

Abstract
Knowledge of the chemical identity of metabolite molecules is critical for the understanding of the complex biological systems they belong to. Since metabolite identities and their concentrations are often directly linked to the phenotype, such information can be used to map biochemical pathways and understand their role in health and disease. A very large number of metabolites however are still unknown, i.e. their spectroscopic signatures do not match those in existing databases, suggesting unknown molecule identification is both imperative and challenging. Although metabolites are structurally highly diverse, the majority shares a rather limited number of structural motifs, which are defined by sets of 1H, 13C chemical shifts of the same spin system. This allows one to characterize unknown metabolites by a divide-and-conquer strategy that identifies their structural motifs first. Here we present the structural motif-based approach “SUMMIT Motif” for the de novo identification of unknown molecular structures in complex mixtures, without the need for extensive purification, using NMR in tandem with two newly curated NMR molecular structural motif metabolomics databases (MSMMDB). In identifying structural motif(s), first the 1H and 13C chemical shifts of all the individual spin systems are extracted from 2D and 3D NMR spectra of the complex mixture. Next, the molecular structural motifs are identified by querying these chemical shifts against the new MSMMDBs. One database, COLMAR MSMMDB, was derived from experimental NMR chemical shifts of known metabolites taken from the COLMAR metabolomics database, while the other MSMMDB, pNMR MSMMDB, is based on empirically predicted chemical shifts of metabolites of several existing large metabolomics databases. For molecules consisting of multiple spin systems, spin systems are connected via a long-range scalar J-coupling NMR experiment. When this motif-based identification method was applied to the hydrophilic extract of mouse bile fluid, two unknown metabolites could be successfully identified. This approach is both accurate and efficient for the identification of unknown metabolites and hence contribute to the understanding of human health and disease.
Graphical Abstract
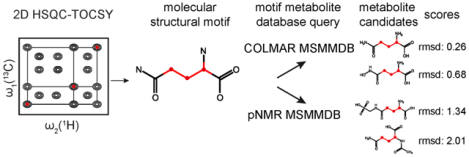
INTRODUCTION
The chemical complexity of living organisms is reflected in the large number of different metabolites they are composed of. The human body alone may contain over 100,000 different metabolites, but the majority still needs to be identified and characterized.1 Such identification is critical to identify potential biomarkers and study new biochemical pathways for the better understanding of biological processes involved in health and disease. The system-wide study of metabolites and pathways, also in relationship to the phenotype, is the subject of the field of metabolomics.2–4
Structure determination of novel organic molecules is a standard task in synthetic organic chemistry and natural product research. Analytical methods, such as infra-red (IR) and UV/Vis spectroscopy, high-resolution mass spectrometry (HRMS) and 1H and 13C NMR spectroscopy are routinely used.5 However, traditional synthetic or natural product characterization requires that a compound has been purified and isolated. In metabolomics, this is often impractical as it can be hard to efficiently isolate a compound at sufficient concentration among hundreds of molecular species. With respect to NMR, important methodological advances now allow routine characterization of known metabolites in a wide range of different complex mixtures with little or no purification.1, 6
Over the recent past, several approaches have been introduced that aim at the de novo characterization and structure determination of metabolites directly in the complex mixture environment.7–13 We recently introduced a protocol for the identification of unknown metabolites in metabolomics samples without the need for purification that combines MS, NMR, and cheminformatics.14, 15 The approach, named SUMMIT MS/NMR, uses accurate mass information, e.g. from Fourier transform ion cyclotron resonance (FT-ICR), to determine the elemental composition of the metabolites present in the sample. A large pool of chemical compounds is then generated, which are consistent with the MS-derived molecular formulas. The candidate compounds are then filtered against multidimensional NMR data, in particular 1H and 13C chemical shifts that belong to individual spin systems. All candidate compounds are rank-ordered by comparing their predicted NMR chemical shifts with experimentally determined chemical shifts of the unknown compounds in the mixture.
SUMMIT MS/NMR requires that two key conditions are fulfilled: (i) the unknown compound must be present in the pool of candidate structures and (ii) the accuracy of the chemical shift predictor must be sufficiently high to identify the correct compound from potentially many others. In practice, both conditions pose specific challenges. For condition (i), the presence of the unknown metabolite in the pool of structures can be difficult to meet depending on the unknown and how the pool of structures has been generated. Instead, it can be easier and less ambiguous to first establish more general molecular structural properties of the unknown, such as its structural motif(s), before attempting to characterize its full molecular structure. For condition (ii), empirical chemical shift predictors, such as Modgraph/Mnova,16, 17 are fast, but their accuracy is limited. The average root-mean-square deviations (RMSD) of empirically predicted chemical shifts for a set of representative metabolites are around 0.292 ppm (1H) and 2.90 ppm (13C), and can be improved to 0.154 ppm (1H) and 1.93 ppm (13C) by quantum-chemical calculations with multiple scaling18 at the cost of largely increased computation time. Due to the currently limited accuracy of the chemical shift prediction, the SUMMIT MS/NMR approach returns a potentially large number of compounds as viable candidates, which makes their final verification in terms of their purchase or chemical synthesis followed by spiking experiments in the complex mixture both time-consuming and expensive.
