

Abstract
Ischemic brain damage is the major cause of mortality in cardiac arrest (CA). However, the molecular mechanism responsible for brain damage is not well understood. We previously found that mitochondrial state-3 respiration, which had been decreased following CA, was recovered in the kidney and liver, but not in the brain following cardiopulmonary bypass (CPB) resuscitation. Examination of mitochondria from these tissues may shed light on why the brain is the most vulnerable. In this study, adult male Sprague-Dawley rats were subjected to asphyxia-induced CA for 30 minutes or 30 min followed by 60 min CPB resuscitation. Mitochondria were then isolated from brain, heart, kidney, and liver tissues for examination using spectrophotometry and mass spectrometry to measure the activities of mitochondrial electron transport complexes and the cardiolipin content. We found significantly decreased complex I activity in mitochondria isolated from all four organs following CA, while complex III and IV activities remained intact. Following CPB resuscitation, complex I activity was normalized in kidney and liver, but unrecovered in brain and heart mitochondria. In addition, complex III activity in brain mitochondria was decreased by 22% with a concomitant decrease in cardiolipin following CPB resuscitation. These results suggest that of the tissues tested only brain mitochondria suffer reperfusion injury in addition to ischemic alterations, resulting in diminished overall mitochondrial respiration following resuscitation.
Keywords: ischemia, reperfusion injury, electron transport chain, LC-MS, monolysocardiolipin
1. Introduction
Cardiac arrest (CA) induces whole-body ischemia and causes damage to multiple organs, (Roberts et al., 2013). The brain is the most vulnerable organ to ischemia/reperfusion injury. Clinical and preclinical evidence suggests that damage in the brain is responsible for high mortality of patients with CA (Stub et al., 2011). However, molecular mechanisms explaining the vulnerability of the brain in CA have yet to be elucidated. Prior studies of post-ischemic dysfunction in isolated organs have described various alterations in the mitochondrial electron transpot chain (ETC) including the decomposition of membrane phospholipid, particularly mitochondria-specific phospholipid, cardiolipin (CL) (Borutaite et al., 2013). Overall these reports suggest that mitochondria may be the key to understanding mechanisms of organ damage in CA.
Asphyxia-induced CA is the most commonly used CA model in small rodents because it is noninvasive and highly reproducible (Vognsen et al., 2017). We combined asphyxia-induced CA with cardiopulmonary bypass (CPB) resuscitation to establish a unique CA+CPB rat model. CPB models extracorporeal membrane oxygenation, which is adopting an increasing role in resuscitation medicine due to improved patient outcomes after CA (Stub et al., 2015). In animal models, CPB delivers a more consistent blood flow to each organ, whereas blood flow in conventional chest compression resuscitation depends heavily on the recovery of heart function. Therefore, the maintenance and control of blood flow through CPB significantly reduces the variability caused by conventional cardiopulmonary resuscitation, providing a compelling platform for studying molecular mechanisms of ischemia/reperfusion injury in individual organs in CA.
Using the CA+CPB model, we previously found that rats did not show any measurable brain function after 30 min of CA and 60 min of CPB, but maintained adequate heart and kidney function (Kim et al., 2014). Consistent with the physiological outcomes, the decrease in mitochondrial state-3 respiration was significantly greater in the brain than other tissues in these resuscitated animals (Kim et al., 2016). This correlation between physiological outcome and mitochondrial respiration activity prompted our current exploration of the mitochondrial ETC for molecular mechanisms of ischemia/reperfusion injury in the brain.
We examined activities of mitochondrial ETC complexes and analyzed CL content. Our data show that mitochondria from the brain, heart, kidney, and liver have common complex I inactivation following CA. After resuscitation, only brain mitochondria undergo additional defects: decreased complex III activity and decreased CL content. These results suggest that brain mitochondria suffer reperfusion injury in addition to ischemic alterations resulting in diminished overall mitochondrial respiration following resuscitation. These defects specific to the brain may be the primary reason the brain is most susceptible to ischemia/reperfusion, leaving brain damage as the major cause for mortality in CA.
