

Abstract
Background
While all clinically translated antibody-drug conjugates (ADCs) contain a single-drug payload, most systemic cancer chemotherapies involve use of a combination of drugs. These regimens improve treatment outcomes and slow development of drug resistance. We here report the generation of an ADC with a dual-drug payload that combines two distinct mechanisms of action.
Methods
Virtual DNA crosslinking agent PNU-159682 and tubulin polymerization inhibitor monomethyl auristatin F (MMAF) were conjugated to a HER2-targeting antibody via site-specific conjugation at engineered selenocysteine and cysteine residues (thio-selenomab).
Results
The dual-drug ADC showed selective and potent cytotoxicity against HER2-expressing cell lines and exhibited dual mechanisms of action consistent with the attached drugs. While PNU-159682 caused S-phase cell cycle arrest due to its DNA-damaging activity, MMAF simultaneously inhibited tubulin polymerization and caused G2/M-phase cell cycle arrest.
Conclusion
The thio-selenomab platform enables the assembly of dual-drug ADCs with two distinct mechanisms of action.
Keywords: ADC, dual-drug payload, MMAF, PNU-159682, cancer
Statement of Significance
A dual-drug ADC with two distinct mechanisms of action was generated and validated in vitro. Such dual-drug ADCs could prove to be effective antibody therapeutics for preventing or overcoming drug resistance in cancer.
INTRODUCTION
Antibody-drug conjugates (ADCs) are a class of antibody therapeutics that utilize the specificity of a monoclonal antibody to deliver highly potent small molecules (the payload) to target cells. The antibody serves as a delivery vehicle with specificity to a certain antigen on the surface of a target cell. Upon binding, the ADC is internalized and subsequently releases the payload to kill the target cell [1, 2]. Five ADCs have been approved by the Food and Drug Administration for cancer therapy and many more are at various stages of preclinical and clinical investigations. Although initial responses to ADC therapies are typically favorable, drug resistance often emerges [3, 4]. Various mechanisms of resistance to ADCs by tumors have been elucidated in recent years, such as down-regulation of the target antigen and up-regulation of drug efflux pumps [5, 6].
Drug resistance is a long-standing problem for systemic cancer chemotherapy. Treatment of cancer with multiple drugs has been attempted to reduce the probability of and/or slow down the rate of drug resistance [7]. In fact, current chemotherapy regimens typically utilize a combination of drugs. Chemotherapy drugs used in such a treatment often have different mechanisms of action (MOA) and are used in combination to provide better outcomes. For example, combinations of cyclophosphamide (a DNA crosslinking agent), doxorubicin (a topoisomerase II inhibitor) and docetaxel (a tubulin polymerization inhibitor) are commonly used as adjuvant chemotherapy for post-surgery early-stage breast cancer (BC) treatment [8].
Considering the benefits of combination chemotherapy, combining ADCs with different drugs or using ADCs with multiple-drug payloads may circumvent ADC-associated drug resistance. Rather than combining two ADCs with different payloads, we rationalized that only a dual-drug ADC would achieve the co-delivery of both payloads at equimolar concentrations. This would ensure that cancer cell survival requires the existence or development of resistance mechanisms to both drugs at the same time. Previous work has explored this possibility. In 2017, Levengood et al. [9] reported a combination of two tubulin polymerization inhibitors, monomethyl auristatin E and F (MMAE and MMAF) on an anti-CD30 antibody. Despite the payloads acting on the same intracellular target, the dual-drug ADC combined advantages of the payload-linkers. While the MMAF payload can retain cytotoxicity against target cells with upregulated efflux pumps, MMAE on a cleavable linker can cause bystander effects and kill co-cultured cells that do not express the cell surface antigen on the target cell. Concurrent with our study, Kumar et al. [10] reported the development of a heterotrifunctional linker to generate a dual-drug ADC with MMAE and a DNA crosslinking payload, pyrrolobenzodiazepine dimer (PBD dimer).
Even though a few dual-drug ADCs have been described, none were shown to simultaneously act on two different targets. Here, we report a dual-mechanistic ADC that combines two payloads with distinct MOA. A previously reported dual conjugation method via engineered selenocysteine (Sec) and cysteine (Cys) residues (thio-selenomab) was utilized to generate the dual-drug ADC [11]. A battery of in vitro studies was performed to demonstrate the dual-mechanistic property of this dual-drug ADC.
MATERIALS AND METHODS
Compound synthesis and characterization
Please see Supplementary Materials and Methods.
