and Longitudinal Evaluation of Familial Frontotemporal Dementia Subjects (LEFFTDS), which were designed to characterize the entire natural history of FTLD. Data from these studies have recently been published in a collection of articles in Alzheimer's and Dementia and its sister journals.
Figure 2.
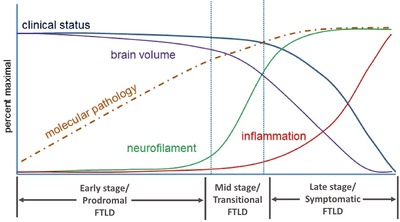
Theoretical model of disease progression in frontotemporal dementia (FTLD), linking theoretical biomarker changes to clinical stages of illness
2. ARTFL, LEFFTDS, AND FAMILIAL FTLD
In recognition of the need to advance toward treatment of FTLD, the National Alzheimer's Project Act (NAPA) Steering Committee on the Alzheimer's Disease‐Related Dementias (ADRD) convened in 2013 and again in 2016, and made recommendations for research.36, 37 The goals included (1) expanding efforts to genotype patients with FTLD and identifying new genes, (2) developing FTLD biomarkers for diagnosis and disease progression, (3) creating an international FTLD clinical trials network, and (4) understanding phenotypic heterogeneity and natural history. In 2014, the U.S. National Institutes of Health (NIH) funded two studies designed to improve our understanding of the natural history of FTLD and to establish a North American network of clinical research centers in preparation for clinical trials in FTLD. ARTFL (NS092089, PI Boxer) was funded by the National Institute for Neurological Diseases and Stroke (NINDS) and the National Center for Advancing Translational Sciences (NCATS) to create a network of 19 centers to study FTLD. LEFFTDS (AG045390, PIs, Boeve, Rosen) was funded by the National Institute on Aging (NIA) and NINDS, and involved a subset of eight of the ARTFL centers (http://www.allftd.org). The aims and methods are reviewed by Boeve et al in the special collection of articles, and the genetic methods and initial findings are reviewed by Ramos et al. Similar efforts are underway in Europe through the Genetic Frontotemporal Dementia Initiative (GENFI38).
One of the main goals for these studies was to characterize the natural history of FTLD, and f‐FTLD is a critical part of this goal. Members of these f‐FTLD families can be recruited based on their family history, and thus, can be identified before symptom onset, providing information on the prodromal, early symptomatic, and fully symptomatic stages. f‐FTLD mutation carriers are also highly attractive candidates for clinical trials because treatment studies that enroll f‐FTLD participants can be assured that every enrollee is affected by the targeted mechanism. In addition, clinical trials in f‐FTLD are currently the only context in which it is possible to demonstrate prevention of symptoms. Thus, ARTFL goals included recruitment of 600 individuals with sporadic FTLD (s‐FTLD) as well as 700 individuals from f‐FTLD families, who had limited longitudinal follow‐up. LEFFTDS was designed to provide more comprehensive longitudinal data in at least 300 members of families affected by mutations in the MAPT, GRN, and C9orf72 mutations, including asymptomatic and symptomatic individuals. As reviewed below, these new studies, combined with other previous and ongoing international studies, are providing critical data on the evolution of FTLD.
3. LATE STAGES OF DISEASE: SYMPTOMATIC FTLD
As highlighted in Figure 2, current models of neurodegeneration propose acceleration into a rapid phase of decline in the symptomatic stage of illness. Several projects have examined longitudinal changes in symptomatic FTLD, and these have clearly demonstrated dramatic changes, well in excess of rates seen in cognitively normal individuals, and in many cases faster than the changes seen in AD.39, 40 Such changes can be quantified using clinical and imaging measures,41, 42, 43, 44, 45, 46, 47 and have provided sample size estimates for clinical trials. Because of variability across individuals, sample size estimates have been fairly large for some variants such as bvFTD (hundreds of people per arm in a double‐blind, placebo‐controlled study), whereas other variants such as svPPA are relatively homogeneous, and thus can be tracked reliably with much lower numbers of subjects. Some studies have shown that sample size estimates can be reduced using methods to identify variables (for instance specific brain regions) that show the most consistent change over time,48 or by enriching studies with patients more likely to decline using baseline predictors.47 Most of this work has been done in s‐FTLD, but results of several longitudinal studies of symptomatic f‐FTLD have also been published,46, 49, 50, 51, 52, 53 which have identified substantial changes, well in excess of normal. Thus, it is clear that the overt symptomatic stages of both s‐FTLD and f‐FTLD are characterized by accelerated rates of decline, similar to other dementias. Additional work is now necessary to identify the best markers for tracking these changes as efficiently as possible while accounting for the wide variation in clinical presentation.
