

Abstract
Adenoviral (Ad) vectors represent promising vaccine platforms for infectious disease. To overcome pre-existing immunity to commonly used human adenovirus serotype 5 (Ad5), vectors based on rare species or non-human Ads are being developed. However, these vectors often exhibit reduced potency compared with Ad5, necessitating the use of innovative approaches to augment the immunogenicity of the encoded antigen (Ag). To achieve this, we engineered model Ag, enhanced green fluorescent protein (EGFP), for targeting to the surface of host-derived extracellular vesicles (EVs), namely exosomes. Exosomes are nano-sized EVs that play important roles in cell-to-cell communication and in regulating immune responses. Directed targeting of Ag to the surface of EVs/exosomes is achieved by “exosome display,” through fusion of Ag to the C1C2 domain of lactadherin, a protein highly enriched in exosomes. Herein, we engineered chimpanzee adenovirus ChAdOx1 and Ad5-based vaccines encoding EGFP, or EGFP targeted to EVs (EGFP_C1C2), and compared vaccine immunogenicity in mice. We determined that exosome display substantially increases Ag-specific humoral immunity following intramuscular and intranasal vaccination, improving the immunological potency of both ChAdOx1 and Ad5. We propose that this Ag-engineering approach could increase the immunogenicity of diverse Ad vectors that exhibit desirable manufacturing characteristics, but currently lack the potency of Ad5.
Keywords: adenovirus, adenoviral, vaccine, exosome, extracellular vesicle, immunogenicity, viral vector, virus
Graphical Abstract
Introduction
The family Adenoviridae comprises double-stranded DNA viruses that include human species A to G, within the genus Mastadenovirus. To date, more than 100 human adenoviruses and more than 200 adenoviruses derived from non-human primates (NHPs) have been identified, the latter of which largely cluster phylogenetically with human adenoviral (Ad) species.1, 2, 3, 4 In the past decade, non-replicating Ad vectors have shown significant promise as vaccine delivery vehicles.5 Ads have an excellent safety profile following intramuscular (i.m.),6,7, intranasal (i.n.),8 or oral9,10 delivery in healthy adults, infants,11 and neonates,12,13 the elderly,7 and the immunocompromised.14 Ad genomes are easy to manipulate and they display desirable vaccine manufacturing attributes, with an estimated production time from selection to formulation and product filling of 11–13 weeks.15 In addition, procedures for clinical development such as regulatory guidance release testing for Ad vectors (e.g., vaccine potency, vector stability) already exist for the US Food and Drug Administration (FDA) and European Medicines Agency (EMA). Furthermore, Ad vaccine formulations maintain stability over time and are thermostable with minimal losses to immunogenicity under cold-chain free conditions.16,17 Collectively, these characteristics highlight the suitability of Ad vaccines for stockpiling, rapid response, and global preparedness efforts against influenza virus5,16,17 and other emerging pathogens.
Ad vaccines are known to elicit robust and sustained frequencies of antigen (Ag)-specific CD8+ T cell responses toward encoded transgene Ag in animal models.18, 19, 20 As a result, vaccines based on Ad vectors have been evaluated in clinical trials for infectious diseases, including influenza,6,7 respiratory syncytial virus (RSV),8 hepatitis C virus (HCV),21 and human immunodeficiency virus (HIV).22 Although human adenovirus serotype 5 (Ad5) is widely considered to be the most immunogenic Ad vector, high seroprevalence from natural infection may limit vaccine efficacy in humans.23,24 As a result, investigators have moved toward the development and evaluation of rare species human Ad vectors or those derived from NHPs. The use of rare species or non-human Ads, as well as fiber-pseudotyped or hexon chimeric Ad vectors, can circumvent pre-existing immunity to Ad5.23, 24, 25, 26, 27, 28 Although selected rare or non-human vectors display comparable immunogenicity to Ad5,28, 29, 30 a large proportion of alternative Ad vectors have substantially reduced immunogenicity,24,30 when compared to Ad5.28 Therefore, efforts to optimize and increase the inherent immunogenicity of Ad vectors, which have otherwise desirable characteristics (i.e., high titer growth), will be important in enabling cost-effective production and dose-sparing usage of commercially manufactured Ad vaccines.
A chimpanzee Ad vector, ChAdOx1, expressing highly conserved internal influenza Ags (nucleoprotein [NP] and matrix protein-1 [M1]) has undergone phase I clinical evaluation in humans as a universal influenza virus vaccine candidate, aimed at boosting Ag-specific cytotoxic T-lymphocyte (CTL) responses.6,7 In agreement with the known capability of Ad vaccines to induce excellent CD8+ T cell responses, ChAdOx1-NP+M1 has been found to elicit robust CD8+ T cells in healthy adults and older adults.6,7 However, induction of Ag-specific antibody (Ab) responses following ChAdOx1 vaccination in mice is impaired when compared with Ad5.1,31 Therefore, in this study, we used a molecular adjuvant approach called “exosome display” to improve the immunogenicity of Ag delivered by ChAdOx1.32
Exosomes are extracellular vesicles (EVs) released by almost all cells, which play critical roles in cell-to-cell communication and in the regulation of immune responses.33, 34, 35 In 1996, it was discovered that exosomes could present Ag to T cells and thereby modulate immune responses.36 As such, there is potential for manipulating their communications with the immune system to improve vaccine responses. Exosome display is the directed targeting of Ags to exosomes,37 achieved by fusion of Ag to a protein domain enriched on exosomes, such as the C1C2 domain from lactadherin.38,39 In animals, exosome display has been shown to increase Ab responses to cancer vaccine Ags ∼100- to 500-fold.40,41 Therefore, we reasoned that this approach could be exploited to increase the immunogenicity of Ag delivered by ChAdOx1 (referred to as ChAd throughout the figures), so that it exhibits a more robust immunogenic profile. Using enhanced green fluorescent protein (EGFP) as a model Ag, we employed this molecular adjuvant approach to tether EGFP_C1C2 fusion protein to the surface of host-derived EVs, including exosomes, comparing the immunogenicity of ChAdOx1 to Ad5 following i.m. and i.n. administration in mice.
Results
Fusion of EGFP to the C1C2 Domain of Lactadherin by Exosome Display Successfully Targets Ag to the Surface of EVs/Exosomes In Vitro
Prior to engineering Ad vector genomes expressing EV-targeted Ag expression cassettes, we wanted to demonstrate in vitro proof of concept for the molecular adjuvant approach using Ag fusion to the exosome-enriched C1C2 domain of lactadherin. We selected monomeric EGFP as a model Ag and generated C1C2-fusion and matched expression plasmids without C1C2 (EGFPΔ), on a pcDNA3.1 backbone (Figure 1A). pcDNA3.1-EGFP_C1C2 or non-targeted pcDNA3.1-EGFPΔ was transfected into Expi293F suspension cells, equivalent EGFP expression was confirmed by western blot (Figure S1A) and EGFP fluorescence-activated cell sorting (FACS) (Table S1), and exosomes were purified from the supernatant (SN) by ultracentrifugation (UC) at 100,000 × g, followed by a commercial size-exclusion chromatography (SEC) approach, validated for purification of exosomes.42
Figure 1.

In Vitro Confirmation of Exosome Display Targeting of Model Ag to Exosomes
(A) Schematic diagram showing expression plasmid design. Enhanced green fluorescent protein (EGFP) was engineered into pcDNA3.1SSmut_C1C2 to generate pcDNA3.1SSmut-EGFP_C1C2 (EGFP_C1C2) or pcDNA3.1SSmut-EGFPΔ (EGFPΔ), respectively. (B and C) Following transfection of Expi293F cells with EGFPΔ (B) or EGFP_C1C2 (C), exosomes were purified by ultracentrifugation (100,000 × g) and size exclusion chromatography (SEC). ELISAs using 20 μL of each SEC fraction confirmed enrichment of exosome markers, tetraspanins CD9, CD63, and CD81 on expected fractions (exosomes are 8–13) for EGFPΔ and EGFP_C1C2, and EGFP on the surface of EGFP_C1C2-transfected cells only. (D) SEC fractions enriched for CD9, CD63, and CD81 were validated using nanoparticle tracking analysis (NTA: Malvern Panalytical) to confirm ∼150-nm size and concentration. The mode value for size is shown, which is the size in nanometers of the majority of particles. (E) ELISA on 1 μg/well of pooled, purified exosomes to confirm the presence of tetraspanins CD9, CD63, and CD81 and the presence of EGFP on the surface of exosomes purified from EGFP_C1C2-transfected cells only. Data shown represent background subtraction of matched isotype control Abs. Positive controls for tetraspanins included exosomes purified from Du145 cells (provided by Prof. A Clayton and Dr. J. Webber, Cardiff University, Cardiff, UK). Wells coated with 1 μg/mL recombinant EGFP protein (Abcam, USA) were used as a positive control for EGFP Ab binding. Dashed line separates exosome samples from control samples. (F) Western blot on purified exosomes (Exo) and cleared cell lysate (CL) from the corresponding transfected cells to detect exosome-enriched endosomal protein ALIX and to show minimal detection of exosome-excluded endoplasmic reticulum protein GRP94.
Tetraspanins such as CD9, CD63, and CD81 are enriched in subpopulations of exosomes43 and can therefore be used to validate exosome-isolation methods.44 In this study, the successful isolation of exosomes in elution fractions 8–13 was confirmed by ELISA for tetraspanins CD9/CD63 and CD81 (Figures 1B and 1C). Analysis of identical fractions confirmed that exosomes purified from EGFP_C1C2-transfected cells had EGFP on the surface (8–13; Figure 1C), whereas exosomes purified from the SN of cells transfected with plasmids expressing EGFPΔ did not (8–13; Figure 1B). Fractions 8–13, corresponding with tetraspanin marker enrichment, were pooled, and purified exosomes were subsequently evaluated by nanoparticle tracking analysis (NTA) to confirm the particle titer and ∼150-nm size (Figure 1D).44 We further validated the pooled, purified exosomes by performing ELISAs for tetraspanins CD9/CD63 and CD81 in parallel with EGFP, again confirming that only exosomes derived from the SN of cells transfected with pcDNA3.1-EGFP_C1C2 had EGFP on the exosome surface (Figure 1E). In addition to confirming the purification of EVs with exosomal-like properties, we further validated these preparations by performing western blots for exosome-associated endosomal protein, ALIX, and the exosome-excluded endoplasmic reticulum marker GRP94 (Figure 1F). As expected, we detected the presence of ALIX in our purified exosome samples, but not in the cleared cell lysate (CL). In contrast, GRP94 was strongly detected in the cell lysate but was hardly detectable on exosomes, in agreement with previous studies.43
In summary, we confirmed that fusion of model Ag to the membrane-associated C1C2 domain of lactadherin could successfully tether EGFP to the surface of EVs, including exosomes, in vitro. These findings supported the engineering of C1C2 fusion Ag expression cassettes into non-replicating Ad vectors for subsequent in vivo expression of exosome display EGFP following vaccination.
EGFP Can Be Targeted to the Surface of EVs In Vitro following Infection of A549 Cells with an Ad Vector Expressing EGFP_C1C2
DNA encoding EGFP_C1C2 and non-targeted control EGFPΔ was engineered into a non-replicating Ad5 or ChAd vectored vaccine to test whether the exosome display approach could improve transgene-specific immunogenicity in vivo. We first confirmed that expression levels of EGFPΔ and EGFP_C1C2 were comparable following transduction of A549 and Chinese hamster ovary (CHO)-coxsackie and adenovirus receptor (CAR) cells with both Ad vectors (Figures S1B–S1E). To support our proof-of-concept data where we confirmed successful targeting of EGFP_C1C2 but not EGFPΔ to the surface of exosomes in vitro using accepted EV purification and validation methodology (Figure 1), we subsequently demonstrated that EGFP could be targeted to the surface of EVs derived from A549 cells following expression of EGFP_C1C2 by an Ad vaccine in vitro (Figure S2). A549 cells were infected with each Ad vector, and using precipitation-based purification, EVs were crudely purified from the SN of A549 cells grown in exosome-depleted fetal bovine serum (FBS) 72 h post-infection. When we performed validation ELISAs on the EV pellet to detect CD9/CD63 and CD81 in parallel with EGFP, we confirmed that only EVs precipitated from the SN of A549 cells infected with Ad5-EGFP_C1C2, but not Ad5-EGFPΔ, had EGFP on the surface of EVs. We were unable to detect an EGFP signal for the ChAd vector (data not shown); however, this would be consistent with its very low transduction efficiency in A549 cells relative to Ad5 (see Figures S1B and S1C). Although the ChAd vector efficiently infects hamster CHO-CAR cells (see Figures S1D and S1E), our validation ELISAs for CD9/CD63 and CD81 are for exosomes derived from human cells, and the Abs are specific for human tetraspanins. Therefore, we were unable to confirm these findings for the ChAd vector in vitro.
It is important to highlight that there are several technical challenges and caveats to using an in vitro system to demonstrate exosome display and release by Ad-infected cells. In order to purify sufficient quantities of exosomes for high-quality analysis that adhere to the minimal guidelines for the International Society for Extracellular Vesicles (ISEV),44,45 we purified exosomes from 100 to 250 mL of serum-free Expi293F SN following transfection with our pcDNA3.1-SS-EGFPΔ and EGFP_C1C2 expression constructs using ultracentrifugation and size-exclusion chromatography (as outlined in Figure 1). However, these large volumes would be impossible to infect with practical quantities of purified Ad viruses (MOI of 250 infectious units [IFU]/cell for 250 mL of suspension at 2 × 106/mL). In addition, we cannot confirm exosome display-based EGFP loading following expression from an Ad vector using Expi293F cells because this cell line supports the replication of E1-deleted Ad vectors, which would confound our results. Therefore, this assay was restricted to using A549 cells grown in media containing exosome-depleted FBS in a six-well plate format with duplicate volumes of 5 mL, resulting in very low yield of EVs/exosomes for analysis. Finally, the precipitation-based method used to isolate concentrated and detectable quantities of EVs from a such a small SN sample purifies a heterogeneous population of vesicles, including non-exosomal particles. Therefore, we think that the transfection of Expi293F cells with pcDNA-based vectors expressing EGFPΔ or EGFP_C1C2 is a more robust and technically feasible means of demonstrating proof of concept for the success of the exosome display approach in vitro.