Here, we present an alternative approach, named SUMMIT Motif, for the de novo determination of molecular structures of unknowns by first focusing on the determination of molecular structural motif(s) (MSM) without the need for any mass spectrometry data. Such information represents a key step toward the determination of the full structure. The approach starts out with the identification of 1H and 13C NMR spin systems to define an unknown metabolite’s backbone or contiguous parts thereof. This step is achieved by querying the experimental chemical shifts of the unknown spin system against those of molecular structural motifs (MSM) in both an experimental and a synthetic MSM chemical shift database. It is shown that this approach is highly effective for MSM identification provided that the MSM of the unknown compound is in fact present in the molecular structural motif metabolomics databases (MSMMDB). As is shown here, this requirement is much easier fulfilled than condition (i) of SUMMIT MS/NMR described above. The power of the approach is demonstrated by determining unknowns present in mouse bile fluid.
METHODS
Definition of spin systems, molecular structural motifs, and COLMAR MSMMDB curation
For any given molecular structure, molecular structural motifs (MSM) are defined using spin systems as a starting point. As customarily defined, for each proton spin system consisting of NH protons each 1H spin is connected to another 1H spin through no more than 3 bonds. A basic molecular structural motif is then defined by the 1H spins together with up to NC carbons (13C or 12C) where each carbon is directly attached to at least one of the 1H atoms (Figure 1). This “0th shell molecular motif” can then be systematically expanded by including additional atoms (N, O, S, P) that are not directly observable in 1H and 13C NMR experiments. When including additional heavy atoms that are exactly one bond away one obtains the “1st shell molecular structural motif” and when heavy atoms are included that are up to two bonds away the “2nd shell molecular structural motif” is obtained (Figure 1). The shell order is analogous to the HOSE code used for the empirical prediction of chemical shifts of small molecules.19 The higher the shell order, the more chemically distinct the structural motif is and the more unique its 1H and 13C chemical shifts are, but the lower the chance that one or several molecules with the same structural motif already exist in a current metabolomics NMR database.
Figure 1.

Definition of 1st and 2nd shell molecular motifs based on a spin system. The proton spin system together with its directly bonded carbon atoms defines the molecular spin system or “0th shell molecular motif” (red). The “1st shell molecular motif” (dashed orange box) is obtained after inclusion of the heavy atoms that are directly bonded to the spin system (orange). The additional inclusion of heavy atoms that are up to two bonds away from the spin system yields the “2nd shell molecular motif” (dashed green box).
In order to recognize molecular structural motifs that are part of an unknown metabolite, a 1st and 2nd shell molecular structural motif database was generated from the COLMAR small molecule database (experimentally measured in aqueous solution) along with 1H, 13C chemical shifts of each motif that were assigned to specific groups within each motif. It is called COLMAR MSM Metabolomics Database or COLMAR MSMMDB. COLMAR MSMMDB stores the molecular structures of 1st and 2nd shell MSM, the parent metabolites of a MSM along with their chemical shifts, their averages and standard deviations. By considering motifs with NC > 1 spin systems only, 623 metabolites in the parent COLMAR metabolomics database share 180 unique 1st shell and 397 unique 2nd shell molecular structural motifs (Table S1). Examples of 2nd shell molecular structural motifs are depicted in Figure 2, along with 1H, 13C chemical shift standard deviations of individual atoms.
Figure 2.

Examples of metabolites with identical molecular structural motifs (in color). The three 2nd shell molecular motifs on the left are highlighted in color. Each row depicts examples of molecules with the same molecular structural motif as the MSM furthest on the left. The experimental NMR chemical shift root-mean-square deviations (RMSDs) at the same C-H positions for all molecules containing the same motif are indicated where the first (second) number is the 1H (13C) RMSD in units of ppm.
The standard deviations of chemical shifts in 2nd shell MSMs are generally well below the average chemical shift errors of NMR chemical shifts prediction programs (see Figure S1). This demonstrates that the chemical shifts of 2nd shell MSMs are in most cases considerably more accurate than computationally predicted chemical shifts, which is the reason why 2nd shell MSMs have a better chance to be successfully identified from experimental chemical shifts of unknown metabolites.
Curation of the empirically predicted pNMR MSMMDB
Since the COLMAR MSMMDB features only a subset of MSMs (currently 180 1st shell and 397 2nd shell MSMs) of all possible metabolite MSMs, it is possible that an unknown spin system does not have a good match. For such cases, a MSM database has been built that covers a larger range of MSMs from empirically predicted, rather than experimental chemical shifts. This database, termed pNMR molecular structural motif metabolomics database (pNMR MSMMDB), consists of MSMs that were extracted from molecules in the HMDB, the Chemical Entities of Biological Interest (ChEBI) database, and the Kyoto Encyclopedia of Genes and Genomes (KEGG) database.20–22 The HMDB is currently the most comprehensive, organism-specific metabolomics database and is the largest collection of human metabolites with their chemical structures and biological roles annotated. ChEBI covers both metabolites produced in biological systems and synthetic products that can intervene with living organisms. The KEGG database is one of the most widely used biochemical pathway databases, containing metabolites involved in human diseases and molecular interactions in various organisms. The pNMR MSMMDB, which currently covers 23,697 metabolites with a molecular weight below 800 Da, focuses on motifs of hydrophilic metabolites, which are defined as metabolites with a predicted lipophilicity logP value smaller than 3.0 (as computed by ALOGP,23, 24 Figure S2). The pNMR MSMMDB contains motifs that overlap with those of COLMAR MSMMDB, but with their chemical shifts predicted rather than experimentally determined.