Antibody cloning, expression and purification
Antibody cloning, expression and purification were performed following previously described methods [11,12] with the following modification: Cloning: (i) Alanine-to-cysteine and serine-to-selenocysteine codon mutations were introduced to trastuzumab’s heavy chain in mammalian expression vector pCEP4 by site-directed mutagenesis using overlap PCR; (ii) His6 tag codons were introduced to facilitate the second purification step required for the selenomab and thio-selenomab. (The tag was kept for consistency in the thiomab construct). (iii) The SECIS element from the 3’-UTR of the cDNA of human TXNRD1 was introduced directly after the canonical stop codon in the selenomab and thio-selenomab heavy chains. Expression: For transient transfection, a total amount of 30 μg expression vector DNA per plate was used (150-mm plate, 15 μg of each heavy and light chain expression vector). Purification: Purified selenomab and thio-selenomab were buffer exchanged to 100-mM NaOAc pH 5.2 for storage instead of PBS.
Antibody conjugation
Sec conjugation [13]: The purified selenomab or thio-selenomab (1 mg/mL in 100-mM NaOAc pH 5.2) was treated with 0.1-mM dithiothreitol (DTT) and incubated at room temperature (RT; 25°C) for 20 min followed by the addition of 10 equivalents (eq) of iodoacetamide reagents (biotin or PNU-159682). The reaction was incubated at RT for 2 h. Using a 30-kDa centrifugal filter device, the biotinylated thio-selenomab was buffer exchanged to 50-mM EDTA, PBS pH 7.4 prior to Cys conjugation. The PNU-159682-conjugated thio-selenomab was purified with a PD-10 desalting column and brought into 50-mM EDTA, PBS pH 7.4 prior to Cys conjugation. Cys conjugation [14]: The purified thiomab or the Sec-conjugated thio-selenomab (1 mg/mL in 50-mM EDTA, PBS pH 7.4) was reduced with 0.33 mM tris(2-carboxyethyl) phosphine (TCEP) and incubated at 37°C for 2 h. TCEP was removed with a 10-kDa centrifugal filter device. The reduced antibody was treated with 4-mM dehydroascorbic acid (DHAA) at 4°C for 16 h before it was added to 20 eq of methylsulfone phenyloxadiazole (MSODA; fluorescein or MMAF). The reaction was incubated at RT for 2 h. Following fluorescein conjugation, the antibody was buffer exchanged into Dulbecco’s PBS (DPBS) pH 7.2 (Thermo Fisher Scientific) using a 30-kDa centrifugal filter device. Following MMAF conjugation, the antibody was purified with a PD-10 desalting column and brought into DPBS pH 7.2. For antibody-biotin and antibody-fluorescein conjugates, concentrations were estimated by molar absorbance at 280 nm using an extinction coefficient of 210 000 M−1cm−1. For ADCs, the concentration was determined by a bicinchoninic acid (BCA) assay (Pierce BCA Assay Kit, Thermo Fisher Scientific).
Mass spectrometry
All samples were enzymatically deglycosylated with PNGase F (New England Biolabs) overnight at 37°C under reducing conditions (50-mM DTT, PBS pH 7.4). Data were obtained on an Agilent Electrospray Ionization Time of Flight (ESI-TOF) mass spectrometer. Deconvoluted masses were obtained using Agilent BioConfirm software.
Reversed-phase HPLC
Reversed-phase (RP)-HPLC was used to determine an average drug-to-antibody ratio (DAR) for the single-MMAF ADC since we were not able to obtain the data by mass spectrometry. Average DAR was calculated from signal integration of the remaining unconjugated heavy chain in the ADC sample. The light chain of the ADC was used as a standard peak. RP-HPLC was performed on a PLRP-S 1000-Å, 5-μm column (Agilent Technologies). As solvent system, solvent A, water with 0.1% TFA (v/v), and solvent B, acetonitrile with 0.1% TFA (v/v), were used. The RP-HPLC was run at a flow rate of 0.25 mL/min over 45 min. The samples were prepared by reducing the ADC (1 mg/mL in 50-mM Tris-HCl pH 8.0) with 50-mM (final) DTT at 37°C for 30 min. A total of 10 μg of the reduced ADC was loaded onto the RP-HPLC column. The absorbance signal was monitored at 280 nm.
Cell lines
Human BC cell lines SK-BR-3, MDA-MB-231, MDA-MB-468 and MDA-MB-453 were obtained from American Culture Type Collection. Human BC cell line KPL-4 was kindly provided by Dr. Naoto T. Ueno on a Material Transfer Agreement (MTA) with University of Texas MD Anderson Cancer Center (Houston, TX) and with permission from Dr. Junichi Kurebayashi (Kawasaki Medical School; Kurashiki, Japan). Human BC cell line MDA-MB-361/DTY2 was kindly provided by Dr. Gregory P. Adams (Fox Chase Cancer Center; Philadelphia, PA) based on an MTA with Georgetown University (Washington DC). All human BC cells were maintained in complete growth medium (DMEM, high glucose, + glutamate, − pyruvate, supplemented with 10% fetal bovine serum and 1% Pen Strep (Thermo Fisher Scientific)) at 37°C in a humidified 5% CO2 atmosphere.