Incorporation of the Exosome Display Model Ag EGFP into Human and Non-human Ad Vaccine Vectors Results in Increased Ag-Specific Humoral Immunity following i.m. Vaccination in Mice
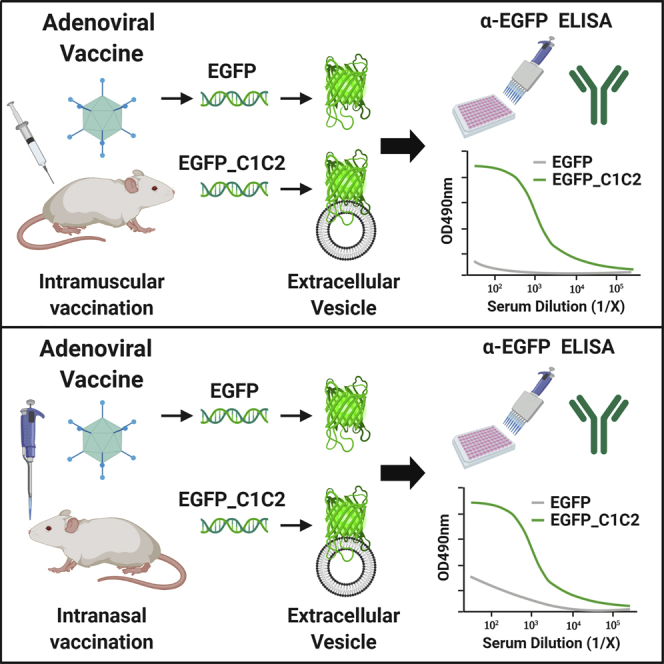
Following vaccination with non-replicating Ad5 or ChAd vectored vaccines encoding EGFP_C1C2 and non-targeted control EGFPΔ, EGFP protein should be expressed within cells infected with Ad5/ChAd following immunization and processed via classical Ag presentation pathways. However, we hypothesized that exosome-bound EGFP_C1C2 could also be released from host cells (via natural, endogenous exosome biogenesis pathways) and taken up by neighboring Ag-presenting cells (APCs), potentially augmenting immunogenicity. Ad5 vaccines are well established for their ability to elicit robust humoral immune responses directed toward encoded transgene Ags.20,30,46, 47, 48 Although the ChAd vaccine used in this study (ChAdOx1) can elicit potent T cell responses in animal models1,31,49 and humans,6,7 its ability to elicit high Ab titers in mice is impaired when compared with Ad5.1,31 We anticipated that the presentation of EGFP on the surface of EVs/exosomes could improve Ab responses to this Ag when encoded within the ChAd vector. Therefore, we performed a head-to-head comparison of the in vivo immunogenicity of ChAd-EGFPΔ and ChAd-EGFP_C1C2, as well as Ad5-EGFPΔ and Ad5-EGFP_C1C2, following i.m. or i.n. vaccination of mice.
We determined that EGFP-specific serum immunoglobulin (Ig)G Ab responses elicited following a single i.m. vaccination with an Ad vaccine were greatly improved when EGFP was fused to C1C2 (Figures 2A–2D, green boxes, green circles). Surprisingly, improvements in immunogenicity were not limited to ChAd, but were also detected for the highly immunogenic vector Ad5 (Figures 2A–2D), with geometric mean endpoint titers for Ad5-EGFP_C1C2 ∼10-fold greater than Ad5-EGFPΔ (Figure 2C, p < 0.0001). Differences in endpoint titers for ChAd were more pronounced than those observed for Ad5, with ChAd-EGFP_C1C2 responses ∼400-fold greater than those for ChAd-EGFPΔ (Figure 2C, p < 0.0001). These increases in immunogenicity were sustained at day 28 (D28) (Figures S3A–S3D), with geometric mean endpoint titers for Ad5-EGFP_C1C2 being ∼9-fold greater than Ad5-EGFPΔ (Figure S3C, p = 0.0079) and 363-fold greater for ChAd-EGFP_C1C2 compared with ChAd-EGFPΔ (Figure S3C, p = 0.0079).
Figure 2.

Expression of EGFP_C1C2 Fusion Ag by an Ad Vaccine Results in Improved Ag-Specific Humoral Immune Responses in Serum following i.m. Delivery in Mice
(A) Mice were vaccinated i.m. with 1 × 108 IFU Ad5-EGFPΔ (gray box), Ad5-EGFP_C1C2 (green box), or PBS (naive) in a final volume of 50 μL. Two weeks later, anti-EGFP IgG responses in sera were measured by ELISA using plates coated with 1 μg/mL recombinant EGFP protein. (B) Mice were vaccinated i.m. with 1 × 108 IFU ChAd-EGFPΔ (gray circle) or ChAd-EGFP_C1C2 (green circle), and ELISA assays were performed exactly as described in (A). Data show mean ± SEM (n = 10 mice/group) of duplicates, and plots are representative of two to three technical repeats and two independent repeat experiments (combined) with n = 5/group. (C) Endpoint titers represent the reciprocal dilution of the x intercept with the baseline (set to the mean plus 3 times the standard deviation [SD] of the mean of naive controls). Solid line indicates the geometric mean. Dashed line indicates starting dilution of sera (1:100), and therefore the lower limit of detection for the assay. Values below this line are estimated at half the input dilution (i.e., 1:50 dilution is estimated to represent the endpoint). (D) The area under the curve (AUC) represents the total peak area calculated from ELISA values, where the baseline was set to the mean plus 3 times the SD of the mean of naive controls. Samples where no AUC could be calculated were set arbitrarily to a value of 1.0. Line indicates the geometric mean. Statistical significance was determined using a non-parametric Mann-Whitney test. ****p < 0.0001. (E–J) Ag-specific isotype subclass Abs IgG1, IgG2a and IgG2b were also measured by ELISA using a starting dilution of 1:33. (E) Ad5 serum IgG1. (F) ChAd serum IgG1. (G) Ad5 serum IgG2a. (H) ChAd serum IgG2a. (I) Ad5 serum IgG2b. (J) ChAd serum IgG2b. Data show mean ± SEM (n = 5 mice/group) representative of two to three technical repeats performed in biological duplicates. Note that where SEM error bars are not visible, this is due to the error bar being shorter than the size of the symbol.
We also compared Ad vaccines expressing EGFPΔ or EGFP_C1C2 using an area under the curve (AUC) analysis (Figure 2D). We determined that AUC values were increased 10-fold for Ad5- and 817-fold for ChAd, respectively, when EGFP was fused to C1C2 compared with EGFPΔ (both p < 0.0001). These increases in immunogenicity were again sustained at D28, with AUC values for Ad5-EGFP_C1C2 being >7-fold greater than Ad5-EGFPΔ and 631-fold greater for ChAd-EGFP_C1C2 compared with ChAd-EGFPΔ (Figure S3D, both p = 0.0079). Although some mice in the day 14 (D14) ChAd-EGFPΔ group had IgG endpoint titers and AUC values that were below the limit of detection, the same mice had robust splenic T cell responses to EGFP (see below), suggesting that the lack of IgG response was not due to ineffective i.m. vaccination. Data presented in Figures 2A–2D and S3A–S3D agree with earlier findings regarding the reduced immunogenicity of ChAd relative to Ad5,1,31 with minimal EGFP-specific Ab responses detected following i.m. vaccination with ChAd-EGFPΔ (gray circles).
Delivery of Exosome Display EGFP by Ad5 or ChAd Vaccine Vectors Leads to Increases in Ag-Specific IgG Subclass Abs following i.m. Vaccination in Mice
In the disease context, it is well established that specific Ab isotypes, and IgG subclasses, elicited through vaccination using different vaccine platforms can contribute to protective efficacy. This can be due to the induction of protective Abs which preferentially engage specific Fcγ receptors (FcγRs) and mediate effector function-based clearance and subsequent protection from pathogen challenge, independently of neutralizing Abs.50, 51, 52, 53 In mice, such Abs include IgG isotype subclasses that have high-affinity interactions with activating FcγRs, namely IgG2a > IgG2b > IgG1.54,55 Although the vaccines described herein express a model Ag EGFP, for which no protective function can be measured, we wanted to investigate whether the exosome display molecular adjuvant approach could broaden or skew the profile of Ag-specific IgG subclasses elicited by vaccination, and to investigate whether the route of vaccine administration impacts this.
We performed EGFP-specific IgG subclass ELISAs, detecting mouse IgG1, IgG2a, and IgG2b Abs in the serum (Figures 2E–2J). Similar ELISAs were performed for isotypes IgG3, IgA, and IgM, but responses were very low or undetectable (data not shown), in agreement with previous studies.56 As anti-EGFP responses elicited by ChAd-EGFPΔ were so low, it is difficult to assess the isotype subclass distribution (Figures 2F, 2H, and 2J). However, we determined that delivery of EGFP_C1C2 by both Ad5 and ChAd vaccines resulted in increased responses to EGFP across all IgG subclasses measured. There were subtle differences in the skewing of responses between the Ad5 and ChAd vaccines. Ad5 induced EGFP-specific responses in the order IgG2a > IgG1 > IgG2b, whereas ChAd induced responses that were IgG2a > IgG2b > IgG1. This differential pattern of IgG subclass skewing for Ad5- and chimpanzee Ad-based vaccines is consistent with previous studies.46,56,57 The predominance of IgG2a responses following i.m. vaccination was reassuring, as it suggested that fusion of a disease-specific Ag to C1C2, combined with Ad-vectored delivery, may result in IgG responses of a Th1 phenotype. Such Abs would preferentially interact with murine activating FcγRs and should therefore be capable of mediating effector functions in vivo. Th1 responses have been shown to be essential for protection from viral respiratory pathogens in animal models,58,59 emphasizing the relevance of this approach for vaccines for viral diseases.
Intranasal Vaccination with Human and Non-human Ad Vaccine Vectors Expressing Exosome Display Model Ag EGFP Results in Increased Ag-Specific Humoral Immunity in Mice
Ad vaccines are gaining interest as vaccine delivery vehicles that might be capable of eliciting both systemic and mucosal immunity for respiratory pathogens such as seasonal and pandemic influenza virus60 and RSV.8,61 Therefore, we wanted to establish whether the exosome display molecular adjuvant approach could also increase the Ag-specific immunogenicity of Ad5 and ChAd following i.n. vaccination in mice.
In agreement with findings following i.m. vaccination, EGFP-specific serum IgG responses were also increased following i.n. administration of Ad5- and ChAd- expressing EGFP-C1C2, when compared with control EGFPΔ (Figures 3A and 3B). Geometric mean endpoint titers were increased ∼45-fold (p = 0.0001) and 30-fold (p = 0.0051) for Ad5 and ChAd, respectively, when EGFP was fused to C1C2 (Figure 3C). Endpoint titers at D28 following i.n. immunization maintained improvements of ∼21-fold for Ad5-EGFP_C1C2 compared with Ad5-EGFPΔ (Figures S4A and S4C, p = 0.0079) and 65-fold for ChAd-EGFP_C1C2 (Figures S4B and S4C, p = 0.0159). Similarly, D14 AUC values were increased ∼45-fold (p = 0.0001) and 40-fold (p = 0.0051) for Ad5/ChAd-EGFP_C1C2 compared with EGFPΔ (Figure 3D). Again, AUC responses at D28 were still increased for Ad5-EGFP_C1C2 compared with Ad5-EGFPΔ (15-fold, p = 0.0079), and responses were more pronounced for ChAd-EGFP_C1C2, which was increased 112-fold over ChAd-EGFPΔ (Figure S4D, p = 0.0079). We identified three mice in the D14 EGFPΔ groups with no detectable IgG ELISA responses following i.n. vaccination, but with a detectable splenic T cell response (see below).
Figure 3.

Expression of EGFP_C1C2 Fusion Ag by an Ad Vaccine Results in Improved Ag-Specific Humoral Immune Responses in Serum following i.n. Delivery in Mice
(A) Mice were vaccinated i.n. with 1 × 108 IFU Ad5-EGFPΔ (gray box), Ad5-EGFP_C1C2 (green box), or PBS (naive) in a final volume of 50 μL of PBS. Two weeks later, anti-EGFP IgG responses in sera were measured by ELISA using plates coated with 1 μg/mL recombinant EGFP protein. (B) Mice were vaccinated i.n. with 1 × 108 IFU ChAd-EGFPΔ (gray circle) or ChAd-EGFP_C1C2 (green circle), and assays were performed exactly as described in (A). Data show mean ± SEM (n = 5–10 mice/group) of duplicates and are representative of two independent experiments (data combined), as well as technical repeats. (C) Endpoint titers represent the reciprocal dilution of the x intercept with the baseline (set to the mean plus 3 times the standard deviation [SD] of the mean of naive controls). Line indicates the geometric mean. Dashed line indicates starting dilution of sera (1:100); values below this line are estimated at half the input dilution (i.e., 1:50 dilution is estimated to represent endpoint). Line indicates the geometric mean. (D) The area under the curve (AUC) represents the total peak area calculated from ELISA values, where the baseline was set to the mean plus 3 times the SD of the mean of naive controls. Samples where no AUC could be calculated were set arbitrarily to a value of 1.0. Solid line indicates the geometric mean. Statistical significance was determined using a non-parametric Mann-Whitney test. **p < 0.01, ***p < 0.001. (E–J) Ag-specific isotype subclass Abs IgG1, IgG2a, and IgG2b were also measured by ELISA. (E) Ad5 serum IgG1. (F) ChAd serum IgG1. (G) Ad5 serum IgG2a. (H) ChAd serum IgG2a. (I) Ad5 serum IgG2b. (J) ChAd serum IgG2b. Data show mean ± SEM (n = 5–10 mice/group) representative of two to three technical repeats performed in biological duplicates. Note that where SEM error bars are not visible, this is due to the error bar being shorter than the size of the symbol.