1H, 13C chemical shifts were computed and stored for each compound in the pNMR MSMMDB using the empirical chemical shift predictor by Modgraph implemented in MestReNova 10.0.1 (Mestrelab Research). The 1H chemical shift prediction is based on the effects of functional groups that were individually parameterized, whereas the 13C chemical shift prediction is achieved with a HOSE code algorithm. The predicted 1H, 13C chemical shifts have been sorted into individual spin systems belonging to unique MSMs so that they can be compared directly with experimental 1H, 13C chemical shifts extracted from experiments. For each MSM, predicted chemical shifts from multiple metabolites are stored separately.
Workflow of MSM-based metabolite identification
The total workflow of metabolite identification based on molecular structural motifs extracted from NMR spin systems is depicted in Figure S3. After identification of a H-C spin system from 2D/3D TOCSY spectra (see details in SI), it is queried against COLMAR MSMMDB. If no hits are returned with RMSD < 2.5 ppm, the spin system information is then queried against pNMR MSMMDB. This two-pronged SUMMIT Motif approach focuses first on the query against the more accurate experimental COLMAR MSMMDB before turning to the larger, but less accurate pNMR MSMMDB. The MSM hits returned by COLMAR MSMMDB (with RMSD < 2.5 ppm) and the top 15 MSM hits returned by pNMR MSMMDB (with RMSD < 5.0 ppm) are subject to structure determination of unknown metabolites with spiking NMR experiments and/or additional experiments (e.g., 2D HSQMBC). Molecular structural motif query based on COLMAR MSMMDB and pNMR MSMMDB are publicly accessible via the COLMAR suite of web servers (http://spin.ccic.ohio-state.edu/index.php/motif).
RESULTS
Evaluation of COLMAR and pNMR MSMMDB in identification of known molecules in bile and E. coli extracts
The strategy for the identification of molecular structural motifs (1st or 2nd shell) in unknown metabolites was first tested on known metabolites in bile and E. coli cell extracts. There are 26 metabolites in bile and 111 metabolites in E. coli cell extract, which could be identified and verified by previously established methods, i.e. 2D HSQC, 2D TOCSY and 2D HSQC-TOCSY via COLMARm web server. Each of these known metabolites contains at least one spin system with two or more 1H spins and their directly attached 13C spins. The 1H, 13C chemical shifts of each spin system were queried against the COLMAR and pNMR MSMMDB (details regarding spin system matching and scoring are described in SI). To be able to treat these spin systems like real unknowns, their true metabolite motifs were intentionally removed from COLMAR MSMMDB. After querying experimental spin systems against COLMAR MSMMDB, both 1st shell and 2nd shell motifs are returned if hits exist within an RMSD cutoff of 5.0 ppm. The motifs with lowest RMSDs are prioritized for further evaluation. For bile metabolites, the 1H, 13C chemical shifts of 19 metabolites matched the correct 1st or 2nd shell MSMs from the COLMAR MSMMDB as the top hit. In some cases, a clear distinction between 1st and 2nd shell hits is difficult. For instance, when querying an unknown spin system with the motif of taurine (SO3HCH2CH2NH2) against the COLMAR MSMMDB, the identified MSM (SO3HCH2CH2NH-CO-) is partially 2nd shell. Overall, the RMSD for the correct hits range between 0.03 and 2.11 ppm (mean value of 0.97 ppm). The RMSD of MSMs of this and other (known) molecules provides a useful benchmark for the range of RMSD values that belong to the correct MSMs. Six metabolites (glycerol, valine, isoleucine, N-alpha-acetyl-L-lysine, leucine, alpha epsilon-diaminopimelic acid) did not return any good hits, because no other COLMAR metabolite contains the same 1st or 2nd shell MSM as these six metabolites. The only spin system whose MSM was misidentified belonged to L-serine where the (incorrect) top hit had a RMSD of 3.21 ppm, confirming that higher RMSDs are generally associated with lower confidence in the returned MSMs. Similarly, after querying the 26 experimental spin systems against the pNMR MSMMDB the motif of the true metabolites ranks as follows: 1st hits for 12 spin systems, 2nd hits for 4 spin systems, 3rd hits for 4 spin systems, 4th hits for 1 spin system, 5th – 15th top hits for 5 spin systems. The RMSD of correct hits range between 0.57 and 2.78 ppm (mean value of 1.78 ppm), which are significantly higher than the RMSD of hits returned by COLMAR MSMMDB. Examples of MSM identification and representative metabolites with the same MSM returned by COLMAR and pNMR MSMMDB are listed in Table 1. Since pNMR MSMMDB covers a much larger pool of metabolites and MSMs and the chemical shift prediction is less accurate than for COLMAR MSMMDB, the top 15 different MSM hits are considered here as viable candidates when identifying an unknown motif.
