

Abstract
Defining the variables that impact the specificity of CRISPR/Cas9 has been a major research focus. Whereas sequence complementarity between guide RNA and target DNA substantially dictates cleavage efficiency, DNA accessibility of the targeted loci has also been hypothesized to be an important factor. In this study, functional data from two genome-wide assays, genome-wide, unbiased identification of DSBs enabled by sequencing (GUIDE-seq) and circularization for in vitro reporting of cleavage effects by sequencing (CIRCLE-seq), have been computationally analyzed in conjunction with DNA accessibility determined via DNase I-hypersensitive sequencing from the Encyclopedia of DNA Elements (ENCODE) Database and transcriptome from the Sequence Read Archive to determine whether cellular factors influence CRISPR-induced cleavage efficiency. CIRCLE-seq and GUIDE-seq datasets were selected to represent the absence and presence of cellular factors, respectively. Data analysis revealed that correlations between sequence similarity and CRISPR-induced cleavage frequency were altered by the presence of cellular factors that modulated the level of DNA accessibility. The above-mentioned correlation was abolished when cleavage sites were located in less accessible regions. Furthermore, CRISPR-mediated edits were permissive even at regions that were insufficient for most endogenous genes to be expressed. These results provide a strong basis to dissect the contribution of local chromatin modulation markers on CRISPR-induced cleavage efficiency.
Keywords: CRISPR, chromatin, bioinformatics, GUIDE-seq, CIRCLE-seq, RNA-seq, DNase-seq
Graphical Abstract
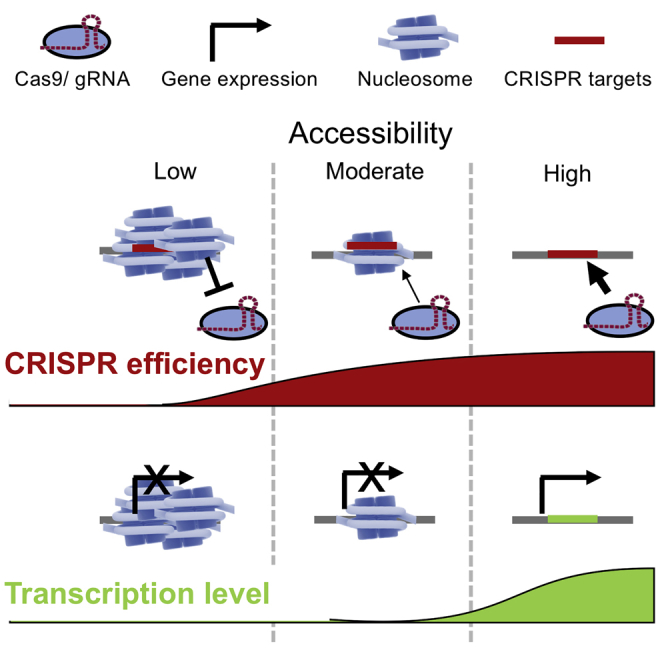
Chromatin configurations that occupy CRISPR targeting sites have been shown to modulate CRISPR-mediated cleavage efficiency. Our computational analyses using integrated genomic data and multiple quantification models have revealed a threshold of DNA accessibility required for detectable CRISPR-mediated cleavage events that is less than what is needed for gene expression.
Introduction
The CRISPR system that was first discovered as a bacterial defense mechanism has recently been re-engineered for genome editing in eukaryotic cells.1,2 The CRISPR system has been shown to recognize and cleave target loci using a guide RNA (gRNA) transcribed from the CRISPR locus and an RNA-guided Cas.3,4 The gene-editing process has been shown to begin with the recognition and binding between the Cas protein and a protospacer adjacent motif (PAM); subsequently, this process is followed by a progressive hybridization between the gRNA and the chromosomal DNA adjacent to the PAM, termed target hereafter. Cas in turn induces double-stranded breaks (DSBs) followed by endogenous DNA repair responses that result in sequence editing at the target locus.1,2,5, 6, 7
The contribution of gRNA:target sequence similarity has been well characterized and is a major determinant of CRISPR-induced cleavage efficiency. Data from screening techniques have suggested that CRISPR-induced cleavage can occur at the target loci with up to seven mismatches across the 20-bp complementary sequence.8,9 The relationship between a mismatch position and cleavage efficiency has been quantified and the resulting data organized into a position-specific penalty matrix (which we will refer to as the MIT matrix, developed by Hsu et al.10 at the Massachusetts Institute of Technology) and with additional functional studies, leading to the development of the cutting frequency determination matrix (known as the CFD matrix) that has defined the contribution of the count, position, and identity of each nucleotide mismatch across gRNA:target pairs10,11 and their relationship to cutting efficiency.
The tolerance of mismatches in each gRNA:target pair has raised some concern that CRISPR may cause unintended sequence modifications at sites other than the designated target in the host genome.10, 12, 13, 14 This has prompted the development of a number of genome-wide CRISPR-induced cleavage screening techniques that detect CRISPR-cleaved loci tagged by molecular markers followed by genome sequencing to locate the modified sites.15, 16, 17, 18 Specifically, the genome-wide, unbiased identification of DSBs enabled by the sequencing (GUIDE-seq) technique introduces oligodeoxynucleotides (ODNs), a 34-bp exogenous DNA marker, into living cells along with plasmids encoding SpCas9 (Cas derived from Streptococcus pyogenes) and the desired gRNAs.19 The ODNs integrate into the DSBs in the chromosomes during the non-homologous end joining (NHEJ) DNA repair process in treated cells integrating into approximately 51% of all DSB events on average across all transfected cells.20 These investigators also repeated this experimental approach to exclude the effect of cellular factors such as nucleosomes and chromatin structures on CRISPR-induced cleavage efficiency. They developed the circularization for in vitro reporting of cleavage effects by sequencing (CIRCLE-seq) technique; this technique uses in vitro-constructed Cas9 and gRNA complexes to cleave specially prepared circles of purified genomic DNA that are then selectively amplified and sequenced using next generation sequencing (NGS).21
Previous studies have shown that sequence complementarity scoring matrices can explain only 20% of CRISPR-induced cleavage efficiency where the gRNA:target pairs have more than two mismatches; however, CFD performed with better specificity and sensitivity than the MIT matrix.11 Therefore, we hypothesize that cellular factors play a substantial role in determining CRISPR-induced cleavage efficiency in these situations. However, the quantitative assessment of higher-order cellular factor complexes on CRISPR-induced cleavage efficiency remains poorly characterized.
Nucleosome occupancy and chromatin structure have been demonstrated to be crucial epigenetic regulators for DNA accessibility and subsequent gene expression.22,23 DNase I-hypersensitive site sequencing (DNase-seq) has been used to measure DNA accessibility of genomic DNA in intact nuclei.24,25 Whole-genome screening of the CRISPR-Cas9 binding landscape has been utilized to correlate the effect of DNA accessibility using chromatin immunoprecipitation (ChIP) with deactivated Cas9 (dCas9) or a lentiviral target-site library in human cell lines.26, 27, 28 Instead of genome-wide screening, reporter cell lines with an inducible system that modulates chromatin states were used to demonstrate the direct impact of DNA accessibility on CRISPR-induced DSB formation.29, 30, 31 In general, previous studies have suggested that the frequency of DSB formation induced by CRISPR-Cas9 was significantly lower in heterochromatic regions compared with euchromatic regions.