2. Material and methods
2.1. Chemicals
Nicotinamide adenine dinucleotide (NADH), decylubiquinone, bovine serum albumin, antimycin a, n-Dodecyl β-D-maltoside, and cytochrome-c were purchased from Sigma-Aldrich (St Louis, Mo). Potassium ferricyanide and the solvents used for HPLC analysis were purchased from Fisher Scientific (Hampton, NH). Dcylubiquinol was synthesized by reducing decylubiquinone with sodium borohydride (Trounce et al., 1996). Reduced cytochrome-c was prepared by reducing cytochrome-c with sodium dithionite (Dixon and McIntosh, 1967). Reagent grade water was prepared using a Milli-Q water system.
2.2. Cardiac Arrest Model
The experimental protocol conformed to the Guide for the Care and Use of Laboratory Animals published by the United States National Institute of Health, and was approved by the Institutional Animal Care and Use Committee. Sprague-Dawley male rats (465 – 530 g, Charles River, Wilmington, MA), housed in a rodent facility with unrestricted access to food and water, were arbitrarily assigned into three groups, control, CA, and CA+CPB. Six animals were included in each group and each animal was used to generate one data point.
The detailed surgical procedures are reported in the Supplementary material. Briefly, rats were anesthetized with 4% isoflurane and ventilated with an intubated catheter with 2% isoflurane. After the injection of heparin (150 U) and vecuronium (1 mg), asphyxia was induced by stopping the ventilator. The isoflurane was discontinued immediately after. Following 30 min of CA, resuscitation was started with the initiation of CPB flow and resumption of ventilation and continued for 60 min. The rats were sacrificed by decapitation and tissue samples collected. Animals in the CA group were sacrificed after 30 min of asphyxia. Control group animals were sacrificed 7 min after administration of isoflurane.
2.3. Mitochondria Isolation
Mitochondria were isolated from the freshly harvested brain, heart, kidney, and liver using differential centrifugation as previously reported (Kim et al., 2016) and stored at −80 °C. The detailed procedure is provided in the supplementary materials. Mitochondrial protein concentration was determined by the BCA assay using bovine serum albumin as a standard. Mitochondrial yields were expressed as, “mg mitochondrial protein/g tissue.” The data from all assays were normalized to the mitochondria protein concentration.
2.4. Measuring Activity of Individual Complexes
Complex I, III, and IV activities were measured using previously frozen mitochondria by a modification of the published method (Rustin, 2008). The amounts of substrates and detergent were determined after multiple test runs to find a concentration that gives maximal activity with linear response for each complex for each tissue. During the method optimization, we used known inhibitors—rotenone, antimycin A, and sodium azide—for complexes I, III, and IV respectively. We found inhibitor-sensitive activity to be minimal compared to non-inhibited activity and elected not to measure inhibitor-sensitive activity in order to minimize the use of these toxic chemicals.
The detailed assay conditions for each complex are presented in Table S1. Briefly, complex I activity was measured by following the decrease in absorbance of NADH at 340 nm. Complex III activity was measured by following the increase in the absorbance of reduced cytochrome-c at 550 nm. Complex IV activity was measured by following the decrease in the absorbance of reduced cytochrome-c at 550 nm. We further measured the activity of NADH dehydrogenase, the first component of complex I, using ferricyanide as the artificial acceptor for electrons (NADH ferricyanide reductase activity) (Chen et al., 2008). The data were obtained every 0.5 s for 60 s. The rate of activity was calculated using the linear region of the slope of the first 50 s (100 data points).
2.5. Measuring Amount of Complex I
The levels of complex I were measured using Rodent Protein Quantity Dipstick Assay Kits containing two monoclonal antibodies (Abcam, Cambridge, MA). The steps of this experiment were followed as shown in the protocol provided by the company. The amount of mitochondria used for the assay was 500 ng of brain and kidney mitochondria, 250 ng of heart mitochondria, and 880 ng of liver mitochondria. The peak intensity was quantified using the Image J software (NIH).