In addition, ongoing work is attempting to identify CSF and blood‐based markers of disease state, and one protein that has emerged as valuable is neurofilament light chain (NfL), a neuronal cytoskeletal protein that is elevated in symptomatic FTLD and other neurodegenerative diseases.54, 55 Longitudinal studies of NfL have indicated that it does not change much over time in patients with symptomatic s‐FTLD,56 but several studies have indicated that baseline levels of NfL can predict rates of future change across s‐FTLD patients, suggesting that NfL is an indicator of the aggressiveness of an individual's disease.56, 57, 58 Similar findings have been shown in f‐FTLD.59 These findings suggest that a reduction in NfL due to a drug could be a powerful indicator that the treatment will alter disease course in continued longitudinal follow‐up. In addition, NfL can be used to enrich clinical trials with patients more likely to progress, thereby improving efficiency. One great advantage of NfL is that measurements from blood correlate well with measurements from CSF, and have similar predictive value.58, 59
Several articles in the special collection of articles in Alzheimer's and Dementia touch on clinical trials in symptomatic FTLD. In particular, Boxer et al discuss recent progress and challenges that were reviewed in a meeting of the academic‐industry collaborative Frontotemporal Dementia Treatment Study Group in 2018. One of the approaches highlighted that there was the concept of the “basket” trial, where patients with several variants of FTLD that share an underlying mechanism can be combined into a single study. For instance, a trial for an anti‐tau antibody could enroll patients with PSP‐RS, CBS, and MAPT mutation carriers. As discussed in the article, such studies would be critically dependent on identification of clinical outcome measures that are relevant to all these variants. In another article, Heuer et al demonstrate that s‐FTLD and f‐FTLD patients with a diagnosis of bvFTD show similar clinical features and patterns of brain atrophy, supporting the idea that genetic and sporadic variants of FTLD could be followed with similar clinical measures. Finally, an article by Lapid et al presents data on quality of life and caregiver burden in f‐FTLD, which is an important patient‐oriented outcome for clinical trials.
4. EARLY STAGES OF DISEASE: ASYMPTOMATIC/PRODROMAL FTLD
Most models of neurodegeneration assume a much slower rate of decline during the prodromal phase. A critical question, however, is whether rates of change in any measures are still increased compared with normal (as depicted in Figure 2), which may indicate that these measures are still sensitive to biological changes associated with disease, and what sample sizes are required to reliably track these changes and detect effects of drugs. Staffaroni et al report on the use of NIH EXAMINER in the ARTFL/LEFFTDS f‐FTLD cohort. EXAMINER is a mostly computerized, psychometrically robust measure of executive function developed for clinical trials.60 They show that longitudinal changes in EXAMINER scores can be identified in the asymptomatic stage of FTLD, and similar findings have been identified for other measures of cognition and behavior by other groups.61 The associated sample size estimates for putative trials, however, are sobering, and this finding underscores the importance of identifying better predictors of future change in f‐FTLD, which can be used for inclusion and stratification (see below). In addition, Olney et al (special collection) provide cross‐sectional characterization of the initial features in the ARTFL/LEFFTDS f‐FTLD cohort and identify specific cognitive and behavioral measures that are abnormal in a percentage of asymptomatic f‐FTLD participants. This finding is consistent with prior studies in f‐FTLD showing that cognitive testing begins to deviate from normal levels of performance in the asymptomatic stage,38 and such measures may be suitable for longitudinal tracking in the setting of clinical trials.
In the absence of overt symptoms, brain imaging is an alternative measure that may track disease state. Several studies have shown that brain volumes,62, 63, 64 white matter integrity,53, 63, 64 functional connectivity,62, 65 perfusion,66 glucose metabolism,67 N‐acetylaspartate/creatinine (NAA/Cr), and NAA/myo‐inositol ratios68 are reduced in asymptomatic f‐FTLD compared with normal. The analysis in Olney et al (special collection) also identified brain regions where a percentage of f‐FTLD family members show reduced brain volumes, and these may be suitable for longitudinal tracking. One of the notable findings, however, was significant variability in which regions were abnormal across participants, which means that choosing one region for all individuals would be difficult. This had been noted previously for the C9orf72 mutation,50 but Olney et al also found considerable variability in GRN and MAPT mutation carriers, with MAPT carriers being the most homogeneous. Few studies have examined longitudinal brain changes in asymptomatic f‐FTLD. Data from Chen et al (special collection) indicate that rates of decline in brain volume are faster in f‐FTLD mutation carriers compared with non‐carrier family members. Similar findings have been identified by other groups,63, 64 but the numbers of cases have thus far been small, and probably not sufficient to generate sample size estimates for clinical trials. Finally Casaletto et al (special collection) provide data showing that rate of decline in cognition in asymptomatic mutation carriers is altered by modifiable lifestyle factors, including intellectual activity and exercise, indicating that even the profound effects of a mutation can be modified by individual choices, and pointing the way to another class of interventions.