In this study, the effect of DNA accessibility on CRISPR-Cas9 cleavage efficiency was quantified in an effort to better estimate CRISPR-induced cleavage efficiency in cells. We assumed that chromosomal sites edited only in naked DNA (CIRCLE-seq), but that remained unedited in intact chromatin (GUIDE-seq), would correspond to chromosomal regions of low DNA accessibility (DNase-seq). Although the cleavage detection assays have often been implemented in the identification of off-target cleavage events, we included all detectable events (desired and undesired cleavage sites) present in the study, to generalize the observation across assays. Overlaying the aforementioned datasets by chromosomal locations with DNase-seq conducted on the same cell lines, we discovered that the local DNA accessibility and gRNA:target sequence similarity are not mutually exclusive processes. Both the degree of sequence complementarity and the level of DNA accessibility dictate the amount of CRISPR-induced cleavage. The observations presented here elucidate the role of DNA accessibility on the CRISPR system and have provided insight into the cleavage process to guide future investigation on CRISPR efficiency in a given target cell population.
Results
The Vast Majority of the Potential Cleavage Sites Were Not Accessible When Cellular Factors Were Present
Chromatin structure has been shown to be one of the cellular factors that affect the cleavage efficiency of CRISPR-Cas9. DNase-seq was used as the measurement of DNA accessibility at the CRISPR-induced cleavage sites with either the presence (GUIDE-seq) or absence (CIRCLE-seq) of cellular factors. We hypothesized that the CRISPR-induced cleavage sites that were identified by only CIRCLE-seq, subsequently designated CS-only subset, possess lower DNA accessibility than those identified by both GUIDE-seq and CIRCLE-seq, subsequently designated GS and CS subsets, respectively (Figures 1 and 2A). GUIDE-seq identified 374 CRISPR-induced cleavage sites among the four gRNAs examined in HEK293T and the six gRNAs examined in U2OS cells, whereas CIRCLE-seq identified 4,138 cleavage sites using the same set of gRNAs and cell lines. Considering the GUIDE-seq-identified cleavage sites, 94.9% (355/374 cleavage sites) were recovered by CIRCLE-seq. However, CIRCLE-seq identified an additional 3,783 cleavage sites (Figure 2A). This has suggested that a vast majority of the potential cleavage sites were not accessible in living cells. The lack of DNA accessibility may be one of the cellular factors that mask the potential CRISPR cleavage sites in living cells.
Figure 1.

The Schematic Diagram Demonstrates the Hypothesis that Presence of Cellular Factors in the GUIDE-Seq Experiment Reduces CRISPR-Induced Cleavage Efficiency
The CRISPR-induced cleavage efficiency could be negatively affected by the lack of DNA accessibility even with high sequence similarity between gRNA and target. The gray shade represents local DNA accessibility. The shade of potential target sites represents the sequence similarity of gRNA:target pairing. Note that the sequence similarity could be implied by any matrices that describe the sequence recognition pattern for the CRISPR-Cas9 system. The thickness of black arrow represents the level of CRISPR-induced cleavage efficiency. The gray shade behind the nucleosome represents the DNase I hypersensitivity detected in the DNase-seq assay system.
Figure 2.

CRISPR-Cas9 Is More Effective at Targeting More Accessible Regions of DNA
(A) The Venn diagram displays the number of combined cleavage sites from four gRNAs in HEK293T cells or six gRNAs in U2OS cells identified by both GUIDE-seq (GS; brown) and CIRCLE-seq (CS; green). (B) The cleavage sites that both GUIDE-seq and CIRCLE-seq identified (designated GS and CS) show higher DNA accessibility than those sites identified by only CIRCLE-seq (CS only). The DNA accessibility of cleavage sites was the DNase-seq read depth per million mapped reads within a 50-bp window flanking the DSB positions (termed RPM). The boxplot shows the distribution of DNA accessibility in both groups. The box represents 50% quantile, and the line inside the box represents the median. The error bar represents the maximum and minimum of data distribution. *p < 0.001, two-tailed t test. (C) The DNA accessibility normalized to the mean DNase-seq RPM of the CS-only subset. *p < 0.001, two-tailed t test.
To quantitatively test the overall impact of DNA accessibility, we quantified the DNase hypersensitivity for each CRISPR-induced cleavage site as a continuous variable by calculating the DNase-seq reads per million mapped reads (RPM) within a 50-bp window centered on the DSB sites induced by CRISPR-Cas9. This analysis showed that the average DNA accessibility of the GS and CS subset was 1.6-fold higher than the average for the cleavage sites in the CS-only subset (Figures 2B and 2C). The phenomenon remained significant when the analysis was performed using individual cell types (Figure S1). It is worth noting that the distributions of DNA accessibility were similar across individual gRNAs (Figure S2). This result indicated that CRISPR-Cas9 could not effectively target regions with low DNA accessibility.
Relationship between Sequence Similarity and CRISPR-Induced Cleavage Frequency Varies with the Level of DNA Accessibility
It has been known that the sequence complementarity between gRNA and target DNA (termed gRNA:target sequence similarity in subsequent analyses) plays a major role in determining cleavage efficiency. A multiple linear regression analysis was performed to test the relationship between CRISPR-induced cleavage frequency (number of cleavage events per million mapped reads [CPM]), gRNA:target sequence similarity (CFD scoring matrix adopted from Doench et al.11), and DNA accessibility (DNase-seq RPM), as well as their interaction terms. The statistical analysis of these results has demonstrated that the CFD score alone accounts for 21.5% of the variation in the CRISPR-induced cleavage frequency in the GS and CS subset, whereas log-transformed DNase-seq RPM alone was not significantly correlated with log-transformed CPM (Table 1). By adding the DNA accessibility to fit an additive model with CFD score, the log-transformed RPM did not significantly contribute to the correlation with log-transformed CPM. However, regressing with an interaction term between CFD and log-transformed RPM was positively correlated with log-transformed CPM using the GS and CS subset, but not the CS-only subset (Table 1; Table S1). These results have suggested that the interaction between gRNA:target sequence similarity and DNA accessibility together impact the cleavage frequency in cells.
Table 1.
The Interaction between CFD Score and DNA Accessibility Impacts the Cleavage Frequency in the GS and CS Subset
| Model | Parameters | p Value | Adjusted R2a |
|---|---|---|---|
| sequence similarity | <0.001b | 0.215 | |
| DNA accessibility | 0.666 | −0.002 | |
| sequence similarity | <0.001b | 0.214 | |
| DNA accessibility | 0.543 | ||
| sequence similarity | 0.563 | 0.222 | |
| DNA accessibility | 0.192 | ||
| sequence similarity × DNA accessibility | 0.029b |
The multiple regression analysis was performed by adding independent variables and interaction of independent variables sequentially to the models. CFD, nucleotide-specific scoring matrix for gRNA:target pair; CPM, number of cleavage events per million mapped reads; RPM, DNase-seq reads per million mapped reads within 50-bp window flanking the DSB positions.
Adjusted R2 was used to account for the number of independent variables each model has.
The beta coefficient is significantly different from zero under the t test with a two-tailed p < 0.05.
