

Abstract
Cholinesterase inhibitors have long been used in the treatment of Alzheimer’s Disease (AD) via the protection of acetylcholine levels. However, recent research has shown that the specific inhibition of butyrylcholinesterase (BChE) could better ameliorate symptoms within patients. In addition, it has recently been shown that selective inhibition of BChE can also significantly attenuate the toxicity and physiological effects of heroin. Currently, there are no specific and potent inhibitors of BChE approved for use in AD or heroin abuse. Through a combined use of in silico and in vitro screening, we have found three compounds with sub-50 nM IC50 values that specifically target BChE. These newly discovered BChE inhibitors can act as the lead scaffolds for future development of the desirably potent and selective BChE inhibitors.
Keywords: Butyrylcholinesterase, Acetylcholinesterase, Inhibitor, Pharmacophore, Docking
Graphical Abstract
Three potent and selective butyrylcholinesterase inhibitors were discovered through a combination of in silico and in vitro screening of the NCI/DTP Open Chemicals Repository.
Alzheimer’s Disease (AD) continues to be a pivotal public health issue.1 In one year, roughly one million people will be diagnosed with AD, nearly one per minute.1 During the progression of AD, cholinesterase activity increases reducing the amount of acetylcholine within the brain.2 The disruption of this cholinergic signaling has profound effects, as this signaling plays an important role in both learning and long-term memory.3, 4 With this pathology, cholinesterase inhibitors have played a vital role in the treatment of Alzheimer’s through the protection of acetylcholine levels.2, 5 These inhibitors provide a relief in symptoms through the reestablishment of cholinesterase levels, thus prolonging the time needed for those afflicted to receive more serious care (i.e. nursing home care).5, 6 Acetylcholinesterase (AChE) and butyrylcholinesterase (BChE) are the cholinesterases primary affected through AD.7 Both proteins are serine esterases with an ability to metabolize acetylcholine.7 While these two proteins share a similar functionality in the pathology of this disease, it does not make them equal as targets for therapeutics against AD. During the progression of AD, AChE activity levels decline by up to 85%, and the ratio of BChE/AChE can dramatically change from 1:5 to 11:1.7, 8 This reversal in activity between the two proteins implies that selective BChE inhibition is a more prudent strategy for raising acetylcholine levels within the brain, as non-specific binding with AChE could sequester drug away from our intended target, for little gain on ameliorating the decreased acetylcholine levels. Additionally, while the AChE in the brain may have lost activity, the enzyme is still present, and is associated with the forming amyloid-beta plaques found within AD affected patients, which could potentially exacerbate the drug sequestering issue.9
While BChE specific inhibition appears to be a potential benefit to AD treatment, physicians are currently not able to take advantage of this difference due to the availability of BChE specific pharmaceuticals. There have been four cholinesterase inhibitors approved by the FDA for treatment of AD: Rivastigmine, Galantamine, Tacrine, and Donepezil.10 Out of these four compounds, none of them display BChE selectivity. All four compounds show either mixed inhibition of the two cholinesterases (i.e. Rivastigmine, Galantamine, and Tacrine) or AChE specificity (i.e. Donepezil).9 Additionally, Tacrine production was discontinued in 2013 due to hepatotoxicity, limiting the choices possible for treating AD.11-13 Considering the possible benefits of BChE inhibition within AD, significant effort has been undergone to find highly specific inhibitors that retain high potency against BChE.9, 14-18
Along with positive results concerning AD, BChE inhibitors have also been recently shown to have protective properties against the toxicity and physiological effects of heroin.19 Due to heroin’s reliance on BChE for metabolism to its active and toxic state, 6-monoacetylmorphine (6-MAM), potent inhibition of this protein has been shown to protect even against LD100 doses of heroin in mice.19-22 This study was primarily done through the use of ethopropazine, a selective BChE inhibitor approved for use within Parkinson’s patients.23 However it is currently not marketed in the United States, due to manufacturer discontinuation.24, 25 Additionally, ethopropazine is a weak inhibitor of BChE with an IC50 of 210 nM.26 Despite its discontinuation, this BChE-specific inhibitor was chosen deliberately for this study, as any AChE inhibition could remove a pathway of removing the exceptionally toxic 6-MAM.20 These two indications make BChE an especially intriguing target, considering the impact of both AD and heroin abuse within the United States.27