Table 1.
Examples of molecular structural motif identification of bile metabolites by COLMAR and pNMR MSMMDB queries
| Chemical shifts of input (1H, 13C) spin pairs | Motif | Hit that contains true motif returned by COLMAR MSMMDB (Hit rank, type of identified true motif, RMSD (ppm)) | Hit that contains true motif returned by pNMR MSMMDB (Hit rank, type of identified true motif, RMSD (ppm)) | True Metabolite |
|---|---|---|---|---|
| (4.120, 86.907) (4.342, 76.337) (4.213, 72.108) (3.802, 63.425) (3.918, 63.385) (5.902, 92.079) |
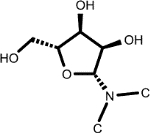 |
5-Methyluridine (1, 2nd shell, 0.16) | Beta-D-3-Ribofuranosyluric acid (2, 2nd shell, 2.08) | Uridine |
| (7.183, 133.486) (6.888, 118.557) |
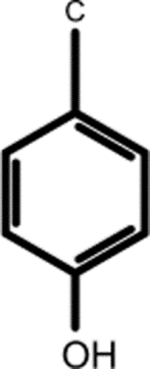 |
3-Chlorotyrosine (1, 2nd shell, 0.20) | N-(1-Deoxy-1-fructosyl)tyrosine (1, 2nd shell, 1.42) | L-tyrosine |
| (3.768, 74.784) (3.632, 65.208) (3.610, 73.570) |
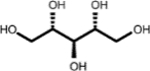 |
Adonitol (1, 2nd shell, 0.79) | Xylitol (1, 2nd shell, 1.80) | Xylitol |
| (1.788, 33.111) (2.996, 41.918) (4.135, 57.765) (1.710, 29.096) (1.398, 24.725) |
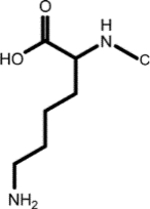 |
Biocytin (1, 1st shell, 1.29) | N6-L-homocysteinyl-N2-L-valyl-L-lysine (3, *2nd shell, 1.29) | Aspartyl-lysine |
| (3.966, 62.877) (3.234, 43.195) |
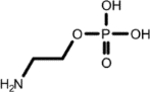 |
Ethanolamine (1, 1st shell, 1.63) | Phosphoethanolamine (1, 2nd shell, 1.44) | Phosphoethanolamine |
| (4.243, 68.598) (3.575, 63.141) (1.318, 22.145) |
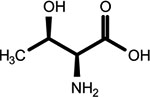 |
O-phosphothreonine (l, *2nd shell, 1.64) | Cyclic N(6)-threonylcarbamoyladenosine (1, 2nd shell, 1.83) | Threonine |
For these molecules, a partial 2nd shell MSM was identified, since in the true molecule a hydrogen terminates the 1st shell MSM.
For E. coli metabolites, the molecular motif of the top hit is 100% correct up to 1st shell atoms when using an RMSD threshold of 2.1 ppm. With respect to 2nd shell atoms, 89 out of 111 top hits contain the correct 2nd shell MSMs (true positives) for an RMSD threshold of 5 ppm, whereas 22 top hits contain incorrect 2nd shell molecular motifs (false positives). The quantitative evaluation of true/false positives/negatives for MSM identification is illustrated in the SI. The number of true and false positive top hits of E. coli metabolites with various RMSD thresholds are summarized in Table S2 with the receiver operating characteristic (ROC) curve shown in Figure S4 and an Area Under Curve (AUC) of 0.851. The number of false positives was reduced when setting the RMSD threshold to 2.1 ppm with 81 true positive 2nd shell MSMs and only 9 false positives. Among the 9 false positives, the true 2nd shell MSMs either ranked as high as 2nd or were not returned at all, because COLMAR MSMMDB did not contain an entry with the same MSM as the true metabolite.
Structure elucidation of unknown metabolites
The determination of MSMs represents a critical step toward the structure elucidation of unknown metabolites, which is demonstrated here for gallbladder bile fluid. We focused on three experimentally extracted spin systems with unknown identity, designated as spin systems A, B, and C. The MSM identification method was applied to identify the unknown molecular motifs belonging to these spin systems followed by identifying the structures of these unknown metabolites. Unknown spin system A has 4 cross-peaks with chemical shifts (δH, δC) of (0.883, 20.032), (0.906, 21.543), (2.090, 33.293) and (4.048, 63.569) ppm. After querying against the COLMAR MSMMDB, it best matches the valine-like MSM with structure CH3(CH3)CHCH(COOH)N–. When querying against the pNMR MSMMDB, the same valine-like motif was returned that is shared by 32 metabolites. By selecting the top 6 hits with the same molecular motif in the return list and performing spiking experiments, L-alanyl-L-valine was found to accurately match the unknown spin system (Figure 3). L-alanyl-L-valine is a dipeptide composed of alanine and valine, which was not previously identified in human tissues or biofluids. Although most dipeptides are relatively short-lived intermediates, some dipeptides are known to have physiological effects, for example, for cell-signaling. The identification of L-alanyl-L-valine in mouse bile provides new information toward the understanding of amino-acid specific pathways for further biological interpretation. The identification and validation of L-alanyl-L-valine is depicted in Figure 3.