5. MID‐STAGES OF DISEASE: TRANSITION FROM ASYMPTOMATIC/PRODROMAL TO SYMPTOMATIC PHASE
A major goal for study of all neurodegenerative disease is to predict and identify the time when an individual transitions from the prodromal to the symptomatic phase of disease (Figure 2). This might be a critical period for starting interventions, which may also be used to delay or prevent this transition (see Boxer et al, special collection). Prediction of this transition is particularly problematic because there are no established measures that help with this prediction. Unlike mutations that cause familial AD and Huntington disease, for which age at onset in the family and genetic information significantly predict each individual's age at onset,69, 70 age at onset in f‐FTLD can vary dramatically, even within a family.71
Recent work from ARTFL/LEFFTDS and other projects has begun to address this transition. One critical goal is to identify clinical features that signify when this transition might be occurring, and ARTFL and LEFFTDS have done significant work in this regard. In most prior studies of f‐FTLD, participants have been described as being “asymptomatic” or “symptomatic.” This categorization ignores that for most patients with neurodegenerative disease, the transition to the symptomatic phase includes a period of mild symptoms that are not sufficiently severe to interfere with function and can be difficult to separate from the normal spectrum of cognition and behavior. As noted earlier, this stage is often referred to as MCI. To categorize participants in a way that captures this early/questionable phase, the ARTFL and LEFFTDS studies use an approach based on the Clinical Dementia Rating Staging Instrument (CDR72), which allows classification of each participant into mild/questionable (CDR = 0.5) or definite (CDR = 1, 2, or 3) impairment based on the participant's daily function. Although the CDR focuses predominantly on memory and executive cognitive domains, additional rating categories for language and behavior42, 43 have been implemented by the National Alzheimer's Coordinating Center (NACC) to improve sensitivity to FTLD (CDR plus NACC FTLD). The Alzheimer's and Dementia special collection includes two articles by Miyagawa et al, which demonstrate the utility of the CDR plus NACC FTLD scale and that these additional categories increase sensitivity to early features in FTLD. Furthermore, Olney et al (special collection) provide data on ARTFL/LEFFTDS f‐FTLD participants rated as CDR plus NACC FTLD = 0.5 on their first visit, and show that this stage is associated with a higher degree of cognitive impairment, and more evidence of neurodegeneration, as measured with volumetric MRI, compared with asymptomatic participants (CDR plus NACC FTLD = 0) and non‐mutation carriers. These findings reinforce that traditional approaches for rating dementia severity, based on changes from the prior level of function, are sensitive to biological changes. Indeed, among those individuals observed to convert to dementia within ARTFL/LEFFTDS who were reported on in another article for the special collection73 (see below), all had a CDR plus NACC FTLD = 0.5 score on the prior visit (personal communication).
In addition, the ARTFL/LEFFTDS team is developing a new instrument, the multidomain impairment rating (MIR), which includes many elements for improving the capture of early symptoms in FTLD, including precise delineation of the basis for a decision (participant report, informant report, neuropsychological testing), explicit criteria for use of objective neuropsychological testing in classification, and inclusion of visuospatial, motor, and sensory impairment. To implement this approach, ARTFL and LEFFTDS use performance on the neuropsychological tasks from the current (third) version of the cognitive testing battery used by the U.S. National Institute on Aging Alzheimer's disease centers program for the Uniform Data Set (UDS‐3). Kornak et al (special collection) present an improved method for generating z‐scores based on UDS‐3 data from cognitively normal controls stored at the NACC (http://www.alz.washington.edu).