Cell viability assay
The cells were seeded in a 96-well plate at 5 × 103/well (except KPL-4, 3 × 103/well). Cells were allowed to adhere overnight before the medium was replaced with serially diluted ADCs in complete growth medium. The cells were incubated with the ADC for 72 h before measure for cell viability by a CellTiter 96 Aqueous One Solution Cell Proliferation Assay (Promega) following the manufacturer’s instructions. Cell viability was calculated as a percentage of untreated cells. Viability curves and absolute EC50 values were calculated from GraphPad Prism software version 6.01.
Western blot
Biotin detection: Antibody samples (67 ng) were incubated with 4× NuPAGE loading buffer (Thermo Fisher Scientific) supplemented with 20% (v/v) 2-mercaptoethanol at 90°C for 10 min. The samples were then electrophoresed on a NuPAGE Bis-Tris 4-12% gradient gel (Thermo Fisher Scientific) and transferred on a PVDF membrane (Millipore). The blot was blocked with Western Blocking Reagent (Roche) and probed with horse radish peroxidase (HRP)-conjugated ExtrAvidin (Sigma-Aldrich) or goat anti-human IgG (H+L) (Invitrogen). Immunoreactive bands were developed with ECL Prime Western Blotting Detection Reagent (GE Healthcare) and detected with CL-XPosure Film (Thermo Fisher Scientific). KPL-4 lysates: KPL-4 cells were seeded in a 6-well plate (1 × 106/well) and allowed to adhere overnight. The medium was replaced with 2-nM ADCs or unconjugated trastuzumab diluted in complete growth medium for 24 h. The medium was removed, washed with PBS, and the cells were harvested (trypsinized and washed twice with DPBS). The cell pellets were lysed with radioimmunoprecipitation assay buffer (RIPA: 50-mM Tris-HCl pH 8.0; 150-mM NaCl; 1% NP40; 0.1% sodium dodecyl sulfate; 0.5% sodium deoxycholate; 1:25 protease inhibitor cocktail and phosphatase inhibitors (Roche Applied Science)). The lysates were quantified for total protein content with a BCA assay. Cell lysate (20 μg) was incubated with 4× NuPAGE loading buffer supplemented with 20% (v/v) 2-mercaptoethanol at 90°C for 10 min. The samples were electrophoresed on a NuPAGE Bis-Tris 4–12% gradient gel. The proteins were transferred on an activated PVDF membrane. The blot was blocked with western blocking reagent, incubated with a primary antibody, washed and then incubated with a secondary antibody. (Primary antibodies: mouse anti-human vinculin (VIN-11-5, Sigma Aldrich) and rabbit anti-human phosphor-Chk1 (Ser345, Cell Signaling Technology); secondary antibodies: goat anti-rabbit IgG and HRP-conjugated horse anti-mouse IgG HRP-linked polyclonal antibodies (Cell Signaling Technology)). Immunoreactive bands were developed with ECL Prime Western Blotting Detection Reagent and detected with a CCD camera (Bio-Rad).
Cell cycle analysis
KPL-4 cells were seeded in a 6-well plate (1.25 × 106/well) and allowed to adhere overnight. The medium was replaced with 10-nM ADCs or unconjugated trastuzumab diluted in complete growth medium for 24 h. The medium was removed, washed with DPBS, and the cells were harvested (trypsinized and washed twice with DPBS). Cell pellets were thoroughly resuspended and fixed in 70% (v/v) ice-cold ethanol. Fixed cells were stained with propidium iodide prior to analysis with a Canto II Flow Cytometer (Becton-Dickinson). For each sample, 10 000 events in the G1 gate were recorded. The data were analyzed with FlowJo software (Tree Star). Cell cycle analysis function was used to calculate the subpopulation distribution from events in G0/G1 to G2/M phase. The Sub G1 population was calculated as a percentage of all recorded events.
RESULTS AND DISCUSSION
Generation of a thio-selenomab in IgG1 format
The study reported here began with the generation of an appropriate HER2-targeting (trastuzumab-based) thio-selenomab in IgG1 format. Compared to our previous thio-selenomab in scFv-Fc format [12], the engineered Sec residue was positioned at heavy chain residue 396 (S396U, Kabat numbering), which conjugates to an iodoacetamide derivative more efficiently than a C-terminal Sec. The engineered Cys residue was positioned at heavy chain residue 114 (A114C, Kabat numbering) to enable production of a serum stable conjugate using MSODA (Supplementary Figure 1) [15]. Thio-selenomab (A114C/S396U) IgG1 and control antibodies were expressed and purified to high purity (Supplementary Figure 2).