DNA Accessibility below a Threshold Completely Abrogates the Effect of the gRNA:Target Similarity on CRISPR-Induced Cleavage Frequency
We performed a stepwise correlation test to understand how the gRNA:target sequence similarity and DNA accessibility interact together to determine the CRISPR-induced cleavage frequency in the GS and CS data subsets. A dot plot in a three-dimensional space was used to visualize the distribution among these three variables (Figure 3A). A surface plot was generated using the nearest-neighbor method described above to depict the spatial relationship among the variables explored in this analysis. The surface plot showed the trend of altering the beta coefficient (β) between CFD and CPM changes across different DNase-seq RPM (Figure 3B). In addition, the top 15% of ranked CFD (N = 53) showed a significant correlation between CPM and RPM, which echoed the impact of DNA accessibility on CRISPR-induced cleavage efficiency (Figure S3).
Figure 3.

Low DNA Accessibility Abrogated the Correlation between gRNA:Target Sequence Similarity and CRISPR-Induced Cleavage Frequency
CFD score, RPM, and CPM of cleavage sites in the GS and CS subset (N = 355) were used in this analysis. (A) The three-dimensional scatterplot of sequence similarity, DNA accessibility, and CRISPR-induced cleavage frequency using the CRISPR-induced cleavage sites listed in the GS and CS subset. Each dot represents a CRISPR-induced cleavage site identified by both GUIDE-seq and CIRCLE-seq. CPM represents the number of cleavage events at a CRISPR-induced cleavage site detected by GUIDE-seq; sequence similarity represents the likelihood of CRISPR cutting based on the sequence between gRNA and target using CFD matrix; RPM represents the DNA accessibility at a CRISPR-induced cleavage site. (B) The surface plot estimated by the nearest-neighbor method described in the Materials and Methods. The sequence similarity is estimated by the position-specific matrix of CFD score [0,1] that describes the cleavage possibility of gRNA:target pair at the detected sites. Red represents high cleavage frequency, whereas blue represents low cleavage frequency identified by the GUIDE-seq technique. (C) Contour map of CRISPR-induced cleavage frequency based on the grids of CFD score and DNase-seq RPM; a top-down view of (B). (D) The beta coefficient between CFD and CRISPR-induced cleavage frequency at given 15% quantile of DNA accessibility. Note that the data point was the lower boundary of a given quantile. The shaded regions represent 95% confidence intervals of the t test. The horizontal dashed line at beta coefficient equal to 0 represents the threshold of the significance of the beta coefficient. The correlation was not significant when the 95% confidence interval covered the horizontal line. (E) The beta coefficient relative to the first quantile that contained the cleavage sites with the top 15% DNA accessibility in the GS and CS subset. The dashed line represents the regions that were not significant in the Wald test (D). The right vertical lines represent the threshold of DNA accessibility that started to affect the significance between CFD and CRISPR-induced cleavage frequency. The left vertical line represents the threshold such that the correlation between homology and CRISPR-induced cleavage efficiency was insignificant anywhere below the DNA accessibility. (F) Correlation between CRISPR-induced cleavage frequency and CFD score of the 15% most accessible sites (left panel) or 15% least accessible sites (right panel) in the GS and CS subset. p value of Wald test for a hypothesis test that the slope is 0. β1, beta coefficient of simple linear regression.
The correlations among different accessible sites were further analyzed to dissect, in greater detail, the role of DNA accessibility on CRISPR activity. A 15% quantile of ranked RPM with a 1% sliding window was used to calculate the stepwise correlations between sequence similarity and CRISPR-induced cleavage frequency in GS and CS subsets as described above (Figure 3C). The beta coefficient between gRNA:target similarity and CRISPR-induced cleavage frequency represents the degree of CPM change when CFD varies; the results showed that the beta coefficients were always positive and yet decreased when the DNA accessibility decreased (Figure 3D). As shown in Figures 3D and 3E, the Wald tests across high DNA accessibility quantiles were always significant until the lower boundary of the quantile approached log-transformed DNase-seq RPM at −1.889. This result suggested that the effect of sequence similarity on CRISPR-induced cleavage efficiency has been modulated by the level of DNA accessibility. More importantly, the correlation became insignificant when DNA accessibility was below log-transformed DNase-seq RPM of −2.240, indicating that DNA accessibility below the threshold abrogated the positive effect of sequence similarity on CRISPR-induced cleavage frequency. The data points at the top 15% and bottom 15% of ranked DNase-seq RPM (N = 53) were selected from the GS and CS subsets to demonstrate the change in correlation between gRNA:target similarity and CRISPR-induced cleavage frequency (Figure 3F). In the top 15% accessible sites, CFD and CRISPR-induced CPM were significantly and positively correlated (adjusted R2 = 0.508; p < 0.001). This type of correlation was not evident in the less accessible regions in the GS and CS subsets, suggesting that DNA accessibility moderates the correlation.
As for the CS-only subsets without the presence of cellular factors, the modulation mediated by DNA accessibility was not observed in the CS-only subsets (Figures S4A–S4E). The correlation between gRNA:target similarity and CRISPR-induced cleavage frequency maintained at a mean of 0.156 ± 0.0267 and was always significant as expected, given this assay does not have cellular factors (Figure S4D), albeit the correlation coefficient is relatively low (Table S1; Figure S4D). The relative beta coefficient in the CS-only subset (Figure S4E) was not reduced when the DNA was less accessible compared with the result of the GS and CS subsets (Figure 3E). For example, the top and bottom 15% quantile of ranked DNA accessibility in the CS-only subset exhibited a similar slope between CFD score and CPM (Figure S4F). These results have indicated that the CIRCLE-seq dataset was not affected by DNA accessibility, which was consistent with the premise that all cellular factors were removed during the catalytic reaction of CRISPR-induced cleavage events in the CIRCLE-seq protocol.
Chromatin Accessibility Required for a CRISPR-Mediated Cleavage Reaction Was Significantly Less Than that Required for Endogenous Gene Expression
Although the data have shown that low DNA accessibility altered the contribution of gRNA:target complementarity to CRISPR-mediated cleavage (Figures 3D and 3E), 26.8% and 44.0% of 355 cleavage sites were observed in low accessible regions below thresholds at log-transformed RPM of −2.240 and −1.889, respectively (Figure S5A). To test whether the thresholds of DNA accessibility mentioned above were comparable with the chromatin environment of transcribing genes, the local DNA accessibility at the promoters of expressed genes was evaluated and compared with local DNA accessibility at CRISPR-induced cleavage sites in the GS and CS subset. The gene expression profiles were positively correlated between untreated HEK293T and U2OS cells (R2 = 0.673; Figure S6), which validated the compatibility of datasets from independent publications. The corresponding DNase-seq RPM of each expressed gene was calculated at a window of 1,000 bp upstream of the transcription start site (TSS) and 200 bp downstream to cover the majority of promoter positions across the human genome, as previously described.32 The mean DNA accessibility flanking the CRISPR-induced cleavage sites in the GS and CS subset was 5.4-fold less than the mean DNA accessibility flanking the TSS of expressed genes (Figure 4A). Furthermore, 47.4% of CRISPR-induced cleavage sites were identified at chromosomal regions with DNA accessibility lower than the log-transformed DNase-seq RPM of −1.889 where the effect of gRNA:target similarity was abrogated (Figure 4B). Conversely, only less than 3.1% of human genes were expressed at the same level of DNA accessibility (Figure S5B). This suggested that the amount of accessibility needed for CRISPR-Cas9 cleavage was typically less than that needed for normal gene expression. This statement holds true when datasets acquired from either HEK293T or U2OS cells were analyzed separately (Figures S7–S9). These results indicated that the CRISPR-Cas9 system will likely not need large, global chromatin rearrangement to effectually cleave its intended target site. However, adequate DNA accessibility was required, but not sufficient, for the completion of transcription. Therefore, we cannot exclude the possible roles of other regulators on transcription. This result allowed us to re-interrogate the necessity of cell activation treatment that may cause undesired gene activation during the CRISPR-based therapy. This is a critical consideration for aiding the development of CRISPR-based therapy in vivo.