We also measured the IgG subclass of the EGFP-specific serum Ab response following i.n. vaccination with Ad5 or ChAd vectors (Figures 3E–3J). Results obtained following i.n. administration of Ad vectors expressing EGFP_C1C2 largely mirrored the subclass distribution observed following i.m. vaccination. Again, Ad5 elicited EGFP-specific IgG responses, which were largely IgG2a > IgG1 > IgG2b, whereas ChAd elicited responses, which were IgG2a > IgG2b > IgG1.
Exosome Display of EGFP Delivered by Ad5- or ChAd-Based Vaccines Elicits Increased Mucosal IgG Responses following i.n. Vaccination in Mice
In addition to serum, we also collected bronchoalveolar lavage (BAL) and lung tissue from mice vaccinated i.n, to quantify local humoral and cellular immune responses to the vaccine Ag EGFP. Ag-specific IgG responses in BAL were measured by ELISA (Figures 4A–4D). In agreement with serum IgG responses following i.m. and i.n. vaccination (see Figures 2A–2D and 3A–3D), we also detected increased IgG responses in the BAL when EGFP was fused to C1C2. Again, the improvement in immunogenicity was observed for both Ad5 and ChAd vectors. IgG endpoint titers were increased ∼42-fold for Ad5-EGFP_C1C2 over Ad5-EGFPΔ (p = 0.0002), and ∼30-fold for ChAd when EGFP was fused to C1C2 (p = 0.0027). When we performed similar ELISAs on BAL at D28 post-immunization (Figures S4E–S4H), geometric mean endpoint titers for Ad5-EGFP_C1C2 were still increased 11-fold when compared with Ad5-EGFPΔ (p = 0.0079). Ag-specific IgG responses in the BAL were also increased 20-fold at D28 for ChAd-EGFP_C1C2 compared with ChAd-EGFPΔ (p = 0.0159). Similarly, AUC at D14 values were increased ∼63-fold (p = 0.0002) and 47-fold (p = 0.002) for Ad5 and ChAd expressing EGFP_C1C2 compared with EGFPΔ, respectively. Again, AUC values at D28 post-immunization were 10-fold greater for Ad5-EGFP_C1C2 compared with Ad5-EGFPΔ (p = 0.0079) and 32-fold greater for ChAd-EGFP_C1C2 versus ChAd-EGFPΔ (p = 0.0159).
Figure 4.

Expression of EGFP_C1C2 Fusion Ag by an Ad Vaccine Results in Improved Ag-Specific Humoral Immune Responses in BAL following i.n. Delivery in Mice
(A) Mice were vaccinated i.n. with 1 × 108 IFU Ad5-EGFPΔ (gray box), Ad5-EGFP_C1C2 (green box), or PBS (naive) in a final volume of 50 μL of PBS. Two weeks later, anti-EGFP IgG responses in BAL were measured by ELISA using plates coated with 1 μg/mL recombinant EGFP protein. (B) Mice were vaccinated i.n. with 1 × 108 IFU ChAd-EGFPΔ (gray circle) or ChAd-EGFP_C1C2 (green circle), and assays were performed exactly as described in (A). Data show mean ± SEM (n = 5–10 mice/group) representative of two independent experiments with biological duplicates (data combined). (C) Endpoint titers represent the reciprocal dilution of the x intercept with the baseline (set to the mean plus 3 times the standard deviation (SD) of the mean of naive sera controls). Solid line indicates the geometric mean. Dashed line indicates starting dilution of sera (1:33); values below this line are estimated at half the input dilution (i.e., 1:17 dilution is estimated to represent endpoint). (D) The area under the curve (AUC) represents the total peak area calculated from ELISA values where the baseline was set to the mean plus 3 times the SD of the mean of naive sera controls. Samples where no AUC could be calculated were set arbitrarily to a value of 1.0. Statistical significance was determined using a non-parametric Mann-Whitney test. **p < 0.01, ***p < 0.001.
In conclusion, the exosome display molecular adjuvant approach, achieved by fusion of EGFP to the C1C2 domain of lactadherin, allowed increases in systemic and mucosal Ag-specific IgG immune responses elicited by ChAd and Ad5. Importantly, this approach resulted in ChAd-EGFP_C1C2 eliciting humoral immune responses to EGFP that were comparable to the baseline immunogenicity of Ad5 (EGFPΔ), a gold-standard benchmark in the field for Ad vector immunogenicity.
Fusion of Model Ag EGFP to C1C2 and Delivery Using Ad5/ChAd Vaccine Vectors Does Not Negatively Impact Systemic Ag-Specific Cellular Immunity in Mice
As Ad vectors are well established for their ability to induce potent T cell responses to transgene Ag,20,28,30 we wanted to assess whether fusion of EGFP to the C1C2 domain affected the induction of Ag-specific cellular immune responses. We measured T cell responses in the spleen by interferon (IFN)-γ enzyme-linked immunospot (ELISpot) following i.m. (Figures 5A and 5B) and i.n. (Figures 5C and 5D) vaccination with each vector, EGFPΔ and EGFP_C1C2. T cell responses to the entire EGFP peptide pool (Figures 5A and 5C) and to the H2-Kd-restricted CD8+ T cell epitope (Figures 5B and 5D) followed a similar trend, and robust responses were detected for both stimulation conditions. Following i.m. vaccination with the ChAd vaccines, we observed an increase in ELISpot responses when comparing EGFP_C1C2 with EGFPΔ; however, this was not statistically significant. Importantly, the robust cellular immune responses associated with Ad vectors were not impaired by targeting EGFP to the surface of EVs/exosomes for either vector, Ad5 or ChAd. As expected, ELISpot responses in the spleen following i.n. vaccination were lower than responses detected following i.m. vaccination. Ag-specific T cell responses to the EGFP200–208 CD8+ T cell epitope were again increased when comparing EGFP_C1C2 with EGFPΔ following delivery by the ChAd vaccine platform relative to Ad5, although this was not statistically significant.
Figure 5.

Ag-Specific T Cell Responses Are Not Impaired following i.m. or i.n. Delivery of Ad Vectors Expressing Exosome Display EGFP in Mice
(A and B) Mice were vaccinated i.m. with 1 × 108 IFU Ad5-EGFPΔ (gray box), Ad5-EGFP_C1C2 (green box), ChAd-EGFPΔ (gray circle), ChAd-EGFP_C1C2 (green circle), or PBS (naive) in a final volume of 50 μL of PBS. Two weeks later, anti-EGFP IFN-γ+ T cell responses in the spleen were measured by ELISpot. Splenocytes were stimulated overnight with an EGFP peptide pool spanning the entire EGFP Ag (EGFP PepMix) (A) or with the H2-Kd-restricted peptide EGFP200–208 (B). Solid line indicates median (n = 5 mice/group). (C and D) Figures show anti-EGFP IFN-γ+ T cell responses in the spleen detected by ELISpot stimulating with EGFP PepMix (C) or EGFP200-208 (D) following i.n. vaccination, using identical vaccines as described above, administered in a 50-μL volume. Line indicates median (n = 5 mice/group). (E–J) Ag-specific T cell responses in the lung (E–G) and BAL (H–J) were quantified using cell surface staining with ICS by flow cytometry. Cells were stimulated with a peptide pool spanning the entire EGFP Ag (EGFP PepMix), DMSO (negative control), or PMA/ionomycin (positive control). Legend indicates cytokine-secreting cells as a percentage of total, live CD8+ T cells. CD4+ responses were below the limit of detection for all cytokines analyzed (data not shown). Solid lines indicates geometric mean and represent n = 3–5 mice/group for lung and BAL, with the exception of n = 1 for BAL PBS due the low numbers of cells present in the BAL of PBS-treated mice. The lower limit of detection frequency was determined by the equation: [(1/minimum CD8+ count across DMSO and GFP stimulations) × 100]. Statistical significance was determined using a non-parametric Kruskal-Wallis test with a post hoc Dunn’s multiple comparison test. *p < 0.05, NS = p > 0.05. IFN-γ, interferon-γ; ELISpot, enzyme-linked immunospot assay; SFU, spot forming unit; DMSO, dimethyl sulfoxide, PMA, phorbol 12-myristate 13-acetate; LLD, lower limit of detection.
Fusion of Model Ag EGFP to C1C2 Improves Local Cellular Immunity in the Lung for ChAd but Not Ad5 following i.n. Vaccination in Mice
As previously stated, Ad vaccines are a very attractive platform vector for mucosal administration, particularly for vaccination against respiratory pathogens.8,60,61 We confirmed that targeting EGFP to the surface of EVs/exosomes through fusion to the C1C2 domain of lactadherin could improve EGFP-specific IgG responses in the BAL following i.n. vaccination (Figures 4A–4D). Therefore, we wanted to assess whether T cell responses in the lung and in the BAL were improved with the EGFP_C1C2 transgene cassette compared with EGFPΔ, when delivered by Ad5 or ChAd vaccines (Figure 5E–5J). Because the ELISpot we performed does not permit distinction between CD4+ or CD8+ T cell responses (with the exception of the EGFP200–208 CD8+ restricted epitope), we performed flow cytometry with intracellular cytokine staining (ICS) to phenotype cells isolated from BAL and whole perfused lung. An example of the gating strategy is presented in Figure S5. To allow detection of CD4+ and CD8+ T cell responses, we stimulated cells with the entire EGFP peptide pool, rather than the CD8+ restricted epitope. We did not detect CD4+ T cell responses (data not shown). CD8+ T cell responses in both the lung and BAL were largely similar for Ad5, with no significant difference in IFN-γ, tumor necrosis factor (TNF)-α or interleukin (IL)-2 production when comparing EGFP_C1C2 and EGFPΔ (Figures 5E–5J). However, we again observed consistent trends toward increased T cell responses with EGFP_C1C2 for ChAd, with significantly increased responses measured for TNF-α production in CD8+ lung T cells (Figure 5G), and IFN-γ production in BAL CD8+ T cells (Figure 5H). In the lung, the TNF-α+ response from CD8+ T cells increased ∼7-fold for ChAd-EGFP_C1C2 compared with ChAd-EGFPΔ (p = 0.045). In the BAL, CD8+ T cells secreting IFN-γ for ChAd-EGFP_C1C2 were increased ∼7.8-fold (p = 0.0475) over ChAd-EGFPΔ.
Discussion
It is well documented that pre-existing anti-vector immunity can negatively impact the immunogenicity of Ad vaccines,25,62, 63, 64 particularly those based on common human serotypes such as Ad5. In an effort to overcome this, there has been an expansion of interest in recent years in developing rare species of human Ad vectors (i.e., Ad26),22 or vectors derived from NHPs, including chimpanzees (i.e., ChAd68,57 ChAdOx1,32 ChAd6365). The potency of Ad-based vectors in eliciting transgene-specific CD8+ T cell responses is well established.20,28,30,66 However, the ability to elicit transgene-specific responses, particularly Abs, can vary from vector to vector, or with route of vaccine administration.56,57 This emphasizes the importance of choosing the optimal Ad vaccine platform and vaccination regimen for a specific disease of interest. Although selected Ad vectors display comparable cellular or humoral immunogenicity to the gold-standard Ad5 vector (i.e., ChAd330 or ChAd6328), the vast majority of new Ad platforms exhibit immunological potencies that are reduced ∼100 to 1,000-fold relative to Ad5.28,30 The precise mechanistic factors that contribute to these inherent differences in immunogenicity are not conclusive, but it is considered that a combination of factors could affect the differential immunogenicity of various Ad vectors. These factors include (1) differences in receptor, co-receptor, and/or in vivo tissue tropism;1,31,67 (2) the efficiency of viral uncoating and trafficking to the nucleus;30,68,69 (3) the route of vaccine administration;57 (4) differences in the innate immune sensing of Ad particles30; as well as (5) the level and persistence of Ag following Ad vaccination.20,47,48,64,70
The receptor usage and in vitro entry mechanism of Ad5, mediated via binding to CAR and internalization via cellular integrins, is well established.71, 72, 73 In addition, the in vivo tropism of Ad5 in mice following intravenous (i.v.) administration has been very well studied, and we have substantial information regarding the interacting cell types that contribute to immune activation.23,74, 75, 76, 77, 78 In contrast, there is a paucity of information in the field regarding the precise cellular interactions that occur following i.m. or i.n. administration with Ad5, and how these interactions contribute to immunogenicity or protective efficacy.5 Consequently, we have extremely limited insight into how rare species or non-human Ad vectors engage with specific APCs or non-lymphoid cells in vivo at immunological priming sites, and how these interactions, in conjunction with innate activation, shape adaptive immune responses.
The in vitro entry receptor for ChAdOx1 has not been characterized. It has been proposed that it can use CAR, in addition to another as yet unidentified receptor on the surface of CHO-K1.1 Evidence suggests that persistent Ag expression both within draining lymph nodes (dLNs) and cells within nonlymphoid compartments,20,47 combined with minimal induction of innate immunity (namely pathways driven by type I IFN and/or STING signaling),30,56 are defining characteristics of potently immunogenic Ad vectors.30,79 In support of this, it has been shown that Ad5 expresses transgene Ag more efficiently in dLNs than does ChAdOx1 following i.m. vaccination,1 potentially offering an explanation for the increased immunogenicity of Ad5 relative to ChAdOx1. As a result, future studies that aim to comprehensively characterize the basic biology of rare species or non-human Ad vectors will be invaluable in deciphering the underlying mechanisms and interactions that result in robust immunogenicity.67 The identification of defined entry receptors, the characterization of Ad virion pathogen-associated molecular patterns (PAMPs) that trigger differential innate signaling pathways, as well as determining the factors that contribute to sustained transgene expression in vivo will enable genetic engineering of optimal platform vectors that elicit a distinct phenotype of protective immunity, tailored to specific disease targets.