To identify novel inhibitors for BChE, we have employed and improved upon several structural design methods previously employed to find BChE inhibitors.14 These methods would allow us to rank out compounds that were unlikely to be specific BChE inhibitors. This was accomplished by determining the binding pose of tacrine, a previously approved inhibitor and determining potential additions that would increase selectivity for BChE.14, 28 While tacrine is potent against BChE, it has no selectivity for the protein. This unselective binding with either protein primarily comes from its smaller size and flatter shape allowing it to fit even within the larger substituted aromatic residues seen within AChE (Fig. 1B). By modifying tacrine’s structure, we can take advantages of these differences in the two protein’s binding sites to introduce a steric clash within AChE, while potentially increasing the binding affinity with BChE through the addition of another hydrophobic interaction. To accomplish this, we employed the AutoDock Vina software to generate several binding poses of tacrine within the BChE crystal structure (RCSB:4BDS).28, 29 One of the generated binding modes displayed relative proximity to a hydrophobic pocket within BChE, that was not present in the AChE crystal structure due to a substituted tyrosine: AChE:Y337 (Fig. 1B) (RCSB: 4EY5).30 We utilized this difference through the introduction of two large hydrophobic features in the form of an additional chlorophenyl group, located on the cyclohexyl moiety of tacrine. The binding pose of this modified tacrine revealed a very similar pose to that of tacrine, however, the additional hydrophobic/aromatic addition had increased the predicted binding energy from −8.1 kcal/mol to −9.6 kcal/mol. Overlaying AChE reveals several clashes with the previously mentioned aromatic residues, specifically AChE:F297 and AChE: Y337 (Fig. 1D).
Fig. 1.

(A) Structure of Tacrine, along with its IC50for both BChE and AChE, denoting its indiscriminate binding with either protein. (B) Binding mode of Tacrine with BChE generated by Autodock Vina, primary interactions include π stacking interactions with W231 and F239 along with a hydrogen bond with G117. AChE:Y337, AChE:Y124 and AChE:F297 in red denoting changes between the two proteins’ binding sites. (C) Modified Tacrine structure includes an additional chlorophenyl group to extend further into the binding pocket, within BChE while simultaneously clashing with one of the previously mentioned aromatic residues within the AChE binding site. Orange toroid indicate aromatics, green spheres represent hydrophobic groups, and light blue spheres indicate hydrogen bond donators. (D) Binding mode of our modified tacrine structure, generated by VINA. This binding mode retains the interactions with W231 and F239 in the structure, but has a severe clash with both AChE:F297 and AChE:Y337, reducing its potential binding energy.31
Once we had validation that our new pharmacophore would filter for structures that would be more selective for BChE, it was then reconstructed using the Pharmit web-service, which was also employed to filter the NCI/DTP Open Chemical Repository to compounds that matched these features.32 Once the NCI/DTP library had been filtered, we were left with 275 compounds that were then docked using a modified version of Autodock Vina (i.e. SMINA) and the crystal structure for BChE (RCSB:4BDS).28 This fork of AutoDock Vina integrates a post-docking energy minimization of the ligand to determine a more accurate energy score of the protein/ligand complex.32, 33 The search area for SMINA was centered on the position of the tacrine binding site within the crystal structure.28 This same process was performed for the AChE crystal structure (RCSB: 4EY5).30 Once scored with SMINA, the affinity of these compounds varied from −11 to −8 kcal/mol for BChE and −9.8 to −6.6 kcal/mol for AChE. The difference between these binding values was then taken to rank these compounds by their selectivity for BChE.