Regarding prediction of this transition, a previous study from GENFI indicated that cognitive performance on specific tests begins to deviate from normal (at the group level) about 5 years before symptom onset in f‐FTLD, whereas brain volumes may become abnormally low about 10 years before symptoms.38 This estimate was based on cross‐sectional analysis of asymptomatic patients and modeling of disease evolution using an estimate of predicted age at onset based on the age at onset in other family members. As noted above, there is reason to doubt that this approach is sufficiently accurate in f‐FTLD, and additional analyses are necessary to quantify its predictive value. One recent article suggested that brain imaging changes may occur closer to symptom onset (between 2 and 4 years) in GRN and MAPT carriers.74 Studies highlighting tests or brain regions that become abnormal at the group level raise the issue of whether it might be possible to choose a single cognitive measure or single brain region for each type of mutation as an early indicator of evolving disease. Several articles in the special collection address the challenge of identifying markers of transition. Olney et al (special collection) examine the distribution of abnormalities on cognitive testing and brain volumes in the CDR plus NACC FTLD = 0 and CDR plus NACC FTLD = 0.5 stages, and show that there are no tests or brain regions where >80% of mutation carriers in these groups are abnormal (defined as a z‐score ≥ 1.5 standard deviations [SD]from normal). This is consistent with prior observations indicating that f‐FTLD mutation carriers may present with a variety of symptoms,71 and suggests that it may be difficult to choose a single test or brain region for all carriers, even when focusing on one mutation. On the other hand, an article by Staffaroni et al73 (special collection) examines the value of brain imaging as a predictor of decline using individualized maps of brain atrophy. They created a classification algorithm from atrophy maps based on CDR plus NACC FTLD = 0 and CDR plus NACC FTLD ≥ 1 mutation carriers and generated atrophy‐based dementia prediction scores for each individual. In an independent sample of FTLD mutation carriers who had been followed longitudinally, they then showed that these atrophy‐based scores improved prediction of conversion to CDR plus NACC FTLD ≥ 1 compared with models that only included age. Other prediction models using multimodal imaging and clinical data have also shown promising results.75 These models were not customized for the type of mutation, an approach that will be tried as larger numbers of subjects are recruited. An additional article by Chen et al (special collection) describing longitudinal changes in MAPT mutation carriers indicates that brain volumes decline more rapidly in the few years before conversion to FTLD (similar to observations from other groups74), although imaging changes using the methodologies described by Chen et al appear earlier in the course than is shown by other groups. Recent work also indicates that changes in white matter integrity, measured with diffusion‐weighted MRI, accelerate prior to development of dementia76 and recent findings from ARTFL/LEFFTDS have also demonstrated a similar pattern in the NAA/Cr ratio.77 Much of the work indicating acceleration in biomarkers as FTLD clinical features emerge has come from small studies of GRN and MAPT. Similar studies of C9orf72 carriers have not yet emerged. There are reasons to believe that pattern of change over time may differ by mutation type. Cases of C9orf72 mutation carriers, in particular, have been described with very long and insidious courses of illness,78, 79 but how representative these cases are of C9orf72‐related disease overall remains to be studied.
Because fluid biomarkers such as NfL are associated with symptomatic FTLD and predict rate of decline, it stands to reason that rises in NfL may mark the transition to the symptomatic phase. Such a finding with NfL recently emerged from a large study of familial AD,80 and cross‐sectional studies have demonstrated that NfL levels are higher in symptomatic than in presymptomatic f‐FTLD.59 This will be examined more in ongoing studies of f‐FTLD.
6. CONCLUSIONS
Data collected over the last decade supports the idea that common models of neurodegenerative disease, which propose low rates of change in clinical measures and biomarkers in the prodromal phase followed by acceleration of disease markers in the symptomatic phase, apply to FTLD. Large ongoing studies of f‐FTLD have provided critical information about the prodromal phase. As these data have emerged, it has become clear that substantial continued development will be necessary to prepare for clinical intervention trials. In the symptomatic phase, despite rapid rates of decline overall, significant variability across people means that large sample sizes (given the relative rarity of FTLD) are required for studies that would use traditional designs and current measures. This problem is even more significant in the asymptomatic, prodromal phase, where rates of change are smaller. These observations mean that new, creative approaches to trial design and more sensitive clinical measures and biomarkers will need to be developed.
A critical goal for treatment in the prodromal phase is to delay or prevent the onset of features, and studies are beginning to demonstrate that a combination of clinical and biomarker measures can help to predict when a person will develop FTLD features, but much additional work is needed to refine these models to the point where they are most useful. Detecting acceleration in clinical measures and/or biomarkers may become an important component of these models, and fluid‐based biomarkers such as NfL may also enhance such models. One would predict that approaches that incorporate multimodal indices of FTLD evolution (ie, biofluid analytes, genetic variation, neuropsychological findings, imaging findings, and so on) will be particularly informative.81
DISCLOSURES
Boxer A receives research support from NIH, the Tau Research Consortium, the Association for Frontotemporal Degeneration, Bluefield Project to Cure Frontotemporal Dementia, Corticobasal Degeneration Solutions, the Alzheimer's Drug Discovery Foundation, and the Alzheimer's Association. He has served as a consultant for Aeton, Abbvie, Alector, Amgen, Arkuda, Ionis, Iperian, Janssen, Merck, Novartis, Passage BIO, Pinteon, Samumed, Toyama, and UCB, and received research support from Avid, Biogen, BMS, C2N, Cortice, Eli Lilly, Forum, Genentech, Janssen, Novartis, Pfizer, Roche, and TauRx.
Rosen H has received research support from Biogen Pharmaceuticals, has consulting agreements with Wave Neuroscience and Ionis Pharmaceuticals, and receives research support from NIH.