Figure 3.

Identification of L-alanyl-L-valine metabolite in mouse bile extracts. Panels a and b: 2D 13C-1H HSQC and 2D 1H-1H TOCSY of the unknown spin system A. Panels c and d: overlay of 2D NMR spectra of L-alanyl-L-valine (blue peaks) and bile extracts (gray peaks). Chemical shift agreement confirms the presence of L-alanyl-L-valine in the mouse bile mixture. Panel e: the chemical structure of L-alanyl-L-valine. Panel f: partial FT-ICR spectrum of the mouse bile extracts. The red peak with m/z 189.12337 of the [M+H]+ adduct is consistent with molecular formula C8H17N2O3 of L-alanyl-L-valine (mass error: 16 ppb).
Unknown spin system B contains two peaks with chemical shifts (δH, δC) of (3.068, 52.507) and (3.558, 37.715) ppm (Figure S5a, b). After querying against the COLMAR MSMMDB, it matches the MSM –NHCH2CH2SO3H, which is also found in taurine. When querying against the pNMR MSMMDB, the same taurine-like MSM is returned that is shared by 96 metabolites. Unknown spin system C contains 6 peaks with chemical shifts (δH, δC) of (0.943 20.558), (1.339 34.342), (1.735 34.345), (1.413 37.477), (2.199 35.428) and (2.296 35.435) ppm (Figure S5c, d). After querying against COLMAR MSMMDB, no hit was returned, indicating that this particular molecular motif does not exist in the COLMAR MSMMDB. By comparison, when querying against pNMR MSMMDB, 33 MSMs were returned that are shared by 49 metabolites. However, the RMSDs of all hits exceeded 2.8 ppm, suggesting that none of the MSM candidates may include the true MSM. The 2D 13C-1H HSQMBC spectrum showed that the two unknown spin systems B and C are part of the same molecule, which were connected via a quaternary carbon peak at 180.820 ppm (Figure S5e). This indicates the unknown compound has at least two spin systems, and the number of potential unknown compound candidates is lowered from 96 to 25 compounds. The 25 compounds were further separated into four groups based on their second MSMs in addition to MSM (–NHCH2CH2SO3H). By selecting and purchasing the individual, isolated compounds of the top hit in each MSM group with the lowest RMSD (total of 4 hits) and performing NMR spiking experiment, taurocholic acid was found to precisely match both unknown spin systems B and C, confirming that they belong to taurocholic acid (Figure S6). The reason that neither COLMAR nor pNMR MSMMDB returned a match for spin system C is that spin system C represents a sub-spin system of a much larger spin system of taurocholic acid, which contains 19 carbons together with their attached hydrogens. During 120 ms TOCSY mixing, magnetization transfer was incomplete and, in addition, a number of cross-peaks overlapped with those of other molecules in the mixture. Therefore, spin system C, which was returned by the maximal clique method, did not correspond to the full spin system, which prevented identification of this motif when querying against pNMR MSMMDB. Although taurocholic acid is a known metabolite of bile, NMR database information for taurocholic acid is available only in DMSO. Because of the dependence of chemical shifts on the solvent (chemical shifts in DMSO are substantially different from those in aqueous condition), taurocholic acid was not part of COLMAR MSMMDB. These examples illustrate how the molecular structural motif-based method for the identification of unknown metabolites successfully works for rather large, real-world metabolites.
Coverage of COLMAR motifs of HMDB
The COLMAR MSMMDB, although established with the motif information from only 632 metabolites, performed remarkably well. The strong performance can be rationalized when comparing the motifs in COLMAR MSMMDB and motifs extracted from the much larger HMDB database. Because a large fraction of the metabolites in the HMDB are hydrophobic metabolites, we focus here on the hydrophilic subset, which includes 13,138 metabolites with a lipophilicity logP < 3 as predicted by ALOGP software.24 All MSMs with 2 or more carbons per spin system were extracted from the hydrophilic HMDB and COLMAR metabolites. The MSM identification results are summarized in Table S1 with 180 COLMAR vs. 1924 HMDB 1st shell MSMs and 397 COLMAR vs. 4912 HMDB 2nd shell MSMs. The frequency of COLMAR MSMs (nodes and their sizes) is depicted as a network graph in Figure 4, which reflects that MSMs are notably unevenly distributed among metabolites. The most common MSMs are unsaturated carbon-carbon bonds that are part of aromatic ring structures. Many MSMs are frequently found in the same molecule together with other MSMs as is indicated by the many edges of the graph. The top 10 most abundant motifs in the COLMAR and HMDB databases are listed in Table S3 and Table S4. For all 1st shell motifs found in the HMDB database, 37 out of the most frequent 50 motifs are covered by COLMAR (Figure S7). Importantly, the 1st shell COLMAR MSMs cover 10,728 out of 12,506 (85.8%) hydrophilic compounds (with NC > 1 spins for each spin system) of the HMDB, which shows that, despite its much smaller size, COLMAR NMR provides very good coverage of the MSMs of HMDB metabolites. 1778 hydrophilic compounds of the HMDB that contain 1042 different 1st shell MSMs are not covered by COLMAR MSMMDB. However, among the 1042 motifs, only 92 MSMs are present in more than 10 compounds, whereas 92 MSMs represent 5 – 10 compounds. 858 MSMs are present in fewer than 5 compounds. This motif analysis shows that the vast majority of hydrophilic metabolite motifs not covered by COLMAR MSMMDB are rare motifs.