Figure 4.

Chromatin Accessibility Required for CRISPR-Mediated Cleavage Reaction Was Significantly Less Than that for an Endogenous Gene to Express
(A) The mean DNA accessibility flanking CRISPR-induced cleavage sites (N = 355) was significantly lower than that flanking the TSS of expressed genes (N = 8,619). The error bar represents the SD of sampling distribution. Gray dots represent the data points in each dataset. ∗p < 0.05, two-tailed t-test. (B) The green curve represents the cumulative percentage of CRISPR-induced cleavage sites identified in the GS and CS subset. The red curve represents the cumulative percentage of expressed genes detected in HEK293T and U2OS cells. The blue curve represents the relative β to the first quantile that contained the cleavage sites with the top 15% DNA accessibility in the GS and CS subset. The gray vertical lines represent the DNA accessibility thresholds at RPM of −2.240 (left) and −1.889 (right), where DNA accessibility abrogated the significance between CFD and CRISPR-induced cleavage frequency. The percentage shown on the plot indicates the cumulative percentage at the intersection of cumulative percentage curves and DNA accessibility thresholds. The blue curve and gray threshold lines are adopted from Figure 3C.
Discussion
Previous studies using dCas9 screening have suggested that the DNA accessibility implicated by DNase I sensitivity was a significant factor for the CRISPR-Cas9 binding efficiency.26,27 However, further studies have demonstrated that there are distinctive features between dCas9 binding efficiency and CRISPR-induced cleavage efficiency using catalytically active Cas9.19,28 In this study, both GUIDE-seq and CIRCLE-seq assays measured cleavage frequency rather than the binding frequency, providing a more useful measure of editing potential. By comparing the cleavage sites identified by the GUIDE-seq platform or not, the results have suggested that low DNA accessibility was a significant cellular factor that protected potential target sites from being cleaved by CRISPR-Cas9 (Figures 2B and 2C). This study has demonstrated the significance of DNA accessibility using datasets across different platforms with true positive (GS and CS subset) and true negative (CS only subset) experimental conditions. It is worth noting, however, that the sensitivity of ODN insertion events in the GUIDE-seq assay could be another hidden variable that may affect the number of detectable cleavage events when compared with results obtained with the CIRCLE-seq technology.19,21
The positive correlation between DNA accessibility and CRISPR-induced cleavage efficiency has been demonstrated in previous studies using either DNase-seq or ATAC-seq in human cell lines and zebrafish embryonic cells.28,31,33,34 In the study presented here, DNA accessibility was assessed by DNase-seq RPM instead of defining enriched regions of DNase activity as reported in previous studies.35, 36, 37, 38, 39, 40 Our results have indicated that levels of DNA accessibility impact CRISPR-Cas9 activity across the cleavage sites that occurred in living cells. The data suggested that the level of DNA accessibility has a gradient effect with respect to CRISPR-induced cleavage frequency (Figures 3C and 3D). The results reported herein support previous observations and have provided a more robust approach and greater statistical rigor.28,31,33,34 More importantly, DNA accessibility below a threshold further abrogated the contribution of gRNA:target sequence similarity to CRISPR-induced cleavage frequency (Figure 3E). In contrast, the impact of DNA accessibility on CRISPR-induced cleavage frequency was not observed in the cleavage sites identified by CIRCLE-seq (CS-only subset; Figures S4C–S4E). As such, the CRISPR-induced cleavage frequency in CIRCLE-seq was significantly correlated with gRNA:target similarity predicted by the CFD score at a constant level regardless of DNA accessibility. These observations are consistent with the premise of CIRCLE-seq and GUIDE-seq with respect to the presence of nucleosomes during CRISPR treatment, which has suggested that the effect of DNA accessibility we described is practical.
The results showed that DNA accessibility should be included in the prediction of CRISPR-induced cleavage efficiency. Singh et al.41 previously integrated the DNase-seq data into the estimation of CRISPR cleaving likelihood in the CROP-IT algorithm. The predicted cleavage efficiency was proportional to the number of cell types that shared particularly hypersensitive sites as a linear function. In this study, we observed a DNA accessibility threshold that fully abrogated the effect of gRNA:target similarity on the CRISPR-Cas9 reaction. This relationship could be illustrated as a rectifier activation function such that the correlation was fully masked when DNA accessibility was below a threshold, whereas the beta coefficient between gRNA:target similarity and observed cleavage efficiency was a function of DNA accessibility above the threshold. However, it should be noted that the DNase-seq RPM thresholds identified by both cell lines combined in this study may not be generalizable to all CRISPR-Cas systems or cell types (Figures S7 and S8). The conclusions of these experimental studies can be further bolstered by examining additional gRNAs and cell lines to determine whether the pattern holds true. Nevertheless, it has provided a preliminary framework to investigate the relationship between chromatin structure and CRISPR-Cas9 specificity in cells for more detailed experiments in the future.
Cellular Factors that May Contribute to the Equation of Cleavage Efficiency
The scoring matrices previously developed have not effectively fit the observed cleavage frequency in living cells, even with the CFD score developed recently using large-scale screening. These matrices correlated significantly better when there was only one base pair mismatch at the gRNA-targeting regions, whereas the correlation coefficient reduced to approximately 20% at the targets that had more than two mismatches.11 This points out the need of better algorithms that could explain the sequence similarity required for the CRISPR-Cas9 system. A recent effort described the process of cleavage involving a sequential order of PAM recognition, R-loop formation, and cleavage within the context of an enzyme kinetic model.42 Again, the DNA accessibility represented only the collective consequence of upstream cellular factors including epigenetic modulation. It will require more advanced studies, however, to understand the underlying mechanisms to which the change of CRISPR cleavage events was attributed. The methylation status of DNA, including methylation at CpG sites, may contribute to cleavage efficiency. Although Hsu et al.10 did not observe a significant impact of DNA methylation status on cleavage efficiency, the dCas9 binding landscape assay conducted by Wu et al.26 suggested a negative correlation between the level of CpG methylation and CRISPR-binding activity at given target sites. These observations were consistent with the evidence that gRNAs that pair with the complementary strand promote R-loop formation after the recognition of the PAM sequence by Cas9,6,43,44 whereas the level of DNA methylation was negatively correlated with R-loop formation observed in general transcription.45,46 The modification of histones has also been correlated with the CRISPR binding efficiency including H3 acetylation (H3ac),29 H3K9me3,29 H3K27me3,30 and H3K4me3.28,31 Based on these studies, it will be crucial to further examine how specific types of acetylation could be quantified as part of the function of DNA accessibility.