After the compounds had been scored and ranked, the top-41 available compounds were ordered from the NCI. In vitro testing of the ordered compounds were performed in line with previously reported cholinesterase inhibition assays.14, 34, 35 Each of these compounds was weighed and diluted into a 500 μM solution with DMSO as the solvent. These solutions were then used in a single-concentration (5 μM) screening test against BChE using the Ellman’s esterase activity assay. To perform this assay, the 500 μM solution of the inhibitor was diluted 10:90 μ with a 5 nM BChE solution. This mixture was then subsequently diluted 15:135 μL with a 1 mM solution of Ellman’s reagent (DTNB/ATC) after a 10-minute incubation time with the inhibitor. This would give a final concentration of 5 μM of inhibitor, 450 pM of the enzyme, and 900 μM of the Ellman’s reagent with a 1% composition of DMSO. Progress of absorbance was measured at 450 nM for 30 minutes via a Tecan GeniosPro microplate reader. Each well’s change in absorbance was measured against control wells containing only the Ellman’s reagent and BChE.
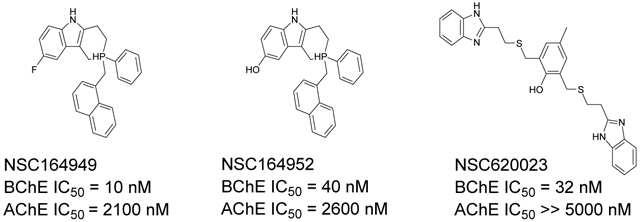
Six compounds with at least 75% inhibition compared to the control wells were selected for IC50 testing against BChE. The IC50 assay was also carried out in accordance with our previously published work.14 These compounds were serially diluted to eight different concentrations, which were then used in the same assay described above. Each compound tested had their IC50 measured in triplicate. The slope of absorbance for each well was then compared to the control containing only BChE and the Ellman’s reagent to determine their percentage of inhibition. For each set of serial dilutions, a non-linear regression was then performed to obtain the IC50 of each compound against each protein. These assays show that three of our top-six compounds, NSC620023, NSC164949, and NSC164952 have a sub-50 nM IC50 potency for BChE, with NSC164949 having the lowest at 10 nM.
To test the selectivity of these compounds, they were tested against AChE using the same IC50 assay. Of these top-six compounds, four showed no activity up to 5 μM while NSC164949 and NSC164952 exhibited an IC50 of approximately 2.1 and 2.6 μM respectively (Fig. 3). This set of IC50 measurements revealed three compounds with at least a 50:1 selectivity for BChE over AChE. Of the compounds tested, NSC620023 appears as a front runner for a BChE inhibitor due to its high selectivity (no inhibition of AChE at all) and high activity (low IC50) against BChE (Figs. 2 & 3, Table 1).
Fig. 3.

IC50 assays for NSC164949 and NSC164952 against AChE. These two compounds show an IC50 above 2 μM. the lowest IC50 values from our top-six compounds tested against AChE.
Fig. 2.

IC50 Ellman’s assays for top-6 compounds from the set of NCI/DTP compounds ordered. NSC164949, 164952, and 620023 show sub-100 nM inhibition making them competitive with the current compounds approved for cholinesterase inhibition.
Table 1:
Potency measurements for six selected compounds.