Boeve B has served as an investigator for clinical trials sponsored by Biogen and Alector. He receives royalties from the publication of a book entitled “Behavioral Neurology of Dementia” (Cambridge Medicine, 2009, 2017). He serves on the Scientific Advisory Board of the Tau Consortium. He receives research support from NIH, the Mayo Clinic Dorothy and Harry T. Mangurian Jr. Lewy Body Dementia Program, and the Little Family Foundation.
ACKNOWLEDGMENTS
We extend our appreciation to Drs. John Hsiao and Dallas Anderson from the National Institute on Aging; Drs. Marg Sutherland and Codrin Lungu from the National Institute of Neurological Disorders and Stroke; the clinicians and coordinators at all ARTFL/LEFFTDS centers; and particularly to our patients and their families for their participation in this research.
Rosen HJ, Boeve BF, Boxer AL. Tracking disease progression in familial and sporadic frontotemporal lobar degeneration: Recent findings from ARTFL and LEFFTDS. Alzheimer's Dement. 2020;16:71–78. 10.1002/alz.12004
For special issue on Frontotemporal Lobar Degeneration
Funding information
This work is supported by the National Institutes of Health (grants AG045390 [LEFFTDS], NS092089 [ARTFL], AG032306, AG021886, AG016976, AG019724, AG038791, AG056749, and AG045333).
REFERENCES
- 1. Bang J, Spina S, Miller BL. Frontotemporal dementia. Lancet. 2015;386:1672‐1682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Rademakers R, Neumann M, Mackenzie IR. Advances in understanding the molecular basis of frontotemporal dementia. Nat Rev Neurol. 2012;8:423‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Ratnavalli E, Brayne C, Dawson K, Hodges JR. The prevalence of frontotemporal dementia. Neurology. 2002;58:1615‐1621. [DOI] [PubMed] [Google Scholar]
- 4. Rosso SM, Donker Kaat L, Baks T, et al. Frontotemporal dementia in the Netherlands: patient characteristics and prevalence estimates from a population‐based study. Brain. 2003;126:2016‐2022. [DOI] [PubMed] [Google Scholar]
- 5. Knopman DS, Petersen RC, Edland SD, Cha RH, Rocca WA. The incidence of frontotemporal lobar degeneration in Rochester, Minnesota, 1990 through 1994. Neurology. 2004;62:506‐508. [DOI] [PubMed] [Google Scholar]
- 6. Onyike CU, Diehl‐Schmid J. The epidemiology of frontotemporal dementia. Int Rev Psychiatry. 2013;25:130‐137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Coyle‐Gilchrist IT, Dick KM, Patterson K, et al. Prevalence, characteristics, and survival of frontotemporal lobar degeneration syndromes. Neurology. 2016;86:1736‐1743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Roberson ED, Hesse JH, Rose KD, et al. Frontotemporal dementia progresses to death faster than Alzheimer disease. Neurology. 2005;65:719‐725. [DOI] [PubMed] [Google Scholar]
- 9. Papageorgiou SG, Kontaxis T, Bonakis A, Kalfakis N, Vassilopoulos D. Frequency and causes of early‐onset dementia in a tertiary referral center in Athens. Alzheimer Dis Assoc Disord. 2009;23:347‐351. [DOI] [PubMed] [Google Scholar]
- 10. Mourik JC, Rosso SM, Niermeijer MF, Duivenvoorden HJ, Van Swieten JC, Tibben A. Frontotemporal dementia: behavioral symptoms and caregiver distress. Dement Geriatr Cogn Disord. 2004;18:299‐306. [DOI] [PubMed] [Google Scholar]
- 11. Merrilees J, Klapper J, Murphy J, Lomen‐Hoerth C, Miller BL. Cognitive and behavioral challenges in caring for patients with frontotemporal dementia and amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2010;11:298‐302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Galvin JE, Howard DH, Denny SS, Dickinson S, Tatton N. The social and economic burden of frontotemporal degeneration. Neurology. 2017;89:2049‐2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Boxer AL, Knopman DS, Kaufer DI, et al. Memantine in patients with frontotemporal lobar degeneration: a multicentre, randomised, double‐blind, placebo‐controlled trial. Lancet Neurol. 2013;12:149‐156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Boxer AL, Gold M, Huey E, et al. Frontotemporal degeneration, the next therapeutic frontier: molecules and animal models for frontotemporal degeneration drug development. Alzheimers Dement. 2013;9:176‐188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Tsai RM, Boxer AL. Therapy and clinical trials in frontotemporal dementia: past, present, and future. J Neurochem. 2016;138(Suppl 1):211‐221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Riboldi G, Zanetta C, Ranieri M, et al. Antisense oligonucleotide therapy for the treatment of C9ORF72 ALS/FTD diseases. Mol Neurobiol. 2014;50:721‐732. [DOI] [PubMed] [Google Scholar]