Implication of Chromatin Accessibility with Respect to CRISPR-Cas9 Activity
The potential of CRISPR-Cas9 in the biomedical science and biotechnology industries has driven numerous studies to characterize and improve the specificity and sensitivity of the CRISPR-Cas9 system. The goal has been to increase gene-editing efficiency and optimize safety, especially in the treatment of human disease. The present analyses in conjunction with previous studies show the significance of chromatin accessibility on CRISPR-induced cleavage frequency. It will now be crucial to understand the change in chromatin states at the intended targets with different cell types and corresponding experimental treatments in order to optimize on-target efficiency. One application of CRISPR-Cas9 with promising therapeutic potential has been the excision and/or mutagenesis of integrated HIV-1 proviral DNA in infected cells.47, 48, 49, 50, 51, 52 Studies have suggested that the transcription from the integrated proviral HIV-1 genome is highly regulated by the nucleosomes nuc-0 or nuc-1 on the long terminal repeat (LTR) and histone modulators interacting with transcription factors during latent infection.53, 54, 55, 56, 57 The provirus-associated nucleosomes that were maintained in highly heterochromatic status have been thought to be one of the mechanisms to keep viral transcription at a low level.58 It is therefore important to know what level of DNA accessibility the CRISPR system may be required to facilitate HIV-1 provirus disruption/excision at the HIV-1 integration loci by using gRNAs that target the HIV-1 LTR regions.59
The results presented here have demonstrated that the CRISPR-Cas9 system was significantly more permissive to low accessibility regions than the eukaryotic transcription machinery (Figure 4). This result has implied that a CRISPR-based therapy could be efficacious with subtherapeutic or no cell activation treatments. For example, T cell activation with PMA/ionomycin was commonly used to make integrated HIV-1 provirus more susceptible to CRISPR-mediated gene editing.49,60 CRISPR-mediated knockout efficiency has been shown to vary across different target genes in human primary T cells activated by anti-CD3/CD28 or PMA/ionomycin for the use of immunotherapy, whereas unstimulated T cells showed poor editing efficiency.61 However, the use of cell activation agents could adversely affect regular cell metabolism and gene expression profiles, thus hindering the development of CRISPR-based therapy in vivo. The experimental approach used in these analyses has provided an opportunity to better control the DNA accessibility that has prevented unnecessary gene activation while preserving effective CRISPR-Cas9 cleavage for the development of CRISPR-based therapy in conjunction with cell activation drugs and/or histone modification drugs in vivo. Hence, ongoing experiments will be of importance to interrogate whether the CRISPR-based therapy could be administered in conjunction with a low amount of exogenous activation agents that optimize DNA accessibility without excessive side effects due to undesired gene activation.
The present study has demonstrated that DNA accessibility and gRNA:target similarity interact with CRISPR-induced cleavage efficiency in human cell lines. The results further suggested that compressed chromatin abrogated the correlation between gRNA:target similarity and CRISPR-induced cleavage frequency, even omitting moderate sequence similarity between the gRNA and its target. More importantly, the CRISPR-Cas9 system required sufficient DNA accessibility to catalyze sequence editing; however, the required level of DNA accessibility for CRISPR-Cas9 reaction was significantly less than that used for endogenous genes to be expressed.
Materials and Methods
Public Dataset Acquisition
The dataset resources analyzed in this study are summarized in Table 2. The raw-read data of previous GUIDE-seq, and CIRCLE-seq runs were graciously shared by Dr. Joung. The DNase-seq and RNA-seq datasets were downloaded from NCBI Sequence Read Archive (SRA) or the Encyclopedia of DNA Elements (ENCODE) database by their indicated accession number. The technique, treatment, number of gRNAs, and cell lines are indicated in Table 2. The total number of detected cleavage sites by GUIDE-seq and CIRCLE-seq are listed in Tables S2–S4. The list of detected cleavage sites for GUIDE-seq, named GS in the manuscript, and CIRCLE-seq, named CS, are listed in Tables S5 and S6, respectively. The assays were all performed with unstimulated cell lines or untreated controls. We acknowledge the possibility that the experimental variation among independent studies may affect the results.
Table 2.
Description of Datasets and Corresponding Techniques Analyzed in This Study
| Technique | Target Detection | Treatment | gRNAs | Cell Line | Data Resource |
|---|---|---|---|---|---|
| GUIDE-seq | unbiased detection of CRISPR-induced cleavage sites in living cells | Cas9/gRNA expression vector transfected by nucleofection | 4 | HEK293T | SRA: SRP050338 and directly supplied19 |
| 6 | U2OS | SRA: SRP050338 and directly supplied19 | |||
| CIRCLE-seq | unbiased detection of CRISPR-induced cleavage sites on purified genomic DNA | RNA-guided nuclease (RGN) complex in vitro | 4 | HEK293T | SRA: SRP103697 and directly supplied21 |
| 6 | U2OS | SRA: SRP103697 and directly supplied21 | |||
| DNase-seq | genome-wide DNA accessibility detecting DNase I hypersensitivity | DNase I digestion on isolated nuclei | N/A | HEK293T | ENCODE: 1ENCFF500HTP72 |
| N/A | U2OS | SRA: SRR441399067 | |||
| RNA-seq | transcriptome | untreated cell culture | N/A | HEK293T | SRA: SRP08096663 |
| N/A | U2OS | SRA: ERP00194862 |
ENCFF500HTP is the accession number in the ENCODE Project. Other data resources with accession number SRP have been stored in Sequence Read Archive (SRA). U2OS, human osteosarcoma epithelial cell(s) or cell line.
Data Preprocessing
GUIDE-Seq
The raw-read data of previous GUIDE-seq runs were processed using the implementation of the guideseq analysis pipeline as previously published (https://github.com/aryeelab/guideseq) using default parameters. In brief, the detected cleavage sites were tabulated by guideseq upon the detection of double-stranded oligodeoxynucleotide (dsODN) breaks induced by CRISPR-Cas9.19 The output of genomic locations indicating CRISPR-induced cleavage sites and corresponding numbers of CPM were used for subsequent analysis.
CIRCLE-Seq
The raw-read data of previous CIRCLE-seq were processed using the implementation of the circleseq analysis pipeline previously published (https://github.com/tsailabSJ/circleseq) using the default parameters. In brief, CIRCLE-seq detects the DSBs on sheared and circularized genomic fragments induced by gRNA-Cas9 RNA-guided nuclease (RGN) complex in vitro. The tabular output of genomic locations as detected by CIRCLE-seq and corresponding numbers of cleavage events per million mapped reads (CPM) were used for subsequent analysis.
RNA-Seq Analysis
Gene expression profiles of HEK293T (human embryonic kidney epithelial cell[s] or cell line) and U2OS (human osteosarcoma epithelial cell[s] or cell line) were collected from SRA: SRP080966 and ERP001948, respectively.62,63 Gene expression level (transcript per million [TPM]) was estimated by kallisto after quality control with FastQC and read trimming with trim_galore.64, 65, 66 The criterion used for an expressed gene was any transcripts that had more than five TPMs. The TPM cutoff was defined under the assumption that a gene estimated to have at least one transcript in each cell when each cell has, on average, expressed 200,000 transcripts. Only expressed genes were selected for subsequent analysis.
DNase-Seq
The pre-aligned DNase-seq data from HEK293T cells were obtained from the ENCODE database (ENCODE Project Consortium, 2004). The DNase-seq data from U2OS cells in raw-read format were obtained from the work of Ibarra et al.67 followed by the alignment using bwa-align due to the short read length in the DNase-seq assay.68 The DNA accessibility for each cleavage site was calculated as the reads per million mapped reads (RPM) of a 50-bp window centered by the DSB position (3 bp upstream of the PAM site for SpCas9) in DNase-seq runs with corresponding cell types. The corresponding RPM of each expressed gene was calculated at a window of 1,000 bp upstream of the TSS and 200 bp downstream to cover the vast majority of promoter positions. It is worth noting that there were no extrinsic manipulations performed to purposefully stimulate the cells in the GUIDE-seq, DNase-seq, and RNA-seq protocols, which allows us to compare the DNase-seq results against other genomic assays.