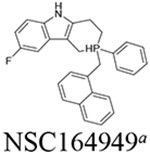
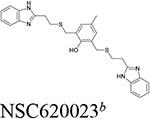
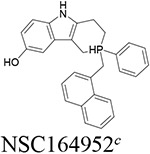
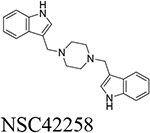
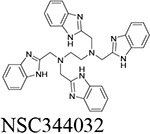
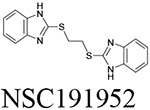
Through our in silico and in vitro studies, we were able to find three compounds with both nanomolar potency as well as high selectivity towards BChE. Both NSC620023 and NSC164949 display a synthesis of the sub-50 nM potency of tacrine and the selectivity of ethopropazine towards BChE. These improvements would allow for a lower dosage needed for BChE inhibition, without resorting to indiscriminate cholinesterase inhibitors. Out of these three compounds, NSC620023 presents as a top candidate to its selectivity, potency towards BChE, and relative ease of synthesis when compared to the phosphine containing NSC164949 and NSC164952. NSC620023 achieves this nanomolar potency using several π-stacking interactions with its dual benzimidazole moieties (Fig. 5A). These two fragments stack with both F329 and Y332. Additionally, the molecule has several favorable hydrophobic interactions with D70, W231, and L286 further within the binding site (Fig. 5A). The other two potent compounds, NSC164949 and NSC164952, are analogues of one another, with only one substituent group different between them. As such, the two compounds largely occupy the same binding pose within BChE (Fig. 5C & E). Both compounds utilize several π-stacking and hydrogen bonding interactions. F329 and W231 both have T-orientation π-stacking interactions with the indole moiety in the center of both compounds. Additionally, S198 and G117 interact with the secondary amine of the indole moiety, with a hydrogen donor bond and a hydrogen receiving bond, respectively (Fig. 5C & 5E). Due to the large size of both compounds, there are several hydrophobic interactions with: W82, W430, and A328 (Fig. 5C & 5E). The major difference between these two compounds comes from the substitution on the indole moiety from a halogen to an alcohol. This substitution in NSC164952 allows it to donate a hydrogen to the carbonyl in the S287 backbone, in addition to the other interactions seen with NSC164949 (Fig. 5E).
Fig. 5.

(A) Binding mode of NSC620023 with BChE, primary interactions with BChE include π-stacking interactions with F329 & Y332. (B) Clashing of NSC620023 with AChE substitutions F297 and Y337. (C) Binding of NSC164949 with BChE, primary interactions include π-stacking with F332 and W231, along with a hydrogen bond interaction with G117 and S198. (D) Clashing of NSC164949 with AChE residues F297 and Y337. (E) Binding of NSC164952 with BChE, primary interactions include π-stacking interactions with F329 and W231 with additional hydrogen bonds with G117, S198 and S287. (F) Clashing of NSC164952 with AChE residues F297 and Y337. All distances are in angstroms.31
The reasoning for the intense selectivity for these compounds for BChE over AChE relies on our previous intuition concerning the AChE binding site. Based upon the binding mode of these three compounds, the binding site of AChE would induce several steric clashes primarily from three large aromatic amino acids: Y124, F297, and Y337 (Figs. 4, 5B, 5D, & 5F). These three bulky amino acids are changed from smaller Q119, V288, and A328 residues in BChE. As we predicted, these changes severely restrict the size of the available binding site within AChE, thus inducing a steric penalty against our compounds (Figs. 4, 5B, 5D, & 5F).
Fig. 4.

Sequence comparison of BChE and AChE binding sites.36
In conclusion, through a combination of in silico/in vitro methodologies we have discovered three new, selective BChE inhibitors (NSC620023, NSC164959, and NSC164952) that exhibit improved potency versus ethopropazine without losing the high selectivity. These inhibitors primarily achieve this greater potency through a variety of different interactions with the protein, including π-stacking interactions and hydrogen bond donors. These compounds also clash with several aromatic residues within the AChE binding site, limiting their binding affinity and consequently enhancing their binding selectivity with BChE. These compounds represent two new potential scaffolds for use in future specific and potent BChE inhibitor development.
Supplementary Material
Acknowledgments
This work was supported in part by the funding of the Molecular Modeling and Biopharmaceutical Center at the University of Kentucky College of Pharmacy, the National Science Foundation (NSF grant CHE-1111761), and the National Institutes of Health via the National Institute on Drug Abuse (T32 DA016176) and National Center for Advancing Translational Sciences (UL1TR001998) grants. The authors also acknowledge the Computer Center at University of Kentucky for supercomputing time on a Dell Supercomputer Cluster consisting of 388 nodes or 4,816 processors.