- 17. Sha SJ, Miller ZA, Min SW, et al. An 8‐week, open‐label, dose‐finding study of nimodipine for the treatment of progranulin insufficiency from GRN gene mutations. Alzheimers Dement (N Y). 2017;3:507‐512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Finger EC, MacKinley J, Blair M, et al. Oxytocin for frontotemporal dementia: a randomized dose‐finding study of safety and tolerability. Neurology. 2015;84:174‐781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Chitramuthu BP, Bennett HPJ, Bateman A. Progranulin: a new avenue towards the understanding and treatment of neurodegenerative disease. Brain. 2017;140:3081‐3104. [DOI] [PubMed] [Google Scholar]
- 20. West T, Hu Y, Verghese PB, et al. Preclinical and clinical development of ABBV‐8E12, a humanized anti‐tau antibody, for treatment of alzheimer's disease and other tauopathies. J Prev Alzheimers Dis. 2017;4:236‐241. [DOI] [PubMed] [Google Scholar]
- 21. Crunkhorn S. Alzheimer disease: antisense oligonucleotide reverses tau pathology. Nat Rev Drug Discov. 2017;16:166. [DOI] [PubMed] [Google Scholar]
- 22. Yanamandra K, Patel TK, Jiang H, et al. Anti‐tau antibody administration increases plasma tau in transgenic mice and patients with tauopathy. Sci Transl Med. 2017;9:pii: eaal2029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Hutton M, Lendon CL, Rizzu P, et al. Association of missense and 5'‐splice‐site mutations in tau with the inherited dementia FTDP‐17. Nature. 1998;393:702‐705. [DOI] [PubMed] [Google Scholar]
- 24. Baker M, Mackenzie IR, Pickering‐Brown SM, et al. Mutations in progranulin cause tau‐negative frontotemporal dementia linked to chromosome 17. Nature. 2006;442:916‐919. [DOI] [PubMed] [Google Scholar]
- 25. DeJesus‐Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p‐linked FTD and ALS. Neuron. 2011;72:245‐256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Renton AE, Majounie E, Waite A, et al. A hexanucleotide repeat expansion in C9ORF72 is the cause of chromosome 9p21‐linked ALS‐FTD. Neuron. 2011;72:257‐268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. van Swieten JC, Rosso SM. Epidemiological aspects of frontotemporal dementia. Handb Clin Neurol. 2008;89:331‐341. [DOI] [PubMed] [Google Scholar]
- 28. Rohrer JD, Guerreiro R, Vandrovcova J, et al. The heritability and genetics of frontotemporal lobar degeneration. Neurology. 2009;73:1451‐1456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Petersen RC. How early can we diagnose Alzheimer disease (and is it sufficient)? The 2017 Wartenberg lecture. Neurology. 2018;91:395‐402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Cohen AD, Landau SM, Snitz BE, Klunk WE, Blennow K, Zetterberg H. Fluid and PET biomarkers for amyloid pathology in Alzheimer's disease. Mol Cell Neurosci. 2019;97:3‐17. [DOI] [PubMed] [Google Scholar]
- 31. Petersen RC, Thomas RG, Aisen PS, et al. Randomized controlled trials in mild cognitive impairment: sources of variability. Neurology. 2017;88:1751‐1758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Jack CR, Jr. , Knopman DS, Jagust WJ, et al. Tracking pathophysiological processes in Alzheimer's disease: an updated hypothetical model of dynamic biomarkers. Lancet Neurol. 2013;12:207‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mills SM, Mallmann J, Santacruz AM, et al. Preclinical trials in autosomal dominant AD: implementation of the DIAN‐TU trial. Rev Neurol (Paris). 2013;169:737‐743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Sperling RA, Rentz DM, Johnson KA, et al. The A4 study: stopping AD before symptoms begin? Sci Transl Med. 2014;6:228fs13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ismail Z, Aguera‐Ortiz L, Brodaty H, et al. The mild behavioral impairment checklist (MBI‐C): a rating scale for neuropsychiatric symptoms in pre‐dementia populations. J Alzheimers Dis. 2017;56:929‐938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Montine TJ, Koroshetz WJ, Babcock D, et al. Recommendations of the Alzheimer's disease‐related dementias conference. Neurology. 2014;83:851‐860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Corriveau RA, Koroshetz WJ, Gladman JT, et al. Alzheimer's Disease‐Related Dementias Summit 2016: national research priorities. Neurology. 2017;89:2381‐2391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Rohrer JD, Nicholas JM, Cash DM, et al. Presymptomatic cognitive and neuroanatomical changes in genetic frontotemporal dementia in the Genetic Frontotemporal Dementia Initiative (GENFI) study: a cross‐sectional analysis. Lancet Neurol. 2015;14:253‐262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Rascovsky K, Salmon DP, Lipton AM, et al. Rate of progression differs in frontotemporal dementia and Alzheimer disease. Neurology. 2005;65:397‐403. [DOI] [PubMed] [Google Scholar]