Bioinformatics Analysis
Data processing was conducted in Python along with open-source programs including bwa, samtools, and sambamba.68, 69, 70 The bwa and samtools were used to map the reads from each sequencing assay to the human reference genome GRCh37/hg19 using the default parameters. The sambamba was used to calculate the RPM that represents the DNA accessibility at either CRISPR-induced cleavage sites or expressing genes with the given sequence windows described above. The figures were generated by Python package matplotlib. All Python scripts have been deposited at https://github.com/DamLabResources/chroCRISPR.
Stepwise Correlation Test
The stepwise linear regression was calculated by 15% quantile of cleavage sites by ranked DNA accessibility along with a sliding step of 1 percentile across the ranked data. The size of the 15% quantile for subsequent analysis was decided by power analysis, using an effect size of 0.25 calculated by Cohen’s d, α = 0.05, β = 0.1, and 1 predictor. It resulted in approximately 53 cleavage sites (335 × 15%) in each 15% quantile for the GS and CS subset and 567 cleavage sites (3,783 × 15%) in each 15% quantile for only the CS-only subset. The beta coefficient between sequence similarity predicted by CFD score and observed CRISPR-induced cleavage frequency within each 15% quantile was plotted.11 A relative beta coefficient was calculated by normalizing the current coefficient to the coefficient acquired from the cleavage sites with the top 15% DNA accessibility.
Estimated CPM for the Three-Dimensional Surface Plot Using a Nearest-Neighbor Function
The estimated CPM in either the GUIDE-seq or CIRCLE-seq datasets for each grid was calculated by the nearest k data points from the grid point using the function described as follows:
where D is the distance between grid point and given data point; k = 15 was used in this study based on the average density of data points in the grids.
Statistical Analysis
The simple linear regression analysis was conducted in Python with the scipy.stats package.71 The multiple regression analysis among CPM, RPM, and sequence similarity was conducted by Python package statsmodels (https://www.statsmodels.org/stable/index.html). All combinations of independent variables including additive and interactive models were proposed and tested. All analysis details are described and reproducible in the Jupyter notebook (https://github.com/DamLabResources/chroCRISPR). In two-tailed unpaired t tests for multiple linear regression, DNA accessibility (DNase-seq RPM) between the GS and CS datasets, or DNA accessibility between CRISPR-induced cleavage efficiency and gene expression levels, the alpha level was set at 5%. In the Wald test for the significance of β coefficient (slope) of simple linear regression analyses, the alpha level was set at 1%.
Author Contributions
C.-H.C., M.R.N., W.D., and B.W. proposed experimental ideas. C.-H.C. and W.D. designed the experiments and conducted data processing and statistical analyses. C.-H.C., A.G.A., A.A., N.T.S., M.R.N., W.D., and B.W. wrote the manuscript and made critical revisions/analyses. All authors approved the final copy.
Conflicts of Interest
The authors declare no competing interests.
Acknowledgments
These studies were funded in part by the NIH through grants from the National Institute of Mental Health (NIMH) R01 MH110360 (contact principal investigator [PI], B.W.); NIMH Comprehensive NeuroAIDS Center (CNAC) P30 MH092177 (PI, Kamel Khalili; PI of the Drexel subcontract involving the Clinical and Translational Research Support Core, B.W.; PI of the Developmental Funding Award, W.D.); and the Ruth L. Kirschstein National Research Service Award (T32 MH079785; (PI of the Drexel University College of Medicine component, B.W.; Dr. Olimpia Meucci was co-director). The contents of the paper were solely the responsibility of the authors and do not necessarily represent the official views of the NIH. A.G.A. was also supported by the Drexel University College of Medicine Deans Fellowship for Excellence in Collaborative or Themed Research (A.G.A., fellow; B.W., mentor).
Footnotes
Supplemental Information can be found online at https://doi.org/10.1016/j.ymthe.2019.10.008.
Contributor Information
Brian Wigdahl, Email: bw45@drexel.edu.
Will Dampier, Email: wnd22@drexel.edu.
Supplemental Information
References
- 1.Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. Multiplex genome engineering using CRISPR/Cas systems. Science. 2013;339:819–823. doi: 10.1126/science.1231143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. RNA-guided human genome engineering via Cas9. Science. 2013;339:823–826. doi: 10.1126/science.1232033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Garneau J.E., Dupuis M.E., Villion M., Romero D.A., Barrangou R., Boyaval P., Fremaux C., Horvath P., Magadán A.H., Moineau S. The CRISPR/Cas bacterial immune system cleaves bacteriophage and plasmid DNA. Nature. 2010;468:67–71. doi: 10.1038/nature09523. [DOI] [PubMed] [Google Scholar]
- 4.Deltcheva E., Chylinski K., Sharma C.M., Gonzales K., Chao Y., Pirzada Z.A., Eckert M.R., Vogel J., Charpentier E. CRISPR RNA maturation by trans-encoded small RNA and host factor RNase III. Nature. 2011;471:602–607. doi: 10.1038/nature09886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sorek R., Lawrence C.M., Wiedenheft B. CRISPR-mediated adaptive immune systems in bacteria and archaea. Annu. Rev. Biochem. 2013;82:237–266. doi: 10.1146/annurev-biochem-072911-172315. [DOI] [PubMed] [Google Scholar]
- 6.Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 2012;337:816–821. doi: 10.1126/science.1225829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jasin M., Haber J.E. The democratization of gene editing: Insights from site-specific cleavage and double-strand break repair. DNA Repair (Amst.) 2016;44:6–16. doi: 10.1016/j.dnarep.2016.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tycko J., Myer V.E., Hsu P.D. Methods for Optimizing CRISPR-Cas9 Genome Editing Specificity. Mol. Cell. 2016;63:355–370. doi: 10.1016/j.molcel.2016.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Koo T., Lee J., Kim J.S. Measuring and Reducing Off-Target Activities of Programmable Nucleases Including CRISPR-Cas9. Mol. Cells. 2015;38:475–481. doi: 10.14348/molcells.2015.0103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hsu P.D., Scott D.A., Weinstein J.A., Ran F.A., Konermann S., Agarwala V., Li Y., Fine E.J., Wu X., Shalem O. DNA targeting specificity of RNA-guided Cas9 nucleases. Nat. Biotechnol. 2013;31:827–832. doi: 10.1038/nbt.2647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Doench J.G., Fusi N., Sullender M., Hegde M., Vaimberg E.W., Donovan K.F., Smith I., Tothova Z., Wilen C., Orchard R. Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol. 2016;34:184–191. doi: 10.1038/nbt.3437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 2013;31:822–826. doi: 10.1038/nbt.2623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Mali P., Aach J., Stranges P.B., Esvelt K.M., Moosburner M., Kosuri S., Yang L., Church G.M. CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 2013;31:833–838. doi: 10.1038/nbt.2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Pattanayak V., Lin S., Guilinger J.P., Ma E., Doudna J.A., Liu D.R. High-throughput profiling of off-target DNA cleavage reveals RNA-programmed Cas9 nuclease specificity. Nat. Biotechnol. 2013;31:839–843. doi: 10.1038/nbt.2673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bolukbasi M.F., Gupta A., Wolfe S.A. Creating and evaluating accurate CRISPR-Cas9 scalpels for genomic surgery. Nat. Methods. 2016;13:41–50. doi: 10.1038/nmeth.3684. [DOI] [PubMed] [Google Scholar]