Figure 4.

Graph-theoretical representation of molecular motif clustering of current COLMAR MSMMDB molecules. Each node denotes a molecular motif where the node area is proportional to the number of molecules in the motif. For molecules containing two molecular motifs, their nodes are connected by an edge. The edge thickness (weight) is proportional to the number of molecules that contain both motifs. Representative molecular structures of some of the most abundant molecular motifs are depicted in the graph, and the spin system backbone is highlighted in yellow.
DISCUSSION
We have established a motif-based method to identify unknown metabolites. The introduction and curation of motif databases is of central importance, whereby the accuracy and precision of the experimental database entries are crucial for the successful identification of MSMs of unknowns. The 1st and 2nd shell atoms of a structural motif sensitively influence the 1H and 13C chemical shifts and, conversely, experimental 1H, 13C chemical shifts can be used for the determination of molecular structural motifs. This information offers a path toward the determination of the structure of unknown metabolites. Based on the finding presented here that 1st and especially 2nd shell MSMs generally have chemical shifts that are remarkably well conserved, an MSM NMR database termed COLMAR MSMMDB was established with experimental chemical shifts measured in aqueous solution. The chemical shift accuracy in COLMAR MSMMDB is much better than that of both quantum-chemical18 and empirical chemical shift prediction used for pNMR MSMMDB. The true MSMs are often found with RMSD < 2.5 ppm and false MSMs typically have RMSDs > 3.0 ppm. If the RMSD of an MSM is between 2.5 ppm and 3.0 ppm, application of the pNMR MSMMDB to check whether the same MSM is returned as one of the top 15 hits further increases confidence. Finally, for MSMs (1st shell or 2nd shell) not present in the COLMAR MSMMDB, the more comprehensive, but less accurate pNMR MSMMDB consisting of 3512 1st shell MSM and 7874 2nd shell MSM with computationally predicted chemical shifts can be used to identify unknown MSMs.
Although the present COLMAR MSMMDB only includes 397 2nd shell MSMs, these MSMs cover a remarkably large number of metabolites. In particular, they represent 10,728 out of 12,506 (85.8%) hydrophilic compounds (logP < 3.0 with NC > 1) of the much larger HMDB, which contains many more metabolites, including many metabolites without experimental NMR chemical shifts and many expected, but still unconfirmed metabolites. An interesting question is how many additional MSMs with chemical shifts would need to be added in order to cover all metabolites in the current HMDB. When limiting the MSMs to hydrophilic metabolites only (logP < 3), this can be accomplished with the addition of another 1042 unique MSMs. This is a relatively small number considering that this would cover the MSMs of additional 1778 HMDB metabolites that are currently not covered by the COLMAR MSMMDB.
The best motifs identified by COLMAR MSMMDB query can be further developed into complete metabolite candidates. For the examples presented here, a database of potential metabolites was created using a wealth of information from existing databases, such as KEGG, ChEBI, and HMDB. The subset of metabolites that emerge with the correct MSMs and good predicted chemical shift scores represent the candidate molecules that can be purchased (or synthesized) for spiking experiments to confirm their authenticity in the mixture. The SUMMIT Motif approach was successfully illustrated for the identification of two “unknown” metabolites L-alanyl-L-valine and taurocholic acid in mouse bile fluid. The feasibility of the approach depends on the total concentration of the unknown metabolites, which should exceed ~50 – 100 μM, otherwise the sample needs to be concentrated first. Because the approach is not designed for high-throughput analysis, it is best applied to few representative samples selected, for example, from a large cohort of samples. Also, for unknown compounds that have titratable groups with pKa ~ 7, even small changes in pH can cause significant changes in chemical shifts that might adversely interfere with motif identification. In principle, NMR pH titration experiments (e.g. via 13C-1H HSQC) can help identify such unknowns.