- 40. Krueger CE, Dean DL, Rosen HJ, et al. Longitudinal rates of lobar atrophy in frontotemporal dementia, semantic dementia, and Alzheimer's disease. Alzheimer Dis Assoc Disord. 2010;24:43‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Diehl‐Schmid J, Grimmer T, Drzezga A, et al. Decline of cerebral glucose metabolism in frontotemporal dementia: a longitudinal 18F‐FDG‐PET‐study. Neurobiol Aging. 2006. [DOI] [PubMed] [Google Scholar]
- 42. Knopman DS, Kramer JH, Boeve BF, et al. Development of methodology for conducting clinical trials in frontotemporal lobar degeneration. Brain. 2008;131:2957‐2968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Knopman DS, Jack CR, Jr. , Kramer JH, et al. Brain and ventricular volumetric changes in frontotemporal lobar degeneration over 1 year. Neurology. 2009;72:1843‐1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Whitwell JL, Jack CR, Jr. , Pankratz VS, et al. Rates of brain atrophy over time in autopsy‐proven frontotemporal dementia and Alzheimer disease. Neuroimage. 2008;39:1034‐1040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Gordon E, Rohrer JD, Kim LG, et al. Measuring disease progression in frontotemporal lobar degeneration: a clinical and MRI study. Neurology. 2010;74:666‐673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Mahoney CJ, Simpson IJ, Nicholas JM, et al. Longitudinal diffusion tensor imaging in frontotemporal dementia. Ann Neurol. 2015;77:33‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Staffaroni AM, Ljubenkov PA, Kornak J, et al. Longitudinal multimodal imaging and clinical endpoints for frontotemporal dementia clinical trials. Brain. 2019;142:443‐459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Binney RJ, Pankov A, Marx G, et al. Data‐driven regions of interest for longitudinal change in three variants of frontotemporal lobar degeneration. Brain Behav. 2017;7:e00675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Whitwell JL, Weigand SD, Gunter JL, et al. Trajectories of brain and hippocampal atrophy in FTD with mutations in MAPT or GRN. Neurology. 2011;77:393‐398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Mahoney CJ, Downey LE, Ridgway GR, et al. Longitudinal neuroimaging and neuropsychological profiles of frontotemporal dementia with C9ORF72 expansions. Alzheimers Res Ther. 2012;4:41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Whitwell JL, Boeve BF, Weigand SD, et al. Brain atrophy over time in genetic and sporadic frontotemporal dementia: a study of 198 serial magnetic resonance images. Eur J Neurol. 2015;22:745‐752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Floeter MK, Bageac D, Danielian LE, Braun LE, Traynor BJ, Kwan JY. Longitudinal imaging in C9orf72 mutation carriers: relationship to phenotype. Neuroimage Clin. 2016;12:1035‐1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Floeter MK, Danielian LE, Braun LE, Wu T. Longitudinal diffusion imaging across the C9orf72 clinical spectrum. J Neurol Neurosurg Psychiatry. 2018;89:53‐60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Scherling CS, Hall T, Berisha F, et al. Cerebrospinal fluid neurofilament concentration reflects disease severity in frontotemporal degeneration. Ann Neurol. 2014;75:116‐126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Zhao Y, Xin Y, Meng S, He Z, Hu W. Neurofilament light chain protein in neurodegenerative dementia: a systematic review and network meta‐analysis. Neurosci Biobehav Rev. 2019;102:123‐138. [DOI] [PubMed] [Google Scholar]
- 56. Ljubenkov PA, Staffaroni AM, Rojas JC, et al. Cerebrospinal fluid biomarkers predict frontotemporal dementia trajectory. Ann Clin Transl Neurol. 2018;5:1250‐1263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Rohrer JD, Woollacott IO, Dick KM, et al. Serum neurofilament light chain protein is a measure of disease intensity in frontotemporal dementia. Neurology. 2016;87:1329‐1336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Rojas JC, Karydas A, Bang J, et al. Plasma neurofilament light chain predicts progression in progressive supranuclear palsy. Ann Clin Transl Neurol. 2016;3:216‐225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Meeter LH, Dopper EG, Jiskoot LC, et al. Neurofilament light chain: a biomarker for genetic frontotemporal dementia. Ann Clin Transl Neurol. 2016;3:623‐636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Kramer JH, Mungas D, Possin KL, et al. NIH EXAMINER: conceptualization and development of an executive function battery. J Int Neuropsychol Soc. 2013:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Jiskoot LC, Dopper EG, Heijer T, et al. Presymptomatic cognitive decline in familial frontotemporal dementia: a longitudinal study. Neurology. 2016;87:384‐391. [DOI] [PubMed] [Google Scholar]