- 16.Martin F., Sánchez-Hernández S., Gutiérrez-Guerrero A., Pinedo-Gomez J., Benabdellah K. Biased and Unbiased Methods for the Detection of Off-Target Cleavage by CRISPR/Cas9: An Overview. Int. J. Mol. Sci. 2016;17:e1507. doi: 10.3390/ijms17091507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tsai S.Q., Joung J.K. Defining and improving the genome-wide specificities of CRISPR-Cas9 nucleases. Nat. Rev. Genet. 2016;17:300–312. doi: 10.1038/nrg.2016.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zhang X.H., Tee L.Y., Wang X.G., Huang Q.S., Yang S.H. Off-target Effects in CRISPR/Cas9-mediated Genome Engineering. Mol. Ther. Nucleic Acids. 2015;4:e264. doi: 10.1038/mtna.2015.37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iafrate A.J., Le L.P. GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 2015;33:187–197. doi: 10.1038/nbt.3117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kleinstiver B.P., Tsai S.Q., Prew M.S., Nguyen N.T., Welch M.M., Lopez J.M., McCaw Z.R., Aryee M.J., Joung J.K. Genome-wide specificities of CRISPR-Cas Cpf1 nucleases in human cells. Nat. Biotechnol. 2016;34:869–874. doi: 10.1038/nbt.3620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tsai S.Q., Nguyen N.T., Malagon-Lopez J., Topkar V.V., Aryee M.J., Joung J.K. CIRCLE-seq: a highly sensitive in vitro screen for genome-wide CRISPR-Cas9 nuclease off-targets. Nat. Methods. 2017;14:607–614. doi: 10.1038/nmeth.4278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Margueron R., Reinberg D. Chromatin structure and the inheritance of epigenetic information. Nat. Rev. Genet. 2010;11:285–296. doi: 10.1038/nrg2752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Li B., Carey M., Workman J.L. The role of chromatin during transcription. Cell. 2007;128:707–719. doi: 10.1016/j.cell.2007.01.015. [DOI] [PubMed] [Google Scholar]
- 24.Song L., Crawford G.E. DNase-seq: a high-resolution technique for mapping active gene regulatory elements across the genome from mammalian cells. Cold. Spring Harb. Protoc. 2010. 2010 doi: 10.1101/pdb.prot5384. pdb.prot5384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.John S., Sabo P.J., Canfield T.K., Lee K., Vong S., Weaver M., Wang H., Vierstra J., Reynolds A.P., Thurman R.E., Stamatoyannopoulos J.A. Genome-scale mapping of DNase I hypersensitivity. Curr. Protoc. Mol. Biol. 2013;103:21.27.1–21.27.20. doi: 10.1002/0471142727.mb2127s103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wu X., Scott D.A., Kriz A.J., Chiu A.C., Hsu P.D., Dadon D.B., Cheng A.W., Trevino A.E., Konermann S., Chen S. Genome-wide binding of the CRISPR endonuclease Cas9 in mammalian cells. Nat. Biotechnol. 2014;32:670–676. doi: 10.1038/nbt.2889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Kuscu C., Arslan S., Singh R., Thorpe J., Adli M. Genome-wide analysis reveals characteristics of off-target sites bound by the Cas9 endonuclease. Nat. Biotechnol. 2014;32:677–683. doi: 10.1038/nbt.2916. [DOI] [PubMed] [Google Scholar]
- 28.Chari R., Mali P., Moosburner M., Church G.M. Unraveling CRISPR-Cas9 genome engineering parameters via a library-on-library approach. Nat. Methods. 2015;12:823–826. doi: 10.1038/nmeth.3473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chen X., Rinsma M., Janssen J.M., Liu J., Maggio I., Gonçalves M.A. Probing the impact of chromatin conformation on genome editing tools. Nucleic Acids Res. 2016;44:6482–6492. doi: 10.1093/nar/gkw524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Daer R.M., Cutts J.P., Brafman D.A., Haynes K.A. The Impact of Chromatin Dynamics on Cas9-Mediated Genome Editing in Human Cells. ACS Synth. Biol. 2017;6:428–438. doi: 10.1021/acssynbio.5b00299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jensen K.T., Fløe L., Petersen T.S., Huang J., Xu F., Bolund L., Luo Y., Lin L. Chromatin accessibility and guide sequence secondary structure affect CRISPR-Cas9 gene editing efficiency. FEBS Lett. 2017;591:1892–1901. doi: 10.1002/1873-3468.12707. [DOI] [PubMed] [Google Scholar]
- 32.Koudritsky M., Domany E. Positional distribution of human transcription factor binding sites. Nucleic Acids Res. 2008;36:6795–6805. doi: 10.1093/nar/gkn752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Chen Y., Zeng S., Hu R., Wang X., Huang W., Liu J., Wang L., Liu G., Cao Y., Zhang Y. Using local chromatin structure to improve CRISPR/Cas9 efficiency in zebrafish. PLoS ONE. 2017;12:e0182528. doi: 10.1371/journal.pone.0182528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Uusi-Mäkelä M.I.E., Barker H.R., Bäuerlein C.A., Häkkinen T., Nykter M., Rämet M. Chromatin accessibility is associated with CRISPR-Cas9 efficiency in the zebrafish (Danio rerio) PLoS ONE. 2018;13:e0196238. doi: 10.1371/journal.pone.0196238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Miga K.H., Eisenhart C., Kent W.J. Utilizing mapping targets of sequences underrepresented in the reference assembly to reduce false positive alignments. Nucleic Acids Res. 2015;43:e133. doi: 10.1093/nar/gkv671. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Koohy H., Down T.A., Spivakov M., Hubbard T. A comparison of peak callers used for DNase-Seq data. PLoS ONE. 2014;9:e96303. doi: 10.1371/journal.pone.0096303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rashid N.U., Giresi P.G., Ibrahim J.G., Sun W., Lieb J.D. ZINBA integrates local covariates with DNA-seq data to identify broad and narrow regions of enrichment, even within amplified genomic regions. Genome Biol. 2011;12:R67. doi: 10.1186/gb-2011-12-7-r67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.John S., Sabo P.J., Thurman R.E., Sung M.H., Biddie S.C., Johnson T.A., Hager G.L., Stamatoyannopoulos J.A. Chromatin accessibility pre-determines glucocorticoid receptor binding patterns. Nat. Genet. 2011;43:264–268. doi: 10.1038/ng.759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Boyle A.P., Guinney J., Crawford G.E., Furey T.S. F-Seq: a feature density estimator for high-throughput sequence tags. Bioinformatics. 2008;24:2537–2538. doi: 10.1093/bioinformatics/btn480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Singh R., Kuscu C., Quinlan A., Qi Y., Adli M. Cas9-chromatin binding information enables more accurate CRISPR off-target prediction. Nucleic Acids Res. 2015;43:e118. doi: 10.1093/nar/gkv575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Klein M., Eslami-Mossallam B., Arroyo D.G., Depken M. Hybridization Kinetics Explains CRISPR-Cas Off-Targeting Rules. Cell Rep. 2018;22:1413–1423. doi: 10.1016/j.celrep.2018.01.045. [DOI] [PubMed] [Google Scholar]