The MSM identification approach presented here is “NMR spin system centric” in the sense that 1H, 13C spin systems form 0th shell MSMs, which are then extended to 1st and 2nd shell heavy atoms. As a consequence, the MSMs are only indirectly identifiable by techniques other than NMR. Still, other types of experimental molecular fragment information can be used, such as metabolite fragments produced by tandem mass spectrometry (MS/MS or MS2), which are routinely used in targeted metabolomics. For the identification of unknown metabolites, MS/MS fragments can be predicted for candidate structures as additional scoring criterion thereby further limiting the number of potential metabolites.25 Moreover, the molecular formula of the parent ions, determined from their accurate mass, can serve as an additional filter to narrow down viable candidate compounds that contain the correct MSMs.14, 15 For instance, if mass information (Figure 3f) were used in addition, L-alanine-L-valine emerges as the only candidate among the six top hits to be further verified by spiking NMR experiments, thereby further speeding up the verification of this unknown metabolite.
CONCLUSIONS
The accurate determination of molecular motifs of unknown metabolites presented here is made possible because of the high quality of NMR chemical shifts of the COLMAR metabolomics database, which was customized primarily using data from the BMRB26 and HMDB.22 Our results suggest that the future addition of a modest number of suitably chosen metabolites, the coverage of COLMAR MSMMDB can be substantially broadened to include most real and putative hydrophilic metabolites of the HMDB.
The new COLMAR MSMMDB and pNMR MSMMDB could pave the way for the systematic and efficient determination of motifs and their associated unknown metabolites in a wide range of metabolomics samples. This information will help fill in critical gaps in our understanding of molecular conversions along new metabolomics pathways and their modulations upon internal and external perturbations. The characterization of metabolites with novel MSMs will not immediately benefit from the new COLMAR MSMMDB. It remains a challenge requiring a traditional and, hence, much slower approach to the determination of new natural products relying on extensive purification and comprehensive characterization by the combination of many different analytical techniques. From an NMR perspective, a long-term goal is a universal chemical shift predictor with a similar performance as COLMAR MSMMDB as it would permit the determination of unknowns at a rate that is comparable to the identification of molecular motifs by COLMAR MSMMDB. An empirical predictor with this property would need to be trained on experimental small-molecule chemical shift databases that are much larger than those currently available or require substantially improved quantum-chemical methods for the calculation of chemical shifts. Until then, the systematic addition of high-quality experimental chemical shifts and spin system information of strategically chosen metabolites to NMR metabolomics databases, such as COLMAR MSMMDB, is a practically feasible although time-consuming undertaking.
Supplementary Material
ACKNOWLEDGMENT
This work was supported by a graduate fellowship from Foods for Health (to C.W.), a focus area of the Discovery Themes Initiative at OSU (to C.W.), and the National Institutes of Health (grants R01GM066041, P30 CA016058, S10 OD018507). All NMR and FT-ICR experiments were performed at the Campus Chemical Instrument Center at the Ohio State University.
Footnotes
Supporting Information
The Supporting Information is available free of charge via the Internet at http://pubs.acs.org/. Experimental section, classification of hydrophilic metabolites, spin system identification, matching and scoring, quantitative metric on evaluation of the MSM identification, workflow of SUMMIT Motif, examples of top abundant MSMs in COLMAR MSMMDB and HMDB.
The authors declare no competing financial interest.
REFERENCES
- (1).Markley JL; Brüschweiler R; Edison AS; Eghbalnia HR; Powers R; Raftery D; Wishart DS, The future of NMR-based metabolomics. Curr. Opin. Biotechnol 2017, 43, 34–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Nicholson JK; Lindon JC; Holmes E, ‘Metabonomics’: understanding the metabolic responses of living systems to pathophysiological stimuli via multivariate statistical analysis of biological NMR spectroscopic data. Xenobiotica 1999, 29, 1181–9. [DOI] [PubMed] [Google Scholar]
- (3).Fiehn O, Metabolomics--the link between genotypes and phenotypes. Plant Mol. Biol 2002, 48, 155–71. [PubMed] [Google Scholar]
- (4).Patti GJ; Yanes O; Siuzdak G, Innovation: Metabolomics: the apogee of the omics trilogy. Nat. Rev. Mol. Cell Biol 2012, 13, 263–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Pretsch E; Bühlmann P; Badertscher M, Structure determination of organic compounds: tables of spectral data. Springer: Berlin Heidelberg, 2009. [Google Scholar]