Nonetheless, in the absence of detailed information regarding the in vivo kinetics of novel Ad platforms, alternative efforts are currently being undertaken to improve the immunogenicity of existing Ad vectors. One way this can be achieved is through the incorporation of genetic adjuvants to increase immune stimulation,80,81 or recognition of the transgene Ag delivered by a non-replicating Ad vector. In this study, we have taken an approach to increase the dissemination of transgene Ag encoded within non-replicating Ad vectors (i.e., EGFP) to improve immune recognition and, subsequently, Ag-specific immune responses. The previously described exosome display approach involves the Velcro-like tethering of Ag to the surface of host-derived EVs/exosomes,37 which we validated in vitro using a model Ag, EGFP. However, by using non-replicating Ad vectors to deliver and express the exosome-targeted EGFP_C1C2 and non-targeted control Ag cassettes in vivo, we anticipated that exosome tethering of EGFP could take place following vaccination. In this scenario, EGFP would be expressed within cells that take up the Ad vaccine following immunization, and expressed Ag would be processed through classical major histocompatibility complex (MHC) I presentation pathways, resulting in potent CD8+ T cell responses. However, exosome surface-bound EGFP_C1C2 could also be released from host cells and taken up by neighboring APCs, thus creating multiple waves of immune stimulation and potentially augmenting humoral and cellular immunogenicity. We hypothesized that this increased Ag dissemination could be used as a general approach to improve the potency of ChAd, which has weaker immunogenicity than does Ad5.
We confirmed that this approach increased humoral immune responses in serum following vaccination, without negatively impacting the potency of cellular immunity. Importantly, the adjuvanting effects were observed following i.m. and i.n. vaccination, for both Ad5 and ChAd, and differences were maintained 28 days following immunization. Many Ad vectors are being investigated for mucosal delivery to provide protection against respiratory pathogens,49,61 including in the development of influenza virus vaccines.60,82 Ideally, such a vaccine should elicit high levels of cross-reactive Ab and T cell responses following a single vaccination. Recombinant Ad vectors are very well suited to the development of a universal influenza virus vaccine, due to their egg-independent production, rapid manufacturing capacity, thermostability, and safety in young, old and immunocompromised individuals.5,15 With this in mind, we confirmed that both systemic (serum) and mucosal (BAL) IgG responses were substantially improved following i.n. vaccination for both Ad5 and ChAd vectors when EGFP was fused to C1C2 (D14 and D28). Importantly, we determined that these Ab responses displayed a predominantly IgG2a response in mice. In the context of influenza virus, Abs of this subclass which recognize conserved epitopes on the stalk domain of hemagglutinin (HA), have been shown to play a crucial role in providing Fc-mediated protection from influenza virus challenge through effector function-based clearance mechanisms.50, 51, 52 Therefore, our data suggest that combining an influenza virus, exosome display fusion Ag with Ad-mediated vaccination in the future may increase the magnitude of Ag-specific responses and enhance protective efficacy. Increases in the magnitude of the Ab response could be exploited to maximize immunogenicity in specific populations, such as the elderly, in which immunosenescence is known to negatively impact vaccine effectiveness.83,84 Improvements in immunological potency could also permit Ad vector dose sparing,79 leading to reduced manufacturing costs and increased availability of vaccine doses during a pandemic.
As stated, recombinant Ad vectors represent a promising platform for development, as vaccines for infectious diseases and are increasingly undergoing clinical evaluation.85 A large number of human and non-human Ad serotypes with low seroprevalence in humans are available. This could facilitate their use in heterologous prime-boost or sequential immunization vaccination regimens, thereby bypassing issues related to pre-existing Ad immunity. This emphasizes the importance of continuing to evaluate and develop novel Ad vectors as a plug-and-play, easily customizable vaccine platform.
In this study, we have shown that fusion of model Ag EGFP to the C1C2 domain of lactadherin can dramatically improve the humoral immune responses of a chimpanzee Ad vector, ChAdOx1, an Ad platform that is currently being evaluated in phase I human clinical testing.6,7 The increased immunogenicity we observed for ChAd-EGFP_C1C2 resulted in serum Ab and mucosal (i.e., BAL) responses that were comparable to, or better than, the potently immunogenic Ad5 vector (EGFPΔ), following both i.m. and i.n. vaccination. We also observed no impairment to CD8+ T cell responses, and noted improvements in cellular immune responses elicited by ChAd in the lung, when EGFP was fused to C1C2. These data represent the first report of using the exosome display molecular adjuvant approach to improve the immunogenicity of a non-human ChAd (or any non-Ad5-based) vector. Therefore, this strategy could represent a general approach to improve the immunogenicity of other rare species or non-human Ad vectors that have weak humoral immunogenicity, but otherwise desirable stability and manufacturing characteristics.
Materials and Methods
Plasmid Construction and DNA Cloning
An expression plasmid containing the lipid-binding C1C2 domain of lactadherin Mfge8 and a C-terminal 6XHIS tag, pcDNA3.1-SS_C1C2,37,39, 40, 41,86 was kindly provided by Dr. Clotilde Théry (INSERM, France). EGFP was amplified from pEGFP-C1 (Clontech) by PCR using primers to introduce engineered restriction sites HindIII at the 5′ terminus and AgeI-HindIII at the 3′ terminus. PCR fragments were digested with HindIII or AgeI/HindIII and ligated into HindIII-digested pcDNA3.1-SS_C1C2 to generate the C1C2-fusion construct pcDNA3.1-SS-EGFP_C1C2, or AgeI/HindIII-digested pcDNA3.1-SS_C1C2 to generate C1C2-deleted pcDNA3.1-SS-EGFPΔ (Figure 1A).
Adenovirus Genome Engineering and Vaccine Production
In order to facilitate the subsequent generation of recombinant Ad vectors for Ad5 and chimpanzee adenovirus ChAdOx16,32 genomes encoding C1C2-fusion EGFP (EGFP_C1C2) and controls lacking the C1C2 domain (EGFPΔ), silent mutations were introduced to remove existing PmeI and PacI sites in the signal sequence (SS) within pcDNA3.1-SS-EGFP constructs to create pcDNA3.1-SSmut derivatives. The entire Ag cassette was then amplified by PCR to introduce terminal NotI restriction sites and the Ag fragment was cloned into NotI-digested entry vector (pENTR4) for subsequent homologous recombination with E1/E3-deleted Ad backbones using Gateway technology (Life Technologies, Carlsbad, CA, USA), as previously described.32 Adenoviruses were linearized by PacI/PmeI to release the infectious viral genome (Ad5/ChAd) and transfected into TRex293 cells. Viruses were amplified, purified by two rounds of cesium chloride banding, and titrated at the Jenner Institute Viral Vector Core Facility (University of Oxford, Oxford, UK), as previously described.6,32,87 To confirm that transgene expression was comparable between EGFPΔ and the fusion construct EGFP_C1C2 when expressed by an Ad vector, A549 or CHO-CAR cells were infected with each Ad5 or ChAd vector at the same multiplicity of infection (MOI) of 250 IFU/cell. EGFP expression was quantified 22 h post-transduction by flow cytometry (Figures S1A–S1D).
Cells and Culture Media
TRex293 cells were grown in high-glucose (4,500 mg/L) Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 4 mM l-glutamine, 1% (v/v) penicillin-streptomycin, and 5 μg/mL blasticidin. The addition of blasticidin allows silencing of Ag expression during Ad vaccine production in TRex293 cells (Life Technologies, Carlsbad, CA, USA). Expi293 cells (Life Technologies, Carlsbad, CA, USA) were grown in serum-free Expi media according to the manufacturer’s instructions. A549 cells were obtained from the American Tissue Culture Collection (ATCC; CCL-185), and CHO-CAR cells were a gift from Dr. Georges Santis (King’s College London, London, UK). A549 cells were grown in ATCC-formulated Kaighn’s modification of Ham’s F-12 (F-12K) medium supplemented with 10% (v/v) FBS and 1% (v/v) penicillin-streptomycin. For work requiring purification of EVs from the SN of Ad-infected A549 cells, F-12K media was made up with exosome-depleted FBS (Thermo Fisher, Waltham, MA, USA; lot #2094828). CHO-CAR cells were maintained in high-glucose DMEM supplemented with 10% (v/v) fetal calf serum, 110 mg/L sodium pyruvate, 4 mM l-glutamine, and 1% (v/v) penicillin-streptomycin.
Murine splenocytes were cultured in minimum essential medium with alpha modification (α-MEM) supplemented with 10% (v/v) FBS, 1% (v/v) penicillin-streptomycin, 4 mM l-glutamine, and 50 μM 2-mercaptoethanol for ex vivo IFN-γ ELISpot. Roswell Park Memorial Institute (RPMI) 1640 medium supplemented with 10% (v/v) FBS, 100 U/mL penicillin, 100 μg/mL streptomycin, and 2 mM l-glutamine (denoted R10) was used for preparing murine lymphocytes for flow cytometry-based immunology assays (described in detail below).
Exosome Purification by Ultracentrifugation and Size-Exclusion Chromatography
To assess the incorporation of the C1C2-fusion Ag into exosomes in vitro, 100–250 mL of Expi293F cells (2 × 106 cells/mL) were transfected with 1.375 μg/mL pcDNA3.1-SSmut expression vectors using 4 μg/mL polyethylenimine (PEI), as previously described.88,89 Transfection efficiency was evaluated by flow cytometry analysis of EGFP expression 48–72 h post-transfection, and cell number was recorded at harvest (Table S1). A sample of the cell lysate was collected for anti-HIS western blot analysis to confirm protein expression following transfection (Figure S1A) or as a control for western blots to detect proteins enriched in, or excluded from, exosomes but present in the cell lysate (Figure 1F). 1 × 107 cells were lysed in 200 μL of cell lysis buffer (300 mM NaCl, 50 mM Tris, 0.5% Triton X-100, pH 7.4), and then incubated on ice for 20 min before centrifugation at 20,000 × g for 20 min at 4°C. The cleared cell lysate was collected, protein concentration was determined by bicinchoninic acid (BCA) assay, and aliquots were frozen at −20°C prior to further analysis.
EVs were purified from serum-free conditioned media using a combination of high-speed ultracentrifugation to enrich the exosome pellet from 250 mL of SN to 1 mL, followed by the use of size-exclusion chromatography columns previously validated for purification of exosomes.42 96 h post-transfection, Expi293F cells were pelleted by centrifugation at 400 × g for 7 min at 4°C. SN was transferred into a new 50-mL conical tube and centrifuged again at 400 × g for 7 min at 4°C. SN was again transferred and centrifuged at 2,000 × g for 15 min at 4°C, and SN was transferred to a new tube and centrifuged at 10,000 × g for 30 min followed by filtration through a 0.22-μm filter as previously described90 before being stored at −80°C prior to ultracentrifugation. A Beckman Coulter Optima LE-80K ultracentrifuge and 70Ti fixed-angle rotor were pre-chilled to 4°C. SNs were divided over 32.4-mL Bell Optiseal ultracentrifugation tubes and centrifuged at 100,000 × g for 1.5 h at 4°C using slow acceleration and deceleration. The exosome pellet was pooled and resuspended in a final volume of 1 mL of low-particle Dulbecco’s PBS (DPBS) (Lonza, Basel, Switzerland).
Exo-spin Midi columns (Cell Guidance Systems, St. Louis, MO, USA) were prepared as described previously42 and loaded with the 1-mL sample obtained following ultracentrifugation of Expi293F cell SN. A total of 24 aliquots of 500 μL were collected and analyzed by ELISA for tetraspanins CD9, CD63, and CD81 and targeted Ag (i.e., EGFP) on the surface of exosome elution fractions (see Table 1 for Ab information). Relevant data from our experiments have been submitted to the EV-TRACK knowledgebase (EV-TRACK: EV190049),91 and methodology complies with guidelines for exosome classification as outlined by position papers from the International Society for Extracellular Vesicles.44,45
Table 1.
Abs for ELISA and Western Blot
| Marker | Species | Ab Clone | Isotype | Working Concentration (μg/mL) | Stock Concentration (mg/mL) | Application | Source |
|---|---|---|---|---|---|---|---|
| CD9 | mouse | 209306 | IgG2b | 1.0 | 0.5 | ELISA | R&D Systems |
| CD63 | mouse | MEM-259 | IgG1 | 1.0 | 1.0 | ELISA | Bio-Rad |
| CD81 | mouse | 1D6 | IgG1 | 1.0 | 1.0 | ELISA | Bio-Rad |
| EGFP | mouse | 9F9.9 | IgG1 | 0.4 | 1.0 | ELISA | Abcam |
| Alix | mouse | G10 | IgG1 | 1.0 | 0.2 | Western blot | Santa Cruz |
| Grp94 | rat | 9G10 | IgG2a | 1.0 | 0.2 | Western blot | Enzo |
| 6XHIS | mouse | HIS.H8 | IgG2b | 0.2 | 1.0 | Western blot | Abcam |
CD9, CD63, CD81, and Anti-EGFP ELISA
High-protein-binding 96-well plates were incubated overnight at 4°C with 20 μL of each size-exclusion chromatography elution fraction in quadruplicate (i.e., for CD9, CD63, CD81, and EGFP). The following day, plates were washed three times with 200 μL of 0.1% (v/v) BSA in DPBS (without calcium or magnesium) and blocked with 200 μL of 1% (v/v) BSA for 2 h at room temperature (RT). BSA dilutions were made up with 10× concentrated reagent diluent solution (R&D Systems, Minneapolis, MN, USA) diluted in DPBS before being filtered through a 0.1-μm filter. Plates were incubated with 100 μL of mouse anti-human CD9/CD63/CD81 or anti-EGFP diluted in DPBS with 0.1% (v/v) BSA for 2 h at RT. Following washing, 100 μL of secondary horseradish peroxidase (HRP)-conjugated Ab was added and plates were incubated at 37°C for 1 h. Plates were washed and development was performed using 100 μL of SigmaFast o-phenylenediamine dihydrochloride (OPD) solution and stopped with 50 μL of 3 M HCl (Thermo Fisher Scientific, Waltham, MA, USA). Plates were read at 490 nm. Fractions that had high expression of CD9/CD63 and/or CD81 were pooled and aliquoted before being stored at −80°C for downstream exosome analysis and validation. A small aliquot was not frozen and was kept at 4°C for quantification of protein content using a BCA assay and for quantification of vesicle titer and particle size using NanoSight (see below). ELISAs for CD9, CD63, CD81, and EGFP were repeated on purified preparations in triplicate, coating plates with 1 μg/well, alongside matched isotype control Abs (which were used to subtract background, non-specific staining). Positive controls for tetraspanins included validated exosomes derived from Du145 cells (provided by Dr. J. Webber and Prof. A. Clayton, Cardiff University),92 and the positive control for detection of EGFP included wells coated with 1 μg/mL recombinant EGFP protein (Abcam, Cambridge, UK).