Footnotes
Supporting Information
An additional table for molecular structures of the 41 compounds and their in vitro activity data obtained from the single-concentration screening.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Thies W, Bleiler L. 2012 Alzheimer's disease facts and figures. Alzheimer's & dementia: the journal of the Alzheimer's Association. 2012. [DOI] [PubMed] [Google Scholar]
- 2.Kihara T, Shimohama S. Alzheimer's disease and acetylcholine receptors. Acta Neurobiol Exp (Wars). 2004;64(1): 99–105. [DOI] [PubMed] [Google Scholar]
- 3.Woolf NT Acetylcholine, cognition, and consciousness. Journal of Molecular Neuroscience. 2006;30(1): 219–222. [DOI] [PubMed] [Google Scholar]
- 4.Woolf NT A structural basis for memory storage in mammals. Progress in neurobiology 1998;55(1): 59–77. [DOI] [PubMed] [Google Scholar]
- 5.Nordberg A, Svensson A-L. Cholinesterase inhibitors in the treatment of Alzheimer’s disease. Drug safety. 1998; 19(6): 465–480. [DOI] [PubMed] [Google Scholar]
- 6.Pepeu G, Giovannini MG. Cholinesterase inhibitors and memory. Chemico-biological interactions. 2010; 187(1-3): 403–408. [DOI] [PubMed] [Google Scholar]
- 7.Greig NH, Utsuki T, Ingram DK, et al. Selective butyrylcholinesterase inhibition elevates brain acetylcholine, augments learning and lowers Alzheimer β-amyloid peptide in rodent. Proceedings of the National Academy of Sciences. 2005; 102(47): 17213–17218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Giacobini E Cholinergic function and Alzheimer's disease. International Journal of Geriatric Psychiatry. 2003;18(S1): S1–S5. [DOI] [PubMed] [Google Scholar]
- 9.Giacobini E Selective inhibitors of butyrylcholinesterase. Drugs & aging. 2001;18(12): 891–898. [DOI] [PubMed] [Google Scholar]
- 10.Birks JS. Cholinesterase inhibitors for Alzheimer's disease. Cochrane Database Syst Rev. 2006(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Blackard WG Jr, Sood GK, Crowe DR, Fallon MBJocg. Tacrine: a cause of fatal hepatotoxicity? 1998;26(1): 57–59. [DOI] [PubMed] [Google Scholar]
- 12.Watkins PB, Zimmerman HJ, Knapp MT, Gracon SI, Lewis KWJJ. Hepatotoxic effects of tacrine administration in patients with Alzheimer's disease. 1994(271(13): 992–998. [PubMed] [Google Scholar]
- 13.Mehta M, Adem A, Sabbagh MJIJoAsd. New acetylcholinesterase inhibitors for Alzheimer's disease. 2012;2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhou S, Yuan Y, Zheng F, Zhan C-G. Structure-based virtual screening leading to discovery of highly selective butyrylcholinesterase inhibitors with solanaceous alkaloid scaffolds. Chemico-biological interactions. 2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.KoĆak U, Bras B, Knez D, et al. The Magic of Crystal Structure-Based Inhibitor Optimization: Development of a Butyrylcholinesterase Inhibitor with Picomolar Affinity and in Vivo Activity. J Med Chem. 2018;61(1): 119–139. [DOI] [PubMed] [Google Scholar]
- 16.Knez D, Coquelle N, PiĆlar A, et al. Multi-target-directed ligands for treating Alzheimer's disease: Butyrylcholinesterase inhibitors displaying antioxidant and neuroprotective activities. Eur J Med Chem. 2018;156: 598–617. [DOI] [PubMed] [Google Scholar]
- 17.KoĆak U, Bras B, Knez D, et al. Development of an in-vivo active reversible butyrylcholinesterase inhibitor. Sci Rep. 2016;6: 39495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bras B, KoĆak U, Turk S, et al. Discovery, biological evaluation, and crystal structure of a novel nanomolar selective butyrylcholinesterase inhibitor. J Med Chem. 2014;57(19): 8167–8179. [DOI] [PubMed] [Google Scholar]