- 43.Sternberg S.H., Redding S., Jinek M., Greene E.C., Doudna J.A. DNA interrogation by the CRISPR RNA-guided endonuclease Cas9. Nature. 2014;507:62–67. doi: 10.1038/nature13011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Szczelkun M.D., Tikhomirova M.S., Sinkunas T., Gasiunas G., Karvelis T., Pschera P., Siksnys V., Seidel R. Direct observation of R-loop formation by single RNA-guided Cas9 and Cascade effector complexes. Proc. Natl. Acad. Sci. USA. 2014;111:9798–9803. doi: 10.1073/pnas.1402597111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mutskov V., Felsenfeld G. Silencing of transgene transcription precedes methylation of promoter DNA and histone H3 lysine 9. EMBO J. 2004;23:138–149. doi: 10.1038/sj.emboj.7600013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ginno P.A., Lott P.L., Christensen H.C., Korf I., Chédin F. R-loop formation is a distinctive characteristic of unmethylated human CpG island promoters. Mol. Cell. 2012;45:814–825. doi: 10.1016/j.molcel.2012.01.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Dampier W., Sullivan N.T., Mell J.C., Pirrone V., Ehrlich G.D., Chung C.H., Allen A.G., DeSimone M., Zhong W., Kercher K. Broad-Spectrum and Personalized Guide RNAs for CRISPR/Cas9 HIV-1 Therapeutics. AIDS Res. Hum. Retroviruses. 2018;34:950–960. doi: 10.1089/aid.2017.0274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Kaminski R., Bella R., Yin C., Otte J., Ferrante P., Gendelman H.E., Li H., Booze R., Gordon J., Hu W., Khalili K. Excision of HIV-1 DNA by gene editing: a proof-of-concept in vivo study. Gene Ther. 2016;23:690–695. doi: 10.1038/gt.2016.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kaminski R., Chen Y., Fischer T., Tedaldi E., Napoli A., Zhang Y., Karn J., Hu W., Khalili K. Elimination of HIV-1 Genomes from Human T-lymphoid Cells by CRISPR/Cas9 Gene Editing. Sci. Rep. 2016;6:22555. doi: 10.1038/srep22555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dampier W., Nonnemacher M.R., Sullivan N.T., Jacobson J.M., Wigdahl B. HIV excision utilizing CRISPR/Cas9 technology: Attacking the proviral quasispecies in reservoirs to achieve a cure. MOJ Immunol. 2014;1:00022. doi: 10.15406/moji.2014.01.00022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Datta P.K., Kaminski R., Hu W., Pirrone V., Sullivan N.T., Nonnemacher M.R., Dampier W., Wigdahl B., Khalili K. HIV-1 Latency and Eradication: Past, Present and Future. Curr. HIV Res. 2016;14:431–441. doi: 10.2174/1570162x14666160324125536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Link R., Nonnemacher M.R., Wigdahl B., Dampier W. Prediction of human immunodeficiency virus type 1 subtype-specific off-target effects arising from CRISPR-Cas9 gene editing therapy. CRISPR J. 2018;1:294–302. doi: 10.1089/crispr.2018.0020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Verdin E. DNase I-hypersensitive sites are associated with both long terminal repeats and with the intragenic enhancer of integrated human immunodeficiency virus type 1. J. Virol. 1991;65:6790–6799. doi: 10.1128/jvi.65.12.6790-6799.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Verdin E., Paras P., Jr., Van Lint C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993;12:3249–3259. doi: 10.1002/j.1460-2075.1993.tb05994.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shah S., Pirrone V., Alexaki A., Nonnemacher M.R., Wigdahl B. Impact of viral activators and epigenetic regulators on HIV-1 LTRs containing naturally occurring single nucleotide polymorphisms. BioMed Res. Int. 2015;2015:320642. doi: 10.1155/2015/320642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kilareski E.M., Shah S., Nonnemacher M.R., Wigdahl B. Regulation of HIV-1 transcription in cells of the monocyte-macrophage lineage. Retrovirology. 2009;6:118. doi: 10.1186/1742-4690-6-118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Shirazi J., Shah S., Sagar D., Nonnemacher M.R., Wigdahl B., Khan Z.K., Jain P. Epigenetics, drugs of abuse, and the retroviral promoter. J. Neuroimmune Pharmacol. 2013;8:1181–1196. doi: 10.1007/s11481-013-9508-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Battistini A., Sgarbanti M. HIV-1 latency: an update of molecular mechanisms and therapeutic strategies. Viruses. 2014;6:1715–1758. doi: 10.3390/v6041715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Dampier W., Sullivan N.T., Chung C.H., Mell J.C., Nonnemacher M.R., Wigdahl B. Designing broad-spectrum anti-HIV-1 gRNAs to target patient-derived variants. Sci. Rep. 2017;7:14413. doi: 10.1038/s41598-017-12612-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hu W., Kaminski R., Yang F., Zhang Y., Cosentino L., Li F., Luo B., Alvarez-Carbonell D., Garcia-Mesa Y., Karn J. RNA-directed gene editing specifically eradicates latent and prevents new HIV-1 infection. Proc. Natl. Acad. Sci. USA. 2014;111:11461–11466. doi: 10.1073/pnas.1405186111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hendel A., Bak R.O., Clark J.T., Kennedy A.B., Ryan D.E., Roy S., Steinfeld I., Lunstad B.D., Kaiser R.J., Wilkens A.B. Chemically modified guide RNAs enhance CRISPR-Cas genome editing in human primary cells. Nat. Biotechnol. 2015;33:985–989. doi: 10.1038/nbt.3290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Akan P., Alexeyenko A., Costea P.I., Hedberg L., Solnestam B.W., Lundin S., Hällman J., Lundberg E., Uhlén M., Lundeberg J. Comprehensive analysis of the genome transcriptome and proteome landscapes of three tumor cell lines. Genome Med. 2012;4:86. doi: 10.1186/gm387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Aktaş T., Avşar Ilık İ., Maticzka D., Bhardwaj V., Pessoa Rodrigues C., Mittler G., Manke T., Backofen R., Akhtar A. DHX9 suppresses RNA processing defects originating from the Alu invasion of the human genome. Nature. 2017;544:115–119. doi: 10.1038/nature21715. [DOI] [PubMed] [Google Scholar]
- 64.Andrews S. 2010. FastQC: a quality control tool for high throughput sequence data.https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ [Google Scholar]
- 65.Bray N.L., Pimentel H., Melsted P., Pachter L. Near-optimal probabilistic RNA-seq quantification. Nat. Biotechnol. 2016;34:525–527. doi: 10.1038/nbt.3519. [DOI] [PubMed] [Google Scholar]
- 66.Krueger F. 2015. Trim Galore!: A wrapper tool around Cutadapt and FastQC to consistently apply quality and adapter trimming to FastQ files. 0.4.https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/ [Google Scholar]
- 67.Ibarra A., Benner C., Tyagi S., Cool J., Hetzer M.W. Nucleoporin-mediated regulation of cell identity genes. Genes Dev. 2016;30:2253–2258. doi: 10.1101/gad.287417.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li H., Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Tarasov A., Vilella A.J., Cuppen E., Nijman I.J., Prins P. Sambamba: fast processing of NGS alignment formats. Bioinformatics. 2015;31:2032–2034. doi: 10.1093/bioinformatics/btv098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., 1000 Genome Project Data Processing Subgroup The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Seabold S., Perktold J. Vol. 57. 2010. Statsmodels: Econometric and statistical modeling with python; p. 61. (Proceedings of the 9th Python in Science Conference). [Google Scholar]
- 72.Consortium E.P., ENCODE Project Consortium An integrated encyclopedia of DNA elements in the human genome. Nature. 2012;489:57–74. doi: 10.1038/nature11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.