- (6).Emwas AH; Roy R; McKay RT; Tenori L; Saccenti E; Gowda GAN; Raftery D; Alahmari F; Jaremko L; Jaremko M; Wishart DS, NMR Spectroscopy for Metabolomics Research. Metabolites 2019, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Sanchon-Lopez B; Everett JR, New Methodology for Known Metabolite Identification in Metabonomics/Metabolomics: Topological Metabolite Identification Carbon Efficiency (tMICE). J. Proteome Res 2016, 15, 3405–19. [DOI] [PubMed] [Google Scholar]
- (8).Hao J; Liebeke M; Sommer U; Viant MR; Bundy JG; Ebbels TMD, Statistical Correlations between NMR Spectroscopy and Direct Infusion FT-ICR Mass Spectrometry Aid Annotation of Unknowns in Metabolomics. Anal. Chem 2016, 88, 2583–2589. [DOI] [PubMed] [Google Scholar]
- (9).Chekmeneva E; Dos Santos Correia G; Gomez-Romero M; Stamler J; Chan Q; Elliott P; Nicholson JK; Holmes E, Ultra-Performance Liquid Chromatography-High-Resolution Mass Spectrometry and Direct Infusion-High-Resolution Mass Spectrometry for Combined Exploratory and Targeted Metabolic Profiling of Human Urine. J. Proteome Res 2018, 17, 3492–3502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Wolfender JL; Nuzillard JM; van der Hooft JJJ; Renault JH; Bertrand S, Accelerating Metabolite Identification in Natural Product Research: Toward an Ideal Combination of Liquid Chromatography-High-Resolution Tandem Mass Spectrometry and NMR Profiling, in Silico Databases, and Chemometrics. Anal. Chem 2019, 91, 704–742. [DOI] [PubMed] [Google Scholar]
- (11).Djoumbou-Feunang Y; Fiamoncini J; Gil-de-la-Fuente A; Greiner R; Manach C; Wishart DS, BioTransformer: a comprehensive computational tool for small molecule metabolism prediction and metabolite identification. J. Cheminform 2019, 11, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Paudel L; Nagana Gowda GA; Raftery D, Extractive Ratio Analysis NMR Spectroscopy for Metabolite Identification in Complex Biological Mixtures. Anal. Chem 2019, 91, 7373–7378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Shen X; Wang R; Xiong X; Yin Y; Cai Y; Ma Z; Liu N; Zhu ZJ, Metabolic reaction network-based recursive metabolite annotation for untargeted metabolomics. Nat Commun 2019, 10, 1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Bingol K; Bruschweiler-Li L; Yu C; Somogyi A; Zhang F; Brüschweiler R, Metabolomics beyond spectroscopic databases: a combined MS/NMR strategy for the rapid identification of new metabolites in complex mixtures. Anal. Chem 2015, 87, 3864–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Wang C; He L; Li DW; Bruschweiler-Li L; Marshall AG; Brüschweiler R, Accurate Identification of Unknown and Known Metabolic Mixture Components by Combining 3D NMR with Fourier Transform Ion Cyclotron Resonance Tandem Mass Spectrometry. J. Proteome Res 2017, 16, 3774–3786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).MestreLab Mnova NMRPredict. http://mestrelab.com/software/mnova-nmrpredict-desktop/
- (17).Modgraph NMRPredict. http://www.modgraph.co.uk/product_nmr.htm
- (18).Hoffmann F; Li DW; Sebastiani D; Brüschweiler R, Improved Quantum Chemical NMR Chemical Shift Prediction of Metabolites in Aqueous Solution toward the Validation of Unknowns. J. Phys. Chem. A 2017, 121, 3071–3078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (19).Bremser W, Hose — a novel substructure code. Anal. Chim. Acta 1978, 103, 355–365. [Google Scholar]
- (20).Kanehisa M; Goto S, KEGG: kyoto encyclopedia of genes and genomes. Nucleic Acids Res. 2000, 28, 27–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (21).Degtyarenko K; de Matos P; Ennis M; Hastings J; Zbinden M; McNaught A; Alcantara R; Darsow M; Guedj M; Ashburner M, ChEBI: a database and ontology for chemical entities of biological interest. Nucleic Acids Res. 2008, 36, D344–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Wishart DS; Feunang YD; Marcu A; Guo AC; Liang K; Vazquez-Fresno R; Sajed T; Johnson D; Li C; Karu N; Sayeeda Z; Lo E; Assempour N; Berjanskii M; Singhal S; Arndt D; Liang Y; Badran H; Grant J; Serra-Cayuela A; Liu Y; Mandal R; Neveu V; Pon A; Knox C; Wilson M; Manach C; Scalbert A, HMDB 4.0: the human metabolome database for 2018. Nucleic Acids Res. 2018, 46, D608–D617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Tetko IV; Tanchuk VY, Application of associative neural networks for prediction of lipophilicity in ALOGPS 2.1 program. J. Chem. Inf. Comput. Sci 2002, 42, 1136–45. [DOI] [PubMed] [Google Scholar]
- (24).Thompson SJ; Hattotuwagama CK; Holliday JD; Flower DR, On the hydrophobicity of peptides: Comparing empirical predictions of peptide log P values. Bioinformation 2006, 1, 237–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Boiteau RM; Hoyt DW; Nicora CD; Kinmonth-Schultz HA; Ward JK; Bingol K, Structure Elucidation of Unknown Metabolites in Metabolomics by Combined NMR and MS/MS Prediction. Metabolites 2018, 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Ulrich EL; Akutsu H; Doreleijers JF; Harano Y; Ioannidis YE; Lin J; Livny M; Mading S; Maziuk D; Miller Z; Nakatani E; Schulte CF; Tolmie DE; Wenger RK; Yao HY; Markley JL, BioMagResBank. Nucleic Acids Res. 2008, 36, D402–D408. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.