2.6. Analysis of Cardiolipin and monolysocardiolipin
Extraction and separation of lipids was performed using the published procedures (Kim and Hoppel, 2013). Briefly, 60 μg of frozen mitochondria was suspended in 50 μL of potassium phosphate buffer (100 mM, pH 7.4) and extracted with 950 μL of a CHCl3:MeOH (2:1, v:v) solution, containing butylated hydroxytoluene (2 mM) as an antioxidant. The lipid mixture was separated using solid-phase extraction and reconstituted in 200 μL of an isopropanol:methyl t- butyl ether:aqueous ammonium formate soulution (34:17:5, v:v:v) and 25 μL of the reconstitute was injected for LC-MS analysis. MS data were obtained with a Thermo LTQ XL spectrometer operated in the negative ion mode. The concentrations of CL and monolysocardiolipin (MLCL) were determined using standard curves (Kim and Hoppel, 2013).
2.7. Statistical analysis
All samples were analyzed together for each assay in order to minimize variation. Data are presented as mean ± standard deviation. Group comparisons were made with one-way analysis of variance using SPSS Statistics Ver. 22 for Mac (IBM Corp., Armonk, NY). Bonferroni’s test was used as a post hoc test; a p value < 0.05 was considered statistically significant.
3. Results
3.1. Asphyxial cardiac arrest and CPB resuscitation
The average time for animals to reach CA was approximately 3 min after the initiation of asphyxia. All animals used in this study achieved return of spontaneous circulation (ROSC) approximately 8 min after the initiation of CPB resuscitation. The success rate of achieving ROSC was over 90%. A representative trace of mean arterial pressure and electrocardiograms at multiple time points during the protocol are shown in supplementary Fig. S1 and S2. These data indicate heart function was not fully recovered. These rats did not respond to any stimuli, such as toe pinching and corneal irritation. Overall, after 30 min of CA and 60 min of CPB resuscitation, these animals displayed no brain function and compromised heart function—the typical condition of patients with severe CA.
3.2. Individual Complex Activity
To understand the mechanism for the decreased state-3 respiration, we measured activities of individual complexes I, III, and IV in brain, heart, kidney, and liver mitochondria (Fig. 1). Complex I activity decreased 30% (p < 0.0001) in brain mitochondria, 33% (p = 0.01) in heart mitochondria, 49% (p < 0.0001) in kidney mitochondria, and 14 % (p = 0.045) in liver mitochondria after 30 minutes of CA (Fig. 1A). Following CA with CPB resuscitation, complex I activity in brain and heart mitochondria was not significantly changed compared to CA without CPB resuscitation, however, the activity was restored to 73% of the control activity in kidney mitochondria and was fully restored in liver mitochondria (Fig. 1A). Also in brain mitochondria, complex III activity was decreased by 28% (p < 0.032) from control activity following CA with subsequent CPB resuscitation (Fig. 1B). Complex III and IV activities in heart, kidney, and liver mitochondria were unchanged following CA or CA with CPB resuscitation (Fig. 1B-C).
Fig. 1.
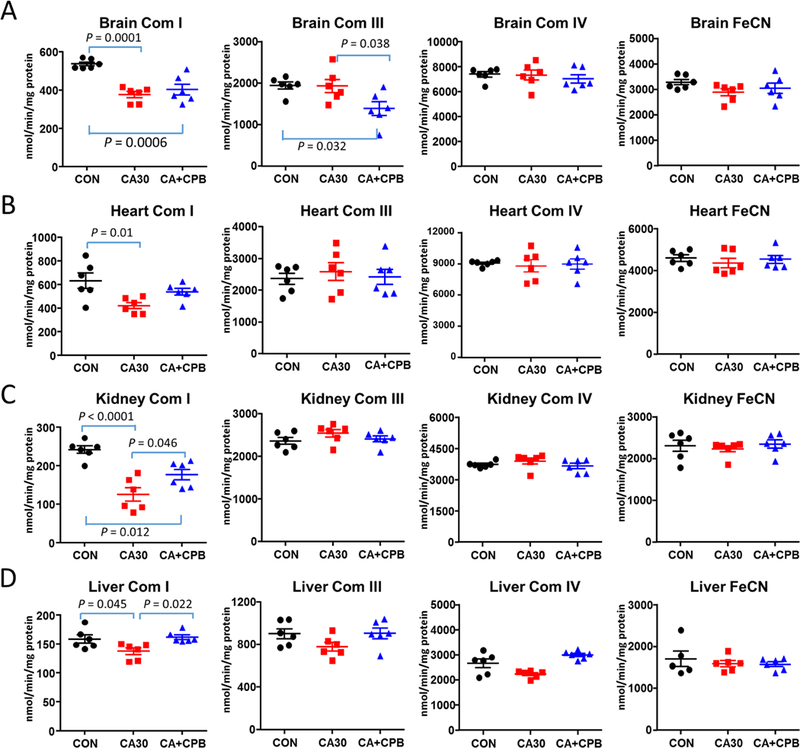
Activity of individual complexes and FeCN in isolated mitochondria following CA or CA and CPB resuscitation in the brain (A), heart (B), kidney (C), and liver (D). (CON, control; CA30, 30 min CA; CA+CPB, 30 min CA +60 min CPB resuscitation; FeCN, NADH ferricyanide reductase activity; n=5 or 6).