- 19.Zhang T, Zheng X, Kim K, Zheng F, Zhan C-G. Blocking drug activation as a therapeutic strategy to attenuate acute toxicity and physiological effects of heroin. Sci Rep. 2018;8(1): 16762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Qiao Y, Han K, Zhan C-G. Reaction pathways and free energy profiles for cholinesterase-catalyzed hydrolysis of 6-monoacetylmorphine. Org Biomol Chem. 2014;12(14): 2214–2227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Qiao Y, Han K, Zhan C-G. Fundamental reaction pathway and free energy profile for butyrylcholinesterase-catalyzed hydrolysis of heroin. Biochemistry. 2013;52(37): 6467–6479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Selley DE, Cao C-C, Sexton T, Schwegel JA, Martin TJ, Childers SR. μ Opioid receptor-mediated G-protein activation by heroin metabolites: evidence for greater efficacy of 6-monoacetylmorphine compared with morphine. Biochem Pharmacol. 2001;62(4): 447–455. [DOI] [PubMed] [Google Scholar]
- 23.Chouinard G, Annable L, Ross-Chouinard A, Kropsky ML. Ethopropazine and benztropine in neuroleptic-induced parkinsonism. The Journal of clinical psychiatry. 1979. [PubMed] [Google Scholar]
- 24.Hansen L The Truven health MarketScan databases for life sciences researchers. Truven Health Ananlytics IBM Watson Health. 2017. [Google Scholar]
- 25.Hilts PJ. Drug Company Pleads Guilty To Deceit in Product Testing The New York Times. New York: The New York Times Company; 1995. [Google Scholar]
- 26.Atack JR, Yu Q-S, Soncrant TT, Brossi A, Rapoport SI. Comparative inhibitory effects of various physostigmine analogs against acetyl-and butyrylcholinesterases. Journal of Pharmacology and Experimental Therapeutics. 1989;249(1): 194–202. [PubMed] [Google Scholar]
- 27.Scholl L, Seth P, Kariisa M, Wilson N, Baldwin G. Drug and opioid-involved overdose deaths—United States, 2013–2017. Morbidity and Mortality Weekly Report. 2019;67(5152): 1419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Nachon F, Carletti E, Ronco C, et al. Crystal structures of human cholinesterases in complex with huprine W and tacrine: elements of specificity for anti-Alzheimer's drugs targeting acetyl-and butyryl-cholinesterase. Biochemical Journal. 2013;453(3): 393–399. [DOI] [PubMed] [Google Scholar]
- 29.Trott O, Olson AJ. AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading. J Comput Chem. 2010;31(2): 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cheung J, Rudolph MJ, Burshteyn F, et al. Structures of human acetylcholinesterase in complex with pharmacologically important ligands. Journal of medicinal chemistry. 2012;55(22): 10282–10286. [DOI] [PubMed] [Google Scholar]
- 31.Salentin S, Schreiber S, Haupt VJ, Adasme MF, Schroeder MJNar. PLIP: fully automated protein-ligand interaction profiler. 2015;43(W1): W443–W447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sunseri J, Koes DR. Pharmit: interactive exploration of chemical space. Nucleic Acids Res. 2016;44(W1): W442–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koes DR, Baumgartner MP, Camacho CJ. Lessons learned in empirical scoring with smina from the CSAR 2011 benchmarking exercise. J Chem Inf Model. 2013;53(8): 1893–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shetab-Boushehri SV. Ellman's method is still an appropriate method for measurement of cholinesterases activities. EXCLI journal. 2018;17: 798. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Ellman GL, Courtney KD, Andres V Jr, Featherstone RM. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol. 1961. ;7(2): 88–95. [DOI] [PubMed] [Google Scholar]
- 36.Waterhouse AM, Procter JB, Martin DM, Clamp M, Barton GJ. Jalview Version 2—a multiple sequence alignment editor and analysis workbench. Bioinformatics. 2009;25(9): 1189–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.