- 62. Dopper EG, Rombouts SA, Jiskoot LC, et al. Structural and functional brain connectivity in presymptomatic familial frontotemporal dementia. Neurology. 2014;83:e19‐e26. [DOI] [PubMed] [Google Scholar]
- 63. Olm CA, McMillan CT, Irwin DJ, et al. Longitudinal structural gray matter and white matter MRI changes in presymptomatic progranulin mutation carriers. Neuroimage Clin. 2018;19:497‐506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Panman JL, Jiskoot LC, Bouts M, et al. Gray and white matter changes in presymptomatic genetic frontotemporal dementia: a longitudinal MRI study. Neurobiol Aging. 2019;76:115‐124. [DOI] [PubMed] [Google Scholar]
- 65. Lee SE, Sias AC, Mandelli ML, et al. Network degeneration and dysfunction in presymptomatic C9ORF72 expansion carriers. Neuroimage Clin. 2017;14:286‐297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Dopper EG, Chalos V, Ghariq E, et al. Cerebral blood flow in presymptomatic MAPT and GRN mutation carriers: a longitudinal arterial spin labeling study. Neuroimage Clin. 2016;12:460‐465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Jacova C, Hsiung GY, Tawankanjanachot I, et al. Anterior brain glucose hypometabolism predates dementia in progranulin mutation carriers. Neurology. 2013;81:1322‐1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Chen Q, Boeve BF, Tosakulwong N, et al. Frontal lobe 1H MR spectroscopy in asymptomatic and symptomatic MAPT mutation carriers. Neurology. 2019;. 93(8):e758‐e765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Lee JM, Ramos EM, Lee JH, et al. CAG repeat expansion in Huntington disease determines age at onset in a fully dominant fashion. Neurology. 2012;78:690‐695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Mendez MF. Early‐onset Alzheimer disease and its variants. Continuum (Minneap Minn). 2019;25:34‐51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Greaves CV, Rohrer JD. An update on genetic frontotemporal dementia. J Neurol. 2019;266(8):2075‐2086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Morris JC. Clinical dementia rating: a reliable and valid diagnostic and staging measure for dementia of the Alzheimer type. Int Psychogeriatr. 1997;9:173‐176; discussion 7‐8. [DOI] [PubMed] [Google Scholar]
- 73. Staffaroni AM, Cobigo Y, Goh SM, et al. Individualized atrophy scores predict dementia onset in familial frontotemporal lobar degeneration. Alzheimers Dement. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74. Jiskoot LC, Panman JL, Meeter LH, et al. Longitudinal multimodal MRI as prognostic and diagnostic biomarker in presymptomatic familial frontotemporal dementia. Brain. 2019;142:193‐208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75. Feis RA, Bouts M, de Vos F, et al. A multimodal MRI‐based classification signature emerges just prior to symptom onset in frontotemporal dementia mutation carriers. J Neurol Neurosurg Psychiatry. 2019;90(11):1207‐1214. [DOI] [PubMed] [Google Scholar]
- 76. Chen Q, Boeve BF, Schwarz CG, et al. LEFFTDS Consortium Tracking white matter degeneration in asymptomatic and symptomatic MAPT mutation carriers. Neurobiol Aging. 2019;83:54‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77. Chen Q, Boeve BF, Tosakulwong N, et al. Brain MR spectroscopy changes precede frontotemporal lobar degeneration phenoconversion in mapt mutation carriers. J Neuroimaging. 2019;29(5):624‐629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78. Khan BK, Yokoyama JS, Takada LT, et al. Atypical, slowly progressive behavioural variant frontotemporal dementia associated with C9ORF72 hexanucleotide expansion. J Neurol Neurosurg Psychiatry. 2012;83:358‐364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79. Llamas‐Velasco S, Garcia‐Redondo A, Herrero‐San Martin A, et al. Slowly progressive behavioral frontotemporal dementia with C9orf72 mutation. Case report and review of the literature. Neurocase. 2018;24:68‐71. [DOI] [PubMed] [Google Scholar]
- 80. Preische O, Schultz SA, Apel A, et al. Serum neurofilament dynamics predicts neurodegeneration and clinical progression in presymptomatic Alzheimer's disease. Nat Med. 2019;25:277‐283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81. Boeve BF, Rosen HJ. Multimodal imaging in familial FTLD: phenoconversion and planning for the future. Brain. 2019;142:8‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]