We also measured NADH ferricyanide reductase activity using ferricyanide as the electron acceptor. As shown in Fig. 1D, the activity does not change following either CA or CA with subsequent CPB resuscitation in any of the tissues tested. Unchanged NADH ferricyanide reductase activity indicates that the decreased complex I activity is not due to the NADH dehydrogenase but in other subunits of complex I. The result also suggest that the amount of complex 1 is not changed after CA or CPB resuscitation.
3.3. Amount of Complex I
To further confirm the amount of complex I was not changed following CA or CA with CPB resuscitation, the concentration of whole complex I was measured using an enzyme kit. The amount of complex I in mitochondria isolated from any of the organs remained unchanged in all of the tissues after CA or CA followed by CPB resuscitation (supplementary Fig. S3). This result is also consistent with the unchanged mitochondria isolation yield.
3.4. Cardiolipin and monolysocardiolipin analysis
The results from MS analysis of CL and MLCL in isolated mitochondria are shown in Fig. 2. Ion chromatograms and MS spectra of CL in brain mitochondria are provided as an example. (Fig. 2A). There is no significant change in the concentration of CL in any of the tissue following CA (Fig. 2B). Following resuscitation, the concentration of CL was decreased by 33% in brain mitochondria, whereas no change in other tissues (Fig. 2B). There were no significant changes in the composition of CL as the MS spectra of CL are similar between baseline, post CA, and post resuscitation (Fig. 2A).
Fig. 2.
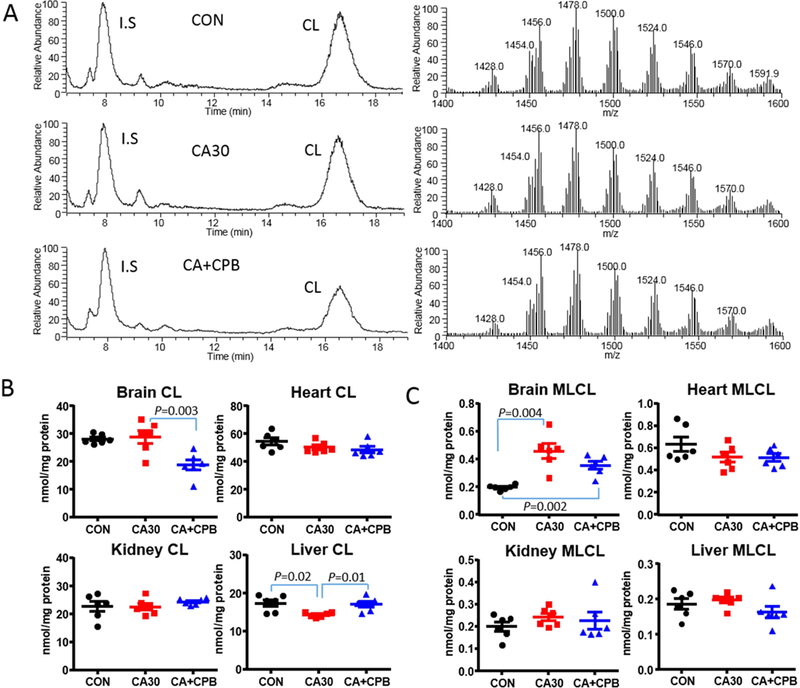
Representative ion chromatograms and mass spectra of brain CL at baseline (top panels), following 30 min of CA (middle panels), and 60 min of CPB resuscitation (bottom panels) (A) and concentrations of CL (left panel) and MLCL (right panel) in brain, heart, kidney, and liver mitochondria (B) (nmol/mg protein; n=6; *, against baseline; †, against CA).