Western Blot
The protein concentrations of cleared lysate and purified exosomes were assessed using the Pierce microBCA protein assay kit (Thermo Fisher, Waltham, MA, USA). Samples (20 μg) were reduced using 6× loading buffer containing dithiothreitol (DTT) and were boiled at 95°C for 10 min. Samples were loaded onto a 10% Mini-Protean TGX pre-cast gel (Bio-Rad, Hercules, CA, USA), and SDS-PAGE was performed at 125 V. Proteins were transferred to a 0.2-μm polyvinylidene fluoride (PVDF) membrane (Bio-Rad, Hercules, CA, USA) using the Trans-Blot Turbo transfer system. Following transfer, the membrane was incubated in blocking buffer (3% BSA in PBS) for 1 h at RT on a shaking platform. After washing three times with PBS with 0.1% Tween-20 (v/v), the membrane was incubated for 1 h at RT with 0.2 μg/mL mouse anti-hexahistidine (Abcam, Cambridge, UK), 1 μg/mL rabbit anti-human Grp94 (Enzo, New York, NY, USA), or 1 μg/mL mouse anti-human ALIX (Santa Cruz, Dallas, TX, USA) diluted in 3% BSA/PBS (Table 1). Following incubation, a 1:3,000 dilution of alkaline phosphatase-conjugated donkey anti-mouse or donkey anti-rat secondary Ab (Jackson Laboratory, Bar Harbor, ME, USA) was added for 1 h. The membrane was washed four times and then incubated with 5-bromo-4-chloro-3-indolyl phosphate (BCIP)/nitroblue tetrazolium (NBT) substrate (Sigma-Aldrich, St. Louis, MO, USA) until clear bands developed. The reaction was stopped using deionized water, and images were scanned and edited using GIMP 2.10.10 photo editing software to uniformly adjust brightness/contrast and to crop images for clear visualization.
NTA
The size and concentration of purified EVs were measured at the Radcliffe Department of Medicine, University of Oxford, using a NanoSight NS300 instrument (Malvern Panalytical, Malvern, UK), sCOMS camera, and NTA software version 2.3, build 0033. Synthetic 100-nm Nanosphere size standard particles (Thermo Fisher Scientific, Loughborough, UK) were used for calibration. EVs purified from 100–250 mL of Expi293F SNs were prepared for NTA analysis by dilution in nanoparticle-free H2O (Fresenius Kabi, Runcorn, UK). Dilutions ranging from 1:100 to 1:1,000 were performed so that more than 1 × 108 EVs/mL were being analyzed. Samples were analyzed according to the following measurement script—prime, 5-s delay, 30-s capture—and measurements were repeated three times per sample. Camera shutter speed was fixed at 19.97 ms and camera gain at 300 ms. Ambient temperature was recorded to be 21.8°C–22.8°C. Video acquisitions were analyzed using proprietary NTA software, with minimal expected particle size, minimum track length, and blur setting set to AUTO.
Expression of Exosome Display EGFP by Ad Vaccines In Vitro
A549 cells were seeded in six-well plates at 0.5 × 106 cells/well and allowed to adhere overnight. The following day, serum-containing F-12K media were removed, cells were washed with PBS, and media were replaced with 1 mL of serum-free media containing Ad virus at a MOI of 250 (IFU/cell). Each test was performed in duplicate. Cells were infected for 3 h at 37°C, after which the virus suspension was removed and media were replaced with 5 mL of F-12K containing 10% exosome-depleted FBS (Thermo Fisher, Waltham, MA, USA). The Ad infection was allowed to proceed for 72 h, after which the cell SNs were harvested for purification of EVs. The SNs from duplicate wells (5 mL) were combined (10 mL) and pelleted by centrifugation at 400 × g for 7 min at 4°C. SN was transferred into a new 50-mL conical tube and centrifuged again at 400 × g for 7 min at 4°C. SN was again transferred and centrifuged at 2,000 × g for 15 min at 4°C, transferred to a new tube, and centrifuged at 10,000 × g for 30 min, followed by filtration through a 0.22-μm filter. The filtered 10 mL of SN was then added to 5 mL of total exosome isolation reagent (Thermo Fisher, Waltham, MA, USA), and the suspension was mixed by vortexing and incubated overnight at 4°C to allow precipitation of vesicles. The following day, SN samples were pelleted by centrifugation at 10,000 × g for 1 h at 4°C, after which the pellet was resuspended in 250 μL of DPBS, as above. Protein concentration was determined using the Pierce microBCA protein assay kit (Thermo Fisher, Waltham, MA, USA). Exosome ELISA assays to quantify CD9, CD63, CD81, and exosome display EGFP were performed exactly as described above with the exception that 4 μg/well purified exosomes was used to coat ELISA plates (Figure S2).
Mice and Vaccination
All animal studies were approved by the Icahn School of Medicine at Mount Sinai Institutional Animal Care and Use Committee (IACUC-2017-0170). Animal studies adhere to the ARRIVE guidelines.93 Female BALB/cJ mice (Jackson Laboratory, Bar Harbor, ME, USA) aged 6–8 weeks (n = 5–10 mice/group) received 108 IFU87 of a non-replicating Ad vaccine vector diluted in sterile PBS, administered either i.m. or i.n. in a total volume of 50 μL. Control animals received 50 μL of PBS (Life Technologies, Carlsbad, CA, USA). For i.n. vaccinations, mice were anesthetized i.m. using ketamine and xylazine diluted in water for injection (WFI; Thermo Fisher Scientific, Waltham, MA, USA). A pre-vaccination blood sample was obtained from each animal by submandibular bleeding at least 2 days prior to vaccination (day −2). Maximal blood sampling throughout the duration of the experiment did not exceed recommended guidelines per total blood volume (TBV), as established by the National Centre for the Replacement, Refinement and Reduction of Animals in Research (NC3Rs): a maximum of <10% on any single occasion and <15% TBV within 28 days. Two to four weeks (D14 or D28) later, mice were euthanized by increasing CO2 concentration, death was confirmed by cervical dislocation, and serum and/or spleens were collected for analysis of humoral and cellular immunogenicity. We also collected the BAL fluid and lungs of mice vaccinated i.n, for analysis of mucosal immune responses. The latter mice were euthanized by increasing CO2 concentration, followed by exsanguination rather than cervical dislocation, to facilitate collection of BAL fluid without disruption of the trachea.
Quantification of EGFP IgG Ab Responses
To measure total Ag-specific IgG responses following vaccination, Immulon 4 HBX flat-bottom 96-well plates (Thermo Fisher Scientific, Waltham, MA, USA) were coated overnight at 4°C with recombinant EGFP protein at 1 μg/mL (Abcam, Cambridge, UK) diluted in 50 μL of 50 mM carbonate Na2CO3 buffer (Sigma-Aldrich, St. Louis, MO, USA). The following day, plates were washed using 1× PBS (Life Technologies, Carlsbad, CA, USA) containing 0.1% (v/v) Tween 20 (Millipore, Burlington, MA, USA) and subsequently blocked with 200 μL of blocking buffer, PBS containing 1% (w/v) BSA (Sigma-Aldrich, St. Louis, MO), for at least 1 h at RT to reduce non-specific binding. After washing, a 1:100 dilution of mouse sera was added to the plate in duplicate, a 3-fold serial dilution in blocking buffer was performed (final volume, 100 μL), and plates were incubated for 2 h at RT on an orbital shaker. A mouse monoclonal Ab to EGFP (Abcam, Cambridge, UK) was used as a positive control, and an isotype control (mouse monoclonal IgG1; Abcam, Cambridge, UK) was used as a negative control. Additional controls included naive unvaccinated mouse sera and secondary Ab-only controls. After washing, 100 μL of goat anti-mouse IgG HRP-conjugated secondary Ab (Millipore, Burlington, MA, USA) diluted 1:5,000 in blocking buffer was added to the plate. After a 1-h incubation at 37°C, the plate was washed and developed using 100 μL of SigmaFast OPD (Sigma-Aldrich, St. Louis, MO, USA) tablets diluted in water and stopped with 50 μL of 3 M HCl. Plates were read at 490 nm. Baseline was defined as the mean response of naive sera plus 3 times the standard deviation (SD) of the mean optical density (OD) values. Endpoint titers were calculated using GraphPad Prism v8.2.1 and represent the x intercept with the baseline (mean of naive sera at 1:100 dilution + 3 times the SD). Values below the limit of detection were estimated to be at half the input dilution. The AUC was calculated in GraphPad Prism v8.2.1 and represents the total peak area, where baseline was set as described above, and was <10% of the y axis minimum to maximum value. When AUC values could not be calculated, an arbitrary value of 1.0 was assigned. Naive sera controls, in addition to a positive control Ab (i.e., mouse anti-EGFP) with matched isotype control, were run on every ELISA plate for quality control.
Anti-EGFP IgG Subclass ELISA
To measure Ag-specific responses across multiple IgG subclasses following vaccination, ELISAs were performed as above with the following modifications. Following incubation with serial dilutions of vaccinated mouse sera starting at 1:33.3–1:100, or control sera/Abs, 50 μL of biotin-conjugated secondary rat Abs specific for each IgG subclass, i.e., anti-mouse IgG1 (BD Biosciences, Franklin Lakes, NJ, USA), anti-mouse IgG2a (BD Biosciences, Franklin Lakes, NJ, USA), and anti-mouse IgG2b (BD Biosciences, Franklin Lakes, NJ, USA), were diluted to 1 μg/mL in blocking buffer and added to the plate. After a 1 h incubation shaking at RT, 50 μL of ExtrAvidin-peroxidase (Sigma-Aldrich, St. Louis, MO, USA) diluted 1:2,500 in blocking buffer was added to each well and incubated for 30 min at RT. Plates were washed prior to development and analysis, exactly as described above.
Quantification of Spleen ELISpot Responses to Vaccination
EGFP-specific IFN-γ-producing cells were quantified using a murine ELISpot kit (Mabtech, Nacka Strand, Sweden). 96-well PVDF plates (Millipore, Burlington, MA, USA) were pre-coated with 5 μg/mL rat anti-mouse IFN-γ capture Ab (clone AN18: Mabtech, Nacka Strand, Sweden) 24–48 h prior to performing the ELISpot. Spleens were homogenized manually through a 40-μm cell strainer, and red blood cells were lysed by incubation for 5 min in ammonium-chloride-potassium (ACK) lysis buffer (0.15 M NH4Cl, 10 mM KHCO3, 100 mM EDTA-Na2 diluted in water). Splenocytes were resuspended at 106 cells/mL in complete α-MEM. A range of dilutions for the input cell number per well was tested, ranging from 1.25 to 5.0 × 105 cells/well. Samples were analyzed in triplicate for each mouse in a final volume of 100 μL. Splenocytes were stimulated with EGFP PepMix, a pool of 57 peptides (15-mer peptides overlapping by 11 aa) spanning the full length of EGFP (JPT Peptides, Berlin, Germany), or with EGFP200–208 (HYLSTQSAL), a H2-Kd-restricted CD8+ T cell epitope (JPT, Berlin, Germany), at a final individual peptide concentration of 1 μg/mL. Stimulated splenocytes were incubated at 37°C with 5% CO2 for 18–20 h. Controls included cells stimulated with concanavalin A from Canavalia ensiformis (ConA; Sigma-Aldrich, St. Louis, MO, USA) at a final concentration of 5 μg/mL as a positive control, or α-MEM media alone as an unstimulated control. Following incubation, plates were washed with PBS to remove cells and incubated with biotinylated rat anti-mouse IFN-γ (Mabtech, Nacka Strand, Sweden) diluted to 1 μg/mL in PBS for 2–4 h at RT. Streptavidin-conjugated alkaline phosphatase (Mabtech, Nacka Strand, Sweden) diluted to 1 μg/mL in PBS was added directly to the plates for 1 h at RT. The plates were washed six times with PBS and responses were detected using an alkaline phosphatase development buffer (Bio-Rad, Hercules, CA, USA). The ELISpot plates were dried before quantification of spots using an AID ELISpot reader (AID Diagnostika, Strassberg, Germany). Results were expressed as spot-forming units (SFU) per million splenocytes and were calculated by subtracting the mean negative control response from the mean peptide response. Responses in negative control wells were <30 SFU/106 splenocytes, and positive control wells (ConA) displayed >500 SFU/106 splenocytes.
Lung Processing and Flow Cytometry with ICS
Mice were humanely euthanized with an increasing concentration of CO2, followed by exsanguination by severing the femoral artery. BAL was obtained from the lung using a 2 mL PBS wash. BAL cell samples were pelleted by centrifugation (500 × g, 5 min), and SN was collected for ELISAs. The cell pellet was resuspended in 500 μL of RBC lysis buffer (QIAGEN, Hilden, Germany) for 5 min, then washed in 5 mL of R10 media.