Dual biotin/fluorescein conjugation
Next, we confirmed the dual conjugation ability of thio-selenomab IgG1 with biotin-iodoacetamide and fluorescein-MSODA (Figure 1). Thio-selenomab IgG1 and control antibodies (thiomab, selenomab and unmutated trastuzumab) were mildly reduced with DTT to uncap the Sec residues (S396U). The antibodies were then treated with biotin-iodoacetamide 1 in 100-mM NaOAc pH 5.2. Next, the antibodies were treated with TCEP to reduce interchain disulfide bonds and uncap the engineered Cys (A114C). The reduced antibodies were partially re-oxidized with DHAA and subsequently treated with fluorescein-MSODA in PBS pH 7.4 supplemented with 50-mM EDTA. The data presented in Figure 1 show that thio-selenomab IgG1 can be dually labeled by both biotin-iodoacetamide and fluorescein-MSODA, as demonstrated by western blot and in-gel fluorescent analyses. The heavy chain of the thio-selenomab (Cys/Sec) showed specific conjugation of both biotin and fluorescein, while the heavy chains of the corresponding thiomab (Cys/WT) and selenomab (WT/Sec) were exclusively conjugated with fluorescein and biotin, respectively. The heavy chain and light chain of wild-type trastuzumab (WT/WT) were not conjugated with either fluorescein or biotin.
Figure 1 .
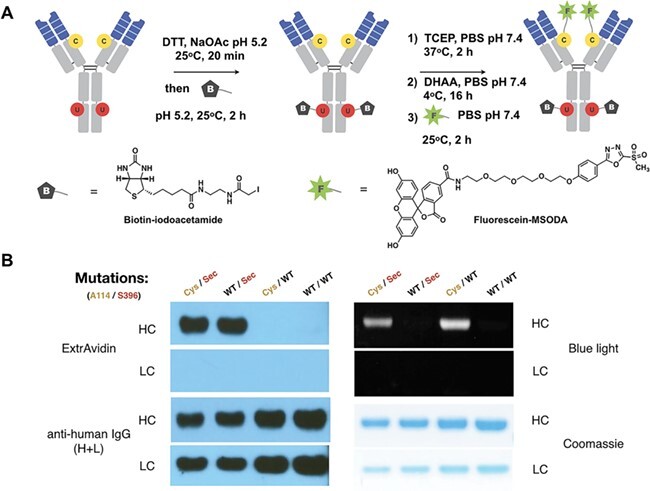
Dual biotin/fluorescein conjugation. (A) Sequential dual labeling of thio-selenomab IgG1. First, the Sec residues (S396U) were reduced and conjugated with biotin-iodoacetamide. The biotinylated antibody was then reduced and partially oxidized prior to the conjugation with fluorescein-MSODA at the engineered Cys residues (A114C). (B) Western blot and in-gel fluorescence revealed the dual site-specific conjugation of the thio-selenomab (Cys/Sec). Only the antibodies with Sec residues were able to conjugate to biotin-iodoacetamide and only the antibodies with engineered Cys residues were able to conjugate with fluorescein-MSODA.
Dual PNU-159682/MMAF conjugation
The HER2-targeting thio-selenomab was utilized to generate a dual-drug ADC with two mechanistically distinct payloads. MMAF, a well-established ADC payload, was selected as a tubulin-targeting agent [1]. PNU-159682 was chosen as a DNA-damaging payload for its potent activity against drug-resistant and non-dividing cancer cells [16–20]. PNU-159682 is considered a virtual DNA crosslinking agent that intercalates and covalently binds to form stable DNA-PNU-159682 adducts [16,17]. PNU-159682 was synthesized and conjugated to an ethylenediamine-triglycine (EDA-Gly3) linker [20]. An iodoacetamide group was then incorporated at the N-terminus of the EDA-Gly3 linker to facilitate the conjugation at S396U (Compound 1, Figure 2A). An MMAF derivative with a C-terminal primary amide was synthesized by solid-phase peptide synthesis and linked to a 6-aminohexanoic acid linker. The MSODA group was coupled to the amino group of the 6-aminohexanoic acid via a glutaric acid spacer (Compound 2, Figure 2B).
Figure 2 .