We also observed changes in the MLCL contents of the brain (Figure 2C). The concentration of MLCL in brain mitochondria was increased two-fold following 30 min of CA. The concentration decreased following resuscitation, however considering the 33% decrease in CL, the decrease in MLCL may be due to the decrease in total CL pool. In fact, the ratio of MLCL/CL was increased following resuscitation compared to CA (0.020 vs. 0.016, p = 0.06), indicating the hydrolysis of CL continues after resuscitation. Overall, only brain mitochondria underwent significant content changes in CL and MLCL following CA and resuscitation.
4. Discussion
We report a unique response of brain mitochondria following CA and CPB resuscitation and this may be the reason for the vulnerability of the brain in CA. Mitochondria from the brain, heart, kidney, and liver share a common ischemic defect, decreased complex I activity, following CA. However, only brain mitochondria decreased complex III activity and CL content following resuscitation. These unique alterations may be the mechanism for impaired mitochondrial respiration and for the absence of brain function, both of which were observed previously using the same model (Kim et al., 2016; Kim et al., 2014).
This decreased complex activity induced after CA, but no additional change by CPB resuscitation, suggests the complex I inhibition is a result of—and specific to—ischemia. To address the reason for the decreased complex I activity, we measured NADH ferricyanide reductase activity. Unchanged NADH ferricyanide reductase activity suggest that there is no significant change in amount of complex I in any of the four tissues either following 30 min CA or CA and CPB and that the change in complex I activity is indeed a result of either dysregulation or dysfunction of the enzyme. The result also indicates that the decreased activity is not due to the NADH dehydrogenase but in other subunits of complex I, which are likely embedded in the membranes. The detailed mechanism for the inhibition of complex I requires further investigations.
Our data indicate that unrecovered mitochondrial respiration in the brain following resuscitation is attributable to unrecovered complex I activity and decreased complex III activity, which are caused by the failure in maintaining CL. In mitochondria, CL is maintained by remodeling enzymes through MLCL (Dudek et al., 2016). The increase in the concentration of MLCL shows that the CL remodeling is disturbed in brain mitochondria following CA. Since decreased complex I activity was found in mitochondria isolated from all four tissues, the defect in CL remodeling may not be responsible for complex I inhibition. However, the increased MLCL observed only in brain mitochondria suggests the ischemic conditions in the brain are unique, or more severe, than those of other organs. In addition, the finding that the ratio of MLCL/CL kept increasing following resuscitation indicates that ischemic alterations responsible for the increased MLCL still remain following resuscitation, providing the rationale for the inability of complex I to regain its activity in brain mitochondria. In contrast, no change in the content of MLCL observed in the heart, kidney and liver is consistent with no changes of CL following CA or resuscitation in these tissues
The loss of CL may be the direct cause of the decreased complex III activity following resuscitation as supported by the findings from studies using tafazzin knock-down mice (Dudek et al., 2016; Kiebish et al., 2013). Tafazzin is an enzyme involved in CL synthesis.
Downregulation of this enzyme resulted in decrease CL content and decreased complex III activity. However, the other ETC complexes remain relatively intact. Therefore, preserving CL may prevent the loss of complex III activity and improve survival. Our recent study showed that the administration of a CL targeting drug, SS-31, during resuscitation significantly increased rat survival time in this CA model (Zhang et al., 2018). The detailed mechanisms for the decrease in CL content and its role in ETC function in CA are under investigation.