Following collection of BAL, lungs were subsequently perfused with 1 mL of PBS by cardiac puncture, then dissected into 5 mL of digestion media (1.5% CaCl2, 1% FBS, 30 μg/mL DNase I, 0.7 mg/mL collagenase type IV in distilled H2O [dH2O]) in C Tubes (Miltenyi Biotec, Bergisch Gladbach, Germany) on ice. Lungs were homogenized using a gentleMACS dissociator (Miltenyi Biotec, Bergisch Gladbach, Germany) and placed in a shaking incubator at 37°C for 45–60 min. The lung homogenate was filtered through a 100-μm sieve, pelleted by centrifugation (300 × g, 8 min), and resuspended in 5 mL of RBC lysis buffer for 5 min. Lung homogenate was washed through a 40-μm sieve using 10 mL of R10 and then pelleted by centrifugation. Cells from BAL and perfused lungs were resuspended in R10 media containing anti-mouse CD28 (1:1,000, BD Biosciences, Franklin Lakes, NJ, USA), brefeldin A (1:1,000, BD Biosciences, Franklin Lakes, NJ), and monensin (1:1,000, BD Biosciences, Franklin Lakes, NJ, USA). Cells from BAL and lung were stimulated for 6 h at 37°C in 5% CO2 with either 1 μg/mL EGFP PepMix, an equivalent volume of DMSO as a negative control, or a positive control stimulation cocktail containing phorbol 12-myristate 13-acetate (PMA; 0.5 μg/mL, Sigma-Aldrich, St. Louis, MO, USA) and ionomycin (1 μg/mL, Sigma-Aldrich, St. Louis, MO, USA). After stimulation, cells were washed in FACS buffer (PBS containing 0.1% BSA and 2 mM EDTA; 500 × g for 5 min) and incubated with Fc block (1:100, eBioscience, Thermo Fisher Scientific, Waltham, MA, USA) for 10 min at 4°C. Cells were washed in FACS buffer and incubated with surface staining cocktail for 30 min at 4°C (see Table 2). Cells were washed in FACS buffer and then incubated in fixation/permeabilization buffer (BD Biosciences, Franklin Lakes, NJ, USA) for 10 min at 4°C. Cells were washed in 1× permeabilization buffer (BD Biosciences), then incubated with the intracellular staining cocktail for 30 min at 4°C (see Table 2). Samples were washed in 1× permeabilization buffer and then FACS buffer and resuspended in FACS buffer for acquisition. Samples were acquired on an LSR II cytometer (BD Biosciences, Franklin Lakes, NJ, USA) using FACSDiva v7.03 (BD Biosciences, Franklin Lakes, NJ, USA), with the relevant single fluorochrome compensation controls and photon multiplier tube voltages set by daily acquisition of Cytometer Setup and Tracking beads (BD Biosciences, Franklin Lakes, NJ, USA).
Table 2.
Abs for Flow Cytometry
| Marker | Staining Cocktail | Fluorochrome | Ab Clone | Dilution | Stock Concentration (mg/mL) | Source |
|---|---|---|---|---|---|---|
| CD3 | surface | Pacific Blue | 17A2 | 1 in 100 | 0.5 | BioLegend |
| CD4 | surface | Brilliant Violet 510 | RM4-5 | 1 in 150 | 0.2 | BioLegend |
| CD8 | surface | Alexa Fluor 700 | 53-6.7 | 1 in 50 | 0.2 | eBioscience |
| LIVE/DEAD | surface | Near Infrared | N/A | 1 in 1,000 | N/A | Thermo Fisher |
| IL-2 | intracellular | PE-Cy7 | JES6-5H4 | 1 in 250 | 0.2 | eBioscience |
| IFN-γ | intracellular | eFluor 660 | XMG1.2 | 1 in 500 | 0.2 | eBioscience |
| TNF-α | intracellular | Alexa Fluor 488 | MP6-XT22 | 1 in 2,000 | 0.5 | eBioscience |
N/A, not applicable; PE, phycoerythrin.
FACS Analysis
The transduction efficiency and comparable transgene expression of Ad vectors Ad5- and ChAd- expressing EGFPΔ and EGFP_C1C2 were confirmed by flow cytometry, exactly as described previously94 (Figures S1B–S1E). Samples were acquired using an LSR II (BD Biosciences, San Jose, CA, USA) acquiring >20,000 gated events, and analysis was performed using FlowJo v10.4.2 (Tree Star, Ashland, OR, USA). The relative fluorescence intensity (RFI) was calculated by multiplying the percentage of EGFP-positive cells by the geometric mean fluorescence intensity.
For the detection of cellular immune responses by ICS, >24,000 CD4+ and >3,800 CD8+ T cells were acquired on lung lymphocyte test samples, and >360 CD4+ and >160 CD8+ T cells were acquired on BAL test samples. Data were analyzed using FlowJo software to gate on size, singlets, live cells, CD3+, and then CD4+ or CD8+, and then assessed for IFN-γ, IL-2, and TNF-α secretion (see Figure S5 for gating strategy). Autologous unstimulated (DMSO) responses were subtracted from EGFP-stimulated responses for lung lymphocytes. BAL samples were gated using an identical gating strategy and the mean cytokine frequency of two representative unstimulated (DMSO) BAL samples subtracted from each EGFP-stimulated sample. Lung lymphocytes and BAL PMA/ionomycin-stimulated samples passed a >1% cytokine response positive control threshold. All BAL samples passed negative quality control. Two lung samples did not pass negative quality control and were excluded from the analysis.
Statistical Analyses and Data Analysis Software
Differences between each Ad-vaccinated group (Δ versus C1C2) were calculated using the non-parametric Mann-Whitney test. For lung and BAL ICS analyses, a Kruskal-Wallis test with Dunn’s correction for multiple comparisons was used to compare EGFPΔ, EGFP_C1C2, and PBS control groups. Statistical significance was determined to be *p < 0.05, **p < 0.01, ***p < 0.001, or ****p < 0.0001. Data were graphed using GraphPad Prism v8.2.1 (GraphPad, San Diego, CA, USA). Flow cytometry data were analyzed using FlowJo v10.4.2 (Tree Star, Ashland, OR, USA).
Author Contributions
Conceptualization, L.C.; Methodology, L.C., C.M.B., R.N., J.P.W., and A.C.; Validation, C.M.B. and L.C.; Investigation, L.C., A.J.P., C.M.B., F.C., R.N., J.R.H., and M.U.; Writing – Original Draft, L.C., C.M.B., and A.J.P.; Writing – Review and Editing, all authors contributed to critical revisions of the manuscript; Writing – Final Draft, C.M.B. and L.C.; Funding Acquisition, L.C. and A.V.S.H.; Resources, L.C., J.P.W., A.C., and A.V.S.H.; Supervision, L.C.
Conflicts of Interest
A.V.S.H. is named as an inventor on a patent covering use of ChAdOx1-vectored vaccines and is a co-founder of, consultant to, and shareholder in Vaccitech plc, which is developing Ad-vectored vaccines. The remaining authors declare no competing interests.
Acknowledgments
We would like to thank Dr. Alison Turner and Louisa Rose (The Jenner Institute Viral Vector Core Facility) for excellent technical assistance in producing recombinant Ad vaccines. We thank Dr. Clotilde Théry (INSERM, France) for providing the pcDNA3.1-SS_C1C2 plasmid. We would also like to acknowledge the staff at the Flow Cytometry Core, Icahn School of Medicine at Mount Sinai (ISMMS), and Chen Wang, our laboratory manager at ISMMS. We thank Dr. Naveed Akbar at the Division of Cardiovascular Medicine, Radcliffe Department of Medicine, University of Oxford, for access and assistance with NTA acquisition and analysis. We would also like to thank Prof. Peter Palese, ISMMS, for helpful and constructive discussion of these data. L.C. is funded by the HC Roscoe Grant 2016 from the British Medical Association Foundation for Medical Research. This research project was supported in part by funding from NIH/NIAID CEIRS (HHSN272201400008C), and by grants awarded to L.C., including the US Graduate Women in Science (GWIS) 2017 Nell Mondy and Monique Braude Fellowship, a UK Royal Society for Tropical Medicine and Hygiene small grant (GR000550), and by a Medical Research Fund pump-priming grant from the University of Oxford (MRF/TT2015/2150), United Kingdom. A.J.P. was in part supported by the pre-doctoral and post-doctoral USPHS Institutional Research Training Award T32-AI07647. The graphical abstract was created with Biorender.com.
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.omtm.2019.12.003.
Supplemental Information
References
- 1.Dicks M.D., Spencer A.J., Coughlan L., Bauza K., Gilbert S.C., Hill A.V., Cottingham M.G. Differential immunogenicity between HAdV-5 and chimpanzee adenovirus vector ChAdOx1 is independent of fiber and penton RGD loop sequences in mice. Sci. Rep. 2015;5:16756. doi: 10.1038/srep16756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Roy S., Gao G., Clawson D.S., Vandenberghe L.H., Farina S.F., Wilson J.M. Complete nucleotide sequences and genome organization of four chimpanzee adenoviruses. Virology. 2004;324:361–372. doi: 10.1016/j.virol.2004.03.047. [DOI] [PubMed] [Google Scholar]
- 3.Roy S., Vandenberghe L.H., Kryazhimskiy S., Grant R., Calcedo R., Yuan X., Keough M., Sandhu A., Wang Q., Medina-Jaszek C.A. Isolation and characterization of adenoviruses persistently shed from the gastrointestinal tract of non-human primates. PLoS Pathog. 2009;5:e1000503. doi: 10.1371/journal.ppat.1000503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Robinson C.M., Seto D., Jones M.S., Dyer D.W., Chodosh J. Molecular evolution of human species D adenoviruses. Infect. Genet. Evol. 2011;11:1208–1217. doi: 10.1016/j.meegid.2011.04.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Coughlan L., Mullarkey C., Gilbert S. Adenoviral vectors as novel vaccines for influenza. J. Pharm. Pharmacol. 2015;67:382–399. doi: 10.1111/jphp.12350. [DOI] [PubMed] [Google Scholar]
- 6.Antrobus R.D., Coughlan L., Berthoud T.K., Dicks M.D., Hill A.V., Lambe T., Gilbert S.C. Clinical assessment of a novel recombinant simian adenovirus ChAdOx1 as a vectored vaccine expressing conserved Influenza A antigens. Mol. Ther. 2014;22:668–674. doi: 10.1038/mt.2013.284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Coughlan L., Sridhar S., Payne R., Edmans M., Milicic A., Venkatraman N., Lugonja B., Clifton L., Qi C., Folegatti P.M. Heterologous two-dose vaccination with simian adenovirus and poxvirus vectors elicits long-lasting cellular immunity to influenza virus A in healthy adults. EBioMedicine. 2018;29:146–154. doi: 10.1016/j.ebiom.2018.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Green C.A., Scarselli E., Sande C.J., Thompson A.J., de Lara C.M., Taylor K.S., Haworth K., Del Sorbo M., Angus B., Siani L. Chimpanzee adenovirus- and MVA-vectored respiratory syncytial virus vaccine is safe and immunogenic in adults. Sci. Transl. Med. 2015;7:300ra126. doi: 10.1126/scitranslmed.aac5745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gurwith M., Lock M., Taylor E.M., Ishioka G., Alexander J., Mayall T., Ervin J.E., Greenberg R.N., Strout C., Treanor J.J. Safety and immunogenicity of an oral, replicating adenovirus serotype 4 vector vaccine for H5N1 influenza: a randomised, double-blind, placebo-controlled, phase 1 study. Lancet Infect. Dis. 2013;13:238–250. doi: 10.1016/S1473-3099(12)70345-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stephenson K.E., Keefer M.C., Bunce C.A., Frances D., Abbink P., Maxfield L.F., Neubauer G.H., Nkolola J., Peter L., Lane C. First-in-human randomized controlled trial of an oral, replicating adenovirus 26 vector vaccine for HIV-1. PLoS ONE. 2018;13:e0205139. doi: 10.1371/journal.pone.0205139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bliss C.M., Drammeh A., Bowyer G., Sanou G.S., Jagne Y.J., Ouedraogo O., Edwards N.J., Tarama C., Ouedraogo N., Ouedraogo M. Viral vector malaria vaccines induce high-level t cell and antibody responses in West African children and infants. Mol. Ther. 2017;25:547–559. doi: 10.1016/j.ymthe.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Afolabi M.O., Tiono A.B., Adetifa U.J., Yaro J.B., Drammeh A., Nébié I., Bliss C., Hodgson S.H., Anagnostou N.A., Sanou G.S. Safety and immunogenicity of ChAd63 and MVA ME-TRAP in West African children and infants. Mol. Ther. 2016;24:1470–1477. doi: 10.1038/mt.2016.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mensah V.A., Roetynck S., Kanteh E.K., Bowyer G., Ndaw A., Oko F., Bliss C.M., Jagne Y.J., Cortese R., Nicosia A. Safety and immunogenicity of malaria vectored vaccines given with routine expanded program on immunization vaccines in Gambian infants and neonates: a randomized controlled trial. Front. Immunol. 2017;8:1551. doi: 10.3389/fimmu.2017.01551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Churchyard G.J., Snowden M.A., Hokey D., Dheenadhayalan V., McClain J.B., Douoguih M., Pau M.G., Sadoff J., Landry B. The safety and immunogenicity of an adenovirus type 35-vectored TB vaccine in HIV-infected, BCG-vaccinated adults with CD4+ T cell counts >350 cells/mm3. Vaccine. 2015;33:1890–1896. doi: 10.1016/j.vaccine.2015.02.004. [DOI] [PubMed] [Google Scholar]