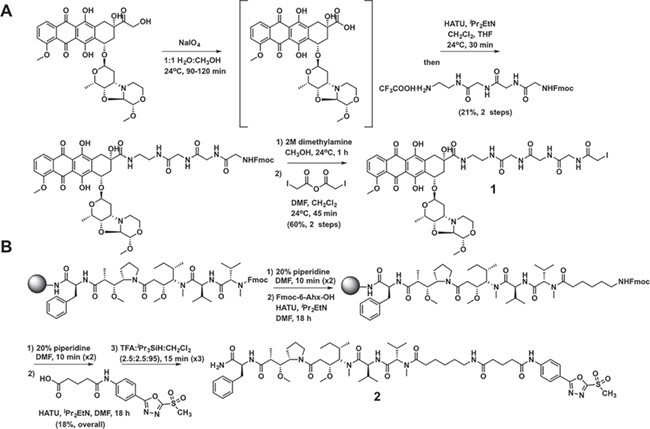
Synthesis of payload-linker compounds. (A) Synthesis of PNU-159682-Gly3-iodoacetamide 1. (B) Synthesis of MMAF-nc-MSODA 2.
The dual-drug ADC was generated by sequentially conjugating compound 1 to S396U and then compound 2 to A114C in a manner analogous (Figure 3) to the biotin/fluorescein dual labeling (Figure 1). The generated anti-HER2 ADCs were characterized by mass spectrometry to reveal a DARPNU-159682 of 1.9 and a DARMMAF of 1.5 (Supplementary Figure 3).
Figure 3 .
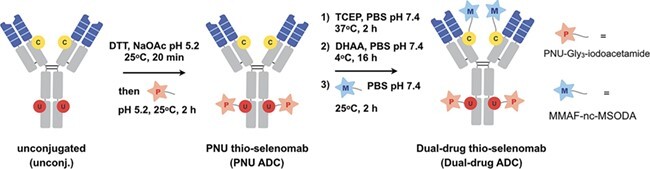
Dual PNU-159682/MMAF conjugation. For sequential dual labeling, S396U was reduced and conjugated with PNU-159682 (PNU)-Gly3-MSODA 1. The antibody was then reduced and partially oxidized to uncap A114C prior to the conjugation with MMAF-nc-MSODA 2.
Cytotoxicity and MOA of dual-drug versus single-drug ADCs
The dual-drug and single-drug ADCs were tested for their cytotoxicity against six BC cell lines (Figure 4 and Supplementary Figure 4). In general, the dual-PNU-159682/MMAF and single-PNU-159682 ADCs exhibited similar potency against HER2-expressing cell lines and were both more potent than the single-MMAF ADC. This suggested that in this experiment, cytotoxicity was primarily mediated by the DNA-damaging payload PNU-159682. Similar findings were recently reported using a dual-PBD dimer/MMAE ADC, which shows dominant cytotoxicity by the PBD-dimer [10]. Domination of DNA-damaging payloads such as PNU-159682 and PBD-dimer could be explained by the fact that these agents are cytotoxic toward both dividing and non-dividing cells. In contrast, a tubulin inhibitor such as MMAF primarily kills actively dividing cells [19].
Figure 4 .
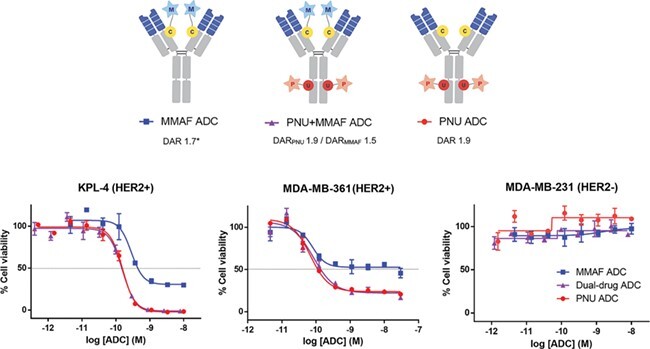
Cytotoxicity of single-drug and dual-drug ADCs. Cytotoxicity graphs for HER2+ BC cell lines KPL-4 and MDA-MB-361/DYT2, and HER2− BC cell line MDA-MB-231 (left to right). Mean ± SD values of triplicates were plotted. *An average DAR of single-MMAF ADC was determined by RP-HPLC.