Mitochondrial respiration is the main source for cellular ATP generation. Restoring this essential function may be the key to the successful resuscitation of each organ. Previous studies examined the responses of individual organs to ischemia/reperfusion and found various biochemical and physiological alterations in mitochondria.(Chen et al., 2008; Paradies et al., 2004). Despite abundant publications on the mechanism for mitochondrial alterations, their application to CA research has been limited. This limitation is mainly due to model-to-model variations. Particularly, the interaction between multiple organs, which is an important factor in CA pathology (Jiang et al., 2007), cannot be replicated in isolated organ studies.
Another unique feature of studying CA is that the primary outcome is not assessed through damage to individual organs, but overall survival. In this context, alterations observed in an organ after unresuscitatable heart damage is not important for therapeutic purposes. Our animal model, resulting in the absence of brain function and certain death within a few hours even with the resuscitated heart, closely mimics the outcome of the majority of CA patients. The unique alterations found in brain mitochondria compared to the alterations observed in other relatively less injured organs may be the key molecular targets in order to rescue brain function, which in turn may lead to enhanced survival in CA.
A limitation is that this study does not fully evaluate mitochondria. Our study focuses on mitochondrial ETC. However, it provides adequate evidence clarifying the role of mitochondria, particularly the ETC complexes and CL, in the pathophysiology of CA. In future direction, it would prove valuable to determine if the elucidated biochemical alterations in CA and CPB are preventable or reversible through the implementation of pharmacologic, or other, therapy. Ruling out the phenomena that are the final and irreversible results of ischemic organ damage would be an important stride in the endeavor.
In conclusion, this CA model is a novel animal survival model that closely and consistently mimics the severe ischemia/reperfusion injury of CA, allowing for detailed biochemical analysis focusing on mitochondria. Using this model, which represents fatally injured patients, we examined activities of mitochondrial ETC complexes and analyzed CL content. Our data show that mitochondria from the brain, heart, kidney, and liver have common complex I inactivation following CA. After resuscitation, only brain mitochondria undergo additional defects: decreased complex III activity and decreased CL content. These results suggest that brain mitochondria suffer reperfusion injury in addition to ischemic alterations resulting in diminished overall mitochondrial respiration following resuscitation. These defects specific to the brain may be the primary reason the brain is most susceptible to ischemia/reperfusion, leaving brain damage as the major cause for mortality in CA.
Supplementary Material
Highlights.
We found unique alterations in brain mitochondria in a rat model of CA.
Decreased complex I activity is a common ischemic defect following CA.
Resuscitation fails to normalize ischemic alteration in brain mitochondria.
Resuscitation causes additional damage to brain mitochondria only.
The unique alterations may be responsible for unrecovered brain damage.
Acknowledgments
Funding: This work was supported by the National Institutes of Health (RO1 HL067630).
Abbreviations:
- CA
cardiac arrest
- CPB
cardiopulmonary bypass
- ETC
electron transport chain
- CL
cardiolipin
- MLCL
monolysocardiolipin
- ROSC
return of spontaneous circulation
Footnotes
Declarations of interest: None
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Borutaite V, Toleikis A, Brown GC, 2013. In the eye of the storm: mitochondrial damage during heart and brain ischaemia. FEBS J 280, 4999–5014. [DOI] [PubMed] [Google Scholar]