- 15.Vemula S.V., Mittal S.K. Production of adenovirus vectors and their use as a delivery system for influenza vaccines. Expert Opin. Biol. Ther. 2010;10:1469–1487. doi: 10.1517/14712598.2010.519332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Alcock R., Cottingham M.G., Rollier C.S., Furze J., De Costa S.D., Hanlon M., Spencer A.J., Honeycutt J.D., Wyllie D.H., Gilbert S.C. Long-term thermostabilization of live poxviral and adenoviral vaccine vectors at supraphysiological temperatures in carbohydrate glass. Sci. Transl. Med. 2010;2:19ra12. doi: 10.1126/scitranslmed.3000490. [DOI] [PubMed] [Google Scholar]
- 17.Afkhami S., LeClair D.A., Haddadi S., Lai R., Toniolo S.P., Ertl H.C., Cranston E.D., Thompson M.R., Xing Z. Spray dried human and chimpanzee adenoviral-vectored vaccines are thermally stable and immunogenic in vivo. Vaccine. 2017;35:2916–2924. doi: 10.1016/j.vaccine.2017.04.026. [DOI] [PubMed] [Google Scholar]
- 18.Yang T.C., Dayball K., Wan Y.H., Bramson J. Detailed analysis of the CD8+ T-cell response following adenovirus vaccination. J. Virol. 2003;77:13407–13411. doi: 10.1128/JVI.77.24.13407-13411.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sullivan N.J., Hensley L., Asiedu C., Geisbert T.W., Stanley D., Johnson J., Honko A., Olinger G., Bailey M., Geisbert J.B. CD8+ cellular immunity mediates rAd5 vaccine protection against Ebola virus infection of nonhuman primates. Nat. Med. 2011;17:1128–1131. doi: 10.1038/nm.2447. [DOI] [PubMed] [Google Scholar]
- 20.Tatsis N., Fitzgerald J.C., Reyes-Sandoval A., Harris-McCoy K.C., Hensley S.E., Zhou D., Lin S.W., Bian A., Xiang Z.Q., Iparraguirre A. Adenoviral vectors persist in vivo and maintain activated CD8+ T cells: implications for their use as vaccines. Blood. 2007;110:1916–1923. doi: 10.1182/blood-2007-02-062117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barnes E., Folgori A., Capone S., Swadling L., Aston S., Kurioka A., Meyer J., Huddart R., Smith K., Townsend R. Novel adenovirus-based vaccines induce broad and sustained T cell responses to HCV in man. Sci. Transl. Med. 2012;4:115ra1. doi: 10.1126/scitranslmed.3003155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Barouch D.H., Tomaka F.L., Wegmann F., Stieh D.J., Alter G., Robb M.L., Michael N.L., Peter L., Nkolola J.P., Borducchi E.N. Evaluation of a mosaic HIV-1 vaccine in a multicentre, randomised, double-blind, placebo-controlled, phase 1/2a clinical trial (APPROACH) and in rhesus monkeys (NHP 13-19) Lancet. 2018;392:232–243. doi: 10.1016/S0140-6736(18)31364-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Coughlan L., Bradshaw A.C., Parker A.L., Robinson H., White K., Custers J., Goudsmit J., Van Roijen N., Barouch D.H., Nicklin S.A., Baker A.H. Ad5:Ad48 hexon hypervariable region substitutions lead to toxicity and increased inflammatory responses following intravenous delivery. Mol. Ther. 2012;20:2268–2281. doi: 10.1038/mt.2012.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Abbink P., Lemckert A.A., Ewald B.A., Lynch D.M., Denholtz M., Smits S., Holterman L., Damen I., Vogels R., Thorner A.R. Comparative seroprevalence and immunogenicity of six rare serotype recombinant adenovirus vaccine vectors from subgroups B and D. J. Virol. 2007;81:4654–4663. doi: 10.1128/JVI.02696-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Roberts D.M., Nanda A., Havenga M.J., Abbink P., Lynch D.M., Ewald B.A., Liu J., Thorner A.R., Swanson P.E., Gorgone D.A. Hexon-chimaeric adenovirus serotype 5 vectors circumvent pre-existing anti-vector immunity. Nature. 2006;441:239–243. doi: 10.1038/nature04721. [DOI] [PubMed] [Google Scholar]
- 26.Duffy M.R., Alonso-Padilla J., John L., Chandra N., Khan S., Ballmann M.Z., Lipiec A., Heemskerk E., Custers J., Arnberg N. Generation and characterization of a novel candidate gene therapy and vaccination vector based on human species D adenovirus type 56. J. Gen. Virol. 2018;99:135–147. doi: 10.1099/jgv.0.000978. [DOI] [PubMed] [Google Scholar]
- 27.Mennechet F.J.D., Paris O., Ouoba A.R., Salazar Arenas S., Sirima S.B., Takoudjou Dzomo G.R., Diarra A., Traore I.T., Kania D., Eichholz K. A review of 65 years of human adenovirus seroprevalence. Expert Rev. Vaccines. 2019;18:597–613. doi: 10.1080/14760584.2019.1588113. [DOI] [PubMed] [Google Scholar]
- 28.Colloca S., Barnes E., Folgori A., Ammendola V., Capone S., Cirillo A., Siani L., Naddeo M., Grazioli F., Esposito M.L. Vaccine vectors derived from a large collection of simian adenoviruses induce potent cellular immunity across multiple species. Sci. Transl. Med. 2012;4:115ra2. doi: 10.1126/scitranslmed.3002925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sayedahmed E.E., Hassan A.O., Kumari R., Cao W., Gangappa S., York I., Sambhara S., Mittal S.K. A bovine adenoviral vector-based H5N1 influenza -vaccine provides enhanced immunogenicity and protection at a significantly low dose. Mol. Ther. Methods Clin. Dev. 2018;10:210–222. doi: 10.1016/j.omtm.2018.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Quinn K.M., Da Costa A., Yamamoto A., Berry D., Lindsay R.W., Darrah P.A., Wang L., Cheng C., Kong W.P., Gall J.G. Comparative analysis of the magnitude, quality, phenotype, and protective capacity of simian immunodeficiency virus gag-specific CD8+ T cells following human-, simian-, and chimpanzee-derived recombinant adenoviral vector immunization. J. Immunol. 2013;190:2720–2735. doi: 10.4049/jimmunol.1202861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dicks M.D., Guzman E., Spencer A.J., Gilbert S.C., Charleston B., Hill A.V., Cottingham M.G. The relative magnitude of transgene-specific adaptive immune responses induced by human and chimpanzee adenovirus vectors differs between laboratory animals and a target species. Vaccine. 2015;33:1121–1128. doi: 10.1016/j.vaccine.2015.01.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dicks M.D., Spencer A.J., Edwards N.J., Wadell G., Bojang K., Gilbert S.C., Hill A.V., Cottingham M.G. A novel chimpanzee adenovirus vector with low human seroprevalence: improved systems for vector derivation and comparative immunogenicity. PLoS ONE. 2012;7:e40385. doi: 10.1371/journal.pone.0040385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Li J., Liu K., Liu Y., Xu Y., Zhang F., Yang H., Liu J., Pan T., Chen J., Wu M. Exosomes mediate the cell-to-cell transmission of IFN-α-induced antiviral activity. Nat. Immunol. 2013;14:793–803. doi: 10.1038/ni.2647. [DOI] [PubMed] [Google Scholar]
- 34.Théry C., Duban L., Segura E., Véron P., Lantz O., Amigorena S. Indirect activation of naïve CD4+ T cells by dendritic cell-derived exosomes. Nat. Immunol. 2002;3:1156–1162. doi: 10.1038/ni854. [DOI] [PubMed] [Google Scholar]
- 35.Théry C., Ostrowski M., Segura E. Membrane vesicles as conveyors of immune responses. Nat. Rev. Immunol. 2009;9:581–593. doi: 10.1038/nri2567. [DOI] [PubMed] [Google Scholar]
- 36.Raposo G., Nijman H.W., Stoorvogel W., Liejendekker R., Harding C.V., Melief C.J., Geuze H.J. B lymphocytes secrete antigen-presenting vesicles. J. Exp. Med. 1996;183:1161–1172. doi: 10.1084/jem.183.3.1161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Delcayre A., Estelles A., Sperinde J., Roulon T., Paz P., Aguilar B., Villanueva J., Khine S., Le Pecq J.B. Exosome display technology: applications to the development of new diagnostics and therapeutics. Blood Cells Mol. Dis. 2005;35:158–168. doi: 10.1016/j.bcmd.2005.07.003. [DOI] [PubMed] [Google Scholar]
- 38.Théry C., Regnault A., Garin J., Wolfers J., Zitvogel L., Ricciardi-Castagnoli P., Raposo G., Amigorena S. Molecular characterization of dendritic cell-derived exosomes. Selective accumulation of the heat shock protein Hsc73. J. Cell Biol. 1999;147:599–610. doi: 10.1083/jcb.147.3.599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sedlik C., Vigneron J., Torrieri-Dramard L., Pitoiset F., Denizeau J., Chesneau C., de la Rochere P., Lantz O., Thery C., Bellier B. Different immunogenicity but similar antitumor efficacy of two DNA vaccines coding for an antigen secreted in different membrane vesicle-associated forms. J. Extracell. Vesicles. 2014;3:24646. doi: 10.3402/jev.v3.24646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rountree R.B., Mandl S.J., Nachtwey J.M., Dalpozzo K., Do L., Lombardo J.R., Schoonmaker P.L., Brinkmann K., Dirmeier U., Laus R., Delcayre A. Exosome targeting of tumor antigens expressed by cancer vaccines can improve antigen immunogenicity and therapeutic efficacy. Cancer Res. 2011;71:5235–5244. doi: 10.1158/0008-5472.CAN-10-4076. [DOI] [PubMed] [Google Scholar]
- 41.Hartman Z.C., Wei J., Glass O.K., Guo H., Lei G., Yang X.Y., Osada T., Hobeika A., Delcayre A., Le Pecq J.B. Increasing vaccine potency through exosome antigen targeting. Vaccine. 2011;29:9361–9367. doi: 10.1016/j.vaccine.2011.09.133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Welton J.L., Webber J.P., Botos L.A., Jones M., Clayton A. Ready-made chromatography columns for extracellular vesicle isolation from plasma. J. Extracell. Vesicles. 2015;4:27269. doi: 10.3402/jev.v4.27269. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kowal J., Arras G., Colombo M., Jouve M., Morath J.P., Primdal-Bengtson B., Dingli F., Loew D., Tkach M., Théry C. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proc. Natl. Acad. Sci. USA. 2016;113:E968–E977. doi: 10.1073/pnas.1521230113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lötvall J., Hill A.F., Hochberg F., Buzás E.I., Di Vizio D., Gardiner C., Gho Y.S., Kurochkin I.V., Mathivanan S., Quesenberry P. Minimal experimental requirements for definition of extracellular vesicles and their functions: a position statement from the International Society for Extracellular Vesicles. J. Extracell. Vesicles. 2014;3:26913. doi: 10.3402/jev.v3.26913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Théry C., Witwer K.W., Aikawa E., Alcaraz M.J., Anderson J.D., Andriantsitohaina R., Antoniou A., Arab T., Archer F., Atkin-Smith G.K. Minimal information for studies of extracellular vesicles 2018 (MISEV2018): a position statement of the International Society for Extracellular Vesicles and update of the MISEV2014 guidelines. J. Extracell. Vesicles. 2018;7:1535750. doi: 10.1080/20013078.2018.1535750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Tatsis N., Ertl H.C. Adenoviruses as vaccine vectors. Mol. Ther. 2004;10:616–629. doi: 10.1016/j.ymthe.2004.07.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Bassett J.D., Yang T.C., Bernard D., Millar J.B., Swift S.L., McGray A.J., VanSeggelen H., Boudreau J.E., Finn J.D., Parsons R. CD8+ T-cell expansion and maintenance after recombinant adenovirus immunization rely upon cooperation between hematopoietic and nonhematopoietic antigen-presenting cells. Blood. 2011;117:1146–1155. doi: 10.1182/blood-2010-03-272336. [DOI] [PubMed] [Google Scholar]
- 48.Yang T.C., Millar J., Groves T., Grinshtein N., Parsons R., Takenaka S., Wan Y., Bramson J.L. The CD8+ T cell population elicited by recombinant adenovirus displays a novel partially exhausted phenotype associated with prolonged antigen presentation that nonetheless provides long-term immunity. J. Immunol. 2006;176:200–210. doi: 10.4049/jimmunol.176.1.200. [DOI] [PubMed] [Google Scholar]
- 49.Stylianou E., Griffiths K.L., Poyntz H.C., Harrington-Kandt R., Dicks M.D., Stockdale L., Betts G., McShane H. Improvement of BCG protective efficacy with a novel chimpanzee adenovirus and a modified vaccinia Ankara virus both expressing Ag85A. Vaccine. 2015;33:6800–6808. doi: 10.1016/j.vaccine.2015.10.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.DiLillo D.J., Palese P., Wilson P.C., Ravetch J.V. Broadly neutralizing anti-influenza antibodies require Fc receptor engagement for in vivo protection. J. Clin. Invest. 2016;126:605–610. doi: 10.1172/JCI84428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.DiLillo D.J., Tan G.S., Palese P., Ravetch J.V. Broadly neutralizing hemagglutinin stalk-specific antibodies require FcγR interactions for protection against influenza virus in vivo. Nat. Med. 2014;20:143–151. doi: 10.1038/nm.3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.He W., Chi-Jene C., Mullarkey C., Hamilton J., Wong C., Leon P., Uccellini M.B., Chromikova V., Henry C., Hoffman K.W. Alveolar macrophages are critical for broadly-reactive antibody-mediated protection against influenza A virus. Nat. Commun. 2017;8:846. doi: 10.1038/s41467-017-00928-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Coughlan L., Palese P. Overcoming barriers in the path to a universal influenza virus vaccine. Cell Host Microbe. 2018;24:18–24. doi: 10.1016/j.chom.2018.06.016. [DOI] [PubMed] [Google Scholar]
- 54.Bruhns P., Jönsson F. Mouse and human FcR effector functions. Immunol. Rev. 2015;268:25–51. doi: 10.1111/imr.12350. [DOI] [PubMed] [Google Scholar]