Next, we sought to understand the MOA of cytotoxicity of the dual-drug ADC. We were particularly interested to probe whether or not MMAF still exhibits its MOA in the dual-drug ADC. MMAF is a tubulin polymerization inhibitor that interferes with cell cycle progression and leads to cell cycle arrest at the G2/M phase [21]. During our cytotoxicity study, we observed that the morphology of KPL-4 cells changed from their typical polygonal shape to a distinctive rounded shape after incubation with MMAF ADC or the dual-drug ADC (Supplementary Figure 5). These round-shaped cells were less prevalent for untreated cells and cells treated with the single-PNU-159682 ADC (Supplementary Figure 5). Such rounded cellular morphology is indicative of arrest at G2/M phase, in contrast to the usual polygonal morphology [22]. This supports the notion that the dual-PNU-159682/MMAF ADC retains the MOA of the MMAF payload despite the apparent dominance of the PNU-159682 payload in the cytotoxicity studies. Cell cycle analysis by flow cytometry was employed to distinguish cells in different phases based on their DNA content. The single-MMAF ADC increased the frequency of cells in G2/M phase in comparison to unconjugated trastuzumab and the single-PNU-159682 ADC, indicating that the MMAF payload caused G2/M cell cycle arrest (Figure 5A and B). The dual-PNU-159682/MMAF ADC also caused a significant increase in G2/M cell population when compared to the single-PNU-159682 ADC sample, confirming that the MOA of MMAF is retained. In addition to G2/M arrest, both single-MMAF ADC and the dual-PNU-159682/MMAF ADC caused a significant increase in sub G1 population. The sub G1 population comprises cells that are undergoing apoptotic DNA fragmentation after exposure to MMAF [21]. The increase in sub G1 population was not observed when cells were exposed to single-PNU-159682 ADC (Supplementary Figure 6). The effect of the PNU-159682 payload can be observed by the same cell cycle analysis. When KPL-4 cells were treated with single-PNU-159682 ADC or dual-PNU-159682/MMAF ADC, a drastic increase in S-phase cell population was observed. This S-phase increase was not observed when cells were treated with either single-MMAF ADC or unconjugated trastuzumab (Figure 5A and C), suggesting that it is associated with the DNA damage activity of PNU-159682. Western blot analysis was then used to correlate the observed S phase arrest and the cellular DNA damage of the PNU-159682 payload. Lysates of KPL-4 cells treated with the single-PNU-159682 ADC or the dual-PNU-159682/MMAF ADC showed that checkpoint kinase 1 (Chk1) is phosphorylated at serine residue 345 (Figure 5D). This phosphorylated Chk1 is associated with DNA damage and is necessary for an intra-S phase cell cycle arrest [23]. It is worth noting that the intensity of pChk1 band in the dual-PNU/MMAF ADC is lower than the one in single-PNU ADC. This difference in pChk1 level may result from the presence of MMAF. In another study, a microtubule-targeting agent was shown to change the level of a DNA damage marker by disrupting intracellular trafficking of DNA repair proteins [24]. We also noted that the intra-S phase arrest from PNU-159682 is more pronounced than the G2/M phase arrest from MMAF in the dual-drug ADC. These data confirm that a DNA-damaging agent dominates a tubulin inhibitor payload.
Figure 5 .
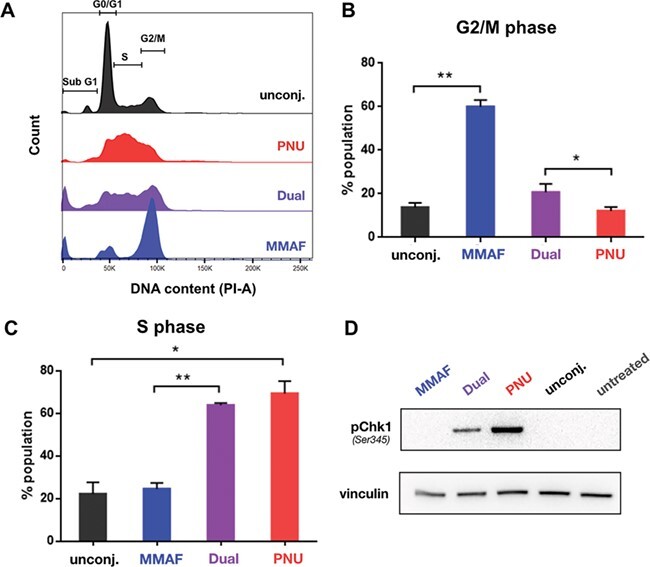
Cell cycle arrest in the presence of single-drug and dual-drug ADCs. (A) Histograms of KPL-4 cells after 24-h treatment with ADCs. (B and C) The single-MMAF ADC (blue) causes significant increase in G2/M phase (G2/M phase arrest), while the single-PNU-159682 (PNU) ADC (red) causes increase in S phase (intra-S phase arrest). The dual-drug ADC (purple) increases the cell population in both S and G2/M phase (intra-S and G2/M phase arrest) when compared to the single-drug ADCs. Cell cycle analysis was performed in three independent experiments. The average number of cells in different phases are plotted as bar graphs (mean ± SD). Statistical significance was calculated with one-way ANOVA, non-parametric (*, P ≤ 0.05 and **, P ≤ 0.01). (D) Western blot analysis of DNA damage markers after 24-h incubation with ADCs and unconjugated trastuzumab. Only the dual-PNU-159682/MMAF and single-PNU-159682 ADCs revealed Ser345-phosphorylated checkpoint kinase 1 (pChk1).