- Chen Q, Moghaddas S, Hoppel CL, Lesnefsky EJ, 2008. Ischemic defects in the electron transport chain increase the production of reactive oxygen species from isolated rat heart mitochondria. Am J Physiol Cell Physiol 294, C460–466. [DOI] [PubMed] [Google Scholar]
- Dixon HB, McIntosh R, 1967. Reduction of methaemoglobin in haemoglobin samples using gel filtration for continuous removal of reaction products. Nature 213, 399–400. [DOI] [PubMed] [Google Scholar]
- Dudek J, Cheng IF, Chowdhury A, Wozny K, Balleininger M, Reinhold R, Grunau S, Callegari S, Toischer K, Wanders RJ, Hasenfuss G, Brugger B, Guan K, Rehling P,2016. Cardiac-specific succinate dehydrogenase deficiency in Barth syndrome. EMBO Molecular Medicine 8, 139–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang H, Meng F, Li W, Tong L, Qiao H, Sun X, 2007. Splenectomy ameliorates acute multiple organ damage induced by liver warm ischemia reperfusion in rats. Surgery 141, 32–40. [DOI] [PubMed] [Google Scholar]
- Kiebish MA, Yang K, Liu X, Mancuso DJ, Guan S, Zhao Z, Sims HF, Cerqua R, Cade WT, Han X, Gross RW, 2013. Dysfunctional cardiac mitochondrial bioenergetic, lipidomic, and signaling in a murine model of Barth syndrome. J Lipid Res 54, 1312–1325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Hoppel CL, 2013. Comprehensive approach to the quantitative analysis of mitochondrial phospholipids by HPLC-MS. J Chromatogr B Analyt Technol Biomed Life Sci 912, 105–114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Perales Villarroel JP, Zhang W, Yin T, Shinozaki K, Hong A, Lampe JW, Becker LB, 2016. The Responses of Tissues from the Brain, Heart, Kidney, and Liver to Resuscitation following Prolonged Cardiac Arrest by Examining Mitochondrial Respiration in Rats. Oxid Med Cell Longev 2016, 7463407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Yin T, Yin M, Zhang W, Shinozaki K, Selak MA, Pappan KL, Lampe JW, Becker LB, 2014. Examination of physiological function and biochemical disorders in a rat model of prolonged asphyxia-induced cardiac arrest followed by cardio pulmonary bypass resuscitation. PLoS One 9, e112012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paradies G, Petrosillo G, Pistolese M, Di Venosa N, Federici A, Ruggiero FM, 2004. Decrease in mitochondrial complex I activity in ischemic/reperfused rat heart: involvement of reactive oxygen species and cardiolipin. Circ Res 94, 53–59. [DOI] [PubMed] [Google Scholar]
- Roberts BW, Kilgannon JH, Chansky ME, Mittal N, Wooden J, Parrillo JE, Trzeciak S, 2013. Multiple organ dysfunction after return of spontaneous circulation in postcardiac arrest syndrome. Crit Care Med 41, 1492–1501. [DOI] [PubMed] [Google Scholar]
- Rustin P, 2008. Mitochondrial Respiratory Chain, In: Blau N, Duran M, Gibson KM(Eds.), Laboratory Guide to the Methods in Biochemical Genetics,Springer Berlin Heidelberg, Berlin, Heidelberg, pp. 265–286. [Google Scholar]
- Stub D, Bernard S, Duffy SJ, Kaye DM, 2011. Post cardiac arrest syndrome: a review of therapeutic strategies. Circulation 123, 1428–1435. [DOI] [PubMed] [Google Scholar]
- Stub D, Bernard S, Pellegrino V, Smith K, Walker T, Sheldrake J, Hockings L, Shaw J, Duffy SJ, Burrell A, Cameron P, Smit de V, Kaye DM, 2015. Refractory cardiac arrest treated with mechanical CPR, hypothermia, ECMO and early reperfusion (the CHEER trial). Resuscitation 86, 88–94. [DOI] [PubMed] [Google Scholar]
- Trounce IA, Kim YL, Jun AS, Wallace DC, 1996. [42]Assessment of mitochondrial oxidative phosphorylation in patient muscle biopsies, lymphoblasts, and transmitochondrial cell lines, Methods in Enzymology. Vol. 264,Academic Press, pp. 484–509. [DOI] [PubMed] [Google Scholar]
- Vognsen M, Fabian-Jessing BK, Secher N, Lofgren B, Dezfulian C, Andersen LW, Granfeldt A, 2017. Contemporary animal models of cardiac arrest: A systematic review. Resuscitation 113, 115–123. [DOI] [PubMed] [Google Scholar]
- Zhang W, Tam J, Shinozaki K, Yin T, Lampe JW, Becker LB, Kim J, 2018. Increased Survival Time With SS-31 After Prolonged Cardiac Arrest in Rats. Heart, Lung and Circulation. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.