- 55.Nimmerjahn F., Ravetch J.V. Divergent immunoglobulin G subclass activity through selective Fc receptor binding. Science. 2005;310:1510–1512. doi: 10.1126/science.1118948. [DOI] [PubMed] [Google Scholar]
- 56.Hensley S.E., Cun A.S., Giles-Davis W., Li Y., Xiang Z., Lasaro M.O., Williams B.R., Silverman R.H., Ertl H.C. Type I interferon inhibits antibody responses induced by a chimpanzee adenovirus vector. Mol. Ther. 2007;15:393–403. doi: 10.1038/sj.mt.6300024. [DOI] [PubMed] [Google Scholar]
- 57.Xiang Z., Gao G., Reyes-Sandoval A., Cohen C.J., Li Y., Bergelson J.M., Wilson J.M., Ertl H.C. Novel, chimpanzee serotype 68-based adenoviral vaccine carrier for induction of antibodies to a transgene product. J. Virol. 2002;76:2667–2675. doi: 10.1128/JVI.76.6.2667-2675.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Moran T.M., Park H., Fernandez-Sesma A., Schulman J.L. Th2 responses to inactivated influenza virus can Be converted to Th1 responses and facilitate recovery from heterosubtypic virus infection. J. Infect. Dis. 1999;180:579–585. doi: 10.1086/314952. [DOI] [PubMed] [Google Scholar]
- 59.Moran T.M., Isobe H., Fernandez-Sesma A., Schulman J.L. Interleukin-4 causes delayed virus clearance in influenza virus-infected mice. J. Virol. 1996;70:5230–5235. doi: 10.1128/jvi.70.8.5230-5235.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Matsuda K., Huang J., Zhou T., Sheng Z., Kang B.H., Ishida E., Griesman T., Stuccio S., Bolkhovitinov L., Wohlbold T.J. Prolonged evolution of the memory B cell response induced by a replicating adenovirus-influenza H5 vaccine. Sci. Immunol. 2019;4:eaau2710. doi: 10.1126/sciimmunol.aau2710. [DOI] [PubMed] [Google Scholar]
- 61.Green C.A., Scarselli E., Voysey M., Capone S., Vitelli A., Nicosia A., Cortese R., Thompson A.J., Sande C.S., de Lara C. Safety and immunogenicity of novel respiratory syncytial virus (RSV) vaccines based on the RSV viral proteins F, N and M2-1 encoded by simian adenovirus (PanAd3-RSV) and MVA (MVA-RSV); protocol for an open-label, dose-escalation, single-centre, phase 1 clinical trial in healthy adults. BMJ Open. 2015;5:e008748. doi: 10.1136/bmjopen-2015-008748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Bliss C.M., Bowyer G., Anagnostou N.A., Havelock T., Snudden C.M., Davies H., de Cassan S.C., Grobbelaar A., Lawrie A.M., Venkatraman N. Assessment of novel vaccination regimens using viral vectored liver stage malaria vaccines encoding ME-TRAP. Sci. Rep. 2018;8:3390. doi: 10.1038/s41598-018-21630-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McCoy K., Tatsis N., Korioth-Schmitz B., Lasaro M.O., Hensley S.E., Lin S.W., Li Y., Giles-Davis W., Cun A., Zhou D. Effect of preexisting immunity to adenovirus human serotype 5 antigens on the immune responses of nonhuman primates to vaccine regimens based on human- or chimpanzee-derived adenovirus vectors. J. Virol. 2007;81:6594–6604. doi: 10.1128/JVI.02497-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lindsay R.W., Darrah P.A., Quinn K.M., Wille-Reece U., Mattei L.M., Iwasaki A., Kasturi S.P., Pulendran B., Gall J.G., Spies A.G., Seder R.A. CD8+ T cell responses following replication-defective adenovirus serotype 5 immunization are dependent on CD11c+ dendritic cells but show redundancy in their requirement of TLR and nucleotide-binding oligomerization domain-like receptor signaling. J. Immunol. 2010;185:1513–1521. doi: 10.4049/jimmunol.1000338. [DOI] [PubMed] [Google Scholar]
- 65.O’Hara G.A., Duncan C.J., Ewer K.J., Collins K.A., Elias S.C., Halstead F.D., Goodman A.L., Edwards N.J., Reyes-Sandoval A., Bird P. Clinical assessment of a recombinant simian adenovirus ChAd63: a potent new vaccine vector. J. Infect. Dis. 2012;205:772–781. doi: 10.1093/infdis/jir850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Casimiro D.R., Chen L., Fu T.M., Evans R.K., Caulfield M.J., Davies M.E., Tang A., Chen M., Huang L., Harris V. Comparative immunogenicity in rhesus monkeys of DNA plasmid, recombinant vaccinia virus, and replication-defective adenovirus vectors expressing a human immunodeficiency virus type 1 gag gene. J. Virol. 2003;77:6305–6313. doi: 10.1128/JVI.77.11.6305-6313.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Baker A.T., Greenshields-Watson A., Coughlan L., Davies J.A., Uusi-Kerttula H., Cole D.K., Rizkallah P.J., Parker A.L. Diversity within the adenovirus fiber knob hypervariable loops influences primary receptor interactions. Nat. Commun. 2019;10:741. doi: 10.1038/s41467-019-08599-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Shayakhmetov D.M., Li Z.Y., Ternovoi V., Gaggar A., Gharwan H., Lieber A. The interaction between the fiber knob domain and the cellular attachment receptor determines the intracellular trafficking route of adenoviruses. J. Virol. 2003;77:3712–3723. doi: 10.1128/JVI.77.6.3712-3723.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Shayakhmetov D.M., Eberly A.M., Li Z.Y., Lieber A. Deletion of penton RGD motifs affects the efficiency of both the internalization and the endosome escape of viral particles containing adenovirus serotype 5 or 35 fiber knobs. J. Virol. 2005;79:1053–1061. doi: 10.1128/JVI.79.2.1053-1061.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Finn J.D., Bassett J., Millar J.B., Grinshtein N., Yang T.C., Parsons R., Evelegh C., Wan Y., Parks R.J., Bramson J.L. Persistence of transgene expression influences CD8+ T-cell expansion and maintenance following immunization with recombinant adenovirus. J. Virol. 2009;83:12027–12036. doi: 10.1128/JVI.00593-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Bergelson J.M., Cunningham J.A., Droguett G., Kurt-Jones E.A., Krithivas A., Hong J.S., Horwitz M.S., Crowell R.L., Finberg R.W. Isolation of a common receptor for Coxsackie B viruses and adenoviruses 2 and 5. Science. 1997;275:1320–1323. doi: 10.1126/science.275.5304.1320. [DOI] [PubMed] [Google Scholar]
- 72.Kirby I., Davison E., Beavil A.J., Soh C.P., Wickham T.J., Roelvink P.W., Kovesdi I., Sutton B.J., Santis G. Identification of contact residues and definition of the CAR-binding site of adenovirus type 5 fiber protein. J. Virol. 2000;74:2804–2813. doi: 10.1128/jvi.74.6.2804-2813.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Coughlan L., Alba R., Parker A.L., Bradshaw A.C., McNeish I.A., Nicklin S.A., Baker A.H. Tropism-modification strategies for targeted gene delivery using adenoviral vectors. Viruses. 2010;2:2290–2355. doi: 10.3390/v2102290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Alba R., Bradshaw A.C., Coughlan L., Denby L., McDonald R.A., Waddington S.N., Buckley S.M., Greig J.A., Parker A.L., Miller A.M. Biodistribution and retargeting of FX-binding ablated adenovirus serotype 5 vectors. Blood. 2010;116:2656–2664. doi: 10.1182/blood-2009-12-260026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Parker A.L., Waddington S.N., Buckley S.M., Custers J., Havenga M.J., van Rooijen N., Goudsmit J., McVey J.H., Nicklin S.A., Baker A.H. Effect of neutralizing sera on factor X-mediated adenovirus serotype 5 gene transfer. J. Virol. 2009;83:479–483. doi: 10.1128/JVI.01878-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Shayakhmetov D.M., Gaggar A., Ni S., Li Z.Y., Lieber A. Adenovirus binding to blood factors results in liver cell infection and hepatotoxicity. J. Virol. 2005;79:7478–7491. doi: 10.1128/JVI.79.12.7478-7491.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Vigant F., Descamps D., Jullienne B., Esselin S., Connault E., Opolon P., Tordjmann T., Vigne E., Perricaudet M., Benihoud K. Substitution of hexon hypervariable region 5 of adenovirus serotype 5 abrogates blood factor binding and limits gene transfer to liver. Mol. Ther. 2008;16:1474–1480. doi: 10.1038/mt.2008.132. [DOI] [PubMed] [Google Scholar]
- 78.Waddington S.N., McVey J.H., Bhella D., Parker A.L., Barker K., Atoda H., Pink R., Buckley S.M., Greig J.A., Denby L. Adenovirus serotype 5 hexon mediates liver gene transfer. Cell. 2008;132:397–409. doi: 10.1016/j.cell.2008.01.016. [DOI] [PubMed] [Google Scholar]
- 79.Vitelli A., Folgori A., Scarselli E., Colloca S., Capone S., Nicosia A. Chimpanzee adenoviral vectors as vaccines—challenges to move the technology into the fast lane. Expert Rev. Vaccines. 2017;16:1241–1252. doi: 10.1080/14760584.2017.1394842. [DOI] [PubMed] [Google Scholar]
- 80.Alejo D.M., Moraes M.P., Liao X., Dias C.C., Tulman E.R., Diaz-San Segundo F., Rood D., Grubman M.J., Silbart L.K. An adenovirus vectored mucosal adjuvant augments protection of mice immunized intranasally with an adenovirus-vectored foot-and-mouth disease virus subunit vaccine. Vaccine. 2013;31:2302–2309. doi: 10.1016/j.vaccine.2013.02.060. [DOI] [PubMed] [Google Scholar]
- 81.Halbroth B.R., Sebastian S., Poyntz H.C., Bregu M., Cottingham M.G., Hill A.V.S., Spencer A.J. Development of a molecular adjuvant to enhance antigen-specific CD8+ T cell responses. Sci. Rep. 2018;8:15020. doi: 10.1038/s41598-018-33375-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Lambe T., Carey J.B., Li Y., Spencer A.J., van Laarhoven A., Mullarkey C.E., Vrdoljak A., Moore A.C., Gilbert S.C. Immunity against heterosubtypic influenza virus induced by adenovirus and MVA expressing nucleoprotein and matrix protein-1. Sci. Rep. 2013;3:1443. doi: 10.1038/srep01443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Aw D., Silva A.B., Palmer D.B. Immunosenescence: emerging challenges for an ageing population. Immunology. 2007;120:435–446. doi: 10.1111/j.1365-2567.2007.02555.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sambhara S., McElhaney J.E. Immunosenescence and influenza vaccine efficacy. Curr. Top. Microbiol. Immunol. 2009;333:413–429. doi: 10.1007/978-3-540-92165-3_20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Sebastian S., Lambe T. Clinical advances in viral-vectored influenza vaccines. Vaccines (Basel) 2018;6:E29. doi: 10.3390/vaccines6020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Zeelenberg I.S., Ostrowski M., Krumeich S., Bobrie A., Jancic C., Boissonnas A., Delcayre A., Le Pecq J.B., Combadière B., Amigorena S., Théry C. Targeting tumor antigens to secreted membrane vesicles in vivo induces efficient antitumor immune responses. Cancer Res. 2008;68:1228–1235. doi: 10.1158/0008-5472.CAN-07-3163. [DOI] [PubMed] [Google Scholar]
- 87.Cottingham M.G., Carroll F., Morris S.J., Turner A.V., Vaughan A.M., Kapulu M.C., Colloca S., Siani L., Gilbert S.C., Hill A.V. Preventing spontaneous genetic rearrangements in the transgene cassettes of adenovirus vectors. Biotechnol. Bioeng. 2012;109:719–728. doi: 10.1002/bit.24342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Durocher Y., Perret S., Kamen A. High-level and high-throughput recombinant protein production by transient transfection of suspension-growing human 293-EBNA1 cells. Nucleic Acids Res. 2002;30:E9. doi: 10.1093/nar/30.2.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tom R., Bisson L., Durocher Y. Transfection of HEK293-EBNA1 cells in suspension with linear PEI for production of recombinant proteins. CSH Protoc. 2008. 2008 doi: 10.1101/pdb.prot4977. pdb prot4977. [DOI] [PubMed] [Google Scholar]
- 90.Thery C., Amigorena S., Raposo G., Clayton A. Isolation and characterization of exosomes from cell culture supernatants and biological fluids. Curr. Protoc. Cell Biol. 2006;Chapter 3 doi: 10.1002/0471143030.cb0322s30. Unit 3.22. [DOI] [PubMed] [Google Scholar]
- 91.Van Deun J., Mestdagh P., Agostinis P., Akay Ö., Anand S., Anckaert J., Martinez Z.A., Baetens T., Beghein E., Bertier L., EV-TRACK Consortium EV-TRACK: transparent reporting and centralizing knowledge in extracellular vesicle research. Nat. Methods. 2017;14:228–232. doi: 10.1038/nmeth.4185. [DOI] [PubMed] [Google Scholar]
- 92.Webber J., Clayton A. How pure are your vesicles? J. Extracell. Vesicles. 2013;2:2. doi: 10.3402/jev.v2i0.19861. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Kilkenny C., Browne W.J., Cuthill I.C., Emerson M., Altman D.G. Improving bioscience research reporting: the ARRIVE guidelines for reporting animal research. PLoS Biol. 2010;8:e1000412. doi: 10.1371/journal.pbio.1000412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Coughlan L., Vallath S., Saha A., Flak M., McNeish I.A., Vassaux G., Marshall J.F., Hart I.R., Thomas G.J. In vivo retargeting of adenovirus type 5 to αvβ6 integrin results in reduced hepatotoxicity and improved tumor uptake following systemic delivery. J. Virol. 2009;83:6416–6428. doi: 10.1128/JVI.00445-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.