CONCLUSIONS
In summary, we employed dual antibody conjugation using an engineered thio-selenomab to generate a dual-drug ADC bearing two payloads with distinct MOA. The dual-drug ADC was generated by combining the tubulin-targeting payload MMAF and the DNA-damaging payload PNU-159682. Close examination of cell cycle arrest revealed the dual-drug ADC exhibited dual MOA mediated by each payload. While the MMAF payload inhibited tubulin polymerization and caused target cells to arrest in the G2/M phase, the PNU payload caused DNA-damaging and disrupted the cell cycle at the intra-S phase. Our in vitro cytotoxicity data suggest that MMAF and PNU-159682 do not antagonize or synergize with each other.
A particularly interesting application for dual-drug ADCs is the delivery of two different drugs that not only interact additively but synergistically. ADC payload synergism has been suggested, but none have been reported to our knowledge. Payload selection for a dual-drug ADC could benefit from a screening effort to find such payload combinations either as small molecules or as ADCs. Even though synergistic payloads might yield an ultra-potent ADC, it might come at greater risk of on-target and off-target toxicity with no net gain of therapeutic index. On the other hand, exploiting synthetic lethality in cancer with drug combinations could be applied to dual-drug ADCs and is anticipated to yield high therapeutic indices [25]. Interestingly, it has been reported that even drug combinations with no observed additive or synergistic interaction could confer beneficial effects in larger populations due to patient-to-patient variability [26].
In addition to payload interactions, physicochemical properties of drug-linkers for dual-drug ADCs should be considered. Similar to other highly loaded ADCs, hydrophobicity of payloads should be managed by appropriate linker modifications. Linkers can be modified to mask or mitigate hydrophobicity of the payloads. For example, highly PEGylated linkers were used for a dual-MMAF/MMAE ADC and a dual-MMAE/PDB-dimer ADC [9,10]. In the current study, we utilize a combination of MMAF, which contains a polar carboxylic group at its C-terminus, and a PNU-159682 derivative, which is relatively hydrophilic (ClogP = 0.61). Both payloads are also linked to a short spacer to minimize linker hydrophobicity.
Even though our in vitro cytotoxicity results suggest that the combination of PNU-159682 and MMAF does not enhance the potency of PNU-159682 alone, the dual-mechanistic property of the ADC might still have therapeutic utility in the highly heterogeneous in vivo tumor environment. The dual-drug ADC also warrants examination in studies on rate of resistance acquisition similar to previously reported single-drug ADC resistance studies [3,6].
AUTHORS’ CONTRIBUTIONS
N.N., W.R.R. and C.R. conceived and designed the study. N.N. conducted and analyzed all experiments. X.L., L.P. and A.R.N. contributed key biological and chemical reagents. N.N. and C.R. wrote the manuscript with edits from all co-authors.
FUNDING
This study was supported by National Institutes of Health grants R01 CA174844 and CA204484. N.N. received a Predoctoral Fellowship from the Royal Thai Government (Bangkok, Thailand). This is manuscript no. 29823 from The Scripps Research Institute.
CONFLICTS OF INTEREST STATEMENT
None declared.
Supplementary Material
REFERENCES
- 1. Beck, A, Goetsch, L, Dumontet, Cet al. . Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov 2017; 16: 315–37. [DOI] [PubMed] [Google Scholar]
- 2. Chari, RVJ, Miller, ML, Widdison, WC. Antibody–drug conjugates: an emerging concept in cancer therapy. Angew Chem Int Ed 2014; 53: 3796–827. [DOI] [PubMed] [Google Scholar]
- 3. Loganzo, F, Tan, X, Sung, Met al. . Tumor cells chronically treated with a trastuzumab–maytansinoid antibody–drug conjugate develop varied resistance mechanisms but respond to alternate treatments. Mol Cancer Ther 2015; 14: 952–63. [DOI] [PubMed] [Google Scholar]
- 4. Chen, R, Hou, J, Newman, Eet al. . CD30 downregulation, MMAE resistance, and MDR1 upregulation are all associated with resistance to brentuximab vedotin. Mol Cancer Ther 2015; 14: 1376–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. García-Alonso, S, Ocaña, A, Pandiella, A. Resistance to antibody–drug conjugates. Cancer Res 2018; 78: 2159–65. [DOI] [PubMed] [Google Scholar]
- 6. Loganzo, F, Sung, M, Gerber, HP. Mechanisms of resistance to antibody–drug conjugates. Mol Cancer Ther 2016; 15: 2825–34. [DOI] [PubMed] [Google Scholar]
- 7. Housman, G, Byler, S, Heerboth, Set al. . Drug resistance in cancer: an overview. Cancers 2014; 6: 1769–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Fujii, T, Le Du, F, Xiao, Let al. . Effectiveness of an adjuvant chemotherapy regimen for early-stage breast cancer: a systematic review and network meta-analysis. JAMA Oncol 2015; 1: 1311–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Levengood, MR, Zhang, X, Hunter, JHet al. . Orthogonal cysteine protection enables homogeneous multi-drug antibody–drug conjugates. Angew Chem Int Ed 2017; 56: 733–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Kumar, A, Kinneer, K, Masterson, Let al. . Synthesis of a heterotrifunctional linker for the site-specific preparation of antibody-drug conjugates with two distinct warheads. Bioorg Med Chem Lett 2018; 28: 3617–21. [DOI] [PubMed] [Google Scholar]
- 11. Li, X, Patterson, JT, Sarkar, Met al. . Site-specific dual antibody conjugation via engineered cysteine and selenocysteine residues. Bioconjug Chem 2015; 26: 2243–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Li, X, Rader, C. Utilization of selenocysteine for site-specific antibody conjugation. Methods Mol Biol 2017; 1575: 145–64. [DOI] [PubMed] [Google Scholar]
- 13. Li, X, Nelson, CG, Nair, RRet al. . Stable and potent selenomab-drug conjugates. Cell Chem Biol 2017; 24: 433–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Tumey, LN, Li, F, Rago, Bet al. . Site selection: a case study in the identification of optimal cysteine engineered antibody drug conjugates. AAPS J 2017; 19: 1123–35. [DOI] [PubMed] [Google Scholar]
- 15. Patterson, JT, Asano, S, Li, Xet al. . Improving the serum stability of site-specific antibody conjugates with sulfone linkers. Bioconjug Chem 2014; 25: 1402–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Quintieri, L, Geroni, C, Fantin, Met al. . Formation and antitumor activity of PNU-159682, a major metabolite of nemorubicin in human liver microsomes. Clin Cancer Res 2005; 11: 1608–17. [DOI] [PubMed] [Google Scholar]
- 17. Scalabrin, M, Quintieri, L, Palumbo, Met al. . Virtual cross-linking of the active nemorubicin metabolite PNU-159682 to double-stranded DNA. Chem Res Toxicol 2017; 30: 614–24. [DOI] [PubMed] [Google Scholar]
- 18. Yu, SF, Zheng, B, Go, Met al. . A novel anti-CD22 anthracycline-based antibody–drug conjugate (ADC) that overcomes resistance to auristatin-based ADCs. Clin Cancer Res 2015; 21: 3298–306. [DOI] [PubMed] [Google Scholar]
- 19. Junttila, MR, Mao, W, Wang, Xet al. . Targeting LGR5+ cells with an antibody-drug conjugate for the treatment of colon cancer. Sci Transl Med 2015; 7: 314ra186. [DOI] [PubMed] [Google Scholar]
- 20. Stefan, N, Gébleux, R, Waldmeier, Let al. . Highly potent, anthracycline-based antibody–drug conjugates generated by enzymatic, site-specific conjugation. Mol Cancer Ther 2017; 16: 879–92. [DOI] [PubMed] [Google Scholar]
- 21. Francisco, JA, Cerveny, CG, Meyer, DLet al. . cAC10-vcMMAE, an anti-CD30–monomethyl auristatin E conjugate with potent and selective antitumor activity. Blood 2003; 102: 1458–65. [DOI] [PubMed] [Google Scholar]
- 22. Park, JH, Shin, YJ, Riew, TRet al. . The indolinone MAZ51 induces cell rounding and G2/M cell cycle arrest in glioma cells without the inhibition of VEGFR-3 phosphorylation: involvement of the RhoA and Akt/GSK3β signaling pathways. PLoS One 2014; 9: e109055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Feijoo, C, Hall-Jackson, C, Wu, Ret al. . Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J Cell Biol 2001; 154: 913–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Poruchynsky, MS, Komlodi-Pasztor, E, Trostel, Set al. . Microtubule-targeting agents augment the toxicity of DNA-damaging agents by disrupting intracellular trafficking of DNA repair proteins. Proc Natl Acad Sci U S A 2015; 112: 1571–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Chan, DA. Giaccia, AJ (2011) Harnessing synthetic lethal interactions in anticancer drug discovery. Nat Rev Drug Discov 2011; 10: 351–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Palmer, AC, Sorger, PK. Combination cancer therapy can confer benefit via patient-to-patient variability without drug additivity or synergy. Cell 2017; 171: 1678–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.